

ABSTRACT
It is increasingly recognized that epigenetic mechanisms play a key role in acclimatization and adaptation to thermal stress in invertebrates. DNA methylation and its response to temperature variation has been poorly studied in insects. Here, we investigated DNA methylation and hydroxymethylation patterns in the viviparous cockroach Diploptera punctata at a global and gene specific level in response to variation in temperature. We specifically studied methylation percentage in the heat shock protein 70 (Hsp70), whose function is linked to thermal plasticity and resistance. We found high levels of DNA methylation in several tissues but only low levels of DNA hydroxymethylation in the brain. Hsp70 methylation patterns showed significant differences in response to temperature. We further found that global DNA methylation variation was considerably lower at 28°C compared to higher or lower temperatures, which may be indicative of the optimal temperature for this species. Our results demonstrate that DNA methylation could provide a mechanism for insects to dynamically respond to changing temperature conditions in their environment.
KEYWORDS: DNA methylation, Hsp70, insect, temperature, MS-AFLPs
Introduction
Epigenetic processes are central to trait evolution because novel phenotypes may be generated in response to environmental cues. This process promotes differences in gene expression, and therefore, it might allow acclimatization to environmental changes and even enhance local adaptation [1,2]. Further, under novel or environmental conditions, epigenetic variation may increase [3]. This epigenetic variation may contribute to heritable variation on which selection can act or create novel selectable phenotypes. Such novel phenotypes may be beneficial if the environment is constant across generations.
Among the main epigenetic processes, DNA methylation is the best studied epigenetic mark, which involves the addition of a methyl group on the fifth position of the cytosine. In mammals, DNA methylation is mainly enriched in regulatory regions and is associated with gene silencing [4,5]. By contrast, in invertebrates DNA methylation is enriched in exons and is associated with gene activation. DNA methylation is highly dynamic as it can vary in response to, for example, environmental factors, requirements of the cell, or developmental stage [6,7]. DNA hydroxymethylation (DNA 5-hmC), by contrast, is a largely unexplored epigenetic mechanism that is presumably involved in gene upregulation and active demethylation processes [8]. DNA hydroxymethylation is mainly found in the nervous system, suggesting important and specific neural and developmental functionality [9,10].
To date, DNA methylation patterns in response to environmental stressors such as thermal stress have been poorly studied, especially in insects. Because of the significant relevance that methylation may have for adaptation to environmental stressors we sought to establish how this key epigenetic mechanism is affected by arguably the most important abiotic stressor currently, temperature.
To investigate the effects of thermal stress on methylation we used two different approaches. First, we focussed on one of the main genes involved in thermal response, the Heat Shock Protein 70 (Hsp70). Apart from performing several important physiological roles, such as secretion, degradation and regulation, this protein also facilitates organismal thermotolerance [11,12]. For example, in the fruit fly, thermotolerance varies according to the amount of Hsp70 present in the cell before heat stress. Usually, Hsp70 will be absent if a cell, or an organism, has not been exposed to thermal stressors [11]. Second, we analysed methylation patterns and methylation variation on a global scale. Several studies propose that methylation profiles are determined by the environment and that individuals living in similar environmental conditions will have similar methylation profiles [13–15]. It has been suggested that high levels of epigenetic variation could help to overcome reduced levels of genetic variation or abrupt changes in the environment, by inducing phenotypic changes that might help organisms to survive in novel environments [16].
It is important to emphasize that the way that methylation may respond to the environment could also be influenced by the genotype. Even though methylation patterns can be determined by the environment and behave as an autonomous system [17], several studies have shown that methylation patterns can also be determined by the genotype. This close link between the genotype and methylation patterns is possible because the latter may have originated from silencing transposable elements or random epimutations [18–20]. Notably, in a variety of organisms collected from the wild, a higher degree of epigenetic variation compared to genetic variation has been recorded [21,22].
In this paper, we present the first investigation of DNA methylation and hydroxymethylation in the cockroach Diploptera punctata and its response to thermal stress. Diploptera punctata is among the very few truly viviparous insects and the only truly viviparous cockroach. It belongs to Blattodea, a group that presents several adaptations to thermal stress [23]. Our first research aim was to investigate if DNA methylation and hydroxymethylation are present in Diploptera punctata. To achieve this, we quantified global methylation and hydroxymethylation levels at a tissue-specific level. Then we sought to establish if methylation patterns are affected by temperature and if organisms sharing similar genotypes will react similarly to thermal stress. This lead to our second research aim, which was to study if methylation of Hsp70 was affected by temperature. We investigated the intragenic region of the Hsp70 gene after we exposed seven genotypes to four different temperature treatments. Our final research aim was to investigate how global methylation patterns respond to thermal stress. To achieve our final aim we use MS-AFLPs to analyse methylation patterns in individuals from seven different genotypes that were exposed to four different temperature treatments.
Methods
Animal maintenance
D. punctata colonies had been established for over 10 years in the laboratory and were maintained in plastic containers (30 × 22 × 20 cm) at 25°C on a 12:12 h light:dark cycle and fed with blended dog food (WAGG Complete Dog Food) and water provided ad libitum.
Global DNA methylation percentage
Dissections
From the main colony we selected females adults, which were dissected in bath saline solution (135 Mm NaCl, 5 mM KCl, 4 mM MgCl 2 6 H 2 O, 2 mM CaCl 2 2 H 2 O, 5 mM TES, 36 mM sucrose). We dissected the legs, head, fat body and embryos. Embryos were obtained between days 45 and 55 of pregnancy (59–75% of total development [24]). At this stage, the embryos are between 4 and 5 mm long. Tissues were immediately frozen in dry ice and stored at −80°C until used.
DNA extraction and methylation analysis
DNA was extracted from 12 samples (three for each of the four tissues, and every sample was done as a technical replicate) using Qiagen DNeasy Blood & Tissue Kit following the manufacturer’s protocol. All DNA samples were precipitated and cleaned using standard ethanol precipitation [25]. After DNA extraction, samples were quantified using Qubit dsDNA HS Assay Kit (ThermoFisher). Methylation and hydroxymethylation global levels were quantified using the MethylFlash Methylated DNA 5-mC Quantification Kit (Colorimetric), and the MethylFlash Hydroxymethylated DNA 5-hmC Quantification Kit (Colorimetric). We used 100 ng of DNA as input and followed the protocol as indicated by the manufacturer (Epigentek). For negative control in both assays we used 100 ng of adult Drosophila melanogaster DNA, as the Drosophila genome does not carry DNA methylation or hydroxymethylation [26,27]. Exclusively for the hydroxymethylation assay, we used mouse brain as a positive control because hydroxymethylation levels within the mouse brain is reported to be 0.2% [9].
Statistical analysis
All data were analysed using linear mixed-effects models in the R environment [28], using the packages lme4 and car [29,30]. The logarithm of global methylation and hydroxymethylation percentages were used as response variables, tissue (five levels and six levels, respectively, see Figure 1) was a fixed factor and individual considered as a random effect. Post hocs test was done using the package emmeans (Tukey test) [31].
Figure 1.
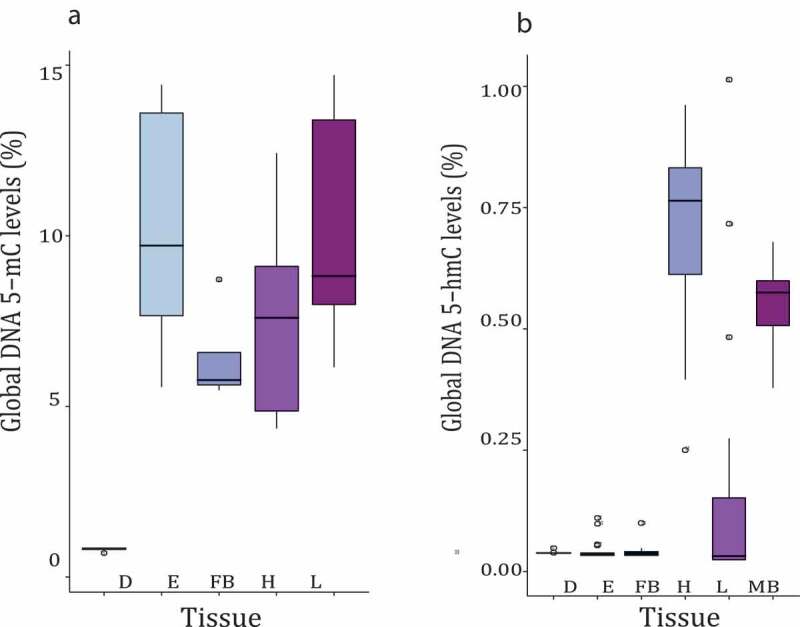
Methylation and hydroxymethylation levels in Diploptera punctata. (a) Global methylation levels from four different tissues (embryo (e), fat body (FB), head (h), and legs (l)). Drosophila DNA (d) was used as a negative control (no DNA methylation). (b) Global DNA hydroxymethylation levels in different tissues. Mouse brain (MB) was used as a positive control and Drosophila DNA a negative control
Temperature manipulation
We isolated pregnant females from the main colony (parental generation) and monitored them daily until they gave birth (F1). We used the first two clutches from the parental generation. During the first week of life, we randomly divided and allocated each clutch (F1) to four different temperature regimes: 26°C, 28°C, 30°C and 32°C. During pregnancy and the offspring’s first week of life, cockroaches were kept at 25°C (see supplementary material Figure 1). To control the temperature we constructed wooden boxes (62 x 40 × 37 cm) and added a ceramic heating element (Exo-Terra), a thermostat, (600 watts, Habitat) a thermometer (Exo-Terra), a humidity metre, and LED light programmed to 12/12 hrs light cycle. Inside the boxes, we kept the cockroaches in plastic boxes (15 x 7 × 15 cm) grouped by family (three to four cockroahes in each box). Water and blended dog food (WAGGS) was provided once weekly. We monitored the cockroaches weekly and measured (Mitutoyo calliper), weighed, and marked any new adult.
DNA extraction
Once the cockroaches had reached adulthood we sacrificed adults (males and females) using liquid nitrogen. We dissected the head in sterile conditions under a UV hood. Tissue was stored at −80°C before usage. We homogenized tissue manually using plastic pellets and extracted DNA using Qiagen tissue and blood extraction kit as indicated by the manufacturer. The cockroach eye pigment precipitates with the DNA and inhibits PCR. To avoid PCR inhibition we cleaned the samples using the Qiagen cleaning kit as indicated by the manufacturer.
Response of Hsp70 gene methylation to thermal stress
Amplification of Hsp70 gene in Diploptera punctata
To obtain the Hsp70 gene body DNA sequence of Diploptera punctata, we first collected Hsp70 available sequences from closely related species. In total we collected three sequences from three species: Blatella germanica (Accession No: PYGN01000002.1:4236897–4238334), Periplaneta americana (Accession No: KY661334.1) and Cryptocercus punctulatus (Accession No: JQ686949.1). The sequences were aligned on Clustal Omega [32]. We then used PriFi [33] (https://services.birc.au.dk/prifi/main.py) to design multiple set of primers. This tool is useful for designing primers from multiple sequence alignments derived from phylogenetically related species, in particular when working with organisms without a reference genome. The sequence of the primers that amplified successfully the desired fragment is: Fw 5ʹ-AAGGGTCATGGAGAACGCAA-3ʹ and Rv 5ʹ-CTCTTCATGTTGAAGCAGTA-3ʹ. For the PCR amplification, we added 2 μl (150ng/μl) of DNA to the PCR mix (Foward primer 1 μl (0.4 μM), Reverse primer 1 μl (0.4 μM), PCR mix 12.5 μl, Nuclease free water 8.5 μl) with the following PCR conditions: 95°C for 4 min, followed by 35 cycles of 95°C for 15 sec, 61°C for 1 min, 72°C for 1.15 min and 72°C for 10 min. To verify that the amplified section corresponds to Hsp70 we performed Sanger sequencing. PCR products were purified using QIAquick PCR Purification Kit (Qiagen) and the purified fragments were sent as premixed samples to the Genomic Technologies Core Facility (GTCF, University of Manchester) for Sanger sequencing. The samples contained either forward or reverse primers, purified templates and nuclease-free water to make a total 10 μl reaction volume.
Bisulphite conversion and pyrosequencing
DNA was bisulphite converted using the EpiTect Bisulphite Kit. To obtain the Hsp70 primers, we aligned the Diploptera punctata Hsp70 sequence with the Hsp70 sequence of B. glabratahe (see suplementary material S2). In the conserved regions we identified and selected for further analysis, a region with four CpGs that was found to be methylated in B. glabratahe [12]. We used PyroMark Assay Design software (Qiagen) to design a set of a forward, reverse and sequencing primers, which are as follow: Fw 5ʹ-ATTTAAGTTTAAGAAGGTGAGAGAGTAATG-3ʹ, Rv 5ʹ-CTCCTTTCCTATTAATTTTTCAACTACTA-3ʹ, sequence 5ʹ-GGTGTTTTATAAATTGAGGTTATT-3ʹ. The reverse primer is biotinylated at the 5ʹ end. PyroMark PCR Kit (Qiagen) was used to carry out methylation-specific PCRs as specified by the manufacturer. From the PCR sample 10 μl was used for each pyrosequencing reaction using the sequencing primer 5ʹ- GGTGTTTTATAAATTGAGGTTATT −3ʹ. We scan in total 31 samples. The PyroMark Q24 (Qiagen) was used for pyrosequencing using PyroMark Q24 Advanced Reagents kit (Qiagen)
Statistical methods
All four positions were analysed independently. Data were analysed using linear mixed-effects models in the R environment [28], using the packages lme4 and car [29,30]. The percentage of methylation at each cytosine was used as a response variable, the genotype (six levels), temperature (four levels) and developmental time were considered as a fixed factor. Post hocs test was done using the package emmeans using tukey test. Post hocs test was done using the package emmeans (Tukey test) [31].
Response of global methylation patterns to thermal stress
Methylation sensitive amplified length polymorphisms
DNA extraction was done as described above. MspI and HpaII are isoenzymes with the same restriction site (CCGG) but with different sensitivities to DNA methylation [22,34]. HpaII activity is blocked when the inner or outer C is methylated at both strands. By contrast, MspI cleavage is not allowed when the outer cytosine is fully methylated. By treating DNA with both enzymes we can identify four different methylation states at each restriction site (Type 1: when both enzymes cut (no methylation) Type 2: when HpaII cleavage is blocked and MspI does cut (methylation present in the internal cytosine) Type 3: when HpaII does cut and MspI activity is blocked (hemimethylated outer C) Type 4: Both of the enzyme activity is blocked (hypermethylation or sequence mutation at the restriction site).
For MS-AFLPS a total of 67 organisms were screened. We processed two technical replicates per sample. We followed the protocol in Amarasinghe et al (2014) [34] with some modifications (see primers in supplementary material table 4). We digested DNA in two separate reactions. The first one used EcoRI (0.05 μl NEB, 20 000 units/ml) + MspI (0.025 μl NEB, 20 000 units/ml). The second one used EcoRI + HpaII (0.5 μl NEB, 20 000 units/ml). We added 5 μl of DNA to the two independent digestion mixtures (EcoRI, MspI/HpaII, 1 μl NEB cut buffer 10X, and 3 μl of ddH 2 O). The reaction was incubated for three hours at 37°C. Immediately after digestion, we added 5 μl of the digested product to the ligation reaction (0.25 μl T4 DNA ligase NEB (400,000 units/ml), 1 μl of NEB ligase buffer, 1 μl of EcoRI adapter (5 pmol), 1 μl HpaII-MspI (50 pmol), and 1.75 ul of ddH 2 O). The ligation reaction was incubated at 37°C for three hours and left overnight at room temperature. Then, we ran a pre-selective PCR (pPCR) by adding 5 μl of the ligated product to the pPCR mix (1.25 μl pre-selective EcoRI primers (0.5 μM), 1.25 μl pre-selective HpaII-MspI primers (0.5 μM), 12.5 μl of PCR master mix Agilent, Paq5000 Hotstart PCR Master Mix) and the following PCR conditions: 94°C for 2 min, followed by 35 cycles of 94°C for 30 sec, 58°C for 1 min, 72°C for 1 min and 72°C for 5 min. Then we ran selective PCR (sPCR) in which three different primers were used. We used 5 μl of the pPCR product as a DNA template, which was added to the following mix (0.5 μl selective EcoRI primer (0.5 μM), 0.5 μl of selective HpaII/MspI primers (0.5 μM) and 5 μl of PCR master mix (Agilent, Paq5000 Hotstart PCR Master Mix), with the following PCR conditions: 94°C for 2 min, followed by 13 cycles of 94°C for 30 sec, 65°C (decreasing 0.7°C per cycle) for 30 sec, followed by 30 cycles of 94°C for 30 sec, 56°C for 30 sec, 72°C for 1 min, and a final extension of 72°C for 5 min. Each of the forward selective primers was marked with a different fluorophore (6-FAM, HEX, ROX). The fluorophore allows the identification of the fragment after capillary electrophoresis. Finally, we mixed 0.5 μl of the sPCR product with 0.4 μl of 500 LIZ dye Size Standard (ThermoFisher) and 9 μl of Hi-Di formamide (ThermoFisher). The samples were then sent to the University of Manchester sequencing facility for fragment analysis.
Methylation analysis
We checked for general quality of the fragments using PeakScanner (Thermofisher) and used the R package Raw Geno [35] to filter low-quality samples and to score the peaks. RawGeno uses the number of peaks per individual as a proxy of reaction quality. We removed all those individuals that fell outside of the 5–95% interval.
Scoring peaks is crucial as it is important to eliminate non-reliable peaks. The objective of this step is to identify homologous peaks between samples relying on the size of the peaks, hence the peaks that share the same size are considered homologous. However, the size of the homologous peaks might not be exactly the same due to technical bias [35]. It is expected that peak sizes vary between 0 and 0.66 bp. To tackle this size variation, the peaks were categorized by bin (i.e. size). Into this bin category, the presence or absence of the peak is recorded. We set the maximum bin width parameter to 1.5 bp, and the minimum to 1bp. Once the bins have been established, they are filtered using the following parameters: 1) Bin size: we only limited the scoring range to the size of the ladder (500 bp). The minimum size we considered was 100 bp. 2) Fluorescence: peaks with higher fluorescence intensity are more likely to be consistent. We selected a minimum threshold of 100 RFU. 3) Reproducibility: this filter evaluates the robustness of the peak across the dataset based on duplicated samples. RawGeno compares each bin on both replicates for which the MS-AFLPs signal was successfully reproduced. Bins where reproducibility did not reach 80% rate across the entire dataset were eliminated.
The selected peaks from RawGeno were classified as 1 if the peak is present and 0 if it is absent. Then we analysed the selected fragments using the Msap R package [36]. The Msap package determines if each fragment is susceptible to methylation or not. If the fragment is susceptible to methylation Msap designates a methylation status according to band presence due to the cut patterns of the EcoRI- MspI/HpaII enzymes. The presence of EcoRI-HpaII/MspI peak (pattern 1/1) is considered unmethylated, only the presence of EcoRI-HpaII (pattern 1/0) or EcoRI-MspI (pattern 0/1) are considered as methylated. Finally, the absence of both enzymes fragments
EcoRI- MspI/HpaII (pattern 0/0) was considered as a hypermethylated state as we know from our previous results, methylation in D. punctata is high (global cytosine methylation percentage of around 9%). There is, also, not much genetic variation expected with this experimental design as we are only working with seven genotypes (cross sibling experimental design [37]).
Statistical analysis
Effect of temperature and family on global methylation patterns
We performed a perMANOVA (Permutational multivariate analysis of variance [38];) using the adonis function from the vegan R package [39] to test the effect of temperature, family and maternal developmental temperature on methylation patterns. We set the permutation number at 1,000,000 and used the Euclidean method to create the distance matrix, which was used as the response variable. We also performed a pairwise perMANOVA using pairwiseAdonis function in R [40]. We considered temperature and family as predictors. However, for the pairwise analysis we tested for temperature effects and controlled for family and vice-versa.
Methylation variation
To determine whether the variation in methylation patterns is influenced by temperature we obtained the distance of each coordinate from the PCoA to the centre of each respective group using the Euclidean distance formula:
| (1) |
where D is the distance between the centroid and a given point, x1 and y1 are the coordinates of interest and x2 and y2 are the centroid coordinates.
To determine whether the observed differences in methylation distance between temperature groups were greater than we would expect to see by chance alone, and therefore statistically significant, we ran a series of pairwise comparisons of the different developmental temperatures (e.g. 26°C vs 28°C, 26°C vs 30°C, etc) and compared them to a null distribution of differences obtained in 10 6 randomly generated permutations of the data. We created permutations by reassigning the observed data points between the temperature groups, subject to the constraint that the number of observations could not change within temperature groups. This constraint is important because there are different numbers of observations in the different temperature groups, and the variance of methylation per temperature group depends on the number of observations. For each comparison, the p-value is the proportion of the null distribution in which the difference between temperatures was as great as, or greater than, in the observed data.
Methylation proportion
We obtained the proportion of methylated sites per sample by counting the number of methylated sites and dividing by the total number of loci. We tested whether family and temperature had an effect on methylation proportion using a linear model with the logarithm of methylation proportion as the response variable and family and temperature as predictors.
Genetic and epigenetic correlation
To analyse the correlation between epigenetic variation and genetic variation we performed a Mantel test using the R vegan package [39]. To run the Mantel test we first created two different distance matrices using the Euclidean method. We created the first matrix using the MSL and another using NML. For the Mantel test, we used the Pearson method and ran 1,000,000 permutations.
Results
Global methylation levels
We found evidence of DNA methylation in all tissues (average DNA methylation level 8.8%), with no significant difference among them (GLM F 3,25 = 0.9535, p > 0.05). We used adult Drosophila melanogaster as a negative control (no DNA methylation), and as expected we did not find evidence of methylation in Drosophila melanogaster. Further, all Diploptera punctata tissues showed significantly higher levels than Drosophila (HSD, Drosophila vs embryo, t − ratio = − 9.81, p < 0.0001, Drosophila vs fat body, t − ratio = − 7.71, p < 0.0001, Drosophila vs head, t − ratio = − 9.71, p < 0.001, Drosophila vs leg, t − ratio = − 9.75, p < 0.001;Figure 1(a)). In contrast to DNA methylation, global DNA hydroxymethylation was found only in the cockroach head. We found that 0.75% of the cytosines were hydroxymethylated (Figure 1(b)). For the hydroxymethylation assay we also used adult individuals of Drosophila melanogaster as a negative control, and mouse brain as a positive control. Interestingly, we found similar percentages of 5-hmCs in the head of D. punctata as those found in the mouse brain (0.22%). The levels of hydroxymethylation in the mouse brain and in the cockroach head were not significantly different from each other (z − value = − 1.51, p > 0.05). The hydroxymethylation levels of Drosophila were not different from the levels of the embryo, fat body and leg (Drosophila vs embryo z − value = 0.123, p > 0.05 Drosophila vs fat body z- value = 0.79, p > 0.05, Drosophilavs leg z - value= 1.29, p > 0.05).
Response of Hsp70 methylation to thermal stress
We analysed four cytocines in the coding sequence region of the Hsp70. The four positions analysed were highly methylated, the first postion being the one that showed a higher methylation percentage (98.806%, 94.838%, 93.774% and 88.483% in the first, second, third and fourth positions respectively). All four CpGs analysed showed significant temperature effects on methylation (Figure 2; Postion 1: F 3,20 = 4.575, p = 0.013; Position 2: F 3,20 = 12.50, p = 7.91e − 05; Position 3: F 3,20 = 5.986, p = 0.004; Position 4: F 3,20 = 9.048, p = 0.0005). The post hoc test showed that methylation percentage at position one varies the most between temperatures, the organisms that developed at 32°C were those with the higher methylation percentage (Figure 2(b)). Family had an effect on methylation percentage at three of the four positions (Figure 3; Postion 1: F 6,20 = 3.922, p = 0.009; Position 2: F 6,20 = 3.041, p = 0.027; Position 3: F 6,20 = 1.598, p = 0.199; Position 4: F 6,20 = 2.641, p = 0.047). The post hoc test showed that most of the families in all the positions were significantly different from each other (Figure 3(b)). Developmental time did not have an effect on methylation percentage at any of the positions (Postion 1: F 1,20 = 0.101, p = 0.753; Position 2: F 1,20 = 3.271, p = 0.085; Position 3: F 1,20 = 1.060, p = 0.315; Position 4: F 1,20 = 1.914, p = 0.181).
Figure 2.
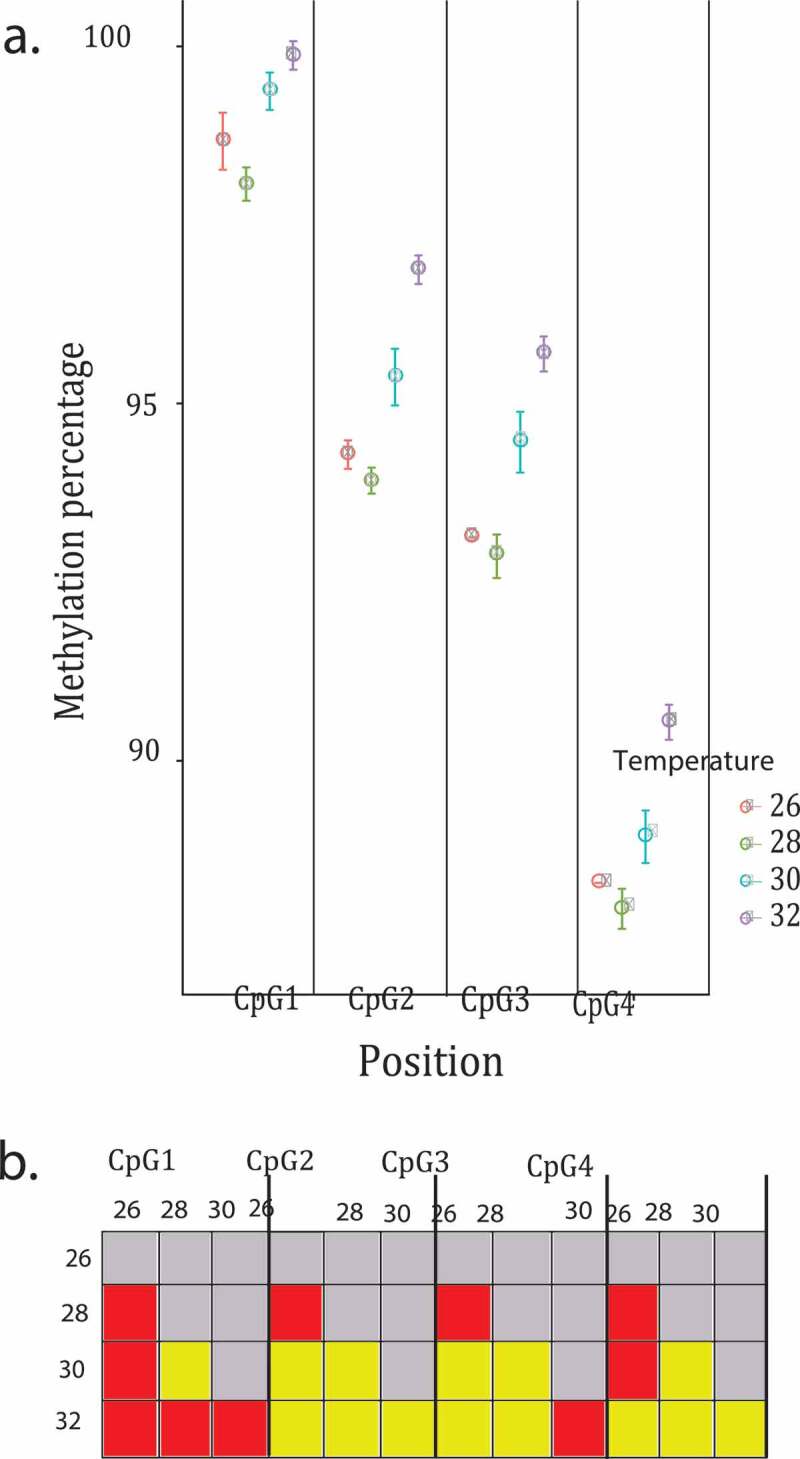
Effect of temperature on the four Hsp70 CpGs analysed. Shown are the raw data with error bars representing the standard error. (a) Methylation percentage of the four Hsp70 CpGs analysed colour coded by temperature treatment. (b) Pairwise post hoc results of each position analysed in the Hsp70, yellow boxes are significant p values, red boxes are non-significant values
Figure 3.
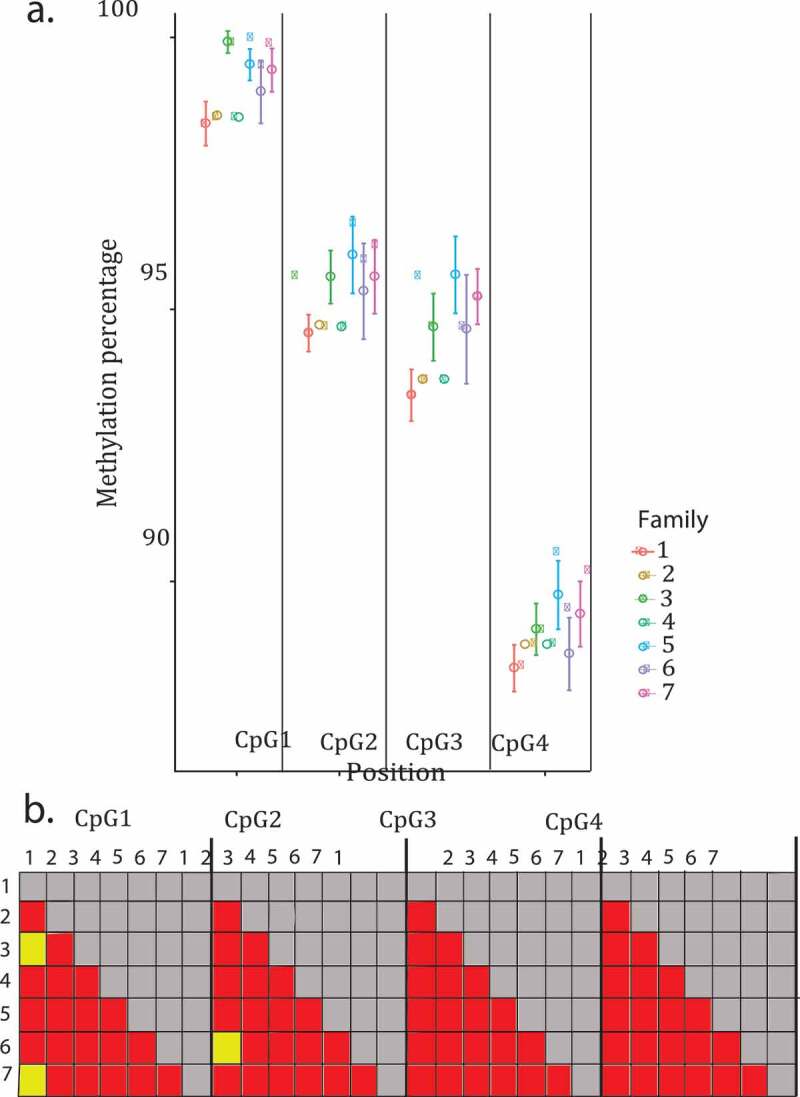
Effect of family on the four Hsp70 CpGs analysed. Shown is the raw data with error bars representing the standard error. (a) Methylation percentage of the four CpGs analysed in Hsp70 colour coded by family. (b) Pairwise post hoc results of each position analysed in Hsp70, yellow boxes are significant p values, red boxes are non-significant values
Response of global methylation patterns to thermal stress
We scanned 67 individuals, and from the three primer combinations we obtained a total of 719 loci, of which 677 were susceptible to methylation (MSL) and 354 were polymorphic (52% of the total MSL). Of the total number of loci, 42 were not susceptible to methylation (NML) and 35 were polymorphic (83% of the total NML). The perMANOVA results showed that developmental temperature (F 3,57 = 3.39, p < 0.001; Figure 4(a)) and family (F 1,57 = 1.45, p < 0.05; Figure 4(b)) had a significant effect on methylation patterns (MSL), however the interaction between these two predictors was not significant (F 14,43 = 0.91, p > 0.05). Using a pairwise perMANOVA we analysed the effect of temperature, controlling for family. The results reveal that methylation patterns of individuals developing at 28°C were significantly different from those at any of the other temperature (26°C vs 28°C F 1,36 = 3.20, p < 0.05, 28°C vs 30°C F 1,35 = 4.68, p = 0.001, 28°C vs 32°C, F 1,28 = 8.15, p = 0.001). The methylation patterns of the individuals at 26°C and 32°C grouped significantly different from each other (F 1,28 = 2.78, p < 0.01) but neither of these temperatures were different to those at 30°C (26°C vs 30°C F 1,35 = 1.27, p > 0.05, 30°vs 32°C F 1,27 = 1.49, p > 0.05). The differences between temperature conditions are given in supplementary material Table 1. We also ran a pairwise per-MANOVA to test for differences between families, controlling for temperature. The results indicate that just a few families differ between each other in their methylation patterns being family five and six the ones that differ from several families. Family six significantly differs from family one and five (1 vs 6 F 1,16 = 2.83, p > 0.05, 6 vs 5 F 1,16 = 3.71, p < 0.01). While family 5 significantly differed from family two, three and six (5 vs 2 F 1,17 = 3.32, p < 0.05, 5 vs 3 F 1,24 = 2.02, p > 0.05). The pairwise comparison are presented in the supplementary material table 3. It is important to mention that sex did not had an effect either on methylation patterns not proportion (see supplementary material S1)
Figure 4.
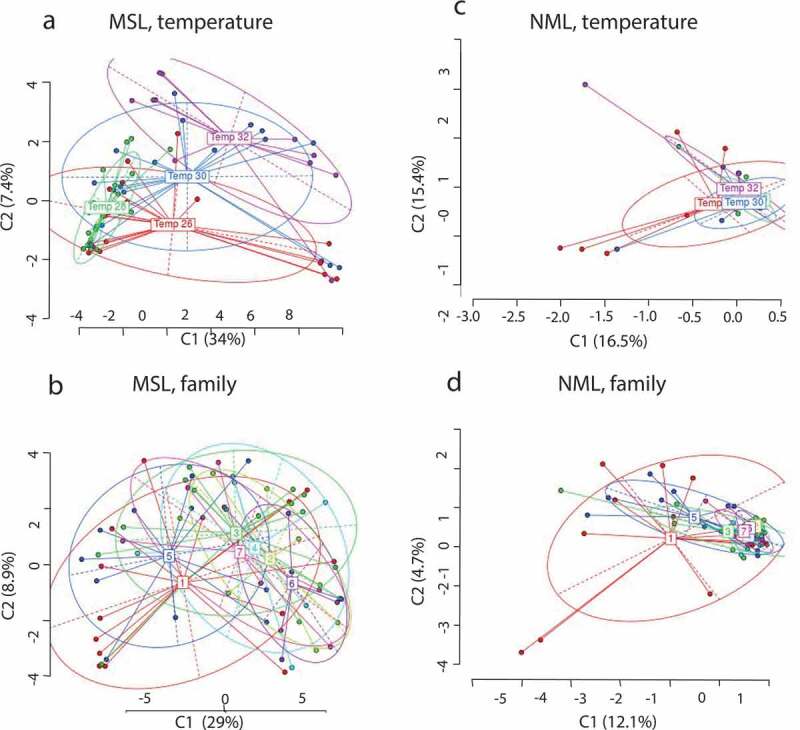
Principal coordinate analysis. The figure shows the principal coordinate analysis for epigenetic (methylation sensitive loci; MSL) and genetic (not methylation sensitive loci; NSL). The two coordinates presented are shown with the percentage of variation explained by them. The points represent the individuals and the group labels the centroid for the individuals in each group. The ellipses represent the mean dispersion of the points around the centroid. The individuals are grouped by temperature (a and c) and family (b and d)
Table 1.
Effect of temperature on methylation pattern variation. Shown are the p values from 1,000,000 random permutations of the individual’s euclidean distance to the centroid of the PCoA. Pairwise comparison was done between the four temperature treatments to which the first generation was exposed to
| 26°C | 28°C | 30°C | |
|---|---|---|---|
| 26°C | |||
| 28°C | < 0 | ||
| 30°C | > 0.05 | < 0 | |
| 32°C | > 0.05 | < 0 | > 0.05 |
Methylation variation
We evaluated the variability of the methylation patterns within each temperature condition. We found that individuals at 28°C have less variability in their methylation patterns. The level of dispersion of this group is significantly different from the individuals at 26°C, 32°C, and 30°C (p-values are given in Table 1).
Not susceptible methylation loci
The not susceptible methylation loci (NML) are those that were not methylated in any of the samples. Because the loci are not susceptible to methylation, the presence or absence of these loci represent genetic mutations. Therefore, these loci are useful to evaluate genetic variation across the samples. We found 42 NML, on which temperature had a significant effect (F 3,57 = 1.41, p < 0.05; Figure 4 (c)) but family did not have an effect (F 6,57 = 1.18, p > 0.05; Figure 4(d)). However, only the individuals from temperature 28°C vs the individuals at 32°C are significantly different (F 1,28 = 1.96, p < 0.01; supplementary material table 2. We found no evidence of a correlation between genetic and epigenetic variation (Mantelr = − 0.12, p > 0.05).
Discussion
After quantifying global methylation percentage in the cockroach genome we confirm an average of 9% of global DNA methylation in all cockroach tissues investigated. By contrast, hydroxymethylation was present in the cockroach brain only, with similar levels to those reported in the mouse brain. Our results further show that methylation is highly sensitive to thermal stress. We found that methylation at all Hsp70 cytosines was sensitive to temperature. However, genotype effects on methylation were detected only at some sites. Finally, we found that global methylation profiles were affected by both temperature and the genotype showing that methylation variation is much lower at 28°C than at other temperatures.
Global methylation
Our first aim was to identify and quantify DNA methylation and hydroxymethylation at tissue specific level in Diploptera punctata. We found overall high levels of methylation in all tissues. Our results are concordant with previous work that reported DNA 5-mC levels between 2% and 14% in
Blattodea [4,41]. Hydroxymethylation, by contrast, was only found in the head supporting the hypothesis that tissue-specific DNA 5-hmC might be implicated in neural development and neural plasticity [9,42]. Hydroxymethylation is an epigenetic mark poorly explored in insects, as it has only been studied in the honeybee. The presence of hydroxymethylation has been linked to neural tissues in mammals and its presence in the brain suggests a link to flexible alterations in the chromatin. It has been hypothesized that neurons need to have a flexible epigenetic mechanism because they cannot divide. Therefore if an epigenetic mark, such as methylation, needs to be rearranged (due to, for example, cellular requirements or environmental stressors) neurons need to rely on a demethylation process that does not require cell duplication [9]. The fact that hydroxymethylation is only present in the brain is especially interesting due to the complex neural and behavioural structure of cockroaches. In fact, it has recently been shown that cockroaches hold the largest chemosensory gene repository known in arthropods [43]. Blattella germanica has the largest family of odorant binding proteins and ionotropic receptor proteins, and the second largest number of gustatory proteins. The large chemosensory repository present in the cockroach suggests that these proteins may play an important role in the chemical ecology of the species, for example in sex and aggregation pheromones or the remarkable evolution of sugar aversive strains [43]. Further studies on the sites where hydroxymethylation is enriched in the brain of the cockroach will be necessary to elucidate whether hydroxymethylation is related in any way with the chemosensory repository.
Methylation level in Hsp70
Our second research aim was to investigate whether DNA methylation in the coding sequence region of Hsp70 was susceptible to thermal stress across seven different genotypes. In this regard, we found that temperature and genotype had an effect on methylation percentage. In several species, it has been observed that thermal stress causes upregulation of Hsp70. The level of upregulated expression is often correlated with thermal stress resistance [44]. For example, a study performed by Hu et al [44] investigated if the divergence in thermal plasticity of two invasive congenic fruit fly species (Bactrocera correcta and Bactrocera dorsalis) is associated with Hsp expression levels. B. dorsalis is a widely distributed species, while B. correcta is narrowly distributed. They found evidence suggesting that Hsp70 may be involved in regulating thermal plasticity, as the more widespread species had greater ability to express Hsp70 [44]. Other studies in invertebrates have corroborated the relation between thermal plasticity and thermal resistance to Hsp gene expression [7,45,46,47]. However, the molecular mechanism that regulates Hsp70 expression is poorly studied in invertebrates. A study performed on the mollusc Biomphalaria glabratahe found that methylation of Hsp70 responded to heat shock [12], proposing methylation as an important regulatory mechanism of Hsp70 in invertebrates. The fact that we found the highest methylation level at 32°C indicates that methylation at these specific regions responds to heat shock. Further, the lowest methylation percetage in all sites was at 28°C. This might indicate that methylation at these positions is not responding to temperature stress. This supports our previous results proposing that 28°C is the optimal temperature for Diploptera punctata. In insects, methylation is enriched in the gene body and is linked to gene activation [5,48]. We found higher levels of methylation at the highest temperature (32°C), which could mean high rates of gene expression. This needs to be confirmed in future work looking at the relation between methylation and gene expression in this specific case. It is also crucial to understand the physiological and biological implications of the observed methylation percentage and investigate if it has any effect on, for example, gene expression, alternative splicing or the phenotype.
Global methylation profiles
Methylation variation
Our third and final aim was to investigate how global DNA methylation profiles respond to thermal stress. We used MS-AFLPs to scan for epigenetic profiles across the genome and found that temperature and genotype affected methylation patterns. Methylation patterns were more similar in organisms that developed at the same temperature. Furthermore, we observed an interesting pattern in the variation of methylation profiles in each treatment. Methylation patterns vary more stochastically in all temperature regimes, except for the 28°C treatment, in which all the samples clustered together. A possible explanation for this might be that 28°C is the closest to the optimal developmental temperature. The results from several studies propose that when organisms are exposed to stressful conditions methylation patterns vary stochastically. Several studies have found a link between DNA methylation and environmental stress, describing higher variability in DNA methylation when organisms are under environmental stress. If the same methylome or phenotype is expressed constantly over generations through transitory methylation patterns, then these patterns are expected to become common and fixed in the population, and therefore may contribute to epigenetic differentiation between populations.
Controlled experiments show that several environmental stressors such as low nutrients, salinity, or pathogen attacks can induce methylation variation [3,37,49,50]. This variability has been recorded in natural and lab conditions. For example, Leung et al described that in Chrosomus neogaeus, the fine-scale dace, under unpredictable environments, stochastic epigenetic variation is induced. However, they reported that this variation will be highly influenced by the genotype [51]. In several experiments on stress related methylation, it has been described that the stability of these marks is highly variable. The marks have been recorded to be stable from several hours up to several generations [49]. Other studies report that stochastic DNA methylation variation occurs just several hours following the exposure to the stressful environment [17]. For instance, a study on three species of coral demonstrated that DNA methylation variation influences their tolerance to thermal stress and ocean acidification [52].
Following our results, for future research, we propose to study the costs associated with high rates of stochastic epimutations and establish for how long these patterns are stable. A wider study focusing on species that differ in their life history would produce interesting findings on the cost of generating epigenetic stochasticity under stressful environments.
Our results also demonstrate that epigenetic variation is greater than genetic variation. This has been widely reported in the literature where especially in natural population epigenetic loci are more variable than genetic loci [17,22,49]. This supports the idea that epigenetic variation can help organisms to cope with environmental changes more rapidly than genetic variation. Indeed, in several invasive species, which are characterized by low genetic variation, methylation variation is higher than genetic variation [53]. This has also been observed in populations with naturally low levels of genetic variation (e.g. clonal species), in which increased epigenetic diversity may help overcome the naturally low amount of genetic variation. We were expecting to find low genetic diversity, as the cockroaches have been kept in the laboratory for over a decade, and indeed we find low genetic variation between families. However, we note that we evaluated genetic variation based on a relatively low number of NML. To further corroborate this finding more exhaustive studies of genetic variation need to be conducted using e.g. AFLPs.
Our study failed to find a correlation between genetic and epigenetic variation. This means that specific genetic profiles are not correlated to specific methylation patterns. However, the lack of correlation between the genetic and epigenetic matrices and the fact that a large amount of epigenetic variation could be explained by the environment, suggests that several epigenetic marks might be independent of the genome in D. punctata. It would then be necessary to examine which methylated regions are correlated with genotype, and which are correlated to the environment to gain an understanding of the function of methylation marks associated with the genotype as opposed to the environment.
The genetic dependency of epigenetic variation is not well described, but it is possible to be species or taxa dependent [18]. It is important to bear in mind that the fact that the environment determines a high amount of epigenetic patterns, does not mean that these patterns have a functional link or that these are under selection. To address the functionality of methylation in response to temperature in D. punctata we would need to explore in more detail wherein the genome methylation changes are occurring.
Supplementary Material
Acknowledgments
Mariana Villalba was funded by CONACYT.
Funding Statement
This work was supported by the Consejo Nacional de Ciencia y Tecnologia (CONACYT) [690129].
Disclosure statement
The authors declare that they have no competing interests.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Metzger DCH, Schulte PM.. Persistent and plastic effects of temperature on DNA methylation across the genome of threespine stickleback (Gasterosteus aculeatus). Proc R Soc B. 2017;284(1864):20171667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Flores KB, Wolschin F, Amdam GV.. The role of methylation of DNA in environmental adaptation. Integr Comp Biol. 2013;53(2):359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Verhoeven KJF, Jansen JJ, Van Dijk PJ, et al. Stress-induced DNA methylation changes and their heritability in asexual dandelions. New Phytol. 2010;185(4):1108–1118. [DOI] [PubMed] [Google Scholar]
- [4].Bewick AJ, Vogel KJ, Moore AJ, et al. Evolution of DNA methylation across insects. Mol Biol Evol. 2016;34(3):654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Field LM, Lyko F, Mandrioli M, et al. DNA methylation in insects. Insect Mol Biol. 2004;13(2):109–115. [DOI] [PubMed] [Google Scholar]
- [6].Greenberg MVC, Bourc’his. D. The diverse roles of dna methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20(10):590–607. [DOI] [PubMed] [Google Scholar]
- [7].Hamdoun AM, Cheney DP, Cherr GN. Phenotypic plasticity of hsp70 and hsp70 gene expression in the pacific oyster (Crassostrea gigas): implications for thermal limits and induction of thermal tolerance. Biol Bull. 2003;205(2):160–169. [DOI] [PubMed] [Google Scholar]
- [8].Münzel M, Globisch D, Brückl T, et al. Quantification of the sixth DNA base hydroxymethylcytosine in the brain. Angew Chem. 2010;49(31):5375–5377. [DOI] [PubMed] [Google Scholar]
- [9].Santiago M, Antunes C, Guedes M, et al. TET enzymes and DNA hydroxymethylation in neural development and function - How critical are they? Genomics. 2014;104(5):334–340. [DOI] [PubMed] [Google Scholar]
- [10].Chaithanya Ponnaluri VK, Ehrlich KC, Zhang G, et al. Association of 5-hydroxymethylation and 5-methylation of DNA cytosine with tissue-specific gene expression. Epigenetics. 2017. February;12(2):123–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Krebs RA. A comparison of hsp70 expression and thermotolerance in adults and larvae of three Drosophila species. Cell Stress Chaperones. 1999;4(4):243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ittiprasert W, Miller A, Knight M, et al. Evaluation of cytosine DNA methylation of the Biomphalaria glabratahe at shock protein 70 locus after biological and physiological stresses. J Parasitology Vector Biol. 2015;7(10):182–193. [Google Scholar]
- [13].Gugger PF, Fitz‐Gibbon S, PellEgrini M, et al. Species-wide patterns of DNA methylation variation in Quercus lobata and their association with climate gradients. Mol Ecol. 2016;25(8):1665–1680. [DOI] [PubMed] [Google Scholar]
- [14].Robertson M, Schrey A, Shayter A, et al. Genetic and epigenetic variation in Spartina alterniflora following the deepwater horizon oil spill. Evol Appl. 2017;10(8):792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Strader ME, Wong JM, Kozal LC, et al. Parental environments alterD NA methylation in offspring of the purple sea urchin, Strongylocentrotus purpuratus. J Exp Mar Biol Ecol. 2019;517:54–64. [Google Scholar]
- [16].Banta JA, Richards CL. Quantitative epigenetics and evolution. Heredity (Edinb). 2018;121(3):210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ping N, Shiguo L, Lin Y, et al. Methylation divergence of invasive Ciona ascidians: significant population structure and local environmental influence. Ecol Evol. 2018;8(20):10272–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Münzbergová Z, Latzel V, Šurinová M, et al. DNA methylation as a possible mechanism affecting ability of natural populations to adapt to changing climate. Oikos. 2019;128(1):124–134. [Google Scholar]
- [19].Dubin MJ, Zhang P, Meng D, et al. DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. elife. 2015;4:e05255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Paszkowski J. Controlled activation of retrotransposition for plant breeding. Curr Opin Biotechnol. 2015;32:200–206. [DOI] [PubMed] [Google Scholar]
- [21].Dai T-M, Lü Z-C, Liu W-X, et al. The homology gene BtDnmt1 is essential for temperature tolerance in invasive Bemisia tabaci mediterranean cryptic species. Sci Rep. 2017;7(1):3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Foust CM, Preite V, Schrey AW, et al. Genetic and epigenetic differences associated with environmental gradients in replicate populations of two salt marsh perennials. Mol Ecol. 2016;25(8):1639–1652. [DOI] [PubMed] [Google Scholar]
- [23].Bell WJ, Roth LM, Nalepa CA. Cockroaches: ecology, behavior, and natural history. JHU Press, Baltimore; 2007. [Google Scholar]
- [24].Stay B, Coop A. Developmental stages and chemical composition in embryos of the cockroach, Diploptera punctata, with observations on the effect of diet. J Insect Physiol. 1973;19(1):147–171. [DOI] [PubMed] [Google Scholar]
- [25].Sambrook J, Russell DW. Standard Ethanol Precipitation of DNA in Microcentrifuge Tubes. Cold Spring Harb Protoc. 2006;2006(1):171. [DOI] [PubMed] [Google Scholar]
- [26].Boffelli D, Takayama S, Martin DIK. Now you see it: genome methylation makes a comeback in Drosophila. BioEssays. 2014;36(12):1138–1144. [DOI] [PubMed] [Google Scholar]
- [27].Takayama S, Dhahbi J, Roberts A, et al. Genome methylation in D. melanogaster is found at specific short motifs and is independent of DNMT2 activity. Genome Res. 2014;24(5):821–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].R Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2018. [Google Scholar]
- [29].Fox J, Weisberg S. An R Companion to Applied Regresion. second ed. SAGE publications, United States of America; 2011. [Google Scholar]
- [30].Bates D, Mächler M, Bolker B, et al. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67(1):1–48. [Google Scholar]
- [31].Lenth R. Emmeans: estimated marginal means, aka Least-Squares Means. R package version 1.4.4; 2020.
- [32].Chojnacki S, Cowley A, Lee J, et al. Programmatic access to bioinformatics tools from embl-ebi update: 2017. Nucleic Acids Res. 2017;45(W1):W550–W553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fredslund J, Schauser L, Madsen LH, et al. Prifi: using a multiple alignment of related sequences to find primers for amplification of homologs. Nucleic Acids Res. 2005;33(suppl_2):W516–W520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Amarasinghe HE, Clayton CI, Mallon EB. Methylation and worker reproduction in the bumble-bee (Bombus terrestris). Proc R Soc B. 2014;281(1780):20132502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Arrigo N, Tuszynski JW, Ehrich D, et al. Evaluating the impact of scoring parameters on the structure of intra-specific genetic variation using rawgeno, an R package for automating AFLP scoring. Bmc Bioinformatics. 2009;10(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Pérez-Figueroa A. msap: a tool for the statistical analysis of methylation-sensitive amplified polymorphism data. Mol Ecol Resour. 2013;13(3):522–527. [DOI] [PubMed] [Google Scholar]
- [37].Morán P, Marco-Rius F, Megías M, et al. Environmental induced methylation changes associated with seawater adaptation in brown trout. Aquaculture. 2013;392:77–83. [Google Scholar]
- [38].Anderson MJ. Permutational multivariate analysis of variance (permanova). In Balakrishnan N, et al, editors. Wiley Online Library. Wiley statsref: statistics reference online; 201. 7. p.1–15. [Google Scholar]
- [39].Oksanen J, Guillaume Blanchet F, Friendly M, et al. vegan: community ecology package. R package version 2.5–4; 2019.
- [40].P Arbizu M. Pairwise multilevel comparison using adonis, r package version 0.3, 2019.
- [41].Hayashi Y, Kiyoto Maekawa CA, Nalepa TM, et al. Transcriptome sequencing and estimation of DNA methylation level in the subsocial wood-feeding cockroach Cryptocercus punctulatus (Blattodea: cryptocercidae). Appl Entomol Zool. 2017. November;52(4):643–651. [Google Scholar]
- [42].Cheng Y, Xie N, Jin P, et al. DNA methylation and hydroxymethylation in stem cells. Cell Biochem Funct. 2015;33(4):161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Robertson HM, Baits RL, Walden KKO, et al. Enormous expansion of the chemosensory gene repertoire in the omnivorous german cockroach Blattella germanica. J Exp Zool Part B. 2018;330(5):265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jun-tao H, Chen B, Zhi-hong L. Thermal plasticity is related to the hardening response of heat shock protein expression in two bactrocera fruit flies. J Insect Physiol. 2014;67:105–113. [DOI] [PubMed] [Google Scholar]
- [45].Sørensen JG, Dahlgaard J, Loeschcke V. Genetic variation in thermal tolerance among natural populations of Drosophila buzzatii: down regulation of hsp70 expression and variation in heat stress resistance traits. Funct Ecol. 2001;15(3):289–296. [Google Scholar]
- [46].Arias MB, Poupin MJ, Lardies MA. Plasticity of life-cycle, physiological thermal traits and hsp70 gene expression in an insect along the ontogeny: effect of temperature variability. J Therm Biol. 2011;36(6):355–362. [Google Scholar]
- [47].Encomio VG, Chu F-LE. Heat shock protein (Hsp70) expression and thermal tolerance in sublethally heat-shocked eastern oysters Crassostrea virginica infected with the parasite Perkinsus marinus. Dis Aquat Organ. 2007;76(3):251–260. [DOI] [PubMed] [Google Scholar]
- [48].Glastad KM, Hunt BG, Goodisman MAD. DNA methylation and chromatin organization in insects: insights from the ant Camponotus floridanus. Genome Biol Evol. 2015;7(4):931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Huang X, Shiguo L, Ping N, et al. Rapid response to changing environments during biological invasions: DNA methylation perspectives. Mol Ecol. 2017;26(23):6621–6633. [DOI] [PubMed] [Google Scholar]
- [50].Platt A, Gugger PF, Pellegrini M, et al. Genome-wide signature of local adaptation linked to variable cpg methylation in oak populations. Mol Ecol. 2015;24(15):3823–3830. [DOI] [PubMed] [Google Scholar]
- [51].Leung C, Breton S, Angers B. Facing environmental predictability with different sources of epigenetic variation. Ecol Evol. 2016;6(15):5234–5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Dimond JL, Roberts SB. Germline DNA methylation in reef corals: patterns and potential roles in response to environmental change. Mol Ecol. 2016;25(8):1895–1904. [DOI] [PubMed] [Google Scholar]
- [53].Hawes NA, Fidler AE, Tremblay LA, et al. Understanding the role of DNA methylation in successful biological invasions: a review. Biol Invasions. 2018;20(9):2285–2300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.