

ABSTRACT
Chromatin reorganization governs the regulation of gene expression during preimplantation development. However, the landscape of chromatin dynamics in this period has not been explored in bovine. In this study, we constructed a genome-wide map of accessible chromatin in bovine oocytes and early embryos using an improved assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) which revealed unique features of the accessible chromatin during bovine early embryo development. We found that chromatin accessibility is low in oocytes and 2-/4-cell embryos, followed by a significant increase in embryos during major embryonic genome activation (EGA), and peaked in elongating day 14 embryos. Genome-wide characteristics of open chromatin showed that ATAC-seq signals in both transcription start sites (TSS) and transcription end sites (TES) were strong. Additionally, the distal ATAC-seq peaks were enriched in repeat elements in a type-specific and stage-specific manner. We further unveiled a series of transcription factor (TF) motifs with distinct variation of enrichment from distal ATAC-seq peaks. By integrated analysis of chromatin accessibility with transcriptomes and DNA methylomes in bovine early embryos, we showed that promoter accessibility was positively correlated with gene expression, especially during major EGA, and was strongly correlated to DNA methylation and CpG density. Finally, we identified the critical chromatin signatures and TFs that differ between in vivo and in vitro derived blastocysts, which provides insights to the potential mechanisms leading to low quality of embryos produced in vitro. Together, this comprehensive analysis revealed critical features of chromatin landscape and epigenetic reprogramming during bovine preimplantation embryo development.
KEYWORDS: Accessible chromatin, ATAC-seq, oocytes, embryos, bovine
Introduction
Preimplantation embryonic development is a complex process, in which a series of developmental milestones, including fertilization, cleavage divisions, and early lineage specification, must be successfully completed for the embryo to maintain viability. The molecular regulatory network controls each of these transitions during early embryo development. The precise activation and establishment of this regulatory network is largely dependent on the epigenetic reprogramming, including chromatin remodelling.
The accessible chromatin is highly dynamic and the extensive chromatin remodelling is essential for transcription and preimplantation embryo development [1]. However, it has been challenging to explore the global chromatin landscape in this period due to limited experimental materials. Most recently, low input or single-cell assays to profile chromatin remodelling have been developed and the genome-wide map of accessible chromatin in gametes and early embryos has been extensively analyzed in both mice and humans [2–9]. Unsurprisingly, these studies have revealed highly active chromatin landscapes in gametes and early embryos. Generally, chromatin has a higher degree of accessibility in embryos after embryonic genome activation (EGA). Putative cis-regulatory elements including transcription factors (TFs) that regulate EGA were also identified [3]. Additionally, by integrating the chromatin accessibility with the transcriptome in early embryos, a regulatory network was constructed, which identified the transcription factors associated with preimplantation development and lineage specification [3]. However, the chromatin dynamics in bovine early embryos remains unknown. It remains to be determined how chromatin reconfigures and regulates the transcription programmes in bovine early embryos. And it is also unclear whether the chromatin dynamics are associated with other epigenomic reprogramming events such as global DNA methylation in the same developmental period.
As an economically valuable livestock species, improvement of pregnancy rates with in vitro produced embryos is a critical problem which remains to be addressed for bovine. The efficiency of producing viable embryos in vitro and the development of such embryos after transferring them to recipients is inferior to their in vivo derived counterparts, especially in cattle [10–13]. In addition, the offspring from in vitro derived embryos, especially those from livestock species, have a high incidence of abnormalities including large offspring syndrome (LOS), severe placental abnormalities, respiratory problems, prolonged gestation, and dystocia [14–16]. The underlying mechanisms are largely unknown at present, but alterations in gene expression and epigenetic modifications, largely DNA methylation, during this critical period are thought to be involved in LOS [17–20].
Here, we examined the accessible chromatin in bovine oocytes and across consecutive stages of early embryos using ATAC-seq. Our data provide a high-resolution map of accessible chromatin during bovine early embryo development. We also performed an integrative analysis of transcriptome, DNA methylome, and chromatin dynamics and describe an essential complement of the regulatory network during bovine early embryo development. Finally, we analyzed the differences of chromatin accessibility by combining the gene expression profiles between in vitro and in vivo produced blastocysts, which sheds light on the mechanisms contributing to embryo defects and relatively low pregnancy rates after embryo transfer when using in vitro produced blastocysts.
Materials and methods
Ethics statement
In vivo produced oocytes and embryos were obtained from cross breed cows housed at the Reproductive Biological Center at the School of Animal Sciences, Louisiana State University Agricultural Center (LSU AgCenter). The animal protocol was approved by the Animal Care and Use Committee of LSU AgCenter (A2017-19).
Bovine in vitro and in vivo embryo production
For in vitro production, germinal vesicle stage oocytes (GV oocytes) were collected as cumulus-oocyte complexes from follicles of 3–5 mm in diameter aspirated from slaughterhouse ovaries. BO-IVM medium (IVF Bioscience) was used for oocyte in vitro maturation. Maturation was conducted in four-well dishes for 22–23 hours at 38.5°C with 6% CO2. Cumulus cells were completely removed and maturation was confirmed by light microscopy examination. Cryopreserved semen from a Holstein bull with proven fertility was diluted with BO-SemenPrep medium (IVF Bioscience) and added to drops containing COCs with a final concentration of 2 × 106 spermatozoa/ml. Gametes were co-incubated in 6% CO2 in air at 38.5°C for 18 hours. Embryos were then washed and cultured in BO-IVC medium (IVF Bioscience) at 38.5°C with 6% CO2.
For in vivo production, ovarian stimulation and embryo retrieval from cross bred cows (n = 10) were performed as previously described [21,22]. Superovulation was achieved using five doses of intramuscular injections of FSH beginning 5 days after insertion of a Controlled Intra-vaginal Drug Release (CIDR) device. Two doses of prostaglandin F2 alpha were given along with the last two FSH treatments, followed by CIDR removal. Standing oestrus (Day 0) was seen approximately 48 h post-prostaglandin injection. GnRH was then administered at oestrus. Each cow was inseminated 12- and 24-h post-standing oestrus. Morulae and blastocysts were collected by routine non-surgical uterine flushing on days 5, 6, and 7. Elongating embryos were collected by uterine flushing on day 14 (D14).
Embryo stages were then evaluated under light microscopy and only Grade 1 embryos by standards of the International Embryo Technology Society were selected for further study. All oocytes and embryos were washed with D-PBS containing 1 mg/ml polyvinylpyrrolidone (PBS-PVP) and transferred into 50 μl droplets of 0.1% protease to remove the zona pellucida. Oocytes and embryos were rinsed three times in PBS-PVP and confirmed to be free of contaminating cells, and then snap frozen in minimal medium and stored at −80°C until ATAC-seq library preparation. D14 elongating embryos were classified based on morphology and length, and embryos with similar length (1–3 mm) were selected for further study. In this study, for biological replicates, we pooled approximately 50 oocytes (GV: n = 6; and MII: n = 5), 20 embryos at 2-, 4-, 8, and 16-cell stage (n = 6), 10 in vitro derived morula (n = 6) and blastocyst (n = 4) stage embryos, 10 in vivo derived morula (n = 4) and blastocyst (n = 4) stage embryos, and individual elongating embryos (n = 6) for the ATAC-seq analysis (Table S1).
ATAC-seq library preparation and sequencing
The ATAC-seq libraries of bovine oocytes and embryos were prepared as previously described with minor modifications [2,3]. Briefly, oocytes or embryos were lysed in ice-cold lysis buffer (10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2 and NP-40 (0.5%)) for 15 minutes on ice to prepare the nuclei. Immediately after lysis, Nuclei were then incubated with the Tn5 transposase (TDE1, Illumina) and tagmentation buffer at 37°C for 30 minutes with shaking on a thermomixer at 500 g. After the tagmentation, 0.5 ul of 10% SDS was added directly into the reaction to end the tagmentation and tagmentated DNA was purified using MinElute Reaction Cleanup Kit (Qiagen). PCR was performed to amplify the ATAC-seq libraries using Illumina TrueSeq primers and multiplex by indexes primers with the following PCR conditions: 72°C for 3 min; 98°C for 30 s; and 12 cycles of 98°C for 15 s, 60°C for 30s, and 72°C for 3 min; followed by 72°C for 5 min. After the PCR reaction, libraries were purified with the 1.1X AMPure beads (Beckman). The concentration of the sequencing libraries was determined by using Qubit dsDNA HS Assay Kit (Life Technologies). The size of the sequencing libraries was determined by means of High Sensitivity D5000 Assay with a Tapestation 4200 system (Agilent). Indexed libraries were then pooled and sequenced on the Illumina HiSeq platform with 150-bp paired-end reads.
ATAC-seq data processing and filtration
Sequencing reads of all samples underwent adapter removal using NGmerge [23], followed by quality assessment using FastQC. Reads were then aligned to the bovine reference genome UMD3.1.1 using Bowtie 2.3 with the following options: – very-sensitive -X 2000 – no-mixed – no-discordant. Only unique alignments within each sample were retained in subsequent analysis (Table S2). Moreover, alignments resulting from PCR duplicates or located in mitochondria were excluded.
The Bovine genome was tiled with consecutive non-overlapping 2,000bp bins. The accessibility of each of the 2,000bp bins was assessed by the number of fragments per million mapped (FPM) that was aligned to the bin. There was a total of 1,328,402 bins with at least one aligned fragment in at least one sample. Pearson correlation between each pair of samples was computed and cluster analysis was performed using hierarchical clustering in R. Five samples including two in vitro morula and one of each in vitro 8-, 16-cell and blastocyst stage embryos were identified as outliers because they were not clustered with the rest of samples at the same developmental stage in the cluster analysis and therefore excluded from subsequent analysis. Similar to cluster analysis, t-distributed stochastic neighbour embedding (t-SNE) was performed on pairwise correlation data in R with functions Rtsne in package Rtsne, respectively.
Peak calling and enrichment of genomic features in peaks
ATAC-seq peaks were called separately for each sample by MACS2 [24] with the following options: – keep-dup all – nolambda – nomodel. Peaks in individual samples from the same development stage were subsequently merged using bedtools (https://bedtools.readthedocs.io/en/latest/). The annotations of genomic features, including transcription start sites (TSS), transcription end sites (TES), exons, introns, and CpG islands and repeat-elements: long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), long terminal repeats (LTRs) and simple sequence repeats (Simple) were downloaded from UCSC genome browser. Promoters were defined as 500bp up- and down-stream from the TSS of each annotated gene (TSS ± 500bp). Intergenic regions were defined as genomic regions before the TSS of the first gene and after the TES of the last gene in each chromosome, and in-between the TES and TSS of two consecutive genes. Peaks that did not overlap with annotated promoters were deemed as distal peaks. To evaluate the enrichment of the above genomic features with identified ATAC-seq peaks, a set of random peaks was first generated by matching the length of ATAC-seq peaks. The enrichment was then assessed by the ratio (or the log of the ratio) between the numbers of ATAC-seq and random peaks that overlapped with the corresponding genetic feature. The enrichment of transcriptional factor motifs in peaks was evaluated using HOMER (http://homer.ucsd.edu/homer/motif/).
Assessment of promoter accessibility
Promoter accessibility in each developmental stage was assessed by the number of ATAC-seq fragments (FPM) mapped to the defined promoter region (i.e., TSS ± 500bp) in all samples from that stage. Z-score for each promoter at each stage was obtained by standardizing the FPM values stage-wisely, having mean 0 and variance 1 within each stage. Promoters with z-scores below zero at all the stages were deemed to have constantly low accessibility; while those with z-scores above zero at all the stages were deemed to have constantly high accessibility. The remaining promoters were deemed to have dynamic accessibility. Pearson correlation was calculated between promoter accessibility and corresponding gene expression across stages. Promoters with accessibility that highly correlates with corresponding gene expression were then identified (with cor ≥ 0.5).
Identifying distal peaks with stage-specific accessibility
ATAC-seq peaks from all individual samples were merged by bedtools. All merged peaks that do not overlap with any annotated promoters were deemed as distal peaks. The accessibility of each of these identified distal peaks in each individual developmental stage was then quantified by the number of sequence fragments (FPM) mapped to the peak region in all samples from the stage. To identify the distal peaks with stage-specific accessibility, we formulated a vector for each developmental stage to represent the stage-specific pattern, which has its size equal to the number of stages and consists of all zeros except having one at the entry corresponding to the stage. Then, the Pearson correlation between these stage pattern vectors and the vector representing the accessibility of each distal peak across stages was calculated. Peaks with an adjusted (FDR, false discovery rate) p-value ≤0.05 against a stage pattern were deemed as the peaks with stage-specific accessibility with respect to the corresponding stage.
RNA sequencing (RNA-seq) data processing
We processed published RNA-seq datasets from embryos derived in vivo [21] and in vitro [25]. Briefly, the sequencing reads that passed filters were trimmed to remove low-quality reads and adaptors by TrimGalore-0.4.3. The quality of reads after filtering was assessed by fastQC, followed by alignment to the bovine genome (UMD3.1.1) by STAR (2.5.3a) with default parameters. Individual mapped reads were adjusted to provide FPKM (fragments per kilobase of exon model per million mapped fragments) values with bovine genome as reference.
Identification of stage specifically expressed genes
To identify stage specifically expressed genes, a binary vector was formulated to represent the stage-specific pattern for each stage. The size of all these vectors is identical and equal to the number of total samples in the data. In other words, each sample has a corresponding entry in each of the vectors. Each vector consists of all zeros except with one at the entries corresponding to the samples from the stage to be represented by this vector. Genes with a FPKM > 10 in more than half of the samples at each individual developmental stage are considered constantly expressed (or housekeeping) genes; thus, they are not stage specifically expressed genes and removed as a preprocessing step to identify stage specifically expressed genes. Pearson correlation was then computed between each pattern vector and the expression level in all samples of each remaining gene. A gene with an adjusted p-value by FDR ≤ 0.05 was deemed as a specifically expressed gene of the stage. Stage specifically expressed genes were identified for oocytes including samples of GV and MII oocytes, minor embryonic genome activation (EGA) including samples at the 4-cell stage, major EGA including samples at 8- and 16-cell stages, and blastocyst.
Identification of EGA genes
To identify minor and major EGA genes, the stage specifically activated genes were first identified for 4-cell, 8-cell and16-cell. For the identification of specifically activated genes of each stage, a binary vector was formulated to represent the desired expression pattern. The size of the vector is equal to the total number of samples at all early stages up to the stage for which stage specifically activated gene is to be identified (i.e., target stage). Each vector consists of all zeros except ones at entries corresponding to samples at the target stage. Genes with a FPKM > 1 in more than half of the samples among either GV or MII oocytes are considered as genes expressed in oocytes, thus are excluded from the identification of EGA genes. Then, Pearson correlation was computed between expression levels of each remaining gene and the pattern vector described above. Genes with p-value ≤0.05 associated with a stage were defined as specifically activated genes for that stage. Genes activated at 4-cell stage were subsequently labelled as minor EGA genes; while the combination of 8-cell and 16-cell activated genes was considered as major EGA genes.
Whole genome bisulfite sequencing (WGBS) data processing
We used WGBS data from our previous studies [22,26]. For the present analysis, only estimated methylation level of CpG sites covered by at least five sequencing reads were used; methylation level of all other CpG sites was considered unknown. The methylation level of each annotated promoter (TSS ± 500bp) was measured by the average methylation level of all CpG sites with data located within the promoter.
Assessment of promoter CpG density
CpG density of annotated promoters (TSS ± 500bp) was assessed by function CpGDensityByRegion in R package BSgenome. In our analysis, when categorization of the promoter CpG density was needed, it was done as follows: low (<25), medium (≥25 and ≤75) and high (>75).
Comparative analysis between in vivo and in vitro derived blastocysts
To perform differential analysis of promoter accessibility between in vivo and in vitro derived embryos, promoters with FPM below 25% quantile in both derivations were excluded for downstream analysis. A total of 3211 and 426 promoters were identified with differential accessibility by using two- or four-fold change thresholds, respectively. Differential gene expression between in vivo [21] and in vitro [25] derived blastocysts were conducted using R functions (lmFit and eBayes). A total of 273 genes were identified with differential expression (adjusted p-value ≤0.05). Integrative analysis on the changes of promoter accessibilities and their corresponding gene expression changes between in vitro and in vivo was conducted using the promoters with differential accessibilities (four-fold change threshold) and the differential expressed genes. The enrichment of transcriptional factor motifs in peaks was evaluated using HOMER as previously described.
Gene ontology and pathway analysis
Gene ontology (GO) and pathway analysis was performed using Ingenuity Pathway Analysis (IPA) (Qiagen). GO and pathways terms with an FDR adjust p-value ≤0.05 were deemed statistically significant.
Results and discussion
Accessible chromatin landscape in bovine oocytes and early embryos
To investigate accessible chromatin in bovine oocytes and preimplantation embryos, we performed ATAC-seq analysis on GV and MII oocytes, in vitro embryos at the 2-cell, 4-cell, 8-cell, 16-cell, morula and blastocyst stages, in vivo embryos at morula and blastocyst, and D14 elongating embryos (Table S1). We sequenced an average of 110 million paired-end reads per sample, which provided the expected genomic coverage (Table S2). The raw FASTQ files are available at Gene Expression Omnibus (GEO) (www.ncbi.nlm.nih.gov/geo) under accession number GSE145040.
We performed t-Distributed Stochastic Neighbour Embedding (t-SNE) and unsupervised hierarchical clustering to validate the reproducibility of ATAC-seq data (Figure 1a,b). All biological replicates showed highly consistent results (Figure 1a,b). Accessible chromatin of oocytes and embryos could be divided into four distinct profiles (Figure 1a-c; Figure S1A): 1) low accessibility in oocytes (GV and MII) and during minor EGA (2-/4-cell stage), 2) high accessibility during major EGA (8-/16-cell, and morula stage), 3) less accessibility in blastocyst stage embryos, and 4) the highest accessibility in elongating embryos. This dynamic and sequential chromatin remodelling is consistent with transcription silencing in oocytes, transcription activation at the minor EGA (oocytes to 4-cell) and major EGA (8-cell to morula), dynamic transcription activation and inhibition during first differentiation events at blastocyst stage and drastic transcription initiation for embryo elongation [21,25], which further validate our ATAC-seq data.
Figure 1.
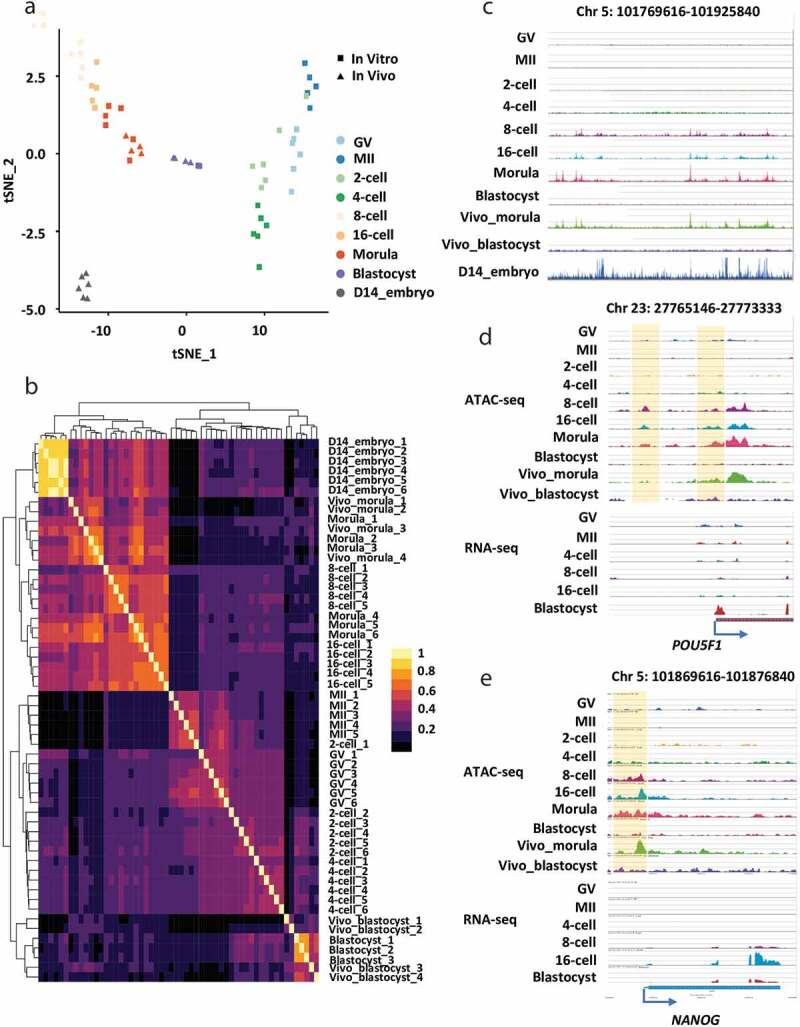
Accessible chromatin landscape in bovine oocytes and early embryos. (a) Distribution of samples in the two-dimensional space, where the embeddings were computed by t-SNE with the data of the pairwise Pearson correlation evaluated using the accessibility of 2,000bp genome bins. (b) Hierarchical clustering of all samples, where the distance among samples was measured by the pairwise Pearson correlation evaluated using the accessibility of 2,000bp genome bins. The colour spectrum, ranging from yellow to black, indicates correlation from high to low. (c) The genome browser view showing the enrichment of ATAC-seq peaks in bovine oocytes and early embryos. The genome browser views showing the ATAC-seq peaks and RNA-seq reads enrichment near POU5F1 (d) and NANOG1 (e)
We found elevated chromatin accessibility at upstream sequences (promoters) of many crucial transcription factors, such as POU5F1, NANOG, and GATA6, which coincides with their gene expression during bovine early embryo development (Figure 1d,e; Figure S1B). Notably, the ATAC-seq peaks of POU5F1 were enriched upstream of TSS and within intronic regions in embryos, especially from the 8-cell to blastocyst stage (Figure 1d). These enrichments have also been reported in mice and humans [2,27]. Specifically, the expression of NANOG was significantly increased from the 8-cell stage, which coincides with their promoter ATAC-seq enrichment within the promoter at the same stage (Figure 1e). Overall, these results show that the ATAC-seq provides reliable and highly sensitive data to detect open chromatin in bovine preimplantation embryos.
Unique features of the accessible chromatin during bovine early embryo development
We next sought to determine the chromatin dynamics in specific genomic regions during bovine early embryo development. ATAC-seq peaks were enriched both at the transcription start (TSS) and transcription end (TES) sites, especially in embryos from the 8-cell to morula stage (Figure 2a,b). The significant enrichment of open chromatin at TSS and TES sites suggests that they might act as promoters and enhancers, respectively, to regulate gene expression during early embryo development [28].
Figure 2.
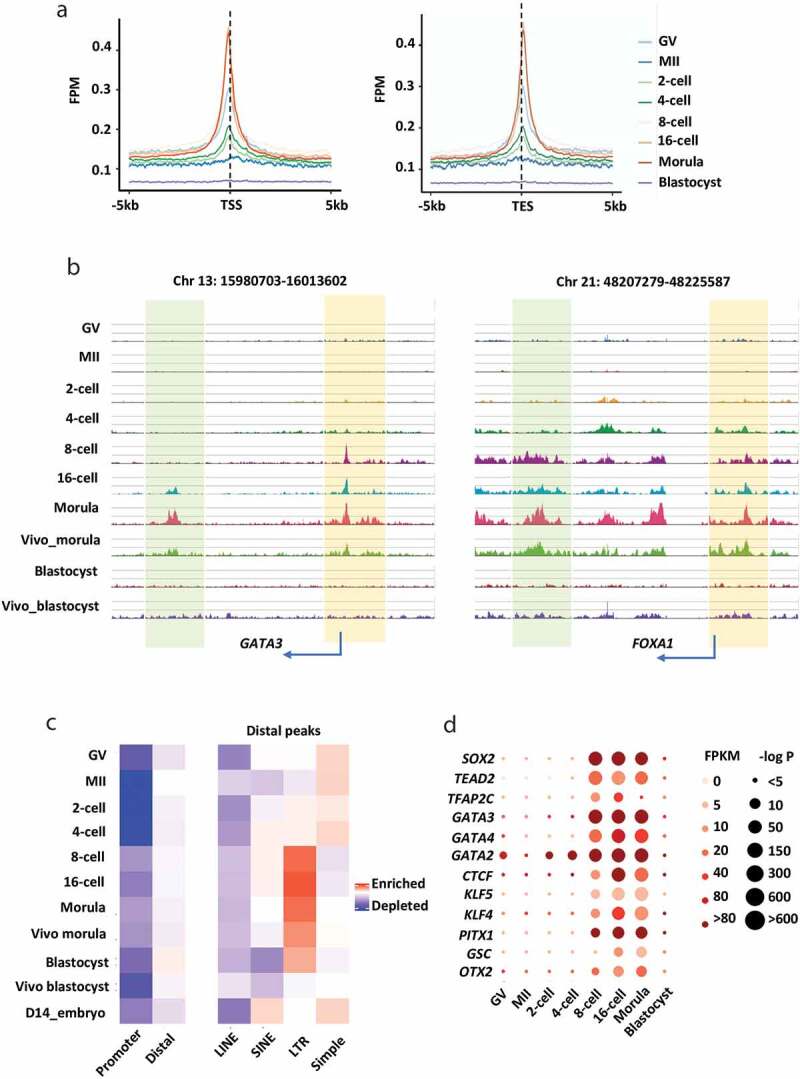
Unique characteristics of accessible chromatin in bovine oocytes and early embryos. (a) The enrichment of ATAC-seq peaks at annotated promoters (TSS ± 5kb) and annotated transcription end sites (TES ± 5kb) (normalized and on average) in oocytes and embryos. The enrichment was measured at individual base pair level by FPM (Fragments per Million mapped reads). (b) The genome browser view showing the enrichment of ATAC-seq peaks near two active genes (GATA3, left panel; FOXA1, right panel) in oocytes and embryos. ATAC-seq peaks near TSSs and TESs were shaded in different colours (TSSs in yellow; TESs in green). (c) Enrichment of ATAC-seq peaks in the promoters and distal peaks (right panel), and repetitive elements (left panel). (d) Transcription factor motifs identified from distal ATAC-seq peaks at each stage during early embryo development
A large fraction of ATAC-seq peaks in bovine oocytes and early embryos overlapped with promoters (Figure S2A). Interestingly, promoters showed less chromatin accessibility in MII oocytes than in GV oocytes (Figure S1A and S2A). The chromatin condensation phenomenon is conserved in various mammalian oocytes including bovine oocytes and regulates the transcription silencing in oocytes, which is one of the hallmarks of oocyte remodelling [29–33]. The majority of ATAC-seq peaks were detected in the intergenic and intron regions (Figure S2B). Although DNA sequences in intergenic are regarded as ‘junk DNA’, studies have shown mutations in such loci can change the chromatin state and DNA conformation [34–36]. In addition, long-range regulatory elements harboured in ‘junk sequence’ have interactions with distal genes and influence their expression [37].
Repetitive elements constitute 46.5% of the bovine genome [38], and the majority of these elements are retrotransposons, including Long Interspersed Elements (LINEs), Short Interspersed Elements (SINEs), long terminal repeats (LTR) and Simple sequence repeats [39]. An emerging body of evidence has shown that they are involved in the regulation of chromatin structure [40–43], transcription [44,45], RNA processing [46,47] and messenger RNA localization and translation [48,49]. We found a large fraction of ATAC-seq peaks in early bovine embryos overlapped with distal regions, and most of the enriched regions mapped to repetitive elements (Figure 2c). It is noteworthy that certain classes of repeats are highly transcribed in early embryos [50,51] and can function as promoters and enhancers [52–54]. We confirmed that distal ATAC-seq peaks are enriched in repeat elements in a type-specific and stage-specific manner, i.e., 1) they were enriched in LINE, SINE, and LTR during major EGA stages but depleted in Simple repeats during the same developmental period, 2) distal ATAC-seq peaks were more enriched in SINE, LRT, and Simple than LINE elements. 3) they were preferentially enriched at LTR during major EGA stages and a similar enrichment was seen in LINE. However, enrichment in Simple repeats was the opposite, i.e., more distal peaks at Simple loci were found in oocytes, 2-/4-cell and D14 embryos. The observed open chromatin in repeat elements provides valuable information to further explore its functions in bovine early embryo development.
We also identified 994 known transcription factor (TF) motifs from distal ATAC-seq peaks in early bovine embryos. Among them, there were 218 motifs for which the transcriptomic data are available for the corresponding genes in bovine embryos (Table S3). By comparing those TFs identified from human embryo distal ATAC-seq peaks [2], 12 TF motifs were found to be present in both bovine and human embryos (Figure 2d). The enrichment within TF motifs is in a highly stage-specific manner, and most of them have important functions in early embryo development. For example, CTCF, KLF, and OTX2 are maternal transcription factors [55–57]. By mining published bovine embryo RNA-seq data [58], we found their expression is constantly high throughout oocyte and early embryo stages, while their binding motifs are not highly enriched until the 8-cell stage. Both the binding motifs and the expression levels of some TFs including TFAP2 C, GATA2, GATA3, and PITX1 were strongly enriched and high at the 8-cell stage, which agrees with previous findings that they are essential during the major EGA and onwards [59,60].
Notably, the motifs of trophectoderm (TE) lineage markers, GATA2 and GATA3 were also strongly enriched in 8-/16-cell and morula stages, suggesting a preparation for the early trophoblast linage emergence. Interestingly, as early as GV and the 2-cell stage, GATA2 motifs became enriched, indicating it might play additional roles during bovine embryonic genome activation. TFAP2 C is a regulator of TE development at late embryonic stages and plays a crucial role in formation and differentiation of the trophoblast lineage in mammals, including bovine [59,61–65]. Intriguingly, we found that, although the expression of TFAP2 C was high in the later stages, the enrichment of its binding motifs decreased dramatically in the morula and blastocyst stages, which is opposite to that observed in humans [66]. We thus speculated that the signalling network directing trophoblast lineage specification may occur much earlier in bovine embryos.
Together, the open chromatin in bovine early embryos was shown near both TSSs and TESs of active genes. Distal ATAC-seq peaks were found to be enriched in repetitive elements in a stage- and type-specific manner, specifically, it was highly enriched in SINE and LTR and depleted in SIMPLE during EGA and later developmental stages. The accessible chromatin enrichment of TF motifs was independent of the corresponding TF transcription level but followed a stage-specific manner.
Regulatory network in bovine oocytes and early development
Chromatin remodelling and DNA methylation are two of the essential epigenetic mechanisms regulating gene expression and embryonic stage transitions during early development. To determine the regulatory networks of accessible chromatin, DNA methylation, and gene expression in bovine preimplantation embryos, we performed integrated analysis using the promoter ATAC-seq dataset, previously published DNA methylation dataset [22,26], and RNA-seq dataset [25] generated from the same developmental stage (Figure 3a). We identified stage-specific expressed genes and examined their promoter chromatin accessibility, as well as DNA methylation and CpG densities at their promotor regions. Among the analysed ATAC-seq peaks, we catalogued promoter chromatin accessibility across all stages and found 219, 125, and 350 genes with constantly low, high, and dynamic accessibility, respectively. From those genes, 160, 4, 276, and 254 genes were identified as specifically expressed genes at oocyte, minor EGA (2-/4-cell), major EGA (8-/16-cell), and blastocyst stages, respectively (Figure 3a; Table S4).
Figure 3.
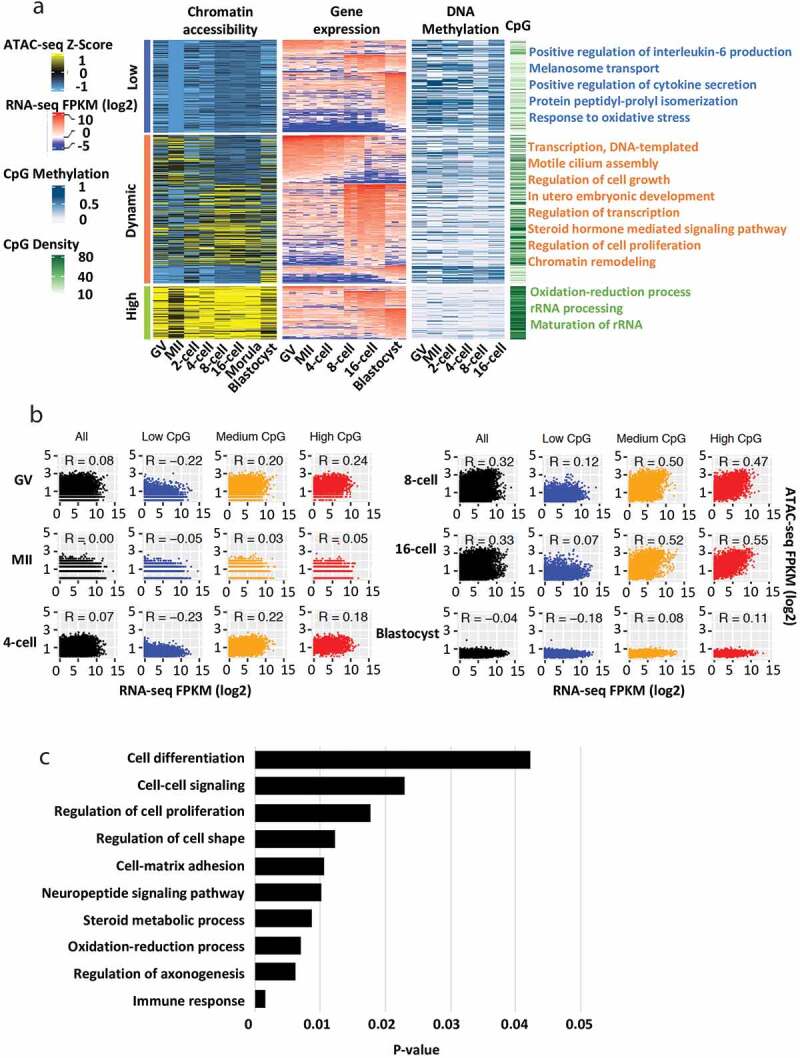
Transcription and promoter regulation in bovine oocytes and early embryos. (a) Heat maps showing promoter chromatin with constantly low, constantly high, or dynamic accessibility (first panel), the expression (FPKM) of stage-specific expressed genes (oocyte: (GV and MII), minor EGA (2-/4-cell), major EGA (8-/16-cell, and morula), and blastocyst) (second panel), promoter DNA methylation (third panel), and promoter CpG densities (fourth panel). (b) Scatter plots showing promoter chromatin accessibilities and gene expression for all genes (black) or genes with promoters of low (blue), medium (yellow), or high (red) CpG densities. Spearman correlation coefficients are shown. (c) Top represented gene ontology (GO) terms for the major EGA stage-specific genes
We found that stage-specific genes had dynamic promoter accessibility which correlated with their gene expression (Figure 3a). These genes preferentially function in transcription, regulation of cell growth, and in utero embryonic development. We also found that promoters with constantly high accessibility were preferentially CpG-rich, as has been reported in both humans and mice [2,3,67], whereas promoters with constantly low accessibility were generally CpG-poor (Figure 3a). Specifically, the majority of open chromatin was located at genes with medium and high CpG content compared to those with low CpG content (Figure 3b). Moreover, DNA methylation was negatively correlated with promoter open chromatin especially between the groups with constantly low and high accessibility across all stages (Figure 3a). Interestingly, a significant correlation between chromatin accessibility and gene expression was found in major EGA stages, suggesting chromatin dynamics plays a dominate role in regulating gene expression during this period. GO analysis showed that genes activated in this stage were involved in the regulation of important functions during embryogenesis, including immune response, steroid metabolic process, cell-matrix adhesion, as well as cell proliferation and differentiation (Figure 3c). Most of these identified GO terms are associated with early embryo differentiation and maternal recognition [68–72], suggesting that the embryo prepares for lineage differentiation and implantation as early as major EGA stages in bovine.
In sum, promoter chromatin accessibility in early bovine embryo development positively correlates with gene activities, CpG densities, and negatively correlates with promoter DNA methylation. Notably, open chromatin in promoter regions is the main factor to impact the transcriptome profiles during bovine major EGA.
Distinct gene network programmes between in vivo and in vitro derived embryos
Here we sought to reveal critical chromatin signatures associated with bovine in vivo and in vitro derived blastocysts, which would help to understand the potential epigenetic mechanisms leading to embryonic loss and pregnancy failure observed with in vitro produced embryos. Although the global accessibility of chromatin between in vitro and in vivo produced embryos did not vary much (Figure 1; Figure S1), the chromatin accessibilities of specific genes or specific loci are dramatically different. A total of 3,211 (>2-fold change) or 426 (>4-fold change) promoters were identified with differential enrichment of ATAC-seq peaks between in vivo and in vitro derived blastocysts. By integrating the published transcriptomes of bovine embryos derived in vivo [21] and in vitro [25], we found that for the majority of the expressed genes promoter accessibility alone has minimal impact on their expression (Figure 4a; Figure S3). We then performed differential gene expression analysis between in vitro and in vivo blastocysts and integrated them into the changes of promoter accessibilities (4-fold change threshold). Interestingly, among 273 identified differentially expressed genes, we identified 124 genes with consistent changes in their promoter accessibilities and expression levels (Figure 4b; Table S5). Genes with consistent change were involved in cell functions determining the quality of blastocysts, such as protein metabolism, macromolecule assembly and localization, organelle organization, and mitochondrion function (Figure 4c). Particularly, a set of genes regulating mitochondrion organization and activities were significantly different between in vitro and in vivo derived blastocysts (Figure S4). During early embryo development, a massive supply of energy is required to support a series of cell divisions and differentiation. This process is largely dependent upon the activities of mitochondria, which could either maintain life or contribute to apoptosis during early development [73]. For example, the accessibility of PRELID1, an encoded protein located in mitochondria affecting mitochondrion organization, mitochondrial function, as well as exerting cytoprotectant properties [74], was much higher in in vivo produced blastocysts than in vitro ones. Other genes including COX20, TIMM21, and PARL are crucial in regulating respiratory chain, cytochrome c release, and mitochondrial structure [75–77]. Interestingly, we found the enrichment of ATAC-seq within these genes were higher in in vitro derived blastocysts than in vivo ones, which further indicates that chromatin dynamics is dysregulated due to the in vitro manipulation during bovine early embryo development.
Figure 4.
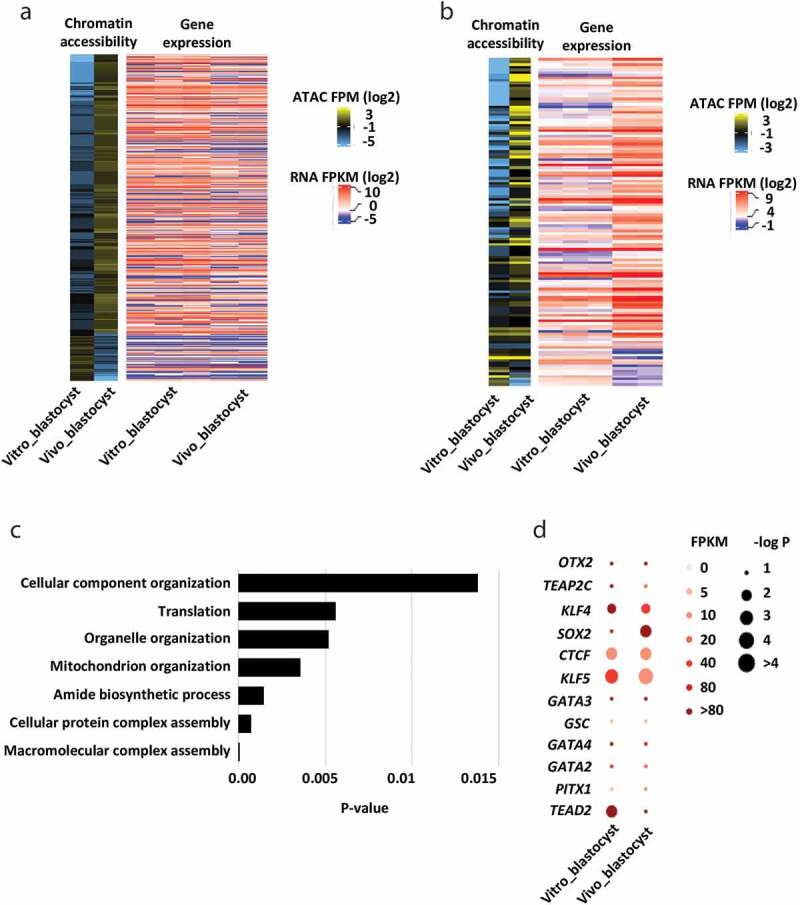
Differences of chromatin accessibilities between in vivo and in vitro derived embryos. (a) Heat map showing the promoter chromatin accessibilities of in vitro and in vivo derived blastocysts (≥4-fold change), together with the corresponding gene expression. (b) Heat map showing the identified genes with consistent changes in the promoter chromatin accessibilities and their expression levels between in vitro and in vivo derived blastocyst (p-value < 0.05). (c) Top represented GO terms for genes with consistent changes in promoter chromatin accessibilities and expression levels between in vitro and in vivo derived blastocysts. (d) Transcription factor motifs identified from distal ATAC-seq peaks between in vitro and in vivo derived blastocysts
We also performed comparative analysis of the TFs motif enrichment in distal peaks and their expression levels between in vivo and in vitro derived blastocysts. We identified numerous differences between the in vivo and in vitro derived blastocysts (Figure 4d). For example, SOX2, an essential regulator of embryo development and a pluripotency marker, had increased expression levels and higher enrichment of binding motifs in in vivo blastocysts compared to their in vivo counterparts. Also, although TEAD2 is mainly involved in the early stage of preimplantation embryo development [78], the enrichment of TEAD2 motifs was significantly higher in in vitro blastocysts than that of in vivo, indicating that TEAD2 might play an important role in later stages of in vitro produced embryos. In addition, slight differences in gene expression were seen in TFAP2 C, KLF4, KLF5, GATA2, GATA3, and GATA4. TFAP2 C expression has been shown to be altered in early preimplantation SCNT embryos [65]. A comprehensive study comparing both global gene expression patterns and the chromatin accessibility of IVF, SCNT-derived bovine embryos to their in-vivo counterparts would be very interesting.
To note, superovulation was used to collect in vivo derived embryos in this study, which may introduce abnormal gene expression and epigenetic profiles. However, study of in vivo produced embryos without superovulation is less feasible considering a cow ovulates one or two oocytes per cycle. Another limitation is that we performed our analyses on blastocysts as a whole. Although it is not easy to cleanly separate ICM and TE cells in bovine embryos, future studies to differentiate the gene expression and chromatin dynamics of these two lineages of the bovine embryos would be of major interest and importance. Collectively, the identified genes and TFs which differ between in vitro and in vivo derived embryos may serve as markers to determine embryo viability. These findings provided insights to the lower pregnancy rates following transfer of in vitro produced blastocysts.
Conclusion
In this study, we provided a genome-wide map of accessible chromatin in bovine oocytes and early embryos, which revealed critical features of accessible chromatin during bovine early embryo development. We also established the regulatory networks by integrating analysis of transcriptome, DNA methylome, and accessible chromatin, and provided a valuable dataset of gene regulation and epigenetic reprogramming during bovine early embryo development. Finally, we presented the conserved and distinct gene network programmes between in vivo and in vitro derived embryos, which provide molecular insights of perturbed development of in vitro produced embryos compared to their in vivo counterparts.
Supplementary Material
Funding Statement
This work was supported by the USDA-NIFA grant (2019-67016-29863), the Audubon Center for Research of Endangered Species, and UDSA-NIFA W4171.
Authors’ contributions
Z.J. conceived the study; H.M., R.P., J.H., L.G., Y.Y., C. P., K.B., R.K. and Z.J collected samples. H.M. and Z.J. performed the experiments; J.S., H.M., and Z.J. performed data analysis; H.M. and Z.J. interpreted the data, assembled the results, and wrote the manuscript with inputs from all authors. All authors revised and approved the final manuscript.
Disclosure statement
The authors declare no competing interests.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Burton A, Torres-Padilla ME.. Chromatin dynamics in the regulation of cell fate allocation during early embryogenesis. Nat Rev Mol Cell Biol. 2014;15(11):723–734. [DOI] [PubMed] [Google Scholar]
- [2].Wu J, Xu J, Liu B, et al. Chromatin analysis in human early development reveals epigenetic transition during ZGA. Nature. 2018;557(7704):256–260 [DOI] [PubMed] [Google Scholar]
- [3].Wu J, Huang B, Chen H, et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature. 2016;534(7609):652–657 [DOI] [PubMed] [Google Scholar]
- [4].Jung YH, Sauria MEG, Lyu X, et al. Chromatin states in mouse sperm correlate with embryonic and adult regulatory landscapes. Cell Rep. 2017;18(6):1366–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jachowicz JW, Bing X, Pontabry J, et al. LINE-1 activation after fertilization regulates global chromatin accessibility in the early mouse embryo. Nat Genet. 2017;49(10):1502–1510. [DOI] [PubMed] [Google Scholar]
- [6].Inoue A, Jiang L, Lu F, et al. Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature. 2017;547(7664):419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lu F, Liu Y, Inoue A, et al. Establishing chromatin regulatory landscape during mouse preimplantation development. Cell. 2016;165(6):1375–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Guo F, Li L, Li J, et al. Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res. 2017;27(8):967–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Liu L, Leng L, Liu C, et al. An integrated chromatin accessibility and transcriptome landscape of human pre-implantation embryos. Nat Commun. 2019;10(1):364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Diskin MG, Morris DG.. Embryonic and early foetal losses in cattle and other ruminants. Reprod Domest Anim. 2008;43(Suppl 2):260–267. [DOI] [PubMed] [Google Scholar]
- [11].Diskin MG, Parr MH, Morris DG. Embryo death in cattle: an update. Reprod Fertil Dev. 2011;24(1):244–251. [DOI] [PubMed] [Google Scholar]
- [12].Diskin MG, Waters SM, Parr MH, et al. Pregnancy losses in cattle: potential for improvement. Reprod Fertil Dev. 2016;28(1–2):83–93. [DOI] [PubMed] [Google Scholar]
- [13].Thompson JG. Comparison between in vivo-derived and in vitro-produced pre-elongation embryos from domestic ruminants. Reprod Fertil Dev. 1997;9(3):341–354. [DOI] [PubMed] [Google Scholar]
- [14].Abnormal Offspring Syndrome . In: Bovine Reproduction. 620–638.
- [15].Young LE, Sinclair KD, Wilmut I. Large offspring syndrome in cattle and sheep. Rev Reprod. 1998;3(3):155–163. [DOI] [PubMed] [Google Scholar]
- [16].Yang X, Smith SL, Tian XC, et al. Nuclear reprogramming of cloned embryos and its implications for therapeutic cloning. Nat Genet. 2007;39(3):295–302. [DOI] [PubMed] [Google Scholar]
- [17].Li T, Vu TH, Ulaner GA. IVF results in de novo DNA methylation and histone methylation at an Igf2-H19 imprinting epigenetic switch. Mol Hum Reprod. 2005;11(9):631–640. [DOI] [PubMed] [Google Scholar]
- [18].Fernandez-Gonzalez R, Ramirez MA, Pericuesta E, et al. Histone modifications at the blastocyst Axin1(Fu) locus mark the heritability of in vitro culture-induced epigenetic alterations in mice. Biol Reprod. 2010;83(5):720–727. [DOI] [PubMed] [Google Scholar]
- [19].Sutcliffe AG, Peters CJ, Bowdin S, et al. Assisted reproductive therapies and imprinting disorders – a preliminary British survey. Hum Reprod. 2006;21(4):1009–1011. [DOI] [PubMed] [Google Scholar]
- [20].Wrenzycki C, Niemann H. Epigenetic reprogramming in early embryonic development: effects of in-vitro production and somatic nuclear transfer. Reprod Biomed Online. 2003;7(6):649–656. [DOI] [PubMed] [Google Scholar]
- [21].Jiang Z, Sun J, Dong H, et al. Transcriptional profiles of bovine in vivo pre-implantation development. BMC Genomics. 2014;15:756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jiang Z, Lin J, Dong H, et al. DNA methylomes of bovine gametes and in vivo produced preimplantation embryos. Biol Reprod. 2018;99(5):949–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gaspar JM. NGmerge: merging paired-end reads via novel empirically-derived models of sequencing errors. BMC Bioinformatics. 2018;19(1):536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Graf A, Krebs S, Zakhartchenko V, et al. Fine mapping of genome activation in bovine embryos by RNA sequencing. Proc Natl Acad Sci U S A. 2014;111(11):4139–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Duan JE, Jiang ZC, Alqahtani F, et al. Methylome dynamics of bovine gametes and in vivo early embryos. Front Genet. 2019;10:512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yeom YI, Fuhrmann G, Ovitt CE, et al. Germline regulatory element of Oct- 4specific for the totipotent cycle of embryonal cells. Development. 1996;122(3):881–894. [DOI] [PubMed] [Google Scholar]
- [28].Randise-Hinchliff CE, Brickner JH. A new direction for gene looping. Dev Cell. 2012;23(5):919–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zuccotti M, Piccinelli A, Giorgi Rossi P, et al. Chromatin organization during mouse oocyte growth. Mol Reprod Dev. 1995;41(4):479–485. [DOI] [PubMed] [Google Scholar]
- [30].Liu H, Aoki F. Transcriptional activity associated with meiotic competence in fully grown mouse GV oocytes. Zygote. 2002;10(4):327–332. [DOI] [PubMed] [Google Scholar]
- [31].Zuccotti M, Ponce RH, Boiani M, et al. The analysis of chromatin organisation allows selection of mouse antral oocytes competent for development to blastocyst. Zygote. 2002;10(1):73–78. [DOI] [PubMed] [Google Scholar]
- [32].De La Fuente R, Eppig JJ. Transcriptional activity of the mouse oocyte genome: companion granulosa cells modulate transcription and chromatin remodeling. Dev Biol. 2001;229(1):224–236. [DOI] [PubMed] [Google Scholar]
- [33].Luciano AM, Franciosi F, Dieci C, et al. Changes in large-scale chromatin structure and function during oogenesis: a journey in company with follicular cells. Anim Reprod Sci. 2014;149(1–2):3–10. [DOI] [PubMed] [Google Scholar]
- [34].Schierding W, Cutfield WS, O’Sullivan JM. The missing story behind genome wide association studies: single nucleotide polymorphisms in gene deserts have a story to tell. Front Genet. 2014;5:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Freedman ML, Monteiro AN, Gayther SA, et al. Principles for the post-GWAS functional characterization of cancer risk loci. Nat Genet. 2011;43(6):513–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cowper-Sal Lari R, Zhang X, Wright JB, et al. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat Genet. 2012;44(11):1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Dryden NH, Broome LR, Dudbridge F, et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Res. 2014;24(11):1854–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].de Koning AP, Gu W, Castoe TA, et al. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011;7(12):e1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Adelson DL, Raison JM, Edgar RC. Characterization and distribution of retrotransposons and simple sequence repeats in the bovine genome. Proc Natl Acad Sci U S A. 2009;106(31):12855–12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Elbarbary RA, Lucas BA, Maquat LE. Retrotransposons as regulators of gene expression. Science. 2016;351(6274):aac7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ichiyanagi K. Epigenetic regulation of transcription and possible functions of mammalian short interspersed elements, SINEs. Genes Genet Syst. 2013;88(1):19–29. [DOI] [PubMed] [Google Scholar]
- [42].Estecio MR, Gallegos J, Dekmezian M, et al. SINE retrotransposons cause epigenetic reprogramming of adjacent gene promoters. Mol Cancer Res. 2012;10(10):1332–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chow JC, Ciaudo C, Fazzari MJ, et al. LINE-1 activity in facultative heterochromatin formation during X chromosome inactivation. Cell. 2016;166(3):782. [DOI] [PubMed] [Google Scholar]
- [44].Sasaki T, Nishihara H, Hirakawa M, et al. Possible involvement of SINEs in mammalian-specific brain formation. Proc Natl Acad Sci U S A. 2008;105(11):4220–4225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Faulkner GJ, Kimura Y, Daub CO, et al. The regulated retrotransposon transcriptome of mammalian cells. Nat Genet. 2009;41(5):563–571 [DOI] [PubMed] [Google Scholar]
- [46].Sela N, Mersch B, Gal-Mark N, et al. Comparative analysis of transposed element insertion within human and mouse genomes reveals Alu’s unique role in shaping the human transcriptome. Genome Biol. 2007;8(6):R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lev-Maor G, Sorek R, Shomron N, et al. The birth of an alternatively spliced exon: 3ʹ splice-site selection in Alu exons. Science. 2003;300(5623):1288–1291. [DOI] [PubMed] [Google Scholar]
- [48].Chen LL, DeCerbo JN, Carmichael GG. Alu element-mediated gene silencing. Embo J. 2008;27(12):1694–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Elbarbary RA, Li W, Tian B, et al. STAU1 binding 3ʹ UTR IRAlus complements nuclear retention to protect cells from PKR-mediated translational shutdown. Genes Dev. 2013;27(13):1495–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Criscione SW, Zhang Y, Thompson W, et al. Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics. 2014;15:583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Peaston AE, Evsikov AV, Graber JH, et al. Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev Cell. 2004;7(4):597–606. [DOI] [PubMed] [Google Scholar]
- [52].Robbez-Masson L, Rowe HM. Retrotransposons shape species-specific embryonic stem cell gene expression. Retrovirology. 2015;12:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Jia G, Preussner J, Chen X, et al. Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement. Nat Commun. 2018;9(1):4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Corces MR, Granja JM, Shams S, et al. The chromatin accessibility landscape of primary human cancers. Science. 2018;362(6413):eaav1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jung YH, Kremsky I, Gold HB, et al. Maintenance of CTCF- and transcription factor-mediated interactions from the gametes to the early mouse embryo. Mol Cell. 2019;75(1):154–171. e155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gao Y, Cao Q, Lu L, et al. Kruppel-like factor family genes are expressed during Xenopus embryogenesis and involved in germ layer formation and body axis patterning. Dev Dyn. 2015;244(10):1328–1346. [DOI] [PubMed] [Google Scholar]
- [57].Pannese M, Polo C, Andreazzoli M, et al. The Xenopus homologue of Otx2 is a maternal homeobox gene that demarcates and specifies anterior body regions. Development. 1995;121(3):707–720. [DOI] [PubMed] [Google Scholar]
- [58].Kues WA, Sudheer S, Herrmann D, et al. Genome-wide expression profiling reveals distinct clusters of transcriptional regulation during bovine preimplantation development in vivo. Proc Natl Acad Sci U S A. 2008;105(50):19768–19773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wei Q, Zhong L, Zhang S, et al. Bovine lineage specification revealed by single-cell gene expression analysis from zygote to blastocyst. Biol Reprod. 2017;97(1):5–17. [DOI] [PubMed] [Google Scholar]
- [60].Tickle C. How the embryo makes a limb: determination, polarity and identity. J Anat. 2015;227(4):418–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Cao Z, Carey TS, Ganguly A, et al. Transcription factor AP-2gamma induces early Cdx2 expression and represses HIPPO signaling to specify the trophectoderm lineage. Development. 2015;142(9):1606–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kuckenberg P, Buhl S, Woynecki T, et al. The transcription factor TCFAP2C/AP-2gamma cooperates with CDX2 to maintain trophectoderm formation. Mol Cell Biol. 2010;30(13):3310–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kuckenberg P, Kubaczka C, Schorle H. The role of transcription factor Tcfap2c/TFAP2C in trophectoderm development. Reprod Biomed Online. 2012;25(1):12–20. [DOI] [PubMed] [Google Scholar]
- [64].Berg DK, Smith CS, Pearton DJ, et al. Trophectoderm lineage determination in cattle. Dev Cell. 2011;20(2):244–255. [DOI] [PubMed] [Google Scholar]
- [65].Aston KI, Li GP, Hicks BA, et al. Genetic reprogramming of transcription factor ap-2gamma in bovine somatic cell nuclear transfer preimplantation embryos and placentomes. Cloning Stem Cells. 2009;11(1):177–186. [DOI] [PubMed] [Google Scholar]
- [66].Pastor WA, Liu W, Chen D, et al. TFAP2C regulates transcription in human naive pluripotency by opening enhancers. Nat Cell Biol. 2018;20(5):553–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Fenouil R, Cauchy P, Koch F, et al. CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters. Genome Res. 2012;22(12):2399–2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Talukder AK, Yousef MS, Rashid MB, et al. Bovine embryo induces an anti-inflammatory response in uterine epithelial cells and immune cells in vitro: possible involvement of interferon tau as an intermediator. J Reprod Dev. 2017;63(4):425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Hinman VF, Burke RD. Embryonic neurogenesis in echinoderms. Wiley Interdiscip Rev Dev Biol. 2018;7(4):e316. [DOI] [PubMed] [Google Scholar]
- [70].Liu M, Xie S, Zhou J. Use of animal models for the imaging and quantification of angiogenesis. Exp Anim. 2018;67(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Adler R. A model of retinal cell differentiation in the chick embryo. Prog Retin Eye Res. 2000;19(5):529–557. [DOI] [PubMed] [Google Scholar]
- [72].Mendieta-Serrano MA, Schnabel D, Lomeli H, et al. Cell proliferation patterns in early zebrafish development. Anat Rec (Hoboken). 2013;296(5):759–773. [DOI] [PubMed] [Google Scholar]
- [73].Dumollard R, Duchen M, Carroll J. The role of mitochondrial function in the oocyte and embryo. Curr Top Dev Biol. 2007;77::21–49. [DOI] [PubMed] [Google Scholar]
- [74].Hall BM, Owens KM, Singh KK. Distinct functions of evolutionary conserved MSF1 and late embryogenesis abundant (LEA)-like domains in mitochondria. J Biol Chem. 2011;286(45):39141–39152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lorenzi I, Oeljeklaus S, Aich A, et al. The mitochondrial TMEM177 associates with COX20 during COX2 biogenesis. Biochim Biophys Acta Mol Cell Res. 2018;1865(2):323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sokol AM, Sztolsztener ME, Wasilewski M, et al. Mitochondrial protein translocases for survival and wellbeing. FEBS Lett. 2014;588(15):2484–2495. [DOI] [PubMed] [Google Scholar]
- [77].Shi G, McQuibban GA. The mitochondrial rhomboid protease PARL is regulated by PDK2 to integrate mitochondrial quality control and metabolism. Cell Rep. 2017;18(6):1458–1472. [DOI] [PubMed] [Google Scholar]
- [78].Kaneko KJ, DePamphilis ML. Soggy, a spermatocyte-specific gene, lies 3.8 kb upstream of and antipodal to TEAD-2, a transcription factor expressed at the beginning of mouse development. Nucleic Acids Res. 2000;28(20):3982–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.