

Abstract
Allogeneic haematopoietic stem cell transplantation (allo-HSCT) was the first successful therapy for patients with haematological malignancies, predominantly owing to graft-versus-tumour (GvT) effects. Dramatic methodological changes, designed to expand eligibility for allo-HSCT to older patients and/or those with co-morbidities, have led to the use of reduced-intensity conditioning regimens, in parallel with more aggressive immunosuppression to better control graft-versus-host disease (GvHD). Consequently, disease relapse has become the major cause of death following allo-HSCT. Hence, the prevention and treatment of relapse has come to the forefront and remains an unmet medical need. Despite >60 years of preclinical and clinical studies, the immunological requirements necessary to achieve GvT effects without promoting GvHD have not been fully established. Herein, we review learnings from preclinical modelling and clinical studies relating to the GvT effect, focusing on mechanisms of relapse and on immunomodulatory strategies that are being developed to overcome disease recurrence after both allo-HSCT and autologous HSCT. Emphasis is placed on discussing current knowledge and approaches predicated on the use of cell therapies, cytokines to augment immune responses and dual-purpose antibody therapies or other pharmacological agents that can control GvHD whilst simultaneously targeting cancer cells.
ToC Blurb
Haematopoietic stem cell transplantation (HSCT) is a potentially curative treatment for several haematological malignancies. Improvements in HSCT methodologies have considerably reduced treatment-related morbidity and mortality, thus broadening eligibility and placing increased emphasis on the prevention of disease relapse. In this Review, the authors discuss approaches to dissecting the biology of HSCT and exploiting the biological insights to enhance the graft-versus-tumour response, in particular with adoptive cell therapies and other immune-directed therapies, whilst minimizing graft-versus-host disease.
Introduction
Allogeneic haematopoietic stem cell transplantation (allo-HSCT) and autologous HSCT (auto-HSCT) have been a cornerstone of cancer therapy, primarily for haematological malignancies, for three decades. Allo-HSCT and, to a much lesser extent, auto-HSCT are predicated on exploiting the graft-versus-tumour (GvT) effect, defined as an immune-mediated reaction by engrafted donor cells against tumour cells. The annual number of such transplantations continues to rise, with >8,000 allo-HSCT and >14,000 auto-HSCT procedures performed in the USA in 20181. In adults, the majority of allo-HSCTs are performed for the treatment of acute leukaemias, approximately two-thirds in patients with acute myeloid leukaemia (AML)1. Other major indications include myelodysplastic syndrome (MDS) or lymphoma (predominantly non-Hodgkin), and to a lesser extent, multiple myeloma (MM), chronic myeloid leukaemia (CML) and chronic lymphocytic leukaemia (CLL)1. With regard to auto-HSCT, two-thirds of recipients have MM; the remaining third mostly have non-Hodgkin or Hodgkin lymphoma1. Allo-HSCT for non-haematological cancers has also been evaluated, although with less robust responses observed2. HSCT is increasingly being offered to older patients, including a growing number aged >70 years1. This trend largely reflects improvements in supportive care, especially infection prophylaxis and treatment, as well as the development of reduced-intensity and/or non-myeloablative conditioning and improved immunosuppressive therapy3. Indeed, over the past three decades, non-relapse mortality (NRM) has been the greatest cause of mortality in the first 200 days after HSCT, and reductions in NRM are the predominant factor responsible for the improvements in outcome1,3. The risk of disease relapse has also declined, albeit less dramatically than NRM, over the past three decades; beyond 200 days after HSCT, relapse is now responsible for the majority (>50%) of deaths after allo-HSCT and >70% of deaths after auto-HSCT1,3. Thus, the dramatic changes in methodologies and indications for HSCT have resulted in the prevention and treatment of disease relapse becoming the major unmet needs in the field, focusing efforts to enhance the GvT effect without increasing the risk of graft-versus-host disease (GvHD).
The source of donor stem cells for allo-HSCT has also changed dramatically over the past 5 years, with haploidentical — that is, 50% human leukocyte antigen (HLA)-matched — family donors increasingly favoured over other alternative sources (for example, umbilical cord blood or HLA-mismatched unrelated donors), owing to the ease of access to such donors and the excellent outcomes achievable in the era of post-transplant cyclophosphamide (PT-Cy)-based immunosuppression4. Indeed, an acceptable donor stem cell source can be found for almost all patients. Age and co-morbidities no longer present strict limitations on eligibility for most patients. Granulocyte colony-stimulating factor (G-CSF)-mobilized peripheral blood stem cells (PBSCs) are used for most allo-HSCT procedures in adults (>85%)1. In paediatric patients, however, transplantation for nonmalignant conditions, typically immunodeficiencies, haemoglobinopathies, anaemias and congenital errors of metabolism, are proportionately higher5, with bone marrow as the preferred source of donor cells. This preference reflects the fact that PBSCs are associated with a higher rate of chronic GvHD (cGvHD, occurring primarily beyond 100 days of transplantation)6, which presents an unnecessary risk in most of these patients. Regardless of the source of donor cells, immunosuppressive therapies for both acute GvHD (aGVHD, occurring primarily within 100 days after transplantation) and cGvHD, probably impair the GvT effects of HSCT, a major issue complicating efforts to delay or prevent disease relapse.
In this Review, we provide an overview of the findings of preclinical and clinical studies that have informed our current understanding of the GvT effect. Emphasis is placed on current knowledge of the mechanisms of disease relapse after HSCT, allogeneic and autologous, and on immunomodulatory therapeutic strategies targeting these mechanisms, which include cell therapies, immunostimulatory cytokines and dual-purpose antibodies or other pharmacological agents that can enhance efficacy without compromising safety.
Antitumour immune effects of auto-HSCT
Auto-HSCT has traditionally been thought of as a means of enabling the use of high-dose (that is, supra-myeloablative) chemoradiotherapy, which can result in complete or near-complete eradication of the cancer cells and necessitates haematopoietic stem cell rescue. However, the effects of auto-HSCT likely extend beyond cytoreduction and lymphodepletion, owing to restoration of a balanced immune system. In MM, disruption of the bone marrow microenvironment after auto-HSCT is likely to have tumour-promoting immunological effects, supporting the exploitation of applying tumour-specific immunotherapies that may invoke T cell-mediated and MM-specific disease control by re-establishing a state of immune equilibrium7. T cell exhaustion and immune escape are implicated in disease relapse after auto-HSCT in preclinical models and patients with MM8,9 and can be countered through immune-checkpoint inhibition (ICI), especially co-targeting of the inhibitory T cell immunoreceptor with Ig and ITIM domains (TIGIT) and programmed cell death 1 (PD-1) pathways10. Understanding and harnessing the immunological properties of auto-HSCT is thus an area of increasing research interest. In addition, the period of profound immunodeficiency following auto-HSCT and states of low disease burden might also provide a window for application of adoptive cell immunotherapy (for example, with T cells engineered to express chimeric antigen receptors (CARs) that bind cell-surface antigens expressed on malignant cells; NCT03455972).
Antitumour immunity after allo-HSCT
Preclinical modelling of GvT effects.
This Review will not be a compendium of strategies that augment the GvT effect because the GvT response, in its truest sense of the tumour-specific effects of donor cells, has received far less attention than GvHD or the efficacy of direct antitumour therapies as the primary readout of preclinical models of allo-HSCT. Herein, we have focused on graft-versus-leukaemia (GvL) effects, which are the most extensively studied GvT responses. Moreover, allo-HSCT is most commonly used to treat patients with leukaemia — although, cell therapy approaches to the treatment of solid cancers are gaining considerable interest, in particular, with the advent of genetically engineered third-party or ‘off-the-shelf’ T cell products expressing CARs and T cell receptor (TCRs) targeting antigens associated with solid tumours.
The predominant preclinical models of allo-HSCT use inbred laboratory mice owing, in part, to low costs, short times to end points and broad accessibility to reagents and strains, as well as the potential for high levels of reproducibility between different laboratories11,12. Preclinical studies that exploit mouse models to examine the GvT effect usually involve engraftment of long-term propagated transplantable tumour cell lines followed by allo-HSCT consisting of both donor HSCs and additional T cells to induce GvHD. Reflecting the focus of most of these studies, however, investigations of the GvL effect are often performed only to ensure that the antitumour activity is not compromised by therapeutic interventions to minimize GvHD. Notably, however, fewer T cells are needed to mount specific GvT responses than result in GvHD; as such, preservation of the GvT effect might reflect the fact that a low threshold number of T cells might be sufficient for antitumour responses, rather than true selectivity of interventions to suppress GvHD (reviewed in13). Thus, preclinical models have been optimized for GvHD analysis, whereas GvL modelling is less developed, thus hindering clinical extrapolation of findings relating to the GvL effect.
In contrast to the clinical scenario, preclinical studies involve well-characterized tumour cell lines transplanted into inbred mice that are healthy, young, lean and are housed within specific-pathogen-free (SPF) facilities, all of which are likely to result in antitumour immune responses that do not accurately mirror those associated with the antigen-experienced immune system of patients with cancer (TABLE 1). Simply altering one preclinical variable, such as age or obesity status, has been demonstrated to result in markedly different outcomes and toxicities, including cytokine dysregulation14. Furthermore, incorporating human-modifying variables such as obesity can sometimes result in parodoxical but clinically validated outcomes, highlighting the importance of considering such variables in preclinical modelling15.
Table 1 |.
Key differences between preclinical modelling of allo-HSCT and the clinical paradigms
| Feature | Mice | Canines | Non-human primates | Humans |
|---|---|---|---|---|
| Genotype | Inbred (genetically homogenous), although genetic drift occurs within colonies and between vendors. Limited strain combinations exist or used, and grafts are rarely haploidentical and usually full MHC mismatched | Outbred, with substantial genetic variation between breeds. Haematopoietic stem cell transplantation (HSCT) studies usually involve laboratory beagles. Client-owned canines are used in studies of cancer, but these rarely involve HSCT. | Outbred, although colonies are often have some degree of inbreeding. | Outbred, requiring extensive HLA typing, with a preference for the use of related haploidentical donors. |
| Age | Predominantly very young (8–12 weeks old, equivalent to early adolescence in humans) | Variable age, often adult | Predominantly young adult | Variable age (heavily skewed to >50 years), depending on cancer type |
| Environment | Highly restricted and specific-pathogen free (SPF) in most facilities. | Similar environmental exposures to those of humans in client-owned canines, but more restricted in laboratory beagles. | Colonies are highly controlled but have similar environmental and pathogen exposures to those of humans. Cytomegalovirus-positive, unless from SPF colony. | Numerous pathogen exposures (acute, chronic and latent) and pre-existing diseases and morbidities |
| Recipient body mass index (BMI) | Predominantly young and very lean mice (20–25g) are used | Variable, but often lean in HSCT studies. | Variable, but not obese | Highly variable, with an increasing obesity population (BMI >30), including in paediatric patients |
| Cancer development and modelling conditions | Established rapidly growing tumor cell lines injected into healthy cancer-naive mice, often presenting with minimal tumour burden at the time of HSCT. | Spontaneous cancer (including lymphoma, sarcoma and melanoma) occurs, with age with breed variations; however, no reliable cancer models exist in laboratory beagles that are typically used for HSCT studies | Minimal cancer occurrence within colonies and often in old age, precluding ability to perform cancer treatment efficacy studies. Accordingly, allogeneic HSCT (allo-HSCT) studies in non-human primates are primarily used to investigate graft-versus-host disease (GvHD) and/or engraftment and immune reconstitution outcomes. | Progressive heterogeneous cancers patients with different tumour burdens, various prior treatments and diverse immune statuses |
| HSCT conditioning regimens | Predominantly total body radiation (lethal single or split doses) | Reduced-intensity chemotherapy conditioning regimens for HSCT studies in laboratory beagles. Client-owned cancer treatments nonmyeloablative as most veterinary facilities are outpatient. | Chemotherapy with or without total body irradiation, including non-myeloablative regimens, with anti-viral and GvHD prophylaxis | Chemotherapy with or without total body irradiation, including non-myeloablative regimens |
| HSCT use | Modelling of HSCT for human cancers, including xenogeneic transplant models (human into mouse) | Use of laboratory beagles to model allo-HSCT. HSCT is used in client-owned canines for treatment of spontaneous cancers. | Studies of HSCT, with particular regard to engraftment and immune reconstitution, as well as GvHD | Treatment of a variety of disease states ranging from cancer (predominantly leukaemias and lymphomas) to haematopoietic disorders. |
| GvHD development | Donor bone marrow used as source of haematopoietic stem cells (HSCs) but additional T cell sources and infusions are required to induce GvHD in SPF mice due lack of infectious agents that augment the GvHD processes. | Donor HSCs are sufficient to cause GvHD, depending on the degree of dog leukocyte antigen (DLA) compatibility; Donor lymphocyte infusion involving DLA-identical siblings could result in no GvHD. | Donor HSCs are sufficient to cause GvHD depending on MHC compatibility and recipient conditioning. | Adult HSC sources, even from related donors or cord blood, are sufficient to cause GvHD depending on the level of HLA compatibility and the conditioning regimen used, although non-myeloablative regimens reduce the risk of GvHD. |
| GvHD type | Usually either acute or chronic, crucially dependent on strain combination used (MHC mismatch or minor MHC mismatch model) and the type of conditioning. Restricted organ involvement in most models. | Acute, chronic or mixed (acute and chronic) | Acute, chronic or mixed, but predominantly severe acute GvHD, depending on MHC compatibility, the conditioning regimen used and the intensiveness of post-HSCT immunosuppressive treatment. | Various immunosuppressive prophylaxis strategies are used and GvHD can be mixed (acute and chronic), with delayed chronic GvHD becoming more prevalent. |
| Graft-versus-tumour (GvT) effect | Numerous studies of approaches to enhance GvT and limit GvHD, although the emphasis is on GvHD prevention or treatment and only short-term results. | Not evaluated | Minimal investigation; however, the immune and myeloid effects and possible off-target toxicities of regimens that could potentially be used to enhance GvT effects have been assessed | Emphasis has been to improve GvT either through adoptive cell therapies or improved immune reconstitution following HSCT, without exacerbating GvHD |
Full major histocompatibility complex (MHC) and multiple minor histocompatibility antigen disparate mouse strain combinations continue to be a mainstay of preclinical GvL models, owing to the fact that GvHD is readily titratable by varying T cell doses and that strains deficient in cytokine, co-stimulatory molecules and other relevant proteins are commonly available, enabling demonstration of true cause-and-effect relationships. Beyond the considerable species differences between the mouse and the human immune system, mouse models of GvHD commonly rely on a limited number of strain combinations associated with induction of a particular GvHD type or pathology, with only aGvHD or cGvHD (but less commonly both), which is not reflective of the frequently observed clinical presentation. Mouse allo-HSCT usually involves conditioning with total body lethal irradiation alone (that is, without chemotherapy), the use of bone marrow as a source of HSCs and typically requires the use of donor T cells (usually from a splenocyte source) to induce GvHD (TABLE 1). The limited range of transplantable mouse tumour lines available are highly aggressive and proliferate rapidly, such that only a minimal tumour burden is treatable. Other GvHD and GvT models consist of human T cells (and typically allogeneic human tumours from either established cell lines or primary tumour cells from patients) grafted into immunodeficient mice, which cause xenogeneic GvHD12. While advantageous in assessing interactions between human cells and the effects of experimental therapies in vivo, these models are limited by species-specific factors (for example, human TCRs cannot recognize mouse MHCs and many human cytokines to do not bind the relevant mouse cytokine receptors) and the unclear mechanisms underlying the pathogenesis of xenogeneic GvHD. Large-animal models, primarily in canines and non-human primates, are also used for studies of allo-HSCT, but predominantly for investigations of GvHD owing to high costs, the outbred nature of the animals that often limits the experimental control of MHC disparities, limited validated immune reagent availability and immune monitoring capabilities, and the lack of established GvT models in non-human primates (TABLE 1). These challenges further are coupled with difficulties in applying unproven or high-risk experimental therapies to client-owned canines with cancers and limited ‘in-patient’ facility capabilities or support (TABLE 1).
Thus, while SPF, inbred mice are the most informative means of initially assessing therapeutics for enhancing GvT activity, clinical GvT responses clearly need to be better reflected in these models, considering that patient outcomes are markedly affected by age, health status, obesity, previous therapies, immune system status and prior exposure to opportunistic pathogens, including latent cytomegalovirus (CMV) and Epstein–Barr virus (EBV), as well as cancer burden (TABLE 1). The clinical allo-HSCT paradigm involves various combinatorial radiotherapy, chemotherapy and immunosuppressive regimens chosen, in part, on the basis of the degree of HLA compatibility, stem cell source, patient age and co-morbidities, and cancer type. In contrast to cancer models involving relatively homogeneous tumour cell lines transplanted to healthy treatment-naive mice, human cancers arise over longer time periods and are heterogeneous with respect to tumour burden, utilization of immune suppressive or evasion pathways and treatment exposure prior to allo-HSCT. Prophylactic GvHD immunosuppressive agents profoundly affect immune cell and haematopoietic cell reconstitution in patients but are rarely applied in mouse models, although they are used routinely in large-animal models11,12. Donor HLA compatibility is crucial in clinical allo-HSCT, with preferred use of related donors, whereas many mouse models have full MHC mismatching on a background of allelic disparities for multiple different genes. These models are immunologically problematic for GvT activity, in which donor T cell responses against the tumour cells might be promoted by some degree of MHC compatibility with the recipient’s cells. Changes in the demographics of the population of patients undergoing allo-HSCT (that is, increasing numbers of older recipients, patients with obesity and those from minority populations with HLA disparate donors1) are likely to affect GvHD and GvT responses. Additionally, the microbiota has been demonstrated to markedly affect the outcomes of allo-HSCT and anticancer immunotherapy, thus warranting re-examination of appropriateness of the existing SPF mouse models, which also do not incorporate systemic antibiotics that alter the microbiota16. In addition, some clinically used immunomodulatory and/or immunosuppressive drugs present difficulties related to species-specific effects and might not be active in the absence of transgenic modification (for example, immunomodulatory imide drugs (IMiDs))17. Finally, some transplant-relevant malignancies (for example, EBV-driven lymphomas) and target antigens (such as the minor histocompatibility antigen HA-1 or NY-ESO) are uniquely human and very difficult to model in preclinical systems, thus necessitating studies in patients from the outset (TABLE 1).
GvL effects after allo-HSCT.
In the context of allo-HSCT, the observation of direct cytostatic or cytotoxic effects on malignant cells does not prove that the graft — rather that the allo-HSCT procedure itself — has anticancer activity, although immune-mediated GvT responses constitute the principal curative pathway associated with this therapeutic approach. Target antigens on leukaemia cells that are recognized by donor T cells are typically alloantigens, and are less frequently haematopoietic or leukaemia-specific antigens, and thus GvL activity is often associated with deleterious GvHD responses. Separating GvL from GvHD responses, while a principal therapeutic goal, has been difficult in clinical practice. With this in mind, the selective generation of GvL responses to recipient haematopoietic alloantigens is a highly attractive approach to avoiding off-target effects on other recipient tissues (that is, GvHD). A number of haematopoietic cell-restricted minor histocompatibility antigens have been identified (such as HA-1 and HA-2) and corresponding adoptive T cell therapies are under development18–20 (BOX 1). While targeting leukaemia and haematopoiesis-specific antigens reduces the risk of GvHD, preclinical modelling is needed to better understand the mechanisms initiating and maintaining GvL responses that are potentially distinct from GvHD and thereby develop strategies to promote immunity in bone marrow rather than parenchymal tissue.
Box 1 |. Potential approaches to augment and sustain GvT responses after allo-HSCT.
T cell-focused therapeutic strategies
Target malignant cells directly and/or enhance their susceptibility to graft-versus-tumour (GvT) responses, for example, using ibrutinib, venetoclax, sorafenib, 5-azacytidine or JAK1 and/or JAK2 inhibitor (such as ruxolitinib, baricitinib and INCB18424).
Promote T cell recognition of the recipient’s leukaemic cells and/or haematopoietic progenitor cells, for example, by infusing isolated cytotoxic CD8+ T lymphocytes (CTLs) specific to leukaemia-associated or haematopoietic cell-restricted minor antigen, such as Wilms tumour protein (WT1) or minor histocompatibility antigen HA-1, respectively.
Prevent T cell exhaustion via immune-checkpoint inhibition, for example, with monoclonal antibodies to programmed cell death 1 (PD-1), cytotoxic T lymphocyte protein 4 (CTLA-4), T cell immunoreceptor with Ig and ITIM domains (TIGIT) and/or lymphocyte activation gene 3 protein (LAG3), although with the potential increased risk of graft-versus-host disease (GvHD) in allogeneic haematopoietic stem cell transplantation (allo-HSCT), but not autologous HSCT (auto-HSCT).
Enhance antigen presentation and recognition using IFNα or IFNβ.
Deplete, incapacitate, destabilize or neutralize immunosuppressive immune cells, including host regulatory T (Treg) cells (for example, using the IL-2–diphtheria toxin conjugate, denileukin diftitox), myeloid-derived suppressor cells (MDSCs, using gemcitabine) and/or monocytes or macrophages (using anti-colony-stimulating factor 1 receptor antibodies).
Counteract cell-mediated immunosuppressive mechanisms in the tumour microenvironment, for example, using inhibitors of indoleamine 2,3-dioxygenase enzymes, transforming growth factor-β (TGFβ) or nitric oxide production, anti-IL 18 antibodies, or cyclooxygenase-2 inhibitors to suppress prostaglandin E2 signalling.
Selective depletion of α/βT cells from the donor graft to enrich donor γ/δT cells.
Infuse CTLs transduced with specific T cell recepters (TCRs) or chimeric antigen receptors (CARs) targeting particular tumour-associated antigen (for example, anti-CD19 CAR T cells for patients with B cell malignancies).
Infuse anti-viral CTLs with cross-reactivity to virus-induced malignancies, such as Epstein–Barr virus (EBV)-associated lymphoma
Administer bi-specific T cell engagers (BiTEs), such as the anti-CD19–CD3 BiTE blinatumomab
Vaccination with tumour-associated antigens, such the PR1 peptide derived from proteinase 3 and neutrophil elastase that are highly expressed in myeloid leukaemia blasts
Taper or stop treatment with GvHD prophylaxis agents, such as cyclosporin A
NK cell and NKT cell-focused therapies
Augment and/or accelerate donor natural killer (NK) cell reconstitution and recovery after allo-HSCT, especially via killer cell immunoglobulin-like receptor (KIR) and KIR ligand profiling of the donor and host, respectively, to ensure optimal mismatch in graft-versus-recipient direction, but also by using IL-15 superagonist or Toll-like receptor agonists that stimulate production of IFNα and IFNβ to augment NK cell function.
In conjunction with adoptive NK cell infusions, increase target recognition or sensitivity to cytolysis (for example, using bortezomib, sorafenib or lenolidamide), or through upregulation of NKG2D stress ligands and/or decreased MHC expression.
Disrupt tumour-intrinsic immunosuppressive pathways affecting NK cell function, for example, using immune-checkpoint inhibitors (as above outlined for T cells)
Genetically modify donor or third-party NK cells, for example, to delete MHC (HLA) class I/II genes and/or overexpress HLA-E, in order to reduce their immunogenicity and thus rejection, thereby leading to sustained engraftment.
Determine the optimal source of NK cell progenitor populations (immortalized NK cell lines, cord blood-derived NK cells, placenta-derived NK cells, induced pluripotent stem cell-derived NK cell or peripheral blood mononuclear cell-derived NK cells) resulting greater engraftment, survival and function.
Determine importance of inducing a memory phenotype (NKG2C+) to the activity of transferred NK cells and whether cytomegalovirus reactivation affects the development of memory NK cell and this protection from disease relapse.
NK cell engineering for expression CARs or TCRs to increase tumour targeting.
Use of tumour-targeting antibodies to mediate antibody-dependent cellular cytotoxicity (ADCC); use of NK cell engineering or use of enzymatic inhibitors, particularly deletion or inhibition of ADAM17, to render activated NK cells resistant to Fc receptor cleavage and thus more efficacious in ADCC.
Infuse natural killer T (NKT) cell subsets, either enriched or ex vivo expanded populations, or molecules that support NKT cells, such as α-galactosylceramide.
Administer bi-specific antibodies or tri-specific killer engagers (TRiKEs) to promote interactions between specific activatory receptors on NK cells (such as CD16) and particular antigens on tumour cells (such as CD19 and/or CD22)
In general, select patients with cancer types most susceptible to NK cell-mediated killing, such as AML and glioblastoma, and those with metastatic versus primary disease based on the inherent ability of NK cells to target and kill metastatic tumour cells.
MHC class I (MHC I)-dependent, CD8+ T cell-mediated GvHD is initiated by recipient haematopoietic antigen-presenting cells (APC)21, which activate and induce the differentiation of naive T cells from the donor22,23; no single APC subset (for example, dendritic cell (DCs), macrophages or B cells) is fundamentally required, indicating substantial functional redundancy24,25. Both MHC I-dependent and MHC II-dependent GvL responses require recipient haematopoietic APCs and cognate interactions with alloantigens on leukaemia cells26 (FIG. 1), while donor APCs are minimally required27,28. No convincing evidence suggests that any one specific subset of APCs is essential to initiate MHC I-dependent GvL responses. Whereas GvHD is dominantly caused by naive T cells, both naive and memory T cells, once primed by recipient APCs, can mediate GvL effects29. Recipient DCs can limit both GvHD and GvL in rodent models by promoting activation-induced cell death of alloantigen-specific donor T cells25,30. Non-haematopoietic APCs (for example, epithelial or stromal cells) seem to have a limited role in initiating MHC I-dependent GvL responses in mice, although they are associated with T cell exhaustion and thus impairment of GvL activity27,31. By contrast, non-haematopoietic APC are involved in the initiation of MHC II-dependent GvHD responses25,32 and, in addition to donor granulocyte–macrophage colony-stimulating factor (GM-CSF)-dependent DCs in the gastrointestinal tract, are an essential factor maintaining and amplifying GvHD33,34. Whether these tissue-resident APCs are important determinants of GvL effects requires further testing, particularly given the increasingly recognized importance of loss of MHC II expression on leukaemia cells at disease relapse35,36. Optimal antitumour immune responses require CD4+ T cell activation by MHC II-presented neoantigens in the tumour microenvironment, even in tumours without inherent MHC II expression on the malignant cells37 (FIG. 1). Myeloid malignancies in bone marrow usually have low expression of MHC II, and thus the role of MHC II-dependent antigen presentation within the bone marrow on GvL activity deserves increased attention in future research.
Fig. 1 |. Mechanisms of leukaemia cell recognition and killing after haematopoietic stem cell transplantation.
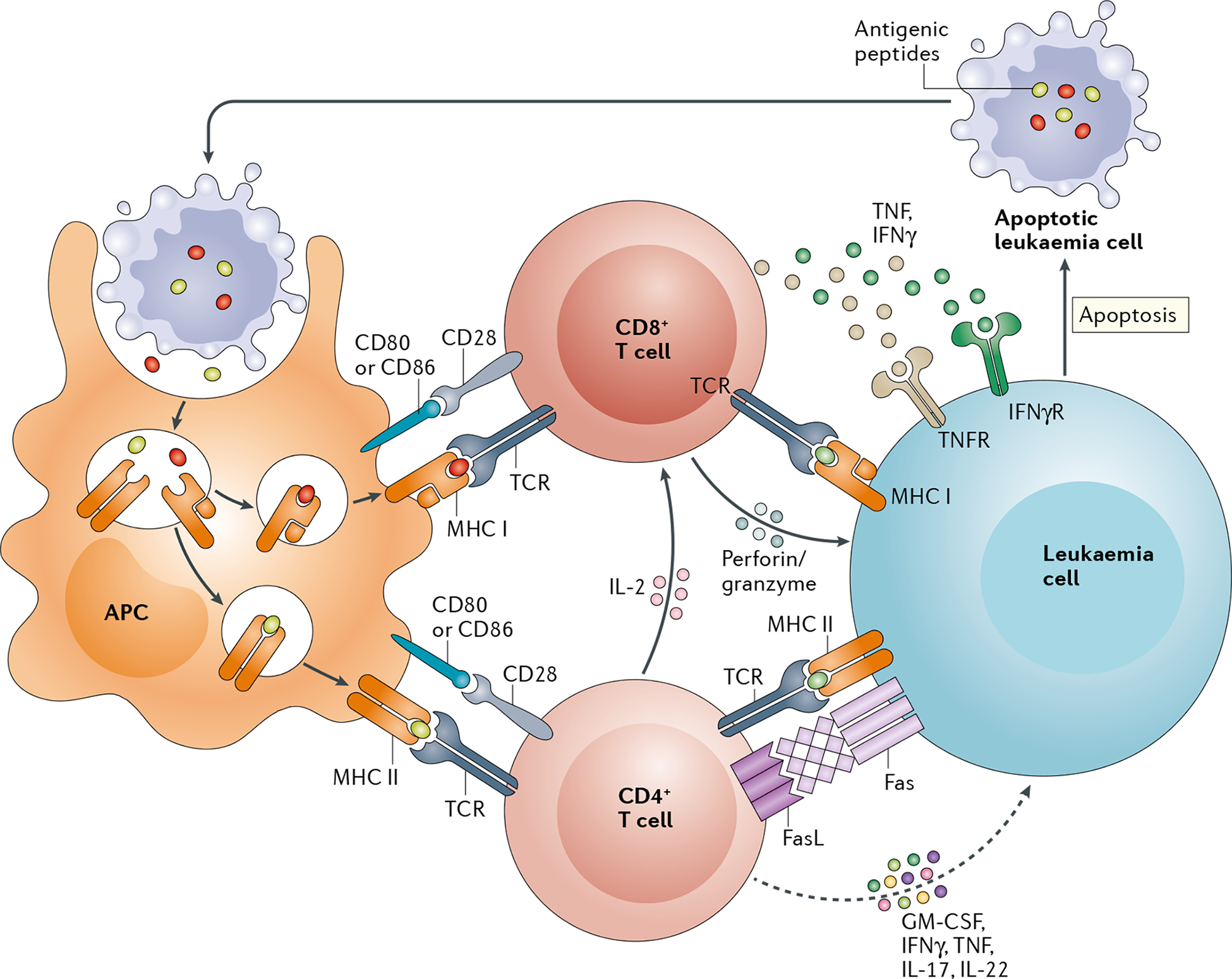
Early after transplantation, allogeneic and leukaemia antigens derived from apoptotic cells of the recipient are presented by host antigen-presenting cells (APCs) in the context of MHC class I (MHC I) and MHC II molecules. Interaction of these peptide–MHC complexes with T cell receptors (TCRs), in conjunction with CD80/CD86–CD28 co-stimulatory signalling, induces the activation and differentiation of donor CD8+ T cells and CD4+ T cells to generate effector T cell responses. These effector T cells mediate MHC I-dependent and MHC II-dependent graft-versus-leukaemia (GvL) effects via cytolytic pathways that involve perforin and granzyme and tumour necrosis factor (TNF) superfamily members, including Fas–Fas ligand (FasL) and TNF–TNF receptors (TNFRs). Secretion of IFNγ by effector T cells enhances antigen presentation by leukaemia cells and thus their recognition during the GvL response. GM-CSF, granulocyte–macrophage colony-stimulating factor; IFNγR, IFNγ receptor.
Antigen presentation by GvHD-initiating APCs is highly regulated by the intestinal microbiota, is absent in germ-free mice and is augmented in dysbiotic mice32,33, correlating with historical clinical data demonstrating that gut decontamination with broad spectrum antibiotics and protective isolation of patients has beneficial effects on GvHD outcomes in some settings38–40. Nevertheless, microbiota components, especially butyrate-producing anaerobic bacteria, can be protective against GvHD41. Associations between microbiota composition and disease relapse are now emerging42, warranting cause-and-effect testing and investigation of the immunological mechanisms in preclinical models.
Mechanisms of relapse after Allo-HSCT.
Disease relapse owing to GvL failure is mediated by tumour intrinsic and extrinsic mechanisms. Immune escape mechanisms (as opposed to leukaemic clonal evolution with acquisition of driver mutations) include loss of MHC expression, increased expression of immune-checkpoint ligands (such as programmed cell death 1 ligand 1 (PD-L1), CD155 or B7 molecules), secretion of inhibitory cytokines (for example, IL-10) and upregulation of immunosuppressive enzymes (including indoleamine 2,3-dioxygenase (IDO) 1 and/or 2, CD73 or CD79), all of which facilitate the malignant cells in evading GvL responses (reviewed in43) (FIG. 2). Concurrent GvHD might contribute to ineffective GvL responses because chronic exposure of T cells to alloantigens increases their expression of cognate immune-checkpoint molecules, such as PD-1 and/or TIGIT on the T cell31. Moreover, while secretion of IFNγ early after allo-HSCT enhances antigen presentation by leukaemia cells44, persistent cytokine secretion by alloresponsive donor T cells invokes immunosuppressive pathways, such as secretion of IDO enzymes and/or stromal and endothelial expression of PD-L1, thereby exacerbating T cell dysfunction31.
Fig. 2 |. Mechanisms of immune escape after haematopoietic stem cell transplantation.
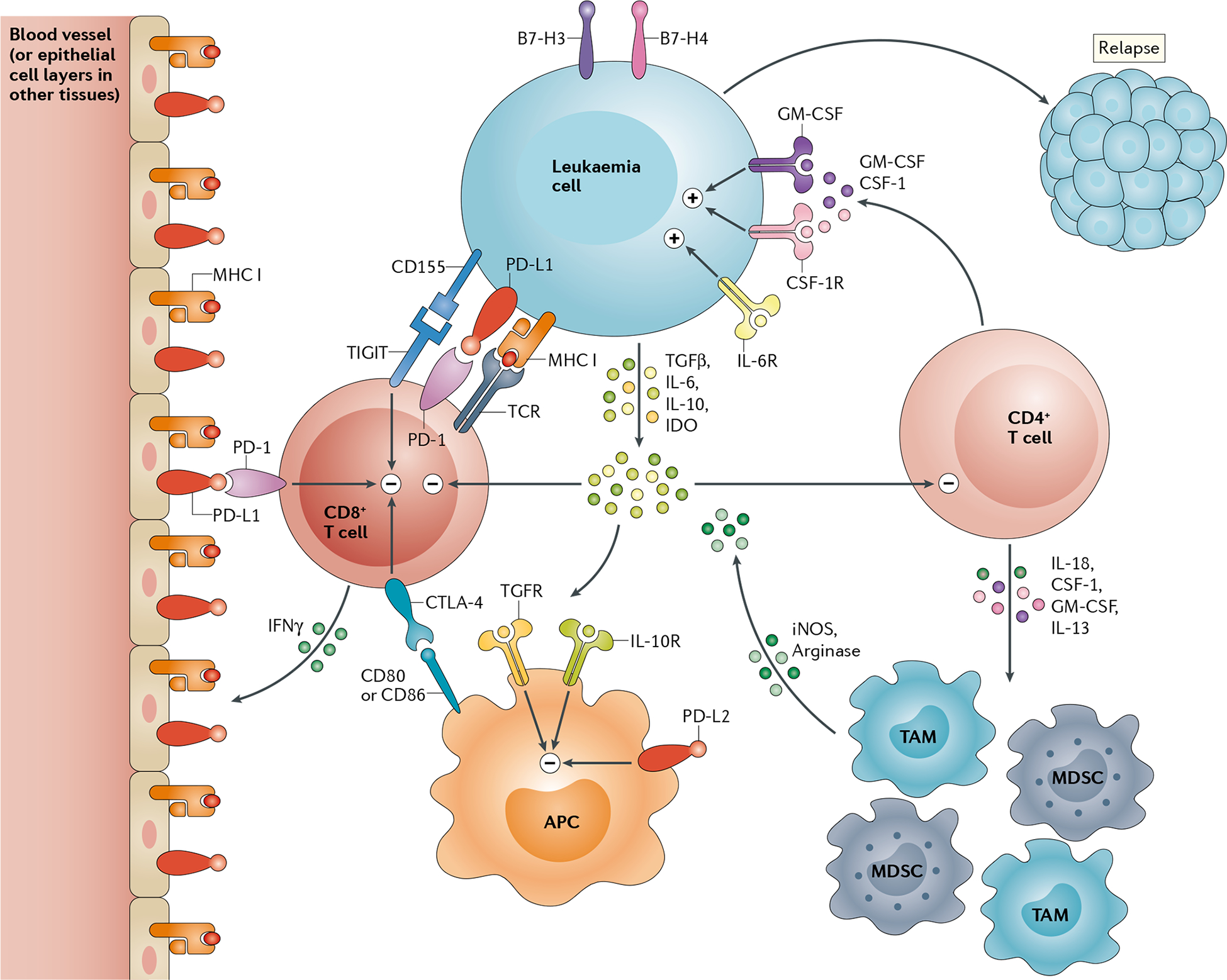
After transplantation, leukaemia cells can lose expression of MHC class II (MHC II) molecules and upregulate expression of inhibitory immune-checkpoint ligands, including programmed cell death 1 ligand 1 (PD-L1), CD155 (also known as poliovirus receptor) and B7–H3 and/or B7–H4. Leukaemia cells can also secrete soluble suppressive factors, such as transforming growth factor-β (TGFβ), IL-10 and produce indoleamine 2,3-dioxygenase (IDO) enzymes resulting immune suppressive metabolites that inhibit donor T cell and antigen-presenting cell (APC) function. Inflammation during an alloimmune response (particularly that mediated by IFNγ) induces MHC I-dependent antigen presentation and expression of immune-checkpoint ligands (such as PD-L1) on non-haematopoietic tissue (such as the endothelium) thus contributing to T cell exhaustion, manifested by broad activation of suppressive immune-checkpoint receptors (such as T-cell immunoreceptor with Ig and ITIM domains (TIGIT), programmed cell death 1 (PD-1) and cytotoxic T lymphocyte protein 4 (CTLA-4)) on these cells, and loss of effector function that prevent cytolysis of leukaemia cells. Inflammatory molecules secreted by stromal cells and CD4+ T cells, such as IL-6, IL-13, IL-18, granulocyte–macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (CSF-1), promote leukaemia growth directly and indirectly via the expansion of myeloid-derived suppressor cells (MDSCs) and tumour-associated macrophages (TAMs). These myeloid cells produce arginase and induce the secretion of nitric oxide synthase (iNOS) that further suppress T cell function and antigen presentation, culminating in immune escape and leukaemia relapse. CSF-1R, macrophage colony-stimulating factor receptor; GM-CSFR, granulocyte–macrophage colony-stimulating factor receptor; IL-6R, IL-6 recepter; IL-10R, IL-10 recepter; PD-L2, programmed cell death 1 ligand 2; TGFR, transforming growth factor-β receptor.
The presence of minimal (or measurable) residual disease (MRD) prior to allo-HSCT correlates with and predicts a higher rate of disease recurrence45. Whether high relapse rates reflect the residual disease burden or and inherently aggressive malignancy, residual leukaemia cells are likely to directly subvert GvL responses via the immunosuppressive pathways detailed above. Hence, optimal GvL activity would be initiated in an MRD-negative state, within the bone marrow, against haematopoietic-restricted antigens and in the absence of GvHD. Indeed, given that the induction of severe aGvHD or cGVHD is unacceptable in composite end points of allo-HSCT studies, approaches to enhancing GvL activity warrant rethinking with a focus on the mechanisms unique to the antitumour immune responses.
Modulating anticancer immunity post HSCT
In patients with haematological cancer, most cases of disease relapse after allo-HSCT or auto-HSCT occur within a year3; therefore, approaches to enhance GvL responses probably need to be initiated within the first 3 months of transplantation, for prevention of disease relapse, or at first detection of MRD. Alloreactivity is key to the GvL effect, and thus immunotherapy approaches are most tractable when alloreactivity is well controlled without substantial ongoing pharmacological immunosuppression. With regard to auto-HSCT, tumour immune escape seems to be driven largely by T cell exhaustion in response to the malignancy itself10,46. With allo-HSCT, approaches to modulating GvL activity are dependent upon augmenting donor T cell alloreactivity and are hindered by T cell-depleting agents47. Accordingly, deliberate use of lower than standard doses of cyclosporine, early or rapid cyclosporine withdrawal, or donor lymphocyte infusions can induce remission in patients with relapse of myeloid malignancies receiving immunosuppression, albeit with an increased risk of GvHD48,49.
ICI for the treatment of relapsed disease after allo-HSCT is problematic, given the propensity for induction of GvHD50, but might be tolerated when used prior to bone marrow transplantation with the use of post-transplant cyclophosphamide based immune suppression51. Cytokines such as type I interferons can enhance GvL activity (BOX 1), but do so at the risk of invoking GvHD52. Similar studies with type II interferon (IFNγ) are logical, owing to the roles of this cytokine in promoting antigen presentation by malignant cells, and their results are highly anticipated35,44. Alternatively, cytokines important in promoting GvHD whilst also providing survival signals to malignant cells, such as GM-CSF34 and macrophage colony-stimulating factor 1 (CSF-1)53 in AML and IL-6 in acute lymphocytic leukaemia (ALL) and MM54, are attractive as targets for neutralization after allo-HSCT.
After auto-HSCT, ICI focused on TIGIT and/or PD-1 might prevent immune escape without increasing the risk of GvHD10 (BOX 1). Moreover, combining T cell-directed ICI and inhibition of cytokine-signalling pathways by targeting, for example, CSF-110,53 or IL-1855 that support immunosuppressive cell populations in the bone marrow, including tumour-associated macrophages and myeloid-derived suppressor cells (MDSCs) (FIG. 2), presents a potentially synergistic strategy to prevent immune escape after auto-HSCT. Rigorous testing of this approach in preclinical models is required before its evaluation as a preventative strategy (as opposed to a treatment for relapsed disease) in clinical studies.
Adoptive cell therapy with T cells genetically modified to express CAR targeting specific tumour-associated antigens, such as CD1956 and BCMA57 on malignant B cells and plasma cells, respectively, is increasingly being deployed for the treatment of relapsed disease58,59. Notably, lymphodepleted and MRD-negative states occurring after auto-HSCT present an advantageous context for CD19-directed or BCMA-directed CAR T cell therapy for B cell lymphomas or MM, respectively (BOX 1). CAR T cells might also be useful prior to allo-HSCT, in order to generate MRD-negative states; however, transplantation might be counterproductive in patients with CAR T cell persistence, owing to CAR T cell depletion by allogeneic donor cells. Preclinical data from mouse models suggest that allogeneic donor-derived CD19-directed CAR T cells, at least those harbouring CD28 co-stimulatory domains, can be utilized after allo-HSCT without exacerbating GvHD, with the CAR T cells expressing alloreactive TCRs having enhanced stimulation relative to those without alloreactive TCRs, leading to clonal deletion of the alloreactive cells60. By contrast, allogeneic T cells expressing 4–1BB-co-stimulated CARs invoked GvHD60, likely necessitating safety measures to eliminate these CAR T cells, such as inducible, dimerization agent-induced caspase-9 suicide switches61.
Similarly, donor T cells genetically modified to express TCRs specific for leukaemia-associated or haematopoietic antigens, such as Wilms tumour protein (WT1)62 or HA-118, respectively, are being studied for the treatment of disease relapse after allo-HSCT, with promising initial findings — including in patients using WT1-targeted T cells. Antibody-based approaches to targeting WT1, and bi-specific T-cell engager (BiTE) antibodies (BOX 1) that link tumor associated proteins such as CD19 on malignant B cells to CD3 on T cells63, are also showing promise. Alternatively, a vaccine-based approach targeting the PR1 peptide derived from proteinase 3 has shown promise in patients with myeloid malignancies, some of whom had previously undergone allo-HSCT64. Notably, adoptive cell transfer of allogeneic EBV-specific T cells to treat EBV-driven malignancies, particularly post-transplant lymphoproliferative disorders and type II latency lymphomas occurring after allo-HSCT, is highly effective, without invoking GvHD, and is now an established standard of care65,66 (BOX 1).
A particularly appealing strategy to treat haematological disease relapse is predicated on the use of small-molecule inhibitors of intracellular signalling pathways or monoclonal antibodies (mAbs) targeting cell-surface antigens with the potential to simultaneously eliminate malignant cells and immune cell populations crucial for deleterious inflammation during GvHD (for example, JAK inhibitors could potentially be used to treat both myeloproliferative diseases and GVHD67) (BOX 1). Other examples include the anti-CD20 mAb rituximab, which is approved for the treatment of CLL and non-Hodgkin lymphoma and has also been used to prevent or treat cGvHD in the clinical setting68,69. Disrupting the Notch pathway using mAbs or γ-secretase inhibitors offers the possibility of both direct anti-leukaemia effects and aGvHD or cGvHD prevention and therapy70,71. Small-molecule inhibitors targeting proteins upregulated or mutated in malignancies, such as BCR–ABL1 in Philadelphia chromosome-positive CML or ALL, FLT3 in AML, BCL-2 in CLL, ALL or AML, and JAK2 in myelofibrosis, are attractive maintenance therapies that can sensitize malignant cells to GvT effects by deprivation of crucial growth signals and/or disruption of pathways involving in resistance to immunological killing. For example, the multi-protein kinase inhibitor sorafenib, which can target the FLT3 internal tandem duplication (FLT3-ITD) associated with a subtype of AML, induces GvL responses possibly via stimulation of IL-15 production by leukaemia cells72 (BOX 1).
Certain kinase or proteasome inhibitors used for cancer therapy have been explored specifically for GvHD prevention and therapy. Ibrutinib targets Bruton tyrosine kinase (BTK) and inducible T cell kinase (ITK) expressed in B cells and T cells, respectively, has direct anticancer effects in patients with CLL and some lymphomas (including small lymphocytic, marginal zone and mantle cell lymphomas) and has also been granted FDA approval for cGvHD therapy73,74. Splenic tyrosine kinase (SYK) inhibitors, which also suppress B cell receptor signalling and have thus been proposed as treatments for CLL and lymphoma, and proteasome inhibitors such as bortezomib, which are approved for the treatment of MM, increase the susceptibility of tumour cells to immune elimination and are in clinical use or testing for cGVHD therapy75–78. Ruxolitinib, a JAK1 and JAK2 inhibitor, is approved for the treatment of myeloproliferative disease (that is, myelofibrosis) and for aGVHD therapy67, and PI3Kδ inhibitors that are approved for patients with CLL or certain lymphomas also have activity in experimental models of GvHD79. These agents offer opportunities to prevent or treat disease relapse after allo-HSCT by directly targeting key survival pathways and indirectly by diminishing the immunosuppression resulting from agents used for GvHD prophylaxis or therapy. However, use of these pharmacological inhibitors, especially prior to a clear GvL response, might complicate interpretation of whether elimination of the disease reflects the contributions of these agents to a true GvL effect or, alternatively, their direct antitumour activity.
Treg cells in GvHD and GvL effects
Thymus-derived CD4+ regulatory T (Treg) cells, which co-express CD4, CD25 and the master transcriptional regulator FOXP3, are essential in maintaining immune homeostasis and inhibiting alloresponses80–83. Infusion of ex vivo expanded Treg cells in high numbers at the time of allo-HSCT, or freshly isolated donor Treg cells in lower numbers several days before donor T cells are transferred, permits in vivo Treg cell expansion in a lymphopenic environment, heightening Treg cell-mediated suppression of GvHD in mice81–83 and patients84,85. Early preclinical studies revealed that Treg cell administration does not overtly dampen GvL responses or lymphoma-cell cytotoxicity by the engrafted mature allogeneic T cells86,87, with only a modest reduction in cytotoxicity against P815 mastocytoma cells reported71. Third-party human Treg cells also have demonstrated efficacy in suppressing GvHD in xenogeneic HSC recipients88 and allogeneic Treg cells reduced the incidence and severity of GvHD in patients undergoing allo-HSCT84. In immunodeficient mouse models, the combined use of clinical-grade ex vivo-expanded Treg cells together with conventional T (Tconv) cells suppressed GvHD while retaining GvL activity against human AML, Philadelphia chromosome-positive ALL and Burkitt-like cell lines88. Interestingly, human Treg cells have been shown to have direct cytotoxicity against glioblastoma cells via the granzyme–perforin pathway in vitro, which can be augmented when combined with bi-specific T cell-engaging antibodies89. Intriguingly, Treg cells can also use cytolytic pathways, such as perforin and granzyme A, to kill autologous immune cells90, which could potentially be exploited for not only direct tumour-cell lysis but also control of immune responses. Thus, infusion of antigen-specific or CAR-expressing Treg cell together with allo-HSCT might not only reduce GvHD and any accompanying immune suppression, but also amplify GvL effects by focusing cytolytic Treg cells responses towards relevant target antigens. Nevertheless, the typically high sensitivity of leukaemia cell lines, especially those derived from mice, to T cell-mediated cytotoxicity and thus GvL effects is a caveat in extrapolating these data to the clinical setting. Furthermore, the finding that GvL effects can be observed in patients without preceding or concurrent GvHD has led to the plausible hypothesis that fewer functional T cells are required for eliminating mouse leukemia cell lines than are needed to cause GvHD of sufficient magnitude to manifest clinically (reviewed in13).
Despite the theoretical concern that the immunosuppressive activities of adoptively transferred Treg cells might lead to increase rates of disease relapse, the results of a study in patients with high-risk ALL (n = 33) or AML (n = 10) indicate significantly lower relapse rates with this approach compared to 114 historical controls (5% versus 21%; P = 0.03), probably owing to superior lymphoid reconstitution and reduced GvHD-induced and/or GvHD treatment-induced immune suppression91. These findings contrast with reports from studies of AML that Treg cell-depletion improves anti-leukaemia cytotoxic T cell responses in mice with established AML92 and that the abundance of Treg cells is inversely correlated with the responsiveness of AML to induction chemotherapy in patients with active disease93, and were observed despite the fact that bone marrow, a predominant site of leukaemia, serves as a Treg cell reservoir94. Differences between the predominant sites of Treg cell trafficking (for example, bone marrow, peripheral blood or lymphoid organs) and metastasis (for example, central nervous system, skin, lung, liver or reproductive organs) might account, in part, for a reported low relapse rate in the aforementioned patients with high risk ALL or AML95.
A second major type of Treg cells comprise induced Treg (iTregs) cells, which are generated from Tconv cell in vitro or can be induced in vivo in the context of low-dose or tolerigenic antigen exposure. iTreg cells generated in mice in vivo after allo-HSCT contribute to ameliorating GvHD96,97, with several reports indicating higher frequencies of CD8+ versus CD4+ iTreg cells96,98. Polyclonal or antigen-specific CD4+ iTreg cells can suppress allogeneic99,100 and human xenogeneic GvHD101 in mouse models. By comparison, CD8+ iTreg cells seem to have a modestly reduced capacity to protect against GvHD, but provided superior GvL effects102. Indeed, CD8+ iTreg cells can overcome CD4+ iTreg cell-mediated abrogation of GvL activity, and the highest degree of GvHD prevention and GvL preservation was observed when both of these cell populations were administered together102.
In lineage-tracing studies in mice, natural thymus-derived Treg (tTreg) cells were seen to lose stability — reflected in loss of FoxP3 expression — in an inflammatory environment, at frequencies ranging from 4% to 15%103,104, with consequent loss of immunosuppressive capacity. These ‘ex-Treg’ cells acquire a pro-inflammatory phenotype and function and can, therefore, contribute to autoimmunity103 and perhaps also to a GvT effect. In contrast to tTreg cells, iTreg cells lack a locked-in gene-expression signature of five transcription factors (Eos, Irf4, Gata1, Lef1 and Satb1) needed for stabilization of FoxP3 activity105. Additionally, methylation of an evolutionary conserved CpG-rich element in the first intron or the conserved non-coding sequence 2 (CNS2) region of FoxP3 renders iTreg cells susceptible to becoming ex-Treg cells. Inflammatory factors can destabilize tTreg cells, iTreg and FoxP3− T regulatory type 1 (TR1) cells in the context of mouse GvHD106, with IL-1 and IL-6 having similar effects on human tTreg cells in the context of xenogeneic GvHD107, which could potentially promote GvT responses. In another mouse model, deletion of the one or both alleles of Card11 in a fraction of tumour-infiltrating Treg cells, thereby disrupting the CARD11–BCL10–MALT1 signalosome complex, was sufficient to increase IFNγ production by these cells, activate macrophages and upregulate MHC I and PD-L1 expression on tumour cells, while avoiding systemic autoimmunity108; when this approach was combined with PD-1-targeted ICI, the mice were able to reject tumours108. By contrast, phenotypic and functional stabilization of iTreg cells has been demonstrated with retinoic acid109–111 and vitamin C112–115, and the stability of these cells is dependent on the chromatin-modifying enzyme EZH2 (potentially providing opportunities to enhance GvT effects using EZH2 inhibitors)116,117. Notably, stabilization of CD8+ iTreg cells does not necessarily lead to impaired GvT responses: GvL activity was retained with vitamin C-treated115 or Jak2-deficient Treg cells118, despite markedly increased levels of FoxP3 expression in these cells compared with untreated or wild-type Treg cells, which is consistent with a higher level of Treg cell stability.
Insufficient published clinical data exist to discuss the effects of IL-10, IL-5, IFNγ and TGFβ producing CD4+ FoxP3− TR1 cells on GvL responses. However, in preclinical studies, TR1-like cells generated by engineering allospecific CD4+ T cells to overexpress IL-10 were cytolytic in a granzyme B-dependent but antigen-independent manner that also required intracellular adhesion molecule 1 (ICAM-1) and nectin-2 (also known as CD112, a modulator of T cell signalling) expression on CD13+ myeloid leukaemia cells119. When transplanted together with peripheral blood mononuclear cells in humanized mouse models, these TR1 cells suppressed xenogeneic GvHD while retaining and contributing to GvL responses119.
On the basis of these findings, it is would highly premature to conclude that Treg cells have no adverse effects on GvL efficacy. Indeed, GvL activity is not uniformly retained upon adoptive transfer of Treg cells in preclinical models87; however, preliminary clinical results with this approach provide encouraging evidence of GvHD suppression without demonstrable evidence thus far of increased relapse rate85,98.
NK cells in GvT responses
NK cells were originally characterized as a radioresistant lymphoid cell population mediating the spontaneous rejection of bone marrow but not solid tissue allografts in lethally irradiated, unsensitized or naive mice120. This rejection occurred even in strain combinations in which irradiated F1 hybrid recipients rejected parental bone marrow cell allografts in a phenomenon termed ‘hybrid resistance’, which was at variance with the classical laws of transplantation, owing to co-dominant expression of MHC121. Subsequently, NK cells were found to be components of the innate immune system that notably mediate spontaneous MHC-unrestricted killing of transformed or virally-infected cells and to lack classical antigen-receptor gene rearrangements with associated diversity and specificity. Unlike T cells, NK cells do not seem to expand clonally and are not considered long-lived, being generated in the bone marrow throughout life, also contrary to the declines in T cell generation in the ageing thymus122,123. The features distinguishing NK cells (innate immunity) from T cells (adaptive immunity) have become blurred owing to their use of common pathways and receptors, as well as the recent identification of potentially long-lived adaptive or memory-like NK cell populations122,124. Human NK cells lack expression of CD3 but express CD56 as well as multiple NK cell-associated receptors, including members of the NKG2 C-type lectin receptor and killer-cell immunologlobulin-like receptor (KIR) families, that can directly bind to MHC and MHC-like molecules, resulting in potent stimulation or inhibition of NK cell activity depending on the nature of the receptor signalling motifs123,125. NK cells also express low-affinity immunoglobulin γ Fc region receptor III (FCγRIII, also known as CD16) and, therefore, participate in antibody-dependent cellular cytotoxicity (ADCC)123,126; whether this characteristic can be exploited clinically is currently being tested by administering NK cells in combination with tumour-targeting mAbs, such a cetuximab (NCT02507154 and NCT03319459) or rituximab (NCT02782546 and NCT0038994) (BOX 1). After NK cells have become activated either through exposure to stimulatory cytokines, such as IL-2, IL-12, IL-15, IL-21 and others, or by engagement of activating receptors such as NKG2D, their cytotoxicity mechanisms are augmented123; NK cells might also undergo a transition to ‘memory-like’ or ‘adaptive’ phenotypes resulting in increased longevity and enable prolonged and rapid-recall responses with the production of a wide array of cytokines and/or chemokines, notably IFNγ and GM-CSF122,124. This memory phenotype is most associated with the role of NKG2C+ NK cells in responses to virally infected cells in individuals with CMV126,127, and the generation and use of memory NK cell populations might result in greater efficacy after adoptive transfer (BOX 1). Given the sizable population of poorly cytolytic CD56hi NK cells in lymph nodes, the dominant effector functions of NK cells might actually be mediated through immunoregulatory cytokines or direct killing of T cells123,127. Paradoxically, NK cells with inhibitory receptors to ‘self’ MHCs develop with increased activation thresholds, possibly as a means to protect against (termed ‘licensing’, ‘education’ or ‘arming’)125, which has stimulated intense investigation on whether this difference in activation is important in responses against cancer in the context of allo-HSCT or after extensive ex vivo activation and expansion.
NK cells seem to be prime candidates to mediate GvT responses after allo-HSCT, owing to their principal localization in haematological tissues, their limited ability to directly attack solid tissue allografts thus being unlikely to initiate GvHD, the fact that they are the first lymphoid cell to reconstitute after HSCT, and their well-described preclinical ability to directly target and kill leukaemia and metastatic tumour cells128. In mouse models of allo-HSCT, additional engraftment of IL-2-activated allogeneic donor NK cells inhibited GvHD, reflecting competition with and suppression or even killing of activated T cells from the graft128,129, or possibly suppression of recipient DCs that fuel GvHD reactions of the donor T cells130, whilst also providing a GvL effect128,130. On extrapolation to clinical studies, adoptive NK cell transfer before or after allo-HSCT caused no marked exacerbation of GvHD131,132, except in one study with donor memory-phenotype NK cells transfer following T cell-depleted allo-HSCT133, suggesting that NK cell culture and activation conditions might be pivotal in determining GVHD effects133–136. Owing to the ability of NK cells to produce numerous pro-inflammatory cytokines, exacerbation of GvHD in T cell-replete grafts will remain a concern associated with infusion of donor NK cells (especially if cytokines such as IL-2 or IL-15 are also administered), whether or not they induce or simply amplify the T cell-driven pathological processes. To date, adoptive cell therapy with donor-type or third-party activated NK cells administered either prior to or following allo-HSCT has been associated with encouraging antitumour effects in paediatric patients with MDS or AML, resulting in both objective responses and reductions in the abundance of high risk clones132; other studies in adults have demonstrated objective responses with negligible toxicities or GvHD131,137,138. Questions still surround the limited persistence of the NK cells after transfer in HSCT, which might in part be attributed to both their cytokine dependence and limited clonal expansion capabilities136, as well as whether the GvT responses occurring after HSCT are solely due to the transferred NK cells.
Preclinically, important species-specific differences in NK cell biology preclude direct extrapolation of findings in mouse models to humans. Notably, the MHC-binding molecules in humans (KIR) and mice (Ly49) are structurally distinct and, as opposed to the dominant presence of CD56hi NK cells in human lymph nodes, mouse NK cells (which also lack expression of CD56) are absent from lymph nodes unless activated123,126. However, an important similarities exist: NK cells of both species are highly diverse, have similar MHC recognition and functional pathways and belong to multiple subpopulations capable of exerting diverse immunological effects123,126.
With regard to clinical application in cancer and HSCT, similar to highly activated T cells, NK cells are capable of mediating considerable ‘off-target effects’ and toxicities, particularly after prior activation. For example, findings from preclinical studies suggest that vascular leak syndrome associated with high-dose IL-2 is attributed, in part, to NK cell responses against endothelial cells expressing NKG2D ligands139. NK cells constitutively express NKG2D, a major activation and recognition receptor for induced self-proteins of the MIC and RAET1/ULBP families that are often upregulated on stressed, rapidly proliferating, infected or neoplastic cells. NKG2D is also expressed by activated T cells and, in this context, has been implicated as mediator of GvHD, but this pathway also likely contributes to GvT effects140. When used to target cancers via NKG2D-CAR T cells, off-target toxicities resembling cytokine-release syndrome occurred in mice139. Thus, broad antitumour effects might be achieved by exploiting NKG2D or even NK cells themselves, although potentially severe toxicities might occur and warrant caution in clinical studies particularly when given with immunostimulatory cytokines and after cytoreductive conditioning of the recipient.
Considerable variability in the contribution of NK cell to the GvT effect has been noted across different haematological malignancies. Among leukaemias, AML has by far the strongest evidence of NK cell-mediated GvL activity, and allo-HSCT is common in patients with this disease, thus generating intense interest in exploiting NK cells in this setting (BOX 1). A pivotal study galvanized the field with the observation that donor–recipient KIR ligand-mismatching during allo-HSCT resulted in substantially greater protection from disease relapse in patients with AML but not adult patients with ALL130. Subsequently, multiple clinical studies revealed similar associations of KIR and KIR ligand incompatibility (that is, the presence or absence of activating KIR ligands on the recipient cells that are capable of binding inhibitory donor NK cell KIRs) can result in greater donor NK cell alloreactivity and GvT141–145. The complexity of these processes are attributable to the inherent complexity of NK cell themselves: NK cells exist as multiple subsets expressing different KIRs, with some being activating and others inhibitory. In addition to KIR on NK cells, other inhibitory molecules, notably NKG2A which is present on activated T cells and resting NK cells, binds HLA-E, impacting NK licensing as well as anti-tumor activities and likely will be an important therapeutic target146. With regard to allo-HSCT, intense interest has been focus on delineating the optimal KIR expression pattern in donors to generate the greatest GvT benefit. Certain KIR combinations do result in improved outcomes, as highlighted in studies using donors with a KIR B haplotype and/or KIR2DS1 genotype, which resulted delays in disease relapse in patients with AML, as well as patients with MM, CLL, lymphomas or even paediatric ALL141–144,147. The not insignificant differences in allo-HSCT protocols between studies have made comparisons difficult, not least because the impact of such differences on the effectiveness of donor NK cell reconstitution can be considerable; however, the associations indicate that application of KIR–KIR ligand-typing protocols to allo-HSCT could be beneficial (BOX 1). Surprisingly, efforts to build on and further augment GvT activity in appropriately mismatched donor–recipient pairs by concurrently promoting or augmenting donor NK cell recovery and/or activation following allo-HSCT have largely been unsuccessful148, and thus the mechanisms underlying the reduced relapse with KIR–KIR ligand mismatching, and how to further exploit this process, are unclear. The complexity in expression of different KIRs in various NK cell subsets and the use of highly activated NK cell products, which might over-ride the effects of the inhibitory KIRs, raise questions regarding the overall ‘net’ impact that these receptors have on GvT responses, especially considering that downmodulation of MHC expression often occurs during cancer progression. Interestingly, similar to the observations relating to KIR expression and GvT effects, intriguing associations exist between with KIR expression status and the efficiency of ADCC, notably with KIR B haplotype donors. For example, differences in KIR expression have been associated with differences in the efficacy of NK cell-mediated ADCC with dinutuximab therapy for neuroblastoma149 indicating that multiple effector functions may be impacted by KIR/KIR ligand interactions. These observations suggest that KIR–KIR ligand typing combined with use of expanded adaptive or memory-like NK cell populations could enhance responses to cell therapies, especially if combined with mAb to induce ADCC (BOX 1). This approach is under clinical investigation using antibodies to a range of targets, including cetuxmimab, transtuzumab, nimotuzumab and rituximab (NCT0398097, NCT02507154, NCT03554889 and NCT00383994).
In contrast to haematopoietic malignancies, clinical NK cell-mediated GvT responses to solid tumours are less robust. NK cell responses to glioblastoma150 and neuroblastoma151 have been correlated with better disease outcomes. However, these associations might be contingent on not only the tumour type and the compatibility of the tumour sites with NK cell trafficking and access, but also how stringently the NK cells were assessed and characterized (NK cells share many markers and properties of T cells). In addition, tumour cell subpopulations might have differential sensitivity to NK cells. For example, multiple reports indicate that NK cells can mediate selective cytotoxicity against various solid cancer cells with a cancer stem cell (CSC) or tumour-initiating cell phenotype, owing to low levels of MHC expression with concurrent NKG2D ligand expression within these cells152,153, which could potentially be exploited clinically. This preferential targeting pattern does not hold true for all cancers, however, given a report that the CSC phenotype in AML is associated a lack of NKG2D ligand expression, which was also correlated with a lower remission rate and an increased risk of relapse in patients with AML154.
Evolutionarily, NK cells have a strong association with protection against viruses, particularly CMV and other herpes viruses; viruses and NK cells have evolved specific evasion and recognition strategies, respectively, highlighting the importance of this relationship123. Clinical studies have revealed that CMV reactivation following allo-HSCT results in increased numbers of activated and memory-like, NKG2C+ NK cells that are correlated with a low risk of disease relapse155,156; however, the mechanisms underlying the effect of CMV reactivation on NK cell recovery and possible GvT responses mediated by these cells remain unclear. Similar to CMV reactivation, greater expansion of memory or adaptive-phenotype NK cell populations, defined either by expression of NKG2C or by transcriptional profiles similar to memory T cells, after allo-HSCT of CMV-negative grafts has been associated with a reduced relapse risk and seems to be determined by host factors (that is, the frequency of these cells mirrors the pattern of NK cell diversity in the recipient prior to transplantation)157. Whether adoptive transfer of adaptive NK cells generated ex vivo, or promotion of their de novo development following allo-HSCT, results in similar effects on cell persistence and GvT activity remains to be determined and is currently under clinical evaluation (NCT01898793 and NCT0289092).
IL-15 is a key cytokine necessary for the development and survival of NK cells123. Importantly, IL-15 is unique in its requirement for trans-presentation, usually by a myeloid cell, for optimal triggering of IL-15 receptor signalling in the NK cell, explaining why optimal signalling and improved efficacy of systemically administered IL-15 is typically achieve though its conjugation with regions of the IL-15 receptor alpha subunit (creating IL-15 ‘superagonists’). Given the relative rarity of NK cells among the haematopoietic cell populations used for HSCT, various ex vivo culture expansion regimens are used needed for clinical application of NK cells and usually involve combinations of cytokines (for example, IL-21, IL-15, IL-2, IL-12 or IL-7) and/or feeder cell lines comprising IL-21-transduced tumour cells126. Unfortunately, upon continuous activation and expansion, like T cells, NK cells upregulate various inhibitory molecules and can possibly become anergic158. For example, increased expression of the immune-checkpoint proteins TIGIT, PD-L1 and lymphocyte activation gene 3 protein (LAG3) results in inhibition of NK cell function, which can potentially be reversed through ICI13,159,160,161, thus, these mechanisms are attractive targets for combination therapies (BOX 1). Many of these proteins are also expressed by T cells and, therefore, ICI following allo-HSCT might result in exacerbation of GvHD; nevertheless, such approaches to circumventing NK cell exhaustion and/or anergy in order to enhance their persistence and function have reached clinical testing162 (NCT03937895, NCT039588097 and NCT02118285). In addition, use of Toll-like receptor agonists (for example, targeting TLR9) is currently under clinical investigation (NCT02452697). Targeting of other pathways that directly augment NK cell function is also being pursued. For example, SLAMF7 targeting with elotuzumab has been demonstrated to not only inhibit MM cells but also activate NK cells163, and is under clinical investigation (NCT03003728).
NK cells can be suppressed extrinsically by Treg cells and MDSCs; strategies to overcome these immunosuppressive interactions enhance NK cell-mediated antitumour effects in patients or patient cells ex vivo164,165. Similarly, the use of exogenous IL-15 or an IL-15 super-agonist, such as ALT-803, constructed to mimic physiological trans-presentation is being tested in patients with AML after allo-HSCT, with or without donor NK cell transfer166 (NCT01898793). Continuous IL-15 signalling has been reported to result in NK cell transformation167; therefore, the use of continuous administration of IL-15, or NK cells genetically engineered to express this cytokine in order to induce autocrine signalling, raises the potential requirement for engineering of donor effector cells with ‘suicide’ vectors to enable termination of their activity.
Blocking interactions between inhibitory KIRs and their ligands using antibodies was viewed as a promising strategy based on preclinical data demonstrating that targeting of inhibitory Ly49 family receptors, which recognize MHC I molecules to distinguish self-cells from non-self-cells, results in heightened NK cell-mediated anti-leukaemia effects in mice168. KIR-blocking antibodies also increase the activity of human NK cells in vitro, but a clinical trial revealed that the anti-KIR2D mAb IPH2101 increased MM progression in association with impairment of NK cell activity attributable to either removal of KIR2D from NK cells by trogocytosis or effects of this agent on NK cell development169. Thus, the effects of any therapeutic intervention to enhance the GvT activity of NK cells — whether via increased stimulatory signaling or blockade of inhibitory pathways — on the overall development and function of NK cells needs to be considered, especially in the setting of lymphopenia and immune reconstitution after allo-HSCT that could adversely affected the developmental pathways.
Next steps in adoptive NK cell immunotherapy.
Several key factors can stifle the antitumour efficacy of NK cells therapies and these revolve around infusing enough NK cells to ensure sufficient engraftment, homing to the tumour, and long-term survival and function. NK cells constitute only a small pool of lymphocytes and need to be extensively expanded in culture in order to generate sufficient numbers of activated cells, although third-party or off-the-shelf sources of NK cells can partially obviate this issue. Pre-allo-HSCT cytoreductive conditioning treatment induces factors such as IL-7 and/or IL-15 that, in a lymphopenic environment, can result in successful, albeit short-term NK cell engraftment166. Considerable research interest has been placed on different sources of NK cells other than bone marrow-derived or peripheral blood mononuclear cell (PBMC)-derived stem cells or progenitors, which might enable enhancements of cell expansion and/or function, particularly as an off-the-shelf, third-party product; however, careful comparisons and assessments of these different approaches have not been performed. The alternative stem cell sources include cord blood170 (NCT00354172), placenta-derived cells (NCT029550) or induced pluripotential stem cells (iPSC)171 (NCT04106167), that might not only have greater expansion and antitumour capabilities but also be more ameniable to genetic manipulation (BOX 1); however, the exact developmental, phenotypic and functional properties of these products relative to those of adult HSC-derived or PBMC-derived NK cells remain unclear. Clinical use of established immortalized NK cell lines (such as NK92 cell) has resulted in transient anticancer effects172,173, and could enable improved access, homogeneity and genome-modification success rates; however, owing to their immortalized nature and sometimes substantial deviations in receptor expression and functional profiles relative to ‘normal’ NK cell, these cells must be irradiated prior to clinical use, which severely limits their in vivo persistence (measured in days) and is likely to substantially alter their biology and function. Such third-party NK cell approaches also facilitate attempts to promote activation of the cell product, for example, through genetic deletion of inhibitory signals or transduction with growth factors such as IL-15, and to improve tumour cell-targeting through CAR expression126 (BOX 1). With regard to the latter approach, whether CAR expression in either NK cells or T cells is most advantageous remains unknown, but warrants consideration of the broad reactivity and extensive inflammatory cascade of NK cells as well as their limited persistence. Whether third-party CAR T cells can induce GvHD is a crucial question for the field, because a primary rationale for using donor or third-party CAR NK cells is their presumed inability to induce GvHD.
Regardless, several CAR NK cell products are under clinical investigation to target not only cancer cells directly174, but also mediators of tumour immune evasion, such as MDSCs175. In addition, disrupting inhibitory factors, such as indolamine (IDO), together with NK cell transfer (NCT02118285), and/or engendering NK cell products with expression of IL-15 to promote sustained autocrine signalling are also being assessed (NCT03056339), with increases in activity and/or persistence being reported in preclinical studies139,170,171,176. Impressive effects of third-party, cord blood-derived, anti-CD19 CAR NK cells transduced with IL-15 and a suicide vector have also been demonstrated in patients with refractory lymphoma, with an overall response rate (ORR) of 73%, no observable CRS or GvHD and long-term CAR NK cell engraftment (for >9 months)177. However, important caveats of this study include the use of secondary treatments in the majority of patients, which might have contributed to the ORR, and the small size of the patient cohort. Additionally, the use of PCR to ascertain product engraftment is particularly useful in patients in whom the level of the abundance of CAR T cells is at or below the limits of sensitivity of flow cytometry-based quantification. Nevertheless, the findings do lend promise to the further development of CAR NK cell products, whether they be autologous or third-party. As with other NK cell products, stringent characterization of the reproducibility of the generated product is needed. Other questions relate to the homogeneity, stability and activity of the cell product, given the complex interactions of the different receptors systems on NK cells and that the cell sources or culture conditions used might not accurately mirror the normal developmental paradigms. Regardless of the source and manipulations, any activated NK cell product used after HSCT must circumvent the issues of cell engraftment and persistence whilst retaining or enhancing antitumour activities and avoiding possible off-target toxicities, including effects on haematopoietic and immune reconstitution. This consideration is particularly germane when using third-party NK cells, which have the potential to target a range of both HSC-donor and recipient cell types.
Increasing the GvT potential of NK cells.
The immunomodulatory activities of currently used anti-neoplastic agents can be exploited to augment NK cell-mediated GvT activity. Lenalidomide, a thalidomide-derivative IMiD, results in increased NK cell activity and ADCC in patients with MM, but also increased GVHD178, possibly owing to effects on MDSCs and PD-1–PD-L1 signalling179. As discussed for T cells, the proteasome inhibitor bortezomib also sensitizes tumour cells, including CSCs, to NK cell-mediated cytotoxicity, in part through upregulation of death receptors and suppression of MHC expression152, and is under clinical investigation in combination with autologous NK cell transfer for CML (NCT00720785)169 (BOX 1). However, immunosuppressive therapies are used after allo-HSCT, and data demonstrate that GvHD can also suppress NK cell function (owing to competition by T cells for IL-15)180; these variables complicate efforts to determine the effects of agents such lenalidomide and bortezomib on NK cell-mediated GvT activity, in addition to the fact that many agents have NK cell-independent antitumour effects.
A new generation of novel molecules that can simultaneously retarget NK cells towards tumour cells and promote NK cell activation are currently under investigation. Originally developed as BiTEs comprising CD19-targeting and CD3-targeting antibody moieties to redirect T cells towards B cells, such constructs have proven clinical activity in patients with ALL181. Variations on this approach, including bi-specific killer cell-engagers (BiKEs) and trispecific killer cell-engagers (TriKEs), are now being pursued to promote NK cell targeting of various tumour-associated markers (such as EGFR in glioblastoma, GD2 in neuroblastoma, CD33 in AML and CD19 in CLL, among others), provide cytokine and/or stimulatory signals (for example, via IL-15 or agonistic anti-FcγRIII moieties, respectively, with the latter being highly specific for NK cells), and/or block inhibitory molecules (such as PD-1, PD-L1 or TGFβ), thereby increasing NK cell GvT effects182–186 (NCT03214666) (BOX 1). The advantages of such agents include their potential to engage and activate both endogenous and engrafted donor NK cell populations, as well as any transferred third-party NK cell products. Thus, approaches to optimize NK cells as an adoptive cell immunotherapy and to augment NK cell reconstitution and activation following HSCT are both likely to improve GvT efficacy.
NKT cells and γδT cells to augment GvT
NKT cells are subset of T cells that express both NK (CD56) and T cell (CD3 and TCR) markers but have restricted TCR diversity and are endowed with prominent immunoregulatory properties. This cell type has been under increased investigation, in part, owing to their reported role in suppressing GvHD in preclinical models and antitumour effects in multiple models187,188. The activity of NKT cells in suppressing GvHD in mice has been shown to be dependent on production of IL-4 by these cells and their migration to germinal centres, which are associated with expansion of Treg cell populations and resultant suppression of germinal centre reactions187. Conditioning regimens that foster the induction of NKT cells, such as total lymphoid irradiation (TLI), are also being applied, as well as adoptive transfer of purified NKT cell populations187. CAR NKT cells are also being assessed in various models of cancer, including solid tumours189. While the limited abundance of NKT cells remains an issue, this cell type might offer advantages in cell therapy compared with broadly lytic NK cell populations, owing to the immunoregulatory effects of NKT cells in suppressing GvHD when used post-allo HSCT as well as their potentially greater clonal expansion capabilities187. Clinical studies involving adoptive transfer of NKT cells are underway (NCT00631072). In preclinical models, NKT cell activation using G-CSF analogues results in potent anti-leukaemia effects via promotion of DC and CD8+ T cell responses190, suggesting indirect pathways leading to suppression of GvHD and enhancement of GvT effects.
γδT cells are another arm of T cell-mediated immunity, and these cells link adaptive and innate responses owing to their MHC-independent specificity for a limited range of target antigens, leading to antitumour effects with lesser GvHD risk188, similar to NK cells. Owing to the positive associations between the abundance of γδT cells and clinical outcomes following HSCT191–195 as well as the ability of activated γδT cells to kill leukaemic cells196, the use of this cell type as an immunotherapy to mediate GvT without exacerbating GvHD is garnering increasing attention193. Similar to NK and NKT cells, the relative rarity of these γδT cells necessitates extensive ex vivo expansion, use of other cell sources (such as iPSCs) and/or genetic manipulation to obtain sufficient numbers for clinical use192. In contrast to NK cells, which rapidly reconstitute after HSCT, the delayed recovery of both NKT and γδT cells likely places more emphasis on the optimizing the characteristics of the donor cells to maximize their persistence and antitumour efficacy, for example, by removing αβT cells from the donor graft197.
Conclusions
Understanding of the immunological mechanisms governing effective GvL responses remains in its infancy. GvL — or GvT — effects can result from the allo-HSCT process itself, tied to GvHD responses by infused donor T cells or selected effector populations, or from the generation of a more balanced and better functioning immune system. Clearly, more robust preclinical models are needed that better reflect the ever-changing clinical allo-HSCT scenario and that enable focus to be placed on GvT processes independently of GvHD. With increased characterization of cancer cells upon disease relapse, new insights can be gleaned on immune evasion pathways and thus the effector cells that are important for GvT efficacy. MHC II-restricted GvL responses, which seem to be highly relevant according to findings from clinical studies, are important to more fully understand, as is GvHD and the contributions of alloreactivity to persistent GvL responses. Currently, the success of allo-HSCT is judged by the absence of marked acute or chronic GvHD, and modern immunosuppression therapies have enabled considerable advances in this regard. Thus, pinpointed focus on inducing immunity against leukaemia and haematopoietic antigens, rather than broad immune responses to alloantigens, will become increasingly vital for prevention of disease relapse. In some malignancies in which GvT responses are limited, such as MM, immunotherapy might be best applied after auto-HSCT. After allo-HSCT, adoptive cell therapy approaches will likely necessitate genome modification of the cell products or the use of novel agents to heighten the specificity and sustainability of antitumour responses. The increasing application of third-party or off-the-shelf cell therapies opens new opportunities to enhance GvT responses, but limited cell persistence and off-target toxicities remain paramount concerns. These advances, which have served to minimize GvHD and transplant-related mortality, thus now seem likely to usher in a new era wherein therapeutic modulation of anticancer immunity becomes pre-eminent and presents the potential to expand the clinical utility of HSCT to cancers other than haematological malignancies.
Key points.
Autologous haematopoetic stem cell transplantation (HSCT) migh present the optimal platform for immunotherapies, particularly for myeloma and lymphoma. Future approaches to improve allogeneic HSCT will require the introduction of innovative immune and/or targeted therapies to prevent relapse, following effective prevention of graft-versus-host disease (GvHD).
In initial clinical trials, freshly isolated or in vitro expanded donor regulatory T (Treg) cell infusions have been associated with low GvHD rates; in preclinical models and in preliminary clinical studies, donor Treg cell infusion does not seem to increase leukaemia relapse rates as had been feared, although more extensive, controlled trials are needed.
Immune system-targeted approaches to treat disease relapse after autologous or allogeneic HSCT now include pharmacological agents that block pathways essential for tumour cell survival and at the same time, for allogeneic HSCT, suppress GvHD.
Immune-checkpoint inhibitors targeting inhibitory signalling pathways in T cells or infusion of T cells genetically modified to express chimeric antigen receptors (CARs) or T cell receptors (TCRs) reactive with tumour cells might prove advantageous in autologous HSCT, but the risk of GvHD warrants careful consideration in allogeneic HSCT.
Preclinical models used to delineate graft-versus-tumour (GvT) from GvHD responses as well as assess potential therapeutic interventions often use inbred laboratory mouse strains, which offer many advantages mechanistically, but crucial caveats beyond immunological differences between species need to be acknowledged during clinical extrapolation.
Nature killer (NK) cells are increasingly being examined for their ability to mediate GvT effects while having minimal effects on GvHD after HSCT, including the use of third-party CAR NK cell products and use of targeted biologic agents to exploit the ability of NK cells to mediate antibody-dependent cellular cytotoxicity.
Footnotes
Competing interests
B.R.B. is a founder of Tmunity Therapeutics, and receives research support and is a member of the Scientific Advisory Board for BlueRock Therapeutics. G.R.H. has received research funding from Compass Therapeutics and Roche. W.J.M. declares no competing interests.
References
- 1.D’Souza A & Fretham C Current Uses and Outcomes of Hematopoietic Cell Transplantation (HCT): CIBMTR Summary Slides, 2018. Available at https://www.cibmtr.org.
- 2.Lundqvist A & Childs R Allogeneic hematopoietic cell transplantation as immunotherapy for solid tumors: current status and future directions. J Immunother 28, 281–288 (2005). [DOI] [PubMed] [Google Scholar]
- 3.McDonald GB, et al. Survival, Nonrelapse Mortality, and Relapse-Related Mortality After Allogeneic Hematopoietic Cell Transplantation: Comparing 2003–2007 Versus 2013–2017 Cohorts. Ann Intern Med (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meybodi MA, et al. HLA-haploidentical vs matched-sibling hematopoietic cell transplantation: a systematic review and meta-analysis. Blood Adv 3, 2581–2585 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Passweg JR, et al. The EBMT activity survey report 2017: a focus on allogeneic HCT for nonmalignant indications and on the use of non-HCT cell therapies. Bone Marrow Transplant 54, 1575–1585 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stem Cell Trialists’ Collaborative, G. Allogeneic peripheral blood stem-cell compared with bone marrow transplantation in the management of hematologic malignancies: an individual patient data meta-analysis of nine randomized trials. J Clin Oncol 23, 5074–5087 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vuckovic S, et al. Bone marrow transplantation generates T cell-dependent control of myeloma in mice. J Clin Invest 129, 106–121 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung DJ, et al. T-cell Exhaustion in Multiple Myeloma Relapse after Autotransplant: Optimal Timing of Immunotherapy. Cancer Immunol Res 4, 61–71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yadav M, et al. Tigit, CD226 and PD-L1/PD-1 Are Highly Expressed By Marrow-Infiltrating T Cells in Patients with Multiple Myeloma. Blood 128, 2102–2102 (2016). [Google Scholar]
- 10.Minnie SA, et al. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood 132, 1675–1688 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Choi SW & Reddy P Current and emerging strategies for the prevention of graft-versus-host disease. Nature reviews Clinical oncology 11, 536 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blazar BR, Murphy WJ & Abedi M Advances in graft-versus-host disease biology and therapy. Nature reviews immunology 12, 443 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Markey KA, MacDonald KP & Hill GR The biology of graft-versus-host disease: experimental systems instructing clinical practice. Blood 124, 354–362 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mirsoian A, et al. Adiposity induces lethal cytokine storm after systemic administration of stimulatory immunotherapy regimens in aged mice. Journal of Experimental Medicine 211, 2373–2383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murphy WJ & Longo DL The Surprisingly Positive Association Between Obesity and Cancer Immunotherapy Efficacy. JAMA 321, 1247–1248 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Stein-Thoeringer CK, et al. Lactose drives Enterococcus expansion to promote graft-versus-host disease. Science 366, 1143–1149 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fink EC, et al. Crbn (I391V) is sufficient to confer in vivo sensitivity to thalidomide and its derivatives in mice. Blood 132, 1535–1544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dossa RG, et al. Development of T-cell immunotherapy for hematopoietic stem cell transplantation recipients at risk of leukemia relapse. Blood 131, 108–120 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bleakley M & Riddell SR Exploiting T cells specific for human minor histocompatibility antigens for therapy of leukemia. Immunol Cell Biol 89, 396–407 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marijt WA, et al. Hematopoiesis-restricted minor histocompatibility antigens HA-1- or HA-2-specific T cells can induce complete remissions of relapsed leukemia. Proc Natl Acad Sci U S A 100, 2742–2747 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shlomchik WD, et al. Prevention of graft versus host disease by inactivation of host antigen presenting cells. Science 285, 412–415 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Bleakley M, et al. Engineering human peripheral blood stem cell grafts that are depleted of naive T cells and retain functional pathogen-specific memory T cells. Biol Blood Marrow Transplant 20, 705–716 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson BE, et al. Memory CD4+ T cells do not induce graft-versus-host disease. J Clin Invest 112, 101–108 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacDonald KP, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood 116, 3955–3963 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Koyama M, et al. Recipient nonhematopoietic antigen-presenting cells are sufficient to induce lethal acute graft-versus-host disease. Nat Med 18, 135–142 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Matte-Martone C, Liu J, Jain D, McNiff J & Shlomchik WD CD8+ but not CD4+ T cells require cognate interactions with target tissues to mediate GVHD across only minor H antigens, whereas both CD4+ and CD8+ T cells require direct leukemic contact to mediate GVL. Blood 111, 3884–3892 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matte CC, et al. Donor APCs are required for maximal GVHD but not for GVL. Nat Med 10, 987–992 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Reddy P, et al. A crucial role for antigen-presenting cells and alloantigen expression in graft-versus-leukemia responses. Nat Med 11, 1244–1249 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Zheng H, et al. Effector memory CD4+ T cells mediate graft-versus-leukemia without inducing graft-versus-host disease. Blood 111, 2476–2484 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Markey KA, et al. Flt-3L Expansion of Recipient CD8alpha(+) Dendritic Cells Deletes Alloreactive Donor T Cells and Represents an Alternative to Posttransplant Cyclophosphamide for the Prevention of GVHD. Clin Cancer Res 24, 1604–1616 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Asakura S, et al. Alloantigen expression on non-hematopoietic cells reduces graft-versus-leukemia effects in mice. J Clin Invest 120, 2370–2378 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koyama M, et al. MHC class II antigen presentation by the intestinal epithelium initiates graft-versus-host disease and is influenced by the microbiota. Immunity 51, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koyama M, et al. Donor colonic CD103+ dendritic cells determine the severity of acute graft-versus-host disease. J Exp Med 212, 1303–1321 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gartlan KH, et al. Donor T-cell-derived GM-CSF drives alloantigen presentation by dendritic cells in the gastrointestinal tract. Blood Adv 3, 2859–2865 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christopher MJ, et al. Immune Escape of Relapsed AML Cells after Allogeneic Transplantation. N Engl J Med 379, 2330–2341 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vago L, et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N Engl J Med 361, 478–488 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Alspach E, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beelen DW, Elmaagacli A, Muller KD, Hirche H & Schaefer UW Influence of intestinal bacterial decontamination using metronidazole and ciprofloxacin or ciprofloxacin alone on the development of acute graft-versus-host disease after marrow transplantation in patients with hematologic malignancies: final results and long term follow-up of an open-label prospective randomized trial. Blood 93, 3267–3275 (1999). [PubMed] [Google Scholar]
- 39.Beelen DW, et al. Evidence that sustained growth suppression of intestinal anaerobic bacteria reduces the risk of acute graft-versus-host disease after sibling marrow transplantation. Blood 80, 2668–2676 (1992). [PubMed] [Google Scholar]
- 40.Storb R, et al. Graft-versus-host disease and survival in patients with aplastic anemia treated by marrow grafts from HLA-identical siblings. Beneficial effect of a protective environment. N.Engl.J.Med 308, 302–307. (1983). [DOI] [PubMed] [Google Scholar]
- 41.Mathewson ND, et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat Immunol 17, 505–513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peled JU, et al. Members of the Intestinal Microbiota Are Associated with Relapse after Allogeneic Hematopoietic Stem Cell Transplantation. Biology of Blood and Marrow Transplant 22, S23–S24. [Google Scholar]
- 43.Zeiser R & Vago L Mechanisms of immune escape after allogeneic hematopoietic cell transplantation. Blood 133, 1290–1297 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Matte-Martone C, et al. Differential requirements for myeloid leukemia IFN-gamma conditioning determine graft-versus-leukemia resistance and sensitivity. J Clin Invest 127, 2765–2776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walter RB, et al. Significance of minimal residual disease before myeloablative allogeneic hematopoietic cell transplantation for AML in first and second complete remission. Blood 122, 1813–1821 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Minnie SA & Hill GR Immunotherapy of multiple myeloma. J Clin Invest (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marmont AM, et al. T-cell depletion of HLA-identical transplants in leukemia. Blood 78, 2120–2130 (1991). [PubMed] [Google Scholar]
- 48.Brandenburg U, Gottlieb D & Bradstock K Antileukemic effects of rapid cyclosporin withdrawal in patients with relapsed chronic myeloid leukemia after allogeneic bone marrow transplantation. Leuk Lymphoma 31, 545–550 (1998). [DOI] [PubMed] [Google Scholar]
- 49.Kolb HJ, et al. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood 86, 2041–2050 (1995). [PubMed] [Google Scholar]
- 50.Soiffer RJ Checkpoint inhibition to prevent or treat relapse in allogeneic hematopoietic cell transplantation. Bone Marrow Transplant 54, 798–802 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Schoch LK, et al. Immune checkpoint inhibitors as a bridge to allogeneic transplantation with posttransplant cyclophosphamide. Blood Adv 2, 2226–2229 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Henden AS, et al. Pegylated interferon-2alpha invokes graft-versus-leukemia effects in patients relapsing after allogeneic stem cell transplantation. Blood Adv 3, 3013–3019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alexander KA, et al. CSF-1-dependant donor-derived macrophages mediate chronic graft-versus-host disease. J Clin Invest 124, 4266–4280 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kennedy GA, et al. Addition of interleukin-6 inhibition with tocilizumab to standard graft-versus-host disease prophylaxis after allogeneic stem-cell transplantation: a phase 1/2 trial. Lancet Oncol 15, 1451–1459 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Nakamura K, et al. Dysregulated IL-18 Is a Key Driver of Immunosuppression and a Possible Therapeutic Target in the Multiple Myeloma Microenvironment. Cancer Cell 33, 634–648. e635 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Garfall AL, et al. Anti-CD19 CAR T cells with high-dose melphalan and autologous stem cell transplantation for refractory multiple myeloma. JCI Insight 4(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pont MJ, et al. gamma-secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.June CH, O’Connor RS, Kawalekar OU, Ghassemi S & Milone MC CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018). [DOI] [PubMed] [Google Scholar]
- 59.Majzner RG & Mackall CL Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med 25, 1341–1355 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Ghosh A, et al. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat Med 23, 242–249 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Di Stasi A, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 365, 1673–1683 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chapuis AG, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med 25, 1064–1072 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kantarjian H, et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N Engl J Med 376, 836–847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qazilbash MH, et al. PR1 peptide vaccine induces specific immunity with clinical responses in myeloid malignancies. Leukemia 31, 697–704 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McLaughlin LP, et al. EBV/LMP-specific T cells maintain remissions of T- and B-cell EBV lymphomas after allogeneic bone marrow transplantation. Blood 132, 2351–2361 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Doubrovina E, et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood 119, 2644–2656 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zeiser R, et al. Ruxolitinib in corticosteroid-refractory graft-versus-host disease after allogeneic stem cell transplantation: a multicenter survey. Leukemia 29, 2062–2068 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cutler C, et al. Rituximab prophylaxis prevents corticosteroid-requiring chronic GVHD after allogeneic peripheral blood stem cell transplantation: results of a phase 2 trial. Blood 122, 1510–1517 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cutler C, et al. Rituximab for steroid-refractory chronic graft-versus-host disease. Blood 108, 756–762 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Y, et al. Notch signaling is a critical regulator of allogeneic CD4+ T-cell responses mediating graft-versus-host disease. Blood 117, 299–308 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Radojcic V, et al. Notch signaling mediated by Delta-like ligands 1 and 4 controls the pathogenesis of chronic GVHD in mice. Blood 132, 2188–2200 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mathew NR, et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nature medicine 24, 282 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jaglowski SM & Blazar BR How ibrutinib, a B-cell malignancy drug, became an FDA-approved second-line therapy for steroid-resistant chronic GVHD. Blood Adv 2, 2012–2019 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dubovsky JA, et al. Ibrutinib treatment ameliorates murine chronic graft-versus-host disease. J Clin Invest 124, 4867–4876 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Flynn R, et al. Targeting Syk-activated B cells in murine and human chronic graft-versus-host disease. Blood 125, 4085–4094 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pai CC, et al. Treatment of chronic graft-versus-host disease with bortezomib. Blood 124, 1677–1688 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koreth J, et al. Bortezomib, tacrolimus, and methotrexate for prophylaxis of graft-versus-host disease after reduced-intensity conditioning allogeneic stem cell transplantation from HLA-mismatched unrelated donors. Blood 114, 3956–3959 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koreth J, et al. Bortezomib-based graft-versus-host disease prophylaxis in HLA-mismatched unrelated donor transplantation. J Clin Oncol 30, 3202–3208 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Paz K, et al. Targeting PI3Kdelta function for amelioration of murine chronic graft-versus-host disease. Am J Transplant 19, 1820–1830 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Taylor PA, Noelle RJ & Blazar BR CD4(+)CD25(+) immune regulatory cells are required for induction of tolerance to alloantigen via costimulatory blockade. J Exp Med 193, 1311–1318 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Taylor PA, Lees CJ & Blazar BR The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft-versus-host disease lethality. Blood 99, 3493–3499 (2002). [DOI] [PubMed] [Google Scholar]
- 82.Hoffmann P, Ermann J, Edinger M, Fathman CG & Strober S Donor-type CD4(+)CD25(+) regulatory T cells suppress lethal acute graft-versus-host disease after allogeneic bone marrow transplantation. J Exp Med 196, 389–399 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cohen JL, Trenado A, Vasey D, Klatzmann D & Salomon BL CD4(+)CD25(+) immunoregulatory T Cells: new therapeutics for graft-versus-host disease. J Exp Med 196, 401–406 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brunstein CG, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood 117, 1061–1070 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brunstein CG, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood 127, 1044–1051 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Edinger M, et al. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med 9, 1144–1150 (2003). [DOI] [PubMed] [Google Scholar]
- 87.Trenado A, et al. Recipient-type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus-leukemia. J Clin Invest 112, 1688–1696 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Del Papa B, et al. Clinical-Grade-Expanded Regulatory T Cells Prevent Graft-versus-Host Disease While Allowing a Powerful T Cell-Dependent Graft-versus-Leukemia Effect in Murine Models. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 23, 1847–1851 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Choi BD, et al. Human regulatory T cells kill tumor cells through granzyme-dependent cytotoxicity upon retargeting with a bispecific antibody. Cancer Immunol Res 1, 163 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Grossman WJ, et al. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 21, 589–601 (2004). [DOI] [PubMed] [Google Scholar]
- 91.Di Ianni M, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 117, 3921–3928 (2011). [DOI] [PubMed] [Google Scholar]
- 92.Zhou Q, et al. Depletion of endogenous tumor-associated regulatory T cells improves the efficacy of adoptive cytotoxic T-cell immunotherapy in murine acute myeloid leukemia. Blood 114, 3793–3802 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Szczepanski MJ, et al. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin Cancer Res 15, 3325–3332 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zou L, et al. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res 64, 8451–8455 (2004). [DOI] [PubMed] [Google Scholar]
- 95.Martelli MF, et al. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood 124, 638–644 (2014). [DOI] [PubMed] [Google Scholar]
- 96.Sawamukai N, et al. Cell-autonomous role of TGFbeta and IL-2 receptors in CD4+ and CD8+ inducible regulatory T-cell generation during GVHD. Blood 119, 5575–5583 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang P, et al. Induced regulatory T cells promote tolerance when stabilized by rapamycin and IL-2 in vivo. J Immunol 191, 5291–5303 (2013). [DOI] [PubMed] [Google Scholar]
- 98.Beres AJ, et al. CD8+ Foxp3+ regulatory T cells are induced during graft-versus-host disease and mitigate disease severity. J Immunol 189, 464–474 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Semple K, Yu Y, Wang D, Anasetti C & Yu XZ Efficient and selective prevention of GVHD by antigen-specific induced Tregs via linked-suppression in mice. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 17, 309–318 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li J, et al. HY-Specific Induced Regulatory T Cells Display High Specificity and Efficacy in the Prevention of Acute Graft-versus-Host Disease. J Immunol 195, 717–725 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hippen KL, et al. Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. Am J Transplant 11, 1148–1157 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Heinrichs J, et al. CD8(+) Tregs promote GVHD prevention and overcome the impaired GVL effect mediated by CD4(+) Tregs in mice. Oncoimmunology 5, e1146842 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhou X, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nature immunology 10, 1000–1007 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rubtsov YP, et al. Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fu W, et al. A multiply redundant genetic switch ‘locks in’ the transcriptional signature of regulatory T cells. Nature immunology 13, 972–980 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang P, et al. Eomesodermin promotes the development of type 1 regulatory T (TR1) cells. Sci Immunol 2(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lu L, et al. Critical role of all-trans retinoic acid in stabilizing human natural regulatory T cells under inflammatory conditions. Proc Natl Acad Sci U S A 111, E3432–3440 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Di Pilato M, et al. Targeting the CBM complex causes Treg cells to prime tumours for immune checkpoint therapy. Nature 570, 112–116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Benson MJ, Pino-Lagos K, Rosemblatt M & Noelle RJ All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. The Journal of experimental medicine 204, 1765–1774 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Coombes JL, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. The Journal of experimental medicine 204, 1757–1764 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhou X, et al. Cutting edge: all-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. J Immunol 185, 2675–2679 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Khafif RA, Gelbfish GA, Attie JN, Tepper P & Zingale R Thirty-year experience with 457 radical neck dissections in cancer of the mouth, pharynx, and larynx. Am J Surg 158, 303–307; discussion 308 (1989). [DOI] [PubMed] [Google Scholar]
- 113.Yue X, et al. Control of Foxp3 stability through modulation of TET activity. The Journal of experimental medicine 213, 377–397 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sasidharan Nair V, Song MH & Oh KI Vitamin C Facilitates Demethylation of the Foxp3 Enhancer in a Tet-Dependent Manner. J Immunol 196, 2119–2131 (2016). [DOI] [PubMed] [Google Scholar]
- 115.Iamsawat S, et al. Vitamin C stabilizes CD8+ iTregs and enhances their therapeutic potential in controlling murine GVHD and leukemia relapse. Blood Adv 3, 4187–4201 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang Y, et al. The polycomb repressive complex 2 governs life and death of peripheral T cells. Blood 124, 737–749 (2014). [DOI] [PubMed] [Google Scholar]
- 117.DuPage M, et al. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity 42, 227–238 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Betts BC, et al. Targeting JAK2 reduces GVHD and xenograft rejection through regulation of T cell differentiation. Proc Natl Acad Sci U S A 115, 1582–1587 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Locafaro G, et al. IL-10-Engineered Human CD4(+) Tr1 Cells Eliminate Myeloid Leukemia in an HLA Class I-Dependent Mechanism. Mol Ther 25, 2254–2269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cudkowicz G & Bennett M Peculiar immunobiology of bone marrow allografts: I. Graft rejection by irradiated responder mice. The Journal of Experimental Medicine 134, 83–102 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cudkowicz G & Bennett M Peculiar immunobiology of bone marrow allografts: II. Rejection of parental grafts by resistant F1 hybrid mice. The Journal of Experimental Medicine 134, 1513–1528 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cerwenka A & Lanier LL Natural killer cell memory in infection, inflammation and cancer. Nature Reviews Immunology 16, 112 (2016). [DOI] [PubMed] [Google Scholar]
- 123.Freud AG, Mundy-Bosse BL, Yu J & Caligiuri MA The broad spectrum of human natural killer cell diversity. Immunity 47, 820–833 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Capuano C, et al. Memory NK cell features exploitable in anticancer immunotherapy. Journal of Immunology Research 2019, Article ID 8795673 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Boudreau JE & Hsu KC Natural killer cell education and the response to infection and cancer therapy: stay tuned. Trends in Immunology 39, 222–239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hodgins JJ, Khan ST, Park MM, Auer RC & Ardolino M Killers 2.0: NK cell therapies at the forefront of cancer control. The Journal of Clinical Investigation 129, 3499–3510 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Waggoner SN, Cornberg M, Selin LK & Welsh RM Natural killer cells act as rheostats modulating antiviral T cells. Nature 481, 394 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Asai O, et al. Suppression of graft-versus-host disease and amplification of graft-versus-tumor effects by activated natural killer cells after allogeneic bone marrow transplantation. The Journal of Clinical Investigation 101, 1835–1842 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Olson JA, et al. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood 115, 4293–4301 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ruggeri L, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 295, 2097–2100 (2002). [DOI] [PubMed] [Google Scholar]
- 131.Lee DA, et al. Haploidentical Natural Killer Cells Infused before Allogeneic Stem Cell Transplantation for Myeloid Malignancies: A Phase I Trial. Biol Blood Marrow Transplant 22, 1290–1298 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Björklund AT, et al. Complete remission with reduction of high-risk clones following haploidentical NK-cell therapy against MDS and AML. Clinical Cancer Research 24, 1834–1844 (2018). [DOI] [PubMed] [Google Scholar]
- 133.Shah NN, et al. Acute GVHD in patients receiving IL-15/4–1BBL activated NK cells following T-cell–depleted stem cell transplantation. Blood 125, 784–792 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Curti A, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood, The Journal of the American Society of Hematology 118, 3273–3279 (2011). [DOI] [PubMed] [Google Scholar]
- 135.Rubnitz JE, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. Journal of Clinical Oncology 28, 955 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Miller JS, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 105, 3051–3057 (2005). [DOI] [PubMed] [Google Scholar]
- 137.Shaffer BC, et al. Phase II study of haploidentical natural killer cell infusion for treatment of relapsed or persistent myeloid malignancies following allogeneic hematopoietic cell transplantation. Biology of Blood and Marrow Transplantation 22, 705–709 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bjorklund AT, et al. Complete Remission with Reduction of High-Risk Clones following Haploidentical NK-Cell Therapy against MDS and AML. Clin Cancer Res 24, 1834–1844 (2018). [DOI] [PubMed] [Google Scholar]
- 139.Sentman M-L, et al. Mechanisms of acute toxicity in NKG2D chimeric antigen receptor T cell–treated mice. The Journal of Immunology 197, 4674–4685 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Karimi MA, et al. NKG2D expression by CD8+ T cells contributes to GVHD and GVT effects in a murine model of allogeneic HSCT. Blood 125, 3655–3663 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Venstrom JM, et al. HLA-C–dependent prevention of leukemia relapse by donor activating KIR2DS1. New England Journal of Medicine 367, 805–816 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sekine T, et al. Specific combinations of donor and recipient KIR-HLA genotypes predict for large differences in outcome after cord blood transplantation. Blood 128, 297–312 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hallner A, et al. The HLA-B− 21 dimorphism impacts on NK cell education and clinical outcome of immunotherapy in acute myeloid leukemia. blood 133, 1479–1488 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bachanova V, et al. Donor KIR B genotype improves progression-free survival of non-Hodgkin lymphoma patients receiving unrelated donor transplantation. Biology of Blood and Marrow Transplantation 22, 1602–1607 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Oevermann L, et al. KIR B haplotype donors confer a reduced risk for relapse after haploidentical transplantation in children with ALL. Blood 124, 2744–2747 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.André P, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 175, 1731–1743. e1713 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shaffer BC & Hsu KC How important is NK alloreactivity and KIR in allogeneic transplantation? Best Practice & Research Clinical Haematology 29, 351–358 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bishara A, et al. The beneficial role of inhibitory KIR genes of HLA class I NK epitopes in haploidentically mismatched stem cell allografts may be masked by residual donor‐alloreactive T cells causing GVHD. Tissue antigens 63, 204–211 (2004). [DOI] [PubMed] [Google Scholar]
- 149.Erbe AK, et al. Neuroblastoma patients’ KIR and KIR-ligand genotypes influence clinical outcome for dinutuximab-based immunotherapy: a report from the Children’s Oncology Group. Clinical Cancer Research 24, 189–196 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Barrow AD, et al. Natural killer cells control tumor growth by sensing a growth factor. Cell 172, 534–548. e519 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tran HC, et al. TGFβR1 blockade with galunisertib (LY2157299) enhances anti-neuroblastoma activity of the anti-GD2 antibody dinutuximab (ch14. 18) with natural killer cells. Clinical Cancer Research 23, 804–813 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ames E, et al. NK cells preferentially target tumor cells with a cancer stem cell phenotype. The Journal of Immunology 195, 4010–4019 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tallerico R, et al. NK cells control breast cancer and related cancer stem cell hematological spread. Oncoimmunology 6, e1284718 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Paczulla AM, et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 572, 254–259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Foley B, et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood 119, 2665–2674 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Litjens NH, van der Wagen L, Kuball J & Kwekkeboom J Potential beneficial effects of cytomegalovirus infection after transplantation. Frontiers in immunology 9, 389 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cichocki F, et al. Adaptive NK cell reconstitution is associated with better clinical outcomes. JCI Insight 4(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Alvarez M, et al. Regulation of murine NK cell exhaustion through the activation of the DNA damage repair pathway. JCI insight (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Dong W, et al. The Mechanism of Anti–PD-L1 Antibody Efficacy against PD-L1–Negative Tumors Identifies NK Cells Expressing PD-L1 as a Cytolytic Effector. Cancer discovery 9, 1422–1437 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ciurea SO, et al. Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood 130, 1857–1868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kim HS & Kim N Targeting checkpoint receptors and molecules for therapeutic modulation of natural killer cells. Frontiers in Immunology 9, 2041 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Haverkos BM, et al. PD-1 blockade for relapsed lymphoma post-allogeneic hematopoietic cell transplant: high response rate but frequent GVHD. Blood 130, 221–228 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pazina T, et al. Enhanced SLAMF7 homotypic interactions by elotuzumab improves NK cell killing of multiple myeloma. Cancer Immunology Research 7, 1633–1646 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bachanova V, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood 123, 3855–3863 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sarhan D, et al. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Advances 2, 1459–1469 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cooley S, et al. First-in-human trial of rhIL-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Advances 3, 1970–1980 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mishra A, et al. Aberrant overexpression of IL-15 initiates large granular lymphocyte leukemia through chromosomal instability and DNA hypermethylation. Cancer Cell 22, 645–655 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Koh CY, et al. Augmentation of antitumor effects by NK cell inhibitory receptor blockade in vitro and in vivo. Blood 97, 3132–3137 (2001). [DOI] [PubMed] [Google Scholar]
- 169.Carlsten M, et al. Checkpoint inhibition of KIR2D with the monoclonal antibody IPH2101 induces contraction and hyporesponsiveness of NK cells in patients with myeloma. Clinical Cancer Research 22, 5211–5222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Liu E, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 32, 520 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Li Y, Hermanson DL, Moriarity BS & Kaufman DS Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell 23, 181–192. e185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Boyiadzis M, et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy 19, 1225–1232 (2017). [DOI] [PubMed] [Google Scholar]
- 173.Han J, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Scientific Reports 5, 11483 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Quintarelli C, et al. Efficacy of third-party chimeric antigen receptor modified peripheral blood natural killer cells for adoptive cell therapy of B-cell precursor acute lymphoblastic leukemia. Leukemia (2019). [DOI] [PubMed] [Google Scholar]
- 175.Parihar R, et al. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors. Cancer Immunol Res 7, 363–375 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Burga RA, et al. Engineering the TGFβ receptor to enhance the therapeutic potential of natural killer cells as an immunotherapy for neuroblastoma. Clinical Cancer Research, clincanres. 3183.2018 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Liu E, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. New England Journal of Medicine 382, 545–553 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wolschke C, et al. Postallograft lenalidomide induces strong NK cell–mediated antimyeloma activity and risk for T cell–mediated GvHD: Results from a phase I/II dose-finding study. Experimental hematology 41, 134–142. e133 (2013). [DOI] [PubMed] [Google Scholar]
- 179.Giuliani M, Janji B & Berchem G Activation of NK cells and disruption of PD-L1/PD-1 axis: two different ways for lenalidomide to block myeloma progression. Oncotarget 8, 24031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bunting MD, et al. GVHD prevents NK-cell-dependent leukemia and virus-specific innate immunity. Blood 129, 630–642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Aldoss I, et al. Redirecting T cells to eradicate B-cell acute lymphoblastic leukemia: bispecific T-cell engagers and chimeric antigen receptors. Leukemia 31, 777–787 (2017). [DOI] [PubMed] [Google Scholar]
- 182.Gleason MK, et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 123, 3016–3026 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wiernik A, et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16× 33 bispecific killer cell engager and ADAM17 inhibition. Clinical Cancer Research 19, 3844–3855 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Van den Bergh JM, et al. Monocyte-derived dendritic cells with silenced PD-1 ligands and transpresenting interleukin-15 stimulate strong tumor-reactive T-cell expansion. Cancer immunology research 5, 710–715 (2017). [DOI] [PubMed] [Google Scholar]
- 185.Tay SS, Carol H & Biro M TriKEs and BiKEs join CARs on the cancer immunotherapy highway. Human vaccines & immunotherapeutics 12, 2790–2796 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hsu J, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. The Journal of clinical investigation 128, 4654–4668 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Du J, et al. Invariant natural killer T cells ameliorate murine chronic GVHD by expanding donor regulatory T cells. Blood 129, 3121–3125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Minculescu L & Sengeløv H The role of gamma delta T cells in haematopoietic stem cell transplantation. Scandinavian journal of immunology 81, 459–468 (2015). [DOI] [PubMed] [Google Scholar]
- 189.Xu X, et al. NKT cells co-expressing a GD2-specific chimeric antigen receptor and IL-15 show enhanced in vivo persistence and antitumor activity against neuroblastoma. Clinical Cancer Research, clincanres. 0421.2019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Morris ES, et al. NKT cell–dependent leukemia eradication following stem cell mobilization with potent G-CSF analogs. The Journal of clinical investigation 115, 3093–3103 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Arruda LCM, Gaballa A & Uhlin M Impact of gammadelta T cells on clinical outcome of hematopoietic stem cell transplantation: systematic review and meta-analysis. Blood Adv 3, 3436–3448 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Straetemans T, et al. GMP-Grade Manufacturing of T Cells Engineered to Express a Defined gammadeltaTCR. Front Immunol 9, 1062 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Godder K, et al. Long term disease-free survival in acute leukemia patients recovering with increased γδ T cells after partially mismatched related donor bone marrow transplantation. Bone Marrow Transplantation 39, 751–757 (2007). [DOI] [PubMed] [Google Scholar]
- 194.Perko R, et al. Gamma delta T cell reconstitution is associated with fewer infections and improved event-free survival after hematopoietic stem cell transplantation for pediatric leukemia. Biology of Blood and Marrow Transplantation 21, 130–136 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Minculescu L, et al. Improved overall survival, relapse-free-survival and less graft-versus-host-disease in patients with high immune reconstitution of TCR gamma delta cells 2 months after allogeneic stem cell transplantation. Frontiers in Immunology 10, Article 1997 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Pistoia V, et al. Human gammadelta T-Cells: From Surface Receptors to the Therapy of High-Risk Leukemias. Front Immunol 9, 984 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Rådestad E, et al. Alpha/beta T-cell depleted grafts as an immunological booster to treat graft failure after hematopoietic stem cell transplantation with HLA-matched related and unrelated donors. Journal of immunology research 2014(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]