

Abstract

A new class of pyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazoles was synthesized for the treatment of hyperproliferative pathologies, including neoplasms. The new compounds were screened in the 60 human cancer cell lines of the NCI drug screen and showed potent activity with GI50 values reaching the nanomolar level, with mean graph midpoints of 0.08–0.41 μM. All compounds were further tested on six lymphoma cell lines, and eight showed potent growth inhibitory effects with IC50 values lower than 500 nM. Mechanism of action studies showed the ability of the new [1,2]oxazoles to arrest cells in the G2/M phase in a concentration dependent manner and to induce apoptosis through the mitochondrial pathway. The most active compounds inhibited tubulin polymerization, with IC50 values of 1.9–8.2 μM, and appeared to bind to the colchicine site. The G2/M arrest was accompanied by apoptosis, mitochondrial depolarization, generation of reactive oxygen species, and PARP cleavage.
Introduction
Microtubules are intracellular polymers involved in the regulation of a large number of cellular processes, including proliferation, division, determination and maintenance of cellular shape, motility, and intracellular transport.1 They are highly dynamic structures composed of multiple heterodimers of α- and β-tubulin, and they undergo alternating polymerization and depolymerization phases.2 Disrupting this dynamic equilibrium interferes with cell division and leads to cell death. Tubulin and its associated structures represent an attractive target in the treatment of cancer.3 Over the past 40 years, a large number of natural and synthetic compounds interfering with microtubule dynamics through interactions with multiple binding sites on tubulin have been described.4 On the basis of their effects on microtubule dynamics, they can be classified as either microtubule-stabilizing agents or microtubule-destabilizing agents.5 Among natural derivatives, taxanes, e.g., paclitaxel and docetaxel, belong to the first group of compounds, while vinca alkaloids, e.g., vinflunine, vinorelbine, vincristine, and colchicine, belong to the second group.6 Despite the large number of new promising drug candidates, no molecules binding at the colchicine site have been approved thus far for the treatment of cancer, leaving the drug discovery process still open.7,8 Since its discovery, combretastatin A-4 (CA-4) is still considered a promising lead compound binding at the colchicine site (Chart 1).9 It inhibits tubulin polymerization with low IC50 values,10 and it presents a potent activity against multiple cancer cell lines, including cells bearing a multidrug resistance (MDR) phenotype.11 In addition to its antimitotic activity, CA-4 can interfere with tumor vasculature, essential for solid tumor survival, leading to necrosis of tumor tissues.12 Nevertheless, due to its poor water solubility, low bioavailability, and rapid clearance, CA-4 exhibits poor activity in vivo, thus leading to the synthesis of different water-soluble prodrugs including CA-4 phosphate disodium (CA-4P) (Chart 1). In different preclinical models, CA-4P reduces blood flow and causes tumor cell death due to changes in the morphology of immature endothelial cells resulting from interference with tubulin polymerization.13 As the cis-configuration of the olefinic double bond is essential for the antiproliferative activity of CA-4, this bond has been fixed through its incorporation into five- or six-membered heterocycle rings.12,14,15 The 4,5-diarylisoxazoles showed potent antitumor activity in inducing cell cycle arrest at the G2/M phase of the cell cycle and potent antitubulin activity (Chart 1).16,17 KRIBB3 (Chart 1), belonging to the same class, displayed antiproliferative activity through inhibition of microtubule polymerization and spindle assembly checkpoint activation. In in vivo models, KRIBB3 caused a 50–70% reduction of tumor growth at a dose of 50–100 mg/kg.18,19
Chart 1. Structures of Tubulin Polymerization Inhibitors.

Isoxazoles or [1,2]oxazoles represent the core structure of many drug candidates. Due to its ability to form multiple noncovalent interactions with a wide number of proteins, this moiety confers different biological activities, such as antitumor, antinflammatory, antidepressant, antiviral, antibacterial, and antitubercolosis activities.20−27 A series of 5-(1H-indol-5-yl)-3-phenylisoxazoles have anticancer activity.28 Several small molecules containing the indole moiety have also been described as potent tubulin polymerization inhibitors.29−32
Our research group has devoted much effort to the synthesis and evaluation of the biological properties of fused tricyclic systems incorporating the pyrrole ring.33−41 Since the [1,2]oxazole system is found as a pharmacophore moiety of several compounds with promising antitumor properties, we started a program investigating different classes of pyrazole- and pyrrole-fused systems of types 1, 2, and 4, incorporating the [1,2]oxazole unit (Figure 1).42−45 In particular, from the class of [1,2]oxazole of type 2, ethyl 8-(3,5-dimethoxybenzyl)-5,8-dihydro-4H-[1,2]oxazolo[4,5-g]indole-7-carboxylate 3 emerged for its in vitro nanomolar growth inhibitory effects across the National Cancer Institute (NCI) cancer cell line panel, with mean graph midpoints (MG_MIDs) of 0.25 μM on the full panel and a GI50 range of 0.03–31.1 μM.
Figure 1.

Structures of [1,2]oxazolo[5,4-e]indazoles (1), [1,2]oxazolo[4,5-g]indoles (2), ethyl 8-(3,5-dimethoxybenzyl)-5,8-dihydro-4H-[1,2]oxazolo[4,5-g]indole-7-carboxylate (3), [1,2]oxazolo[5,4-e]isoindoles (4), and pyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazoles (5).
The potent antitumor activity made the class of compounds worth further evaluation, encouraging the synthesis of new [1,2]oxazolo derivatives with the aim of obtaining more potent antiproliferative agents. For a better insight into the structure–activity relationship (SAR) of the tricyclic scaffold, containing also the pyrrole moiety, we started a drug discovery program aimed at understanding the optimal structural requirements of this class of small molecules. Thus, we first identified [1,2]oxazolo[5,4-e]isoindole system 4, which highlighted the potential of this group of compounds as tubulin polymerization inhibitors.44 This class of compounds also displayed potent growth inhibitory activity on the NCI panel (GI50 = 0.01–27.00 μM).44 Moreover, some derivatives significantly impaired the growth of human cancer cell lines of different histological origin, including experimental models of diffuse malignant peritoneal mesothelioma (DMPM), without interfering with normal cell proliferation. Their antiproliferative activity was found to derive from their ability to impair microtubule assembly during mitosis, with a consequent cell cycle arrest at the G2/M phase and induction of caspase-dependent apoptosis. In addition, selected derivatives, at well-tolerated doses, significantly reduced tumor volume in a DMPM xenograft model.44,45
Bearing in mind the polycyclic structure of colchicine, which includes two cyclohepta rings, and that this structural feature is recurrent in other examples reported in the literature46,47 as potent inhibitors of tubulin assembly, we planned the expansion of the cyclohexyl central ring by one member while maintaining the [1,2]oxazole and pyrrole moieties. Thus, the new tricyclic derivatives pyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazoles 5 (Figure 1) were synthesized in order to investigate the effects of this structural modification on the biological properties of these compounds. This ring system was unexplored so far as a chemical entity, and because of the close correlation with the parent structure of type 2, in this set of derivatives we decided to retain some structural features, specifically the carboxyester and methoxy-substituted benzyl groups, that had emerged as crucial for biological activity.
Chemistry
The synthetic strategy optimized by us to obtain the title ring system is outlined in Scheme 1. We started from cyclohepta[b]pyrrol-8-one ketones of type 6–8,48,49 as the α position to the carbonyl is appropriate for the introduction of the second electrophilic site, essential for the subsequent cyclization with dinucleophiles.
Scheme 1. Synthesis of Pyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazoles 51–76.

Reagents and conditions: (a) NaOH, ethanol, reflux, 3 h, 80%; (b) NaH, DMF, 0 °C to rt, 1.5 h, then alkyl or aralkyl halide, 0 °C to rt, 2–24 h, 80–96%; (c) DMFDMA (1:1), DMF, MW (PW = 50 W,T = 100 °C), 40 min to 1.50 h, 70–87% (method A), or TBDMAM (1:1.5), DMF, MW (PW = 50 W, T = 100 °C), 20–40 min, 92–99% (method B), or DMFDMA (1:10), DMF, MW (PW = 150 W, T = 130 °C), 40 min to 2 h, 86–96% (method C); (d) t-BuOK, toluene, 0 °C to rt, 2 h, then HCOOEt, rt, 4 h, 60–90%; (e) NH2OH·HCl, ethanol, reflux, 50 min, 60–90%; (f) N-chlorosuccinimide, DMF, rt, 50–75%.
Derivatives 7 and 8 were subjected to nitrogen functionalization with alkyl or aralkyl halides in N,N-dimethylformamide (DMF) and NaH, giving compounds 9–13 and 17–21 in 84–96% yields48,49 and 14–16 and 22–24 in 80–90% yields. Enaminoketones 25–33, useful key intermediates for the final cyclization of the [1,2]oxazole ring, were obtained by reaction of ketones 6 and 9–16 with an excess of N,N-dimethylformamide dimethyl acetal (DMFDMA) using DMF as solvent under microwave (MW) conditions (method C, 86–96%). Derivatives 34–41 can be obtained by reaction of ketones 17–24 with stoichiometric amounts of DMFDMA (method A, 70–87%) or 1.5 equiv of tert-butoxy bis(dimethylamino)methane (TBDMAM) (method B, 92–99%) in DMF. Reaction of N-benzyl derivative 18, belonging to the ethoxycarbonyl series, with an excess of DMFDMA led to the isolation of enaminoketone 42 (96%), in which a transesterification reaction was also observed, yielding the 2-carboxymethyl derivative.48 Alternatively, we explored a formylation step from ketones 17–24, using ethyl formate and potassium tert-butoxide, leading to the corresponding hydroxymethyl derivatives 43–50 (60–90%).
Reaction of intermediates 25–42 and 43–50 with hydroxylamine hydrochloride, as a 1,3-dinucleophile, and a stoichiometric amount of acetic acid in refluxing ethanol furnished [1,2]oxazole derivatives 51–68 in 60–90% yields (Table 1). Selected [1,2]oxazoles were then subjected to smooth chlorination with N-chlorosuccinimide to afford the corresponding chloro[1,2]oxazoles. In particular, derivatives 69–75, belonging to the ethoxycarbonyl series, were obtained in good yield (60–75%). For [1,2]oxazoles 51–59, which bear two pyrrole positions for potential chlorination, a mixture of the 7- and 8-halo substituted derivatives was detected by NMR analysis, and it was not possible to isolate either component as a pure compound. Only in the case of the N-methyl derivative 52 was the 8-chloro substituted derivative 76 (50%) recovered as a pure compound from the reaction mixture.
Table 1. Pyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazoles 51–76.
| [1,2]oxazole | R | R1 | R2 | yielda (%) |
|---|---|---|---|---|
| 51 | SO2Ph | H | H | 60 |
| 52 | Me | H | H | 75 |
| 53 | Bn | H | H | 83 |
| 54 | 2-OMeBn | H | H | 70 |
| 55 | 3-OMeBn | H | H | 83 |
| 56 | 4-OMeBn | H | H | 68 |
| 57 | 2,5-(OMe)2Bn | H | H | 78 |
| 58 | 3,5-(OMe)2Bn | H | H | 76 |
| 59 | 3,4,5-(OMe)3Bn | H | H | 74 |
| 60 | Me | COOEt | H | 90b,86c |
| 61 | Bn | COOEt | H | 70b,68c |
| 62 | 2-OMeBn | COOEt | H | 74,b 70c |
| 63 | 3-OMeBn | COOEt | H | 82,b 80c |
| 64 | 4-OMeBn | COOEt | H | 84,b 82c |
| 65 | 2,5-(OMe)2Bn | COOEt | H | 74,b 72c |
| 66 | 3,5-(OMe)2Bn | COOEt | H | 82,b 80c |
| 67 | 3,4,5-(OMe)3Bn | COOEt | H | 76,b 75c |
| 68 | Bn | COOMe | H | 78 |
| 69 | Me | COOEt | Cl | 60 |
| 70 | Bn | COOEt | Cl | 75 |
| 71 | 2-OMeBn | COOEt | Cl | 74 |
| 72 | 3-OMeBn | COOEt | Cl | 65 |
| 73 | 4-OMeBn | COOEt | Cl | 70 |
| 74 | 2,5-(OMe)2Bn | COOEt | Cl | 68 |
| 75 | 3,5-(OMe)2Bn | COOEt | Cl | 70 |
| 76 | Me | Cl | H | 50 |
Values represent the yield obtained at the final reaction step.
These yields were obtained starting from (dimethylamino)methylidene ketones 34–41.
These yields were obtained starting from hydroxymethylidene ketones 43–50.
Results and Discussion
Antiproliferative Activity in the NCI Panel
All the synthesized compounds 51–76 were tested at a 10–5 M concentration for their antitumor activity on the full NCI-60 panel comprising cancer cell lines derived from nine human cancer cell types (leukemia, non-small-cell lung, colon, central nervous system, melanoma, ovarian, renal, prostate, and breast).50 On the basis of these results, six compounds (62, 63, 66, 67, 70, 75) were selected for further screening on the same panel at five concentrations at 10-fold dilutions (10–4–10–8 M). Almost all compounds showed antiproliferative activity against all tested human tumor cell lines, with nM to μM GI50 values (Table 2).
Table 2. Overview of the Results of the NCI in Vitro Human Tumor Cell Line Screening for Derivatives 62, 63, 66, 67, 70, 75.
| compd | Na | Nb | GI50c | MG_MIDd |
|---|---|---|---|---|
| 62 | 56 | 50 | 0.30–46.2 | 4.47 |
| 63 | 56 | 56 | 0.15–18.7 | 1.45 |
| 66 | 56 | 55 | 0.01–13.4 | 0.08 |
| 67 | 55 | 55 | 0.01–64.9 | 0.20 |
| 70 | 55 | 46 | 1.29–5.89 | 7.08 |
| 75 | 57 | 57 | 0.03–27.0 | 0.41 |
Number of cell lines investigated.
Number of cell lines giving positive GI50 values.
GI50 = concentration that inhibits 50% net cell growth (μM).
MG_MID = mean graph midpoint (μM); the arithmetic mean value for all tested cancer cell lines. If the indicated effect was not attainable under the concentration range used, the highest tested concentration was used for the calculation.
From a SAR point of view, the presence of an ethoxycarbonyl group at position 8 was crucial for activity. The most potent compound was 66, which has a 3,5-dimethoxybenzyl substitutent at the pyrrole nitrogen and a mean graph midpoint (MG_MID) of 0.08 μM on the full NCI panel. From analysis of the GI50 values listed in Table 3, 66 was particularly effective against the melanoma (GI50 = 0.09–0.01 μM), prostate (GI50 = 0.04 μM), and renal (GI50 = 0.07–0.02 μM) cancer subpanels (Figures S1 and S2), maintaining nanomolar activity against all the tested cell lines. The calculated MG_MID value for each subpanel was 0.04 μM, much lower than the overall cell line MG_MID value. Notably, the best activity was observed for the MDA-MB-435 cell line of the melanoma subpanel, with a GI50 of 10 nM. Moreover, the colon and CNS cancers had mean values of 0.06 μM, again lower than the average mean value.
Table 3. In Vitro GI50 (μM) Values of Compounds 62, 63, 66, 67, 70, and 75 in Individual Tumor Cell Lines.
| cell line | 62 | 63 | 66 | 67 | 70 | 75 | cell line | 62 | 63 | 66 | 67 | 70 | 75 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Leukemia | M14 | 3.67 | 0.54 | 0.05 | 0.15 | 3.78 | 0.26 | ||||||
| CCRF-CEM | 2.87 | 2.17 | 0.05 | 0.30 | 2.21 | 0.25 | MDA-MB-435 | 0.35 | 0.15 | 0.01 | 0.02 | 1.40 | 0.03 |
| HL-60(TB) | 3.04 | 0.53 | 0.03 | 0.19 | 3.32 | 0.31 | SK-MEL-2 | - | 0.56 | 0.04 | - | - | 0.22 |
| K-562 | 1.59 | 0.39 | 0.04 | 0.13 | 3.68 | 0.29 | SK-MEL-28 | >100 | 5.12 | 0.09 | 0.30 | - | 0.66 |
| MOLT-4 | 5.68 | 2.30 | 0.26 | 0.39 | 3.58 | 0.58 | SK-MEL-5 | 2.61 | 0.75 | 0.04 | 0.17 | 3.53 | 0.49 |
| RPMI-8226 | 4.11 | 1.89 | 0.05 | 0.38 | 3.27 | 0.39 | UACC-257 | - | 7.61 | - | - | - | - |
| SR | 0.43 | 0.28 | 0.03 | 0.04 | 1.80 | 0.07 | UACC-62 | 3.37 | 0.51 | 0.07 | 0.05 | 4.01 | 0.11 |
| Non-Small-Cell Lung Cancer | Ovarian Cancer | ||||||||||||
| A549/ATCC | - | 0.76 | 0.06 | - | - | - | IGROV1 | 1.99 | 1.16 | 0.06 | 0.06 | 4.39 | 0.34 |
| EKVX | - | - | - | - | - | - | OVCAR-3 | 0.87 | 0.30 | 0.02 | 0.03 | 3.13 | 0.19 |
| HOP-62 | 1.89 | 4.34 | - | 0.17 | 4.35 | 0.29 | OVCAR-4 | 9.35 | 18.7 | 13.4 | 0.72 | - | 0.85 |
| HOP-92 | 0.30 | 4.21 | - | - | - | 0.46 | OVCAR-5 | >100 | 5.94 | 0.18 | 0.53 | >100 | - |
| NCI-H226 | >100 | 21.0 | 7.98 | 1.21 | >100 | 9.71 | OVCAR-8 | 3.85 | 3.13 | 0.08 | 0.24 | - | 0.41 |
| NCI-H23 | 8.52 | - | - | 0.37 | - | 0.68 | NCI/ADR-RES | 1.36 | 0.43 | 0.03 | 0.06 | 2.83 | 0.20 |
| NCI-H322M | 5.23 | - | - | 0.32 | >100 | 0.51 | SK-OV-3 | 2.70 | 2.83 | 0.05 | 0.13 | >100 | 0.24 |
| NCI-H460 | 3.88 | 3.53 | 0.04 | 0.23 | 3.62 | 0.35 | Renal Cancer | ||||||
| NCI-H522 | 0.35 | 0.23 | 0.02 | 0.01 | 1.29 | 0.03 | 786–0 | >100 | 6.74 | 0.05 | 0.97 | - | 4.24 |
| Colon Cancer | A498 | 3.46 | 0.44 | 0.02 | 0.11 | 4.38 | 0.17 | ||||||
| COLO 205 | 0.87 | 0.58 | 0.03 | 0.04 | 3.53 | 0.17 | ACHN | 16.6 | 3.40 | 0.06 | 0.29 | 5.89 | 0.94 |
| HCC-2998 | >100 | 4.18 | 0.23 | 0.30 | - | - | CAKI-1 | 3.51 | 2.31 | 0.05 | 0.07 | 3.51 | 0.28 |
| HCT-116 | 3.79 | 0.47 | 0.04 | 0.19 | 4.02 | 0.43 | RXF 393 | 2.08 | 0.67 | 0.02 | 0.12 | - | 0.25 |
| HCT-15 | 2.24 | 0.45 | 0.04 | 0.16 | 3.39 | 0.37 | SN12C | >100 | 3.51 | 0.07 | 0.76 | - | 0.83 |
| HT29 | 0.91 | 0.36 | 0.03 | 0.07 | 2.95 | 0.27 | TK-10 | 67.8 | 11.2 | >100 | 64.9 | >100 | 10.3 |
| KM12 | 2.10 | 0.46 | 0.03 | 0.05 | 3.70 | 0.32 | UO-31 | 6.53 | 3.37 | 0.05 | 0.08 | - | 0.78 |
| SW-620 | 2.02 | 0.48 | 0.04 | 0.14 | 3.81 | 0.32 | Prostate Cancer | ||||||
| CNS Cancer | PC-3 | 2.48 | 2.35 | 0.04 | 0.17 | 3.16 | 0.29 | ||||||
| SF-268 | 46.2 | 6.71 | 0.05 | 0.52 | >100 | 1.91 | DU-145 | 5.27 | 2.18 | 0.04 | 0.28 | >100 | 0.35 |
| SF-295 | 1.38 | 0.62 | 0.03 | 0.44 | >100 | 0.06 | Breast Cancer | ||||||
| SF-539 | 2.48 | 1.14 | 0.03 | 0.12 | - | 0.24 | MCF7 | 0.71 | 0.37 | 0.03 | 0.04 | 3.21 | 0.10 |
| SNB-19 | 64.7 | 8.23 | 0.15 | 0.56 | >100 | 0.52 | MDA-MB-231/ATCC | 9.12 | 1.84 | 0.24 | 0.48 | 3.51 | 0.84 |
| SNB-75 | 1.97 | 1.60 | 0.03 | 0.07 | - | 0.22 | HS 578T | 3.38 | 1.60 | 0.04 | 0.34 | - | 0.45 |
| U251 | 3.78 | - | - | 0.14 | - | 0.31 | BT-549 | 7.73 | 0.98 | 1.72 | 0.29 | - | 27.0 |
| Melanoma | T-47D | 2.46 | 2.00 | - | 0.10 | 2.69 | 0.44 | ||||||
| LOX IMVI | 7.64 | 1.75 | 0.05 | 0.41 | 5.26 | 0.88 | MDA-MB-468 | 2.95 | 0.34 | 0.03 | 0.74 | - | 0.73 |
| MALME-3M | 3.08 | 0.89 | - | 0.05 | 3.68 | 0.20 | |||||||
Compound 67, a 3,4,5-trimethoxybenzyl substituted derivative, was the second best in potency and demonstrated high selectivity against the leukemia (GI50 = 0.39–0.04 μM), colon cancer (GI50 = 0.30–0.04 μM), CNS cancer (GI50 = 0.56–0.07 μM), melanoma (GI50 = 0.41–0.02 μM), ovarian cancer (GI50 = 0.72–0.03 μM), and breast cancer (GI50 = 0.74–0.04 μM) subpanels, with GI50 values at submicromolar to nanomolar levels (Figures S3 and S4). Isoxazole 67, even if it was 1 order of magnitude less potent than the dimethoxy substituted analogue 66, reached nanomolar GI50 values in each subpanel, and it also had a 10 nM GI50 against the NCI-H522 non-small-cell lung cancer cells.
Overall, the presence of a methoxy group at position 4 of the 3,5-dimethoxybenzyl substituent caused a significant loss of activity (compare 66, MG_MID = 0.08 μM, with 67, MG_MID = 0.20 μM), while removal of one methoxy group (62, MG_MID = 4.47 μM; 63, MG_MID = 1.45 μM) produced an even larger (up to 2 logs) decrease in activity. Introduction of chlorine in the 7 position generally reduced activity relative to the corresponding parent compound (compare 66, MG_MID = 0.08 μM, with 75, MG_MID = 0.41 μM). In contrast, the reverse was observed for 70, which is more effective than the parent compound 61.
Compared to the previously reported classes of compounds,43 pyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazoles had strong antiproliferative effect with a 3-fold improvement in overall activity on the NCI panel. The 3,5-dimethoxybenzyl and the ethoxycarbonyl functionalities were preferred substituents for this activity, as was the case with the parent cyclohexyl analogues 2. Although [1,2]oxazoloisoindoles represent positional isomers, the same substitutions yielded ineffective compounds, thus indicating the high correlation between the pyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole 5 and the [1,2]oxazolo[4,5-g]indole system 2.
Screening Results in Lymphoma Models
All compounds were further tested at the concentration of 1 μM on four cell lines derived from distinct lymphoma histotypes, plus two with secondary resistance to the PI3Kδ inhibitor idelalisib51 or to the BTK inhibitor ibrutinib.52 After a 72 h incubation, compounds 57, 66, 67, 71, 74, and 75 showed potent growth inhibitory effects against all tested cell lines, with the percentage of proliferating cells reduced to 9–60% of the untreated cells (Table 4). For comparison, the same experiments were conducted using compound 3, but the response to it was minimal, and thus it was not considered further.
Table 4. Proliferating Cells (%) after Treatment with the Indicated Pyrrolocyclohepta[1,2-d][1,2]oxazolesa.
| compd | VL51 (MZL) | VL51 idelalisib-resistant | VL51 ibrutinib-resistant | MINO (MCL) | HBL1 (ABC DLBCL) | SU-DHL-10 (GCB DLBCL) |
|---|---|---|---|---|---|---|
| 3 | 93.5 | 106.9 | 123.1 | 94.5 | 95.6 | 80.0 |
| 51 | 88.1 | 118.2 | 120.8 | 104.5 | 118.1 | 110.2 |
| 52 | 87.4 | 106.7 | 100.3 | 104.3 | 101.4 | 70.2 |
| 53 | 103.1 | 128.0 | 125.0 | 109.3 | 108.9 | 70.2 |
| 54 | 119.9 | 135.7 | 112.0 | 112.1 | 122.2 | 141.4 |
| 55 | 112.3 | 133.0 | 124.6 | 115.2 | 127.3 | 80.3 |
| 56 | 110.3 | 126.8 | 109.8 | 108.9 | 123.0 | 74.6 |
| 57 | 36.5 | 34.3 | 37.0 | 8.5 | 29.6 | 11.0 |
| 58 | 84.0 | 78.4 | 73.0 | 101.4 | 113.6 | 72.4 |
| 59 | 97.6 | 111.5 | 92.1 | 99.0 | 114.0 | 94.6 |
| 60 | 107.7 | 115.2 | 125.0 | 111.5 | 146.2 | 61.7 |
| 61 | 93.3 | 116.8 | 117.7 | 106.3 | 123.9 | 65.2 |
| 62 | 71.3 | 63.6 | 80.3 | 103.8 | 127.6 | 51.1 |
| 63 | 66.7 | 62.3 | 64.9 | 86.3 | 117.1 | 51.5 |
| 64 | 87.4 | 116.4 | 102.0 | 112.3 | 125.1 | 66.3 |
| 65 | 86.2 | 96.4 | 94.0 | 107.0 | 111.0 | 71.6 |
| 66 | 40.1 | 34.3 | 40.5 | 9.1 | 24.1 | 11.3 |
| 67 | 40.5 | 36.6 | 40.4 | 8.9 | 24.2 | 11.2 |
| 68 | 94.5 | 122.7 | 120.4 | 98.8 | 109.1 | 78.9 |
| 69 | 93.8 | 121.4 | 109.7 | 115.9 | 102.6 | 78.5 |
| 70 | 108.1 | 111.4 | 104.4 | 104.1 | 104.2 | 69.5 |
| 71 | 58.8 | 52.2 | 56.2 | 55.9 | 97.0 | 58.2 |
| 72 | 106.8 | 127.8 | 102.9 | 110.2 | 105.1 | 76.9 |
| 73 | 113.3 | 126.2 | 101.5 | 99.5 | 106.9 | 87.4 |
| 74 | 40.4 | 37.7 | 44.4 | 9.8 | 29.0 | 13.3 |
| 75 | 38.1 | 40.2 | 40.8 | 9.5 | 20.7 | 11.5 |
MZL indicates marginal zone lymphoma; MCL indicates mantle cell lymphoma; ABC DLBCL indicates activated B-cell-like diffuse large B cell lymphoma; and GCB DLBCL indicates germinal center B-cell type diffuse large B cell lymphoma.
Compounds showing some activity, plus compound 70 based on the NCI panel data, were tested with a wider range of concentrations. Some presented potent growth growth inhibitory effects on some or all of the lymphoma cell lines, with IC50 values lower than 500 nM (Table 5).
Table 5. IC50 (μM) Values of Selected Pyrrolocyclohepta[1,2-d][1,2]oxazolesa.
| compd | VL51 (MZL) | MINO (MCL) | HBL1 (ABC DLBCL) | SU-DHL-10 (GCB DLBCL) |
|---|---|---|---|---|
| 62 | 3.2 | 4 | >5 | 3.6 |
| 63 | 2.3 | 2.6 | >5 | 3 |
| 66 | 0.25 | 0.25 | 0.3 | 0.25 |
| 67 | 0.5 | 0.6 | 0.9 | 0.6 |
| 57 | 0.2 | 0.4 | 0.6 | 0.3 |
| 70 | >10 | >10 | >10 | 9 |
| 71 | 2.4 | 3.4 | >5 | 2.2 |
| 74 | 0.5 | 0.8 | 1 | 0.7 |
| 75 | 0.8 | 0.9 | 0.9 | 0.9 |
MZL indicates marginal zone lymphoma; MCL indicates mantle cell lymphoma; ABC DLBCL indicates activated B-cell-like diffuse large B cell lymphoma; and GCB DLBCL indicates germinal center B-cell type diffuse large B cell lymphoma.
Effects of Test Compounds in Human Peripheral Blood Lymphocytes (PBLs)
To obtain an initial idea of whether the compounds described here had activity against normal cells, 63, 66, and 75 were examined for cytotoxicty against PBLs from healthy donors. As shown in Table 6, these three compounds were practically devoid of any activity both in quiescent and in lymphocytes induced to proliferate by the mitogenic stimulus phytohematoaglutinin (PHA). In all cases, we obtained a GI50 > 100 μM, demonstrating low toxicity for these healthy human cells.
Table 6. Cytotoxicity of Compounds for Human Peripheral Blood Lymphocytes.
| GI50 (μM)a |
||
|---|---|---|
| compd | PBLrestingb | PBLPHAc |
| 63 | >100 | >100 |
| 66 | >100 | >100 |
| 75 | >100 | >100 |
| vincristine | 7.5 ± 3.1 | 1.2 ± 0.6 |
Compound concentration required to inhibit cell growth by 50%. Values are the mean ± SEM for three separate experiments.
PBLs not stimulated with PHA.
PBLs stimulated with PHA.
In this context, we point out that in other studies other molecules that bind in the colchicine site were shown to have low toxicity toward lymphocytes from healthy subjects.53−56 Although at present the reason for this low toxicity is unclear, it is nevertheless interesting that even healthy lymphocytes induced to actively replicate with a mitogenic stimulus respond in the same way as quiescent lymphocytes.
Tubulin Assays
To assess if pyrrolocyclohepta[1,2]oxazoles were able to bind to tubulin, seven compounds (Table 7) were tested for their antitubulin activity in comparison with reference compound CA-4, which potently inhibits both tubulin assembly and colchicine binding to tubulin.57 Moreover, compound 3 was also evaluated as a comparison between the two scaffolds.
Table 7. Inhibition of Tubulin Assembly and Colchicine Binding by Compounds 56, 57, 58, 63, 66, 67, and 75.
| inhibition of tubulin assembly | inhibition of colchicine binding | |
|---|---|---|
| compd | IC50 ± SD (μM) | % inhibition ± SD |
| 5 μM inhibitor | ||
| CA-4 | 1.2 ± 0.08 | 97 ± 0.9 |
| 3 | 9.2 ± 0.4 | |
| 56 | 8.2 ± 0.5 | |
| 57 | 3.2 ± 0.06 | 38 ± 0.7 |
| 58 | 5.7 ± 1 | 25 ± 3 |
| 63 | 6.3 ± 0.06 | |
| 66 | 2.6 ± 0.03 | 62 ± 2 |
| 67 | 4.6 ± 0.8 | 25 ± 0.01 |
| 75 | 1.9 ± 0.1 | 42 ± 5 |
The colchicine assay was performed on compounds that yielded IC50 values of <6 μM in the assembly assay. Reaction mixtures in the assembly assay contained 9 μM (0.9 mg/mL) tubulin in the assembly assay, and in the colchicine assay they contained 0.5 μM tubulin, 5.0 μM [3H]colchicine, and 5.0 μM inhibitor.
In the assembly assay, three compounds had IC50 values of >6 μM. These were compounds 3, 56, and 63. In addition, we examined several other compounds shown in Table 1, and they were uniformly minimally active in the tubulin assembly assay. The five other compounds (57, 58, 66, 67, and 75) were more active in the assembly assay, with 66 and 75 the most active, with IC50 values of 2.6 and 1.9 μM, respectively. A value of 1.2 μM was obtained for CA-4. The five pyrrolocyclohepta[1,2]oxazoles most active as assembly inhibitors inhibited colchicine binding by 25–62% versus 97% for CA-4. Overall, the most powerful compound was 66, which inhibited tubulin polymerization with an IC50 of 2.6 μM and displayed 62% inhibition of colchicine binding. No compound was as active as CA-4 in any assay.
The effects of compounds 66 and 75 on tubulin assembly are shown in Figure 2, panels A and B, respectively. These data were obtained in computer-driven recording spectrophotometers equipped with electronic temperature controllers that rapidly change the temperature in the reaction mixtures in the cuvettes. The assembly reaction was measured by following turbidity development at 350 nM. After a minute’s equilibration at 0 °C, the temperature was jumped to 30 °C and assembly was followed for 20 min. At 21 min, the temperature was jumped backward to 0 °C, and the reaction mixtures were followed for another 8 min. Several compound concentrations were evaluated in each experimental sequence, and the IC50 for inhibition of turbidity development was defined as the compound concentration, obtained by interpolation, that inhibited the extent of turbidity development by 50% after 20 min at 30 °C. The 30–0 °C transition was included to distinguish inhibition of microtubule assembly from aberrant assembly reactions induced by numerous compounds. Typically, the aberrant assembly reaction products either are cold stable or have different temperature stability properties as compared to microtubules.
Figure 2.

Inhibition of tubulin assembly by 66 (A) and 75 (B). Reaction mixtures (0.25 mL, final volume) contained 0.8 M monosodium glutamate (adjusted to pH 6.6 in a 2 M stock solution), 0.9 μM (0.9 mg/mL) tubulin, 4% dimethyl sulfoxide, compounds at the indicated concentrations, and following a 15 min preincubation in 0.24 mL, 0.2 mM GTP (added in a 10 μL volume). The reaction mixtures, following the preincubation, were kept on ice and transferred to cuvettes held at 0 °C in a recording spectrophotometer. After baselines were established, the reactions were initiated. At 1 min, the electronic temperature controller automatically increased the temperatures in the cuvettes to 30 °C, and at 21 min, the temperatures in the cuvettes were returned to 0 °C (the temperature transitions take about 30 and 60 s, respectively). (A) The reaction mixtures contained the following concentrations of compound 66: curve 1, none; curve 2, 2.0 μM; curve 3, 3.0 μM; curve 4, 4.0 μM; curve 5, 5.0 μM; curve 6, 7.5 μM. (B) The reaction mixtures contained the following concentrations of compound 75: curve 1, none; curve 2, 1.0 μM; curve 3, 1.5 μM; curve 4, 2.0 μM; curve 5, 3.0 μM; curve 6, 4.0 μM.
Molecular Modeling
Compound 3 and all the compounds belonging to the new class of pyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazoles 5 were docked into the colchicine and vinblastine binding sites, by selecting for each of them the pose with the best G-Score (kcal/mol). A better affinity for the colchicine site (Table S2) was observed for all ligands, further confirming their specificity for this binding pocket. Moreover, most of the newly synthesized compounds had a better affinity than the parent compound 3.
To further investigate the binding mode of the best active compounds (57, 58, 63, 66, 67, and 75) in the biological assays with respect to 3, molecular modeling studies were performed on the 3N2G model, which displays two additional neighboring pockets (zones 2 and 3) in the tubulin colchicine domain. As was the case with their parent compound 3, compounds 57, 58, 63, 66, 67, and 75 had a better G-Score toward the main site (zone 1) of the colchicine domain (Table S3). As shown in Figure S5, our compounds had unfavorable steric contacts with an additional hydrophobic pocket of the β subunit, formed by residues E200, L255, A316, A317, A354, C241, and T179.
On the other hand, the best docking poses of active compounds with tubulin structure 4O2B, containing zones 1 and 2 of the colchicine site, showed strong hydrophobic interactions with β-tubulin residues L248, A250, A354, I318, A316, and L255 (Figure 3). In particular, 66 and 75, which are the compounds with the best biological activity, displayed a binding geometry similar to that of colchicine in zones 1 and 2 of the pocket, by directing their methoxybenzyl groups toward the C241 residue (Figure 3D and Figure 3F). Moreover, 75 also established a halogen bond between its chlorine and the backbone of V181 and an H-bond between the oxazole moiety and the backbone of N249. Conversely, compound 3 and the less active compounds (57, 58, 63, and 67) showed a different binding orientation, with the tricyclic portion steered toward residue C241 (Figure 3A,B,C,E,G).
Figure 3.

Best docked poses of (A) 57, (B) 58, (C) 63, (D) 66, (E) 67, (F) 75, and (G) 3 with the 4O2B crystal structure of tubulin, depicting zones 1 and 2 of the colchicine site. Tubulin is shown in a faded blue surface, while ligands and residues, involved in the most important interactions, are shown as sticks. Halogen bond and π–cation interactions are indicated as dashed violet and dark-green lines, respectively.
In particular, for compound 3 we observed a π–cation between its pyrrole and β-tubulin K352, while hydrophobic interactions were much weaker than with the other derivatives.
The best docking poses of 57, 58, 63, 66, 67, 75, and 3 against the 4O2B model were submitted to explicit water solvent molecular dynamics (MD) simulations, with the aims to add depth to our analysis and to investigate the possibility of induced-fit phenomena in the tubulin recognition process of our ligands. As a reference, the X-ray model of 4O2B, containing colchicine in its binding pocket, was included in similar calculations. In the Supporting Information we reported the geometric behavior of all MD simulations and the analysis of their most representative structures, by computing the related binding free energy and the global number of contacts (Table S4).
With the respect to its docking pose (Figure 3G), the most representative MD structure of 3 showed the establishment of three H-bonds (Figure 4G). In particular, the two methoxy groups interact with β-tubulin N101 and N249, while the carbonyl group establishes an H-bond with S318. However, the formation of these hydrogen bonds does not allow stability of the bonding mode and does not bring about an energy gain, due to the lower ability to establish hydrophobic interactions.
Figure 4.

Most representative MD structure of tubulin (PDB code 4O2B) complexed with (A) 57, (B) 58, (C) 63, (D) 66, (E) 67, (F) 75, and (G) 3. Tubulin is depicted as a pale blue surface, and ligands and residues with the most critical interactions are in stick format. Hydrogen bond and π–cation interactions are indicated as dashed black and dark-green lines, respectively.
Regarding 66, the most representative MD structure showed that the tubulin molecule adjusted its residues to allow establishment of an H-bond between its methoxy group and the side chain of C241 and to permit a π–cation between its oxazole ring and K254 (Figure 3D). Further hydrophobic interactions with β-tubulin K254, A250, L255, A316, and A354 stabilized the complex. Likewise, 75 showed a binding mode similar to that of 66, by engaging an H-bond between its 4-methoxy group and C241 and several hydrophobic interactions with L248, A250, L255, I318, and A354 (Figure 4F). With respect to its docking pose (Figure 3F), we observed the absence of the halogen bond and the H-bond with N249, both of which can be considered useful interactions in the recognition process but not in complex stabilization. In fact, although 67 and 75 differ only by the chlorine atom, the docking simulation proposed different binding poses for them. Thus, the absence of chlorine seems responsible for the different orientation of 67 in the tubulin pocket, preventing also the establishment of the H-bond with C241 during MDs (Figure 4E), which seems very important for the stabilization of the most active compounds. Finally, during MDs 57 and 58 increased their hydrophobic interactions with the L255, A316, I318, K352, and A354 residues (Figure 4A,B).
In conclusion, compared to the parent molecule 3, the most active compounds of the new class of pyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazoles displayed an improved interaction with tubulin because of the greater contribution of the lipophilic energy components. Moreover, 66 and 75 showed a peculiar binding mode, characterized by the methoxybenzyl portion placed similarly to colchicine.
Compounds 63, 66, and 75 Induced Mitotic Arrest of the Cell Cycle
To evaluate the effects of compounds 63, 66, and 75 on cell cycle progression, we first treated HeLa cells for 24 h.The cells were then labeled with propidium iodide (PI) and analyzed by flow cytometry. As shown in Figure 5 (panel B), compound 66 induced arrest of the cell cycle in the G2/M phase. This effect occurred in a concentration-dependent manner, and partial arrest in G2/M occurred at the lowest concentration examined (0.125 μM), while at the highest concentration (0.5 μM) over 80% of the cells were arrested in G2/M. Compound 75 (panel C) exhibited a similar behavior, although the first appearance of G2/M arrest occurred at 0.25 μM. Only modest activity was observed with compound 63 (panel A), as the metaphase arrest occurred only at the highest concentration (1 μM) examined, as compared to the other two compounds, and furthermore, at this concentration there was only a slight reduction in the proportion of S phase cells observed.
Figure 5.

Percentage distribution in the three phases of the cell cycle of HeLa cells treated with 63 (A), 66 (B), or75 (C) at the indicated concentrations for 24 h. Cells were analyzed by flow cytometry after labeling with PI as described in the Experimental Section. Data are presented as the mean ± SEM of three experiments.
Compound 66 Induced Alteration of Cell Cycle Checkpoint Proteins
We studied the effects of 66 on the expression of various checkpoint proteins that play roles in cell cycle regulation. Cells that enter mitosis do so through the involvement of cyclin B1 complexed to cdc2. This complex is activated through the dephosphorylation of phospho-cdc2, which is a cdc25c-dependent process that ultimately leads to the phosphorylation of cyclin B1. This phosphorylated enzyme triggers cells to enter mitosis.58,59
Figure 6 demonstrates a substantial increase of cyclin B1 expression after a 24 h treatment with 0.5 μM 66. In contrast, total cdc25c expression was strongly reduced, and in good agreement, the expression of phosphorylated cdc2 was strongly decreased after both 24 and 48 h. Dephosphorylation of this protein is needed to activate the cdc2/cyclin B complex, and this effect is stimulated by cdc25c.58,59
Figure 6.

Effects of 66 on G2/M checkpoint proteins. HeLa cells were treated with 0.1 or 0.5 μM compound 66 for 24 or 48 h. The cells were harvested and lysed for the detection of cdc25c, p-cdc2Y15, and cyclin B expression by Western blot analysis. To confirm equal protein loading, each membrane was stripped and reprobed with anti-GAPDH antibody. The relative expression of proteins was analyzed by scanning densitometry using ImageJ software and reported as a ratio protein/GAPDH.
These data demonstrate that cdc2/cyclin B1 complexes were not activated, thus blocking cells from exiting mitosis and leading to apoptotic cell death.
Compound 66 Induced Apoptosis
To analyze the mode of cell death induced by compound 66 in HeLa cells, we utilized a double labeling assay of the cells with annexin-V and PI. This procedure, through a flow cytometric analysis, distinguishes four different cell populations: live cells (annexin-V–/PI–), early apoptotic cells (annexin-V+/PI–), late apoptotic cells (annexin-V+/PI+), and necrotic cells (annexin-V–/PI+).
As Figure 7 denonstrates, HeLa cells treated with 66 showed a significant increase in apoptotic cells after a 24 h treatment at 0.5 μM. The percentage of apoptotic cells increased further after 48 h, when there was a substantial increase in necrotic cells, indicating that the compound ultimately induced cell death by necrosis as well as apoptosis.
Figure 7.

Flow cytometric analysis of apoptotic cells after treatment of HeLa cells with 66 at the indicated concentrations after incubation for 24 or 48 h. The cells were harvested and labeled with annexin-V-FITC and PI and analyzed by flow cytometry. Data are represented as the mean ± SEM of three independent experiments.
Compound 66 Induced Apoptosis through the Mitochondrial Pathway
In the initial stages of induction of apoptosis, the mitochondrial transmembrane potential (Δψmt) is altered and leads to to a reduction of Δψmt and release of cytochrome c into the cytoplasm.60,61 Moreover, this effect occurs with many antimitotic agents and in a variety of cell lines.62−64 As shown in Figure 8 (panel A), compound 66 at both concentrations used (0.1 and 0.5 μM) induced in a time- and concentration-dependent manner a significant increase in the percentage of cells with low Δψmt.
Figure 8.
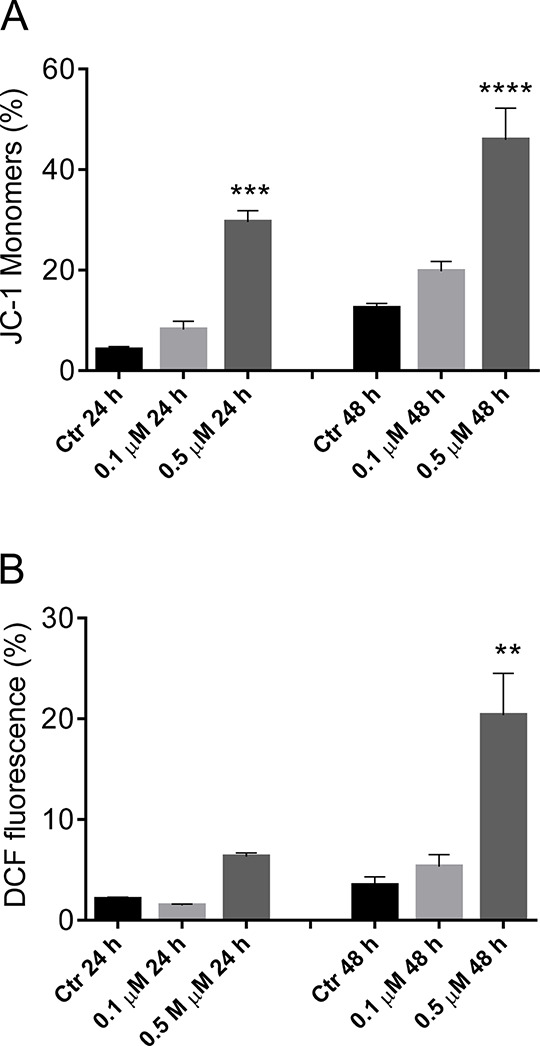
Evaluation of mitochondrial membrane potential (Δψmt) and ROS production after treatment of HeLa cells with compound 66 (panels A and B, respectively). Cells were treated with the indicated concentration of compound for 24 or 48 h and then stained with the fluorescent probe JC-1 (A) or H2-DCFDA as described in the Experimental Section. Data are presented as the mean ± SEM of three independent experiments: **p < 0.01, ***p < 0.001, ****p < 0.0001 vs control.
One consequence of mitochondrial depolarization caused by the release of cytochrome c into the cytoplasm is the increase in reactive oxygen species (ROS).65 Therefore, we wanted to evaluate whether ROS production increased following treatment with compound 66. To do this, we used the fluorescent probe 2,7-dichlorodihydrofluorescein diacetate (H2-DCFDA), which is oxidized to the fluorescent compound dichlorofluorescein (DCF) upon ROS production. The results of the cytofluorimetric analysis are presented in Figure 8 (panel B), which demonstrates that 66 induced the production of ROS in HeLa cells after a 48 h treatment at 0.5 μM, in agreement with the reduction of Δψmt. Note that the increase in ROS is only detectable after mitochondrial depolarization, indicating that ROS production results from mitochondrial damage.
Compound 66 Induced PARP Cleavage and Reduced the Expression of Mcl-1 and XIAP Proteins
To study in greater detail the apoptotic process induced by 66, we evaluated the expression of the cleaved fragment of poly(ADP)ribose polymerase (PARP), a common marker of apoptosis,66 by Western blot analysis. HeLa cells were treated with compound 66 at 0.1 or 0.5 μM for 24 or 48 h. The cleavage fragment of PARP appeared at 24 h after beginning treatment with only 0.1 μM 66. The expression of two antiapoptotic proteins, Mcl-1 and XIAP, was also studied. Mcl-1, a member of the Bcl-2 family, is highly expressed in many types of tumors and takes part in the apoptotic response to multiple stimuli. Specifically, sensitivity to antimitotic drugs is regulated by Mcl-1 levels,67,68 and we found that compound 66 treatment of HeLa cells resulted in a reduction in Mcl-1 levels (Figure 9). Similarly, expression of Xiap, a member of the family of inhibitors of apoptosis proteins, was reduced (at 24 h) and diappeared (at 48 h) after HeLa cell treatment with 66 (Figure 9). The functions of this protein are to inhibit the activity of caspase-3, caspase-7, and caspase-9 through a direct interaction with these enzymes. Following this interaction, the entire apoptotic process is inhibited.69 Thus, treatment of HeLa cells with 66 resulted in downregulation of Mcl-1 and Xiap and impairment of their antiapoptotic functions.
Figure 9.

Western blot analysis of Mcl-1 XIAP and PARP after treatment of HeLa cells with 66 at the indicated concentrations and for the indicated times. To confirm equal protein loading, each membrane was stripped and reprobed with anti-GAPDH antibody. The relative expression of proteins was analyzed by scanning densitometry using ImageJ software and reported as a ratio protein/GAPDH.
Conclusions
Among anticancer agents, colchicine site inhibitors still attract much attention in medicinal chemistry because of their potential to overcome disadvantages encountered by other antitubulin agents binding at other sites. Our study indicates that pyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazoles can be considered a novel class of antimitotic compounds binding at the colchicine site, with antitumor activity in multiple cancer cell lines and with improved features with respect to the previously identified cyclohexyl analogue 3.
We show here that expanding the central ring to seven members, in part to mimic the seven-member rings of colchicine, resulted in enhanced antiproliferative activities in multiple cell lines. This was based on increased antitubulin activity, which in turn caused cell cycle arrest at G2/M, with resultant apoptosis. Molecular modeling rationalized the improvement in activity by central ring expansion, probably caused by an improvement in binding affinity for the colchicine binding pocket because of a greater contribution of the lipophilic energy components.
Among these compounds, five derivatives (62, 63, 66, 67, and 75) showed promising antiproliferative effects, and in particular, 66 and 67, bearing a methoxysubstituted N-benzyl moiety and an ethoxycarbonyl group, reached nanomolar growth inhibitory effects against solid and liquid tumor cells and submicromolar activity against lymphoma cell lines. Their mechanism of action is probably through inhibition of tubulin assembly by binding in the colchicine site, and this mechanism was particularly marked for 66, which inhibited tubulin polymerization with an IC50 of 2.6 μM and inhibited colchicine binding by 62% under the conditions examined.
Investigation of the mechanism of action showed the ability of the new [1,2]oxazoles to impair cell cycle progression and induce apoptosis through the mitochondrial pathway. The most active compound 66 was able to arrest HeLa cells in the G2/M phase of the cell cycle in a concentration dependent manner. This effect was accompanied by apoptosis, mitochondrial depolarization, generation of ROS, and activation of PARP cleavage. These results indicate that the cellular actions of these agents involved mitotic arrest, due to interference with the functions of the mitotic spindle, and an apoptotic cell death. Taken together, the biological results collected so far indicate that our class of [1,2]oxazoles might find an important place in the set of molecules of interest for the development of pharmaceutical strategies against cancer. Further evolution of this class in terms of ADMET profile will be considered to establish the best trade-off between biological activity and drug-like properties for further preclinical studies.
Experimental Section
Chemistry
Synthesis and Characterization
MW irradiation was performed using a CEM Discover Labmate apparatus. All melting points were taken on a Büchi melting point M-560 apparatus. IR spectra were determined in bromoform with a Shimadzu FT/IR 8400S spectrophotometer. 1H and 13C NMR spectra were measured at 200 and 50.0 MHz, respectively, in DMSO-d6 or CDCl3 solution using a Bruker Avance II series 200 MHz spectrometer. Column chromatography was performed with Merck silica gel (230–400 mesh ASTM) or a Büchi Sepacor chromatography module (prepacked cartridge system). Elemental analyses (C, H, N) were within ±0.4% of theoretical values and were performed with a VARIO EL III elemental analyzer. The purity of all the tested compounds was >95%, determined by HPLC (Agilent 1100 series).
Compounds 6–13, 17–21, 25–30, 34–38, and 42 were prepared according to our published procedures.48,49
General Procedure for the Preparation of 1-Substituted-4,5,6,7-tetrahydrocyclohepta[b]pyrrol-8(1H)-one (14–16) and Ethyl 1-Substituted-8-oxo-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (22–24)
To a solution of 7, 8 (9 mmol) in anhydrous DMF (17 mL), NaH (0.24 g, 10 mmol) was added at 0 °C, and the reaction mixture was stirred at room temperature for 1.5 h. Then the suitable alkyl or aralkyl halide (13.5 mmol) was added at 0 °C, and the reaction mixture was stirred at room temperature until the reaction was complete (TLC). Then the reaction mixture was poured onto crushed ice. The precipitate was removed by filtration and dried. If there was no precipitate, the solution was extracted with dichloromethane (3 × 50 mL). The organic layer was dried over Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography, with dichloromethane as eluting solvent.
1-(2,5-Dimethoxybenzyl)-4,5,6,7-tetrahydrocyclohepta[b]pyrrol-8(1H)-one (14)
This compound was obtained from reaction of 8 with 2,5-dimethoxybenzyl chloride after 2 h. White solid; yield 90%; mp 85.0–85.4 °C; IR (cm–1) 1634 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.79–1.85 (m, 4H, 2 × CH2), 2.62 (t, 2H, J = 6.1 Hz, CH2), 2.81 (t, 2H, J = 6.1 Hz, CH2), 3.67 (s, 3H, CH3), 3.81 (s, 3H, CH3), 5.52 (s, 2H, CH2), 5.99 (d, 1H, J = 2.6 Hz, H-3), 6.35 (d, 1H, J = 2.5 Hz, H-6′), 6.73 (d, 1H, J = 2.6 Hz, H-2), 6.75–6.78 (m, 1H, H-4′), 6.80–6.84 (m, 1H, H-3′); 13C NMR (CDCl3, 50 MHz) δ 21.5, 24.9, 26.4, 41.6, 47.4, 55.5, 55.9, 109.5, 111.0, 112.3, 114.4, 128.5, 128.9, 129.4, 136.5, 150.9, 153.7, 192.7. Anal. Calcd for C18H21NO3: C, 72.22; H, 7.07; N, 4.68. Found: C, 72.07; H, 6.93; N, 4.77.
1-(3,5-Dimethoxybenzyl)-4,5,6,7-tetrahydrocyclohepta[b]pyrrol-8(1H)-one (15)
This compound was obtained from reaction of 8 with 3,5-dimethoxybenzyl chloride after 2 h. Gray solid; yield 90%; mp 60.8–61.7 °C; IR (cm–1) 1625 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.78–1.84 (m, 4H, 2 × CH2), 2.61 (t, 2H, J = 6.0 Hz, CH2), 2.81 (t, 2H, J = 6.0 Hz, CH2), 3.73 (s, 6H, 2 × CH3), 5.48 (s, 2H, CH2), 6.01 (d, 1H, J = 2.5 Hz, H-3), 6.22 (d, 2H, J = 2.2 Hz, H-2′ and H-6′), 6.32 (t, 1H, J = 2.2 Hz, H-4′), 6.79 (d, 1H, J = 2.5 Hz, H-2); 13C NMR (CDCl3, 50 MHz) δ 21.5, 24.9, 26.4, 41.6, 52.4, 55.2, 99.0, 104.9, 109.8, 128.8, 129.1, 136.7, 141.1, 160.9, 192.8. Anal. Calcd for C18H21NO3: C, 72.22; H, 7.07; N, 4.68. Found: C, 72.38; H, 7.22; N, 4.54.
1-(3,4,5-Trimethoxybenzyl)-4,5,6,7-tetrahydrocyclohepta[b]pyrrol-8(1H)-one (16)
This compound was obtained from reaction of 8 with 3,4,5-trimethoxybenzyl chloride after 16 h. White solid; yield 89%; mp 64.6–65.3 °C; IR (cm–1) 1635 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.79–1.85 (m, 4H, 2 × CH2), 2.63 (t, 2H, J = 6.1 Hz, CH2), 2.82 (t, 2H, J = 6.1 Hz, CH2), 3.79 (s, 6H, 2 × CH3), 3.81 (s, 3H, CH3), 5.47 (s, 2H, CH2), 6.03 (d, 1H, J = 2.5 Hz, H-3), 6.33 (s, 2H, H-2′ and H-6′), 6.81 (d, 1H, J = 2.5 Hz, H-2); 13C NMR (CDCl3, 50 MHz) δ 21.5, 25.0, 26.4, 41.6, 52.5, 56.0, 60.8, 104.0, 109.8, 128.9, 129.0, 134.3, 136.9, 137.0, 153.3, 192.9. Anal. Calcd for C19H23NO4: C, 69.28; H, 7.04; N, 4.25. Found: C, 69.13; H, 6.86; N, 4.45.
Ethyl 1-(2,5-Dimethoxybenzyl)-8-oxo-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (22)
This compound was obtained from reaction of 7 with 2,5-dimethoxybenzyl chloride after 16 h. White solid; yield 81%; mp 92.8–93.4 °C; IR (cm–1) 1711 (CO), 1647 (CO); 1H NMR (DMSO-d6, 200 MHz) δ 1.20 (t, 3H, J = 7.1 Hz, CH3), 1.65–1.81 (m, 4H, 2 × CH2), 2.59 (t, 2H, J = 5.6 Hz, CH2), 2.84 (t, 2H, J = 5.6 Hz, CH2), 3.54 (s, 3H, CH3), 3.78 (s, 3H, CH3), 4.16 (q, 2H, J = 7.1 Hz, CH2), 5.53 (d, 1H, J = 2.9 Hz, H-6′), 5.90 (s, 2H, CH2), 6.72 (dd, 1H, J = 8.8, 2.9 Hz, H-4′), 6.89 (d, 1H, J = 8.8 Hz, H-3′), 6.90 (s, 1H, H-3); 13C NMR (DMSO-d6, 50 MHz) δ 13.9, 20.7, 24.4, 24.7, 41.3, 44.8, 55.0, 55.6, 60.3, 110.3, 110.8, 111.6, 117.5, 126.5, 129.1, 133.3, 133.6, 149.9, 153.1, 159.9, 194.0. Anal. Calcd for C21H25NO5: C, 67.91; H, 6.78; N, 3.77. Found: C, 67.84; H, 6.62; N, 3.89.
Ethyl 1-(3,5-Dimethoxybenzyl)-8-oxo-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (23)
This compound was obtained from reaction of 7 with 3,5-dimethoxybenzyl chloride after 16 h. White solid; yield 83%; mp 71.8–72.0 °C; IR (cm–1) 1707 (CO), 1647 (CO); 1H NMR (DMSO-d6, 200 MHz) δ 1.22 (t, 3H, J = 7.1 Hz, CH3), 1.72 (m, 4H, 2 × CH2), 2.61 (t, 2H, J = 6.9 Hz, CH2), 2.82 (t, 2H, J = 6.9 Hz, CH2), 3.65 (s, 6H, 2 × CH3), 4.19 (q, 2H, J = 7.1 Hz, CH2), 5.94 (s, 4H, CH2, H-2′ and H-6′), 6.32 (s, 1H, H-4′), 6.88 (s, 1H, H-3); 13C NMR (DMSO-d6, 50 MHz) δ 14.1, 20.8, 24.4, 24.8, 41.4, 48.2, 55.0, 60.4, 97.8, 103.7, 117.8, 126.3, 133.1, 133.9, 141.9, 160.1, 160.5, 194.3. Anal. Calcd for C21H25NO5: C, 67.91; H, 6.78; N, 3.77. Found: C, 68.07; H, 6.90; N, 3.62.
Ethyl 1-(3,4,5-Trimethoxybenzyl)-8-oxo-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (24)
This compound was obtained from reaction of 7 with 3,4,5-trimethoxybenzyl chloride after 24 h. White solid; yield 80%; mp 70.0–70.2 °C; IR (cm–1) 1709 (CO), 1646 (CO); 1H NMR (DMSO-d6, 200 MHz) δ 1.24 (t, 3H, J = 7.1 Hz, CH3), 1.66–1.80 (m, 4H, 2 × CH2), 2.64 (t, 2H, J = 5.6 Hz, CH2), 2.82 (t, 2H, J = 5.6 Hz, CH2), 3.60 (s, 3H, CH3), 3.64 (s, 6H, 2 × CH3), 4.22 (q, 2H, J = 7.1 Hz, CH2), 5.97 (s, 2H, CH2), 6.16 (s, 2H, H-2′ and H-6′), 6.88 (s, 1H, H-3); 13C NMR (DMSO-d6, 50 MHz) δ 14.0, 20.8, 24.3, 24.7, 41.4, 48.1, 55.6, 59.9, 60.4, 102.9, 117.8, 126.3, 133.1, 133.9, 135.0, 136.2, 152.8, 160.2, 194.4. Anal. Calcd for C22H27NO6: C, 65.82; H, 6.78; N, 3.49. Found: C, 65.96; H, 6.65; N, 3.35.
General Procedure for the Preparation of 7-[(Dimethylamino)methylidene]-1-substituted-4,5,6,7-tetrahydrocyclohepta[b]pyrrol-8(1H)-one (31–33) and Ethyl 7-[(Dimethylamino)methylidene]-1-substituted-8-oxo-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (39–41). Method A
To a solution of ketones 22–24 (1.3 mmol) in anhydrous DMF (2.5 mL), DMFDMA (0.19 mL, 1.4 mmol) was added, and the reaction mixture was irradiated under MW conditions (power 50 W; pressure (max) 100 psi; temperature (max) 100 °C) until the reaction was complete (TLC).
Method B
To a solution of ketones 22–24 (1.3 mmol) in anhydrous DMF (2.5 mL), TBDMAM (0.41 mL, 2 mmol) was added, and the reaction mixture was irradiated under MW conditions (power 50 W; pressure (max) 100 psi; temperature (max) 100 °C) until the reaction was complete (TLC).
Method C
To a solution of ketones 14–16 (1.3 mmol) in anhydrous DMF (2.5 mL), DMFDMA (1.73 mL, 13 mmol) was added, and the reaction mixture was irradiated under MW conditions (power 150 W; pressure (max) 150 psi; temperature (max) 130 °C) until the reaction was complete (TLC).
In all cases, the reaction mixtures were poured onto crushed ice. The precipitate was removed by filtration and dried. If there was no precipitate, the solution was extracted with ethyl acetate (3 × 30 mL). The organic layer was dried over Na2SO4, and the solvent was removed under reduced pressure.
7-[(Dimethylamino)methylidene]-1-(2,5-dimethoxybenzyl)-4,5,6,7-tetrahydrocyclohepta[b]pyrrol-8(1H)-one (31)
This compound was obtained from reaction of 14 under MW conditions (method C, 40 min). Brown oil; yield 93%; IR (cm–1) 1633 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.76–1.90 (m, 2H, CH2), 2.44 (t, 2H, J = 6.7 Hz, CH2), 2.65 (t, 2H, J = 6.7 Hz, CH2), 3.06 (s, 6H, 2 × CH3), 3.64 (s, 3H, CH3), 3.79 (s, 3H, CH3), 5.52 (s, 2H, CH2), 5.94 (d, 1H, J = 2.4 Hz, H-3), 6.31 (d, 1H, J = 2.4 Hz, H-2), 6.65–6.81 (m, 3H, H-3′, H-4′, and H-6′), 7.39 (s, 1H, CH); 13C NMR (CDCl3, 50 MHz) δ 24.2, 24.4, 30.3, 43.2, 46.4, 55.6, 55.8, 107.4, 108.2, 110.9, 112.3, 114.0, 126.7, 129.3, 130.0, 130.5, 148.6, 150.8, 153.7, 188.8. Anal. Calcd for C21H26N2O3: C, 71.16; H, 7.39; N, 7.90. Found: C, 71.31; H, 7.53; N, 7.77.
7-[(Dimethylamino)methylidene]-1-(3,5-dimethoxybenzyl)-4,5,6,7-tetrahydrocyclohepta[b]pyrrol-8(1H)-one (32)
This compound was obtained from reaction of 15 under MW conditions (method C, 40 min). Brown oil; yield 90%; IR (cm–1) 1635 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.76–1.90 (m, 2H, CH2), 2.42 (t, 2H, J = 6.7 Hz, CH2), 2.65 (t, 2H, J = 6.7 Hz, CH2), 3.06 (s, 6H, 2 × CH3), 3.71 (s, 6H, 2 × CH3), 5.46 (s, 2H, CH2), 5.95 (d, 1H, J = 2.5 Hz, H-3), 6.22 (d, 2H, J = 2.2 Hz, H-2′ and H-6′), 6.29 (t, 1H, J = 2.2 Hz, H-4′), 6.72 (d, 1H, J = 2.5 Hz, H-2), 7.39 (s, 1H, CH); 13C NMR (CDCl3, 50 MHz) δ 24.1, 24.3, 30.3, 43.2, 51.3, 55.2, 99.1, 104.7, 107.3, 108.3, 126.5, 130.2, 130.5, 141.9, 148.6, 160.8, 188.8. Anal. Calcd for C21H26N2O3: C, 71.16; H, 7.39; N, 7.90. Found: C, 71.02; H, 7.55; N, 8.11.
7-[(Dimethylamino)methylidene]-3,4,5-trimethoxybenzyl)-4,5,6,7-tetrahydrocyclohepta[b]pyrrol-8(1H)-one (33)
This compound was obtained from reaction of 16 under MW conditions (method C, 120 min). Brown oil; yield 86%; IR (cm–1) 1653 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.77–1.90 (m, 2H, CH2), 2.41 (t, 2H, J = 6.6 Hz, CH2), 2.66 (t, 2H, J = 6.6 Hz, CH2), 3.08 (s, 6H, 2 × CH3), 3.77 (s, 6H, 2 × CH3), 3.79 (s, 3H, CH3), 5.46 (s, 2H, CH2), 5.97 (d, 1H, J = 2.5 Hz, H-3), 6.31 (s, 2H, H-2′ and H-6′), 6.74 (d, 1H, J = 2.5 Hz, H-2), 7.41 (s, 1H, CH); 13C NMR (CDCl3, 50 MHz) δ 24.2, 24.3, 30.3, 43.2, 51.4, 55.9, 60.8, 103.7, 107.2, 108.3, 126.4, 130.4, 130.5, 135.2, 136.8, 148.7, 153.2, 188.8. Anal. Calcd for C22H28N2O4: C, 68.73; H, 7.34; N, 7.29. Found: C, 68.59; H, 7.51; N, 7.08.
Ethyl 7-[(Dimethylamino)methylidene]-1-(2,5-dimethoxybenzyl)-8-oxo-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (39)
This compound was obtained from reaction of 22 under MW conditions and used in the next step without purification.
Ethyl 7-[(Dimethylamino)methylidene]-1-(3,5-dimethoxybenzyl)-8-oxo-1,4,5,6,7,8- hexahydrocyclohepta[b]pyrrole-2-carboxylate (40)
This compound was obtained from reaction of 23 under MW conditions (method A, 110 min; method B, 20 min). Pale yellow solid; yield 77% (method A), 94% (method B); mp 139.2–140 °C; IR (cm–1) 1714 (CO), 1698 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.28 (t, 3H, J = 7.1 Hz, CH3), 1.73–1.87 (m, 2H, CH2), 2.39 (t, 2H, J = 6.8 Hz, CH2), 2.62 (t, 2H, J = 6.8 Hz, CH2), 3.07 (s, 6H, 2 × CH3), 3.67 (s, 6H, 2 × CH3), 4.22 (q, 2H, J = 7.1 Hz, CH2), 6.06 (s, 2H, CH2), 6.16 (s, 2H, H-2′ and H-6′), 6.24 (s, 1H, H-4′), 6.77 (s, 1H, H-3), 7.55 (s, 1H, CH); 13C NMR (CDCl3, 50 MHz) δ 14.4, 16.6, 23.7, 30.3, 43.3, 48.6, 55.1, 60.0, 99.0, 104.3, 107.1, 117.0, 125.2, 127.6, 136.6, 142.8, 150.3, 160.9, 161.0, 188.7. Anal. Calcd for C24H30N2O5: C, 67.59; H, 7.09; N, 6.57. Found: C, 67.71; H, 6.88; N, 6.39.
Ethyl 7-[(Dimethylamino)methylidene]-8-oxo-1-(3,4,5-trimethoxybenzyl)-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (41)
This compound was obtained from reaction of 24 under MW conditions (method A, 80 min; method B, 40 min). Brown oil; yield 75% (method A), 99% (method B); IR (cm–1) 1707 (CO), 1635 (CO); 1H NMR (DMSO-d6, 200 MHz) δ 1.23 (t, 3H, J = 7.0 Hz, CH3), 1.70–1.79 (m, 2H, CH2), 2.30 (t, 2H, J = 6.8 Hz, CH2), 2.51 (t, 2H, J = 6.8 Hz, CH2), 2.91 (s, 6H, 2 × CH3), 3.59 (s, 3H, CH3), 3.62 (s, 6H, 2 × CH3), 4.19 (q, 2H, J = 7.0 Hz, CH2), 5.90 (s, 2H, CH2), 6.20 (s, 2H, H-2′ and H-6′), 6.77 (s, 1H, H-3), 7.51 (s, 1H, CH); 13C NMR (DMSO-d6, 50 MHz) δ 14.2, 22.7, 23.1, 29.8, 43.1, 47.2, 55.6, 59.8, 59.9, 103.2, 104.9, 116.8, 123.6, 126.3, 135.5, 136.2, 136.7, 150.6, 152.7, 160.3, 186.5. Anal. Calcd for C25H32N2O6: C, 65.77; H, 7.07; N, 6.14. Found: C, 65.62; H, 6.89; N, 6.31.
General Procedure for the Preparation of Ethyl 7-(Hydroxymethylidene)-1-substituted-8-oxo-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (43–50)
To a suspension of t-BuO–K+ (13.5 mmol) in anhydrous toluene (12 mL), at 0 °C and under a N2 atmosphere, a solution of the appropriate ketone 17–24 (4.5 mmol) in anhydrous toluene (40 mL) was added, and the reaction mixture was stirred at room temperature for 1.5 h. Then a solution of ethyl formate (1.09 mL, 13.5 mmol) in anhydrous toluene (12 mL) was added at 0 °C, and the reaction mixture was stirred until the reaction was complete (1.5–4 h). The solvent was removed under reduced pressure, and water (50 mL) was added to the residue. The aqueous phase was acidified with 3 N HCl and extracted with dichloromethane (2 × 60 mL). The organic phase was dried over Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography with dichloromethane as eluting solvent.
Ethyl 7-(Hydroxymethylidene)-1-methyl-8-oxo-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (43)
This compound was obtained from reaction of 17. Brown oil; yield 80%; IR (cm–1) 3415 (OH), 1704 (CO), 1620 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.36 (t, 3H, J = 7.1 Hz, CH3), 1.84–1.97 (m, 2H, CH2), 2.22 (t, 2H, J = 6.9 Hz, CH2), 2.69 (t, 2H, J = 6.9 Hz, CH2), 4.11 (s, 3H, CH3), 4.31 (q, 2H, J = 7.1 Hz, CH2), 6.70 (s, 1H, H-3), 7.76 (d, 1H, J = 7.9 Hz, CH), 14.93 (d, 1H, J = 7.9 Hz, OH); 13C NMR (CDCl3, 50 MHz) δ 14.3, 25.0, 25.9, 28.5, 35.0, 60.5, 114.0, 117.0, 127.5, 129.7, 132.8, 160.9, 172.3, 186.3. Anal. Calcd for C14H17NO4: C, 63.87; H, 6.51; N, 5.32. Found: C, 63.99; H, 6.43; N, 5.49.
Ethyl 7-(Hydroxymethylene)-1-benzyl-8-oxo-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (44)
This compound was obtained from reaction of 18. Brown oil; yield 90%; IR (cm–1) 3391 (OH), 1702 (CO), 1619 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.29 (t, 3H, J = 7.1 Hz, CH3), 1.84–1.95 (m, 2H, CH2), 2.13 (t, 2H, J = 6.8 Hz, CH2), 2.69 (t, 2H, J = 6.8 Hz, CH2), 4.24 (q, 2H, J = 7.1 Hz, CH2), 5.99 (s, 2H, CH2), 6.83 (s, 1H, H-3), 6.92–7.00 (m, 2H, H-2′ and H-6′), 7.16–7.29 (m, 3H, H-3′, H-4′ and H-5′), 7.71 (d, 1H, J = 8.0 Hz, CH), 14.77 (d, 1H, J = 8.0 Hz, OH); 13C NMR (CDCl3, 50 MHz) δ 14.2, 24.5, 25.7, 28.8, 49.2, 60.5, 113.9, 117.9, 126.1, 126.9, 127.0, 128.3, 130.2, 132.7, 139.2, 160.6, 172.5, 186.5. Anal. Calcd for C20H21NO4: C, 70.78; H, 6.24; N, 4.13. Found: C, 70.91; H, 6.09; N, 3.97.
Ethyl 7-(Hydroxymethylene)-1-(2-methoxybenzyl)-8-oxo-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (45)
This compound was obtained from reaction of 19. Brown oil; yield 80%; IR (cm–1) 3400 (OH), 1711 (CO), 1638 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.26 (t, 3H, J = 7.2 Hz, CH3), 1.86–1.99 (m, 2H, CH2), 2.16 (t, 2H, J = 6.4 Hz, CH2), 2.70 (t, 2H, J = 6.4 Hz, CH2), 3.82 (s, 3H, CH3), 4.21 (q, 2H, J = 7.2 Hz, CH2), 5.94 (s, 2H, CH2), 6.40 (d, 1H, J = 6.7 Hz, H-3′), 6.77–6.81 (m, 2H, H-5′ and H-6′), 6.83 (s, 1H, H-3), 7.16 (t, 1H, J = 6.7 Hz, H-4′), 7.70 (d, 1H, J = 8.1 Hz, CH), 14.72 (d, 1H, J = 8.1 Hz, OH); 13C NMR (CDCl3, 50 MHz) δ 14.2, 24.5, 25.7, 28.9, 45.6, 55.2, 60.4, 109.8, 113.8, 117.6, 120.3, 125.8, 127.6, 127.8, 128.1, 129.8, 133.2, 156.3, 160.5, 172.3, 186.4. Anal. Calcd for C21H23NO5: C, 68.28; H, 6.28; N, 3.79. Found: C, 68.37; H, 6.16; N, 3.91.
Ethyl 7-(Hydroxymethylene)-1-(3-methoxybenzyl)-8-oxo-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (46)
This compound was obtained from reaction of 20. Colorless oil; yield 68%; IR (cm–1) 3389 (OH), 1707 (CO), 1622 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.30 (t, 3H, J = 7.1 Hz, CH3), 1.88–1.98 (m, 2H, CH2), 2.15 (t, 2H, J = 6.7 Hz, CH2), 2.69 (t, 2H, J = 6.7 Hz, CH2), 3.73 (s, 3H, CH3), 4.24 (q, 2H, J = 7.1 Hz, CH2), 5.97 (s, 2H, CH2), 6.52 (d, 1H, J = 2.3 Hz, H-6′), 6.57 (s, 1H, H-2′), 6.72 (dd, 1H, J = 8.2, 2.3 Hz, H-4′), 6.82 (s, 1H, H-3), 7.15 (t, 1H, J = 8.2 Hz, H-5′), 7.72 (s, 1H, CH), 14.76 (s, 1H, OH); 13C NMR (CDCl3, 50 MHz) δ 14.2, 24.6, 25.8, 28.8, 49.1, 55.1, 60.5, 111.8, 112.2, 113.9, 117.9, 118.4, 127.1, 129.4, 130.2, 132.7, 140.9, 159.6, 160.6, 172.5, 186.5. Anal. Calcd for C21H23NO5: C, 68.28; H, 6.28; N, 3.79. Found: C, 68.40; H, 6.46; N, 3.58.
Ethyl 7-(Hydroxymethylene)-1-(4-methoxybenzyl)-8-oxo-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (47)
This compound was obtained from reaction of 21. Pale yellow oil; yield 73%; IR (cm–1) 3399 (OH), 1706 (CO), 1619 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.31 (t, 3H, J = 7.1 Hz, CH3), 1.83–1.97 (m, 2H, CH2), 2.12 (t, 2H, J = 6.6 Hz, CH2), 2.67 (t, 2H, J = 6.6 Hz, CH2), 3.75 (s, 3H, CH3), 4.23 (q, 2H, J = 7.1 Hz, CH2), 5.91 (s, 2H, CH2), 6.74 (s, 1H, H-3), 6.76–6.82 (m, 2H, H-3′ and H-5′), 6.91–7.00 (m, 2H, H-2′ and H-6′), 7.73 (d, 1H, J = 7.8 Hz, CH), 14.80 (d, 1H, J = 7.8 Hz, OH); 13C NMR (CDCl3, 50 MHz) δ 14.3, 24.4, 25.7, 28.9, 48.6, 55.2, 60.5, 113.7, 113.9, 117.9, 126.9, 127.7, 130.2, 131.4, 132.6, 158.5, 160.7, 172.5, 186.6. Anal. Calcd for C21H23NO5: C, 68.28; H, 6.28; N, 3.79. Found: C, 68.14; H, 6.41; N, 3.67.
Ethyl 1-(2,5-Dimethoxybenzyl)-7-(hydroxymethylene)-8-oxo-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (48)
This compound was obtained from reaction of 22. Pale yellow oil; yield 76%; IR (cm–1) 3398 (CO), 1709 (CO), 1623 (CO); 1H NMR (CDCl3, 200 MHz) δ (1.27 (q, 3H, J = 7.1 Hz, CH2), 1.85–1.99 (m, 2H, CH2), 2.17 (t, 2H, J = 6.6 Hz, CH2), 2.70 (t, 2H, J = 6.6 Hz, CH2), 3.64 (s, 3H, CH3), 3.78 (s, 3H, CH3), 4.22 (q, 2H, J = 7.1 Hz, CH2), 5.92 (s, 2H, CH2), 6.02 (d, 1H, J = 2.7 Hz, H-6′), 6.63–6.77 (m, 2H, H-3′ and H-4′), 6.83 (s, 1H, H-3), 7.70 (d, 1H, J = 8.2 Hz, CH), 14.73 (d, 1H, J = 8.2 Hz, OH); 13C NMR (CDCl3, 50 MHz) δ 14.2, 24.5, 25.8, 28.8, 45.6, 55.5, 55.8, 60.4, 110.6, 111.1, 113.0, 113.8, 117.7, 127.5, 129.4, 129.8, 133.1, 150.6, 153.5, 160.5, 172.2, 186.4. Anal. Calcd for C22H25NO6: C, 66.15; H, 6.31; N, 3.51. Found: C, 66.29; H, 6.42; N, 3.38.
Ethyl 1-(3,5-Dimethoxybenzyl)-7-(hydroxymethylene)-8-oxo-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (49)
This compound was obtained from reaction of 23. Pale yellow oil; yield 68%; IR (cm–1) 3398 (CO), 1706 (CO), 1609 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.30 (t, 3H, J = 7.1 Hz, CH3), 1.85–1.94 (m, 2H, CH2), 2.15 (t, 2H, J = 6.7 Hz, CH2), 2.69 (t, 2H, J = 6.7 Hz, CH2), 3.70 (s, 6H, 2 × CH3), 4.25 (q, 2H, J = 7.1 Hz, CH2), 5.95 (s, 2H, CH2), 6.11 (d, 2H, J = 2.2 Hz, H-2′ and H-6′), 6.28 (t, 1H, J = 2.2 Hz, H-4′), 6.82 (s, 1H, H-3), 7.71 (d, 1H, J = 7.9 Hz, CH), 14.78 (d, 1H, J = 7.9 Hz, OH); 13C NMR (CDCl3, 50 MHz) δ 14.3, 24.5, 25.8, 28.8, 49.1, 55.2, 60.5, 98.8, 104.1, 113.9, 118.0, 127.1, 130.2, 132.7, 141.8, 160.6, 160.8, 172.4, 186.6. Anal. Calcd for C22H25NO6: C, 66.15; H, 6.31; N, 3.51. Found: C, 65.93; H, 6.15; N, 3.77.
Ethyl 7-(Hydroxymethylene)-8-oxo-1-(3,4,5-trimethoxybenzyl)-1,4,5,6,7,8-hexahydrocyclohepta[b]pyrrole-2-carboxylate (50)
This compound was obtained from reaction of 24. Yellow oil; yield 60%; IR (cm–1) 3410 (OH), 1710 (CO), 1647 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.33 (t, 3H, J = 7.1 Hz, CH3), 1.88–1.98 (m, 2H, CH2), 2.13 (t, 2H, J = 6.8 Hz, CH2), 2.68 (t, 2H, J = 6.8 Hz, CH2), 3.74 (s, 6H, 2 × CH3), 3.78 (s, 3H, CH3), 4.28 (q, 2H, J = 7.1 Hz, CH2), 5.94 (s, 2H, CH2), 6.28 (s, 2H, H-2′ and H-6′), 6.81 (s, 1H, H-3), 7.74 (d, 1H, J = 8.0 Hz, CH), 14.83 (d, 1H, J = 8.0 Hz, OH); 13C NMR (CDCl3, 50 MHz) δ 14.3, 24.4, 25.7, 28.9, 48.9, 55.9, 60.6, 60.8, 100.0, 103.7, 114.0, 118.0, 126.7, 130.3, 134.9, 136.9, 153.1, 160.8, 172.5, 186.9. Anal. Calcd for C23H27NO7: C, 64.32; H, 6.34; N, 3.26. Found: C, 64.21; H, 6.48; N, 3.09.
General Procedure for the Preparation of Pyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole (51–68)
To a solution of the suitable enaminoketones 25–42 or hydroxymethylene ketones 43–50 (1.5 mmol) in ethanol (6 mL) and acetic acid (3 mL), hydroxylamine hydrochloride (1.65 mmol) was added, and the reaction mixture was heated at reflux for 1 h. Then the reaction mixture was poured onto crushed ice. The precipitate was removed by filtration and dried. If there was no precipitate, the solution was extracted with dichloromethane (3 × 20 mL). The organic layer was dried over Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography with dichloromethane/ethyl acetate 95:5 as eluting solvent.
9-(Phenylsulfonyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole (51)
This compound was obtained from reaction of 25. Orange solid; yield 60%; mp 140.3–141.0 °C; 1H NMR (CDCl3, 200 MHz) δ 1.88–1.98 (m, 2H, CH2), 2.65–2.77 (m, 4H, 2 × CH2), 6.20 (d, 1H, J = 3.3 Hz, H-7), 7.49–7.62 (m, 4H, H-8, H-3′, H-4′, and H-5′), 7.96 (s, 1H, H-3), 8.06 (d, 2H, J = 7.9 Hz, H-2′ and H-6′); 13C NMR (CDCl3, 50 MHz) δ 23.5, 23.7, 28.0, 113.1, 113.9, 118.9, 125.6, 128.1, 128.9, 133.1, 133.8, 138.9, 151.5, 156.1. Anal. Calcd for C16H14N2O3S: C, 61.13; H, 4.49; N, 8.91. Found: C, 61.31; H, 4.40; N, 9.10.
9-Methyl-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole (52)
This compound was obtained from reaction of 26. White solid; yield 75%; mp 60.4 °C; 1H NMR (CDCl3, 200 MHz) δ 1.89–2.00 (m, 2H, CH2), 2.74 (t, 2H, J = 6.0 Hz, CH2), 2.87 (t, 2H, J = 6.0 Hz, CH2), 3.95 (s, 3H, CH3), 5.95 (d, 1H, J = 2.5 Hz, H-7), 6.58 (d, 1H, J = 2.5 Hz, H-8), 8.01 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 23.2, 24.4, 28.8, 37.6, 99.9, 109.3, 111.1, 125.5, 127.3, 151.4, 159.7. Anal. Calcd for C11H12N2O: C, 70.19; H, 6.43; N, 14.88. Found: C, 70.39; H, 6.23; N, 14.63.
9-Benzyl-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole (53)
This compound was obtained from reaction of 27. Pale yellow solid; yield 83%; mp 61.3 °C; 1H NMR (CDCl3, 200 MHz) δ 1.88–2.00 (m, 2H, CH2), 2.72 (t, 2H, J = 6.2 Hz, CH2), 2.88 (t, 2H, J = 6.2 Hz, CH2), 5.52 (s, 2H, CH2), 6.02 (d, 1H, J = 2.6 Hz, H-7), 6.67 (d, 1H, J = 2.6 Hz, H-8), 7.12–7.33 (m, 5H, Ar), 7.94 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 23.1, 24.3, 28.8, 53.0, 110.2, 111.4, 119.1, 125.2, 126.9, 127.3, 127.7, 128.5, 138.3, 151.4, 159.3. Anal. Calcd for C17H16N2O: C, 77.25; H, 6.10; N, 10.60. Found: C, 77.60; H, 5.75; N, 10.25.
9-(2-Methoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole (54)
This compound was obtained from reaction of 28. Pale yellow solid; yield 70%; mp 80.4 °C; 1H NMR (CDCl3, 200 MHz) δ 1.91–2.02 (m, 2H, CH2), 2.74 (t, 2H, J = 5.5 Hz, CH2), 2.90 (t, 2H, J = 5.5 Hz, CH2), 3.87 (s, 3H, CH3), 5.54 (s, 2H, CH2), 6.01 (d, 1H, J = 2.6 Hz, H-7) 6.65 (d, 1H, J = 2.6 Hz, H-8), 6.71–6.89 (m, 3H, Ar), 7.15–7.23 (m, 1H, Ar), 7.95 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 23.2, 24.4, 28.8, 48.2, 55.3, 109.9, 110.0, 111.2, 119.2, 120.6, 125.3, 127.0, 127.3, 127.5, 128.4, 151.3, 156.5, 159.5. Anal. Calcd for C18H18N2O2: C, 73.45; H, 6.16; N, 9.52. Found: C, 73.63; H, 5.85; N, 9.60.
9-(3-Methoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole (55)
This compound was obtained from reaction of 29. Yellow solid; yield 83%; mp 67.6 °C; 1H NMR (CDCl3, 200 MHz) δ 1.89–2.00 (m, 2H, CH2), 2.73 (t, 2H, J = 5.5 Hz, CH2), 2.88 (t, 2H, J = 5.5 Hz, CH2), 3.74 (s, 3H, CH3), 5.50 (s, 2H, CH2), 6.02 (d, 1H, J = 2.6 Hz, H-7) 6.68 (d, 1H, J = 2.6 Hz, H-8), 6.71–6.78 (m, 3H, H-2′, H-4′ and H-6′), 7.20 (t, 1H, J = 7.9 Hz, H-5′), 7.95 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 23.2, 24.4, 28.8, 52.9, 55.1, 110.2, 111.4, 112.5, 112.7, 119.1, 119.2, 125.2, 127.7, 129.6, 140.0, 151.4, 159.3, 159.7. Anal. Calcd for C18H18N2O2: C, 73.45; H, 6.16; N, 9.52. Found: C, 73.31; H, 6.24; N, 9.50.
9-(4-Methoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole (56)
This compound was obtained from reaction of 30. Yellow solid; yield 68%; mp 101 °C; 1H NMR (CDCl3, 200 MHz) δ 1.88–1.99 (m, 2H, CH2), 2.73 (t, 2H, J = 5.8 Hz, CH2), 2.88 (t, 2H, J = 5.8 Hz, CH2), 3.76 (s, 3H, CH3), 5.45 (s, 2H, CH2), 6.01 (d, 1H, J = 2.5 Hz, H-7) 6.68 (d, 1H, J = 2.5 Hz, H-8), 6.82 (d, 2H, J = 8.5 Hz, H-3′ and H-5′), 7.12 (d, 2H, J = 8.5 Hz, H-2′ and H-6′), 7.95 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 23.2, 24.4, 28.8, 52.5, 55.2, 110.0, 111.5, 113.9, 118.9, 124.9, 127.7, 128.5, 130.2, 151.9, 158.8, 156.9. Anal. Calcd for C18H18N2O2: C, 73.45; H, 6.16; N, 9.52. Found: C, 73.55; H, 6.04; N, 9.44.
9-(2,5-Dimethoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole (57)
This compound was obtained from reaction of 31. White solid; yield 78%; mp 116.6 °C; 1H NMR (CDCl3, 200 MHz) δ 1.90–2.01 (m, 2H, CH2), 2.74 (t, 2H, J = 6.2 Hz, CH2), 2.89 (t, 2H, J = 6.2 Hz, CH2), 3.63 (s, 3H, CH3), 3.83 (s, 3H, CH3), 5.51 (s, 2H, CH2), 6.00 (d, 1H, J = 2.6 Hz, H-7), 6.29 (d, 1H, J = 2.6 Hz, H-8), 6.67–6.82 (m, 3H, H-3′, H-4′ and H-6′), 7.95 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 23.2, 24.4, 28.8, 48.2, 55.5, 55.9, 110.6, 110.9, 111.2, 112.1, 114.0, 119.2, 125.4, 127.4, 128.2, 150.8, 151.3, 153.6, 159.4. Anal. Calcd for C19H20N2O3: C, 70.35; H, 6.21; N, 8.64. Found: C, 70.49; H, 6.38; N, 8.47.
9-(3,5-Dimethoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole (58)
This compound was obtained from reaction of 32. Yellow solid; yield 76%; mp 118.6 °C; 1H NMR (CDCl3, 200 MHz) δ 1.89–2.00 (m, 2H, CH2), 2.73 (t, 2H, J = 6.0 Hz, CH2), 2.88 (t, 2H, J = 6.0 Hz, CH2), 3.72 (s, 6H, 2 × CH3), 5.46 (s, 2H, CH2), 6.01 (d, 1H, J = 2.6 Hz, H-7), 6.27 (d, 2H, J = 2.2 Hz, H-2′ and H-6′), 6.32 (t, 1H, J = 2.2 Hz, H-4′), 6.67 (d, 1H, J = 2.6 Hz, H-8), 7.95 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 23.2, 24.3, 28.8, 53.0, 55.2, 99.1, 104.8, 110.2, 111.4, 119.1, 125.2, 127.7, 140. 8, 151.4, 159.3, 160.9. Anal. Calcd for C19H20N2O3: C, 70.35; H, 6.21; N, 8.64. Found: C, 70.55; H, 6.36; N, 8.49.
9-(3,4,5-Trimethoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole (59)
This compound was obtained from reaction of 33. White solid; yield 74%; mp 104.5 °C; 1H NMR (CDCl3, 200 MHz) δ 1.89–2.00 (m, 2H, CH2), 2.74 (t, 2H, J = 6.0 Hz, CH2), 2.88 (t, 2H, J = 6.0 Hz, CH2), 3.77 (s, 6H, 2 × CH3), 3.80 (s, 3H, CH3), 5.45 (s, 2H, CH2), 6.02 (d, 1H, J = 2.6 Hz, H-7), 6.39 (s, 2H, H-2′ and H-6′), 6.69 (d, 1H, J = 2.6 Hz, H-8), 7.97 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 23.3, 24.4, 28.8, 53.2, 56.0, 60.8, 104.1, 110.2, 111.5, 119.1, 125.1, 127.9, 133.8, 137.1, 151.5, 153.3, 159.3. Anal. Calcd for C20H22N2O4: C, 67.78; H, 6.26; N, 7.90. Found: C, 67.87; H, 6.17; N, 7.81.
Ethyl 9-Methyl-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (60)
This compound was obtained from reaction of 34 (yield 90%) or 43 (yield 86%). White solid; mp 91.7 °C; IR (cm–1) 1699 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.36 (t, 3H, J = 7.1 Hz, CH3), 1.89–2.00 (m, 2H, CH2), 2.74–2.85 (m, 4H, 2 × CH2), 4.24–4.35 (m, 5H, CH2 and CH3), 6.76 (s, 1H, H-7), 8.08 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 14.4, 23.4, 24.5, 28.1, 35.2, 60.1, 114.8, 114.9, 118.0, 124.2, 126.1, 151.8, 158.3, 161.1. Anal. Calcd for C14H16N2O3: C, 64.60; H, 6.20; N, 10.76. Found: C, 64.45; H, 5.96; N, 11.00.
Ethyl 9-Benzyl-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (61)
This compound was obtained from reaction of 35 (yield 70%) or 44 (yield 68%). Gray solid; mp 149.4 °C; IR (cm–1) 1695 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.30 (t, 3H, J = 7.1 Hz, CH3), 1.92–2.00 (m, 2H, CH2), 2.71–2.89 (m, 4H, 2 × CH2), 4.24 (q, 2H, J = 7.1 Hz, CH2), 6.17 (s, 2H, CH2), 6.87 (s, 1H, H-7), 6.99–7.07 (m, 2H, H-2′ and H-6′), 7.13–7.30 (m, 3H, H-3′, H-4′ and H-5′), 8.00 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 14.3, 23.4, 24.5, 28.2, 50.1, 60.2, 115.1, 119.0, 124.2, 125.5, 126.1, 126.7, 126.8, 128.3, 139.0, 151.7, 157.8, 160.8. Anal. Calcd for C20H20N2O3: C, 71.41; H, 5.99; N, 8.33. Found: C, 71.26; H, 6.14; N, 8.31.
Ethyl 9-(2-Methoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (62)
This compound was obtained from reaction of 36 (yield: 74%) or 45 (yield: 70%). White solid; mp 137.5–138.3 °C; IR (cm–1) 1700 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.26 (t, 3H, J = 7.1 Hz, CH3), 1.89–2.06 (m, 2H, CH2), 2.77 (t, 2H, J = 6.1 Hz, CH2), 2.86 (t, 2H, J = 6.1 Hz, CH2), 3.89 (s, 3H, CH3), 4.20 (q, 2H, J = 7.1 Hz, CH2), 6.11 (s, 2H, CH2), 6.29 (d, 1H, J = 7.5 Hz, H-3′), 6.68–6.88 (m, 3H, H-7, H-5′ and H-6′), 7.13 (t, 1H, J = 8.1 Hz, H-4′), 7.97 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 14.2, 23.5, 24.5, 28.2, 46.5, 55.3, 60.1, 109.8, 114.9, 118.8, 120.3, 124.7, 125.0, 125.8, 126.5, 127.5, 128.0, 151.6, 156.2, 158.0, 160.6. Anal. Calcd for C21H22N2O4: C, 68.84; H, 6.05; N, 7.65. Found: C, 68.96; H, 5.78; N, 7.53.
Ethyl 9-(3-Methoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (63)
This compound was obtained from reaction of 37 (yield 82%) or 46 (yield 80%). Pale yellow solid; mp 98–98.4 °C; IR (cm–1) 1696 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.30 (t, 3H, J = 7.1 Hz, CH3), 1.91–2.00 (m, 2H, CH2), 2.72–2.87 (m, 4H, 2 × CH2), 3.71 (s, 3H, CH3), 4.24 (q, 2H, J = 7.1 Hz, CH2), 6.15 (s, 2H, CH2), 6.54–6.73 (m, 3H, H-2′, H-4′ and H-6′), 6.86 (s, 1H, H-7), 7.14 (t, 1H, J = 7.9 Hz, H-5′), 8.00 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 14.3, 23.4, 24.5, 28.2, 49.9, 55.0, 60.2, 111.8, 112.0, 115.0, 118.4, 119.0, 124.3, 125.5, 126.7, 129.3, 140.7, 151.7, 157.8, 159.6, 160.8. Anal. Calcd for C21H22N2O4: C, 68.84; H, 6.05; N, 7.65. Found: C, 68.53; H, 6.36; N, 7.41.
Ethyl 9-(4-Methoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (64)
This compound was obtained from reaction of 38 (yield 84%) or 47 (yield 82%). White solid; mp 93.9 °C; IR (cm–1) 1690 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.32 (t, 3H, J = 7.1 Hz, CH3), 1.88–1.99 (m, 2H, CH2), 2.72–2.86 (m, 4H, 2 × CH2), 3.74 (s, 3H, CH3), 4.26 (q, 2H, J = 7.1 Hz, CH2), 6.09 (s, 2H, CH2), 6.76 (d, 2H, J = 6.8 Hz, H-3′ and H-5′), 6.85 (s, 1H, H-7), 7.01 (d, 2H, J = 8.6 Hz, H-2′ and H-6′), 8.02 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 14.3, 23.4, 24.5, 28.2, 49.4, 55.1, 60.2, 113.7, 115.0, 119.0, 124.3, 125.5, 126.7, 127.6, 131.1, 151.8, 157.8, 158.4, 160.9. Anal. Calcd for C21H22N2O4: C, 68.84; H, 6.05; N, 7.65. Found: C, 68.61; H, 6.20; N, 7.87.
Ethyl 9-(2,5-Dimethoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (65)
This compound was obtained from reaction of 39 (yield 74%) or 48 (yield 72%). White solid; mp 94.9 °C; IR (cm–1) 1690 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.26 (t, 3H, J = 7.1 Hz, CH3), 1.93–2.02 (m, 2H, CH2), 2.74–2.88 (m, 4H, 2 × CH2), 3.60 (s, 3H, CH3), 3.85 (s, 3H, CH3), 4.20 (q, 2H, J = 7.1 Hz, CH2), 5.91 (d, 1H, J = 2.6 Hz, H-6′), 6.08 (s, 2H, CH2), 6.63 (dd, 1H, J = 8.7, 2.6 Hz, H-4′), 6.76 (d, 1H, J = 8.7 Hz, H-3′), 6.87 (s, 1H, H-7), 7.97 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 14.2, 23.5, 24.5, 28.2, 46.5, 55.4, 55.9, 60.1, 110.4, 110.6, 112.6, 114.9, 118.9, 124.5, 125.6, 126.5, 129.5, 150.6, 151.6, 153.5, 157.9, 160.6. Anal. Calcd for C22H24N2O5: C, 66.65; H, 6.10; N, 7.07. Found: C, 66.49; H, 6.36; N, 6.81.
Ethyl 9-(3,5-Dimethoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (66)
This compound was obtained from reaction of 40 (yield 82%) or 49 (yield 80%). White solid; mp 114 °C; IR (cm–1) 1701 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.31 (t, 3H, J = 7.1 Hz, CH3), 1.92–2.00 (m, 2H, CH2), 2.64–2.86 (m, 4H, 2 × CH2), 3.70 (s, 6H, 2 × CH3), 4.24 (q, 2H, J = 7.1 Hz, CH2), 6.07–6.28 (m, 4H, CH2, H-2′ and H-6′), 6.26 (s, 1H, H-4′), 6.86 (s, 1H, H-7), 8.00 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 14.3, 23.4, 24.5, 28.2, 50.0, 55.2, 60.2, 98.6, 100.0, 104.1, 115.1, 119.1, 124.2, 126.7, 141.6, 151.7, 157.8, 160.7, 160.8. Anal. Calcd for C22H24N2O5: C, 66.65; H, 6.10; N, 7.07. Found: C, 66.84; H, 6.42; N, 6.75.
Ethyl 9-(3,4,5-Trimethoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (67)
This compound was obtained from reaction of 41 (yield 76%) or 50 (yield 75%). Pale yellow solid; mp 112.9–113.1 °C; IR (cm–1) 1696 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.33 (t, 3H, J = 7.1 Hz, CH3), 1.88–1.99 (m, 2H, CH2), 2.73–2.87 (m, 4H, 2 × CH2), 3.72 (s, 6H, 2 × CH3), 3.77 (s, 3H, CH3), 4.28 (q, 2H, J = 7.1 Hz, CH2), 6.11 (s, 2H, CH2), 6.29 (s, 2H, H-2′ and H-6′), 6.86 (s, 1H, H-7), 8.04 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 14.4, 23.6, 24.5, 28.1, 49.8, 55.9, 60.3, 60.8, 100.0, 103.5, 115.1, 119.1, 124.2, 125.5, 126.9, 134.6, 151.8, 153.1, 157.8, 161.0. Anal. Calcd for C23H26N2O6: C, 64.78; H, 6.15; N, 6.57. Found: C, 64.44; H, 6.05; N, 6.67.
Methyl 9-Benzyl-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (68)
This compound was obtained from reaction of 42. White solid; yield 78%; mp 150.3 °C; IR (cm–1) 1698 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.89–2.00 (m, 2H, CH2), 2.77 (t, 2H, J = 6.2 Hz, CH2), 2.84 (t, 2H, J = 6.2 Hz, CH2), 3.78 (s, 3H, CH3), 6.17 (s, 2H, CH2), 6.86 (s, 1H, H-7), 6.98 (s, 2H, J = 9.7 Hz, H-2′ and H-6′), 7.12–7.28 (m, 3H, H-3′, H-4′ and H-5′), 8.01 (s, 1H, H-3); 13C NMR (CDCl3, 200 MHz) δ 23.3, 24.5, 28.1, 50.0, 51.3, 115.1, 119.1, 123.8, 125.6, 126.0, 126.7, 126.8, 128.3, 138.9, 151.7, 157.7, 161.2. Anal. Calcd for C19H18N2O3: C, 70.79; H, 5.63; N, 8.69. Found: C, 70.65; H, 5.77; N, 8.58.
General Procedure for the Preparation of Chloropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole (69–76)
To a solution of 60–66, 52 (1 mmol) in anhydrous DMF (5 mL) a solution of N-chlorosuccinimide (1.5 mmol) in anhydrous DMF (2 mL) was added, and the reaction mixture was stirred for 16 h at room temperature. Then, the reaction mixture was poured onto crushed ice. The precipitate was removed by filtration and dried. In the absence of a precipitate, the solution was extracted with dichloromethane (3 × 30 mL). The organic layer was dried over Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography with dichloromethane/ethyl acetate 98:2 as eluting solvent.
Ethyl 7-Chloro-9-methyl-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (69)
This compound was obtained from reaction of 60. White solid; yield 60%; mp 88.2 °C; IR (cm–1) 1695 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.43 (t, 3H, J = 7.1 Hz, CH3), 1.94–2.04 (m, 2H, CH2), 2.77–2.89 (m, 4H, 2 × CH2), 4.29 (s, 3H, CH3), 4.40 (q, 2H, J = 7.1 Hz, CH2), 8.08 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 14.4, 23.3, 24.6, 25.4, 36.2, 60.7, 115.7, 119.5, 121.4, 124.1, 124.5, 151.7, 157.7, 160.6. Anal. Calcd for C14H15ClN2O3: C, 57.05; H, 5.13; N, 9.50. Found: C, 57.16; H, 5.00; N, 9.57.
Ethyl 7-Chloro-9-benzyl-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (70)
This compound was obtained from reaction of 61. White solid; yield 75%; mp 141.8–142.4 °C; IR (cm–1) 1696 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.32 (t, 3H, J = 7.1 Hz, CH3), 1.90–2.02 (m, 2H, CH2), 2.73–2.89 (m, 4H, 2 × CH2), 4.30 (q, 2H, J = 7.1 Hz, CH2), 6.12 (s, 2H, CH2), 6.99 (d, 2H, J = 7.2 Hz, H-2′ and H-6′), 7.14–7.26 (m, 3H, H-3′, H-4′ and H-5′), 8.02 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 14.2, 23.1, 24.4, 25.3, 50.8, 60.8, 116.1, 120.4, 121.2, 124.1, 124.8, 126.0, 127.0, 128.4, 138.5, 151.7, 157.0, 160.3. Anal. Calcd for C20H19ClN2O3: C, 64.78; H, 5.16; N, 7.55. Found: C, 64.98; H, 5.06; N, 7.65.
Ethyl 7-Chloro-9-(2-methoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (71)
This compound was obtained from reaction of 62. Yellow solid; yield 74%; mp 159.8 °C; IR (cm–1) 1696 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.26 (t, 3H, J = 7.1 Hz, CH3), 1.92–2.03 (m, 2H, CH2), 2.77 (t, 2H, J = 5.8 Hz, CH2), 2.87 (t, 2H, J = 5.8 Hz, CH2), 3.85 (s, 3H, CH3), 4.26 (q, 2H, J = 7.1 Hz, CH2), 6.05 (s, 2H, CH2), 6.41 (d, 1H, J = 7.4 Hz, H-3′), 6.71–6.85 (m, 2H, H-5′ and H-6′), 7.14 (t, 1H, J = 7.4 Hz, H-4′), 8.00 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 14.0, 23.2, 24.4, 25.3, 42.3, 55.3, 60.7, 109.8, 115.8, 120.0, 120.4, 121.5, 124.3, 124.5, 125.4, 127.4, 127.8, 151.6, 156.2, 157.2, 160.1. Anal. Calcd for C21H21ClN2O4: C, 62.92; H, 5.28; N, 6.99. Found: C, 62.72; H, 5.51; N, 6.91.
Ethyl 7-Chloro-9-(3-methoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (72)
This compound was obtained from reaction of 63. Green solid; yield 65%; mp 92.8–93.5 °C; IR (cm–1) 1695 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.33 (t, 3H, J = 7.1 Hz, CH3), 1.90–2.02 (m, 2H, CH2), 2.73–2.89 (m, 4H, 2 × CH2), 3.72 (s, 3H, CH3), 4.30 (q, 2H, J = 7.1 Hz, CH2), 6.10 (s, 2H, CH2), 6.53–6.61 (m, 2H, H-2′ and H-6′), 6.70 (dd, 1H, J = 7.9, 2.3 Hz, H-4′), 7.15 (t, 1H, J = 7.9 Hz, H-5′), 8.03 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 14.2, 23.1, 24.4, 25.3, 50.6, 55.1, 60.8, 111.9, 112.1, 116.1, 118.3, 120.4, 121.1, 124.7, 129.4, 138.4, 140.2, 151.7, 157.0, 159.6, 160.3. Anal. Calcd for C21H21ClN2O4: C, 62.92; H, 5.28; N, 6.99. Found: C, 62.84; H, 5.17; N, 7.08.
Ethyl 7-Chloro-9-(4-methoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (73)
This compound was obtained from reaction of 64. White solid; yield 70%; mp 118.1 °C; IR (cm–1) 1695 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.34 (t, 3H, J = 7.1 Hz, CH3), 1.89–2.01 (m, 2H, CH2), 2.73–2.87 (m, 4H, 2 × CH2), 3.73 (s, 3H, CH3), 4.32 (q, 2H, J = 7.1 Hz, CH2), 6.05 (s, 2H, CH2), 6.76 (d, 2H, J = 8.7 Hz, H-3′ and H-5′), 6.97 (d, 2H, J = 8.7 Hz, H-2′ and H-6′), 8.04 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 14.2, 23.1, 24.4, 25.3, 50.1, 55.2, 60.8, 113.8, 116.1, 120.3, 121.1, 123.9, 124.8, 127.6, 130.6, 151.8, 151.1, 158.6, 160.4. Anal. Calcd for C21H21ClN2O4: C, 62.92; H, 5.28; N, 6.99. Found: C, 62.99; H, 5.39; N, 6.93.
Ethyl 7-Chloro-9-(2,5-dimethoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (74)
This compound was obtained from reaction of 65. White solid; yield 68%; mp 147.2 °C; IR (cm–1) 1696 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.28 (t, 3H, J = 7.1 Hz, CH3), 1.92–2.03 (m, 2H, CH2), 2.77 (t, 2H, J = 6.0 Hz, CH2), 2.86 (t, 2H, J = 6.0 Hz, CH2), 3.62 (s, 3H, CH3), 3.82 (s, 3H, CH3), 4.27 (q, 2H, J = 7.1 Hz, CH2), 5.98–6.06 (m, 3H, CH2 and H-6′), 6.65 (dd, 1H, J = 8.8, 2.6 Hz, H-4′), 6.76 (d, 1H, J = 8.8 Hz, H-3′), 8.01 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 14.0, 23.1, 24.4, 25.3, 47.2, 55.5, 55.8, 60.7, 110.5, 110.7, 113.0, 115.9, 120.2, 121.3, 124.2, 124.5, 128.8, 150.6, 151.6, 153.4, 157.1, 160.0. Anal. Calcd for C22H23ClN2O5: C, 61.32; H, 5.38; N, 6.50. Found: C, 61.02; H, 5.52; N, 6.62.
Ethyl 7-Chloro-9-(3,5-dimethoxybenzyl)-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole-8-carboxylate (75)
This compound was obtained from reaction of 66. White solid; yield 75%; mp 165.5 °C; IR (cm–1) 1701 (CO); 1H NMR (CDCl3, 200 MHz) δ 1.26 (t, 3H, J = 7.1 Hz, CH3), 1.94–2.03 (m, 2H, CH2), 2.79 (t, 2H, J = 6.0 Hz, CH2), 2.87 (t, 2H, J = 6.0 Hz, CH2), 3.58 (s, 3H, CH3), 3.87 (s, 3H, CH3), 4.21 (q, 2H, J = 7.1 Hz, CH2), 5.50 (d, 1H, J = 2.6 Hz, H-2′), 6.13 (s, 2H, CH2), 6.34 (d, 1H, J = 2.6 Hz, H-6′), 6.91 (s, 1H, H-4′), 7.99 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 14.2, 23.4, 24.5, 28.2, 49.1, 55.2, 56.2, 60.2, 97.2, 102.9, 111.7, 115.1, 119.1, 124.5, 125.7, 126.6, 139.2, 151.6, 155.7, 157.6, 158.9, 160.5. Anal. Calcd for C22H23ClN2O5: C, 61.32; H, 5.38; N, 6.50. Found: C, 61.36; H, 5.39; N, 6.29.
8-Chloro-9-methyl-4,5,6,9-tetrahydropyrrolo[2′,3′:3,4]cyclohepta[1,2-d][1,2]oxazole (76)
This compound was obtained from reaction of 52. White solid; yield 62%; mp 66.9–67.2 °C; 1H NMR (CDCl3, 200 MHz) δ 1.87–1.99 (m, 2H, CH2), 2.71–2.83 (m, 4H, 2 × CH2), 3.92 (s, 3H, CH3), 5.95 (s, 1H, H-7), 8.02 (s, 1H, H-3); 13C NMR (CDCl3, 50 MHz) δ 23.2, 24.3, 28.6, 33.6, 99.9, 108.4, 111.4, 120.2, 126.2, 151.5, 158.9. Anal. Calcd for C11H11ClN2O: C, 59.33; H, 4.98; N, 12.58. Found: C, 59.28; H, 5.09; N, 12.71.
Biology
Cell Lines and Compounds
Established human cell lines derived from germinal center B-cell (GCB) (SU-DHL-10), activated B-cell (ABC) (HBL1) diffuse large B-cell lymphoma (DLBCL), mantle cell lymphoma (MCL) (MINO), splenic marginal zone lymphoma (SMZL) (parental VL51, VL51 idelalisib resistant clone, VL51 ibrutinib resistant clone) were cultured in culture RPMI-1640 media supplemented with fetal bovine serum (10%), penicillin–streptomycin–neomycin (∼5,000 units of penicillin, 5 mg of streptomycinm and 10 mg of neomycin/mL, Sigma), and l-glutamine (1%). Vincristine sulfate was purchased from Sigma-Aldrich. Cell line identities were confirmed by CellCheck test (IDEXX, BioResearch, Ludwigsburg, Germany). All compounds were dissolved in dimethyl sulfoxide to obtain a stock concentration of 10 mM.
Cell Proliferation Analysis
The antiproliferative activity of all compounds was assessed by using the 3-(4.5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) test. Cells were seeded in 96-well plates (nontissue culture treated) at a density of 1 × 105 cells/mL and treated with a single concentration of 1 μM for 72 h. Selected compounds that reached proliferation inhibition below 60% were further tested in order to calculate IC50 values. In this case, cells were treated in triplicate with serially diluted compounds in the appropriate tissue culture medium at a range of 40–10 000 nM. Cells were incubated for 72 h at 37 °C, 5% CO2. Wells containing medium only were included on each plate and served as blanks for absorbance readings. MTT (Sigma, Buchs, Switzerland) was prepared as a 5 mg/mL stock solution in phosphate buffered saline (PBS) and filter-sterilized. MTT solution (22 μL) was added to each well, and tissue culture plates were incubated at 37 °C for 4 h. Cells were then lysed with 25% sodium dodecyl sulfate lysis buffer, and absorbance was read at 570 nm using a Beckman Coulter-AD340 plate reader.
Evaluation of Cytotoxicity in PBLs
PBLs were obtained from human peripheral blood (leucocyte rich plasma-buffy coats) from healthy volunteers using the Lymphoprep (Fresenius KABI Norge AS) gradient density centrifugation.
Buffy coats were obtained from the Blood Transfusion Service, Azienda Ospedaliera of Padova and provided at this institution for research purposes. Therefore, no further informed consent was needed. In addition, buffy coats were provided without identifiers. The experimental procedures were carried out in strict accordance with approved guidelines.
After extensive washing, cells were resuspended (1.0 × 106 cells/mL) in RPMI-1640 with 10% fetal bovine serum and incubated overnight. For cytotoxicity evaluations in proliferating PBL cultures, nonadherent cells were resuspended at 5 × 105 cells/mL in growth medium, containing 2.5 μg/mL PHA (Irvine Scientific). Different concentrations of the test compounds were added, and viability was determined 72 h later by the MTT test. For cytotoxicity evaluations in resting PBL cultures, nonadherent cells were resuspended (5 × 105 cells/mL) and treated for 72 h with the test compounds.
Tubulin Studies
Electrophoretically pure bovine brain tubulin was obtained as described previously.70 Analysis of effects on tubulin polymerization was performed by turbidimetry at 350 nm in recording spectrophotometers equipped with electronic temperature controllers as described in detail elsewhere.71 The tubulin used in these studies was more active than that used in ref (71), and so the concentration of tubulin was reduced from 10 to 9 μM (1.0 to 0.9 mg/mL) and the concentration of GTP from 0.4 to 0.2 mM. This was done to obtain an IC50 for CA-4, the reference compound, similar to that obtained in ref (71). The binding of [3H]colchicine to tubulin was perfomed as described in detail previously72 except that the tubulin concentration was reduced from 0.1 mg/mL to 0.05 mg/mL and only one, instead of two, DEAE-cellulose filter was used for each reaction mixture.
Flow Cytometric Analysis of Cell Cycle Distribution
5 ×105 HeLa cells were treated with different concentrations of the test compounds for 24 h. After the incubation period, the cells were collected, centrifuged, and fixed with ice-cold ethanol (70%). The cells were treated with lysis buffer containing RNase A and 0.1% Triton X-100 and stained with PI. Samples were analyzed on a Cytomic FC500 flow cytometer (Beckman Coulter). DNA histograms were analyzed using MultiCycle for Windows (Phoenix Flow Systems).
Apoptosis Assay
Cell death was determined by flow cytometry of cells double stained with annexin V/FITC and PI. The Coulter Cytomics FC500 (Beckman Coulter) was used to measure the surface exposure of phosphatidylserine on apoptotic cells according to the manufacturer’s instructions (Annexin-V Fluos, Roche Diagnostics).
Assessment of Mitochondrial Potential and ROS
The mitochondrial membrane potential was measured with the lipophilic cationic dye 5,5′,6,6′ tetrachlo-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine (JC-1) (Molecular Probes), as described.73 The method is based on the ability of this fluorescent probe to enter selectively into mitochondria since it changes reversibly its color from green to red as membrane potential increases. This property is due to the reversible formation of JC-1 aggregates upon membrane polarization that causes a shift in the emitted light from 530 nm (i.e., emission of JC-1 monomeric form) to 590 nm (emission of JC-1-aggregate) when excited at 490 nm.
The production of ROS was measured by flow cytometry using H2DCFDA (Molecular Probes), as previously described.59 Briefly, after different times of treatment, cells were collected by centrifugation and resuspended in PBS containing H2DCFDA at the concentration of 0.1 μM. The cells were then incubated for 30 min at 37 °C, centrifuged, and resuspended in PBS. The fluorescence was directly recorded with the flow cytometer, using as excitation wavelength 488 nm and emission at 530 nm.
Western Blot Analysis
HeLa cells were incubated in the presence of the test compound and, after different times, were collected, centrifuged, and washed two times with ice cold PBS. The pellet was resuspended in lysis buffer. After the cells were lysed on ice for 30 min, lysates were centrifuged at 15 000g at 4 °C for 10 min. The protein concentration in the supernatant was determined using the BCA protein assay reagents (Pierce, Italy). Equal amounts of protein (10 μg) were resolved using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Criterion Precast, BioRad, Italy) and transferred to a PVDF Hybond-P membrane (GE Healthcare). Membranes were blocked with a bovine serum albumin solution (5% in Tween PBS 1×), and the membranes were gently rotated overnight at 4 °C in the albumin solution. Membranes were then incubated with primary antibodies against PARP cleaved fragment, cdc25c, cyclin B, p-cdc2Tyr15, XIAP, and Mcl-1 (all from Cell Signaling) or GAPDH (Sigma-Aldrich) for 2 h at room temperature. Membranes were next incubated with peroxidase labeled secondary antibodies for 1 h. All membranes were visualized using ECL Select (GE Healthcare), and images were acquired using an Uvitec-Alliance imaging system (Uvitec, Cambridge, U.K.). To ensure equal protein loading, each membrane was stripped and reprobed with anti-GAPDH antibody. To obtain relative quantitative data, ImageJ software (NIH, USA) was used for scanning densitometry analysis of Western blots.
Molecular Modeling
Docking Studies
All molecular modeling simulations were carried out using the Schrödinger Suite version 2018.74 In particular, the LigPrep tool75 was used to model the 3D structure of each ligand, to calculate and to energy minimize their protonation state at pH 7.4 using OPLS_2005 as force field.76 First, docking studies of new derivatives were performed by using three different crystal structures of tubulin, characterized by two dimers of α–β tubulin heterodimers, downloaded from the Protein Data Bank (PDB).77 Models having PDB codes 4O2B and 1Z2B were selected as colchicine-bound78 and vinblastine-bound79 cocrystal structures, respectively. According to literature data, the tubulin colchicine domain consists of the main site, where colchicine binds (zone 1), and two additional neighboring pockets (zones 2 and 3).80 To better discriminate the most likely binding area of the new derivatives, in addition to the 4O2B structure, representing the colchicine-like binding site area (zones 1 and 2), the model with PDB code 3N2G,80 cocrystallized with the inhibitor G2N, was also used in docking studies to represent the binding zones 2 and 3. Each X-ray model was preprocessed using the Protein Preparation Wizard tool and the OPLS_2005 force field, in order to add hydrogen atoms, to assign partial charges and to build missing atoms, side chains, and loops. The nucleotides (GTP and GDP) and the metals (Mg2+ and Zn2+) were retained during the docking calculations, while all water molecules were removed. Thus, the docking grids were prepared using as centroid the cocrystallized ligands (i.e., colchicine for 1SA0, vinblastine for 1Z2B, and G2N for 3N2G), while box size and position were generated automatically. Docking studies were performed by using the software Glide version 7.881 and by applying the Glide Extra-Precision (XP) protocol, selected after redocking analysis (for details, see Supporting Information paragraph “Redocking Analysis” and Table S1). Ten poses per ligand were taken into account, and the default docking scoring function was used for selecting the best binding mode for each ligand. To perform the following computational studies, for each tubulin crystal structure only one α-tubulin and one β-tubulin structure were selected, specifically the C and D chains for 4O2B, the B and C chains for 1Z2B, and the A and B chains for 3N2G, with respect to the redocking analysis.
Molecular Dynamics Simulations (MDs)
To better characterize the binding mode of our best active compounds (57, 58, 63, 66, 67, 75, and 3), their complexes with the 4O2B model were subjected to molecular dynamics simulations (MDs). The Desmond package82 was used for MDs, employing OPLS_2005 as force field in an explicit solvent (TIP3 water model).83 The best docking pose for each single compound was taken as initial coordinates for the MDs. An orthorhombic water box was built for the solvation of the system, ensuring a buffer distance of approximately 10 Å between each box side and the complex atoms. The system was neutralized by adding K+ counterions, and it was minimized and pre-equilibrated using the default relaxation routine implemented in Desmond. Simulation time was set to 20 ns, under NPT conditions at 1 atm and 300 K, with a recording interval equal to 40 ps. The time step was set to 2 fs. MD analyses were performed using the Simulation Event Analysis tool of Desmond, while visualization of each protein–ligand complex was carried out using Maestro. For each compound, average ligand RMSD value was calculated on their heavy atoms by first aligning the complex on the protein backbone of the reference structure. Moreover, by use of the Desmond Trajectory Clustering tool, the best representative structure of the whole MDs was generated in order to examine the possibility of induced-fit binding events of our compounds. Finally, these selected structures were submitted to the calculation of the ΔGbind value by using the MM/GBSA method as implemented in the Prime module84 from Maestro using the default settings.
Statistical Analysis
The differences between different treatments were analyzed, using the two-sided Student’s t test. P values lower than 0.05 were considered significant.
Acknowledgments
This work was financially supported by the Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR). This research was supported in part by the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute, which includes federal funds under Contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The authors also thank the Developmental Therapeutic Program of the National Cancer Institute for performing cytoxicity studies with selected compounds in the 60 cancer cell line screen. The authors also acknowledge the Italian Association for Cancer Research (AIRC) research project “Small molecule-based targeting of lncRNAs 3D structure: a translational platform for the treatment of multiple myeloma” (Code 21588), the PRIN 2017 research project “Novel anticancer agents endowed with multi-targeting mechanism of action” (Code 201744BN5T), and the PRIN 2017 research project “Selective mGlu3 metabotropic glutamate receptor ligands as new potential therapeutic agents in experimental models of parkinsonism” (Code 2017XZ7A37) funded by the Italian MIUR.
Glossary
Abbreviations Used
- NCI
National Cancer Istitute
- FDA
Food and Drug Administration
- CA-4
combretastatin A-4
- CA-4P
combretastatin A-4 phosphate disodium
- MDR
multidrug resistance
- MG_MID
mean graph midpoint
- DMPM
diffuse malignant peritoneal mesothelioma
- DMF
N,N-dimethylformamide
- DMFDMA
N,N-dimethylformamide dimethyl acetal
- MW
microwave
- TBDMAM
tert-butoxy bis(dimethylamino)methane
- PI3Kδ
phosphatidylinositol 3-kinase-δ
- BTK
Bruton’s tyrosine kinase
- MZL
marginal zone lymphoma
- MCL
mantle cell lymphoma
- ABC DLBCL
activated B-cell-like diffuse large B cell lymphoma
- GCB DLBCL
germinal center B-cell type diffuse large B cell lymphoma
- PBL
peripheral blood lymphocyte
- PHA
phytohematoaglutinin
- RMSD
root-mean-square deviation
- MDs
molecular dynamics simulations
- PI
propidium iodide
- ROS
reactive oxygen species
- H2-DCFDA
2,7-dichlorodihydrofluorescein diacetate
- DCF
dichlorofluorescein
- PARP
poly(ADP)ribose polymerase
- DMSO
dimethyl sulfoxide
- s
singlet
- d
doublet
- t
triplet
- q
quartet
- m
multiplet
- TIC
total ion current.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01315.
Figures S1–S4 showing dose–response curves and mean graph midpoints of compounds 66 and 67; Table S1 listing RMSD (Å) and G-Score (kcal/mol) values, obtained by means of Glide Extra-Precision (XP), for each chain in the dimer used in the crystallographic structures used in computational simulations; Table S2 listing G-Score (kcal/mol) values for the best poses of all compounds complexed with both the 4O2B and 1Z2B crystallographic structures; Table S3 listing G-Score values, expressed as kcal/mol, of the best active compounds 57, 58, 63, 66, 67, 75, and 3 against both the 4O2B and 3N2G crystallographic structures; Figure S5 showing the best-docked poses of 57, 58, 63, 66, 67, 75, and 3 against the crystal structure of tubulin with the PDB code 3N2G, depicting zones 2 and 3 of the colchicine site; geometric and energetic analysis of molecular dynamics simulations (MDs); Table S4 listing representative structures of MDs, number of contacts, and binding energy; Figures S6–S31 showing NMR spectra of compounds 51–76; HPLC traces of compounds 66 and 67 (PDF)
PDB files (ZIP)
Accession Codes
The PDB code for cocrystal structure of tubulin with colchicine is 4O2B. The PDB code for cocrystal structure of tubulin with vinblastine is 1Z2B. The PDB code for cocrystal structure of tubulin with inhibitor G2N is 3N2G.
Author Contributions
◇ V.S., R.R., and M.B. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Preti D.; Romagnoli R.; Rondanin R.; Cacciari B.; Hamel E.; Balzarini J.; Liekens S.; Schols D.; Estévez-Sarmiento F.; Quintana J.; Estevez F. Design, synthesis, in vitro antiproliferative activity and apoptosis-inducing studies of 1-(3′,4′,5′-trimethoxyphenyl)-3-(2′-alkoxycarbonylindolyl)-2-propen-1-one derivatives obtained by a molecular hybridisation approach. J. Enzyme Inhib. Med. Chem. 2018, 33 (1), 1225–1238. 10.1080/14756366.2018.1493473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmanova A.; Steinmetz M. O. Control of microtubule organization and dynamics: two ends in the limelight. Nat. Rev. Mol. Cell Biol. 2015, 16 (12), 711–726. 10.1038/nrm4084. [DOI] [PubMed] [Google Scholar]
- Jordan M. A.; Wilson L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Seddigi Z. S.; Malik M. S.; Saraswati A. P.; Ahmed S. A.; Babalghith A. O.; Lamfon H. A.; Kamal A. Recent advances in combretastatin based derivatives and prodrugs as antimitotic agents. MedChemComm 2017, 8 (8), 1592–1603. 10.1039/C7MD00227K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Xu F.; Shuai W.; Sun H.; Yao H.; Ma C.; Xu S.; Yao H.; Zhu Z.; Yang D. H.; Chen Z.-S.; Xu J. Discovery of novel quinoline-chalcone derivatives as potent antitumor agents with microtubule polymerization inhibitory activity. J. Med. Chem. 2019, 62 (2), 993–1013. 10.1021/acs.jmedchem.8b01755. [DOI] [PubMed] [Google Scholar]
- Prasad B.; Lakshma Nayak V.; Srikanth P. S.; Baig M. F.; Subba Reddy N. V.; Babu K. S.; Kamal A. Synthesis and biological evaluation of 1-benzyl-N-(2-(phenylamino)pyridin-3-yl)-1H-1,2,3-triazole-4-carboxamides as antimitotic agents. Bioorg. Chem. 2019, 83, 535–548. 10.1016/j.bioorg.2018.11.002. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Chen J.; Xiao M.; Li W.; Miller D. D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29 (11), 2943–2971. 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H.; Li X.; Guo Y.; Zhen L. An overview of tubulin modulators deposited in Protein Data Bank. Med. Chem. Res. 2019, 28 (7), 927–937. 10.1007/s00044-019-02352-2. [DOI] [Google Scholar]
- Tron G. C.; Pirali T.; Sorba G.; Pagliai F.; Busacca S.; Genazzani A. A. Medicinal chemistry of combretastatin A4: present and future directions. J. Med. Chem. 2006, 49 (11), 3033–3044. 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- Pettit G. R.; Singh S. B.; Boyd M. R.; Hamel E.; Pettit R. K.; Schmidt J. M.; Hogan F. Antineoplastic Agents 291. Isolation and synthesis of combretastatins A-4, A-5, and A-6. J. Med. Chem. 1995, 38 (10), 1666–1672. 10.1021/jm00010a011. [DOI] [PubMed] [Google Scholar]
- Subba Rao A. V.; Swapna K.; Shaik S. P.; Lakshma Nayak V.; Srinivasa Reddy T.; Sunkari S.; Shaik T. B.; Bagul C.; Kamal A. Synthesis and biological evaluation of cis-restricted triazole/tetrazole mimics of combretastatin-benzothiazole hybrids as tubulin polymerization inhibitors and apoptosis inducers. Bioorg. Med. Chem. 2017, 25 (3), 977–999. 10.1016/j.bmc.2016.12.010. [DOI] [PubMed] [Google Scholar]
- Pérez-Pérez M. J.; Priego E. M.; Bueno O.; Martins M. S.; Canela M. D.; Liekens S. Blocking blood flow to solid tumors by destabilizing tubulin: an approach to targeting tumor growth. J. Med. Chem. 2016, 59 (19), 8685–8711. 10.1021/acs.jmedchem.6b00463. [DOI] [PubMed] [Google Scholar]
- Young S. L.; Chaplin D. J. Combretastatin A4 phosphate: background and current clinical status. Expert Opin. Invest. Drugs 2004, 13 (9), 1171–1181. 10.1517/13543784.13.9.1171. [DOI] [PubMed] [Google Scholar]
- Bailly C.; Bal C.; Barbier P.; Combes S.; Finet J. P.; Hildebrand M. P.; Peyrot V.; Wattez N. Synthesis and biological evaluation of 4-arylcoumarin analogues of combretastatins. J. Med. Chem. 2003, 46 (25), 5437–5444. 10.1021/jm030903d. [DOI] [PubMed] [Google Scholar]
- Rappl C.; Barbier P.; Bourgarel-Rey V.; Grégoire C.; Gilli R.; Carre M.; Combes S.; Finet J. P.; Peyrot V. Interaction of 4-arylcoumarin analogues of combretastatins with microtubule network of HBL100 cells and binding to tubulin. Biochemistry 2006, 45 (30), 9210–9218. 10.1021/bi060476g. [DOI] [PubMed] [Google Scholar]
- Sun C. M.; Lin L. G.; Yu H. J.; Cheng C. Y.; Tsai Y. C.; Chu C. W.; Din Y. H.; Chau Y. P.; Don M. J. Synthesis and cytotoxic activities of 4,5-diarylisoxazoles. Bioorg. Med. Chem. Lett. 2007, 17 (4), 1078–1081. 10.1016/j.bmcl.2006.11.023. [DOI] [PubMed] [Google Scholar]
- Kaffy J.; Pontikis R.; Carrez D.; Croisy A.; Monneret C.; Florent J. C. Isoxazole-type derivatives related to combretastatin A-4, synthesis and biological evaluation. Bioorg. Med. Chem. 2006, 14 (12), 4067–4077. 10.1016/j.bmc.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Shin K. D.; Yoon Y. J.; Kang Y. R.; Son K. H.; Kim H. M.; Kwon B. M.; Han D. C. KRIBB3, a novel microtubule inhibitor, induces mitotic arrest and apoptosis in human cancer cells. Biochem. Pharmacol. 2008, 75 (2), 383–394. 10.1016/j.bcp.2007.08.027. [DOI] [PubMed] [Google Scholar]
- Lee S.; Kim J. N.; Lee H. K.; Yoon K. S.; Shin K. D.; Kwon B. M.; Han D. C. Biological evaluation of KRIBB3 analogs as a microtubule polymerization inhibitor. Bioorg. Med. Chem. Lett. 2011, 21 (3), 977–979. 10.1016/j.bmcl.2010.12.044. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Mo J.; Lin H.; Chen Y.; Sun H. The recent progress of isoxazole in medicinal chemistry. Bioorg. Med. Chem. 2018, 26 (12), 3065–3075. 10.1016/j.bmc.2018.05.013. [DOI] [PubMed] [Google Scholar]
- Yoon H.; Kwak Y.; Choi S.; Cho H.; Kim N. D.; Sim T. A pyrazolo[3,4-d]pyrimidin-4-amine derivative containing an isoxazole moiety is a selective and potent inhibitor of RET gatekeeper mutants. J. Med. Chem. 2016, 59 (1), 358–373. 10.1021/acs.jmedchem.5b01522. [DOI] [PubMed] [Google Scholar]
- Sharma R.; Jain A.; Sahu B.; Singh D.; Mali S.. Heterocyclyl Compounds and Uses Thereof. WO2014181287, 2014.
- Yu L. F.; Tückmantel W.; Eaton J. B.; Caldarone B.; Fedolak A.; Hanania T.; Brunner D.; Lukas R. J.; Kozikowski A. P. Identification of novel Α4β2-nicotinic acetylcholine receptor (NAChR) agonists based on an isoxazole ether scaffold that demonstrate antidepressant-like activity. J. Med. Chem. 2012, 55 (2), 812–823. 10.1021/jm201301h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yermolina M. V.; Wang J.; Caffrey M.; Rong L. L.; Wardrop D. J. Discovery, synthesis, and biological evaluation of a novel group of selective inhibitors of filoviral entry. J. Med. Chem. 2011, 54 (3), 765–781. 10.1021/jm1008715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng B. L.; Hartman T. L.; Buckheit R. W.; Pannecouque C.; De Clercq E.; Cushman M. Replacement of the metabolically labile methyl esters in the alkenyldiarylmethane series of non-nucleoside reverse transcriptase inhibitors with isoxazolone, isoxazole, oxazolone, or cyano substituents. J. Med. Chem. 2006, 49 (17), 5316–5323. 10.1021/jm060449o. [DOI] [PubMed] [Google Scholar]
- Patel N. C.; Schwarz J.; Hou X. J.; Hoover D. J.; Xie L.; Fliri A. J.; Gallaschun R. J.; Lazzaro J. T.; Bryce D. K.; Hoffmann W. E.; Hanks A. N.; McGinnis D.; Marr E. S.; Gazard J. L.; Hajós M.; Scialis R. J.; Hurst R. S.; Shaffer C. L.; Pandit J.; O’Donnell C. J. Discovery and characterization of a novel dihydroisoxazole class of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor potentiators. J. Med. Chem. 2013, 56 (22), 9180–9191. 10.1021/jm401274b. [DOI] [PubMed] [Google Scholar]
- Velaparthi S.; Brunsteiner M.; Uddin R.; Wan B.; Franzblau S. G.; Petukhov P. A. 5-Tert-butyl-N-pyrazol-4-yl-4,5,6,7-tetrahydrobenzo[d]isoxazole-3-carboxamide derivatives as novel potent inhibitors of mycobacterium tuberculosis pantothenate synthetase: initiating a quest for new antitubercular drugs. J. Med. Chem. 2008, 51 (7), 1999–2002. 10.1021/jm701372r. [DOI] [PubMed] [Google Scholar]
- Sysak A.; Obmińska-Mrukowicz B. Isoxazole ring as a useful scaffold in a search for new therapeutic agents. Eur. J. Med. Chem. 2017, 137, 292–309. 10.1016/j.ejmech.2017.06.002. [DOI] [PubMed] [Google Scholar]
- Mahboobi S.; Pongratz H.; Hufsky H.; Hockemeyer J.; Frieser M.; Lyssenko A.; Paper D. H.; Bürgermeister J.; Böhmer F. D.; Fiebig H. H.; Burger A. M.; Baasner S.; Beckers T. Synthetic 2-aroylindole derivatives as a new class of potent tubulin-inhibitory, antimitotic agents. J. Med. Chem. 2001, 44 (26), 4535–4553. 10.1021/jm010940+. [DOI] [PubMed] [Google Scholar]
- Beckers T.; Reissmann T.; Schmidt M.; Fiebig H. H.; Burger A. M.; Vanhoefer U.; Mahboobi S.; Pongratz H.; Hockemeyer J.; Frieser M.; Mahboobi S. 2-Aroylindoles, a novel class of potent, orally active small molecule tubulin inhibitors. Cancer Res. 2002, 62 (11), 3113–3119. [PubMed] [Google Scholar]
- Putey A.; Popowycz F.; Do Q. T.; Bernard P.; Talapatra S. K.; Kozielski F.; Galmarini C. M.; Joseph B. Indolobenzazepin-7-ones and 6-, 8-, and 9-membered ring derivatives as tubulin polymerization inhibitors: synthesis and structure-activity relationship studies. J. Med. Chem. 2009, 52 (19), 5916–5925. 10.1021/jm900476c. [DOI] [PubMed] [Google Scholar]
- Hadimani M. B.; MacDonough M. T.; Ghatak A.; Strecker T. E.; Lopez R.; Sriram M.; Nguyen B. L.; Hall J. J.; Kessler R. J.; Shirali A. R.; Liu L.; Garner C. M.; Pettit G. R.; Hamel E.; Chaplin D. J.; Mason R. P.; Trawick M. L.; Pinney K. G. Synthesis of a 2-aryl-3-aroyl indole salt (OXi8007) resembling combretastatin A-4 with application as a vascular disrupting agent. J. Nat. Prod. 2013, 76 (9), 1668–1678. 10.1021/np400374w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana P.; Stagno A.; Barraja P.; Montalbano A.; Carbone A.; Parrino B.; Cirrincione G. Synthesis of the new ring system pyrrolizino[2,3-b]indol-4(5H)-one. Tetrahedron 2011, 67 (19), 3374–3379. 10.1016/j.tet.2011.03.060. [DOI] [Google Scholar]
- Diana P.; Stagno A.; Barraja P.; Carbone A.; Parrino B.; Dall’Acqua F.; Vedaldi D.; Salvador A.; Brun P.; Castagliuolo I.; Issinger O. G.; Cirrincione G. Synthesis of triazenoazaindoles: a new class of triazenes with antitumor activity. ChemMedChem 2011, 6 (7), 1291–1299. 10.1002/cmdc.201100027. [DOI] [PubMed] [Google Scholar]
- Barraja P.; Spanò V.; Patrizia D.; Carbone A.; Cirrincione G.; Vedaldi D.; Salvador A.; Viola G.; Dall’Acqua F. Pyrano[2,3-e]isoindol-2-ones, new angelicin heteroanalogues. Bioorg. Med. Chem. Lett. 2009, 19 (6), 1711–1714. 10.1016/j.bmcl.2009.01.096. [DOI] [PubMed] [Google Scholar]
- Spanò V.; Montalbano A.; Carbone A.; Scudieri P.; Galietta L. J. V.; Barraja P. An overview on chemical structures as ΔF508-CFTR correctors. Eur. J. Med. Chem. 2019, 180, 430–448. 10.1016/j.ejmech.2019.07.037. [DOI] [PubMed] [Google Scholar]
- Pojero F.; Poma P.; Spanò V.; Montalbano A.; Barraja P.; Notarbartolo M. Targeting multiple myeloma with natural polyphenols. Eur. J. Med. Chem. 2019, 180, 465–485. 10.1016/j.ejmech.2019.07.041. [DOI] [PubMed] [Google Scholar]
- Barraja P.; Diana P.; Lauria A.; Montalbano A.; Almerico A. M.; Dattolo G.; Cirrincione G.; Viola G.; Dall’Acqua F. Pyrrolo[2,3-h]quinolinones: synthesis and photochemotherapic activity. Bioorg. Med. Chem. Lett. 2003, 13 (16), 2809–2811. 10.1016/S0960-894X(03)00529-8. [DOI] [PubMed] [Google Scholar]
- Diana P.; Barraja P.; Lauria A.; Montalbano A.; Almerico A. M.; Dattolo G.; Cirrincione G. Pyrrolo[2,1-d][1,2,3,5]tetrazine-4(3H)-ones, a new class of azolotetrazines with potent antitumor activity. Bioorg. Med. Chem. 2003, 11 (11), 2371–2380. 10.1016/S0968-0896(03)00145-7. [DOI] [PubMed] [Google Scholar]
- Whatmore J. L.; Swann E.; Barraja P.; Newsome J. J.; Bunderson M.; Beall H. D.; Tooke J. E.; Moody C. J. Comparative study of isoflavone, quinoxaline and oxindole families of anti-angiogenic agents. Angiogenesis 2002, 5 (1–2), 45–51. 10.1023/A:1021528628524. [DOI] [PubMed] [Google Scholar]
- Almerico A. M.; Mingoia F.; Diana P.; Barraja P.; Montalbano A.; Lauria A.; Loddo R.; Sanna L.; Delpiano D.; Setzu M. G.; Musiu C. Pyrrolo[1,2-f]phenanthridines and related non-rigid analogues as antiviral agents. Eur. J. Med. Chem. 2002, 37 (1), 3–10. 10.1016/S0223-5234(01)01289-2. [DOI] [PubMed] [Google Scholar]
- Barraja P.; Spanò V.; Giallombardo D.; Diana P.; Montalbano A.; Carbone A.; Parrino B.; Cirrincione G. Synthesis of [1,2]oxazolo[5,4-e]indazoles as antitumour agents. Tetrahedron 2013, 69 (31), 6474–6477. 10.1016/j.tet.2013.05.083. [DOI] [Google Scholar]
- Barraja P.; Caracausi L.; Diana P.; Spanò V.; Montalbano A.; Carbone A.; Parrino B.; Cirrincione G. Synthesis and antiproliferative activity of the ring system [1,2]oxazolo[4,5-g]indole. ChemMedChem 2012, 7 (11), 1901–1904. 10.1002/cmdc.201200296. [DOI] [PubMed] [Google Scholar]
- Spano V.; Pennati M.; Parrino B.; Carbone A.; Montalbano A.; Cilibrasi V.; Zuco V.; Lopergolo A.; Cominetti D.; Diana P.; Cirrincione G.; Barraja P.; Zaffaroni N. Preclinical activity of new [1,2]oxazolo[5,4-e]isoindole derivatives in diffuse malignant peritoneal mesothelioma. J. Med. Chem. 2016, 59 (15), 7223–7238. 10.1021/acs.jmedchem.6b00777. [DOI] [PubMed] [Google Scholar]
- Spanò V.; Pennati M.; Parrino B.; Carbone A.; Montalbano A.; Lopergolo A.; Zuco V.; Cominetti D.; Diana P.; Cirrincione G.; Zaffaroni N.; Barraja P. [1,2]Oxazolo[5,4-e]isoindoles as promising tubulin polymerization inhibitors. Eur. J. Med. Chem. 2016, 124, 840–851. 10.1016/j.ejmech.2016.09.013. [DOI] [PubMed] [Google Scholar]
- Sriram M.; Hall J. J.; Grohmann N. C.; Strecker T. E.; Wootton T.; Franken A.; Trawick M. L.; Pinney K. G. Design, synthesis and biological evaluation of dihydronaphthalene and benzosuberene analogs of the combretastatins as inhibitors of tubulin polymerization in cancer chemotherapy. Bioorg. Med. Chem. 2008, 16 (17), 8161–8171. 10.1016/j.bmc.2008.07.050. [DOI] [PubMed] [Google Scholar]
- Tanpure R. P.; George C. S.; Strecker T. E.; Devkota L.; Tidmore J. K.; Lin C. M.; Herdman C. A.; Macdonough M. T.; Sriram M.; Chaplin D. J.; Trawick M. L.; Pinney K. G. Synthesis of structurally diverse benzosuberene analogues and their biological evaluation as anti-cancer agents. Bioorg. Med. Chem. 2013, 21 (24), 8019–8032. 10.1016/j.bmc.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanò V.; Frasson I.; Giallombardo D.; Doria F.; Parrino B.; Carbone A.; Montalbano A.; Nadai M.; Diana P.; Cirrincione G.; Freccero M.; Richter S. N.; Barraja P. Synthesis and antiproliferative mechanism of action of pyrrolo[3′,2′:6,7]cyclohepta[1,2-d]pyrimidin-2-amines as singlet oxygen photosensitizers. Eur. J. Med. Chem. 2016, 123, 447–461. 10.1016/j.ejmech.2016.07.051. [DOI] [PubMed] [Google Scholar]
- Spanò V.; Giallombardo D.; Cilibrasi V.; Parrino B.; Carbone A.; Montalbano A.; Frasson I.; Salvador A.; Richter S. N.; Doria F.; Freccero M.; Cascioferro S.; Diana P.; Cirrincione G.; Barraja P. Pyrrolo[3′,2′:6,7]cyclohepta[1,2-b]pyridines with potent photo-antiproliferative activity. Eur. J. Med. Chem. 2017, 128, 300–318. 10.1016/j.ejmech.2017.02.008. [DOI] [PubMed] [Google Scholar]
- Monks A.; Scudiero D.; Skehan P.; Shoemaker R.; Paull K.; Vistica D.; Hose C.; Langley J.; Cronise P.; Vaigro-Wolff A.; Gray-Goodrich M.; Campbell H.; Mayo J.; Boyd M. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83 (11), 757–766. 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- Arribas A. J.; Napoli S.; Gaudio E.; Cascione L.; Di Veroli A.; Tarantelli C.; Spriano F.; Zucchetto A.; Rossi F. M.; Rinaldi A.; Stathis A.; Stuessi G.; Gattei V.; Cruciani G.; Zucca E.; Rossi D.; Bertoni F. Secreted factors determine resistance to idelalisib in marginal zone lymphoma models of resistance. Blood 2019, 134, 2569. 10.1182/blood-2019-124299. [DOI] [Google Scholar]
- Arribas A. J.; Napoli S.; Gaudio E.; Cascione L.; Di Veroli A.; Tarantelli C.; Spriano F.; Zucchetto A.; Rossi F.; Sartori G.; Rinaldi A.; Stathis A.; Stussi G.; Gattei V.; Cruciani G.; Zucca E.; Rossi D.; Bertoni F. Abstract A127: Secretion of IL16 is associated with resistance to ibrutinib in pre-clinical models of lymphoma. Mol. Cancer Ther. 2019, 18, A127. 10.1158/1535-7163.TARG-19-A127. [DOI] [Google Scholar]
- Romagnoli R.; Baraldi P. G.; Kimatrai Salvador M.; Preti D.; Aghazadeh Tabrizi M.; Bassetto M.; Brancale A.; Hamel E.; Castagliuolo I.; Bortolozzi R.; Basso G.; Viola G. Synthesis and biological evaluation of 2-(alkoxycarbonyl)-3-anilinobenzo[b]thiophenes and thieno[2,3-b]pyridines as new potent anticancer agents. J. Med. Chem. 2013, 56 (6), 2606–2618. 10.1021/jm400043d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli R.; Baraldi P. G.; Salvador M. K.; Prencipe F.; Bertolasi V.; Cancellieri M.; Brancale A.; Hamel E.; Castagliuolo I.; Consolaro F.; Porcù E.; Basso G.; Viola G. Synthesis, antimitotic and antivascular activity of 1-(3′,4′,5′-trimethoxybenzoyl)-3-arylamino-5-amino-1,2,4-triazoles. J. Med. Chem. 2014, 57 (15), 6795–6808. 10.1021/jm5008193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta D.; Bortolozzi R.; Hamel E.; Basso G.; Moro S.; Viola G.; Ferlin M. G. Novel 3-substituted 7-phenylpyrrolo[3,2-f]quinolin-9(6H)-ones as single entities with multitarget antiproliferative activity. J. Med. Chem. 2015, 58 (20), 7991–8010. 10.1021/acs.jmedchem.5b00805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morigi R.; Locatelli A.; Leoni A.; Rambaldi M.; Bortolozzi R.; Mattiuzzo E.; Ronca R.; Maccarinelli F.; Hamel E.; Bai R.; Brancale A.; Viola G. Synthesis, in vitro and in vivo biological evaluation of substituted 3-(5-imidazo[2,1-b]thiazolylmethylene)-2-indolinones as new potent anticancer agents. Eur. J. Med. Chem. 2019, 166, 514–530. 10.1016/j.ejmech.2019.01.049. [DOI] [PubMed] [Google Scholar]
- Lin C. M.; Singh S. B.; Chu P. S.; Dempcy R. O.; Schmidt J. M.; Pettit G. R.; Hamel E. Interactions of tubulin with potent natural and synthetic analogs of the antimitotic agent combretastatin, a structure-activity study. Mol. Pharmacol. 1988, 34, 200–208. [PubMed] [Google Scholar]
- Clarke P. R.; Allan L. A. Cell-cycle control in the face of damage - a matter of life or death. Trends Cell Biol. 2009, 19 (3), 89–98. 10.1016/j.tcb.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Donzelli M.; Draetta G. F. Regulating mammalian checkpoints through cdc25 inactivation. EMBO Rep. 2003, 4 (7), 671–677. 10.1038/sj.embor.embor887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y.; Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 2007, 12 (5), 835–840. 10.1007/s10495-006-0525-7. [DOI] [PubMed] [Google Scholar]
- Xiong S.; Mu T.; Wang G.; Jiang X. Mitochondria-mediated apoptosis in mammals. Protein Cell 2014, 5 (10), 737–749. 10.1007/s13238-014-0089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovini A.; Savry A.; Braguer D.; Carré M. Microtubule-targeted agents: when mitochondria become essential to chemotherapy. Biochim. Biophys. Biochim. Biophys. Acta, Bioenerg. 2011, 1807 (6), 679–688. 10.1016/j.bbabio.2011.01.001. [DOI] [PubMed] [Google Scholar]
- Romagnoli R.; Baraldi P. G.; Salvador M. K.; Preti D.; Aghazadeh Tabrizi M.; Brancale A.; Fu X. H.; Li J.; Zhang S. Z.; Hamel E.; Bortolozzi R.; Porcù E.; Basso G.; Viola G. Discovery and optimization of a series of 2-aryl-4-amino-5-(3′,4′,5′-trimethoxybenzoyl)thiazoles as novel anticancer agents. J. Med. Chem. 2012, 55 (11), 5433–5445. 10.1021/jm300388h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli R.; Baraldi P. G.; Salvador M. K.; Prencipe F.; Bertolasi V.; Cancellieri M.; Brancale A.; Hamel E.; Castagliuolo I.; Consolaro F.; Porcù E.; Basso G.; Viola G. Synthesis, antimitotic and antivascular activity of 1-(3′,4′,5′-trimethoxybenzoyl)-3-arylamino-5-amino-1,2,4-triazoles. J. Med. Chem. 2014, 57 (15), 6795–6808. 10.1021/jm5008193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami N.; Marchetti P.; Castedo M.; Decaudin D.; Macho A.; Hirsch T.; Susin S. A.; Petit P. X.; Mignotte B.; Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 1995, 182 (2), 367–377. 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aredia F.; Scovassi A. I. Poly(ADP-Ribose): a signaling molecule in different paradigms of cell death. Biochem. Pharmacol. 2014, 92 (1), 157–163. 10.1016/j.bcp.2014.06.021. [DOI] [PubMed] [Google Scholar]
- Haschka M. D.; Soratroi C.; Kirschnek S.; Häcker G.; Hilbe R.; Geley S.; Villunger A.; Fava L. L. The NOXA-MCL1-BIM axis defines lifespan on extended mitotic arrest. Nat. Commun. 2015, 6, 6891. 10.1038/ncomms7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz I. E.; Kusam S.; Lam C.; Okamoto T.; Sandoval W.; Anderson D. J.; Helgason E.; Ernst J. A.; Eby M.; Liu J.; Belmont L. D.; Kaminker J. S.; O’Rourke K. M.; Pujara K.; Kohli P. B.; Johnson Ad. R.; Chiu M. L.; Lill J. R.; Jackson P. K.; Fairbrother W. J.; Seshagiri S.; Ludlam M. J. C.; Leong K. G.; Dueber E. C.; Maecker H.; Huang D. C. S.; Dixit V. M. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471 (7336), 110–114. 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- Kocab A. J.; Duckett C. S. Inhibitor of apoptosis proteins as intracellular signaling intermediates. FEBS J. 2016, 283 (2), 221–231. 10.1111/febs.13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel E.; Lin C. M. Separation of active tubulin and microtubule-associated proteins by ultracentrifugation and isolation of a component causing the formation of microtubule bundles. Biochemistry 1984, 23 (18), 4173–4184. 10.1021/bi00313a026. [DOI] [PubMed] [Google Scholar]
- Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem. Biophys. 2003, 38 (1), 1–21. 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- Verdier-Pinard P.; Lai J. Y.; Yoo H. D.; Yu J.; Marquez B.; Nagle D. G.; Nambu M.; White J. D.; Falck J. R.; Gerwick W. H.; Day B. W.; Hamel E. Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol. Pharmacol. 1998, 53 (1), 62–76. 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- Salvioli S.; Ardizzoni A.; Franceschi C.; Cossarizza A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess ΔΨ changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 1997, 411 (1), 77–82. 10.1016/S0014-5793(97)00669-8. [DOI] [PubMed] [Google Scholar]
- Schrödinger Suite; Schrödinger LLC: New York, NY, U.S., 2018.
- LigPrep; Maestro; Schrödinger Suites; Schrödinger, LLC: New York, NY, U.S., 2018.
- Shivakumar D.; Harder E.; Damm W.; Friesner R. A.; Sherman W. Improving the prediction of absolute solvation free energies using the next generation OPLS force field. J. Chem. Theory Comput. 2012, 8 (8), 2553–2558. 10.1021/ct300203w. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28 (1), 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prota A. E.; Danel F.; Bachmann F.; Bargsten K.; Buey R. M.; Pohlmann J.; Reinelt S.; Lane H.; Steinmetz M. O. The novel microtubule-destabilizing drug BAL27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization. J. Mol. Biol. 2014, 426 (8), 1848–1860. 10.1016/j.jmb.2014.02.005. [DOI] [PubMed] [Google Scholar]
- Gigant B.; Wang C.; Ravelli R. B. G.; Roussi F.; Steinmetz M. O.; Curmi P. A.; Sobel A.; Knossow M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435 (7041), 519–522. 10.1038/nature03566. [DOI] [PubMed] [Google Scholar]
- Barbier P.; Dorléans A.; Devred F.; Sanz L.; Allegro D.; Alfonso C.; Knossow M.; Peyrot V.; Andreu J. M. Stathmin and interfacial microtubule inhibitors recognize a naturally curved conformation of tubulin dimers. J. Biol. Chem. 2010, 285 (41), 31672–31681. 10.1074/jbc.M110.141929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glide; Schrödinger, LLC: New York, NY, 2018.
- Bowers K. J.; Chow D. E.; Xu H.; Dror R. O.; Eastwood M. P.; Gregersen B. A.; Klepeis J. L.; Kolossvary I.; Moraes M. A.; Sacerdoti F. D.; Salmon J. K.; Shan Y.; Shaw D. E.. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE SC’06 Conference (SC’06), Tampa, FL; IEEE, 2006; pp 43–43. [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Prime; Schrödinger LLC: New York, NY, 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.