

Abstract

Developing and optimizing small-molecule biosensors is a central goal of synthetic biology. Here we incorporate additional cellular components to improve biosensor sensitivity by preventing target molecules from diffusing out of cells. We demonstrate that trapping erythromycin within Escherichia coli through phosphorylation increases the sensitivity of its biosensor (MphR) by approximately 10-fold. When combined with prior engineering efforts, our optimized biosensor can detect erythromycin concentrations as low as 13 nM. We show that this strategy works with a range of macrolide substrates, enabling the potential usage of our optimized system for drug development and metabolic engineering. This strategy can be extended in future studies to improve the sensitivity of other biosensors. Our findings further suggest that many naturally evolved genes involved in resistance to multiple classes of antibiotics may increase intracellular drug concentrations to modulate their own expression, acting as a form of regulatory feedback.
Small-molecule induction of gene expression has long been used in biochemistry, yet over the past decade the development of new small-molecule biosensors has rapidly improved and matured.1 Existing biosensors enable cells to detect and respond to a wide variety of chemical signals and have numerous diverse applications within metabolic engineering and synthetic biology.2,3 Biosensors exhibiting a linear range of response have also been used to provide quantitative measurements of increasing substrate levels.4 Inducible transcription factors constitute one of the largest classes of biosensors, and most work by modulating the transcription levels of regulated genes in response to a small-molecule ligand. The efficacy of a biosensor is described by several parameters, including its specificity for a given chemical ligand input, its sensitivity toward low ligand concentrations, and the signal-to-noise ratio of its transcriptional output.
Although numerous biosensors have previously been discovered or constructed, poor efficacy often precludes their usage for many practical applications.1,5 To date, several strategies have been developed to improve transcription-based biosensors. Many methods improve the dynamic range and signal-to-noise ratio of a biosensor by optimizing the expression level of the biosensor itself or the gene it regulates. Expressional-level optimization is typically accomplished by modifying promoters, ribosome binding sites, or plasmid origins.6−8 Other approaches involve engineering of the biosensor protein and are further capable of altering the biosensor specificity in addition to each of the aforementioned biosensor parameters.9−11 While engineering efforts can significantly improve biosensor properties, current strategies largely leave the available cellular concentrations of small-molecule ligands unaltered.
In this work, we sought to incorporate additional biological components into the cell to improve biosensor detection by concentrating small-molecule ligands within cells. To demonstrate the feasibility and utility of this approach, we used the macrolide-responsive biosensor MphR, which induces transcription of genes under the control of the promoter PmphA with high signal-to-noise ratio in response to erythromycin A (EryA).7,8 Prior studies have used MphR in multiple applications, including construction of engineered genetic circuits,8 environmental detection of antibiotics,12 and pharmaceutical screening to identify new macrolide-producing microbes.13 In nature, MphR responds to the antibiotic EryA by derepressing the expression of the macrolide resistance gene mphA.14 Expression of MphA in turn mediates resistance to EryA by phosphorylation of the drug, thereby preventing translation inhibition.14 As the relatively hydrophobic drug EryA typically enters and exits the cell by diffusing across the lipid membrane, we noted that the addition of a charged phosphate group would greatly reduce its diffusion out of the cell.15−17 This effect would serve to improve detection by MphR by increasing the intracellular concentration of EryA (Figure 1), with mphR and mphA thus acting as a regulatory feedback loop in response to EryA. We hypothesized that constitutive expression of mphA (as opposed to expression within a feedback loop) could be used to improve the sensitivity of laboratory biosensors.
Figure 1.

Cellular substrate trapping of EryA. (A) In cells containing our biosensor construct, MphR responds to the addition of the antibiotic EryA by derepressing expression of GFP. Under normal conditions, EryA freely diffuses in and out of the cell. (B) In the presence of macrolide resistance protein MphA, intracellular EryA is phosphorylated, trapping the compound within the cell. This greatly increases the intracellular concentration of the macrolide drug, leading to a stronger biosensor response at lower concentrations of added EryA. (C) Biosensor constructs used in this work. Differences in their genetic contents as well as the proposed resulting effect are indicated.
We first tested our intracellular trapping strategy using the previously engineered biosensor construct pJKR-H-mphR8 as a scaffold (Table S1). This plasmid responds to EryA by increasing the expression of superfolder green fluorescent protein (sfGFP) and also encodes the macrolide resistance gene ermC. In contrast to MphA, ErmC mediates resistance to EryA by modifying the intracellular target of macrolide antibiotics.18 We selected this plasmid as our scaffold after testing it alongside an alternative construct (pMLGFP7) and finding that pJKR-H-mphR exhibited a significantly greater response to EryA (Figure S1). As cells containing pJKR-H-mphR are not expected to trap EryA within cells, we replaced ermC with mphA within this plasmid (constructing vector pCM54; see Table S1) with the goal of producing the highest possible response at the lowest possible EryA concentration (for genetic circuit diagrams showing the effects of resistance genes in this study, see Figure S2). In tests of cells carrying these plasmids using a fluorescent-plate-reader assay, pCM54 cells exhibited ∼10-fold greater EryA detection sensitivity than pJKR-H-mphR cells (Figure 2A).
Figure 2.

Optimization of EryA detection using MphR. (A) Two different macrolide resistance genes were tested within the same plasmid scaffold. Plasmid pCM120, containing both resistance genes, exhibited increased sensitivity at low EryA concentrations without loss of signal at high concentrations. (B) Sensitivity values for the response curves tested in (A). (C) Two different mphR variants were compared, with pCM121 encoding mphR mutant T17R, which has previously been shown to improve EryA sensitivity. (D) Sensitivity values for the response curves tested in (C). Error bars represent the standard deviation across three biological replicates. Small error bars are not visible for some samples exhibiting small standard deviations.
The above findings appear to validate our strategy, as cellular trapping mediated by mphA allows cells carrying pCM54 to respond to lower concentrations of EryA. However, we separately observed that the response of pCM54 was impaired at high EryA concentrations compared with pJKR-H-mphR, which serves as a drawback for the utility of pCM54. After testing the minimum inhibitory concentration (MIC) of EryA in pCM54 cells containing each of the aforementioned plasmids, we found that the loss of signal observed at high EryA concentrations could be explained by the inhibitory effects of the drug (Table 1), as expression of mphA mediated significantly lower levels of drug resistance compared with ermC. This led us to clone a new vector encoding both resistance genes, termed pCM120. Our experiments revealed that cells carrying pCM120 exhibited improved sensitivity toward low concentrations of EryA while also exhibiting strong signals at high concentrations due to improved drug resistance (Figure 2A). While the presence of both resistance genes in pCM120 improves its signal at most EryA concentrations, we observed that plasmid pJKR-H-mphR produced greater sfGFP signal at the highest concentration tested (1000 μM). This might be attributed to either marginally weaker binding of phosphorylated EryA to mphR or a more nuanced effect on protein translation arising from perturbations to cellular osmotic conditions due to high intracellular concentrations of phosphorylated EryA.
Table 1. MIC Values of E. coli Cells Containing Various Plasmids Used in This Studya.
| plasmid | MIC (μM) |
|---|---|
| no plasmid | 80 ± 40 |
| pJKR-H-mphR | >5120 |
| pCM54 | 160 ± 80 |
| pCM98 | 160 ± 80 |
| pCM120 | >5120 |
| pCM121 | >5120 |
Agar dilution MIC tests were performed in triplicate for each sample. All plasmids were tested following transformation into E. coli strain NEB Turbo cells.
To quantify the sensitivity in this work, we used a parameter that we have termed the “two-fold increase parameter” (Figure 2B,D). In previous studies, biosensor sensitivity has often been quantified by calculating the half-maximal parameter.7,8 However, in view of our observation that the presence of different EryA resistance genes alters the maximal biosensor response (see Figure 2A), the half-maximal parameter is unsuitable for comparing the sensitivities of our constructs. For each construct, two-fold increase parameters were calculated by determining the concentration of inducer that results in a 2-fold increase in fluorescence relative to the fluorescence measured in the absence of inducer (see Methods). Tabulated two-fold increase parameters for each construct in this study are also shown in Table S2.
We next sought to further improve the sensitivity of our biosensor system by incorporating mphR engineering efforts from another prior study by Kasey et al.(7) We thus introduced the T17R mutation into the mphR coding sequence in pCM120 to construct pCM121, since this mutation was previously shown to increase the EryA sensitivity.7 Consistent with prior work, the addition of this mutation increased the EryA sensitivity by approximately 3-fold (Figure 2D). To identify the lower limit of detectable response to EryA, we calculated the concentration at which a 2-fold sfGFP response is observed for each construct (Table S2). Our optimized construct pCM121 showed the highest degree of sensitivity, producing a 2-fold signal increase at 13.1 nM EryA.
We subsequently tested our combined biosensor system using other macrolide drugs. While MphR has previously been shown to respond to multiple EryA derivatives,7,19 our method of improving biosensor function also requires recognition and phosphorylation of the ligands by MphA. We first used our fluorescence-based assay to separately test cells containing pJKR-H-mphR and pCM120 for detection of three macrolide compounds: clarithromycin (Clr), roxithromycin (Rxr), and azithromycin (Azr). These macrolides are all derivatives of EryA, with Clr and Rxr containing a 14-membered ring (like EryA) and Azr containing a 15-membered ring (for macrolide structures, see Figure S3). Our experiments found that cells containing pCM120 (expressing mphA) exhibited significantly greater detection sensitivity for all three drugs compared with cells containing pJKR-H-mphR (lacking mphA) (Figure 3). As shown in Figures 2 and Figure 3 and Table S2, the addition of mphA reduced the two-fold increase parameter by 9.9-fold for EryA, 187-fold for Clr, 73-fold for Azr, and 9.9-fold for Rxr. While the reasons underpinning the significantly greater degree of improvement observed for Clr and Azr are unclear, it may be due to lower amounts of cell accumulation of these two compounds compared with EryA and Rxr in the absence of phosphorylation.
Figure 3.

Detection of macrolide compounds using biosensor constructs pCM120 (expressing mphA and ermC) and pJKR-H-mphR (expressing ermC). Dose–response curves are shown alongside bar graphs of detection sensitivity values for (A, B) roxithromycin (Rxr), (C, D) azithromycin (Azr), and (E, F) clarithromycin (Clr) (see Figure S3 for macrolide structures). For each compound, cells containing pCM120 exhibited significantly greater detection sensitivity than cells containing pJKR-H-mphR. Tests were performed using three biological replicates; standard deviations across these three replicates are shown as error bars for each point. Small error bars are not visible for some samples exhibiting small standard deviations.
We further observed that Clr and Azr produced a response at drug dosages comparable to that of EryA, while detection of Rxr required significantly higher drug concentrations (Figure 3). The increased sensitivity observed using pCM120 indicates that each of the macrolides tested serves as a phosphorylation substrate by MphA, with the reduced sensitivity toward Rxr stemming from substrate recognition properties of MphR. These results are in agreement with prior in vitro ELISA experiments performed using purified MphR, which also showed that recognition of Rxr requires higher concentrations.12,19 We also tested recognition of these compounds using plasmid pCM54, which lacks the resistance gene ermC. While cells containing pCM54 produced a similar fluorescence response compared with pCM120 cells at low concentrations of each derivative, the response of pCM54 cells was impaired at high drug concentrations (Figure S4). A similar pattern was observed for the response of pCM54 cells to EryA and can largely be attributed to the effects of these macrolide drugs on cell fitness (see earlier discussion).
We next sought to characterize the recognition parameters of mphR together with mphA toward EryA precursors both to evaluate the utility of this biosensor system for metabolic engineering applications and to improve our understanding of the function of these genes within their natural context. We thus tested the fluorescence response of our combined system toward two 14-membered EryA precursors—erythromycin B (EryB) and erythromycin C (EryC). EryB and EryC each lack different moieties that are present in EryA (Figure S3), and both compounds immediately precede EryA within the forking biosynthetic pathway (see Figure S5). As shown in Figure S6, EryB produced a similar response as EryA, but reduced signal was observed in response to EryC. However, when preparing drug solutions we observed that EryC exhibited incomplete solubility (see Methods), which likely accounts for its signal discrepancy relative to EryA. While macrolides that occur earlier in the biosynthetic pathway are commercially unavailable, we infer that EryD would likely also be recognized by MphR and MphA, as EryD lacks both groups that are separately absent from EryC and EryB. While macrolides preceding EryD may be recognized by MphR, they cannot be phosphorylated by MphA, as they lack the desosamine group (the sugar group attached at C5 in EryA) that serves as the site of phosphorylation. As the absence of phosphorylation precludes the possibility of trapping these macrolides within the cell, the detection sensitivity would be reduced using our combined system. The additional absence of a cladinose group (the sugar group attached at C3 in EryA) from the earlier intermediates erythronolide B and 6-deoxyerithronolide B (6-dEB) makes their detection unlikely, considering the importance of this group to MphR recognition (see discussion below). Thus, while our combined biosensor system is capable of detecting the late EryA precursors EryB and EryC and likely also EryD, detection of earlier precursors is expected to be significantly impaired.
To better understand the structural determinants of substrate specificity in our combined biosensor system, we next examined available cocrystal structures of EryA in complex with MphR (PDB entry 3FRQ) and MphA (PDB entry 5IGP)20,21 (for separate depictions of EryA in complex with MphR and MphA from previously solved structures, see Figure S7). As shown in Figure 4, we initially identified regions of EryA that contain polar contacts either with MphA (shown in red) or MphR (shown in blue). For MphA, contacts with EryA were almost entirely hydrophobic, with the single exception being the site of phosphorylation within the desosamine group. This desosamine moiety is conserved across all of the macrolide compounds tested here, which may serve to explain why MphA was capable of recognizing each compound. While the majority of contacts between EryA and MphR were also hydrophobic, MphR exhibited several polar contacts. We first noted that while the macrolides Clr and EryC lack different polar groups found in EryA (see Figure S3), these moieties did not form contacts with MphR, consistent with our findings that MphR recognition of these compounds is on par with that of EryA. Interestingly, Azr and EryB each lack a different polar group (C9 carbonyl and C12 hydroxyl, respectively) that would otherwise serve as an MphR contact, yet our experiments also show recognition of these compounds to be comparable to that of EryA. This suggests that the absence of these moieties, as well as the increased macrolide ring size of Azr, does not significantly affect MphR recognition. We last noted that compared with EryA, Rxr has replaced the typical C9 carbonyl group with a bulky N-oxime side chain. As the C9 group faces the interior of the MphR binding pocket,20 this N-oxime side chain is expected to cause a steric clash, which may explain the reduced recognition of Rxr observed in Figure 3. These findings indicate that the absence of multiple macrolide groups that form polar protein contacts does not significantly alter the response of MphR, as ligand recognition is largely mediated by hydrophobic interactions. However, the addition of bulky ligand moieties can disrupt MphR recognition, whereas MphA recognition appears to be more tolerant of such alterations.
Figure 4.
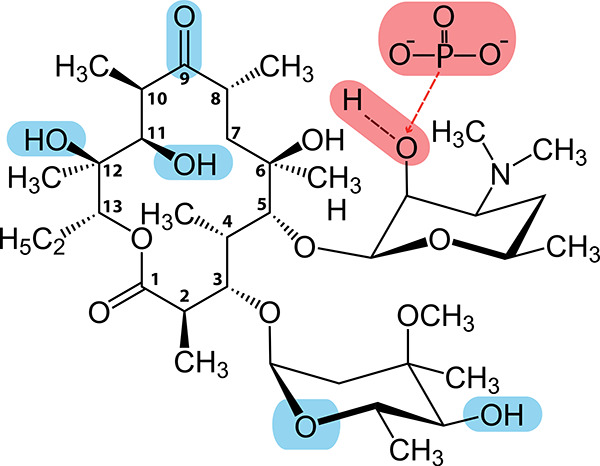
Polar moieties involved in biosensor recognition of EryA. Atoms that contact with MphR are highlighted in blue; contacts with MphA are highlighted in red. Numbering is shown for each carbon within the macrolide ring. Contacts were identified using previously published cocrystal structures of EryA in complex with MphR (PDB entry 3FRQ)20 and MphA (PDB entry 5IGP).21 Notably, both proteins exhibit significant nonpolar contacts, indicating that substrate recognition is primarily mediated by shape complementarity toward the core macrolide structure.
We next considered prior biochemical studies to further advance our understanding of substrate recognition by MphR and MphA. Previously, MphR was shown to exhibit poor recognition of the 14-membered macrolides narbomycin and picromycin using a luminescence-based assay.13 Poor recognition of these compounds is likely due largely to the absence of the C3 cladinose group, which exhibits two polar contacts as well as significant nonpolar contacts with MphR. Reduced recognition may also be attributed to the absence of the C12 hydroxyl (in narbomycin) and the C11 hydroxyl (in narbomycin and picromycin). Another study tested MphR recognition of several 16-membered macrolides (midecamycin, acetylspiramycin, josamycin, meleumycin, and kitsamycin) using an ELISA-based assay19 and found that MphR was unable to recognize any of the 16-membered drugs. Each of these 16-membered compounds contains a different core structure compared with 14-membered EryA derivatives, not only exhibiting an altered macrolide ring but also lacking a C3 cladinose group, a C11 hydroxyl group, and a C12 hydroxyl group, akin to narbomycin. While MphA type I (the variant used here) also exhibits poor activity against 16-membered macrolides,22 this enzyme efficiently phosphorylates a wide variety of 14- and 15-membered macrolide drugs.22,23 Taken together, these results indicate that the addition of MphA in our biosensor system is unlikely to reduce the scope of MphR substrate detection, as our combined system exhibits substrate specificity similar to that of MphR used in isolation.
We next looked for other examples in nature of the use of similar mechanisms of cellular substrate trapping for biosensor detection, noting that MphR and MphA are found together within their natural context.14 Upon reviewing the literature, we found that two previously characterized aminoglycoside-responsive riboswitches separately regulate expression of aminoglycoside adenyltransferase (AAD) and aminoglycoside acetyltransferase (AAC) enzymes.24 Like macrolides, aminoglycosides are relatively hydrophobic antibiotics that enter and exit the cell by diffusing through the membrane.16 Thus, the addition of a polar adenyl or acetyl group is also likely to prevent these drugs from exiting the cell. This operon structure allows these resistance genes to increase their own expression at low concentrations of antibiotic, producing more rapid and “switchlike” gene expression in response to antibiotic exposure through a feedback mechanism. Additional similarly functioning feedback loops likely exist elsewhere in nature, apart from the well-characterized examples given here.
In addition to the natural example given above, our cellular substrate trapping strategy can be further applied in the laboratory to improve the sensitivity of additional biosensors. Antibiotic biosensors provide the clearest targets for improvement, considering the preponderance of known resistance enzymes that mediate covalent modifications of their structures. Numerous examples of antimicrobial biosensors have been identified within the TetR protein family.25 In addition to MphR, example biosensors within this group include the tetracycline biosensor TetR,25 the actinorhodin biosensor ActR,26 the streptogramin biosensor Pip,12 the ethionamide biosensor EthR,27 and the aminoglycoside biosensor IcaR.28 Two-component antibiotic biosensors have also been observed, with the best-described example being the vancomycin-responsive vanS/vanR system.29 Antibiotic-responsive RNA elements have similarly been found, including the aforementioned aminoglycoside riboswitch24 as well as numerous riboregulators that use a complex mechanism to respond to translation inhibitor drugs.30 While these biosensors regulate a wide variety of resistance genes within their natural contexts, they can be rationally paired with antibiotic-modifying genes to increase their sensitivity. Enzymatic modification of drugs is a remarkably common drug resistance mechanism, with a wide variety of antibiotic classes being targeted for the transfer of polar groups from numerous antibiotic resistance enzymes, including phosphotransferases, adenyltransferases, acetyltransferases, glycosyltransferases, S-transferases, and ADP-ribosyltransferases.31 While some modifications may not cause cellular trapping of targeted antibiotics in every instance, the addition of polar (and particularly charged) groups is expected to inhibit traversal of most drugs across bacterial membranes.15,16 As we have illustrated in this study, our trapping strategy can be expected to increase the detection sensitivity by an order of magnitude and can readily be combined with other engineering approaches to improve biosensor efficacy.
In contrast to the positive feedback loop between MphR and MphA in their natural context, previous studies have shown that TetR and the tetracycline resistance efflux pump TetA are naturally present in a negative feedback loop. Prior work has shown that when TetA is present in a feedback loop, it alters the response curve of TetR but does not significantly change its detection sensitivity.32 However, when TetA is constitutively expressed, the sensitivity of TetR decreases.33 Thus, while we have shown that constitutive expression of MphA improves MphR sensitivity, the presence of MphA within a feedback loop may instead result in a more complex change toward the MphR response curve.
In this work, we have appropriated a cell trapping strategy from nature and combined it with prior MphR engineering efforts to produce a more sensitive macrolide biosensor system. Our optimized construct (pCM121) is able to respond to a wide variety of macrolide compounds without loss of signal at high concentrations. A potential future use for our optimized construct lies in metabolic engineering, as this biosensor could be used to screen or select for strains producing engineered non-natural macrolide compounds34 or improved yields of EryA in E. coli.35,36 Other potential applications include screening libraries of producer strains to identify naturally occurring macrolide drugs13 as well as detecting macrolides within environmental samples.12 For each of the above applications, the concentrations of macrolides to be detected are typically very small, making sensitivity a key parameter for an effective biosensor. While this cellular trapping effect is not likely to affect rates of macrolide degradation,37 it may lead to an altered dilution effect as macrolide concentrations become linked to cell growth rather than the total volume of the cell solution, which may be further explored in future studies. This work thus opens several future directions in synthetic biology, biomedicine, and environmental engineering.
Methods
Molecular Cloning
All constructs were prepared using Gibson assembly;38 mutations were introduced by QuikChange mutagenesis (New England Biolabs, cat. no. E0554S). Plasmids pMLGFP and pJZ12 were given as generous gifts by the Williams lab at North Carolina State University. Plasmid pJKR-H-mphR was obtained from the Addgene repository, to which it was originally provided by the Church lab at Harvard University.
Fluorescence-Based Macrolide Detection Assay
We performed a fluorescence-based mphR assay as previously reported.8 We prepared E. coli strain NEB Turbo cells containing a reporter vector encoding constitutively expressed mphR and one or more macrolide resistance genes and also containing sfGFP under the control of the PmphA promoter. Reporter plasmids tested are shown in Table S2. Cells were grown to an optical density at 600 nm (OD600) of 0.2–0.5 in 96-well plates and then induced with varying concentrations of macrolides. Cells were grown for 3–18 h. The fluorescence (Fl) intensity (excitation at 488 nm, emission at 509 nm) was measured and normalized by the OD600 for each well. Experiments were performed on biological triplicates, and background correction was performed by subtracting the Fl/OD value of the strain transformed with an empty vector. For tests using EryA, testing substrate concentrations beyond 1 mM resulted in a significant fitness cost due to solvent effects, resulting in impaired cell growth and inconsistent results.
Preparation of Macrolide Compound Stocks
All of the macrolide compounds were commercially sourced from Sigma-Aldrich. Stock solutions of azithromycin, clarithromycin, roxithromycin, erythromycin B, and erythromycin C were prepared using dimethyl sulfoxide (DMSO). For detection assays involving comparison with other macrolide compounds, EryA solutions were prepared using DMSO. For all other detection assays, stock solutions of EryA were prepared using ethanol. All of the compounds were observed to completely dissolve in DMSO with the exception of erythromycin C.
Two-Fold Increase Parameter Calculations
To determine each parameter, linear regressions were first calculated using Microsoft Excel for the linear regions of each macrolide response curve. Linear regressions were then used to calculate the EryA concentration at which a 2-fold increase in sfGFP signal was observed relative to the fluorescence observed in the absence of inducer.
Acknowledgments
We thank Professor Gavin Williams at NC State University for the gift of plasmids pMLGFP and pJZ12 and Professor George Church at Harvard University for the gift of plasmid pJKR-H-mphR (obtained via Addgene). This work was funded by the National Institutes of Health (Grant R01GM117138), the National Science Foundation (Grant MCB-1936774), and the Welch Foundation (Grant C-1729).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.0c00572.
Figures S1–S7 and Tables S1 and S2 (PDF)
Author Contributions
§ C.A.M. and J.M.H. contributed equally. C.A.M., J.M.H., and M.R.B. conceived and designed the experiments and wrote the paper. C.A.M., J.M.H., and S.E.P. carried out the experiments. All of the authors discussed and analyzed the data.
The authors declare no competing financial interest.
Supplementary Material
References
- Carpenter A. C.; Paulsen I. T.; Williams T. C. (2018) Blueprints for Biosensors: Design, Limitations, and Applications. Genes 9, 375. 10.3390/genes9080375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Liu Y.; Wang M. (2017) Design, Optimization and Application of Small Molecule Biosensor in Metabolic Engineering. Front. Microbiol. 8, 2012. 10.3389/fmicb.2017.02012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner A. P. F. (2013) Biosensors: sense and sensibility. Chem. Soc. Rev. 42, 3184–3196. 10.1039/c3cs35528d. [DOI] [PubMed] [Google Scholar]
- Szenk M.; Yim T.; Balazsi G. (2020) Multiplexed Gene Expression Tuning with Orthogonal Synthetic Gene Circuits. ACS Synth. Biol. 9, 930–939. 10.1021/acssynbio.9b00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneshvar S.; Sudhakumari C. C.; Senthilkumaran B.; Prakash H. (2016) Recent Advances in Biosensor Technology for Potential Applications - An Overview. Front. Bioeng. Biotechnol. 4, 11. 10.3389/fbioe.2016.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Ho J. M. L.; Shis D. L.; Gupta C.; Long J.; Wagner D. S.; Ott W.; Josić K.; Bennett M. R. (2018) Tuning the dynamic range of bacterial promoters regulated by ligand-inducible transcription factors. Nat. Commun. 9, 64. 10.1038/s41467-017-02473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasey C. M.; Zerrad M.; Li Y.; Cropp T. A.; Williams G. J. (2018) Development of Transcription Factor-Based Designer Macrolide Biosensors for Metabolic Engineering and Synthetic Biology. ACS Synth. Biol. 7, 227–239. 10.1021/acssynbio.7b00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J. K.; Guzman C. D.; Taylor N. D.; Raman S.; Anderson K.; Church G. M. (2015) Synthetic biosensors for precise gene control and real-time monitoring of metabolites. Nucleic Acids Res. 43, 7648–7660. 10.1093/nar/gkv616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor N. D.; Garruss A. S.; Moretti R.; Chan S.; Arbing M. A.; Cascio D.; Rogers J. K.; Isaacs F. J.; Kosuri S.; Baker D.; Fields S.; Church G. M.; Raman S. (2016) Engineering an allosteric transcription factor to respond to new ligands. Nat. Methods 13, 177–183. 10.1038/nmeth.3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y.; Katsuyama Y.; Ueno K.; Ohnishi Y. (2019) Switching the Ligand Specificity of the Biosensor XylS from meta to para-Toluic Acid through Directed Evolution Exploiting a Dual Selection System. ACS Synth. Biol. 8, 2679–2689. 10.1021/acssynbio.9b00237. [DOI] [PubMed] [Google Scholar]
- Snoek T.; Chaberski E. K.; Ambri F.; Kol S.; Bjorn S. P.; Pang B.; Barajas J. F.; Welner D. H.; Jensen M. K.; Keasling J. D. (2020) Evolution-guided engineering of small-molecule biosensors. Nucleic Acids Res. 48, e3 10.1093/nar/gkz954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C. C.; Link N.; Fux C.; Zisch A. H.; Weber W.; Fussenegger M. (2005) Broad-spectrum protein biosensors for class-specific detection of antibiotics. Biotechnol. Bioeng. 89, 9–17. 10.1002/bit.20224. [DOI] [PubMed] [Google Scholar]
- Mohrle V.; Stadler M.; Eberz G. (2007) Biosensor-guided screening for macrolides. Anal. Bioanal. Chem. 388, 1117–1125. 10.1007/s00216-007-1300-5. [DOI] [PubMed] [Google Scholar]
- Noguchi N.; Takada K.; Katayama J.; Emura A.; Sasatsu M. (2000) Regulation of transcription of the mph(A) gene for macrolide 2′-phosphotransferase I in Escherichia coli: characterization of the regulatory gene mphR(A). J. Bacteriol. 182, 5052–5058. 10.1128/JB.182.18.5052-5058.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M. F.; Drown B. S.; Riley A. P.; Garcia A.; Shirai T.; Svec R. L.; Hergenrother P. J. (2017) Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 545, 299–304. 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcour A. H. (2009) Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta, Proteins Proteomics 1794, 808–816. 10.1016/j.bbapap.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy G.; Wolloscheck D.; Weeks J. W.; Croft C.; Rybenkov V. V.; Zgurskaya H. I. (2016) Breaking the Permeability Barrier of Escherichia coli by Controlled Hyperporination of the Outer Membrane. Antimicrob. Agents Chemother. 60, 7372–7381. 10.1128/AAC.01882-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisblum B. (1995) Erythromycin resistance by ribosome modification. Antimicrob. Agents Chemother. 39, 577–585. 10.1128/AAC.39.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.; Yang S.; Jia M.; Zhao L.; Hou C.; You X.; Zhao J.; Chen A. (2016) Comparative study between macrolide regulatory proteins MphR(A) and MphR(E) in ligand identification and DNA binding based on the rapid in vitro detection system. Anal. Bioanal. Chem. 408, 1623–1631. 10.1007/s00216-015-9270-5. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Sagar V.; Smolinsky A.; Bourke C.; LaRonde-LeBlanc N.; Cropp T. A. (2009) Structure and function of the macrolide biosensor protein, MphR(A), with and without erythromycin. J. Mol. Biol. 387, 1250–1260. 10.1016/j.jmb.2009.02.058. [DOI] [PubMed] [Google Scholar]
- Fong D. H.; Burk D. L.; Blanchet J.; Yan A. Y.; Berghuis A. M. (2017) Structural Basis for Kinase-Mediated Macrolide Antibiotic Resistance. Structure 25, 750–761. 10.1016/j.str.2017.03.007. [DOI] [PubMed] [Google Scholar]
- O’Hara K.; Kanda T.; Ohmiya K.; Ebisu T.; Kono M. (1989) Purification and characterization of macrolide 2′-phosphotransferase from a strain of Escherichia coli that is highly resistant to erythromycin. Antimicrob. Agents Chemother. 33, 1354–1357. 10.1128/AAC.33.8.1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura A.; Nakazawa K.; Miyakozawa I.; Mizukoshi S.; Tsurubuchi K.; Nakagawa M.; O’Hara K.; Sawai T. (2000) Macrolide esterase-producing Escherichia coli clinically isolated in Japan. J. Antibiot. 53, 516–524. 10.7164/antibiotics.53.516. [DOI] [PubMed] [Google Scholar]
- Jia X.; Zhang J.; Sun W.; He W.; Jiang H.; Chen D.; Murchie A. I. (2013) Riboswitch control of aminoglycoside antibiotic resistance. Cell 152, 68–81. 10.1016/j.cell.2012.12.019. [DOI] [PubMed] [Google Scholar]
- Cuthbertson L.; Nodwell J. R. (2013) The TetR family of regulators. Microbiol Mol. Biol. Rev. 77, 440–475. 10.1128/MMBR.00018-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems A. R.; Tahlan K.; Taguchi T.; Zhang K.; Lee Z. Z.; Ichinose K.; Junop M. S.; Nodwell J. R. (2008) Crystal structures of the Streptomyces coelicolor TetR-like protein ActR alone and in complex with actinorhodin or the actinorhodin biosynthetic precursor (S)-DNPA. J. Mol. Biol. 376, 1377–1387. 10.1016/j.jmb.2007.12.061. [DOI] [PubMed] [Google Scholar]
- Willand N.; Dirie B.; Carette X.; Bifani P.; Singhal A.; Desroses M.; Leroux F.; Willery E.; Mathys V.; Deprez-Poulain R.; Delcroix G.; Frenois F.; Aumercier M.; Locht C.; Villeret V.; Deprez B.; Baulard A. R. (2009) Synthetic EthR inhibitors boost antituberculous activity of ethionamide. Nat. Med. 15, 537–544. 10.1038/nm.1950. [DOI] [PubMed] [Google Scholar]
- Jeng W. Y.; Ko T. P.; Liu C. I.; Guo R. T.; Liu C. L.; Shr H. L.; Wang A. H. (2008) Crystal structure of IcaR, a repressor of the TetR family implicated in biofilm formation in Staphylococcus epidermidis. Nucleic Acids Res. 36, 1567–1577. 10.1093/nar/gkm1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H. J.; Hutchings M. I.; Buttner M. J. (2008) Biotechnology, and Biological Sciences Research Council, U. K. Vancomycin resistance VanS/VanR two-component systems. Adv. Exp. Med. Biol. 631, 200–213. 10.1007/978-0-387-78885-2_14. [DOI] [PubMed] [Google Scholar]
- Dar D.; Sorek R. (2017) Regulation of antibiotic-resistance by non-coding RNAs in bacteria. Curr. Opin. Microbiol. 36, 111–117. 10.1016/j.mib.2017.02.005. [DOI] [PubMed] [Google Scholar]
- Egorov A. M.; Ulyashova M. M.; Rubtsova M. Y. (2018) Bacterial Enzymes and Antibiotic Resistance. Acta Naturae 10, 33–48. 10.32607/20758251-2018-10-4-33-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J. C.; Charlebois D. A.; Nevozhay D.; Bodi Z.; Pal C.; Balazsi G. (2016) Efflux Pump Control Alters Synthetic Gene Circuit Function. ACS Synth. Biol. 5, 619–631. 10.1021/acssynbio.5b00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman S.; Rogers J. K.; Taylor N. D.; Church G. M. (2014) Evolution-guided optimization of biosynthetic pathways. Proc. Natl. Acad. Sci. U. S. A. 111, 17803–17808. 10.1073/pnas.1409523111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. R.; Han A. R.; Ban Y. H.; Yoo Y. J.; Kim E. J.; Yoon Y. J. (2010) Genetic engineering of macrolide biosynthesis: past advances, current state, and future prospects. Appl. Microbiol. Biotechnol. 85, 1227–1239. 10.1007/s00253-009-2326-8. [DOI] [PubMed] [Google Scholar]
- Rogers J. K.; Taylor N. D.; Church G. M. (2016) Biosensor-based engineering of biosynthetic pathways. Curr. Opin. Biotechnol. 42, 84–91. 10.1016/j.copbio.2016.03.005. [DOI] [PubMed] [Google Scholar]
- Fang L.; Guell M.; Church G. M.; Pfeifer B. A. (2018) Heterologous erythromycin production across strain and plasmid construction. Biotechnol. Prog. 34, 271–276. 10.1002/btpr.2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassanzadeh A.; Barber J.; Morris G. A.; Gorry P. A. (2007) Mechanism for the degradation of erythromycin A and erythromycin A 2 ‘-ethyl succinate in acidic aqueous solution. J. Phys. Chem. A 111, 10098–10104. 10.1021/jp073030y. [DOI] [PubMed] [Google Scholar]
- Gibson D. G.; Young L.; Chuang R. Y.; Venter J. C.; Hutchison C. A.; Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–U341. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.