

Abstract
The stability of mRNAs is fundamental to determining expression level and dynamics. Nonetheless, current approaches for measuring transcript half-lives (e.g., transcription shutoff) are generally toxic or technically complex. Here we describe an alternative strategy for targeted measurements of endogenous mRNA stability that is simple, inexpensive, and nontoxic. Cells are first metabolically labeled with the nucleoside analog 4-thiouridine (4sU). Extracted mRNA can then be treated with the thiol-reactive compound N-ethylmaleimide. This compound modifies 4sU nucleotides and sterically interferes with reverse transcription of 4sU-containing transcripts, disrupting their conversion into cDNA. The decay rate of non-4sU-containing preexisting mRNA can then be monitored by quantitative PCR (qPCR). Importantly, this approach avoids the biochemical isolation of 4sU-labeled transcripts and/or RNA-seq analysis required for other metabolic-labeling strategies. In summary, our method combines the simplicity of “transcription shutoff” strategies with the accuracy of metabolic-labeling strategies for measurements of mRNA stability across a wide range of half-lives.
Keywords: 4-thiouridine, mRNA stability
INTRODUCTION
The stabilities of mammalian mRNAs vary from minutes to hours and are important in shaping gene expression over time (Friedel et al. 2009). Short-lived transcripts can be rapidly adapted to changing conditions or signals. Long-lived transcripts can maximize the quantity of protein produced per mRNA. mRNA stabilities can also change in response to cellular signals. Mechanisms include micro-RNAs (miRNAs), RNA-binding proteins, covalent modifications, and quality control mechanisms (e.g., nonsense-mediated decay, ribosome quality control, etc.) (Lee and Lykke-Andersen 2013; Bartel 2018). Knowing how an mRNA's stability changes under varying conditions can be key to understanding the mechanisms that control its expression.
A common approach for measuring the stability of endogenous mRNAs is “transcription shutoff.” In this case, pharmacological (e.g., Actinomycin D, α-amanitin, 5,6-dichloro-1-beta-D-ribofuanosylbenzimidazole) or genetic tools are used to block transcription. Levels of mRNAs can then be monitored to calculate the time required to fall by 50%, i.e., their half-life. However, these approaches are limited by several drawbacks. First, inhibiting all Pol II-mediated transcription causes vast changes to the transcriptome and disrupts cell physiology in ways that likely trigger changes in mRNA regulatory mechanisms (e.g., cell cycle arrest) (Airoldi et al. 2016). Second, transcription and decay processes are often interconnected, such that interfering with one often perturbs the other (Sun et al. 2013). As a consequence, measurements of mRNA stabilities using transcription shutoff may not reflect mRNA dynamics in unperturbed cells.
A second strategy for measuring mRNA stability instead uses metabolic labeling. In this case, cells are exposed to nucleoside analogs, such as 4sU or 5′ bromouridine (BrU) (Dolken et al. 2008; Friedel et al. 2009; Tani et al. 2012). Both analogs are often reported to be nontoxic and are readily incorporated into nascent mRNA. Newly transcribed transcripts can then be isolated through either chemical modification with biotin-conjugated reactants (4sU) or immuno-purification (BrU) and compared to input mRNA to estimate synthesis rate and steady state levels (Dolken et al. 2008; Friedel et al. 2009; Tani et al. 2012). Decay rates can be inferred from these parameters using a simple kinetic model (Friedel et al. 2009). The major advantage of this strategy is that mRNA dynamics are not strongly perturbed. Its primary limitation is that the biochemical separation of newly transcribed from total mRNA can be cumbersome and introduces experimental noise. Several newer strategies (TimeLapse-seq and SLAM-seq) instead chemically modify these nucleoside analogs in a manner that can be detected by deep sequencing (Herzog et al. 2017; Schofield et al. 2018). These powerful tools provide robust measurements of mRNA stabilities on a transcriptome-scale without biochemical separation of newly transcribed transcripts but are expensive and impractical when the stabilities of only a few transcripts are needed.
Here, we describe a simple inexpensive approach for directly monitoring the stability of endogenous mRNAs. Cells are cultured with 4sU and mRNA is then sampled at several timepoints. After isolation, the mRNA is treated with the thiol-reactive compound N-ethylmaleimide, which readily forms covalent bonds with mRNA-incorporated 4sU. This bulky modification interferes with reverse transcription (RT) of newly transcribed 4sU-containing transcripts, preventing the synthesis of full-length cDNA. Subsequent analysis using qPCR therefore only reflects levels of preexisting mRNA and tracks its decline over time. This approach avoids the global disruptions to cell physiology caused by transcription-shutoff strategies and the technical complexity of strategies that require biochemical separation of newly transcribed from preexisting mRNA or the preparation of RNA-seq libraries. Here, we validate this strategy and offer additional insight into potential artifacts of this and other approaches that use 4sU.
RESULTS
NEM-modification of 4sU interferes with reverse transcription
We set out to develop a method for measuring the half-lives of selected mRNAs without perturbing global mRNA dynamics. Recent methods such as SLAM-seq and TimeLapse-seq achieve this on a genome-wide scale using metabolic labeling with 4sU (Herzog et al. 2017; Schofield et al. 2018). The key to both strategies is to chemically modify 4sU nucleotides that are incorporated into mRNA. This modification introduces a base change in cDNA produced from the RNA template, which can be detected and quantified by deep sequencing. We wondered whether 4sU modifications might also be capable of blocking reverse transcription completely. This would limit full-length cDNA production to only mRNA transcripts existing prior to 4sU incubation. The decline in mRNA levels for any single gene could then be monitored using qPCR (Fig. 1A).
FIGURE 1.

Modification of 4-thiouridine (4sU) disrupts reverse transcription. (A) Overview of the strategy for Roadblock-qPCR. (B) Modification of 4-thiouracil using N-ethylmaleimide (NEM) and iodoacetamide (IAA). (C) NEM and IAA interfere with the reverse transcription of RNA. A 121 nt in vitro transcribed RNA was synthesized with either UTP or 4sUTP. RNAs were treated with EtOH, DMSO, NEM, or IAA (see Materials and Methods) and reverse transcribed using a FAM6-labeled primer with the Protoscript II reverse transcriptase (NEB). cDNA products were then analyzed by PAGE. (D) Length distribution and base conversions in cDNA from NEM-treated RNA. (Top panel) cDNA from NEM-treated RNA prepared in C was analyzed by capillary electrophoresis (fragment analysis). Approximate fragment sizes are shown above. Locations of U nucleotides in the RNA sequence are highlighted as indicated. (Bottom panel) Partial sequences (nucleotides 26–77 of the + strand) of three full-length cDNAs from C. T-to-C base conversions in sequence are highlighted in red.
The basis of our strategy, which we call “Roadblock-qPCR,” is that modification of 4sU with a bulky thiol-reactive compound blocks reverse transcription of the target transcript, and consequently detection (Fig. 1A). Two widely available thiol-reactive compounds that are normally used for probing cysteine reactivity in proteins are iodoacetamide and N-ethylmaleimide (Fig. 1B). To determine the efficiency with which these compounds react with RNA-incorporated 4sU, we synthesized two short RNA fragments of 121 nt. One contained uridine, while the other was synthesized with 4sU. A segment at the 3′ end was left free of Us to ensure that 4sU modification would not interfere with primer binding. Both U and 4sU-containing RNA fragments were then treated with iodoacetamide (IAA) or N-ethylmaleimide (NEM) (see Materials and Methods). The efficiency of 4sU modification was then monitored by UV spectroscopy, as the sulfhydryl group of 4sU exhibits a specific absorbance peak at ∼330 nM.
We next tested whether any of these modifications impacted reverse transcription. U and 4sU-containing RNAs were reverse transcribed using the MMLV Protoscript II reverse transcriptase (New England Biolabs) and a fluorophore-labeled primer, and then analyzed by PAGE (Fig. 1C). Neither incorporation of 4sU alone or treatment of U-containing RNA with NEM or IAA impacted reverse transcription, but the combination strongly interfered with the production of the full-length cDNA. NEM blocked reverse transcription slightly more than IAA, yielding overall shorter fragments. We suspect this is due to the increased steric interference caused by the larger NEM molecule. We therefore chose to proceed with NEM rather than IAA but predict that modification with even larger thiol-reactive compounds might increase the efficiency of RT interference. Other reverse transcriptases tested, including other M-MLV variants (Superscript II, Invitrogen), avian myeloblastosis virus RT (New England Biolabs), and the group II intron RT (InGex) produced similar results. We note that a single IAA-modified 4sU nucleotide was previously shown to have little effect on RT (Herzog et al. 2017). Our results indicate that this is not always a safe assumption. However, RT disruption by IAA may depend on sequence context and is expected to become increasingly likely with each IAA-modified nucleotide that is encountered.
Although modification of 4sU with NEM strongly interfered with RT, it was not 100% efficient and many intermediate cDNA fragments were produced (Fig. 1C). To determine whether RT disruption was affected by the sequence context of the modified nucleotide, we analyzed cDNA products using capillary electrophoresis, which provides better resolution on cDNA fragment lengths (Fig. 1D). These traces confirmed that RT is disrupted early on in the transcript. However, we failed to identify synergistic inhibition at either sequential Us or other specific positions. Assuming that 100% of incorporated 4sU nucleotides are modified with NEM, roughly 90% of the primer extension products terminate in the first half of the RNA, which includes 12 uridines. The probability of RT disruption is therefore roughly 0.17 for each modified nucleotide, meaning that an RT enzyme must encounter 24 modified 4sU nucleotides to reach a 99% chance of drop-off. This value sets some limits on endogenous mRNAs that can be monitored using this approach.
Despite this strong interference, a small amount of full-length transcript was produced. Nucleotide modifications that fail to disrupt reverse transcription often interfere with reverse transcription fidelity. To determine whether this was the case for NEM-modified 4sU as well, we cloned and sequenced several full-length cDNA products. Similar to recent reports using 4sU modification with IAA treatment (SLAM-seq), these also contained stereotypic U to C misreads (Fig. 1D; Herzog et al. 2017). Given the structural differences between NEM and IAA, this common effect on RT fidelity suggests that a wide variety of 4sU modifications introduce U to C misreads.
Optimization of 4sU-labeling in cell culture
Our next step was to test whether this strategy could be applied to endogenous mRNAs in living cells. 4sU is commonly considered nontoxic, but several reports have noted decreases in cell viability even after relatively short (12 h) durations (Burger et al. 2013). To determine concentrations of 4sU that minimized toxicity but were sufficient to induce RT drop-off, we treated HEK-293T cells with increasing concentrations of 4sU for 24 h. Somewhat surprisingly, even relatively low (50 µM) concentrations significantly (although moderately) reduced cell viability (Fig. 2A). Concentrations above 50 µM only minimally increased toxicity. To test whether mRNA had incorporated sufficient 4sU to efficiently block reverse transcription, we isolated mRNA from cells grown for 4 h in increasing concentrations of 4sU (Fig. 2B). Isolated mRNA was treated with NEM and reverse transcribed. We then analyzed levels of several mRNAs using qPCR. Even low (25 µM) concentrations blocked detection of each mRNA tested, validating our strategy for at least short-lived endogenous mRNAs (Fig. 2B). Nonetheless, because higher concentrations of 4sU caused no increased toxicity and might better interfere with reverse transcription of mRNAs with low U content, we chose instead to proceed with 400 µM.
FIGURE 2.

Optimization of 4sU-labeling of mRNA in cells. (A) Effect of increasing 4sU concentrations on cell viability. HEK-293T cells were grown 24 h in the indicated concentrations of 4sU. Viability was measured using CellTiter-Glo. Error bars are SD, n = 3. (B) Concentrations of 4sU required to impede reverse transcription of endogenous mRNAs. RNA was isolated from HEK-293T cells treated with the indicated concentrations of 4sU for 4 h, and then treated with NEM and reverse transcribed. Levels of the indicated mRNAs were monitored by qPCR analysis of cDNA. Error bars are SD, n = 3.
Benchmarking against a doxcycline-repressible reporter
We next sought to assess the accuracy of Roadblock-qPCR for measuring mRNA decay rates. As a reference, we generated a reporter encoding Renilla luciferase (Rluc) and controlled by a doxycycline-repressible (dox-off) promoter. This system avoids the indirect effects of general “transcription-shutoff” strategies because it affects only a single gene. Analysis of doxycycline-treated HEK-293T cells stably expressing this reporter established a 1.1 h half-life for this mRNA (Fig. 3A). In parallel, cells were treated with 400 µM 4sU, and RNA was sampled at various timepoints. Isolated mRNA was treated with NEM, reverse transcribed, and then analyzed by qPCR with primers targeting the 5′ end of the transcript (Fig. 3B). This approach yielded a half-life of 1.4 h, slightly longer but nonetheless similar to the measurement made with doxycycline. We noted that levels of the Renilla reporter in NEM-treated mRNA appear to plateau slightly above 0. This may reflect incomplete disruption of reverse transcription even when the mRNA is fully saturated with 4sU. Nonetheless, our approach yielded a half-life estimate that was very close to the “true” value.
FIGURE 3.

Roadblock-qPCR yields accurate measurements of mRNA half-life. (A) Reporter mRNA stability analysis using transcription shutoff with doxycycline. HEK-293T cells stably expressing a dox-off Renilla were treated with 1 µg/mL doxycycline for 0–8 h. Reporter mRNA levels were measured at the indicated times using qPCR. Error bars are SD, n = 3. (B) Reporter mRNA stability analysis using Roadblock-qPCR. RNA was isolated from the same HEK-293T cells expressing Renilla mRNA from A, now treated with 4sU for 0–8 h, then treated with NEM and reverse transcribed. Reporter mRNA levels were measured using qPCR. Error bars are SD, n = 3.
Roadblock-qPCR avoids artifacts associated with actinomycin D
Our next step was to determine whether Roadblock-qPCR maintained its advantage when measuring half-lives of endogenously expressed mRNAs with a wide range of stabilities. Levels of five endogenous mRNAs chosen to cover a range of stabilities were monitored from either actinomycin D (ActD)- or 4sU/NEM-treated cells using qPCR (Fig. 4A). Declining levels of mRNAs were fit to single exponential decay models to estimate half-lives. Each of the mRNAs selected here was efficiently masked by 4sU/NEM treatment at long timepoints, confirming efficient interference with RT. In contrast, ActD caused levels of all but the shortest-lived mRNAs (i.e., Myc) to plateau well above 0, interfering with calculations of decay kinetics (Fig. 4A). Why this occurs is unclear but may reflect a “buffering” mechanism that acts to preserve cytoplasmic mRNA when transcription declines (Sun et al. 2013).
FIGURE 4.

Comparison of approaches for measuring mRNA decay kinetics. (A) mRNA decay measurements using Roadblock-qPCR and transcriptional shutoff with ActD on endogenous mRNAs. RNA was isolated from HEK-293T cells treated with either 2 µg/mL Actinomycin D or 400 µM 4sU for the indicated times. mRNA from 4sU-exposed cells was treated with NEM before reverse transcription. Levels of the indicated mRNAs at each timepoint were determined by qPCR. Error bars are SD, n = 3. (B) Half-life calculations using Roadblock-qPCR and 4sU-biotin are similar. Roadblock-qPCR was performed on HEK-293T cells for 11 mRNAs using 2, 4 and 8 h timepoints of 4sU (400 µM) treatment. The calculated half-lives were compared to those obtained previously using affinity purification of biotinylated 4sU-containing mRNA (Friedel and Dolken 2009). Note: Correlation calculations for B-cell and combined data sets exclude SLC7A11, an extreme outlier. (C) Half-life calculations obtained from Roadblock-qPCR and TimeLapse-seq are similar. Half-lives calculated in B were compared to those reported previously with TimeLapse-seq on K562 cells (Schofield et al. 2018).
We also compared Roadblock-qPCR to other strategies for measuring mRNA half-lives using 4sU. One of these is to biotinylate newly synthesized 4sU-containing mRNA, followed by affinity purification and mRNA quantification. Friedel et al. used this strategy to measure mRNA half-lives in human BL41 cells and mouse 3T3 cells (Friedel et al. 2009). Half-life calculations for human BL41 cells showed good correlation with Roadblock-qPCR results for 10 mRNAs tested (R2 = 0.58), with the exception of one outlier (SLC7A11) that appears to be uniquely stable in BL41 cells (Fig. 4B; Supplemental Table 1). Half-life calculations from mouse 3T3 cells were, surprisingly, even more closely correlated (R2 = 0.84), suggesting that the stabilities of these mRNAs are well conserved between human and mouse cells (Fig. 4B). A more recently developed strategy is TimeLapse-seq, which modifies 4sU nucleotides in a manner that can be detected by RNA-seq analysis (Schofield et al. 2018). Half-lives determined by TimeLapse-seq were also highly correlated with half-lives obtained by Roadblock-qPCR (R2 = 0.99) (Fig. 4C). These findings argue that Roadblock-qPCR avoids the artifacts that occur with transcription-shutoff strategies, and yields half-life estimates in good agreement with other transcriptome-scale strategies.
Caveats of using 4sU for metabolic labeling
One caveat of using 4sU for metabolic labeling is that its incorporation can diminish the levels of some short-lived mRNAs. For instance, we found that Myc mRNA levels continually decline over a period of 8 h in the presence of even low (50 µM) concentrations of 4sU. Although 4sU is widely reported to be mostly inert, multiple studies have noted its potential toxicity and effect on levels of some mRNAs, similar to what we report here (Fig. 2A; Burger et al. 2013; Schofield et al. 2018). In our experience, mRNAs with half-lives greater than 1–2 h seem resistant to these perturbations. The underlying mechanism remains unclear. Nonetheless, the effect of 4sU appears to be minimal for most mRNAs, as levels are highly correlated between control and 4sU-treated samples (Schofield et al. 2018). Moreover, this effect is most likely due to incorporation into new mRNAs following 4sU addition. In Roadblock-qPCR, newly synthesized transcripts containing 4sU are anyways masked by NEM treatment, and so we expect this phenomenon to have only minor effects on half-life measurements.
DISCUSSION
We have described a simple, inexpensive, and noninvasive strategy for targeted measurements of mRNA stability. The basis of our strategy is to label newly transcribed mRNA with the nucleoside analog 4sU. Following mRNA isolation, incorporated 4sU is modified with the thiol-reactive compound NEM, which interferes with reverse transcription. This masks newly transcribed mRNAs from detection by qPCR, allowing declining levels of preexisting mRNA to be monitored directly. mRNA half-life measurements using this approach robustly correlate with those of orthogonal transcriptome-scale strategies (Schofield et al. 2018).
Our approach has several advantages over existing strategies for measuring half-lives of single mRNAs. First, 4sU-labeling of mRNA is relatively nontoxic when compared to transcription inhibitors. ActD, alpha-amanitin, and DRB all profoundly impact cell health over time, and likely perturb mechanisms that control mRNA stability. Transcription shutoff approaches may also trigger “RNA buffering” mechanisms that maintain a minimum concentration of mRNA in the cell (Sun et al. 2013). This compensatory response may explain the unusual decay patterns of longer-lived mRNAs seen in Figure 4A. 4sU labeling has only minimal effects on bulk mRNA synthesis and therefore avoids these complications.
Second, our approach is technically simple and can be easily incorporated into existing qPCR workflows. It also avoids the technical complexity of approaches that rely on biochemical separation of newly transcribed mRNAs (e.g., BRIC) (Dolken et al. 2008; Friedel et al. 2009; Tani et al. 2012). This reduces the number of steps and consequently the potential for introducing experimental noise. Moreover, Roadblock-qPCR directly measures the decline in preexisting mRNA levels rather than inferring it from multiple parameters, simplifying the analysis of mRNA decay kinetics.
Some aspects of Roadblock-qPCR could still be optimized. First, it is difficult to obtain accurate half-life estimates for mRNAs that are particularly stable, such as ribosomal protein mRNAs. This is partly because qPCR analysis requires a “reference” mRNA whose levels remain constant throughout the experiment. This is a safe assumption when queried mRNAs are much less stable than the reference mRNA. It is more problematic for long-lived mRNAs (half-lives > 10 h), as detectable levels of the reference mRNA may also begin to decline. Identification of other well-expressed reference mRNAs that are more stable that the ones used here (RPLP2 and GAPDH) would mitigate this limitation. An additional difficulty with measuring the stabilities of long-lived mRNAs is that 4sU becomes increasingly toxic over time. Whether this toxicity affects the stability of the mRNA of interest would need to be considered.
A second opportunity for improvement is to increase the RT drop-off frequency induced by NEM. Under the conditions described here, the reverse transcriptase must traverse multiple modified 4sU nucleotides to ensure robust drop-off. Although this is unlikely to be a problem for most mRNAs, it may interfere with the analysis of those that are particularly short or have low U content. Further analysis of other thiol-reactive compounds may identify modifications that more completely disrupt reverse transcription. Reverse transcriptases other than the one used here (Protoscript II from New England Biolabs) or altered reaction conditions may also improve rates of drop-off. Increased drop-off rates would not only improve the performance of Roadblock-qPCR as a targeted application. It may also be the basis of a transcriptome-scale approach. For instance, mRNA could be fragmented and then appended with both 3′ and 5′ linkers. The failure of the reverse transcriptase to traverse the entire fragment would produce fragments lacking the 5′ PCR primer binding site, and therefore fail to amplify. Fragments from newly transcribed mRNAs would therefore be masked from the resulting library.
In summary, Roadblock-qPCR offers a simple approach for directly measuring the stabilities of targeted mRNAs. It avoids the artifacts of “transcription shutoff” strategies, while reducing the technical complexity and expense of other approaches that use metabolic labeling. The low cost and ease of implementation make it an easily adoptable tool for investigators interested in the post-transcriptional regulation of mRNAs.
MATERIALS AND METHODS
Materials
Reagents were obtained from the following sources: 4-thiouridine (4sU), N-ethylmaleimide (NEM), Iodoacetamide (IAA), Actinomycin-D, and X-tremeGENE 9 DNA Transfection Reagent from Sigma-Aldrich; iTaq Universal SYBR Green Supermix from Bio-Rad; RNeasy Plus Mini Kit from Qiagen; Protoscript II Reverse Transcriptase, NotI-HF, and T7 RNA Polymerase from New England Biolabs; CellTiter-Glo from Promega; SUPERase•In RNase Inhibitor from ThermoFisher Scientific; RNA Clean & Concentrator-5 from Zymo Research.
Synthesis and modification of 4sU-containing RNA
A 121 bp sequence (see below) from human ATF4 was cloned into the pRK5 plasmid behind a T7 promoter using Sal1 and Not1 restriction sites. 5 µg of plasmid was then digested with 2 µL NotI-HF in 50 µL with 5 µL 10× CutSmart buffer and digested for 2 h at 37°C and purified by spin column. In vitro transcription was then performed in a reaction containing 2.5 µg plasmid, 10 µL 5× transcription buffer, 0.5 µL 1 M DTT, 1.5 µL SUPERase•In, 1 µL 100 mM ATP, 1 µL 100 mM GTP, 1 µL 100 mM CTP, 10 µL 10 mM UTP (New England Biolabs) or 4sUTP (Trilink Biotechnologies), and 2.5 µL T7 RNA polymerase at 37°C for 2 h. Following transcription, DNase I was added and incubated at 37°C for 15 min and RNA was then extracted with RNeasy columns.
Sequence of the synthesized RNA: 5′-GAAGGAGGAUGCCUUCUCCGGGACAGAUUGGAUGUUGGAGAAAAUGGAUUUGAAGGAGUUCGACUUGGAUGCCCUGUUGGGUAUAGAUGACCUGGAAACCAAGCCAGAAGACCAACAGACC-3′.
Modification of RNA with NEM and IAA
RNA was treated with 48 mM N-Ethylmaleimide (NEM; in ethanol) with 50 mM Tris-HCl (pH 8) and 1 mM EDTA at 42°C for 90 min, or 10 mM iodoacetamide (IAA; in 50% DMSO, 50 mM sodium phosphate buffer (pH 8.0)) for 15 min at 50°C. The reaction was stopped with 20 mM DTT. Modified RNA was then purified using a Zymo RNA Clean and Concentrator column and resuspended in 20 µL. A total of 6 µL RNA was reverse-transcribed using Protoscript II (NEB) according to manufacturer's instructions and a 6-FAM-labeled primer (5′-6-FAM- GGTCTGTTGGTCTTCTGGCT-3′). For PAGE-analyzed samples, RNA was additionally hydrolyzed using 2.2 µL 1 M NaOH for 20 min at 98°C followed by addition of 2.2 µL 1 M HCl to neutralize. A total of 10 µL of the reverse transcribed products was run on an 8% polyacrylamide denaturing gel. For fragment analysis, 6-FAM-labeled cDNA was analyzed using a Thermo Fisher 3730xl DNA Analyzer and Genescan Liz-500 size standard at the Yale Keck Biotechnology Resource Laboratory. Approximate fragment sizes were estimated using the Thermo Fisher Peak Scanner software included in the Thermo Fisher Connect suite.
Measurement of cell viability
Cells were seeded to a 96-well plate at 50,000 cells/well and grown overnight. 0–400 µM 4sU was then added to wells. Cell viability was tested 24 h later using the CellTiter-Glo (Promega) assay per the manufacturer's instructions except that 50 µL of reagent was used per well. Luminescence was measured using a Tecan Infinite M1000 plate reader.
Generation of HEK-293T cells stably expressing doxycycline-repressible reporter mRNA
HEK-293T cells were infected with a lentiviral construct encoding Renilla luciferase and controlled by a doxycycline-repressible (Dox-off) promoter. To produce virus, 2 million HEK-293T cells seeded in 6 cm plates were cultured in high-glucose DMEM medium supplemented with 10% (v/v) heat-inactivated FBS and penicillin-streptomycin overnight. The next day, cells were transfected with 0.1 µg VSVG plasmid, 0.9 µg psPAX2 plasmid, and 1 µg pCW57.1 TTA-Renilla luciferase plasmid using XtremeGene 9. Virus was harvested 48 h later, filtered, and used to infect 2 million HEK-293T cells. After 2 d of incubation, cells infected with virus were selected using 2.5 µg/mL puromycin for 2 d. Cultured cells were plated onto six-well plates at 0.5 million/well and grown overnight. The next day, cells were treated with 1 µg/mL doxycycline for 0, 2, 4, and 8 h and then harvested for total RNA isolation.
Analysis of mRNA levels by qPCR
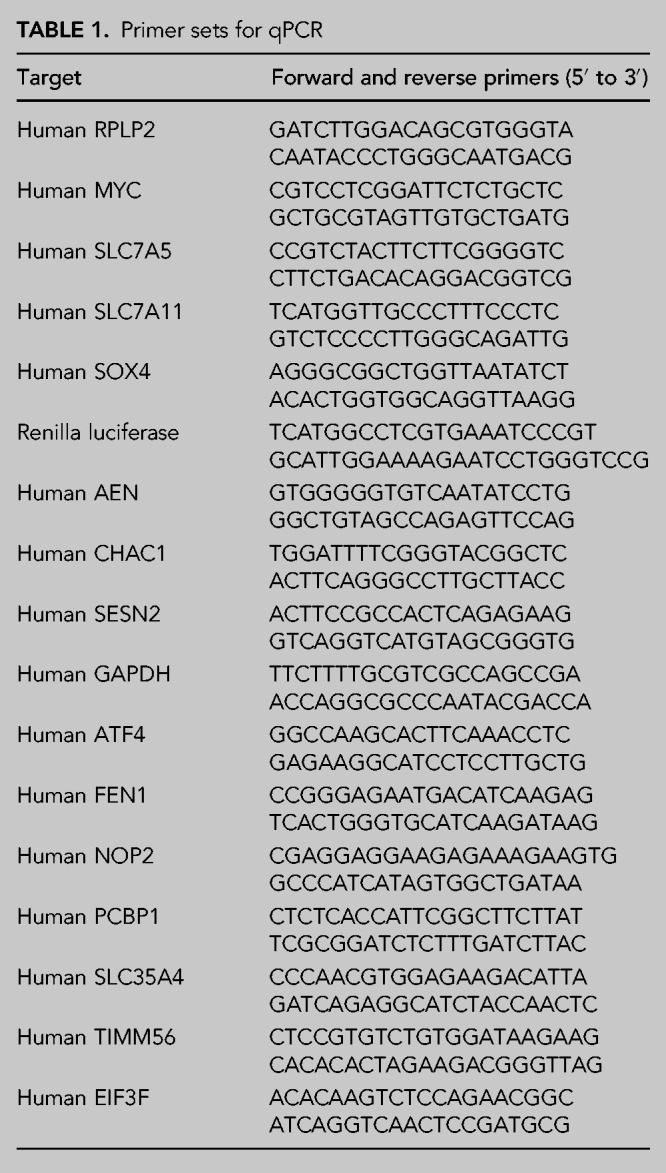
Total RNA was isolated using the RNeasy-Plus Mini Kit per the manufacturer's instructions. cDNA was synthesized using 1 µg RNA, oligo dT18, and Protoscript II reverse transcriptase per the manufacturers instruction and then diluted 1:10. We recommend using oligo dT18 for reverse transcription, as it is expected to maximize the likelihood of RT disruption and in practice yields more sensitive results. qPCR was performed using iTaq Universal SYBR Green Supermix using 300 nM of each primer and 100 ng cDNA (Table 1). Samples were analyzed using an Agilent Stratagene Mx3005P machine, with the following program: 2 min at 50°C; 2 min at 95°C; 40 cycles of 15 sec at 95°C, 20 sec at 58°C, 30 sec at 72°C. Ct values were calculated using the MxPro software set to the default parameters (auto-baseline). Relative mRNA levels were calculated using the ΔΔCt method normalized to either RPLP2 or GAPDH. Decay parameters were estimated by fitting the data to a single-phase exponential decay model using GraphPad Prism 8.4.1. When possible, qPCR primers should be chosen to target regions toward the 5′ end of the mRNA. This maximizes the likelihood that the reverse transcription of 4sU-containing mRNAs will terminate prior to traversing the region targeted by qPCR primers.
TABLE 1.
Primer sets for qPCR
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Lucas Philippe and Antonia van den Elzen for helpful discussions. This work was supported by the Yale Top Scholar Award (C.C.T.) and the National Institutes of Health (GM125955 to C.C.T.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.076885.120.
REFERENCES
- Airoldi EM, Miller D, Athanasiadou R, Brandt N, Abdul-Rahman F, Neymotin B, Hashimoto T, Bahmani T, Gresham D. 2016. Steady-state and dynamic gene expression programs in Saccharomyces cerevisiae in response to variation in environmental nitrogen. Mol Biol Cell 27: 1383–1396. 10.1091/mbc.E14-05-1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2018. Metazoan microRNAs. Cell 173: 20–51. 10.1016/j.cell.2018.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger K, Muhl B, Kellner M, Rohrmoser M, Gruber-Eber A, Windhager L, Friedel CC, Dolken L, Eick D. 2013. 4-thiouridine inhibits rRNA synthesis and causes a nucleolar stress response. RNA Biol 10: 1623–1630. 10.4161/rna.26214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolken L, Ruzsics Z, Radle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P, et al. 2008. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14: 1959–1972. 10.1261/rna.1136108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel CC, Dolken L. 2009. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol Biosyst 5: 1271–1278. 10.1039/b911233b [DOI] [PubMed] [Google Scholar]
- Friedel CC, Dolken L, Ruzsics Z, Koszinowski UH, Zimmer R. 2009. Conserved principles of mammalian transcriptional regulation revealed by RNA half-life. Nucleic Acids Res 37: e115 10.1093/nar/gkp542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog VA, Reichholf B, Neumann T, Rescheneder P, Bhat P, Burkard TR, Wlotzka W, von Haeseler A, Zuber J, Ameres SL. 2017. Thiol-linked alkylation of RNA to assess expression dynamics. Nat Methods 14: 1198–1204. 10.1038/nmeth.4435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SR, Lykke-Andersen J. 2013. Emerging roles for ribonucleoprotein modification and remodeling in controlling RNA fate. Trends Cell Biol 23: 504–510. 10.1016/j.tcb.2013.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield JA, Duffy EE, Kiefer L, Sullivan MC, Simon MD. 2018. TimeLapse-seq: adding a temporal dimension to RNA sequencing through nucleoside recoding. Nat Methods 10.1038/nmeth.4582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Schwalb B, Pirkl N, Maier KC, Schenk A, Failmezger H, Tresch A, Cramer P. 2013. Global analysis of eukaryotic mRNA degradation reveals Xrn1-dependent buffering of transcript levels. Mol Cell 52: 52–62. 10.1016/j.molcel.2013.09.010 [DOI] [PubMed] [Google Scholar]
- Tani H, Mizutani R, Salam KA, Tano K, Ijiri K, Wakamatsu A, Isogai T, Suzuki Y, Akimitsu N. 2012. Genome-wide determination of RNA stability reveals hundreds of short-lived noncoding transcripts in mammals. Genome Res 22: 947–956. 10.1101/gr.130559.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.