

ABSTRACT
Coronavirus disease 2019 (COVID-2019) outbreak originating in December 2019 in Wuhan, China has emerged as a global threat to human health. The highly contagious SARS-CoV-2 infection and transmission presents a diversity of human host and increased disease risk with advancing age, highlighting the importance of in-depth understanding of its biological properties. Structural analyses have elucidated hot spots in viral binding domains, mutations, and specific proteins in the host such as the receptor angiotensin-converting enzyme 2 (ACE2) and the transmembrane protease serine 2 (TMPRSS2) to be implicated in cell entry and viral infectivity. Furthermore, epigenetic changes that regulate chromatin structure have shown a major impact in genome stabilization and maintenance of cellular homoeostasis and they have been implicated in the pathophysiology of the virus infection. Epigenetic research has revealed that global DNA methylation along with ACE2 gene methylation and post-translational histone modifications may drive differences in host tissue-, biological age- and sex-biased patterns of viral infection. Moreover, modulation of the host cells epigenetic landscape following infection represents a molecular tool used by viruses to antagonize cellular signalling as well as sensing components that regulate the induction of the host innate immune and antiviral defence programmes in order to enhance viral replication and infection efficiency. In this review, we provide an update of the main research findings at the interface of epigenetics and coronavirus infection. In particular, we highlight the epigenetic factors that interfere with viral replication and infection and may contribute to COVID-19 susceptibility, offering new ways of thinking in respect to host viral response.
KEYWORDS: COVID-19, DNA methylation, histone PTMs, virus infection, ACE2, X-linked genes
Introduction
Coronaviruses (CoV) include a major family of human and animal pathogens such as severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) strains. The outbreak of COVID-2019 emerged in Wuhan, China in December 2019 due to a highly contagious coronavirus capable of human infection and transmission, responsible for 543,902 deaths worldwide as of 8 July 2020 according to World Health Organization. The rapid spread and the host diversity of SARS-CoV-2 highlights the need for in-depth understanding of its structure and biological function. Emerging experimental evidence reveals an interplay of genetic and epigenetic alterations regulating host response.
Epigenetic changes that regulate chromatin structure have a major impact in genome stabilization and maintenance of cellular homoeostasis and they have been implicated in the pathophysiology of the virus infection.
The epigenetic regulation of gene expression relies on post-translational chemical changes that occur at the level of chromatin as well as on RNA and DNA, including primary methylation but also acetylation, phosphorylation, ubiquitination and sumoylation. This type of regulation bridges genotype and phenotype by altering the function of the gene locus without changing the underlying DNA sequence.
DNA methylation refers to the inheritable epigenetic process by which methyl groups are added to the C-5 position of the cytosine ring of DNA by DNA methyltransferases (DNMTs). Five family members have been determined including DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3 L. DNMT1 preferentially acts on hemimethylated DNA and plays an important role during the replication process. In contrast, DNMT3A and DNMT3B, function on unmethylated CpG dinucleotides and mediate de novo methylation during development [1]. DNA methylation is highly involved in cell development and ageing process, playing a key role in gene imprinting, X inactivation, and silencing of the expression of repeated elements and transposons, ensuring genomic stability [1,2].
Furthermore, the DNA in the nucleus is packed in the form of chromatin fibre wrapped around the nucleosome, the functional unit of the chromatin consisting of histone octamers (2 dimers H3-H4 and 2 dimers H2A-H2B). Post-translational modifications on the histones (PTMs) have been established to interfere with the accessibility of the chromatin and as a consequence of gene expression. The most common PTMs occur at the N-terminal part of H3, H4 and consist mainly of methylation, acetylation, and phosphorylation on different amino acid residues. Combination and synergy of these PTMs at the same and/or different nucleosomes are defining the so-called histone code and determine chromatin accessibility and transcription factor binding [3–7].
Recent studies in the field of gene regulation and RNA have revealed a role of chemical modifications occurring at RNA level, in gene transcription, translational efficiency, and RNA stability. Novel technologies in nucleic acid sequencing and mass spectrometry have enabled scientists to study with high resolution and precision these modifications [8–10]. Nowadays, more than 160 modifications have been identified in different organisms and tissues, implicated in basic molecular mechanisms such as RNA interference [11]. The emerging role of these modifications in the area of cancer, place them as ideal candidates for drug targeting and development [12,13].
Systematic mapping of RNA modifications across the transcriptome of different species and tissues by antibody pull-down or chemical labelling coupled to sequencing, nanopore sequencing, and novel targeted Mass Spec technologies have revealed a series of RNA modifications that are not only abundant in housekeeping, non-coding RNAs, including transfer RNAs (tRNA) and ribosomal RNAs (rRNA) but are also commonly found within functional mRNAs [14,15]. Surprisingly, some of the mapped modifications showed dynamic patterns and tissue-specific distribution supporting elucidation of a completely new field of RNA-epigenetics or epitranscriptomics [16].
Transcriptomic and epitranscriptomic analysis has proved of primary importance in the study of viruses by determining their architecture and function, thus providing important information in relation to host infection. Recent data from the Centre for RNA Research and their collaborators at the Korean National institute of Health (KNHI) within the Korea Centres for Disease Control and Prevention (KCDC) have delineated a high-resolution map of SARS-CoV-2 [17]. Nanopore-based direct RNA sequencing was applied to identify at least 41 RNA modifications sites on viral transcripts with the most frequently observed motif being the AAGAA present throughout the viral genome. The poly(A) tail plays an important role in RNA turnover and it has been suggested that this internal modification may contribute to viral RNA stability and translation efficiency, thus indicating a mechanism to evade host immune response [17,18].
Herein, we discuss recent evidence on the genetic and epigenetic interplay underlying SARS-CoV-2 infection in order to explain differences in tissue/cell-, age- and sex-biased patterns and comorbidities associated with viral infection.
Host genetics and SARS-CoV-2 infection
SARS-CoV-2 is a β-genus coronavirus, containing a genome of a positive-sense, single strand enveloped RNA that is 29 Kb long. It exhibits 50% homology with Middle East respiratory syndrome coronavirus (MERS-CoV) and 80% with SARS-CoV. Although originally thought as an enzootic infection of birds and mammals, cross-species transmission is possible resulting in human outbreaks.
The virus entering point at the host cell is the Angiotensin-converting Enzyme 2 (ACE2) type I membrane receptor present in arterial and venous endothelial cells, lung type II alveolar cells, enterocytes of the small intestine, and arterial smooth muscle cells in most organs, including cerebral cortex and brainstem [18]. ACE2 is a metalloenzyme containing zinc with a collectrin renal amino acid transporter domain and a peptidase M2 domain, which counterbalances ACE by cleaving the angiotensin I hormone into the vasoconstricting angiotensin II. ACE2 in turn cleaves the phenylalanine residue from the carboxyl-terminal of angiotensin II and generates the vasodilator angiotensin [19–21].
Once the virus enters the host cell, it replicates a genomic RNA that consists of nine experimental validated open reading frames (ORFs). There are nine subgenomic RNAs transcribed from the genomic RNA which encode conserved structural proteins, namely, the spike protein (S), envelope protein (E), membrane protein (M), and nucleocapsid protein (N) that synthetize the viral particle [17–19]. Data have shown that SARS-CoV proteins are cleaved by proteases such as the Transmembrane Serine Protease 2 (TMPRSS2), Cathepsin L (CTSL) and Furin, Paired Basic Amino Acid Cleaving Enzyme (FURIN) into two subunits, S1 and S2, respectively (Figure 1a,b).
Figure 1.
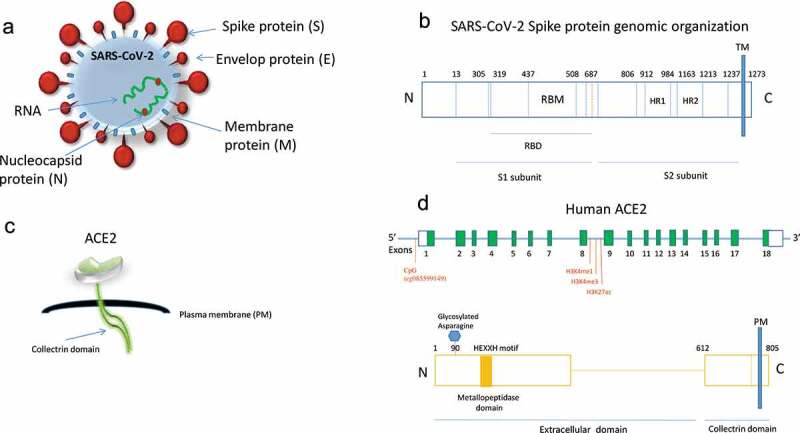
(a) Schematic representation of the coronavirus structure indicating the viral surface proteins (spike, membrane, envelop) embedded in lipid bilayer envelop, the RNA and the nucleocapsid ribonucleoproteins. (b) Genomic organization of SARS-CoV-2 spike protein indicating the receptor-binding domain (RBD), the receptor-binding motif (RBM) and heptad repeats 1, 2 (HR1, 2). The protease site (at 687) is also shown that can be cleaved to generate the S1 and S2 subunits. TM: transmembrane. (c) Schematic representation of the ACE2 protein indicating the peptidase and collectrin domains. (d) Upper panel: Linear schematic of ACE2 gene (not shown to scale) showing exons in green boxes and introns as a horizontal blue line. The location of CpG (cg085599149) near the ACE2 transcription start site that is hypomethylated during ageing [30] and the marks of histone methylation and acetylation [22] are shown. Bottom panel: ACE2 protein indicating the zinc metallopeptidase (HEXXH motif) and collectrin domains along with the key glycan (glycosylated asparagine at position 90) that is implicated in SARS-CoV-2 spike binding
SARS-CoV-2 is using the receptor-binding domain (RBD) of the spike (S1) protein that contains a core structure and a receptor-binding motif (RBM) to bind the outer surface of the receptor and enter the host [19]. Human ACE2 contains two hot spots for virus binding and mutations at the RBM occurring naturally near these hot spots, determine the host range of the virus [20] (Figure 1b,c). According to structural data analysis, SARS-CoV-2 binds with high affinity to human ACE2. A single N501 T mutation at the position 501 has been detected to significantly enhance the SARS-CoV-2 RBD binding capacity. This mutation has been frequently detected in patients and it is therefore important to closely monitor residue 501 of ACE2 for novel mutations in the future [20].
Previous studies in the related virus SARS-CoV have shown that undifferentiated epithelial cells expressing low levels of ACE2 were poorly infected, highlighting the strong correlation of ACE2 expression with the rate of coronavirus infection [21].
Furthermore, a series of comorbidities have been detected in patients who died of COVID-19 infection, including hypertension, lupus, diabetes, cancer, and chronic obstructive lung disease [22,23]. Although the molecular mechanism underlying the increased disease severity in patients with these comorbidities is not so clear, several studies speculate the implication of ACE2.
A recent integrative cross-tissue analysis demonstrated that cells positive for ACE2 co-express either the protease TMPRSS2 or CTSL in diverse organs, including epithelial cells in the liver, kidney, pancreas, and olfactory epithelium, cardiomyocytes, pericytes, and fibroblasts in the heart, and oligodendrocytes in the brain, which have been associated with severe disease. Thus, ACE2 presents the entry point for the virus and it is likely that these comorbidities influence ACE2 levels, which in turn directly affect viral transmission [22,23].
Furthermore, ACE2 has been found up-regulated in airway basal and multiciliated epithelial cells in smokers [24]. Interestingly, single-cell RNA-Seq studies have revealed that ACE2 expression is significantly lower in paediatric samples, reflecting the lack of chromatin accessibility in the ACE2 locus [24]. Furthermore, hypermethylation of ACE2 gene may occur in children lungs, reducing its expression [25].
Of note, ACE2 expression was significantly elevated in endometrial carcinoma and renal papillary cell carcinoma tissues, been positively associated with immune cell infiltration and prognosis, thus suggesting a possible susceptibility to SARS-CoV-2 infection that needs further investigation [26].
However, it should be noted that the regulation of ACE2 expression in different tissues is complex and can be affected by infection as well as by common therapies, including ACE inhibitors or angiotensin receptor blockers.
Host epigenetics and SARS-CoV-2 infection
Association of X inactivation with COVID-19
Of importance, the receptor of SARS-CoV-2, the ACE2 gene has been located on the X chromosome and points towards a closer look of the epigenetically regulated developmental process of X inactivation (XCI).
It is established that one of the two X chromosomes in mammalian female cells undergoes an inactivation (XCI) process during differentiation in order to achieve dosage compensation between males and females. The female-specific non-coding RNA Xist is transcribed from the X–inactivation centre and spreads over the entire X chromosome creating an inactive landscape through the recruitment of histone-modifying proteins, the polycomb repressive 1 (PRC1) and PRC2 complexes. PRC1 catalyzes histone H2A mono-ubiquitination on lysine 119 (H2AK119ub) while PRC2 tri-methylates histone H3 on lysine 27 (H3K27me3) and mainly maintain gene silencing during XCI process, providing an intermediate epigenetic regulation stage and propagating the silent state induced by Xist RNA [27].
However, XCI is incomplete in humans with one-third of X-chromosomal genes been expressed from both active (Xa) and inactive (Xi) X chromosomes in female cells with the genes that escape X inactivation been called ‘escapers’ [27,28].
A recent study mapped the X chromosome inactivation landscape across several human tissues [28]. They systematically analysed 5,500 transcriptomes spanning 29 tissues from 449 individuals of the Genotype Tissue Expression (GTEx) project (v6p release). By investigating male-female differences in the expression of X-chromosomal protein-coding and long non-coding RNA (lncRNA) genes and single-cell transcriptomes, along with genomics data, it was detected that although XCI is mainly uniform across human tissues, some heterogeneity can also occur between cells, tissues, and individuals. Their data showed that incomplete XCI regulated at least 23% of X-chromosomal genes and 7 escaper genes. Of importance, escape from XCI was shown to result in sex biases in gene expression and thus in phenotypic diversity [29]. ACE2 was found among the genes that escape X inactivation in female lung tissue, showing a more heterogeneous sex bias. Moreover, ACE2 expression was slightly higher in male than in female tissues and open chromatin marks were found associated with its expression profile, possibly explaining the evidence of higher respiratory infection severity in male than in female patients [29].
The ACE2 gene was shown located in the evolutionary older region of the chromosome, the Xq and the genes at this region exhibit lower expression and higher tissue specificity features while been associated with higher protein evolutionary rates [29].
In this regard, an evolutionary study compared the ACE2 proteins from humans, rats, and mice and detected deferential SARS-CoV binding affinities. Human SARS-CoV S1-CTD was shown to bind tightly to human ACE2 through two virus-binding hot spots at the residues Lys31 and Lys353. Rat proteins, on the other hand, contained two different residues, a His at the position 353 (instead of Lys) and an Asn at the position 82 which introduces an N-linked glycan and disfavours SARS-CoV binding. Mouse proteins have the His353 residue but do not possess the N-linked glycan at the position 82 and present poor receptors. Thus, rat ACE2 is not a receptor for SARS-CoV while mouse ACE2 is a poor receptor compared to human ACE2 [30].
DNA methylation and histone modifications on ACE2 gene locus – Mechanisms to subvert host immune responses
Research studies have recently implicated DNA methylation in the regulation of the ACE2 gene, indicating that the host epigenome may represent a risk factor for COVID-19 infection (Table 1). By analysing DNA methylation profiles from four different public databases for lung tissues, they were able to screen for differences between genders in DNA methylation at two CpGs sites of the ACE2 gene. Moreover, age-dependent DNA methylation was detected close to the transcription start site of the ACE2 gene in airway epithelial cells. Scientists have used Illumina DNA methylation array data from samples of different biological ages and detected that one CpG (cg085599149) near the ACE2 transcription start site showed decreased methylation level during ageing (Figure 1). Whole genome bisulphite sequencing is therefore highly demanded to map with precision more differential methylated CpG islands regulating ACE2 expression in an age-related manner [31].
Table 1.
Epigenetic regulation of human ACE2 gene in different cell types/tissues
| Cell type/Tissue | Epigenetic mark | Effect on ACE expression | Reference |
|---|---|---|---|
| Airway epithelial cells | Decreased DNA methylation of CpG (cg085599149) near the ACE2 transcription start site (TSS200 region) | Increased ACE2 expression | [31,32] |
| Lung tissue/Elder/Smoker | Hypomethylation of ACE2 gene | Increased ACE2 expression | [24] |
| Lung tissue (E096) | Histone modifications (H3K4me1, H3K4me3, H3K27Ac) in ACE2 gene | Increased ACE2 expression | [22] |
| Lung tissue (E096) | Upregulation of NAD-dependent histone deacetylase Sirtuin 1 (SIRT1) | Increased ACE2 expression | [22] |
| Lung/Children | Hypermethylation of ACE2 gene | Low ACE2 expression | [24,25] |
| Lung tissue/Systemic Lupus Erythematosus (SLE) | Decreased DNA methylation of ACE2 promoter (hypomethylation) | Increased ACE2 expression | [23] |
| Uterine Corpus Endometrial carcinoma | Decreased DNA methylation of ACE2 promoter (hypomethylation) | Increased ACE2 expression | [26] |
| Breast carcinoma | Increased KDM5B histone demethylase | Increased ACE2 expression | [22] |
| Kidney Renal Papillary Cell Carcinoma | Decreased DNA methylation of ACE2 promoter (hypomethylation) | Increased ACE2 expression | [26] |
Previous studies had reported that global DNA methylation levels are reduced during ageing, leading to differential methylation patterns of genes related to inflammation, ageing and immune response. In combination with these observations, it was revealed that DNA methylation is at least one of the main mechanisms highjacked by MERS-CoV to antagonize antigen presentation genes in infected cells in vitro and modulate host adaptive immune response. These data could potentially explain the higher morbidity of the virus infection in older individuals and in patients with underlying medical conditions [31,32]. Furthermore, ACE2 promoter has been found hypomethylated in endometrial carcinoma and renal papillary cell carcinoma tissues which present increased ACE2 expression suggesting that it may activate itself and induce its expression levels [26]. When these patients were affected by SARS-CoV-2, ACE2 levels were reduced and immune infiltration was impaired affecting their prognosis.
The higher expression of ACE2 in the lungs of patients with comorbidities has been correlated with severe COVID-19. Systematic experimental and network analyses revealed many potential regulators of ACE2 in the human lung, including genes that are expressing epigenetic enzymes such as histone acetyltransferase 1 (HAT1), histone deacetylase 2 (HDAC2) and lysine demethylase 5B (KDM5B). Pathway enrichment analysis demonstrated that genes that were positively associated with ACE2 were regulated by KDM5B, and by specific histone methylation (H3K4me1 and H3K4me3) and histone acetylation (H3K27ac) marks (Figure 1). In fact, KDM5B is regulating chromatin accessibility by removing active chromatin marks such as di- and trimethylation from the lysine 4 of histone H3 (H3K4) involved in the regulation of gene transcription and DNA repair. These modifications were further identified in ACE2 locus, suggesting the epigenetic regulation of ACE2 in the lung that drives ACE2 overexpression and disease severity [22]. Furthermore, KDM5B inhibition in breast cancer cells was shown to trigger an interferon response leading to resistance to infection by DNA and RNA viruses, thus suggesting that KDM5B may present a potential target for COVID-19 prevention [22].
Another epigenetic enzyme, the NAD-dependent histone deacetylase Sirtuin 1 (SIRT1) that can also regulate ACE2 under conditions of cell energy stress has been found upregulated in the lung of patients with severe COVID-19 comorbidities and it is worthy to be investigated further [22].
Targeting SARS-CoV-2 infection
Apart from ACE2, the transmembrane protease TMPRSS2 is also a critical regulator of the infective potential of SARS-CoV-2. Upon infection, SARS-CoV-2 has been detected to bind to ACE2 via the Spike proteins (S) on its surface. The S protein is primed by the cellular membrane serine protease TMPRSS2 which allows its cleavage at the S1/S2 while the S2 site allows the fusion of viral and cellular membranes [17–20].
Aside, from neutralizing antibodies raised against the Spike protein of the virus, a series of chemical compounds are being tested in order to block the activity and the binding capacity of TMPRSS2 and ACE2 proteins. A clinically approved serine protease inhibitor camostat mesylate has been revealed to inhibit TMPRSS2 activity and block SARS-2-S driven entry suggesting its potential as an efficient infection treatment [33].
Up to date, very few studies have elucidated the importance of drugs targeting the ACE2 gene expression. A recent study, screening the two databases of genome-wide transcriptional expression data, the Connectivity Map (CMap), and the JeaMoon Map (JMap) detected a number of candidate agents (Pirinixic Acid, Fulvestrant, Staurosporine, AG-013608, and Azathioprine) that decrease ACE2 expression in respiratory tract epithelium with the immunosuppressant azathioprine being the most promising [34].
Furthermore, an important role for epigenetic and epitranscriptomic mechanisms was proposed in regulating the ACE2 X-linked gene expression in the context of COVID-19. DNA methylation and H3K4 methylation at the promoter of the gene have been found to influence its expression profile. Moreover, analysis of the Roadmap Epigenomics Project database revealed that the p300 acetyltransferase and H3K27ac were found located at the promoter region of the ACE2 gene.
The highly homologous HATs, cyclic AMP (cAMP) response element-binding protein (CREB) binding protein (CBP) and p300 are well-established markers of active enhancers. It has been shown that inhibition of the p300 HAT activity reduces the levels of H3K27Ac, mainly at the enhancer regions. A series of small molecules, natural products have been detected to bind the catalytic active site of p300, influencing gene expression on target genes [35–37].
In a recent study, a Mass Spec-based assay was used to identify novel interaction partners of 26 SARS-CoV-2 proteins in human cells (PPIs) [35]. Surprisingly, they observed that the transmembrane E protein of the virus interacts with Bromodomain Containing 2 (BRD2) and BRD4 transcription factors from the host cell. The family of bromodomain (BRD) and bromodomain and extra terminal domain family (BET) are epigenetic readers that recognize acetylated chromatin. It consists of four members (BRD2, BRD3, BRD4, and BRDT) with BRD2 and BRD4 revealed to bind acetylated histones at the N-terminal part and regulate the transcription of target genes. Upon viral infection, they are interacting with the C-terminal of the protein E that mimics the N-part of H3 histone tail. Treatment with BET inhibitors is suggested to interfere with this interaction and affect the host cell transcriptional machinery along with that of the virus [38].
Some natural compounds that may induce epigenetic silencing of ACE2 gene include the DNA methyltransferase inhibitor curcumin, 8-hydroxyquinolones and sulforaphane, exhibiting a preventive potential towards SARS-CoV-2 infection [39].
Conclusion
Taken altogether, an emerging role of epigenetic enzymes and modifications is revealed along with their potential activities in COVID-19 treatment. Future studies should focus on the elucidation of the functional role of these chemical modifications in viral life, infection efficiency, and severity of the disease as well as towards the ability of the virus to avoid the attack from the host cell. Candidate drugs against chromatin accessibility, RNA and DNA modifications, may prove of great help in the future for blocking the infection activity of the virus and novel studies are highly demanded to elucidate their specificity and mechanisms of action.
Funding Statement
There is no funding associated with this manuscript.
Authors’ contributions
SC performed the literature survey and wrote the first draft of the manuscript. AGP revised and edited the manuscript. CP expanded the literature search, revised and wrote the final manuscript. All authors read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Jin B, Li Y, Robertson KD.. DNA methylation: superior or subordinate in the epigenetics hierarchy. Genes Cancer. 2011;2(6):607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Batham J, Lim PS, Rao S.. How to silence an X chromosome. Nature. 2020;578(7795):365–366. [DOI] [PubMed] [Google Scholar]
- [3].Orphanides G, Reinberg D. A unified theory of gene expression. Cell. 2002;108:439–451. [DOI] [PubMed] [Google Scholar]
- [4].Bannister A, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA. 1964;51:786–794.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Luger K, Mader AW, Richmond RK, et al. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. [DOI] [PubMed] [Google Scholar]
- [7].Xhemalce B, Dawson MA, Bannister AJ. Histone modifications. In: Meyers R, editor. Encyclopedia of molecular cell biology and molecular medicine. John Wiley and Sons; 2011. [Google Scholar]
- [8].Shubert C. Epitranscriptomics: RNA revisited. Science. 2019;364:693–695. [Google Scholar]
- [9].Roundtree IA, Evans ME, Pan T, et al. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169:1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li S, Mason CE. The pivotal regulatory landscape of RNA modifications. Annu Rev Genomics Hum Genet. 2014;15:127–150. [DOI] [PubMed] [Google Scholar]
- [11].Shelton SB, Reinsborough C, Xhemalce B. Who watches the watchmen: roles of RNA modifications in the RNA interference pathway. PLoS Genet. 2016;12:e1006139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Esteller M, Pandolfi PP. The epitranscriptome of noncoding RNAs in cancer. Cancer Discov. 2017;7:359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Barbieri I, Kouzarides T. Role of RNA modifications in cancer. Nat Rev Cancer. 2020;20:303–322. [DOI] [PubMed] [Google Scholar]
- [14].He S, Wurtzel O, Singh K, et al. Validation of two ribosomal RNA removal methods for microbial metatranscriptomics. Nat Methods. 2010;7:807–812. [DOI] [PubMed] [Google Scholar]
- [15].Boccaletto P, Machnicka MA, Purta E, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46(D1):D303‐D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Saletore Y, Meyer K, Korlach J, et al. The birth of the epitranscriptome: deciphering the function of RNA modifications. Genome Biol. 2012;13(10):175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kim D, Lee JY, Yang JS, et al. The architecture of SARS-CoV-2 transcriptome. Cell. 2020;181(4):914–921.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kim JM, Chung YS, Jo HJ, et al. Identification of coronavirus isolated from a patient in Korea with COVID-19. Osong Public Health Res Perspect. 2020;11:3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Verdecchia P, Cavallini C, Spanevello A, et al. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. 2020;S0953–6205(20):30151–30155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wan Y, Shang J, Graham R, et al. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. 2020;94(7):e00127–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jia HP, Look DC, Shi L, et al. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J Virol. 2005;79(23):14614–14621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pinto BGG, Oliveira AER, Singh Y, et al. ACE2 expression is increased in the lungs of patients with comorbidities associated with severe COVID-19. J Infect Dis. 2020;jiaa332. DOI: 10.1093/infdis/jiaa332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sawalha AH, Zhao M, Coit P, et al. Epigenetic dysregulation of ACE2 and interferon-regulated genes might suggest increased COVID-19 susceptibility and severity in lupus patients. Clin Immunol. 2020;215:108410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Muus C, Luecken MD, Eraslan G, et al. Integrated analyses of single-cell atlases reveal age, gender, and smoking status associations with cell type-specific expression of mediators of SARS-CoV-2 viral entry and highlights inflammatory programs in putative target cells. bioRxiv. 2020. DOI: 10.1101/2020.04.19.049254. [DOI] [Google Scholar]
- [25].Holmes L Jr, Lim A, Comeaux CR, et al. DNA methylation of candidate genes (ACE II, IFN-γ, AGTR 1, CKG, ADD1, SCNN1B and TLR2) in essential hypertension: a systematic review and quantitative evidence synthesis. Int J Environ Res Public Health. 2019. December 1;16(23):4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yang J, Li H, Hu S, et al. ACE2 correlated with immune infiltration serves as a prognostic biomarker in endometrial carcinoma and renal papillary cell carcinoma: implication for COVID-19. Aging (Albany NY). 2020;12(8):6518‐6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Galupa R, Heard E. X-chromosome inactivation: a crossroads between chromosome architecture and gene regulation. Annu Rev Genet. 2018;52:535‐566. [DOI] [PubMed] [Google Scholar]
- [28].Żylicz JJ, Bousard A, Žumer K, et al. The implication of early chromatin changes in X chromosome inactivation. Cell. 2019;176(1–2):182‐197.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tukiainen T, Villani AC, Yen A, et al. Landscape of X chromosome inactivation across human tissues. Nature. 2017;550(7675):244‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol. 2016;3(1):237‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Corley MJ, Ndhlovu LC. DNA methylation analysis of the COVID-19 host cell receptor, angiotensin i converting enzyme 2 gene (ACE2) in the respiratory system reveal age and gender differences. Preprints. 2020;2020030295. DOI: 10.20944/preprints202003.0295.v1 [DOI] [Google Scholar]
- [32].Menachery VD, Schäfer A, Burnum-Johnson KE, et al. MERS-CoV and H5N1 influenza virus antagonize antigen presentation by altering the epigenetic landscape. Proc Natl Acad Sci U S A. 2018;115(5):E1012‐E1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271‐280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cui Q, Huang C, Ji X, et al. Possible inhibitors of ACE2, the receptor of 2019-nCoV. Preprints. 2020;2020020047. DOI: 10.20944/preprints202002.0047.v1 [DOI] [Google Scholar]
- [35].Raisner R, Kharbanda S, Jin L, et al. Enhancer activity requires CBP/P300 bromodomain-dependent histone H3K27 acetylation. Cell Rep. 2018;24(7):1722–1729. [DOI] [PubMed] [Google Scholar]
- [36].Lasko LM, Jakob CG, Edalji RP, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2017;550:128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Karamouzis MV, Konstantinopoulos PA, Papavassiliou AG. Roles of CREB-binding protein (CBP)/p300 in respiratory epithelium tumorigenesis. Cell Res. 2007;17(4):324–332. [DOI] [PubMed] [Google Scholar]
- [38].Gordon DE, Jang GM, Bouhaddou M, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pruimboom L. Methylation pathways and SARS-CoV-2 lung infiltration and cell membrane-virus fusion are both subject to epigenetics. Front Cell Infect Microbiol. 2020. May 26;10:290. [DOI] [PMC free article] [PubMed] [Google Scholar]