

Abstract
Endosomal trafficking has emerged as a defective biological pathway in Alzheimer’s disease (AD), and the pathway is a source of cerebrospinal fluid (CSF) protein accumulation. Nevertheless, the identity of the CSF proteins that accumulate in the setting of defects in AD’s endosomal trafficking pathway remains unknown. Here, we performed a CSF proteomic screen in mice with a neuronal-selective knockout of the core of the retromer complex VPS35, a master conductor of endosomal traffic that has been implicated in AD. We then validated three of the most relevant proteomic findings: the amino terminus of the transmembrane proteins APLP1 and CHL1, and the mid-domain of tau, which is known to be unconventionally secreted and elevated in AD. In patients with AD dementia, the concentration of amino-terminal APLP1 and CHL1 in the CSF correlated with tau and phosphorylated tau. Similar results were observed in healthy controls, where both proteins correlated with tau and phosphorylated tau and were elevated in about 70% of patients in the prodromal stages of AD. Collectively, the mouse-to-human studies suggest that retromer-dependent endosomal trafficking can regulate tau, APLP1, and CHL1 CSF concentration, informing on how AD’s trafficking pathway might contribute to disease spread and how to identify its trafficking impairments in vivo.
INTRODUCTION
The abnormal accumulation of extracellular proteins is a hallmark feature of Alzheimer’s disease (AD), as typified by the cleaved fragments of the amyloid precursor protein (APP) and the microtubule binding protein tau. APP is cleaved in the endosome (1, 2), and its fragments are secreted through the endosomal pathway, representing an example of so-called unconventional secretion. Although it is now understood that tau is unconventionally secreted from neurons via direct translocation across the plasma membrane (3, 4), the mechanisms underlying active secretion of tau fragments (5) remain largely unknown.
Recent genomic studies have identified endosomal trafficking as a biological pathway pathogenic in AD, and subsequent data have established that the defective pathway manifests as endosomal traffic jams (6). Some of the best evidence in support of this conclusion is provided by molecules related to retromer, a multimodular protein assembly that is considered the “master conductor” of sorting and trafficking cargo out of the endosome (7). For example, mutations in SORL1, encoding a key retromer receptor in the brain (1, 8), are causally pathogenic in AD (9, 10). In addition, the core retromer protein VPS35 is deficient in regionally vulnerable AD brains (11), and a rare de novo mutation in VPS35 has been identified in AD (12), as have variants in a range of other retromer-related genes (13). Retromer-dependent endosomal trafficking has been linked to many of AD’s core features (14)—including amyloid pathology (11, 14–17), tau pathology (18), glial pathology (19, 20), and the enlargement of neuronal endosomes (15, 21–23).
Although the retromer-dependent endosomal trafficking pathway is strongly linked to AD, and the pathway can accelerate endosomal secretion, the identity of the proteins that accumulate in the cerebrospinal fluid (CSF) caused by impairing the pathway is unclear. Because these secreted proteins can have neurotoxic effects, such identification might inform on how intracellular trafficking can link to disease spread. Moreover, identified proteins that accumulate in the CSF might serve as potential biomarkers of AD’s endosomal traffic jams. Therapeutic interventions are now being developed, targeting AD’s endosomal trafficking pathway (17), and such biomarkers could potentially accelerate drug discovery.
With these goals in mind, we genetically engineered a mouse model in which Vps35 is knocked out selectively in forebrain neurons. VPS35 is the molecular backbone of retromer’s heterotrimeric core, the scaffold to which all other retromer modules and retromer receptors bind (7). Accordingly, numerous studies have established that, among other retromer-related proteins, depleting Vps35 is one of the most reliable ways to impair retromer-dependent endosomal traffic and recapitulate many of AD’s core pathological features [as reviewed in (7)]. We performed a proteomic analysis of CSF collected from the conditional Vps35 knockout (cKO) mice compared with CSF from control littermates, and then validated the three reliable findings with the greatest potential relevance to our goals—the N terminus of two BACE1 substrates, APLP1 and CHL1, and the mid-domain of tau. Guided by the mouse findings, we then developed assays to reliably measure the proteins in human CSF and investigated their relationships in a large-scale human study in patients with AD dementia, in patients with prodromal AD, and in healthy controls (HCs). In addition, because recent studies have suggested that elevations in tau phosphorylation at its threonine-217 site (p-tau217) represents one of the earliest and most specific CSF “signature” of AD (24, 25), we also examined the relationship of APLP1 and CHL1 with p-tau217. Collectively, the results suggest that retromer-dependent endosomal trafficking can regulate CSF tau, APLP1, and CHL1, informing on how endosomal trafficking pathway in AD can contribute to disease spread and how to identify the trafficking impairments in vivo.
RESULTS
Screening the CSF proteome in neuronal-selective Vps35 KO mice
Mice expressing loxP-flanked Vps35 (Vps35fl/fl) on a C57BL/6 background were crossed with mice expressing Cre recombinase under the Camk2α promoter to KO Vps35 in forebrain neurons (Fig. 1A, top left). In all experiments, Vps35fl/fl; Camk2α-Cre (Vps35 cKO) were compared with Vps35fl/fl (control). We began by characterizing this mouse model using confocal microscopy and Western blot analysis. Immunofluorescence studies revealed a clear depletion of Vps35 in CA1 pyramidal neurons of Vps35 cKO mice compared with their control littermates (Fig. 1A, top right). We next harvested the hippocampus from 6-month-old mice and found by Western blot analysis a concomitant reduction in VPS35 (P = 0.0002) and other retromer core proteins, VPS26a (P = 0.0286) and VPS29 (P = 0.0008) (Fig. 1A, bottom). Proteomic analysis (illustrated in Fig. 1B and detailed in Materials and Methods) identified 1505 proteins in the CSF by one unique peptide (1048 proteins by two unique peptides) (data file S1).
Fig. 1. Proteomic CSF screen in Vps35-depleted mice.
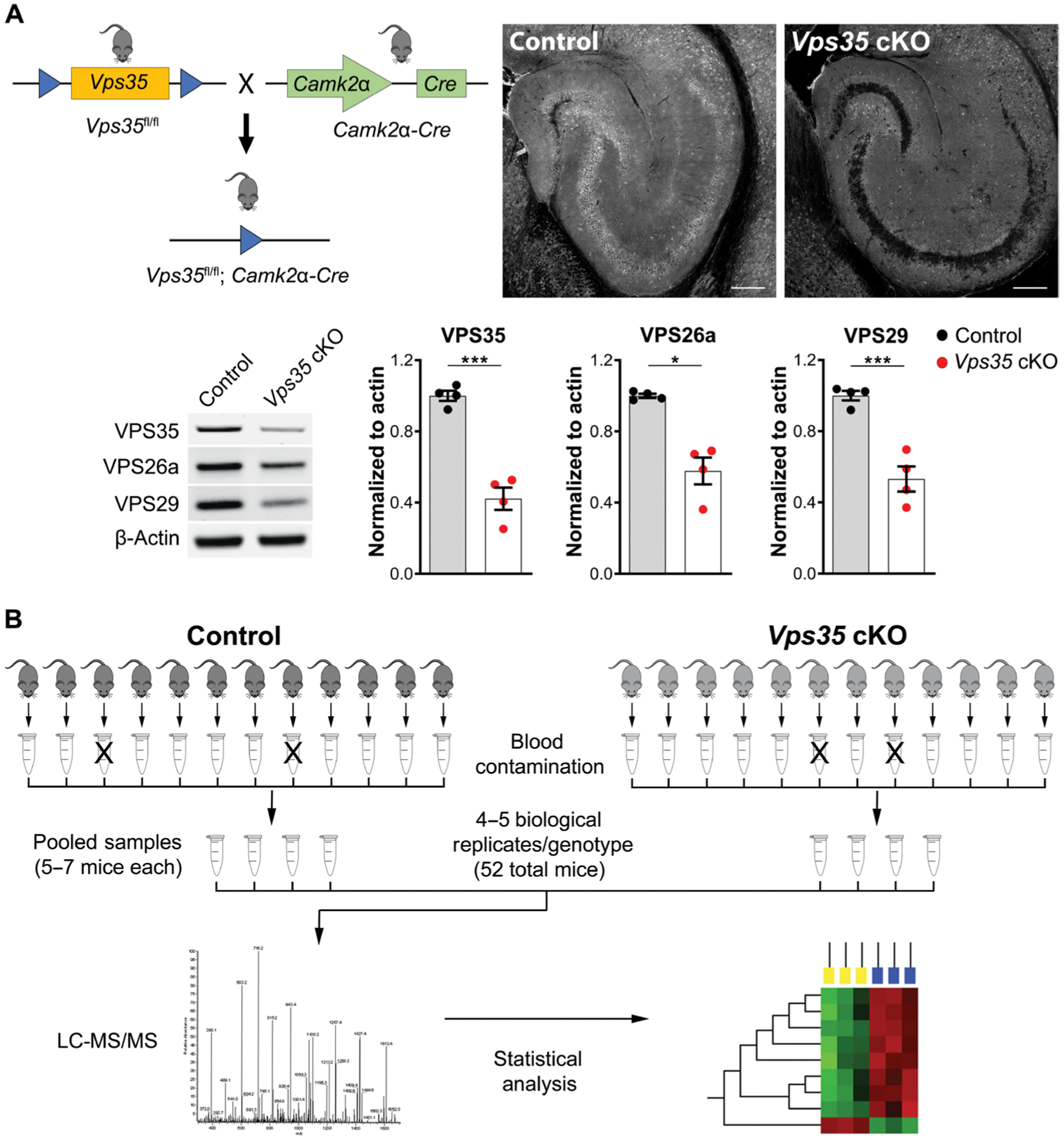
(A) Neuronal-selective Vps35 KO mice were generated by crossing mice expressing loxP-flanked Vps35 (Vps35fl/fl) with mice expressing Cre recombinase under the Camk2α promoter (upper left). Representative confocal images of 6-month-old mice brain sections stained for VPS35. Immunofluorescent images of the hippocampus show depletion of Vps35 in the neuron-selective KO mice (top right). Scale bars, 200 μm. Western blot analysis of hippocampal tissue from Vps35 cKO and control littermates (control) revealed for the retromer core proteins, VPS35, VPS26a, and VPS29. n = 4 animals per genotype. Data are shown as means ± SEM (bottom). (B) The CSF proteomic workflow is illustrated. Individual CSF samples (free of blood contamination) were pooled according to genotype, and the pooled samples were then analyzed by LC-MS/MS in technical duplicates. All data processing and statistical analysis were conducted as described in Materials and Methods. Significance is indicated as *P < 0.05, **P < 0.01, and ***P < 0.005.
Identifying alterations in CSF proteins in neuronal-selective Vps35 KO mice
Different analytic approaches were applied to the CSF proteomics data to identify CSF proteins that abnormally accumulate as a result of depleting neuronal Vps35. A parametric approach was first applied, using analysis of variance (ANOVA) to compare the difference of mean expression in detected proteins between the Vps35 cKOs and controls. Fifty-two proteins were found significantly altered in the CSF of the Vps35 cKO mice (P < 0.05, Fig. 2A). Among the class of elevated proteins, four are type I transmembrane proteins that are established BACE1 substrates: APLP1, APLP2, APP, and CHL1 (Fig. 2B). BACE1 functions primarily at endosomal membranes where it cleaves its substrates (Fig. 2C) (26), liberating their N-terminal fragments into the endosomal lumen from where these fragments are secreted to the extracellular space. This group of substrates has been reported as among the top proteins that were found reduced in a CSF proteomic screen of Bace1 KO mice (27). Vps35 deficiency has been shown to increase BACE1 activity by increasing the resident time of the enzyme and its substrates in endosomal membranes (15, 28). By investigating the peptides identified by tandem mass spectrometry (MS/MS), we found that they mapped onto the protein’s N-terminal domains, consistent with accelerated retromer-dependent endosomal secretion (data file S2).
Fig. 2. Identifying alterations in CSF proteins in neuronal-selective Vps35 KO mice.
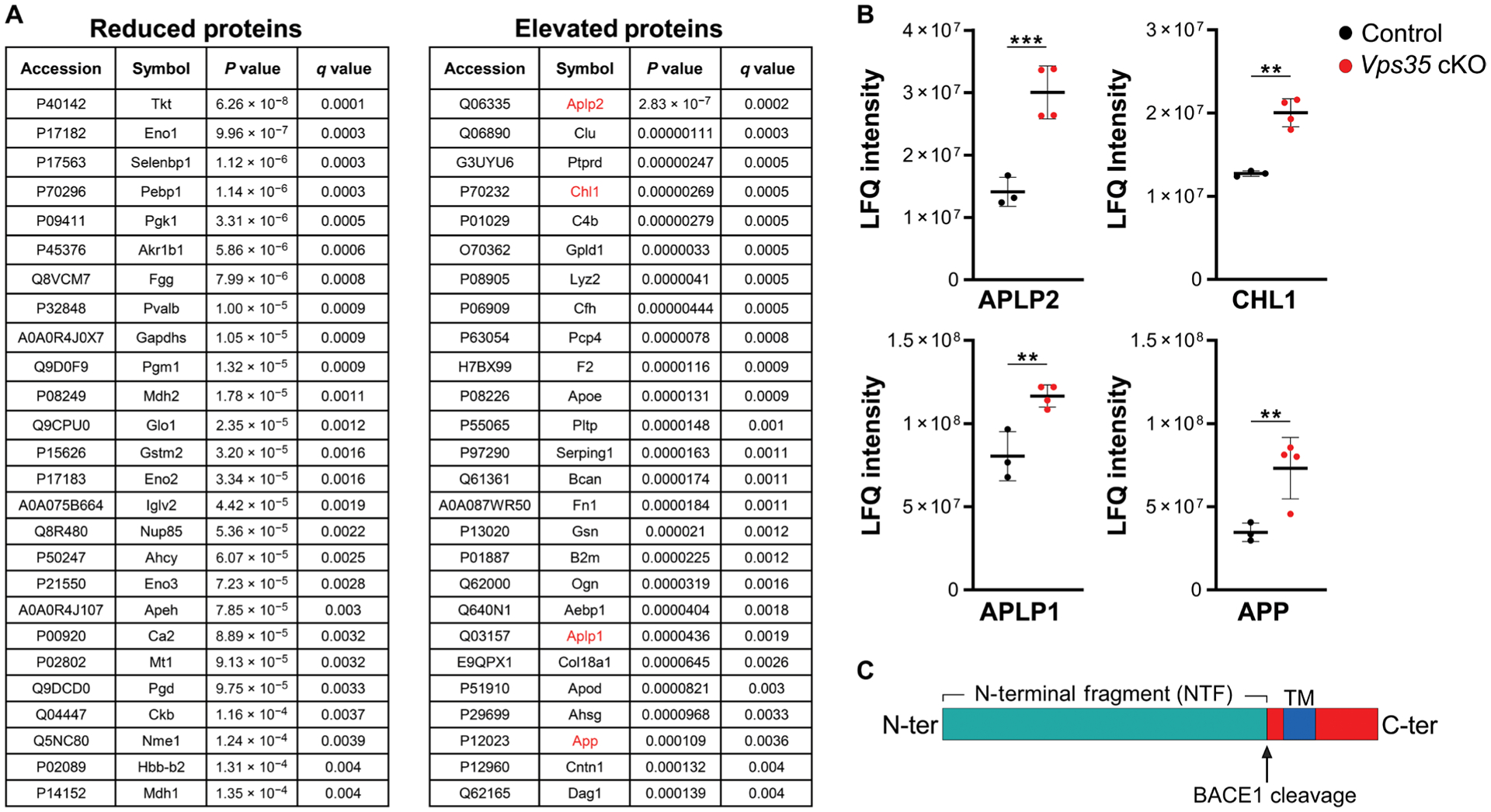
(A) Listed proteins are those that are significantly reduced or elevated in the CSF of Vps35 cKO mice compared with controls. The four BACE1 substrates are indicated in red. (B) Scatterplots show the distribution of the four identified BACE1 substrates: APLP2, CHL1, APLP1, and APP. Values are reported as label-free quantification (LFQ) intensity and represent the mean of technical duplicates. Bars indicate the means ± SD; Limma-calculated q values are reported (n = 3 to 4 per genotype; ***q < 0.001 and **q < 0.05). (C) As illustrated, these substrates are cleaved at the endosomal membrane by BACE1, liberating N-terminal fragments into the endosomal lumen.
A second nonparametric analysis was performed to identify proteins detected in CSF from one genotype but not the other. Eight proteins were detected in the CSF of controls and not the Vps35 cKOs, whereas 11 proteins were detected in the CSF of the Vps35 cKOs and not the controls (Fig. 3A). The microtubule-associated protein tau (MAPT) was one of the proteins detected only in the CSF of Vps35 cKO mice. By investigating the sequence of the tau peptide identified by MS/MS—SGYSSPGSPGTPGSR—we find that it mapped onto the protein’s mid-protein region, a domain contained within the N-terminal tau fragment most commonly found to accumulate in the CSF of patients with AD (Fig. 3B) (5, 29–32).
Fig. 3. Nonparametric and pathway analysis of CSF proteomic abnormalities caused by Vps35 depletion.
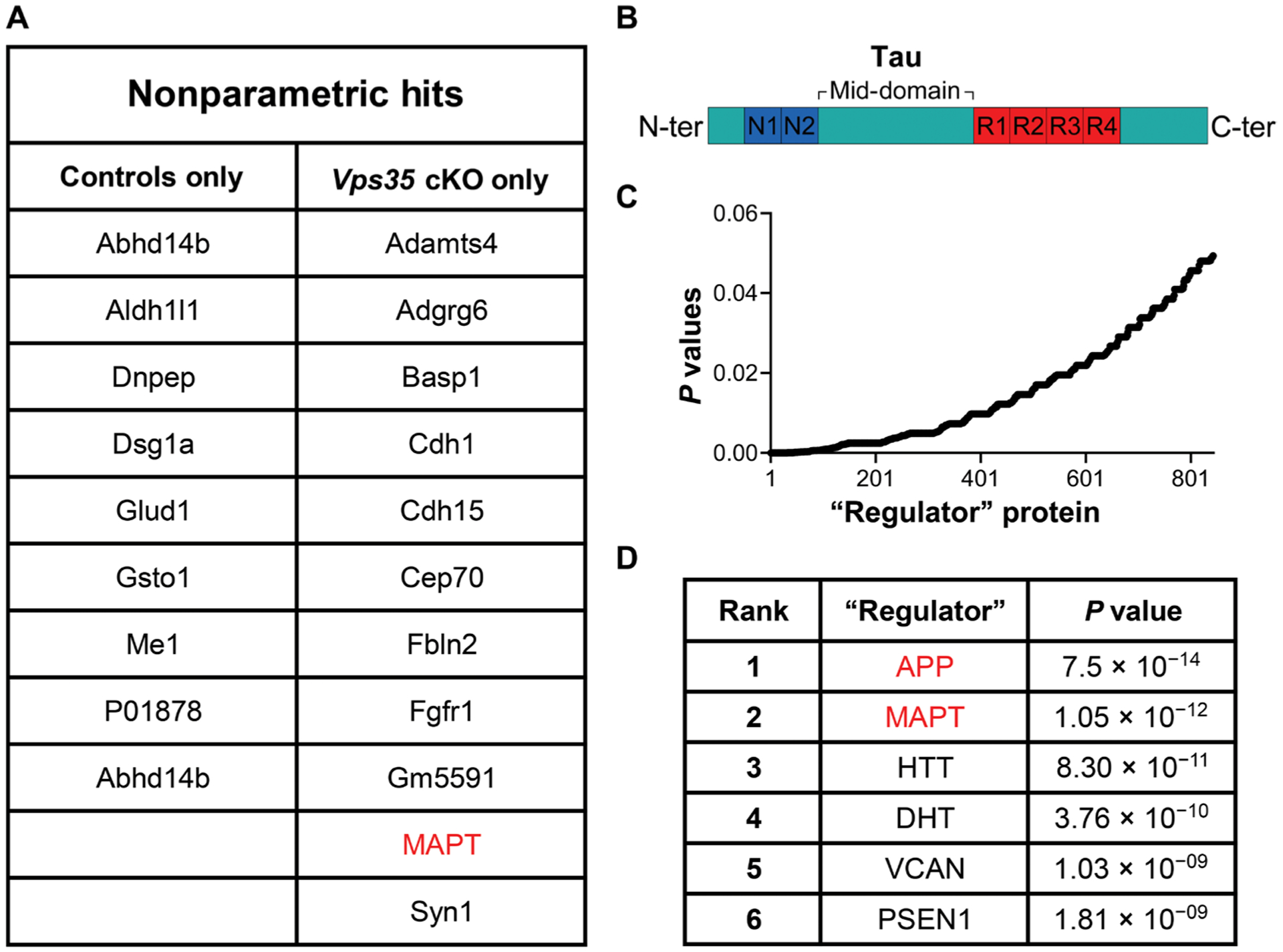
(A) Listed proteins are those detected only in the controls or in the Vps35 cKO mice, with microtubule-associated protein tau (MAPT) indicated in red. (B) Tau protein structural features are shown in the linear diagram. Peptide region identified by MS/MS is indicated with an arrow. (C) Scatterplot shows the P value distribution of over 800 proteins identified by pathway analysis as “regulator” proteins. (D) Listed proteins are the top six regulators, with lowest P values.
The final approach relied on the ingenuity pathway analysis (IPA) (33) to identify molecules that might be “regulating” the global changes in the CSF of Vps35 cKO mice. This software uses established transcriptional interactions to predict upstream molecular convergence of global changes in large datasets (33). Over 800 proteins were identified as potential regulator proteins in this dataset (Fig. 3C). The two top regulators with the lowest P values (>1 × 10−11) were APP and MAPT (Fig. 3D), further supporting the first two analyses performed.
Validating BACE1 substrates and Tau accumulation in mouse CSF
In anticipation of the human CSF studies, we decided to further confirm and validate the n-APLP1 and n-CHL1 findings because these two proteins are highly expressed in the brain (www.proteinatlas.org/). We collected CSF from a separate cohort of Vps35 cKO and control mice and measured n-CHL1 and n-APLP1 via immunoblotting.
Replicating and validating the CSF screen, both n-CHL1 (P = 0.02) and n-APLP1 (P = 0.02) were significantly elevated in the new cohort of Vps35 cKO CSF as compared with control littermates (Fig. 4A, left and middle). To investigate the relationships among CSF n-APLP1 and n-CHL1, we revisited the mouse CSF data and observed a strong correlation between CSF n-APLP1 and n-CHL1 (n = 14, β = 0.89, P < 0.0001) (Fig. 4A, right). In individual genotypes and collectively as a group, the relationship was nearly collinear across mice, with the Vps35 cKO mice simply shifting this relationship to the right. These observations suggest that the concentration of these proteins in CSF seem to be regulated by the same endosomal-based source. The same relationship was observed when a correlation analysis was performed using the MS/MS peak intensity measurements (fig. S1A). We next aimed at identifying neuronal proteins which release into the CSF was not affected by neuronal Vps35 depletion, as another way of validating our proteomic findings. Two candidates were identified: the synaptic adhesion molecule 4 CADM4, highly expressed at the cell surface of neurons, and the neuronal tubulin beta 3 (TUBB3). Correlation analysis using the MS/MS raw peak intensity measurements (fig. S2, B and C) revealed no relationships between these proteins and n-APLP1 or n-CHL1. CSF concentrations of tubulin 2a (TUBB2a), another cytoskeleton protein generally found in CSF, were not affected by Vps35 depletion (fig. S2D).
Fig. 4. Validating BACE1 substrates and Tau accumulation in mouse CSF.
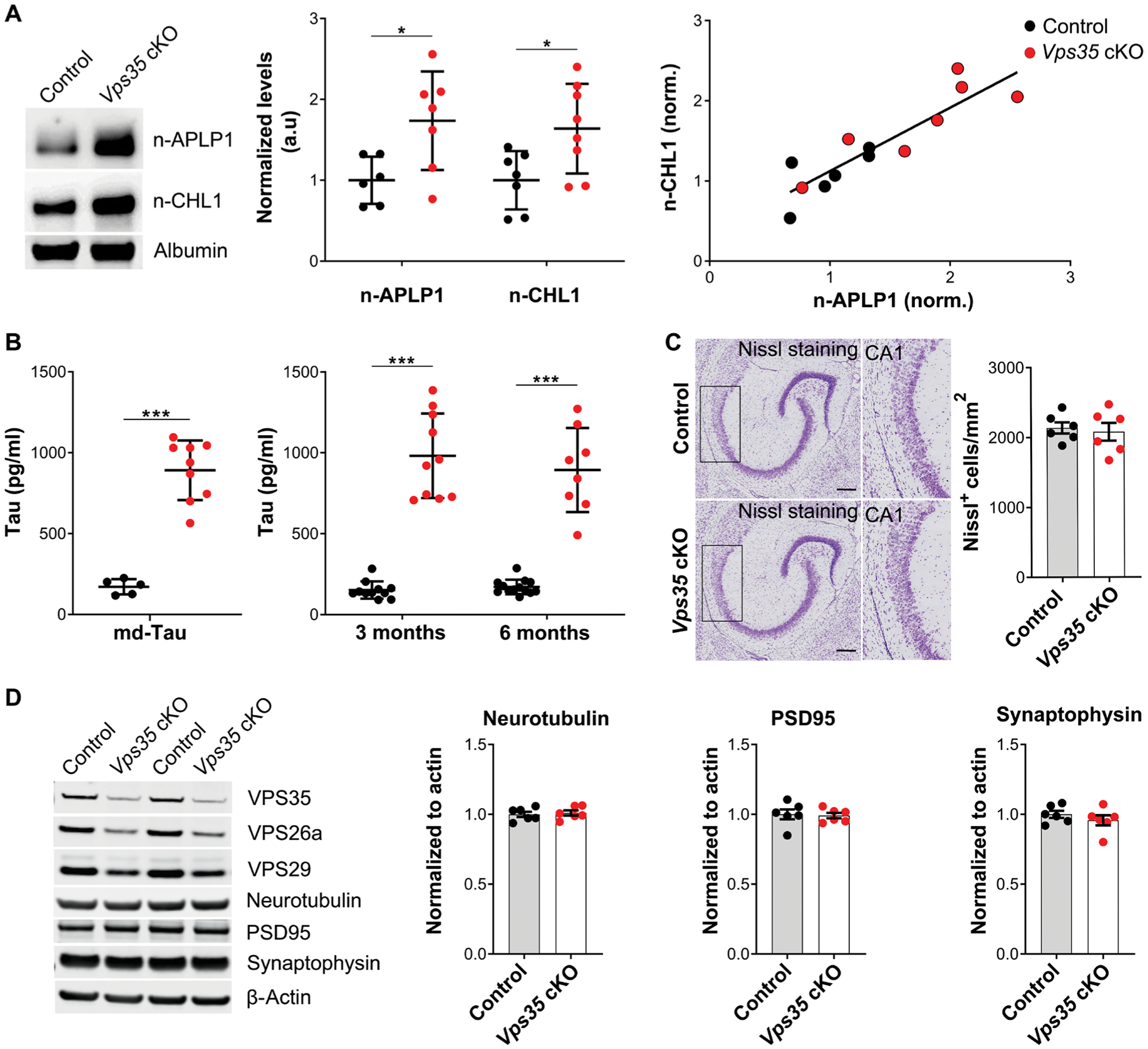
(A) Immunoblotting with antibodies directed against the N-terminal fragments of APLP1 (n-APLP1) and CHL1 (n-CHL1) in the CSF of a mixed sex cohort of Vps35 cKO mice (red circles) versus controls (black circles) (n = 6 to 8 per genotype; *P < 0.05). Representative immunoblots are shown on the left. Data are shown as means ± SD (middle). Scatterplots showing the relationship between n-APLP1 and n-CHL1 in the CSF of Vps35 cKO mice (red circles) and controls (black circles) (n = 14, β = 0.89, P < 0.0001; right). (B) Simoa assay was used to measure md-Tau in postmortem CSF in a mixed sex cohort of Vps35 cKO mice (red circles) versus controls (black circles) (n = 5 to 9 per genotype; P < 0.00001). Two-tailed unpaired Student’s t test was used for the statistical analysis (left). Antemortem CSF collected from females and males at 3 months (n = 11 to 13 per genotype, P < 0.0001) and 6 months (n = 8 to 10 per genotype, P < 0.0001) of age was analyzed using an md-Tau Simoa assay. A two-way ANOVA Sidak’s multiple comparisons test was used for the statistical analysis (right). Data are shown as means ± SD. (C) Representative Nissl staining analysis in the hippocampus of 3-month-old Vps35 cKO mice compared with their control littermates. n = 6 animals per genotype (P = 0.8182, in a nonparametric Mann-Whitney test). Hippocampal CA1 region is shown at higher magnification. Scale bars, 200 μm. (D) Western blot analysis of hippocampal tissue from 3-month-old Vps35 cKO and control littermates (control) showing expression of neurotubulin (P = 0.0001), presynaptic (PSD95, P = 0.8319), or postsynaptic (synaptophysin, P = 0.3558) markers. Data are shown as means ± SEM. n = 6 animals per genotype. Two-tailed unpaired Student’s t test was used for the statistical analysis (*P < 0.05). Significance is indicated as *P < 0.05, **P < 0.01, and ***P < 0.005.
Although a growing body of evidence suggests that tau might also be secreted via the endosomal pathway (34–36), in contrast to BACE1 substrates, the link between tau and the endosomal pathway, in general, and to retromer trafficking, in particular, is still unclear. Quantitation of endogenous tau in mouse CSF is challenging because CSF concentrations are roughly 50,000-fold lower than that of brain tissue (37). Therefore, we turned to single-molecule-array (Simoa) technology, a highly sensitive assay (38) that has been used by prior studies to reliably detect murine CSF tau (39). The assay used antibodies directed at the mid-domain epitope of tau (md-Tau), which are commonly used to measure total tau in human patients (29, 40). We reliably detected md-Tau in as little as 2 to 5 μl of murine antemortem CSF. Furthermore, the Simoa measurement of CSF tau in a new cohort of mice revealed a notable increase in the Vps35 cKOs, replicating and validating the initial nonparametric finding (P = 0.00002) (Fig. 4B, left). In addition, studies in patients (41) and animal models (42) have established that neuronal cell death is, by itself, neither necessary nor sufficient to cause elevation in CSF tau. Nevertheless, we performed biochemical and histological analyses in two additional cohorts of Vps35 cKO mice and control littermates to address this issue. We started by replicating our findings of elevated CSF tau at 3 and 6 months using antemortem CSF (Fig. 4B, right). A semiquantitative analysis by Nissl staining further revealed no signs of neuronal loss in the hippocampus of 3-month-old Vps35 cKO mice, compared with their control littermates (Fig. 4C), which was further supported by NeuN immunofluorescence staining analysis (fig. S2A). Furthermore, protein expression of pre- and postsynaptic markers was also unaffected in Vps35 cKO mice compared with their control littermates (Fig. 4D and fig. S2B). Together, these studies suggest that the elevation of CSF tau observed in Vps35 cKO mice is not driven by neuronal cell death.
Developing and validating n-APLP1 and n-CHL1 Simoa-based assays
Whereas validated assays for human md-Tau already exist, to measure APLP1 and CHL1 concentrations in humans, we developed and validated Simoa assays for n-APLP1 and n-CHL1. We began by identifying eight commercially available antibodies directed against the N terminus of APLP1 and six commercially available antibodies directed against the N terminus of CHL1. Using the Simoa platform, antibody pairs were screened for compatibility via signal with recombinant peptides of either n-APLP1 or n-CHL1 (fig. S3, A and D). The pairs with highest sensitivity were then evaluated using titrations of HC CSF pool, ultimately yielding several APLP1 and CHL1 assays with good precision, sensitivity, and dilution linearity. The best-performing n-APLP1 assay exhibited mean intratest precision of 4.6%, dilution linearity of CSF in the range of 1:32 to 1:512, and reported concentrations of ~700 pg/ml in HC CSF (fig. S3, B and C). The best-performing n-CHL1 assay exhibited mean intratest precision of 7.6%, dilution linearity of CSF in the range of 1:32 to 1:512, and reported concentrations of ~15 ng/ml in HC CSF (fig. S3, E and F). Together, these studies generate Simoa assays that are suitable for reliably identifying n-APLP1 and n-CHL1 in human CSF.
n-APLP1 and n-CHL1 are correlated with Tau in the CSF of patients with dementia
We next set out to determine the relationship of the three validated hits in the CSF of patients with AD with mild to moderate dementia. We used the Simoa-based assays of n-APLP1 and n-CHL1 to measure their concentrations in the CSF of over 316 patients with mild to moderate dementia (79% amyloid positive). To compare with traditional AD biomarkers, the CSF was also assayed for md-Tau and Aβ42. A correlation was observed between the CSF concentrations of n-APLP1 and n-CHL1 (β = 0.72, P = 9.1 × 10−48), between md-Tau and n-APLP1 (β = 0.6, P = 1.3 × 10−30), and between md-Tau and n-CHL1 (β = 0.53, P = 1.7 × 10−33) (Fig. 5A).
Fig. 5. The relationship of n-APLP1, n-CHL1, and md-Tau is selectively correlated in the CSF of patients with Alzheimer’s dementia.
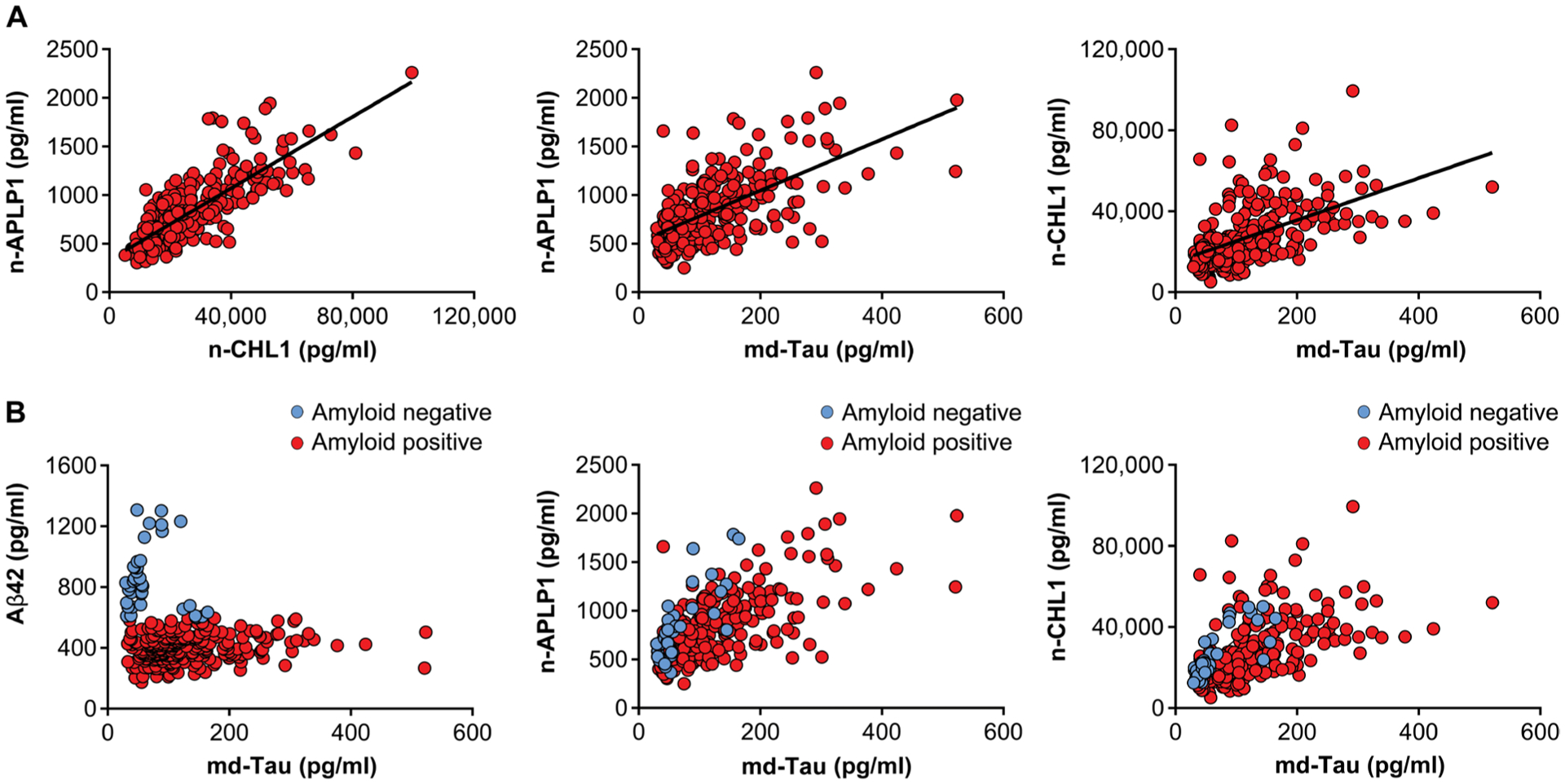
(A) Scatterplots showing the correlations between n-APLP1 and n-CHL1 (left; n = 316; β = 0.72, P = 9.1 × 10−48), n-APLP1 and md-Tau (middle; n = 316; β = 0.6, P = 1.3 × 10−30), and n-CHL1 and md-Tau (right; n = 316; β = 0.53, P = 1.7 × 10−33) in the CSF of patients with mild to moderate AD. (B) Scatterplots showing the relationships in amyloid-negative (blue circles) and amyloid-positive (red circles) individuals.
Many extracellular peptides, including fragments of APP and APLP1, are known to bind extracellular amyloid plaques (43), and we therefore stratified the group into those who, based on established CSF Aβ42 cutoffs, were amyloid plaque positive (Aβ42, <600 pg/ml) or negative (Aβ42, >600 pg/ml). Confirming the interpretation that amyloid plaques can confound relationships, we observed a much stronger relationship in amyloid-negative patients between md-Tau and n-APLP1 (β = 0.85, P = 3.7 × 10−10) and between md-Tau and n-CHL1 (β = 0.91, P = 1.8 × 10−13), whereas the relationship between n-APLP1 and n-CHL1 was found to be plaque independent (Fig. 5B).
n-APLP1 and n-CHL1 are correlated with Tau in the CSF of HCs and elevated in patients with mild cognitive impairment
We used banked CSF from 40 healthy subjects who were clinically determined to be cognitively unimpaired (mean age = 68.3, range = 55 to 87; 53% female) and CSF from 21 subjects who were clinically determined to have mild cognitive impairment (MCI) (mean age = 67.0, range = 52 to 87; 34% female). Strong linear correlations in HCs were observed between CSF concentrations of n-APLP1 and n-CHL1 (β = 0.97, P = 2.4 × 10−24), between md-Tau and n-APLP1 (β = 0.86, P = 1.7 × 10−12), and between md-Tau and n-CHL1 (β = 0.87, P = 2.1 × 10−17) (Fig. 6A).
Fig. 6. The relationship of n-APLP1, n-CHL1, and md-Tau in the CSF of healthy controls and patients with MCI.
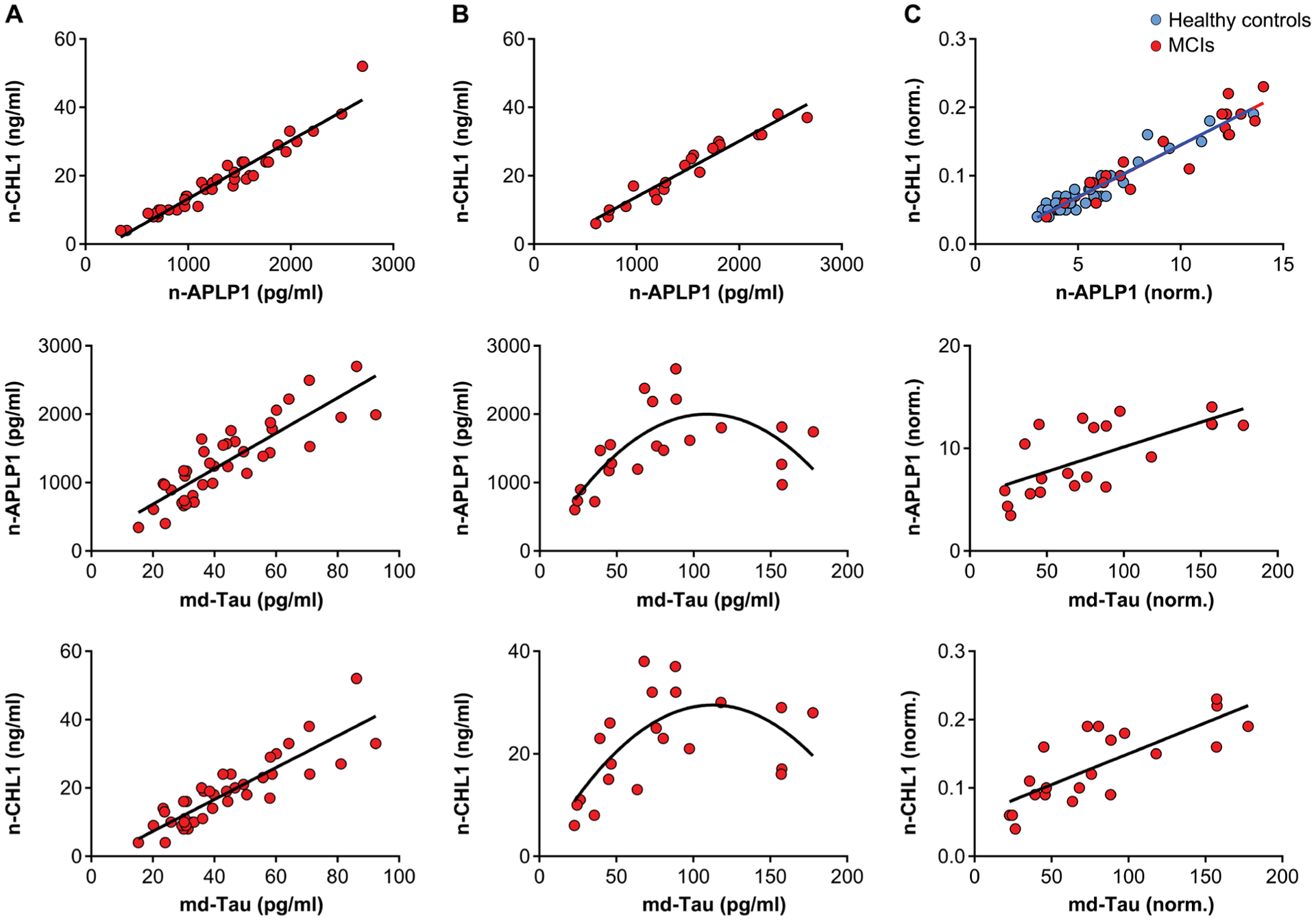
(A) Scatterplots showing the relationship between n-APLP1 and n-CHL1 (top; n = 40; β = 0.97, P = 2.4 × 10−24), n-APLP1 and md-Tau (middle; n = 40; β = 0.86, P = 1.7 × 10−12), and n-CHL1 and md-Tau (bottom; n = 40; β = 0.87, P = 2.1 × 10−17) in the CSF of healthy controls. (B) Scatterplot showing the relationship between n-APLP1 and n-CHL1 (top; n = 21; β = 0.97, P = 6.5 × 10−13) in the CSF of patients with MCI. (C) Scatterplot showing relationships between n-APLP1 and n-CHL1 (top: healthy control, blue circles; MCIs, red circles) after adjustment for CSF Aβ42. Scatterplot showing relationships between n-APLP1 and md-Tau (middle; n = 21; β = 0.67, P = 0.001) and n-CHL1 and md-Tau (bottom; n = 21; β = 0.79, P = 0.00002) after adjustment for CSF Aβ42.
In patients with MCI, we observed a strong linear correlation between n-APLP1 and n-CHL1 (β = 0.97, P = 6.5 × 10−13), whereas the relationship between md-Tau and n-APLP1 (β = 0.53, P = 0.001) or between md-Tau and n-CHL1 (β = 0.50, P = 0.002) were found to be curvilinear (Fig. 6B). To test the assumption that this curvilinear relationship reflected the confound of amyloid plaques, when adjustment was made for CSF Aβ42, the relationship between md-Tau and n-APLP1 (β = 0.67, P = 0.001) or between md-Tau and n-CHL1 (β = 0.79, P = 0.00002) was now linear (Fig. 6C).
Next, to determine who among this cohort had evidence of AD, we used a tau/Aβ42 ratio cutoff of ≥0.15 as indicative of AD based on a sample of 82 CSF for which this cutoff provided an optimal AUC (area under the curve) of 0.87 with specificity (91%) and sensitivity (75%). Most subjects who were clinically unimpaired were biomarker negative for AD, and most subjects who were clinically diagnosed as MCI were biomarker positive for AD and thus presumably represented prodromal AD (χ2 = 24.0, P = 9.6 × 10−7) (data file S1). To test for between-group differences, a significant elevation was found in the corrected measures of n-APLP1 (F = 84.2, P = 9.4 × 10−13) and n-CHL1 (F = 78.2, P = 3.2 × 10−12) in the patients with prodromal AD versus controls (Fig. 7). Of note, on the basis of the distributions, about 70% of patients with prodromal AD were 2 SDs above the means of the controls (data file S1).
Fig. 7. n-APLP1, n-CHL1, and md-Tau in the CSF of healthy controls and patients with prodromal AD.
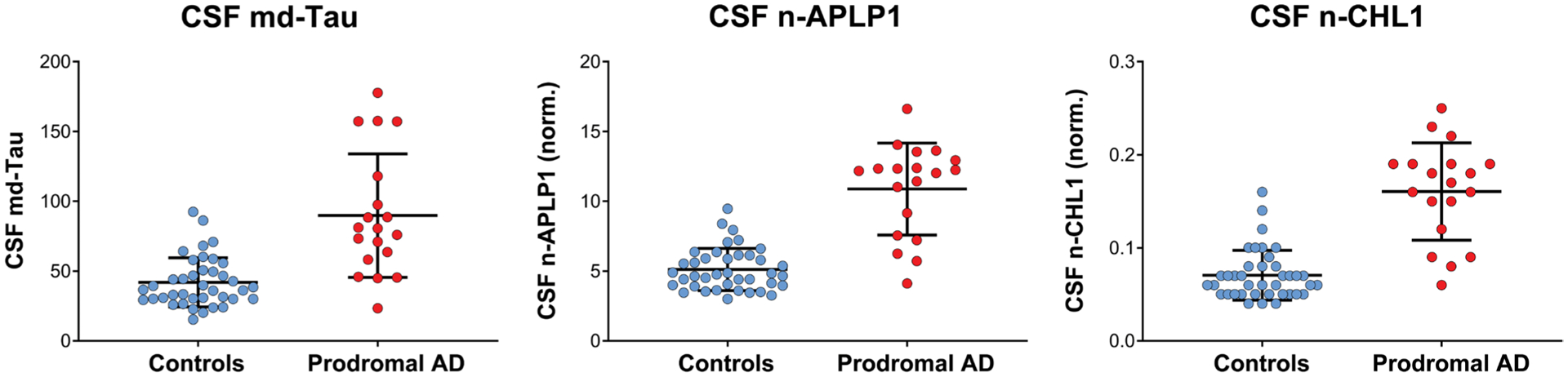
A set of 39 healthy controls and 19 prodromal AD classified on the basis of Tau/Aβ42 cutoff for AD (≥0.15) were analyzed using the n-APLP1, n-CHL1, and md-Tau assays. Shown is an in-between group comparison analysis in the corrected concentrations of n-APLP1 (F = 84.2, P = 9.4 × 10−13) and n-CHL1 (F = 78.2, P = 3.2 × 10−12) in patients with prodromal AD versus healthy controls.
n-APLP1 and n-CHL1 are correlated with phosphorylated Tau in the CSF of HCs and patients
Recent studies have reported that compared with other tau species found in the CSF, tau phosphorylation at its threonine-217 site (p-tau217) is the CSF tau signature that is most sensitive to AD, detected in the earliest preclinical stages of the disease before the onset of neurodegeneration, and the one most specific to AD compared with other tauopathies (24, 25). Although reagents are not currently available to reliably measure mouse CSF p-tau217, we were able to examine the relationship of n-APLP1 and n-CHL1 with p-tau217 in most of the human CSF samples. As with md-Tau, we found that p-tau217 correlates with n-APLP1 (β = 0.36, P = 6.5 × 10−11) and n-CHL1 (β = 0.37, P = 6.4 × 10−11) in the dementia cohort, that p-tau217 correlates with n-APLP1 (β = 0.72, P = 5.1 × 10−7) and n-CHL1 (β = 0.62, P = 5.0 × 10−6) in the HCs, and that p-tau217 correlates with normalized n-APLP1 (β = 0.63, P = 0.002) and n-CHL1 (β = 0.71, P = 0.0003) in the MCI cohort (Fig. 8).
Fig. 8. n-APLP1 and n-CHL1 are correlated with phosphorylated Tau in the CSF of healthy controls and patients with AD.
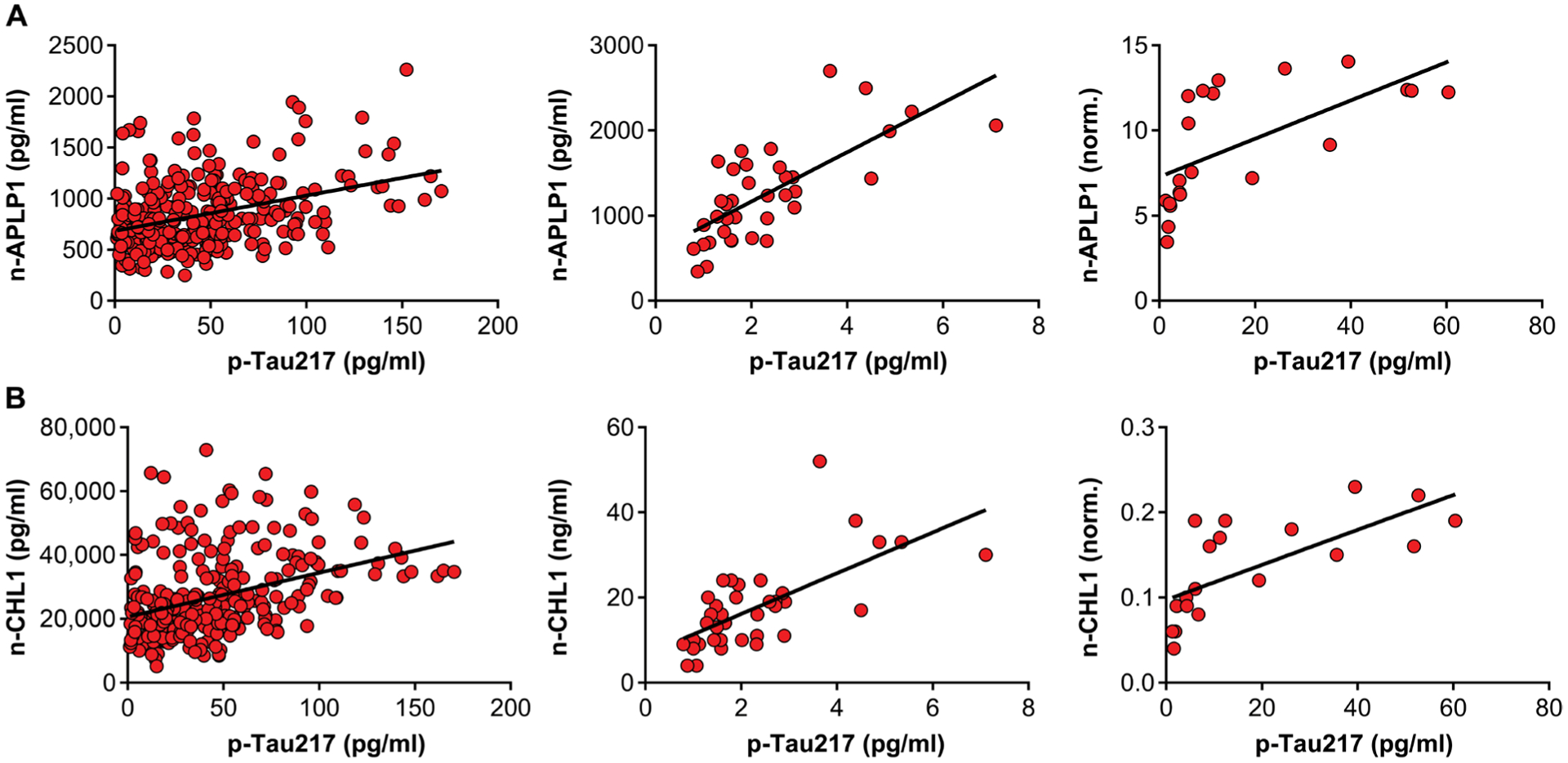
(A) Scatterplots showing the relationship between n-APLP1 and p-tau217 in the CSF of patients with mild to moderate AD (left; n = 316; β = 0.36, P = 6.5 × 10−11), healthy controls (middle; n = 37; β = 0.72, P = 5.1 × 10−7), and MCIs adjusted for CSF Aβ42 (right; n = 21; β = 0.63, P = 0.002). (B) Scatterplots showing relationship between n-CHL1 and p-tau217 in the CSF of patients with mild to moderate AD (left; n = 316; β = 0.37, P = 6.4 × 10−11), healthy controls (middle; n = 37; β = 0.62, P = 5.0 × 10−6), and MCIs adjusted for CSF Aβ42 (right; n = 21; β = 0.71, P = 0.0003).
DISCUSSION
The N-terminal fragments of APLP1, CHL1, APLP2, and APP have already been reported to be found in CSF (27), and fragments of APP and, particularly, APLP1 have been found elevated in AD CSF (44, 45). An independent series of studies clarifies why we find them elevated in the CSF of Vps35 cKO mice. All these type I transmembrane proteins are cleaved by BACE1 at endosomal membranes, and as best illustrated for APP, their cleaved products enter the endosomal lumen from where they are unconventionally secreted (46), mainly from neurons (47). BACE1 (28), which too is a type I transmembrane protein, APP (15), and presumably other transmembrane substrates are trafficked out of endosomes via retromer, and impairing retromer-dependent traffic is known to accelerate BACE1 substrate cleavage. Among these four proteins, we decided to complete additional validation studies on n-APLP1 and n-CHL1 because, as the two are brain enriched, they are most suitable for human CSF biomarkers (45). More than validating an increase in CSF n-APLP1 and n-CHL1, we also observed a relationship between the two, in both controls and Vps35 cKO mice. In individual genotypes and collectively as a group, the relationship was nearly collinear across mice, with the Vps35 cKO mice simply shifting this relationship to the right. These relationships support the interpretation that when elevation in CSF n-APLP1 and n-CHL1 co-occur, they are likely reflective of retromer-dependent endosomal traffic dysfunction.
Fragments of the tau protein are also known to be found in the CSF (29–32) and elevated in AD, but in this case, the mechanisms that regulate CSF tau accumulation have undergone a major shift in understanding. A number of converging lines of evidence have up-ended the original formulation that high CSF tau observed in AD is caused by cell death or by the formation of neurofibrillary tangles. CSF tau, for example, is not typically elevated in most other neurodegenerative diseases that display similar aggressive cell death and tangle formation (41). At the same time, other studies have shown that CSF tau observed in healthy subjects and found increased in AD is mediated by active neuronal secretion (5). A number of mechanisms have been proposed for its secretion, but it is now clear that tau is not secreted by the conventional secretory pathway. As with the fragments of APP and its homologs, tau is also secreted via unconventional pathways. Besides translocation across the plasma membrane (3, 4), a few previous studies have provided clues that tau might be actively secreted from the endosomal pathway. For example, tau is known to gain access to the endosomal pathway (48, 49), and retromer can influence intraneuronal tau pathology (18). Other studies have shown a tight linear correlation between CSF tau and amyloid in plaque-free human subjects, suggesting that they are secreted from the same pathway (50). Our results corroborate this hypothesis by providing indications implicating the retromer-dependent endosomal pathway as one by which tau can be secreted from neurons.
By developing and validating a set of reagents that can assay n-APLP1 and n-CHL1 in human CSF with high precision, observations from our human CSF studies suggest that the conclusions drawn from the Vps35 cKO mice are, at least partly, true in humans. First, in a large-scale study in patients with mild to moderate AD dementia, we found a selective cross-correlation among n-APLP1, n-CHL1, and md-Tau. Moreover, because this was a large-scale study, we were able to perform a stratification analysis, whose results support the prediction that the existence of amyloid plaques, which are known to bind many extracellular peptides, influences the relationship among these three CSF proteins. This interpretation was further supported by the CSF study in HCs and patients with MCI. In controls, we found a nearly collinear relationship among the three proteins, suggesting that in humans as in mice, all are secreted via the same unconventional pathway. In patients with MCI, a curvilinear relationship was observed between md-Tau and n-APLP1 or n-CHL1, and the relationship linearized when plaque correction was performed on the basis of CSF Aβ42 concentrations. By comparing the corrected n-APLP1 and n-CHL1 measures between HCs and patients with MCI, we find a CSF profile that phenocopies what was found when comparing Vps35 cKO mice and their control littermates: a tight linear relationship between n-APLP1 and n-CHL1 that are identical in slopes, but with a shift to the right in patients with MCI. Last, compared with HCs, patients who met the criteria for prodromal AD were found to have substantial, and concordant, elevations in CSF tau and plaque-corrected concentrations of n-APLP1 and n-CHL1.
Our final analysis in human CSF further supports the interpretation that tau is, at least in part, actively released into the CSF via endosomal secretion. Recent reports have shown that compared with other CSF tau species, tau phosphorylation at its threonine-217 site (p-tau217) is the CSF tau signature that is most sensitive to AD, detected in the earliest preclinical stages of AD and before the onset of neurodegeneration (24), and the one most specific to AD compared with other tauopathies (25). Although reagents are not currently available to reliably measure mouse CSF p-tau217, we were able to show that both n-APLP1 and n-CHL1 are associated with p-tau217 in all human CSF cohorts.
The potential for mechanistic insight was one motivation for identifying CSF proteins that accumulate as a result of impairments in retromer-dependent endosomal traffic. The evidence on tau achieved this goal because an increase in extracellular tau is thought to mediate its anatomical spread (51, 52). The second goal was to identify a CSF biomarker of AD-associated impairments in retromer-dependent trafficking. Our findings suggest that such a biomarker can be developed from a joint increase in CSF tau together with either n-APLP1 or n-CHL1.
Collectively, these studies provide evidence that CSF tau is intimately linked to the endosomal system and that the elevation of CSF tau in AD is, at least in part, mediated by impairments in retromer-dependent endosomal traffic. A main limitation of our mouse studies is that currently there are no reagents that can assay CSF p-tau217, which can be addressed once these reagents become available. A limitation of our human studies is that without a much larger number of prodromal patients and controls, we cannot establish precise diagnostic cutoffs for n-APLP1 or n-CHL1, as they currently exist for tau.
The most important limitation of the findings in patients is that, in contrast to our mouse model, we do not know the precise mechanism for retromer-dependent endosomal traffic dysfunction in most patients who have concordant increases in tau, n-APLP1, and n-CHL1. In these patients, retromer-dependent dysfunction might be directly caused by AD-associated endosomal trafficking genes (6), by comorbid conditions such type 2 diabetes and obesity (53), or by an interaction between these genetic and environmental factors. Alternatively, dysfunction can be indirectly caused by intraneuronal misprocessing of APP, which is found to cause endosomal traffic jams (54), retromer deficiency (55), and elevations in CSF tau (42). With the recent identification of retromer-enhancing pharmacological agents (17), future clinical trials can potentially address this limitation by testing whether and by how much retromer-enhancing drugs reduce CSF tau, n-APLP1, and n-CHL1.
MATERIALS AND METHODS
Study design
We performed an unbiased screen of neuronal proteins that accumulate in murine CSF in the setting of endosomal dysfunction using a mouse model deficient for the retromer core protein VPS35. The potential benefits of such a study are multiple. First, identification of abnormal CSF proteins that accumulate as result of neuronal endosomal dysfunction can inform on which of the hundreds of proteins that flow through the endosomal pathway might be particularly relevant to neurons. Second, because these secreted proteins can have neurotoxic effects, such a screen might also provide insight regarding possible mechanisms of disease spread. Third, identified proteins that accumulate in the CSF could serve as biomarkers of endosomal dysfunction, thus illuminating its prevalence and time course in AD and other neurological disorders. Last, as therapeutic interventions are now being developed against endosomal dysfunction, a biomarker would accelerate drug discovery. The experimenter was blinded in all mouse and human studies. Male and female mice used in this study were randomly selected for the respective group. Determination of sample sizes was based on previous experiences with mouse studies. About 40 mice per genotype were used for MS experiments. CSF was collected in a postmortem procedure from 6-month-old Vps35 cKO males and littermate controls. After blood contamination assessment via hemoglobin enzyme-linked immunosorbent assay (ELISA), contamination-free samples from a total of 52 mice were pooled into biological replicates of 30 μl each (n = 4 to 7 per genotype) for proteomic liquid chromatography-MS/MS (LC-MS/MS) analysis. We confirmed that using this approach, the technical replicates were highly correlated as were the mean scores for biological replicates of the same genotype, indicating robustness of the technique. Outliers were identified via Spearman correlation of technical replicates, and one sample from each genotype was removed before subsequent analysis. Further validation of the proteomic hits was performed by Western blot and Simoa analysis using two additional cohorts of mice at 3 and 6 months of age (the n for each experimental group is specified in the figure legend). We next set out to determine the relationship of three proteomic candidates—n-APLP1, n-CHL1, and md-Tau—in the CSF of 316 human AD patients with mild to moderate dementia, 40 cognitively HCs, and 21 subjects who were clinically determined to have MCI. Three outliers of n-APLP1 and n-CHL1 were identified by the SPSS Statistics software, using the program’s boxplot analysis. In Fig. 7, three patients who did not meet the current criteria for “prodromal AD” (subjects clinically diagnosed with MCIs who are biomarker positive) and “controls” (cognitively healthy individuals who are biomarker negative) were excluded from the analysis. Three patients had missing md-Tau measurements. In Fig. 8, in the HC group, three outliers were identified for p-tau217 and excluded from the analysis.
Vps35fl/fl; Camk2α-Cre KO mice
All mice were housed in a barrier facility and used in accordance with the National Institutes of Health and Columbia University Institutional Animal Care and Use Committee (IACUC) regulations. Vps35 floxed mice were generated at the Center for Mouse Genome Modification at UConn Health. Homologous recombination was performed in mouse embryonic stem cells (mES) targeting the Vps35 gene at exons 2 to 6. The recombined gene had LoxP sites before exon 3 and after exon 5. G418 and ganciclovir selection and nested long-range polymerase chain reaction were used to identify targeted clones. Targeted ES cells were aggregated into morula to generate chimeric mice. Next, the chimeric mice were bred with ROSA26-Flpe to remove the Frt-flanked PGKneo cassette. Neuronal-selective Vps35 KO mice were generated by crossing mice expressing loxP-flanked Vps35 (exons 3 to 5) (Vps35fl/fl) with mice expressing Cre recombinase under the Camk2α promoter. Camk2a-CRE mice were obtained from the Jackson laboratory (stock no, 005359). In all experiments, littermate Vps35fl/fl; Camk2α-Cre (Vps35 cKO) were compared to Vps35fl/fl (control). Females and males of 3 and 6 months of age were used for all the experiments, with the exception of the initial proteomic screen performed with 6-month-old males.
Murine CSF collection and proteomic screen
Because of the well-known circadian rhythm fluctuations in CSF proteins (56), all CSF collections were performed within a specific 4-hour time window (generally in the afternoon) that did not vary by cohort or experiment method. For proteomic studies, murine CSF was collected in a postmortem procedure after CO2 overdose in accordance with Columbia University IACUC guidelines. Briefly, euthanized mice were placed in a prone position, and the skin covering the back of the neck was shaved. A cotton swab containing 70% ethanol was used to remove any hair from the exposed skin. Then, a 27-gauge sterile needle (SV*27EL, Terumo Medical Products) attached to a 1-ml syringe (329650, BD Biosciences) was inserted into the cisterna magna, allowing flow of CSF into the butterfly needle. After 10 to 15 s, the needle was removed, and the CSF was aspirated into microcentrifuge tubes (1605–0000, USA Scientific), followed by a brief centrifugation at 600g for 6 min at 4°C. Supernatant was transferred to a new tube, immediately placed on dry ice, and further stored at −80°C. Roughly 5 to 10 μl of fluid was collected per mouse. CSF visibly contaminated with blood (pelleted residual erythrocytes) was discarded. All remaining samples underwent more stringent assessment for blood contamination via hemoglobin ELISA (ab157715, Abcam) using 0.5 μl of CSF with a 1:200 dilution. Samples below 0.01% blood contamination were retained for biochemical analyses and pooled into biological replicates of 30 μl each for proteomic analysis. Four biological replicates of control samples and five Vps35 cKO samples (each a composite of CSF from four to seven mice) were generated. The protein concentration of each pooled sample was determined via Qubit Protein Quantification Assay (Q33211, Life Technology), with sample concentrations ranging from 0.10 to 0.16 mg/ml. Each sample of 4 g of protein was then loaded onto an S-Trap column (PROFITI, NY), digested with trypsin, eluted with trifluoroacetic acid, and dried. Two technical replicates (each containing 1.5 μg of tryptic peptides) were run per sample. Antemortem CSF samples used for the validation of the proteomic hits were collected from anesthetized animals in accordance with Columbia University IACUC guidelines. Mice were anesthetized via intraperitoneal injection with a mixture containing ketamine (100 mg/kg body weight) and xylazine (10 mg/kg body weight) before CSF collection as described above.
LC-MS/MS analysis
The concentrated peptide mix was reconstituted in a solution of 2% acetonitrile (ACN) and 2% formic acid for MS analysis. Peptides were eluted from the ES802 column (75 μm × 25 cm; Thermo Fisher) at 300 nl/min using a Dionex UltiMate 3000 nano LC system with a 90-min gradient from 2 to 35% buffer B (100% acetonitrile and 0.1% formic acid). The gradient was switched from 35 to 85% buffer B over 1 min and held constant for 2 min. Last, the gradient was changed from 85% buffer B to 98% buffer A (100% water and 0.1% formic acid) over 1 min, and then held constant at 98% buffer A for five more minutes. The application of a 2.0-kV distal voltage electrosprayed the eluting peptides directly into the Thermo Fusion Tribrid mass spectrometer equipped with an EASY-Spray source (Thermo Scientific). Mass spectrometer-scanning functions and HPLC (high-performance LC) gradients were controlled by the Xcalibur data system (Thermo Fisher Scientific). MS data were acquired in the Orbitrap Fourier Transform (FT) at 120,000 resolutions from m/z (mass/charge ratio) 400 to 1600. Collision-induced dissociation MS/MS was acquired in the ion trap on 2+ and higher charge state ions for 3-s duty cycles.
Database search and interpretation of MS/MS data
The Proteome Discoverer application extracts relevant tandem mass spectra from the raw file and determines the precursor charge state and the quality of the fragmentation spectrum. The Proteome Discoverer probability-based scoring system rates the relevance of the best matches found by the SEQUEST algorithm. The human protein database was downloaded as FASTA-formatted sequences from UniProt protein database (released on July 2017). The peptide mass search tolerance was set to 10 ppm (parts per million). A minimum sequence length of seven amino acid residues was required. Only fully tryptic peptides were considered. To calculate confidence levels and false discovery rates (FDRs), the Proteome Discoverer generates a decoy database containing reverse sequences of the nondecoy protein database and performs the search against this concatenated database (nondecoy and decoy). FDR (1%) was used to generate the quantitative list for statistical analysis. Total amount of peptides was used to normalize among samples. A total of 1505 mouse proteins were detected and included in the final dataset. For each protein, quantification was determined by averaging the peak area of the three most abundant and distinct peptides (if available) and normalized to the total area. Qlucore Omics Explorer and Minitab 17 Software were used by the Proteomics Core to perform correlation and statistical analyses. Spearman correlations identified one outlier per genotype, removed before downstream analysis. Proteomics data have been deposited and accepted by the MassIVE public repository site and can be accessed by ftp://MSV000085709@massive.ucsd.edu (username for FTP access: MSV000085709; password: a).
Antibodies and other reagents for mouse studies
The following antibodies were used for immunoblot: VPS35 (1:1000; ab57632, mouse, Abcam), actin (1:10,000; NB600–535, mouse, Novus Biologicals), albumin (1:1000; NB600–41532, goat, Novus Biologicals), albumin (1:10,000; ab19194, goat, Abcam), CHL1 (1:500; AF2147-SP, goat, Novus Biologicals), CHL1 (1:1000; 25250–1-AP, rabbit, ProteinTech), APLP1 (1:500; AF3179-SP, rabbit, R&D Systems), Tau (1:500; ab80579, mouse, Abcam), beta-III tubulin (1:5000; ab107216, chicken, Abcam), PSD95 (1:1000; MAB1596, mouse, Millipore), synaptophysin (1:30,000; MAB5258, mouse, Millipore), anti-mouse horseradish peroxidase (HRP) (170–6516, Bio-Rad), anti-goat HRP (1:3000; 172–1034, rabbit, Bio-Rad), anti-rabbit HRP (1706515, goat, Bio-Rad), anti-chicken 800CW (1:20,000; 925–32218, donkey, LI-COR), anti-mouse 680LT (1:20,000; 925–68022, donkey, LI-COR), anti-rabbit 800CW (1:20,000; 925–32211, goat, LI-COR), protein G-HRP (1:10,000; 18–161, Sigma-Aldrich), and protein A-HRP (1:10,000; 101023, Thermo Fisher). The following antibodies were used for immunohistochemistry: NeuN (1:200; ABN78, rabbit, Millipore) and VPS35 (1:300; ab10099, goat, Abcam). The following ELISA kit was used: Hemoglobin Mouse ELISA Kit (ab157715, Abcam). Roche Lumi-Light substrate (12015200001, Sigma-Aldrich) was used for chemiluminescence.
Immunoblotting
Samples were prepared in LDS sample buffer (NP0007, Thermo Fisher) and reducing agent (161–0792, Bio-Rad), boiled for 5 min at 90°C, and run via electrophoresis in 4 to 12% bis-tris gels (NP0336 or NP0335, Thermo Fisher) using MES buffer (NP0002–02, Thermo Fisher). For CSF and media experiments, equal volumes (5 to 15 μl) were loaded in each lane; for lysate, equal masses were loaded in each lane. Proteins were then transferred to nitrocellulose membranes (IB3010–31, Invitrogen) via iBlot (P3; 7:40) and blocked for 1 hour in 3 to 5% bovine serum albumin (A30075–100gm, Research Products International) or Odyssey blocking buffer (927–40000, LI-COR) diluted 1:1 in phosphate-buffered saline with Tween 20 (PBS-T). Membranes were then probed with primary antibodies overnight at 4°C, washed in PBS-T, and probed with secondary antibodies (HRP conjugated or infrared conjugated) for 1 hour at room temperature. Membranes were again washed in PBS-T and imaged in Roche substrate (12015196001, Sigma-Aldrich) for enhanced chemiluminescence (ECL) or alone with the LI-COR Odyssey system. Densitometric values for all proteins were calculated using Image Studio Lite software; reported values are normalized to albumin and β-actin.
Immunohistochemistry and histological analysis
For immunohistochemistry studies, mice (females and males) were anesthetized, rinsed by cardiac perfusion with 0.9% saline buffer, and further fixed with 4% paraformaldehyde (PFA). Whole brains were harvested and postfixed in 4% PFA overnight, followed by cryoprotection treatment in 30% sucrose (w/v) in PBS for 16 hours. Tissue blocks were then embedded in optimal cutting temperature (OCT) compound and stored at −80°C. Briefly, 30-μm free-floating horizontal sections were washed three times with PBS and incubated at 4°C overnight on a rotator in 1 ml of primary antibody (VPS35 or NeuN) diluted in PBS containing 0.3% Triton X-100 (v/v) and 5% normal serum (v/v). After three washes with PBS-T (0.1% Triton X-100), the sections were incubated for 30 min with secondary antibodies (Molecular Probes). After three washes with PBS-T and one wash with PBS, sections were mounted on slides. Fluorescent images were collected using a Zeiss LSM 700 META confocal microscope equipped with a 63× Plan-Apochromat objective and HeNE1, HeNe2, and argon lasers. A semiquantitative count of NeuN-positive neurons in the CA1 hippocampal region was performed in six Vps35 cKO mice and six control littermates. Four to five spaced NeuN-stained horizontal sections, starting from bregma −2.28 to bregma −3.60, were analyzed per animal using an automated cell counting plugin (http://imagej.net/Particle_Analysis) on ImageJ software (version 1.48, U.S. National Institutes of Health). Briefly, evaluation of cell density was made on z projections of 10 consecutive confocal scans (total thickness, 10 μm). A fixed threshold value was applied to all sections to identify the NeuN-positive neurons. Number of cells was counted in the CA1 region manually delineated on each individual section, and area of analysis was measured. The density of immunolabeled neurons was expressed as the mean number of positive cells per square millimeter (number/mm2). All analyses were performed by a scientist blinded to the genotype status of the animal.
Histological Nissl stainings were performed according to the manufacturer’s instructions (IW-3007, NovaUltra Nissl Staining kit, IHC WORLD). Quantification of the neuronal density was performed on four to five sections per animal (n = 6 mice per group) starting from bregma −2.28 mm to bregma −3.60 mm. As for the NeuN quantification, the hippocampal cell layer CA1 was delineated on each cresyl violet-stained sections. Slides were scanned using an MPSR Leica SCN400 slide scanner (40× objective) and further analyzed using the built-in “Multi-point Tool” on ImageJ to manually count the stained cells. Briefly, cell density values were obtained by dividing the number of immunoreactive neurons by the total area of the corresponding hippocampal CA1 region (number/mm2).
md-Tau quantification in murine CSF
In Fig. 4B (left), postmortem CSF tau was quantified by Simoa Technology using the mouse “total-tau assay” (102209, Quanterix), which is directed against the mid-protein epitope of the tau holo-protein. Individual CSF samples (5 μl) were diluted 1:80 in sample buffer and then split into technical duplicates. Standards and samples were run according to the manufacturer’s instructions and read on the HD-1 analyzer (Quanterix). Replication of CSF tau elevation in 3- and 6-month-old mice shown in Fig. 4B (right) was performed using antemortem CSF samples collected from two different cohorts (22 females and 20 males) of anesthetized animals in accordance with Columbia University IACUC guidelines. For the latter, individual CSF samples (2 μl) were diluted 1:100 in sample buffer and then split into technical duplicates and read on the SR-X analyzer (Quanterix). CSF samples from Tau KO mice were used as negative controls.
n-APLP1 and n-CHL1 quantification in human CSF Assay development
The following eight APLP1 antibodies were screened for compatibility: 1 = #354008 (Thermo Fisher), 2 = AF3129 (Novus Biologicals), 3 = AF3179 (Novus Biologicals), 4 = MAB3908 (Novus Biologicals), 5 = 12305–2-AP (ProteinTech), 6 = MAB3129 (ProteinTech), 7 = ab210557 (Abcam), and 10 = ab192481 (Abcam) using extracellular domain APLP1 recombinant protein #AG2951 (ProteinTech). The following six CHL1 antibodies were screened for compatibility: 8 = MABN229 (Millipore), 9 = AF2147 (Novus Biologicals), 11 = MAB2126 (Novus Biologicals), 12 = 25250–1-AP (ProteinTech), 13 = MAB2147 (Novus Biologicals), and 14 = LS-C485419 (LSBio) using membrane proximal extracellular domain CHL1 recombinant protein #AG19263 (ProteinTech) using Simoa homebrew kit (catalog no. 101354, Quanterix) and standard assay definitions. The top six assays for each analyte (most sensitive with recombinant protein) were used to measure titrations of low tau (HC) CSF pool to confirm recognition of endogenous signal and dilution linearity. The top two assays for each analyte from the CSF screen were then optimized to improve sensitivity. The final assay chosen for each analyte (APLP1:2×10 and CHL1:12×9) were used for subsequent clinical cohort measurements. CSF was diluted to 1:80 for n-APLP1 measurement, and 1:50 for n-CHL1 measurement.
Human CSF studies
CSF from HCs and MCI subjects were requested from the Columbia University Alzheimer’s Disease Research Center (ADRC) CSF bank. This CSF bank includes CSF obtained by lumbar puncture and banked for research according to protocols approved by the Columbia University Institutional Review Board. CSF was stored frozen in 400 μl of aliquots in polypropylene tubes, at −80°C. Frozen CSF aliquots were later thawed and analyzed for Aβ42 and total tau by the ADRC Clinical Core using a multiplex fluorimetric microsphere xMAP bead-based sandwich immunoassay consisting of monoclonal antibodies covalently coupled to spectrally specific fluorescent beads. This assay (INNO-BIA AlzBio3, Fujirebio US Inc.) was performed on a Luminex 100 machine. Thresholds implemented to confirm amyloid positivity in patients with AD were CSF total tau/Aβ42 ≥ 0.15. CSF from subjects with mild to moderate dementia was obtained from Janssen clinical trial ELN115727–301/302. These lumbar puncture samples, obtained with informed consent for use in AD biomarker research, were stored in 500 μl of aliquots in polypropylene tubes at −80°C until use. All measurements shown (INNOTEST Aβ42: plaque positivity, Aβ42 = 600 pg/ml) and in-house Simoa n-APLP1, n-CHL1, md-Tau, and p-tau217 that were performed at Janssen R&D are described in (57).
Statistical methods
Formal boxplot analysis was applied to each dataset to exclude outliers. Student’s t test was used for all biochemical experiments with a two-tailed distribution with equal variance (P < 0.05) or as stated in the figure legend. GraphPad Prism version 7.00 for Windows (GraphPad Software) was used for all graph representation and the statistical analysis, except those shown in Figs. 5 to 8. ANOVA was used for parametric analysis of CSF proteomics, and Benjamini and Hochberg FDR-corrected P and q values are reported. Nonparametric analysis identified proteins that were present in at least four technical replicates from one genotype and absent from all replicates of the other genotype. Significance is indicated as *P < 0.05, **P < 0.01, and ***P < 0.005. The SPSS Statistics software was used for human correlation studies. All data are shown as means ± SEM or SD. “n” for each experimental design is indicated in the figure legends.
Supplementary Material
Data file S3. Individual-level data (provided as a separate Excel file).
Data file S1. Completed list of proteins identified in murine CSF (provided as a separate Excel file).
Data file S2. List of peptides detected by MS/MS analysis (provided as a separate Excel file).
Fig. S1. n-APLP1 and n-CHL1 correlation analysis based on MS/MS peak intensity measurements.
Fig. S2. NeuN immunostaining and quantification of retromer protein expression in 3-month-old Vps35 cKOs and control littermates.
Fig. S3. Development and qualification of Simoa APLP1 and CHL1 assays.
Fig. S4. Extended data of Western blots cropped in main Fig. 1.
Fig. S5. Extended data of Western blots cropped in main Fig. 4A.
Fig. S6. Extended data of Western blots cropped in main Fig. 4D.
Acknowledgments:
We would like to acknowledge Quanterix for assistance with tau Simoa, and M. S. Kang for Luminex CSF assays. We would like to acknowledge J. R. Slemmon of Janssen R&D for the critical review of experimental data and analysis. Last, we would also like to thank the Alzheimer’s Disease Research Center (ADRC) at Columbia University Irving Medical Center for providing the human CSF used in Figs. 6 to 8.
Funding: This study was partly supported by NIH grants R01AG034618, R01AG035015, and P50AG008702 to S.A.S.
Footnotes
Competing interests: The authors declare competing financial interests: S.A.S. holds equity in Denali Therapeutics. S.S., Y.H.Q., and S.A.S. are coinventors on provisional patents covering retromer therapies and biomarkers. G.T.-B., S.M., and H.K. are employees of Janssen Research and Development. A patent has been applied entitled “Methods of detecting endosomal trafficking dysfunction and neurodegenerative diseases and disorders” (application no. 63/053,805).
Data and materials availability: All the data associated with this study are present in the paper or the Supplementary Materials. The Vps35 line is available upon request sent to the corresponding author.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Small SA, Gandy S, Sorting through the cell biology of Alzheimer’s disease: Intracellular pathways to pathogenesis. Neuron 52, 15–31 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Udayar V, Buggia-Prévot V, Guerreiro RL, Siegel G, Rambabu N, Soohoo AL, Ponnusamy M, Siegenthaler B, Bali J; AESG M Simons, J. Ries, M. A. Puthenveedu, J. Hardy, G. Thinakaran, L. Rajendran, A paired RNAi and RabGAP overexpression screen identifies Rab11 as a regulator of β-amyloid production. Cell Rep 5, 1536–1551 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katsinelos T, Zeitler M, Dimou E, Karakatsani A, Müller H-M, Nachman E, Steringer JP, de Almodovar CR, Nickel W, Jahn TR, Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep. 23, 2039–2055 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Merezhko M, Brunello CA, Yan X, Vihinen H, Jokitalo E, Uronen R-L, Huttunen HJ, Secretion of tau via an unconventional non-vesicular mechanism. Cell reports 25, 2027–2035.e4 (2018). [DOI] [PubMed] [Google Scholar]
- 5.SStti C, Sullivan M, Crisp MJ, Kasten T, Kirmess KM, Kanaan NM, Yarasheski KE, Baker-Nigh A, Benzinger TLS, Miller TM, Karch CM, Bateman RJ, Tau kinetics in neurons and the human central nervous system. Neuron 98, 861–864 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Small SA, Simoes-Spassov S, Mayeux R, Petsko GA, Endosomal traffic jams represent a pathogenic hub and therapeutic target in Alzheimer’s disease. Trends Neurosci. 40, 592–602 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Small SA, Petsko GA, Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nat. Rev. Neurosci 16, 126–132 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, Chen F, Shibata N, Lunetta KL, Pardossi-Piquard R, Bohm C, Wakutani Y, Cupples LA, Cuenco KT, Green RC, Pinessi L, Rainero I, Sorbi S, Bruni A, Duara R, Friedland RP, Inzelberg R, Hampe W, Bujo H, Song Y-Q, Andersen OM, Willnow TE, Graff-Radford N, Petersen RC, Dickson D, Der SD, Fraser PE, Schmitt-Ulms G, Younkin S, Mayeux R, Farrer LA, St George-Hyslop P, The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat. Genet 39, 168–177 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vardarajan BN, Zhang Y, Lee JH, Cheng R, Bohm C, Ghani M, Reitz C, Reyes-Dumeyer D, Shen Y, Rogaeva E, St George-Hyslop P, Mayeux R, Coding mutations in SORL1 and Alzheimer disease. Ann. Neurol 77, 215–227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Verheijen J, Van den Bossche T, van der Zee J, Engelborghs S, Sanchez-Valle R, Lladó A, Graff C, Thonberg H, Pastor P, Ortega-Cubero S, Pastor MA, Benussi L, Ghidoni R, Binetti G, Clarimon J, Lleó A, Fortea J, de Mendonça A, Martins M, Grau-Rivera O, Gelpi E, Bettens K, Mateiu L, Dillen L, Cras P, De Deyn PP, Van Broeckhoven C, Sleegers K, A comprehensive study of the genetic impact of rare variants in SORL1 in European early-onset Alzheimer’s disease. Acta Neuropathol. 132, 213–224 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, Honig L, Vonsattel J-P, Kim T-W, Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Ann. Neurol 58, 909–919 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Rovelet-Lecrux A, Charbonnier C, Wallon D, Nicolas G, Seaman MNJ, Pottier C, Breusegem SY, Mathur PP, Jenardhanan P, Le Guennec K, Mukadam AS, Quenez O, Coutant S, Rousseau S, Richard A-C, Boland A, Deleuze J-F, Frebourg T, Hannequin D, Campion D; CNR-MAJ collaborators, De novo deleterious genetic variations target a biological network centered on Aβ peptide in early-onset Alzheimer disease. Mol. Psychiatry 20, 1046–1056 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Vardarajan BN, Bruesegem SY, Harbour ME, Inzelberg R, Friedland R, St George-Hyslop P, Seaman MNJ, Farrer LA, Identification of Alzheimer disease-associated variants in genes that regulate retromer function. Neurobiol. Aging 33, 2231.e15–2231.e30 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muhammad A, Flores I, Zhang H, Yu R, Staniszewski A, Planel E, Herman M, Ho L, Kreber R, Honig LS, Ganetzky B, Duff K, Arancio O, Small SA, Retromer deficiency observed in Alzheimer’s disease causes hippocampal dysfunction, neurodegeneration, and Aβ accumulation. Proc. Natl. Acad. Sci. U.S.A 105, 7327–7332 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhalla A, Vetanovetz CP, Morel E, Chamoun Z, Di Paolo G, Small SA, The location and trafficking routes of the neuronal retromer and its role in amyloid precursor protein transport. Neurobiol. Dis 47, 126–134 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wen L, Tang F-L, Hong Y, Luo S-W, Wang C-L, He W, Shen C, Jung J-U, Xiong F, D.-h. Lee, Q.-G. Zhang, D. Brann, T.-W. Kim, R. Yan, L. Mei, W.-C. Xiong, VPS35 haploinsufficiency increases Alzheimer’s disease neuropathology. J. Cell Biol 195, 765–779 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mecozzi VJ, Berman DE, Simoes S, Vetanovetz C, Awal MR, Patel VM, Schneider RT, Petsko GA, Ringe D, Small SA, Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol 10, 443–449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Young JE, Fong LK, Frankowski H, Petsko GA, Small SA, Goldstein LSB, Stabilizing the retromer complex in a human stem cell model of Alzheimer’s disease reduces TAU phosphorylation independently of amyloid precursor protein. Stem Cell Rep. 10, 1046–1058 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yin J, Liu X, He Q, Zhou L, Yuan Z, Zhao S, Vps35-dependent recycling of Trem2 regulates microglial function. Traffic 17, 1286–1296 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Lucin KM, O’Brien CE, Bieri G, Czirr E, Mosher KI, Abbey RJ, Mastroeni DF, Rogers J, Spencer B, Masliah E, Wyss-Coray T, Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron 79, 873–886 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA, Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer’s disease and Down syndrome: Differential effects of APOE genotype and presenilin mutations. Am. J. Pathol 157, 277–286 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LSB, Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raja WK, Mungenast AE, Lin Y-T, Ko T, Abdurrob F, Seo J, Tsai L-H, Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer’s disease phenotypes. PLOS ONE 11, e0161969 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barthélemy NR, Li Y, Joseph-Mathurin N, Gordon BA, Hassenstab J, Benzinger TLS, Buckles V, Fagan AM, Perrin RJ, Goate AM, Morris JC, Karch CM, Xiong C, Allegri R, Mendez PC, Berman SB, Ikeuchi T, Mori H, Shimada H, Shoji M, Suzuki K, Noble J, Farlow M, Chhatwal J, Graff-Radford NR, Salloway S, Schofield PR, Masters CL, Martins RN, O’Connor A, Fox NC, Levin J, Jucker M, Gabelle A, Lehmann S, Sato C, Bateman RJ, McDade E; the Dominantly Inherited Alzheimer Network, A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat. Med 26, 398–407 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barthélemy NR, Bateman RJ, Hirtz C, Marin P, Becher F, Sato C, Gabelle A, Lehmann S, Cerebrospinal fluid phospho-tau T217 outperforms T181 as a biomarker for the differential diagnosis of Alzheimer’s disease and PET amyloid-positive patient identification. Alzheimers Res. Ther 12, 26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis J-C, Collins F, Treanor J, Rogers G, Citron M, β-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Dislich B, Wohlrab F, Bachhuber T, Müller SA, Kuhn P-H, Hogl S, Meyer-Luehmann M, Lichtenthaler SF, Label-free quantitative proteomics of mouse cerebrospinal fluid detects β-site APP cleaving enzyme (BACE1) protease substrates in vivo. Mol. Cell. Proteomics 14, 2550–2563 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang C-L, Tang F-L, Peng Y, Shen C-Y, Mei L, Xiong W-C, VPS35 regulates developing mouse hippocampal neuronal morphogenesis by promoting retrograde trafficking of BACE1. Biol. Open 1, 1248–1257 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cicognola C, Brinkmalm G, Wahlgren J, Portelius E, Gobom J, Cullen NC, Hansson O, Parnetti L, Constantinescu R, Wildsmith K, Chen H-H, Beach TG, Lashley T, Zetterberg H, Blennow K, Höglund K, Novel tau fragments in cerebrospinal fluid: Relation to tangle pathology and cognitive decline in Alzheimer’s disease. Acta Neuropathol. 137, 279–296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meredith JE Jr., Sankaranarayanan S, Guss V, Lanzetti A, Berisha F, Neely RJ, Slemmon JR, Portelius E, Zetterberg H, Blennow K, Soares H, Ahlijanian M, Albright CF, Characterization of novel CSF Tau and ptau biomarkers for Alzheimer’s disease. PLOS ONE 8, e76523 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barthélemy NR, Fenaille F, Hirtz C, Sergeant N, Schraen-Maschke S, Vialaret J, Buée L, Gabelle A, Junot C, Lehmann S, Becher F, Tau protein quantification in human cerebrospinal fluid by targeted mass spectrometry at high sequence coverage provides insights into its primary structure heterogeneity. J. Proteome Res 15, 667–676 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Barthélemy NR, Gabelle A, Hirtz C, Fenaille F, Sergeant N, Schraen-Maschke S, Vialaret J, Buée L, Junot C, Becher F, Lehmann S, Differential mass spectrometry profiles of tau protein in the cerebrospinal fluid of patients with Alzheimer’s disease, progressive supranuclear palsy, and dementia with Lewy bodies. J. Alzheimer Dis 51, 1033–1043 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Kramer A, Green J, Pollard J Jr., S. Tugendreich, Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chai X, Dage JL, Citron M, Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol. Dis 48, 356–366 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Karch CM, Jeng AT, Goate AM, Extracellular Tau levels are influenced by variability in Tau that is associated with tauopathies. J. Biol. Chem 287, 42751–42762 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mohamed N-V, Plouffe V, Rémillard-Labrosse G, Planel E, Leclerc N, Starvation and inhibition of lysosomal function increased tau secretion by primary cortical neurons. Sci. Rep 4, 5715 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barten DM, Cadelina GW, Hoque N, De Carr LB, Guss VL, Yang L, Sankaranarayanan S, Wes PD, Flynn ME, Meredith JE, Ahlijanian MK, Albright CF, Tau transgenic mice as models for cerebrospinal fluid tau biomarkers. J. Alzheimer Dis 24 (suppl 2), 127–141 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Rissin DM, Kan CW, Campbell TG, Howes SC, Fournier DR, Song L, Piech T, Patel PP, Chang L, Rivnak AJ, Ferrell EP, Randall JD, Provuncher GK, Walt DR, Duffy DC, Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol 28, 595–599 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schelle J, Häsler LM, Göpfert JC, Joos TO, Vanderstichele H, Stoops E, Mandelkow E-M, Neumann U, Shimshek DR, Staufenbiel M, Jucker M, Kaeser SA, Prevention of tau increase in cerebrospinal fluid of APP transgenic mice suggests downstream effect of BACE1 inhibition. Alzheimers Dement. 13, 701–709 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Chen Z, Mengel D, Keshavan A, Rissman RA, Billinton A, Perkinton M, Percival-Alwyn J, Schultz A, Properzi M, Johnson K, Selkoe DJ, Sperling RA, Patel P, Zetterberg H, Galasko D, Schott JM, Walsh DM, Learnings about the complexity of extracellular tau aid development of a blood-based screen for Alzheimer’s disease. Alzheimers Dement. 15, 487–496 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zetterberg H, Review: Tau in biofluids - relation to pathology, imaging and clinical features. Neuropathol. Appl. Neurobiol 43, 194–199 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Maia LF, Kaeser SA, Reichwald J, Hruscha M, Martus P, Staufenbiel M, Jucker M, Changes in amyloid-β and Tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Sci. Transl. Med 5, 194re2 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Bayer TA, Cappai R, Masters CL, Beyreuther K, Multhaup G, It all sticks together––the APP-related family of proteins and Alzheimer’s disease. Mol. Psychiatry 4, 524–528 (1999). [DOI] [PubMed] [Google Scholar]
- 44.Yanagida K, Okochi M, Tagami S, Nakayama T, Kodama TS, Nishitomi K, Jiang J, Mori K, Tatsumi S-I, Arai T, Ikeuchi T, Kasuga K, Tokuda T, Kondo M, Ikeda M, Deguchi K, Kazui H, Tanaka T, Morihara T, Hashimoto R, Kudo T, Steiner H, Haass C, Tsuchiya K, Akiyama H, Kuwano R, Takeda M, The 28-amino acid form of an APLP1-derived Aβ-like peptide is a surrogate marker for Aβ42 production in the central nervous system. EMBO Mol. Med 1, 223–235 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Begcevic I, Brinc D, Brown M, Martinez-Morillo E, Goldhardt O, Grimmer T, Magdolen V, Batruch I, Diamandis EP, Brain-related proteins as potential CSF biomarkers of Alzheimer’s disease: A targeted mass spectrometry approach. J. Proteomics 182, 12–20 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Rajendran L, Annaert W, Membrane trafficking pathways in Alzheimer’s disease. Traffic 13, 759–770 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Tüshaus J, Müller SA, Kataka ES, Zaucha J, Monasor LS, Su M, Güner G, Jocher G, Tahirovic S, Frishman D, Simons M, Lichtenthaler SF, Quantitative secretome analysis establishes the cell type-resolved mouse brain secretome. bioRxiv 2020.05.22.110023, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caballero B, Wang Y, Diaz A, Tasset I, Juste YR, Stiller B, Mandelkow E-M, Mandelkow E, Cuervo AM, Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 17, e12692 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vaz-Silva J, Gomes P, Jin Q, Zhu M, Zhuravleva V, Quintremil S, Meira T, Silva J, Dioli C, Soares-Cunha C, Daskalakis NP, Sousa N, Sotiropoulos I, Waites CL, Endolysosomal degradation of Tau and its role in glucocorticoid-driven hippocampal malfunction. EMBO J. 37, e99084 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, Finn MB, Manis M, Geerling JC, Fuller PM, Lucey BP, Holtzman DM, The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 363, 880–884 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K, Trans-synaptic spread of tau pathology in vivo. PLOS ONE 7, e31302 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT, Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron 73, 685–697 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morabito MV, Berman DE, Schneider RT, Zhang Y, Leibel RL, Small SA, Hyperleucinemia causes hippocampal retromer deficiency linking diabetes to Alzheimer’s disease. Neurobiol. Dis 65, 188–192 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kwart D, Gregg A, Scheckel C, Murphy EA, Paquet D, Duffield M, Fak J, Olsen O, Darnell RB, Tessier-Lavigne M, A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP β-CTFs, not Aβ. Neuron 104, 256–270.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 55.Chu J, Pratico D, The retromer complex system in a transgenic mouse model of AD: Influence of age. Neurobiol. Aging 52, 32–38 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Bateman RJ, Wen G, Morris JC, Holtzman DM, Fluctuations of CSF amyloid-β levels: Implications for a diagnostic and therapeutic biomarker. Neurology 68, 666–669 (2007). [DOI] [PubMed] [Google Scholar]
- 57.Triana-Baltzer G, Van Kolen K, Theunis C, Moughadam S, Slemmon R, Mercken M, Galpern W, Sun H, Kolb H, Development and validation of a high sensitivity assay for measuring p217+tau in cerebrospinal fluid. J. Alzheimers Dis 77, 1417–1430 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data file S3. Individual-level data (provided as a separate Excel file).
Data file S1. Completed list of proteins identified in murine CSF (provided as a separate Excel file).
Data file S2. List of peptides detected by MS/MS analysis (provided as a separate Excel file).
Fig. S1. n-APLP1 and n-CHL1 correlation analysis based on MS/MS peak intensity measurements.
Fig. S2. NeuN immunostaining and quantification of retromer protein expression in 3-month-old Vps35 cKOs and control littermates.
Fig. S3. Development and qualification of Simoa APLP1 and CHL1 assays.
Fig. S4. Extended data of Western blots cropped in main Fig. 1.
Fig. S5. Extended data of Western blots cropped in main Fig. 4A.
Fig. S6. Extended data of Western blots cropped in main Fig. 4D.