

Abstract
Burn is an under-appreciated trauma that is associated with unacceptably high morbidity and mortality. Although the survival rate after devastating burn injuries has continued to increase in previous decades due to medical advances in burn wound care, nutritional and fluid resuscitation and improved infection control practices, there are still large numbers of patients at a high risk of death. One of the most common complications of burn is sepsis, which is defined as “severe organ dysfunction attributed to host's disordered response to infection” and is the primary cause of death in burn patients. Indeed, burn injuries are accompanied by a series of events that lead to sepsis and multiple organ dysfunction syndrome, such as a hypovolaemic state, immune and inflammatory responses and metabolic changes. Therefore, clear diagnostic criteria and predictive biomarkers are especially important in the prevention and treatment of sepsis and septic shock. In this review, we focus on the pathogenesis of burn wound infection and the post-burn events leading to sepsis. Moreover, the clinical and promising biomarkers of burn sepsis will also be summarized.
Keywords: Burn, Infection, Sepsis, Septic shock, Multiple organ dysfunction syndrome, Immune dysregulation, Hypermetabolism, Trauma, Biomaker, Inflammation
Highlights.
Sepsis is one of the most common and severe complications of severe burns.
Immunosuppression and hypermetabolism play key roles in the development of burn sepsis.
Recent diagnostic tools and potential biomarkers are discussed.
Background
Burn injuries cause unpredictable and devastating trauma and are associated with high morbidity and mortality. There are numerous causative mechanisms, including physical (friction, high temperature, cold, radiation and electricity) and chemical factors [1]. Nevertheless, thermal injury caused by hot liquids, solids or fire makes up the majority of burn injuries [2]. According to a report from the World Health Organization in 2018, about 11 million burn cases occur annually worldwide, with burn injuries claiming as many as 180,000 lives [3]; looking back to almost a decade ago, mortality from burns has decreased from the 300,000 deaths recorded in 2011 [4]. The significant improvement in the survival rate of burn patients is in part attributed to advances in intensive care unit treatment and improved wound management, infection control practices and control of hemodynamic disorders [5, 6]. The mortality rate, however, remains unacceptably high, particularly in patients with severe burns. The severity and prognosis of burn injuries depends principally on the depth (Figure 1) and size (Figure 2) of the burn site. Most patients who suffer from severe burn injuries require rapid and specialized emergency burn care to reduce morbidity and mortality. The high fatality rate of severe burns is due to not only hypovolaemic shock and vascular leak, but also abnormal body responses, including immunosuppression [6, 7], excessive inflammation [8] and hypermetabolism [9]. These responses that accompany severe burn injury will result in increased incidence of infection, sepsis and multiple organ dysfunction syndrome (MODS), which are the leading causes of death in severe burn patients [10].
Figure 1.
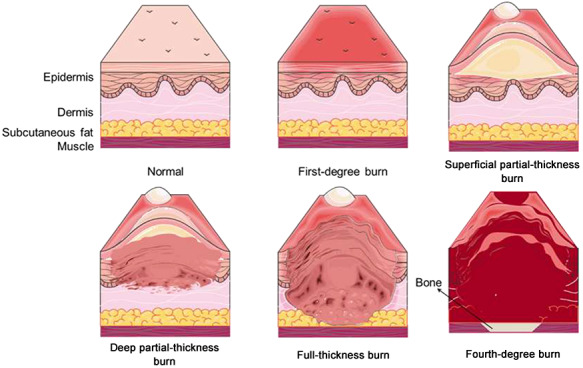
Zones of burn injury for different depths. First-degree burns only involve the epidermis (the uppermost layer of the skin); the skin becomes red and painful, but this is limited in duration. Burns that affect the dermis (the underlying skin layer) are classed as partial-thickness burns, which are frequently accompanied by the formation of painful blisters that increase the risk of infection. Partial-thickness burns can be divided into superficial partial-thickness burns, which are painful, moist, hyperemic and blanch, and deep partial-thickness burns which are less sensate, drier and do not blanch. Full-thickness burns extend through the full dermis and require surgical management due to high risk of infection. Burns extending into deeper tissues (such as muscle or even bone) are defined as fourth-degree burns and are usually blackened and often result in loss of the burned tissues
Figure 2.
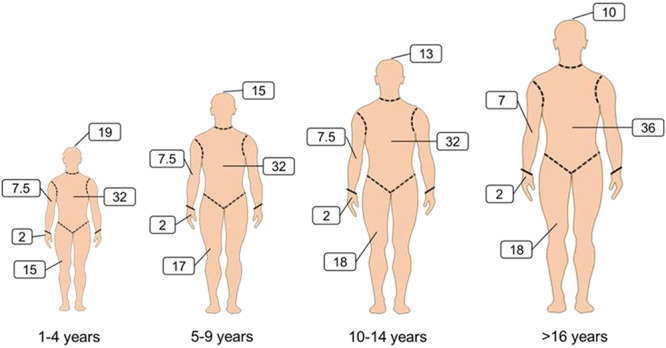
Lund and Browder diagrams for estimation of total burned surface area (TBSA). The “rule of nines” (using multiples of 9) is frequently used to assess the proportion of TBSA affected in adults and to help guide immediate treatment decisions based on burn size. However, the rule of nines is inaccurate in children due to different head-to-body size ratios at different ages. Lund and Browder diagrams are therefore more suitable for assessing of the proportion of TBSA affected in both children and adults. The body areas are separated into different regions (including anterior and posterior) by dashed lines and the numbers are percentages of the TBSA. For instance, 19 in the diagram for children aged 1–4 years relates to the face, neck and head that make up 19% of theTBSA
Burn wound infection is one of the most common and severe complications of severe burns, and occurs due to profound hypermetabolic response and the loss of skin, which is considered the first line of defense against microbial invasion in hosts [11]. Under the conditions of a dysregulated host response to an infection, burn patients may develop sepsis syndrome characterized by fever, increased fluid requirements, decreased urinary output and even MODS [12–14]. In 2016, the Third International Consensus Definition for Sepsis and Septic Shock (Sepsis-3) redefined sepsis as life-threatening organ dysfunction caused by a dysregulated host response to infection [15]. This new sepsis definition places more emphasis on the process of organ dysfunction, and the Sequential Organ Failure Assessment score (Table 1) is used to define sepsis severity, including septic shock [15, 16]. Indeed, the incidence of sepsis in burn patients can range between 3–30% for burns of more than 20% of the total body surface area (TBSA) [17]. Even more concerning is that approximately 54% of burn-related deaths in modern burn units occur due to septic shock and MODS instead of osmotic shock and hypovolemia [18, 19]. A recent autopsy study showed that over 60% of deaths in burn patients resulted from infectious complications and MODS, which is a direct consequence and poor outcome of sepsis [20]. Therefore, the early diagnosis and effective treatment of sepsis would benefit burn patients, especially those with severe burns. Although the Surviving Sepsis Campaign has put in immense effort to drive the improvement of survival in sepsis and septic shock patients, burn wound sepsis is distinguished from general sepsis because of skin loss that suggests the risk of infection is present as long as the burn wounds have not healed [21, 23].
Table 1.
Sequential organ failure assessment (SOFA) scoring system [37]
| Six organ systems | SOFA score 0 | SOFA score 1 | SOFA score 2 | SOFA score 3 | SOFA score 4 |
|---|---|---|---|---|---|
| Respiratory system: PaO2/FiO2 (kPa) | ≥53.3 | <53.3 | <40 | <26.7 | <13.3 |
| Coagulation system: platelets (× 103/μL) | ≥150 | <150 | <100 | <50 | <20 |
| Hepatic system: bilirubin (μmol/L) | <20 | 20–32 | 33–101 | 102–204 | >204 |
| Cardiovascular systema | MAP>70 mm Hg | MAP<70 mm Hg | Dopamine ≤5 or dobutamine (any dose) | Dopamine >5 or epinephrine ≤0.1 or norepinephrine ≤0.1 | Dopamine >15 or epinephrine >0.1 or norepinephrine >0.1 |
| Central nervous system: Glasgow Coma Scale | 15 | 13–14 | 10–12 | 6–9 | <6 |
| Renal system Creatinine (μmol/l) Urine output (ml/day) | <100 | 111–170 | 171–299 | 300–440< 500 | >440< 200 |
Adrenergic agents administered for at least 1 h (doses given are in μg/kg. min) PaO2 partial pressure of arterial oxygen, FiO2 fraction of inspired oxygen, MAP mean arterial pressure
In this review, we seek to address the major pathogenesis of current categories of infection, sepsis and septic shock in patients with burn injury. In addition, the recent diagnostic tools and potential biomarkers, including C-reactive protein (CRP), procalcitonin (PCT) and cytokines, are intensively discussed.
Review
Burn wound infections
Burn wound infection, one of the most important causes of sepsis, is associated with high fatality rates in patients with burn injury. The occurrence of burn wound infection often surfaces during the acute post-injury period and exhibits considerable differences among burn patients of different ages [21, 24]. Young children (under the age of 4 years) and elderly adults (over the age of 55 years) have a higher risk of being infected, with higher fatality rates compared with other age groups [25, 26]. A possible cause is that infants, young children and the elderly have an increased inclination for deep burn injury due to their much thinner dermal layer [27]. Another reason may be the poor compliance of these patients with early medical care and drug regimens. Apart from the above, some special populations, including obese adults, diabetes patients and AIDS patients, have a higher incidence of burn wound infection and have also been shown to have more complications related to infection [28, 29]. For example, AIDS patients have a higher incidence of sepsis and a longer period of hospitalization than HIV-negative patients, although data on the reported outcome are limited due to the small number of AIDS patients [30].
Indeed, burn wound infection can be considered a series of dynamic pathophysiological processes, including microbial colonization, biofilm formation and invasive burn wound infection. Microorganisms can rapidly colonize the burn wounds due to thermal destruction of the skin barrier. Burn eschar (avascular necrotic tissue) caused by deep partial-thickness and full-thickness burns provides a protein-rich niche for bacterial colonization and proliferation [31, 32]. In addition, burn eschar may also increase the risk of infection by inhibiting early healing via basic fibroblast growth factor-induced endothelial cell proliferation and sprouting [33]. However, eschar factors can inhibit hypertrophic scar formation of full-thickness burn wounds by preventing excessive granulation tissue formation [33].
Once planktonic (free-living) organisms form aggregates and attach to burn wounds, the formation of biofilm is initiated. Biofilms are defined as structured communities encased in a self-produced extracellular polysaccharide matrix, or slime [34, 35]. A mature biofilm provides efficient barriers for microorganisms against the host immune system and antimicrobial agents, including biocides, antibiotics, oxidizing agents and nano-drugs [36]. For example, the microorganisms within biofilms have an increased capacity to tolerate and survive stressful environments (such as nutrient deprivation, hypoxia, dehydration and pH changes) compared to planktonic microorganisms [38]. All these resistances pose huge challenges for eliminating antibiotic-resistant microbes and preventing burn wound sepsis. In fact, biofilm formation is a sequential cyclic process comprising at least 5 phenotypically distinct stages, including attachment of planktonic cells, aggregation, biofilm maturation, establishment of cells with the biofilm subpopulation and biofilm dispersion. The biofilm dispersion refers to the process by which cells with biofilm subpopulation escape from the biofilm structure [38]. Although biofilm dispersal leads to the loss of multiple survival advantages, dispersion facilitates the formation of channels on the biofilm surface. These channels contribute to the removal of metabolic waste and the intake of nutrient resources and oxygen [39, 40]. Notably, Kennedy and colleagues detected biofilms not only in ulcerated areas of the burn wound, but also in bacterial wounds invaded with mixed organisms [41]. Therefore, the formation of biofilms may promote the development of an invasive wound infection and, to some extent, sepsis, although the mechanisms remain unclear.
The risk of invasive burn wound infection depends on the surface area and depth of the burn wounds, host immunity and the types (virulence difference) and amount of microbial flora colonizing the burn wounds. Microorganisms causing burn wound infection include gram-positive bacteria, gram-negative bacteria, fungi and viruses (Table 2). During the initial stage of burn injury, some gram-positive bacteria (such as the Staphylococci spp. derived from endogenous skin, gastrointestinal and respiratory flora, or the external environments) are vanguard microbes colonizing the burn wounds [31, 42]. Globally, the major cause of early burn wound infection is by Staphylococcus aureus (S. aureus), which also plays an important role in invasive burn wound infection and sepsis [43]. There are various genes that encode molecules associated with virulence factors, including cell-surface virulence factors (capsular polysaccharides, cell wall anchored proteins and lipoteichoic acids) [44, 45] and secreted virulence factors (superantigens [46], cytotoxins, exoenzymes and miscellaneous proteins [47]) in the S. aureus genome, including the core genome and accessory genome. Although knockouts of these genes have no identifiable effect on the growth of organisms in vitro, their pathogenicity may be diminished in vivo [48, 49]. In addition, these virulence factors can facilitate the adherence of organisms to host tissues, invasion of host cells and tissues and evasion of the host immune system [50]. For example, clumping factor A, a member of the microbial surface components recognizing adhesive matrix molecules family, has been demonstrated to play key roles in sepsis in the murine model [51, 52].
Table 2.
Common microorganisms causing invasive burn wound infection
| Group | Species | References |
|---|---|---|
| Gram-positive organisms |
Staphylococcus aureus
Methicillin-resistant Staphylococcus aureus Coagulase-negative Staphylococci |
[27] [161] [162] |
| Gram-negative organisms |
Pseudomonas aeruginosa
Escherichia coli Klebsiella pneumoniae |
[53] [163] [164] |
| Fungi |
Candida spp. Aspergillus spp. Mucor spp. |
[43] [165] [166] |
| Viruses | Herpes simplex virus Cytomegalovirus Varicella-zoster virus |
[167] [168] [169] |
In the first 5–7 days after injury, burn wounds are occupied by other microorganisms, such as gram-negative bacteria, fungi and viruses. For example, Pseudomonas aeruginosa (P. aeruginosa), a gram-negative organism, is a common culprit of burn wound infection in the intensive care unit due to their multi drug resistances and multiple virulence factors [53, 54]. In a study of P. aeruginosa prevalence in Chinese burn wards from 2007 to 2014, Dou and coworkers showed that the detection rate of P. aeruginosa in hospitalized burn patients increased from 10.20% in 2007 to 26.16% in 2014 [55]. The main cause of this growing trend may be the metabolic versatility of P. aeruginosa, its ability to colonize of a wide range of ecological niches and its low outer membrane permeability, which can resist antiseptics and antibiotics [56]. Similar to S. aureus, P. aeruginosa also has quite a lot of virulence factors, including adhesins, lipopolysaccharides, elastases, exoenzyme S, exotoxin A, leukocidins and proteases. These make P. aeruginosa a major cause of bloodstream invasion, sepsis and poor prognosis in severely burned patients [57, 58]. It remains unclear whether the formation of biofilm or invasive burn wound infection is the important inducer for sepsis. Therefore, the prevention of burn wound infection remains the better choice to diminish the incidence of sepsis and septic shock.
Events leading to sepsis and septic shock following burn injury
Sepsis and MODS are common complications of invasive burn infection and are responsible for a significant proportion of the mortality in patients with burn injuries, particularly severe burns. Williams et al. performed statistical analysis on patients with burn injury and found that 55% of males and 54% of females died from sepsis and infections between 1989 to 2009, but the data short of highlighting the global prevalence of this trend [59]. The limitation is attributed mainly by neglecting low-income and middle-income countries, which may have a higher incidence of sepsis in burn patients. Therefore, clear diagnostic criteria for burn sepsis are necessary to minimize and prevent septic complications. The updated criterion in the Sepsis-3 consensus definition established in 2016 focuses more on multiple organ dysfunction than on signs of inflammation [15], compared with the American Burn Association (ABA) sepsis criteria (2007) [60] and the Mann-Salinas novel burn-specific sepsis predictors (2013) [61, 62] (Table 3). This is particularly discerning, considering that a series of pathophysiological events can lead to sepsis and multiple organ failure, including inflammatory response, hypovolaemic shock and vascular leak, immune dysregulation and hypermetabolism (Figure 3), with inflammation present almost throughout the whole process from initial injury to burn wound healing. Inflammation behaves like a double-edged sword in burn injuries: immediately following minor burn injuries, inflammatory responses are initiated to activate the cascade of signals required for wound healing [8]; however, in patients with severe burns, the inflammatory response (Figure 4) is uncontrolled and leads to vascular endothelium dysfunction, delayed healing, immune suppression and systemic inflammatory response syndrome [63]. Metabolically, inflammation also causes an enhanced catabolic state that is associated with an increased incidence of sepsis and multiple organ failure. Compared with patients with only burns, the level of catabolism in septic burn patients is more than doubled, as measured using stable isotope perfusion [64].
Table 3.
Different criteria for sepsis
| Consensus definitions | Criteria | Predictors |
| ABA Sepsis Criteria [59] | At least one or more of the following | 1) Positive culture 2) Pathologic tissue source identified 3) Clinical response to antimicrobial agents |
| AND at least three of the following predictors | 1) Temperature >39 °C or <36.5 °C 2) Progressive tachycardia (>110 bpm) 3) Progressive tachypnoea 4) Thrombocytopenia 5) Hyperglycaemia 6) Inability to continue enteral feedings 24 hours |
|
| Mann-Salinas et al. Novel burn-specific sepsis predictors [60, 61] | Predictors | 1) Tachycardia >130 bpm 2) MAP <60 mmHg 3) Base deficit <–6 mEq/l 4) Hypothermia <36 °C 5) Use of vasoactive medications 6) Hyperglycaemia >150 mg/dl |
| Sepsis-3 Consensus definition for sepsisa [15] | qSOFA score ≥ 2 | 1) Altered mental status (Glasgow Coma Scale <13) 2) Systolic blood pressure ≤100mmHg 3) Respiratory rate 22 ≥ breaths/min |
| SOFA variables ≥ 2 | 1) PaO2/FiO2 ratio 2) Platelet count 3) Bilirubin 4) Mean arterial pressure 5) Glasgow Coma Scale 6) Vasopressor requirement 7) Serum creatinine or urine output |
|
| Septic shock predictors (sepsis and both predictors) |
1) Vasopressors required to maintain MAP >65mm Hg 2) Lactate >2 mmol/L (after adequate fluid resuscitation) |
aSuspected or documented infection and qSOFA ≥ 2 and/or SOFA ≥ 2 ABA American Burn Association, MAP mean arterial pressure, bpm beats per minute, SOFA Sequential Organ Failure Assessment, qSOFA quick SOFA, PaO2 partial pressure of arterial oxygen, FiO2 fraction of inspired oxygen
Figure 3.
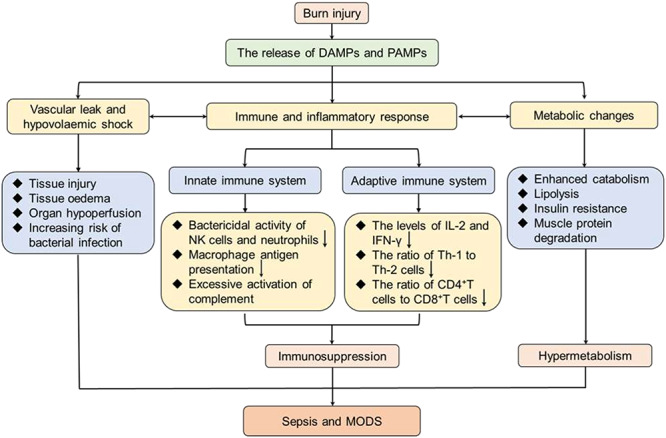
A series of pathogenic events responsible for sepsis post burn injury. Following severe burn injury, damaged tissues lead to the release of endogenous DAMPs (such as double-stranded RNA and mitochondrial DNA) and exogenous PAMPs (such as lipopolysaccharides and peptidoglycans). Subsequently, PAMPs can result in vascular leak and hypovolemic shock, immune and inflammatory responses and metabolic changes. Vascular leak causes tissue edema, organ hypoperfusion and increasing risk of bacterial infection. Meanwhile, the excessive inflammatory response leads to immunosuppression by inhibition of the innate and adaptive immune systems. Moreover, hypermetabolism emerges in the form of enhanced catabolism, lipolysis, insulin resistance and muscle protein degradation. These events contribute to the susceptibility of the burn patients to sepsis and MODS. DAMPs damage-associated molecular patterns, PAMPs pathogen-associated molecular pattern molecules, NK natural killer, IL-2 interleukin 2, IFN-γ interferon γ, Th-1 helper T lymphocyte 1, Th-2 helper T lymphocyte 2, MODS multiple organ dysfunction syndrome
Figure 4.
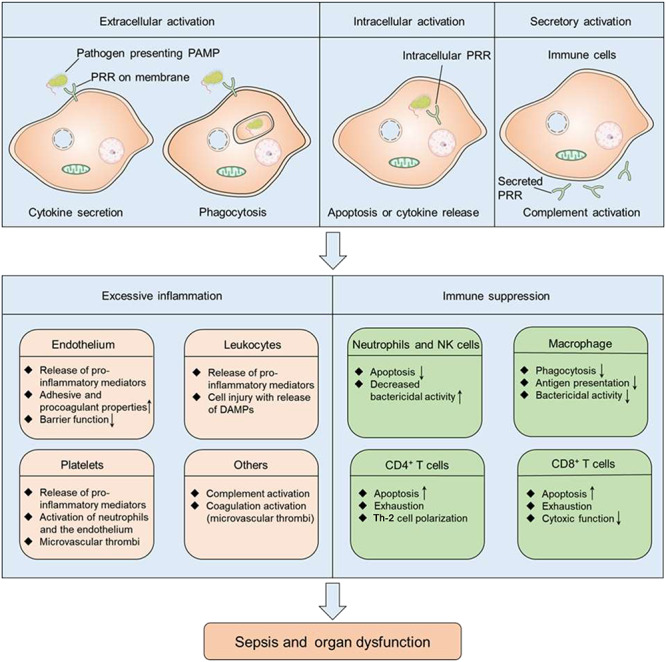
The host response to infection and during sepsis. After infection, PAMPs are released and interact with cell-surface, intracellular and even secreted PRRs, including toll-like receptors, nucleotide-binding oligomerization domain-like receptors, retinoic acid-inducible gene-like receptors and C-type lectin receptors. The interaction between PAMPs and PRRs can result in cytokine secretion, immune cell apoptosis and the activation of the complement system. In some burn patients, these events lead to the simultaneous imbalanced activation of proinflammatory response (excessive inflammation) and anti-inflammatory response (immune suppression). Excessive inflammation can result in the dysfunction of the endothelial barrier, microvascular thrombi and further injuries. Immunosuppression causes decreased bactericidal activity of neutrophils and NK cells, decreased phagocytosis and antigen presentation of macrophages and impaired innate immune system response. Taken together, excessive inflammation and immunosuppression contribute to a greatly increased risk for sepsis and organ dysfunction. PAMPs pathogen-associated molecular pattern molecules, PRR pattern recognition receptors, DAMPs damage-associated molecular patterns, NK natural killer, Th-2 helper T lymphocyte 2
Vascular leak and hypovolemic state
Interconnected microvessels are crucial for substance exchange (nutrients, oxygen and metabolic waste) between blood and surrounding tissues through the regulation of local hydrostatic and oncotic pressures [65]. Under the regulation of physical structure and chemical messenger, the vascular barrier function maintains tissue perfusion and homeostasis to adapt to physiological stimuli. The vascular barrier function maintains tissue perfusion and homeostasis to adapt to physiological stimuli under the regulation of physical structure and chemical messenger. However, in some traumatic injuries, particularly burn injuries, structural disruption and inflammatory mediators lead to increased vascular permeability, which contributes to leakage of the intravascular fluids into the interstitial space, leading to further profound tissue edema and even hypovolemic shock [66].
Multiple mediators of barrier function, including histamine, bradykinin, platelet-activating factor, leukotrienes, vascular endothelial growth factor (VEGF) and VEGF receptors, affect vascular hyperpermeability by inducing cellular signaling and structural alterations. Histamine is released mainly from mast cells and increases vascular permeability through vascular dilation, increasing of blood flow and endothelial barrier disruption. There is increasing evidence that nitric oxide (NO) and RhoA/Rho-associated protein kinase (ROCK) play an important role in histamine-induced hyperpermeability. Ashina and coworkers found that inhibiting NO synthesis with a nitric oxide synthase (NOS) inhibitor, Nitro-L-arginine methyl ester (L-NAME) could alleviate the histamine-induced blood flow increase and hyperpermeability [67]. In addition, the authors also found that histamine disrupted the endothelial barrier due to the localization of vascular endothelial cadherin (VE-cadherin) (a cadherin maintaining the junction of adjacent endothelial cells) at the endothelial cell junction being changed [67]. Consistent with this observation, Mikelis et al. suggested that histamine induced the localization of VE-cadherin adhesion complexes to focal adherens junctions through Rho/ROCK and further led to the formation of gaps in the endothelial barrier [68].
Bradykinin, like histamine, activates RhoA and induces the rearrangement of the cytoskeleton and disassembly of tight junction (TJ) proteins. These changes can cause vascular leakage, although vascular hyperpermeability was observed in the blood–tumor barrier of rat brain microvascular endothelial cells [69]. Furthermore, previous studies have shown that bradykinin downregulates the expression of the TJ-associated proteins zonula occluden-1, occludin and caludin-5, but the regulatory mechanism might involve the cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) signal transduction system rather than the RhoA/ROCK-dependent mechanism [70].
Surprisingly, cysteinyl leukotrienes also directly regulate vascular permeability through the activation of ROCK to promote endothelial contraction and gap formation [71]. Taken together, these studies suggest that RhoA signaling circuitry plays a key role in vascular permeability and RhoA/ROCK is a potential pharmacological target for burn-induced vascular leakage. Indeed, in most cases, leukotrienes act as an upstream regulator to influence vascular permeability by regulating other vasoactive mediators, typically VEGF [72]. VEGF signaling regulates vascular permeability by a variety of mechanisms, including reductions in TJ-associated proteins [73], damage of VE-cadherin junctional contacts [74] and the formation of vesicular vacuolar organelles [75]. Vesicular vacuolar organelles allow endothelial cells to form transendothelial cell pores, which are considered an additional transcellular pathway for large molecules and fluid extravasation [76]. VEGF has been used as a biomarker for the diagnosis of sepsis in patients with severe burn injuries [77].
Apart from vascular leakage, burn injuries also result in cardiac dysfunction due to cardiac mitochondrial damage [78] and the production of inflammatory mediators, including macrophage migration inhibitor factor [79] and tumor necrosis factor (TNF) [80]. In rat models of burn, oxidative stress impairs cardiac function due to a 30–50% increase in lipid peroxidation in cardiac mitochondria [78]. Migration inhibitor factor is released by the skin and cardiomyocytes and is considered a critical mediator of persistent cardiac dysfunction [79]. The cardiac dysfunction and vascular leakage-induced hypovolemia have serious implications for the perfusion of tissues and organs (lungs, liver, kidney and gastrointestinal tract) and may even lead to sepsis and multiple organ failure.
Immune dysregulation
As discussed above, patients with burns have a greater risk of infection, not exclusively due to the loss of the natural barriers function of the skin. Recent studies have demonstrated that burn injuries can also influence other elements of both the innate immune system (including immune cells and complement) and the adaptive immune system, which disrupts coordination of the immune response. Some immune cells, including monocytes, macrophages, dendritic cells, natural killer (NK) cells and neutrophils, are among the first immune cells to respond to wounds and coordinate the wider immune response. Following burn injuries, the antimicrobial actions of neutrophils and NK cells are impaired [81–83]. Coincidentally, the phagocytic capacity of macrophages is also diminished in severe burn [84]. In addition, increased apoptosis of conventional and plasmacytoid dendritic cells has been observed in patients with sepsis [85]. The complement system represents an evolutionarily conserved and important element of the innate immune system [85]. According to the severity of burns, the levels of complement decrease to different extents at the beginning of burn injury and subsequently rise to unprecedented levels [87]. The increased complements, such as C3a, C3b and C5a, may suppress immune response directly by impairing the function of leukocytes and lymphocytes [88, 89]. Intriguingly, multiple interleukins (IL, a kind of lymphokine), such as IL-4 and IL-10, can significantly inhibit the antigen presentation of macrophage and the bactericidal activity of NK cells and neutrophils [90–92].
In addition to impairing the function of the innate immune system, severe burns reduce the total numbers of T lymphocytes, which play dominant roles in the adaptive immune system [93, 94]. Surprisingly, not all T lymphocytes are diminished, with helper T lymphocyte 2 (Th-2) present in increased numbers due to the increased levels of IL-4 and IL-10 [95]. Moreover, the reduced levels of IL-2 and interferon-γ also cause the increase of Th-2 following burn injuries [96]. The depressed levels of IL-2 and interferon-γ and high levels of IL-4 and IL-10 simultaneously inhibit the activity of helper T lymphocyte (Th-1) that support cell-mediated immune responses [95]. The decreased ratio of Th-1 to Th-2 is an important etiologic factor in the suppression of adaptive immune responses [27]. Furthermore, the ratio of CD4-positive T helper cells to CD8-positive T suppressor cells also declines after severe burn [97]. Similarly, burn injury results in immune dysregulation by destabilizing the balance between helper T lymphocyte 17 (Th-17) and regulatory T cell, which plays prominent roles in protection against bacterial infections. On the one hand, Th-17 responses have been shown to be elicited in murine models of burn injury [98]. The perturbation of Th-17 cytokines IL-17 and IL-22 may further delay wound healing and promote burn sepsis [99]. On the other hand, the proportion of Treg cells is increased in patients with burn injury and this may decrease effector T cell function and further contribute to sepsis [100]. Taken together, the compromised alterations in innate and adaptive immune responses result in enhanced susceptibility to infection, sepsis and multiple organ failure.
Hypermetabolic state in bury injury
In addition to hypovolemic response and immune dysfunction, the hypermetabolic state following burn trauma is another primary contributor to multiple organ failure and sepsis. Hypermetabolism (increased metabolic rate) is characterized by an elevated (>10% above normal) resting energy expenditure [101] and is more likely to occur in severe burns (>20% TBSA). Studies have demonstrated that there is an increase of 40–80% in resting energy expenditure during the acute phase of post-burn injury in patients with burns of more than 40% TBSA [102, 103]. Generally, the metabolic level is attenuated in the early stage of burn (<48 hours) owing to diminished cardiac output and oxygen consumption, but metabolism will enhance dramatically after the “ebb” phase [104]. Transient hypermetabolic state has proven to be beneficial to burn patients. For example, hypermetabolism can provide more energy for vital organs (brain, lungs, heart and immune organs) to maintain their functional levels as closely as possible to normal physiological conditions. Nevertheless, as distinguished from other trauma, major burns provoke profound hypermetabolism and the resultant hypermetabolic state can persist for up to and beyond 36 months after the initial insult [9]. Persistent hypermetabolism aggrandizes the rates of glycolysis, lipolysis and proteolysis, and subsequently results in muscle wasting and loss of body weight that can significantly impact the immune response and wound healing [8]. The mechanisms underlying burn-induced hypermetabolism are associated with many factors, including pro-inflammatory cytokines, stress hormones, mitochondrial dysfunction, endoplasmic reticulum stress and the browning of white adipose tissue (WAT).
Pro-inflammatory cytokines not only contribute to immune dysfunction in the burn patients but also induce hypermetabolism. Levels of cytokines, including TNF-α and IL-1β, do not increase sustainedly, but are restricted to the acute phase of burn [9, 63, 105]. The mechanism of TNF-α-induced hypermetabolism may be that TNF-α promotes the production of reactive oxygen species, adipose catabolism and the release of free fatty acids in patients suffering from thermal injuries [106]. In addition to TNF-α, recent studies have postulated that IL-1β contributes to the hypermetabolic state following burn injuries because it interferes with insulin sensitivity by inhibiting the expression of insulin receptor substrate-1 interfere with insulin sensitivity [104, 107]. Compared with pro-inflammatory cytokines, stress hormones (such as epinephrine, norepinephrine, glucagon and cortisol) have profound and enduring impacts on hypermetabolism. Incremental levels of these stress hormones have pleiotropic hypermetabolic effects that enhance lipolysis, proteolysis and glucose metabolism by acting on several target organs, such as adipose tissue, skeletal muscle and the liver [63, 108–110]. Moreover, accumulating evidence indicates that the functional changes of some organelles, such as mitochondria, can contribute to the hypermetabolic response to burns. Several adenosine triphosphate (ATP)-consuming reactions are enhanced in response to burn injury, including protein synthesis, hepatic gluconeogenesis and cycling of glucose and fatty acids [111]. About 57% of the increase of energy expenditure in severely burned patients is attributed to these ATP consuming reactions [111]. Mitochondria, as the powerhouse of cells, play an important role in ATP production, mainly via the coupling of oxidative phosphorylation. However, the coupling of mitochondrial respiration to adenosine diphosphate phosphorylation is significantly attenuated in patients with burns [101]. On the contrary, the uncoupling of oxidative phosphorylation, which contributes to proton conductance via uncoupling proteins for mitochondrial thermogenesis rather than generation of ATP, is enhanced post burn [112]. Hypermetabolism induced by burn injuries imposes an immense burden on the endoplasmic reticulum (ER) and leads to the accumulation of unfolded proteins, which are responsible for ER stress through protein synthesis demand and extracellular signaling [104, 113]. Interestingly, ER stress in turn inhibits WAT browning, which is the emerging culprit of hypermetabolic response, although the mechanism remains unclear [114, 115]. The browning of WAT refers to the conversion of WAT into brown adipose tissue, characterized by higher rates of lipolysis. Following a thermal injury, the browning of WAT is enhanced and increases the circulation of free fatty acids implicated in the hypermetabolic state [116]. The browning of WAT can also be induced by IL-6 [117] and uncoupling protein 1 of mitochondria [118]. Taken together, the browning of WAT is a valuable therapeutic target and needs more mechanistic studies associated with browning and burn-induced hypermetabolism to further understand its function.
Septic shock in burn injury
In cases of burn wound infection, a hypovolemic state, dysregulated inflammatory response and hypermetabolism are the primary risk factors for the development of sepsis. If not diagnosed in time and treated appropriately, burn patients with sepsis may progress to severe sepsis or septic shock, which is defined as sepsis with intravascular hypovolemia and hypotension resistant to fluid resuscitation, accompanied by worsening systemic signs, including oliguria, lactic acidosis and even changes in mental status [119, 120]. Under normal physiological conditions, there is a delicate balance between procoagulant and anticoagulant mediators within the vasculature. In severe sepsis, however, several proinflammatory cytokines (including IL-1, IL-6 and TNF-α), induced by endotoxins, lipopolysaccharides and other infectious mediators, promote the generation and release of procoagulant tissue factor of endothelial cells [121]. The increase of procoagulant tissue factor disrupts vascular homeostasis and results in a procoagulant state that causes thrombin formation and fibrin deposition. Thrombosis and vascular leaking are implicated in hypoperfusion of multiple organs and subsequent MODS, which is the initiating event of septic shock. Moreover, endotoxins and proinflammatory cytokines can also interact with endothelial cells and result in the generation and release of NO and prostacyclin, which play vital roles in vasodilation [122–124]. Vasodilation will further aggravate hypotension and accelerate the occurrence of septic shock. Both thrombosis and hypotension also impair tissue oxygenation and aggravate organ dysfunction. In addition, mitochondrial dysfunction, caused by oxidative stress and the uncoupling mitochondria respiration, impairs cellular oxygen use and the normal operation of vital organs [125]. For instance, in a septic mouse model, there is overproduction of reactive oxygen species and reactive nitrogen species, which have been shown to impact negatively on myocardial mitochondrial function and cardiomyocyte contractility [126]. The contractility of cardiomyocytes is also impaired by other mechanisms, such as the cell-surface adhesion molecule ICAM-1 [127], small calcium-regulated molecules (S100A8 and S100A9) [128] and inappropriate mitochondrial autophagy (mitophagy) [129]. In short, septic cardiomyopathy exacerbates hypoperfusion of other organs and accelerates the progression of sepsis, ultimately resulting in septic shock.
Diagnosis of sepsis after burn trauma
Due to the high lethality of sepsis, it is particularly important to choose an appropriate definition and criteria for early diagnosis and prediction of sepsis in burn patients. Yan and coworkers compared the sensitivity of the sepsis criteria of the ABA definition, the Mann-Salinas definition, and the new Sepsis-3 consensus definition in patients with burn and found Sepsis-3 to have a higher sensitivity (85%) than the ABA (60%) and Mann-Salinas (20%) definitions. The major advantage of Sepsis-3 is that it argues that the multiple organ dysfunction is more sepsis-specific than inflammation [15]. Despite this, it remains challenging to differentiate sepsis from systemic inflammatory response syndrome because they have similar clinical manifestations in multiple aspects, including core body temperature, respiratory rate, heart rate and hyperglycemia [101, 130]. Thus, the importance of more specific diagnostic and prognostic tools of burn sepsis cannot be overemphasized. Several utilized clinical and promising predictors associated with burn sepsis are presented in this section.
C-reactive protein
CRP is an evolutionarily conserved protein and is produced primarily by hepatocytes following induction by inflammatory cytokines, such as IL-6 [131]. This biomarker of inflammation in acute-phase responses has been widely adopted in clinical settings. In healthy individuals, the levels of CRP in plasma are almost undetectable, while more than 500 mg/l can be observed in patients with burn trauma [132]. Its levels may further increase in burn patients with infection or sepsis [133], thus previous studies have suggested CRP as a good predictor of sepsis in burn patients. However, recent evidence has shown that CRP has drawbacks in the specific diagnosis of sepsis in severely burned patients [134, 135]. Taken together, CRP may not be a specific biomarker of sepsis, but its levels have important reference value in conjunction with other tools, such as PCT and some cytokines.
Procalcitonin
PCT, as a popular biomarker in bacterial infections and sepsis, has been studied extensively and utilized clinically [136, 137]. Several studies have compared PCT to CRP in the diagnosis of sepsis, and most of the evidence suggests that PCT is superior to CRP [138–140]. PCT, the prohormone of calcitonin, is a 116-amino acid polypeptide encoded by the CALC-1 gene [141]. PCT is mainly produced by neuroendocrine cells of the thyroid and its expression is inhibited in non-endocrine tissues under normal physiological conditions [142]. Bacterial infection facilitates the transcription of CALC-1 gene in non-endocrine cells and increases PCT levels to a peak during the first 20 hours after infection [143]. The increasing serum levels of PCT in patients with burn was first reported by Assicot and colleagues, who conjectured that levels of PCT are associated with the progression of infections, sepsis and septic shock [136]. Consistent of this hypothesis, Brunkhorst et al. showed that levels of PCT were proportional to the severity of sepsis in critically ill patients [144]. Conversely, recent studies by Seoane et al. and Paratz et al. found no association between PCT levels and sepsis in adult burn patients [145, 146]. Therefore, like CRP, PCT is not specific in early diagnosis of sepsis in burn patients.
Cytokines
Major burn injuries are often accompanied by an inflammatory response that results in the activation of inflammatory pathways and the augmentation of various cytokines, including proinflammatory cytokines (TNF-α, IL-6, IL-8) and anti-inflammatory cytokines (IL-10) [147]. Recently, the potential of these cytokines in the early diagnosis of sepsis post burn injury has been investigated. Compared with the burn patients without signs of sepsis, higher levels of TNF-α were observed in burn patients with sepsis [148]. This difference also appears in serum IL-6 values between the burn patients with and without sepsis [147]. In addition, a clinical study, in which 468 children with burn injuries were divided into 2 groups based on IL-8 levels, has shown the positive correlation between the serum levels of IL-8 and sepsis in pediatric patients with elevated IL-8 [149]. Interestingly, IL-10, an anti-inflammatory cytokine, has a negative impact on the production of proinflammation cytokines, whereas the elevation of serum IL-10 levels is also correlated with the development of sepsis and even the risk of mortality in burn patients [150, 151]. Taken together, these findings indicate that cytokines hold great early diagnostic potential in sepsis and further studies will be needed to verify this.
Promising biomarkers
Presepsin, a glycoprotein fragment produced by monocytes and macrophages, is a soluble subtype of the cluster of differentiation 14 [152]. This glycoprotein recognizes and interacts with endotoxin complexes for the activation of systemic inflammatory signaling pathways [153]. There is mounting evidence to indicate that presepsin is a promising biomarker for diagnosing sepsis in burn patients, although it cannot be used alone to confirm or exclude the presence of sepsis in burn patients [154–157]. Mid-regional pro-atrial natriuretic peptide is another promising biomarker, and Gille et al., in a prospective observational study of 42 burn patients, found that burn patients with sepsis have higher levels of this peptide and PCT [158]. Moreover, Hampson et al. found that neutrophil function, immature granulocyte count and plasma cell-free DNA levels showed significant potential for the early diagnosis of sepsis in burn patients [159]. Especially interesting w that micro RNA can also serve as a diagnostic biomarker. An example of this is miR-495, which is significantly downregulated in patients with sepsis and negatively correlated with CRP and PCT [160]. Although numerous promising biomarkers of sepsis have been discovered, none of them alone can diagnose sepsis post burn injury, and their values must be interpreted with caution to ensure accurate diagnosis.
Conclusions
Sepsis and septic complications not only account for the poor outcomes in burn patients, but also prolonged hospital stays and higher medical costs. Burn wound infection is a major cause of sepsis development in patients with severe burns. Moreover, other events following burn injury play important roles in the occurrence of sepsis, such as vascular leak, hypovolemia, hypermetabolism and immune dysregulation. Integrated management, including, but not limited to, fluid resuscitation, nutritional support, antimicrobial therapy and vasoactive medications is beneficial for the prevention and prognosis of sepsis by targeting the events leading to sepsis following burn injury. However, the prediction and diagnosis of sepsis or infection remains an ongoing challenge in burn patients, although numerous predictors for burn sepsis have been reported. More investigations are needed to explore novel diagnostic tools of burn sepsis due to the unreliability and limitation of the established biomarkers (CRP, PCT and cytokines). Meanwhile, Sepsis-3, which can be applied for analysis or research purposes, appeared to be a better definition of sepsis. Based on this definition and patient-specific molecular and biochemical profiles, clinicians can design an individualized management strategy which may improve the prognosis of burn patients with sepsis.
Acknowledgements
We are grateful to Dr Yew Mun Lee for his helpful discussion and suggestions.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81821002, 81790251 and 81672381), the Guangdong Basic and Applied Basic Research Foundation (2019B030302012), the Science and Technology Department of Sichuan Province (2018RZ0133) and the Chengdu Science and Technology Program (2019-YF05–00715-SN).
Conflicts of interest
The authors declare no conflict of interest.
Abbreviations
MODS: multiple organ dysfunction syndrome; TBSA: total body surface area; Sepsis-3: Third International Consensus Definition for Sepsis and Septic Shock; CRP: C-reactive protein; PCT: procalcitonin; VEGF: vascular endothelial growth factor; ROCK: Rho-associated protein kinase; NO: nitric oxide; VE-cadherin: vascular endothelial cadherin; TNF: tumor necrosis factor; NK: natural killer; WAT: white adipose tissue; ABA: American Burn Association; ABA: American Burn Association; ATP: adenosine triphosphate; CRP: C-reactive protein; DAMPs: damage-associated molecular patterns; ER: endoplasmic reticulum; IL: interleukins; MAP: mean arterial pressure; MODS: multiple organ dysfunction syndrome; NK: natural killer; NO: nitric oxide; PAMPs: pathogen-associated molecular pattern molecules; PCT: procalcitonin; ROCK: Rho-associated protein kinase; Sepsis-3: Third International Consensus Definition for Sepsis and Septic Shock; SOFA: Sequential Organ Failure Assessment; TBSA: total body surface area; Th-1: helper T lymphocyte; Th-2: helper T lymphocyte 2; Th-17: helper T lymphocyte 17; TNF: tumor necrosis factor; TJ: tight junction; VE-cadherin: vascular endothelial cadherin; VEGF: vascular endothelial growth factor; WAT: white adipose tissue.
References
- 1. Jeschke MG, Baar ME, Choudhry MA, Chung KK, Gibran NS, Logsetty S. Burn injury. Nat. Rev. Dis. Primers. 2020. 10.1038/s41572-020-0145-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. National Burn Repository 2019 Update , Report of data from 2009–2018 ameriburn.site- ym.com [Internet]. 2019. Available from: https://ameriburn.site-ym.com/store/ViewProduct.aspx?id=14191872.
- 3. Pereira RF, Barrias CC, Granja PL, Bartolo PJ. Advanced biofabrication strategies for skin regeneration and repair. Nanomedicine (Lond.) 2013;8:603–21. [DOI] [PubMed] [Google Scholar]
- 4. Peck MD. Epidemiology of burns throughout the world. Part I: distribution and risk factors. Burns 2011;37:1087–100. [DOI] [PubMed] [Google Scholar]
- 5. Cioffi WG. deLemos RA, Coalson JJ, Gerstmann DA, Pruitt BA Jr. decreased pulmonary damage in primates with inhalation injury treated with high-frequency ventilation. Ann. Surg. 1993;218:328–35discussion 35-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Finnerty CC, Herndon DN, Jeschke MG. Inhalation injury in severely burned children does not augment the systemic inflammatory response. Crit. Care 2007. 10.1186/cc5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A genomic storm in critically injured humans. J. Exp. Med. 2011;208:2581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stanojcic M, Abdullahi A, Rehou S, Parousis A, Jeschke MG. Pathophysiological response to burn injury in adults. Ann. Surg. 2018;267:576–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jeschke MG, Gauglitz GG, Kulp GA, Finnerty CC, Williams FN, Kraft R, et al. Long-term persistence of the pathophysiologic response to severe burn injury. PLoS One 2011;6:e21245. 10.1371/journal.pone.0021245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 2003;348:1546–54. [DOI] [PubMed] [Google Scholar]
- 11. Rodríguez-Luna A, Ávila-Román J, González-Rodríguez ML, Cózar MJ, Rabasco AM, Motilva V, et al. Fucoxanthin-containing cream prevents epidermal hyperplasia and UVB-induced skin erythema in mice. Mar. Drugs 2018. 10.3390/md16100378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bone RC, Sibbald WJ, Sprung CL. The ACCP-SCCM consensus conference on sepsis and organ failure. Chest 1992;101:1481–3. [DOI] [PubMed] [Google Scholar]
- 13. Neely AN, Fowler LA, Kagan RJ, Warden GD. Procalcitonin in pediatric burn patients: an early indicator of sepsis? J. Burn Care Rehabil. 2004;25:76–80. [DOI] [PubMed] [Google Scholar]
- 14. Stanojcic M, Vinaik R, Jeschke MG. Status and challenges of predicting and diagnosing sepsis in burn patients. Surg. Infect. 2018;19:168–75. [DOI] [PubMed] [Google Scholar]
- 15. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016;315:801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li W, Wang M, Zhu B, Zhu Y, Xi X. Prediction of median survival time in sepsis patients by the SOFA score combined with different predictors. Burns Trauma 2020;8:tkz006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jeschke MG, Patsouris D, Stanojcic M, Abdullahi A, Rehou S, Pinto R, et al. Pathophysiologic response to burns in the elderly. EBioMedicine 2015;2:1536–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chai J, Sheng Z, Diao L, Yang H, Gao J, Xu M. Effect of extensive excision of burn wound with invasive infection on hypermetabolism in burn patients with sepsis. Zhonghua Wai Ke Za Zhi [Chinese journal of surgery]. 2000;38:405–8. [PubMed] [Google Scholar]
- 19. Fitzwater J, Purdue GF, Hunt JL, O'Keefe GE. The risk factors and time course of sepsis and organ dysfunction after burn trauma. J. Trauma 2003;54:959–66. [DOI] [PubMed] [Google Scholar]
- 20. Gomez R, Murray CK, Hospenthal DR, Cancio LC, Renz EM, Holcomb JB, et al. Causes of mortality by autopsy findings of combat casualties and civilian patients admitted to a burn unit. J. Am. Coll. Surg. 2009;208:348–54. [DOI] [PubMed] [Google Scholar]
- 21. Greenhalgh DG. Sepsis in the burn patient: a different problem than sepsis in the general population. Burns Trauma. 2017. 10.1186/s41038-017-0089-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Atiyeh BS, Gunn SW, Hayek SN. State of the art in burn treatment. World J. Surg. 2005;29:131–48. [DOI] [PubMed] [Google Scholar]
- 23. Ren C, Yao RQ, Ren D, Li Y, Feng YW, Yao YM. Comparison of clinical laboratory tests between bacterial sepsis and SARS-CoV-2-associated viral sepsis. Mil Med Res 2020;7:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Warner PM, Coffee TL, Yowler CJ. Outpatient burn management. Surg. Clin. North Am. 2014;94:879–92. [DOI] [PubMed] [Google Scholar]
- 25. Lionelli GT, Pickus EJ, Beckum OK, Decoursey RL, Korentager RA. A three decade analysis of factors affecting burn mortality in the elderly. Burns 2005;31:958–63. [DOI] [PubMed] [Google Scholar]
- 26. Thombs BD, Bresnick MG. Mortality risk and length of stay associated with self-inflicted burn injury: evidence from a national sample of 30,382 adult patients. Crit. Care Med. 2008;36:118–25. [DOI] [PubMed] [Google Scholar]
- 27. Church D, Elsayed S, Reid O, Winston B, Lindsay R. Burn wound infections. Clin. Microbiol. Rev. 2006;19:403–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kraft R, Herndon DN, Williams FN, Al-Mousawi AM, Finnerty CC, Jeschke MG. The effect of obesity on adverse outcomes and metabolism in pediatric burn patients. Int J Obes (Lond). 2012;36:485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Duke JM, Randall SM, Fear MW, Boyd JH, Rea S, Wood FM. Diabetes mellitus after injury in burn and non-burned patients: a population based retrospective cohort study. Burns 2018;44:566–72. [DOI] [PubMed] [Google Scholar]
- 30. Salehi SH, As'adi K, Tabatabaeenezhad SA, Naderan M, Shoar S. Prevalence of HIV infection among burn patients: is there a relationship with patients' outcomes? Int. Wound J. 2017;14:85–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barret JP, Herndon DN. Effects of burn wound excision on bacterial colonization and invasion. Plast. Reconstr. Surg. 2003;111:744–50. [DOI] [PubMed] [Google Scholar]
- 32. Evdokiou A, Kanisicak O, Gierek S, Barry A, Ivey MJ, Zhang X, et al. Characterization of burn eschar pericytes. J. Clin. Med. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Monsuur HN, Broek LJ, Jhingoerie RL, Vloemans A, Gibbs S. Burn eschar stimulates fibroblast and adipose mesenchymal stromal cell proliferation and migration but inhibits endothelial cell sprouting. Int. J. Mol. Sci. 2017. 10.3390/ijms18081790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science (New York, N.Y.) 1999;284:1318–22. [DOI] [PubMed] [Google Scholar]
- 35. Flemming HC, Wingender J, Szewzyk U, Steinberg P, Rice SA, Kjelleberg S. Biofilms: an emergent form of bacterial life. Nat. Rev. Microbiol. 2016;14:563–75. [DOI] [PubMed] [Google Scholar]
- 36. Koo H, Allan RN, Howlin RP, Stoodley P, Hall-Stoodley L. Targeting microbial biofilms: current and prospective therapeutic strategies. Nat. Rev. Microbiol. 2017;15:740–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vincent JL, Moreno R, Takala J, Willatts S, De Mendonça A, Bruining H, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med 1996;22:707–10. [DOI] [PubMed] [Google Scholar]
- 38. Rumbaugh KP, Sauer K. Biofilm dispersion. Nat. Rev. Microbiol. 2020;18:571–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Edwards R, Harding KG. Bacteria and wound healing. Curr. Opin. Infect. Dis. 2004;17:91–6. [DOI] [PubMed] [Google Scholar]
- 40. Harrison-Balestra C, Cazzaniga AL, Davis SC, Mertz PM. A wound-isolated Pseudomonas aeruginosa grows a biofilm in vitro within 10 hours and is visualized by light microscopy. Dermatol Surg. 2003;29:631–5. [DOI] [PubMed] [Google Scholar]
- 41. Kennedy P, Brammah S, Wills E. Burns, biofilm and a new appraisal of burn wound sepsis. Burns 2010;36:49–56. [DOI] [PubMed] [Google Scholar]
- 42. Erol S, Altoparlak U, Akcay MN, Celebi F, Parlak M. Changes of microbial flora and wound colonization in burned patients. Burns 2004;30:357–61. [DOI] [PubMed] [Google Scholar]
- 43. Norbury W, Herndon DN, Tanksley J, Jeschke MG, Finnerty CC. Infection in Burns. Surg. Infect. 2016;17:250–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vollmer W, Blanot D, Pedro MA. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008;32:149–67. [DOI] [PubMed] [Google Scholar]
- 45. Scott JR, Barnett TC. Surface proteins of gram-positive bacteria and how they get there. Annu. Rev. Microbiol. 2006;60:397–423. [DOI] [PubMed] [Google Scholar]
- 46. Salgado-Pabón W, Breshears L, Spaulding AR, Merriman JA, Stach CS, Horswill AR, et al. Superantigens are critical for Staphylococcus aureus infective endocarditis, sepsis, and acute kidney injury. MBio 2013;4. 10.1128/mBio.00494-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin YC, Peterson ML. New insights into the prevention of staphylococcal infections and toxic shock syndrome. Expert. Rev. Clin. Pharmacol. 2010;3:753–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dayan GH, Mohamed N, Scully IL, Cooper D, Begier E, Eiden J, et al. Staphylococcus aureus: the current state of disease, pathophysiology and strategies for prevention. Expert Rev. Vaccines 2016;15:1373–92. [DOI] [PubMed] [Google Scholar]
- 49. Maresso AW, Schneewind O. Sortase as a target of anti-infective therapy. Pharmacol. Rev. 2008;60:128–41. [DOI] [PubMed] [Google Scholar]
- 50. Foster TJ, Geoghegan JA, Ganesh VK, Höök M. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014;12:49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kwiecinski J, Jin T, Josefsson E. Surface proteins of Staphylococcus aureus play an important role in experimental skin infection. APMIS. 2014;122:1240–50. [DOI] [PubMed] [Google Scholar]
- 52. McAdow M, Kim HK, Dedent AC, Hendrickx AP, Schneewind O, Missiakas DM. Preventing Staphylococcus aureus sepsis through the inhibition of its agglutination in blood. PLoS Pathog. 2011. 10.1371/journal.ppat.1002307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. MacVane SH. Antimicrobial resistance in the intensive care unit: a focus on gram-negative bacterial infections. J. Intensive Care Med. 2017;32:25–37. [DOI] [PubMed] [Google Scholar]
- 54. Kaye KS, Pogue JM. Infections caused by resistant gram-negative bacteria: epidemiology and management. Pharmacotherapy 2015;35:949–62. [DOI] [PubMed] [Google Scholar]
- 55. Dou Y, Huan J, Guo F, Zhou Z, Shi Y. Pseudomonas aeruginosa prevalence, antibiotic resistance and antimicrobial use in Chinese burn wards from 2007 to 2014. J. Int. Med. Res. 2017;45:1124–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chevalier S, Bouffartigues E, Bodilis J, Maillot O, Lesouhaitier O, Feuilloley MGJ, et al. Structure, function and regulation of Pseudomonas aeruginosa porins. FEMS Microbiol. Rev. 2017;41:698–722. [DOI] [PubMed] [Google Scholar]
- 57. Tredget EE, Shankowsky HA, Rennie R, Burrell RE, Logsetty S. Pseudomonas infections in the thermally injured patient. Burns 2004;30:3–26. [DOI] [PubMed] [Google Scholar]
- 58. Dzvova N, Colmer-Hamood JA, Griswold JA, Hamood AN. Heparinase is essential for Pseudomonas aeruginosa virulence during thermal injury and infection. Infect. Immun. 2018. 10.1128/IAI.00755-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Williams FN, Herndon DN, Hawkins HK, Lee JO, Cox RA, Kulp GA, et al. The leading causes of death after burn injury in a single pediatric burn center. Crit. Care 2009. 10.1186/cc8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Greenhalgh DG, Saffle JR, Holmes JH, Gamelli RL, Palmieri TL, Horton JW, et al. American burn association consensus conference to define sepsis and infection in burns. J Burn Care Res. 2007;28:776–90. [DOI] [PubMed] [Google Scholar]
- 61. Mann-Salinas EA, Baun MM, Meininger JC, Murray CK, Aden JK, Wolf SE, et al. Novel predictors of sepsis outperform the American burn association sepsis criteria in the burn intensive care unit patient. J Burn Care Res. 2013;34:31–43. [DOI] [PubMed] [Google Scholar]
- 62. Yan J, Hill WF, Rehou S, Pinto R, Shahrokhi S, Jeschke MG. Sepsis criteria versus clinical diagnosis of sepsis in burn patients: a validation of current sepsis scores. Surgery 2018;164:1241–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jeschke MG, Chinkes DL, Finnerty CC, Kulp G, Suman OE, Norbury WB, et al. Pathophysiologic response to severe burn injury. Ann. Surg. 2008;248:387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hart DW, Wolf SE, Mlcak R, Chinkes DL, Ramzy PI, Obeng MK, et al. Persistence of muscle catabolism after severe burn. Surgery 2000;128:312–9. [DOI] [PubMed] [Google Scholar]
- 65. Kottke MA, Walters TJ. Where's the leak in vascular barriers? A review. Shock (Augusta, Ga) 2016;46:20–36. [DOI] [PubMed] [Google Scholar]
- 66. Arbuthnot MK, Garcia AV. Early resuscitation and management of severe pediatric burns. Semin. Pediatr. Surg. 2019;28:73–8. [DOI] [PubMed] [Google Scholar]
- 67. Ashina K, Tsubosaka Y, Nakamura T, Omori K, Kobayashi K, Hori M, et al. Histamine induces vascular hyperpermeability by increasing blood flow and endothelial barrier disruption in vivo. PLoS One 2015. 10.1371/journal.pone.0132367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mikelis CM, Simaan M, Ando K, Fukuhara S, Sakurai A, Amornphimoltham P, et al. RhoA and ROCK mediate histamine-induced vascular leakage and anaphylactic shock. Nat. Commun. 2015. 10.1038/ncomms7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ma T, Liu L, Wang P, Xue Y. Evidence for involvement of ROCK signaling in bradykinin-induced increase in murine blood-tumor barrier permeability. J. Neuro-Oncol. 2012;106:291–301. [DOI] [PubMed] [Google Scholar]
- 70. Liu LB, Xue YX, Liu YH, Wang YB. Bradykinin increases blood-tumor barrier permeability by down-regulating the expression levels of ZO-1, occludin, and claudin-5 and rearranging actin cytoskeleton. J. Neurosci. Res. 2008;86:1153–68. [DOI] [PubMed] [Google Scholar]
- 71. Duah E, Adapala RK, Al-Azzam N, Kondeti V, Gombedza F, Thodeti CK, et al. Cysteinyl leukotrienes regulate endothelial cell inflammatory and proliferative signals through CysLT2 and CysLT1 receptors. Sci. Rep. 2013. 10.1038/srep03274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lee KS, Kim SR, Park HS, Jin GY, Lee YC. Cysteinyl leukotriene receptor antagonist regulates vascular permeability by reducing vascular endothelial growth factor expression. J. Allergy Clin. Immunol. 2004;114:1093–9. [DOI] [PubMed] [Google Scholar]
- 73. Antonetti DA, Barber AJ, Hollinger LA, Wolpert EB, Gardner TW. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J. Biol. Chem. 1999;274:23463–7. [DOI] [PubMed] [Google Scholar]
- 74. Chen XL, Nam JO, Jean C, Lawson C, Walsh CT, Goka E, et al. VEGF-induced vascular permeability is mediated by FAK. Dev. Cell 2012;22:146–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vasile E, Qu H, Dvorak HF, Dvorak AM. Caveolae and vesiculo-vacuolar organelles in bovine capillary endothelial cells cultured with VPF/VEGF on floating Matrigel-collagen gels. J. Histochem. Cytochem. 1999;47:159–67. [DOI] [PubMed] [Google Scholar]
- 76. Feng D, Nagy JA, Hipp J, Dvorak HF, Dvorak AM. Vesiculo-vacuolar organelles and the regulation of venule permeability to macromolecules by vascular permeability factor, histamine, and serotonin. J. Exp. Med. 1996;183:1981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Flier M, Leeuwen HJ, Kessel KP, Kimpen JL, Hoepelman AI, Geelen SP. Plasma vascular endothelial growth factor in severe sepsis. Shock (Augusta, Ga) 2005;23:35–8. [DOI] [PubMed] [Google Scholar]
- 78. Zang Q, Maass DL, White J, Horton JW. Cardiac mitochondrial damage and loss of ROS defense after burn injury: the beneficial effects of antioxidant therapy. J Appl Physiol. 2007;102:103–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Willis MS, Carlson DL, Dimaio JM, White MD, White DJ, Adams GA, et al. Macrophage migration inhibitory factor mediates late cardiac dysfunction after burn injury. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H795–804. [DOI] [PubMed] [Google Scholar]
- 80. Nielson CB, Duethman NC, Howard JM, Moncure M, Wood JG. Burns: pathophysiology of systemic complications and current management. J Burn Care Res. 2017;38:e469–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Griswold JA. White blood cell response to burn injury. Semin. Nephrol. 1993;13:409–15. [PubMed] [Google Scholar]
- 82. Grogan JB. Altered neutrophil phagocytic function in burn patients. J. Trauma 1976;16:734–8. [DOI] [PubMed] [Google Scholar]
- 83. Haar NM, Oswald M, Jeyaratnam J, Anton J, Barron KS, Brogan PA, et al. Recommendations for the management of autoinflammatory diseases. Ann. Rheum. Dis. 2015;74:1636–44. [DOI] [PubMed] [Google Scholar]
- 81. Schildt BE. Function of the RES after thermal and mechanical trauma in mice. Acta Chir. Scand. 1970;136:359–64. [PubMed] [Google Scholar]
- 85. Qiu T, Li M, Tanner MA, Yang Y, Sowers JR, Korthuis RJ, et al. Depletion of dendritic cells in perivascular adipose tissue improves arterial relaxation responses in type 2 diabetic mice. Metab. Clin. Exp. 2018;85:76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Vignesh P, Rawat A, Sharma M, Singh S. Complement in autoimmune diseases. Clinica Chimica Acta. 2017;465:123–30. [DOI] [PubMed] [Google Scholar]
- 87. Hammad A, Westacott L, Zaben M. The role of the complement system in traumatic brain injury: a review. J. Neuroinflammation 2018. 10.1186/s12974-018-1066-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hugli TE. Complement and cellular triggering reactions. Introductory remarks. Federation proceedings. 1984;43:2540–2. [PubMed] [Google Scholar]
- 89. Conde P, Rodriguez M, Touw W, Jimenez A, Burns M, Miller J, et al. DC-SIGN(+) macrophages control the induction of transplantation tolerance. Immunity 2015;42:1143–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Donnelly RP, Fenton MJ, Kaufman JD, Gerrard TL. IL-1 expression in human monocytes is transcriptionally and posttranscriptionally regulated by IL-4. J. Immunol. 1991;146:3431–6. [PubMed] [Google Scholar]
- 91. Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A. IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 1991;147:3815–22. [PubMed] [Google Scholar]
- 92. Oswald IP, Wynn TA, Sher A, James SL. Interleukin 10 inhibits macrophage microbicidal activity by blocking the endogenous production of tumor necrosis factor alpha required as a costimulatory factor for interferon gamma-induced activation. Proc. Natl. Acad. Sci. U. S. A. 1992;89:8676–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Heideman M, Bengtsson A. The immunologic response to thermal injury. World J. Surg. 1992;16:53–6. [DOI] [PubMed] [Google Scholar]
- 94. Sheridan RL, Weber JM, Pasternak MM, Mulligan JM, Tompkins RG. A 15-year experience with varicella infections in a pediatric burn unit. Burns 1999;25:353–6. [DOI] [PubMed] [Google Scholar]
- 95. Gosain A, Gamelli RL. A primer in cytokines. J. Burn Care Rehabil. 2005;26:7–12. [DOI] [PubMed] [Google Scholar]
- 96. Schwacha MG. Macrophages and post-burn immune dysfunction. Burns 2003;29:1–14. [DOI] [PubMed] [Google Scholar]
- 97. Burleson DG, MasonAD, Jr, PruittBA, Jr. Lymphoid subpopulation changes after thermal injury and thermal injury with infection in an experimental model. Ann. Surg. 1988;207:208–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Rendon JL, Choudhry MA. Th17 cells: critical mediators of host responses to burn injury and sepsis. J. Leukoc. Biol. 2012;92:529–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Deitch EA, Bridges RM, Dobke M, McDonald JC. Burn wound sepsis may be promoted by a failure of local antibacterial host defenses. Ann. Surg. 1987;206:340–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Venet F, Pachot A, Debard AL, Bohe J, Bienvenu J, Lepape A, et al. Human CD4+CD25+ regulatory T lymphocytes inhibit lipopolysaccharide-induced monocyte survival through a Fas/Fas ligand-dependent mechanism. J. Immunol. 2006;177:6540–7. [DOI] [PubMed] [Google Scholar]
- 101. Porter C, Tompkins RG, Finnerty CC, Sidossis LS, Suman OE, Herndon DN. The metabolic stress response to burn trauma: current understanding and therapies. Lancet (London, England) 2016;388:1417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Porter C, Herndon DN, Børsheim E, Bhattarai N, Chao T, Reidy PT, et al. Long-term skeletal muscle mitochondrial dysfunction is associated with hypermetabolism in severely burned children. J Burn Care Res. 2016;37:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Porter C, Herndon DN, Børsheim E, Chao T, Reidy PT, Borack MS, et al. Uncoupled skeletal muscle mitochondria contribute to hypermetabolism in severely burned adults. Am. J. Physiol. Endocrinol. Metab. 2014;307:E462–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Auger C, Samadi O, Jeschke MG. The biochemical alterations underlying post-burn hypermetabolism. Biochim. Biophys. Acta Mol. basis Dis. 2017;1863:2633–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wieser V, Moschen AR, Tilg H. Inflammation, cytokines and insulin resistance: a clinical perspective. Arch. Immunol. Ther. Exp. 2013;61:119–25. [DOI] [PubMed] [Google Scholar]
- 106. Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006;440:944–8. [DOI] [PubMed] [Google Scholar]
- 107. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011;17:179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Herndon DN, Hart DW, Wolf SE, Chinkes DL, Wolfe RR. Reversal of catabolism by beta-blockade after severe burns. N. Engl. J. Med. 2001;345:1223–9. [DOI] [PubMed] [Google Scholar]
- 109. Wang Y, Viscarra J, Kim SJ, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015;16:678–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Jeschke MG, Herndon DN, Wolf SE, DebRoy MA, Rai J, Thompson JC, et al. Hepatocyte growth factor modulates the hepatic acute-phase response in thermally injured rats. Crit. Care Med. 2000;28:504–10. [DOI] [PubMed] [Google Scholar]
- 111. Yu YM, Tompkins RG, Ryan CM, Young VR. The metabolic basis of the increase of the increase in energy expenditure in severely burned patients. JPEN J. Parenter. Enteral Nutr. 1999;23:160–8. [DOI] [PubMed] [Google Scholar]
- 112. Matthias A, Ohlson KB, Fredriksson JM, Jacobsson A, Nedergaard J, Cannon B. Thermogenic responses in brown fat cells are fully UCP1-dependent. UCP2 or UCP3 do not substitute for UCP1 in adrenergically or fatty acid-induced thermogenesis. J. Biol. Chem. 2000;275:25073–81. [DOI] [PubMed] [Google Scholar]
- 113. Jeschke MG, Finnerty CC, Herndon DN, Song J, Boehning D, Tompkins RG, et al. Severe injury is associated with insulin resistance, endoplasmic reticulum stress response, and unfolded protein response. Ann. Surg. 2012;255:370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wang C, Huang Z, Du Y, Cheng Y, Chen S, Guo F. ATF4 regulates lipid metabolism and thermogenesis. Cell Res. 2010;20:174–84. [DOI] [PubMed] [Google Scholar]
- 115. Kraft R, Herndon DN, Finnerty CC, Hiyama Y, Jeschke MG. Association of postburn fatty acids and triglycerides with clinical outcome in severely burned children. J. Clin. Endocrinol. Metab. 2013;98:314–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Abdullahi A, Jeschke MG. White adipose tissue browning: a double-edged sword. Trends Endocrinol Metab 2016;27:542–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Meier JA, Larner AC. Toward a new STATe: the role of STATs in mitochondrial function. Semin. Immunol. 2014;26:20–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Porter C, Herndon DN, Chondronikola M, Chao T, Annamalai P, Bhattarai N, et al. Human and mouse brown adipose tissue mitochondria have comparable UCP1 function. Cell Metab. 2016;24:246–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM consensus conference committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 1992;101:1644–55. [DOI] [PubMed] [Google Scholar]
- 120. Yarema TC, Yost S. Low-dose corticosteroids to treat septic shock: a critical literature review. Crit. Care Nurse 2011;31:16–26. [DOI] [PubMed] [Google Scholar]
- 121. Burns JP. Septic shock in the pediatric patient: pathogenesis and novel treatments. Pediatr. Emerg. Care 2003;19:112–5. [DOI] [PubMed] [Google Scholar]
- 122. Vincent JL, Zhang H, Szabo C, Preiser JC. Effects of nitric oxide in septic shock. Am. J. Respir. Crit. Care Med. 2000;161:1781–5. [DOI] [PubMed] [Google Scholar]
- 123. Zardi EM, Zardi DM, Dobrina A, Afeltra A. Prostacyclin in sepsis: a systematic review. Prostaglandins Other Lipid Mediat. 2007;83:1–24. [DOI] [PubMed] [Google Scholar]
- 124. Araújo AV, Ferezin CZ, Rodrigues GJ, Lunardi CN, Vercesi JA, Grando MD, et al. Prostacyclin, not only nitric oxide, is a mediator of the vasorelaxation induced by acetylcholine in aortas from rats submitted to cecal ligation and perforation (CLP). Vasc. Pharmacol. 2011;54:44–51. [DOI] [PubMed] [Google Scholar]
- 125. Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. Br. J. Anaesth. 2011;107:57–64. [DOI] [PubMed] [Google Scholar]
- 126. Alvarez S, Evelson PA. Nitric oxide and oxygen metabolism in inflammatory conditions: sepsis and exposition to polluted ambients. Front Biosci. 2007;12:964–74. [DOI] [PubMed] [Google Scholar]
- 127. Hattori Y, Kasai K. Induction of mRNAs for ICAM-1, VCAM-1, and ELAM-1 in cultured rat cardiac myocytes and myocardium in vivo. Biochem. Mol. Biol. Int. 1997;41:979–86. [DOI] [PubMed] [Google Scholar]
- 128. Li Y, Chen B, Yang X, Zhang C, Jiao Y, Li P, et al. S100a8/a9 Signaling causes mitochondrial dysfunction and cardiomyocyte death in response to ischemic/reperfusion injury. Circulation 2019;140:751–64. [DOI] [PubMed] [Google Scholar]
- 129. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N. Engl. J. Med. 2013;368:1845–6. [DOI] [PubMed] [Google Scholar]
- 130. Abdullahi A, Chen P, Stanojcic M, Sadri AR, Coburn N, Jeschke MG. IL-6 signal from the bone marrow is required for the browning of white adipose tissue post burn injury. Shock (Augusta, Ga) 2017;47:33–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999;340:448–54. [DOI] [PubMed] [Google Scholar]
- 132. Thomas S, Wolf SE, Chinkes DL, Herndon DN. Recovery from the hepatic acute phase response in the severely burned and the effects of long-term growth hormone treatment. Burns 2004;30:675–9. [DOI] [PubMed] [Google Scholar]
- 133. Pruchniewski D, Pawlowski T, Morkowski J, Mackiewicz S. C-reactive protein in management of children's burns. Ann. Clin. Res. 1987;19:334–8. [PubMed] [Google Scholar]
- 134. Lavrentieva A, Kontakiotis T, Lazaridis L, Tsotsolis N, Koumis J, Kyriazis G, et al. Inflammatory markers in patients with severe burn injury. What is the best indicator of sepsis? Burns 2007;33:189–94. [DOI] [PubMed] [Google Scholar]
- 135. Jeschke MG, Finnerty CC, Kulp GA, Kraft R, Herndon DN. Can we use C-reactive protein levels to predict severe infection or sepsis in severely burned patients? Int J Burns Trauma. 2013;3:137–43. [PMC free article] [PubMed] [Google Scholar]
- 136. Assicot M, Gendrel D, Carsin H, Raymond J, Guilbaud J, Bohuon C. High serum procalcitonin concentrations in patients with sepsis and infection. Lancet (London, England) 1993;341:515–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Long B, Koyfman A. Ready for prime time? Biomarkers in sepsis. Emerg. Med. Clin. North Am. 2017;35:109–22. [DOI] [PubMed] [Google Scholar]
- 138. Brunkhorst FM, Eberhard OK, Brunkhorst R. Discrimination of infectious and noninfectious causes of early acute respiratory distress syndrome by procalcitonin. Crit. Care Med. 1999;27:2172–6. [DOI] [PubMed] [Google Scholar]
- 139. Uzzan B, Cohen R, Nicolas P, Cucherat M, Perret GY. Procalcitonin as a diagnostic test for sepsis in critically ill adults and after surgery or trauma: a systematic review and meta-analysis. Crit. Care Med. 2006;34:1996–2003. [DOI] [PubMed] [Google Scholar]
- 140. Wacker C, Prkno A, Brunkhorst FM, Schlattmann P. Procalcitonin as a diagnostic marker for sepsis: a systematic review and meta-analysis. Lancet Infect. Dis. 2013;13:426–35. [DOI] [PubMed] [Google Scholar]
- 141. Nunez Lopez O, Cambiaso-Daniel J, Branski LK, Norbury WB, Herndon DN. Predicting and managing sepsis in burn patients: current perspectives. Ther. Clin. Risk Manag. 2017;13:1107–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Meisner M. Pathobiochemistry and clinical use of procalcitonin. Clin Chim Acta. 2002;323:17–29. [DOI] [PubMed] [Google Scholar]
- 143. Schneider HG, Lam QT. Procalcitonin for the clinical laboratory: a review. Pathology 2007;39:383–90. [DOI] [PubMed] [Google Scholar]
- 144. Brunkhorst FM, Wegscheider K, Forycki ZF, Brunkhorst R. Procalcitonin for early diagnosis and differentiation of SIRS, sepsis, severe sepsis, and septic shock. Intensive Care Med. 2000;26:S148–52. [DOI] [PubMed] [Google Scholar]
- 145. Seoane L, Pértega S, Galeiras R, Astola I, Bouza T. Procalcitonin in the burn unit and the diagnosis of infection. Burns 2014;40:223–9. [DOI] [PubMed] [Google Scholar]
- 146. Paratz JD, Lipman J, Boots RJ, Muller MJ, Paterson DL. A new marker of sepsis post burn injury?*. Crit. Care Med. 2014;42:2029–36. [DOI] [PubMed] [Google Scholar]
- 147. Finnerty CC, Herndon DN, Przkora R, Pereira CT, Oliveira HM, Queiroz DM, et al. Cytokine expression profile over time in severely burned pediatric patients. Shock (Augusta, Ga) 2006;26:13–9. [DOI] [PubMed] [Google Scholar]
- 148. Abdel-Hafez NM, Saleh Hassan Y, El-Metwally TH. A study on biomarkers, cytokines, and growth factors in children with burn injuries. Ann. Burns Fire Disasters 2007;20:89–100. [PMC free article] [PubMed] [Google Scholar]
- 149. Kraft R, Herndon DN, Finnerty CC, Cox RA, Song J, Jeschke MG. Predictive value of IL-8 for sepsis and severe infections after burn injury: a clinical study. Shock (Augusta, Ga) 2015;43:222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Pileri D, Accardo Palombo A, D'Amelio L, D'Arpa N, Amato G, Masellis A, et al. Concentrations of cytokines IL-6 and IL-10 in plasma of burn patients: their relationship to sepsis and outcome. Ann. Burns Fire Disasters 2008;21:182–5. [PMC free article] [PubMed] [Google Scholar]
- 151. Csontos C, Foldi V, Pálinkas L, Bogar L, Röth E, Weber G, et al. Time course of pro- and anti-inflammatory cytokine levels in patients with burns--prognostic value of interleukin-10. Burns 2010;36:483–94. [DOI] [PubMed] [Google Scholar]
- 152. Yaegashi Y, Shirakawa K, Sato N, Suzuki Y, Kojika M, Imai S, et al. Evaluation of a newly identified soluble CD14 subtype as a marker for sepsis. J Infect Chemother. 2005;11:234–8. [DOI] [PubMed] [Google Scholar]
- 153. Wu J, Hu L, Zhang G, Wu F, He T. Accuracy of presepsin in sepsis diagnosis: a systematic review and meta-analysis. PLoS One 2015. 10.1371/journal.pone.0133057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Bellos I, Fitrou G, Pergialiotis V, Thomakos N, Perrea DN, Daskalakis G. The diagnostic accuracy of presepsin in neonatal sepsis: a meta-analysis. Eur. J. Pediatr. 2018;177:625–32. [DOI] [PubMed] [Google Scholar]
- 155. Memar MY, Baghi HB. Presepsin: a promising biomarker for the detection of bacterial infections. Biomed Pharmacother 2019;111:649–56. [DOI] [PubMed] [Google Scholar]
- 156. Miyoshi M, Inoue Y, Nishioka M, Ikegame A, Nakao T, Kishi S, et al. Clinical evaluation of presepsin considering renal function. PLoS One 2019. 10.1371/journal.pone.0215791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Yoon SH, Kim EH, Kim HY, Ahn JG. Presepsin as a diagnostic marker of sepsis in children and adolescents: a systemic review and meta-analysis. BMC Infect. Dis. 2019. 10.1186/s12879-019-4397-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Gille J, Schmidt J, Kremer T, Sablotzki A. Evaluation of MR-proANP and copeptin for sepsis diagnosis after burn injury. J. Crit. Care 2019;52:149–55. [DOI] [PubMed] [Google Scholar]
- 159. Hampson P, Dinsdale RJ, Wearn CM, Bamford AL, Bishop JRB, Hazeldine J, et al. Neutrophil dysfunction, immature granulocytes, and cell-free DNA are early biomarkers of sepsis in burn-injured patients: a prospective observational cohort study. Ann. Surg. 2017;265:1241–9. [DOI] [PubMed] [Google Scholar]
- 160. Guo H, Tang L, Xu J, Lin C, Ling X, Lu C, et al. MicroRNA-495 serves as a diagnostic biomarker in patients with sepsis and regulates sepsis-induced inflammation and cardiac dysfunction. Eur. J. Med. Res. 2019. 10.1186/s40001-019-0396-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Nishiguchi T, Ito I, Lee JO, Suzuki S, Suzuki F, Kobayashi M. Macrophage polarization and MRSA infection in burned mice. Immunol. Cell Biol. 2017;95:198–206. [DOI] [PubMed] [Google Scholar]
- 162. Lourtet-Hascoët J, Félicé MP, Bicart-See A, Bouige A, Giordano G, Bonnet E. Species and antimicrobial susceptibility testing of coagulase-negative staphylococci in periprosthetic joint infections. Epidemiol. Infect. 2018;146:1771–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Shen WC, Wang X, Qin WT, Qiu XF, Sun BW. Exogenous carbon monoxide suppresses Escherichia coli vitality and improves survival in an Escherichia coli-induced murine sepsis model. Acta Pharmacol. Sin. 2014;35:1566–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Chadha P, Katare OP, Chhibber S. Liposome loaded phage cocktail: enhanced therapeutic potential in resolving Klebsiella pneumoniae mediated burn wound infections. Burns 2017;43:1532–43. [DOI] [PubMed] [Google Scholar]
- 165. Denning DW, Cadranel J, Beigelman-Aubry C, Ader F, Chakrabarti A, Blot S, et al. Chronic pulmonary aspergillosis: rationale and clinical guidelines for diagnosis and management. Eur. Respir. J. 2016;47:45–68. [DOI] [PubMed] [Google Scholar]
- 166. Katz T, Wasiak J, Cleland H, Padiglione A. Incidence of non-candidal fungal infections in severe burn injury: an Australian perspective. Burns 2014;40:881–6. [DOI] [PubMed] [Google Scholar]
- 167. Sobouti B, Momeni M, Masalegooyan N, Ansari I, Rahbar H. Herpes simplex virus infection in minor burn injury: a case report. Int J Burns Trauma. 2018;8:149–52. [PMC free article] [PubMed] [Google Scholar]
- 168. Gibbs JT, Zieger M, Sood R. Cytomegalovirus colitis in a burn patient. J Burn Care Res. 2016;37:e298–300. [DOI] [PubMed] [Google Scholar]
- 169. Kubota Y, Kosaka K, Hokazono T, Yamaji Y, Tezuka T, Akita S, et al. Disseminated zoster in an adult patient with extensive burns: a case report. Virol. J. 2019;16:68. [DOI] [PMC free article] [PubMed] [Google Scholar]