

Abstract
Brain aging is characterized by a time-dependent decline of tissue integrity and function, and it is a major risk for neurodegenerative diseases and brain cancer. Chaperone-mediated autophagy (CMA) is a selective form of autophagy specialized in protein degradation, which is based on the individual translocation of a cargo protein through the lysosomal membrane. Regulation of processes such as proteostasis, cellular energetics, or immune system activity has been associated with CMA, indicating its pivotal role in tissue homeostasis. Since first studies associating Parkinson’s disease (PD) to CMA dysfunction, increasing evidence points out that CMA is altered in both physiological and pathological brain aging. In this review article, we summarize the current knowledge regarding the impact of CMA during aging in brain physiopathology, highlighting the role of CMA in neurodegenerative diseases and glioblastoma, the most common and aggressive brain tumor in adults.
Keywords: CMA, LAMP2, physiological aging, neurodegenerative diseases, glioblastoma
Introduction to Autophagy
The term “autophagy” comes from the Ancient Greek meaning “self-eating,” which refers to the catabolic processes that use the cell to recycle its own constituents within the lysosome (Yang and Klionsky, 2010; Boya et al., 2013). The first discoveries were based on the bulk trapping of the cargo in double-membrane autophagosomes that fuse with lysosomes, which afterward was named “macroautophagy” (De Duve, 1963). Further studies demonstrated also the existence of another type of autophagy named “microautophagy,” which involves the uptake of soluble or membrane-bound material directly into the lysosome by invagination (De Duve and Wattiaux, 1966; Marzella et al., 1981). However, it was not until Dice et al. (1986) described a protein-selective form of autophagy, identifying a pentapeptide region on ribonuclease A required for its enhanced degradation during serum deprivation. This form of autophagy, nowadays known as “chaperone-mediated autophagy” (CMA), was at that time the unique selective form of autophagy. Since then, selective forms of macroautophagy and microautophagy have been described (Klionsky et al., 2016), with a special interest in “chaperone-assisted selective autophagy” (CASA), which participates in the selective trapping of ubiquitin-positive protein aggregates via autophagosomes (Arndt et al., 2010) and “endosomal-microautophagy,” in which cytosolic proteins enter endosomal compartments inside vesicles generated at the surface of the late endosomes (Sahu et al., 2011).
However, among all forms of autophagy, CMA is still the only process that enables the direct translocation of an individual cargo protein across the lysosomal membrane for its degradation (Kaushik and Cuervo, 2018). Thus, herein we will review the principal components and mechanisms involved in CMA and highlight its impact on physiological aging of the brain, with special emphasis on neurodegenerative disorders and glioblastoma.
Cma Process
Mechanism of Action
Biochemical and genetic approaches have characterized CMA as a rigorous process where the cargo protein must overcome several stages to reach proper lysosomal degradation (Figure 1).
Figure 1.

General scheme of the chaperone-mediated autophagy (CMA) process. (1) The KFERQ-like motif of a cargo protein is detected by HSC70, (2) which is the one targeting it into the lysosomal membrane protein lysosome-associated membrane protein type 2A (LAMP2A). Then, (3) LAMP2A monomers are assembled into multimeric structures, forming the translocation complex. Once this structure is correctly formed, (4) protein is translocated through the lysosomal membrane, while lysosomal HSC70 (lys-HSC70) pulls it into the lumen and avoid the return out of the membrane. Lysosomal HSP90 (lys-HSP90) is important in LAMP2A stabilization since it masks lysosomal protease binding sequences, and thus, prevents its degradation during this transition. Finally, (5) protein is degraded inside the lysosome, and (6) translocation complex is disassembled.
Cargo Protein Selection
For a specific protein to be degraded via CMA, it first has to bind to the heat shock cognate 71 kDa protein (HSC70), a chaperone that is going to target it to the lysosomal membrane (Dice, 1990). For the recognition of this cargo protein, a “KFERQ” motif is needed (Kaushik and Cuervo, 2018), which comprises a sequence of amino acids with specific charge and hydrophobicity. Overall, it is known that approximately 40% of the mammalian proteome could contain this canonical motif (Kaushik and Cuervo, 2018). However, post-translational modifications, such as phosphorylation, acetylation, or even ubiquitination, within incomplete motifs can confer specific properties, recreating a complete KFERQ-like motif (Kaushik and Cuervo, 2018). Not only inside the motif, but post-translational modifications outside the motif can even expose or mask an existing one, by conformational changes of the protein (Ferreira et al., 2015; Kaushik and Cuervo, 2018). Thus, CMA substrates can vary substantially. In this line, a free web-based resource called “KFERQ finder” has been developed, which directly identifies KFERQ-like motifs in any protein sequence (Kirchner et al., 2019).
HSC70 Binding and Targeting to Lysosomal Membrane
Hidden KFERQ motifs become accessible to HSC70 after: (I) protein conformational changes; (II) multiprotein complex disassembly; or (III) releasing from the subcellular membranes (Cuervo and Wong, 2014). Once bound to the chaperone, the substrate is delivered to the surface of the lysosome where it interacts with the cytosolic tail of the lysosome-associated membrane protein type 2A (LAMP2A; Cuervo and Dice, 1996). It is important to highlight that HSC70’s role in autophagy has been assigned not only to CMA but also to CASA (Arndt et al., 2010) and endosomal-microautophagy (Sahu et al., 2011).
LAMP2A Targeting and Substrate Translocation
LAMP2A is one of the three splicing variants of the LAMP2 gene, which contains a cytosolic tail that differs from the other isoforms and is crucial for CMA to occur (Cuervo and Dice, 2000b). Substrates can bind to LAMP2A in a folded state, but they must be unfolded to be translocated to the lumen of the lysosome (Salvador et al., 2000). In this aspect, HSC70 and other co-chaperones located in the membrane of the lysosome are thought to be mediating this process (Cuervo and Wong, 2014; Rout et al., 2014). During this step, LAMP2A monomers are multimerized, while the heat shock cognate 90 kDa (HSC90) chaperone stabilizes the complex from the luminal part, hiding the protease-sensitive regions of LAMP2A (Bandyopadhyay et al., 2008). When the translocation complex, which comprises 700 kDa, is formed and the cargo protein is unfolded, the substrates enter the lumen of the lysosome. Moreover, there is a form of HSC70 located within the lysosome (lys-HSC70), which reinforces the translocation of the substrate pulling it and preventing its return into the cytosol (Agarraberes and Dice, 2001). A recently published bioinformatic study demonstrated that several domains of the LAMP2 gene present positive selective pressure in placental mammals, giving new insights regarding the molecular evolution of the key protein of CMA (Jalali and Parvaz, 2020). Future work should define the functional impact of those selective substitutions in CMA and autophagy, particularly in the brain. Nevertheless, dependence on LAMP2A is the best criteria to determine whether a specific protein is degraded by CMA (Kaushik and Cuervo, 2018).
Regulation of CMA Function
The CMA process is tightly regulated and this facilitates the maintenance of cell and organism homeostasis. As LAMP2A is the main effector of CMA, its expression, trafficking, and stabilization are key aspects for CMA to occur. Next, we summarize the regulators of these processes and we provide recent data regarding epigenetic regulation of chaperones and CMA (Figure 2).
Figure 2.

Main levels of regulation of CMA. (1) LAMP2A de novo expression can be regulated by the binding of NFAT1 to the promoter of LAMP2 gene, binding of NRF2 and PI3K/AKT/mTOR pathway; (2) LAMP2A trafficking into the lysosomal membrane is altered by cystinosin and Rab11 and RILP protein dysfunctions; (3) in resting conditions, LAMP2A monomers are located in lipid microdomains, where LAMP2A is more sensitive to cathepsin A-mediated degradation. SNX-10 deficiency upregulates LAMP2A expression, probably by dysregulation of cathepsin A trafficking into the lysosomes; (4) Rac1/PHLPP1 complex inhibits Akt, enabling glial fibrillary acidic protein (GFAP)-mediated translocation complex stabilization. On the contrary, this complex is destabilized by GTP-mediated release of Elongation factor 1α (EF1α) from the lysosomal membrane and mTORC2/Akt phosphorylation of GFAP, promoting GFAP self-association. Humanin, a mitochondria-associated peptide, interacts with HSP90 and stabilizes the binding of this chaperone to CMA substrates; (5) recent studies demonstrate that METT21c and potentially HDAC10 regulate HSC70 action by post-transcriptional regulation. HDAC6 regulates HSP90 acetylation and activity.
De novo LAMP2A Synthesis
The first transcription factor identified in the regulation of CMA was the Nuclear Factor of Activated T Cells 2 (NFATC2), also known as NFAT1, which, after activation by calcium-calcineurin, is recruited to the Lamp2 promoter to induce its expression, mediating the response to reactive oxygen species (ROS) in activated murine T cells (Valdor et al., 2014). Together with NFAT1, nuclear factor erythroid 2-related factor 2 (NRF2) transcription factor has been also described as a modulator of LAMP2A expression and consequently, CMA activity in both human and mouse cell types, as the LAMP2 gene presents binding sequences for NRF2 (Pajares et al., 2018). Interestingly, the study of de novo LAMP2A expression modulation has been extended from specific transcription factors to other signaling pathways. Among them, the PI3K/AKT/mTOR signaling pathway should be pointed out, since it has been described as a negative regulator of LAMP2A and HSC70 expression (Li et al., 2017b), but also macroautophagy activity (Heras-Sandoval et al., 2014). However, the detailed mechanism of how this pathway modulates LAMP2A expression remains to be elucidated.
LAMP2A Trafficking to the Lysosome
Different subcellular distributions of LAMP2 splicing variants have been assigned largely by the COOH-terminal amino acid residue (Gough and Fambrough, 1997). Besides this regulatory level, other key effectors in LAMP2A trafficking have been discovered in a cystinosis model. Herein, cystinosin (CTNS) has been associated with the regulation of LAMP2A trafficking, as Ctns−/− cellular model presents reduced co-localization of lysosomal membrane protein LAMP1 and LAMP2A, and a significantly increased number of LAMP2A-positive vesicles with motility restriction (Zhang et al., 2017). Indeed, up-regulation of Rab11 or Rab7/RILP leads to the correction of the LAMP2A mislocalization in Ctns−/− cells, which shows their regulatory role in LAMP2A trafficking, at least, in this model (Zhang et al., 2017). Future studies should evaluate the role of CTNS, Rab11, and RILP in LAMP2A trafficking under physiological conditions.
LAMP2A Stability in the Lysosomal Membrane
Levels of LAMP2A at the lysosomal membrane are mainly modulated by changes in its distribution and half-life. A percentage of LAMP2A is located in lipid-enriched membrane microdomains during resting conditions, and its organization into multimeric complexes for substrate translocation only occurs outside these regions (Kaushik et al., 2006). Loading of lysosomes with cholesterol significantly reduces CMA, mainly due to the increased susceptibility of LAMP2A to be cleaved by proteases such as cathepsin A and posterior degradation in the lysosomal lumen (Cuervo et al., 2003). In this line, sorting nexin (SNX)-10 deficiency upregulates LAMP2A and so, CMA activation, probably due to a dysregulation of cathepsin A trafficking into lysosomes and thus, protecting LAMP2A from its degradation (You et al., 2018).
Translocation Complex Stability
GTP presents an inhibitory effect on CMA by altering the stability of the translocation complex, mediated by glial fibrillary acidic protein (GFAP) and elongation factor 1α (EF1α), in mouse and rat models (Bandyopadhyay et al., 2010). In the absence of GTP stimulus, GFAP stabilizes the multimeric translocation complex, whereas GTP-mediated release of EF1α from the membrane of the lysosome promotes self-association of GFAP, reducing the stabilization of the complex and consequently diminishing CMA activity (Bandyopadhyay et al., 2010). An additional study shows that the interaction between Akt and mTORC2/PHLPP1 also controls the dynamics of the assembly and disassembly of the translocation complex (Arias et al., 2015). In particular, when associated with the lysosomal membrane, the phosphatase PHLPP1 and the kinase mTORC2 regulate the phosphorylation state of Akt, modulating thus the stability of the translocation complex (Arias et al., 2015). Moreover, Humanin, a mitochondria-associated peptide, interacts with HSP90, and stabilizes the binding of this chaperone to CMA substrates (Gong et al., 2018).
Post-translational Regulation of HSC Chaperones
Several studies have recently shown new functions of deacetylase and methyltransferase enzymes on the activity of chaperones within CMA. An initial study revealed that histone deacetylase 6 (HDAC6) regulates HSP90 acetylation, thus controlling its interaction with LAMP2A and HSC70, under the hypoxia-ischemia-stress induced by acute spinal cord injury (Su et al., 2016). Other studies showed that histone deacetylase 10 (HDAC10) deacetylates HSC70 (Oehme et al., 2013) and that HDAC10 knock-out accumulates LAMP2A positive lysosomes around the nucleus, activating CMA (Obayashi et al., 2020). In the same line, an additional study described that the newly classified methyltransferase-like 21c (METTL21c) trimethylates HSP70 at Lys-561 to enhance its stability, inducing its function in CMA in mice (Wang et al., 2019a). All these recent data exhibit the potential that epigenetic enzymes may have in CMA regulation.
Crosstalk With Other Types of Protein Degradation Systems
Although three isoforms of the LAMP2 gene present a common luminal domain, their different cytosolic and transmembrane domains (Cuervo and Dice, 2000b) give them unique properties that hamper compensation of all functions; while LAMP2A is the only isoform that can work in CMA, LAMP2B is associated to macroautophagy (Nishino et al., 2000; Chi et al., 2019) and LAMP2C to a selective type of autophagy called “RNautophagy” (Fujiwara et al., 2013).
Compensatory mechanisms between different proteolytic systems within the cell have been widely described for the correct proteostasis of the tissue (Park and Cuervo, 2013; Wu et al., 2015; Dong and Cui, 2018). Indeed, a recent proteome-wide study identified a globally harmonized rhythm for basal macroautophagy, CMA, and proteasomal activity for proteolysis during the daytime (Ryzhikov et al., 2019).
Regarding macroautophagy, it can be upregulated in CMA-defective conditions to maintain normal rates of long-lived protein degradation, although is unable to compensate for all CMA features (Massey et al., 2006). Supporting this original evidence, a study showed that the upregulation of CMA mediated by the stress caused by hepatitis C, caused selective degradation of beclin1, thus impairing autophagosome-endosome fusion, a key step in the macroautophagic process (Aydin et al., 2018). In the same way, the blockage of macroautophagy leads to upregulation of CMA (Kaushik et al., 2008), which has been associated with resistance to death induced by oxidative stress from menadione or UV light (Singh and Czaja, 2008). Another evidence that supports this mutual regulation of macroautophagy and CMA is that protein kinase Cα (PKCα) mediated phosphorylation of ULK1, on the one hand, prevents autolysosome formation, but on the other hand, enhances its interaction with HSC70 and increases its degradation through CMA (Wang et al., 2018a). However, macroautophagy and CMA can also work together in a coordinated way, as it is demonstrated in a study where, to occur hydrolysis of lipid droplets (LD) during starvation, CMA degradation of PLIN2 and PLIN3 was enhanced, concurrent with elevated levels of macroautophagy proteins on LDs (Kaushik and Cuervo, 2015). The ubiquitin-proteasome system (UPS) is the main non-lysosomal degradative pathway for ubiquitinated proteins (Pandey et al., 2007). Notably, it has been described as the upregulation of CMA (Koga et al., 2011b) and an accumulation of proteasomal subunits in lysosomes in response to inhibition of the proteasome (Cuervo et al., 1995; Goebel et al., 2020). On the contrary, in the first stages of CMA blockage, the activity of both macroautophagy and UPS has been identified to be impaired, precipitating the intracellular accumulation of altered components and making the cell unable to eliminate them even after the subsequent restoration of normal UPS and activation of macroautophagy (Massey et al., 2008).
Interestingly, some of these mechanisms have been described in the brain. Firstly, ubiquilins, highly conserved ubiquitin-binding proteins, have been recently described as integrators of UPS and lysosomal degradation for neuronal maintenance, via mTOR signaling (Šentürk et al., 2019). Indeed, a previous study demonstrated that ubiquilin is present in autophagosomes from brain tissue of mice and that it is degraded during both macroautophagy and CMA (Rothenberg et al., 2010). Not only ubiquilins but also HDAC6, which interacts with polyubiquitinated proteins, has been identified as an essential mechanistic link between UPS and autophagy in a neurodegenerative model (Pandey et al., 2007).
Cma During Physiological Aging
The important regulatory role of CMA highlights its relevance in organ functionality and tissue maintenance. Indeed, a functional decline of CMA during physiological aging has been found in the last decades (Cuervo and Dice, 2000a; Zhou et al., 2017). Interestingly, it has been described that not its transcriptional rate, but the stability of LAMP2A within the lysosomal membrane is altered with age (Kiffin et al., 2007), which indeed can be further degenerated after chronic exposure to a high-fat diet (Rodriguez-Navarro et al., 2012). On the contrary, mice with an inducible exogenous extra copy of LAMP2A in the liver displayed reduced damaged proteins and improved organ function (Zhang and Cuervo, 2008), reinforcing its action on physiological aging. In line with this, resveratrol, an agent with anti-aging activity, upregulates LAMP2 in hepatocytes (Lee et al., 2019). Liver lysosomes from long-lived both Pou1f1/Pit1 mutant (Snell) mice and growth hormone receptor (ghr) knockout mice also present increased CMA substrate uptake activity (Endicott et al., 2020), further associating the beneficial impact of maintaining CMA with healthspan and longevity.
Cma During Physiological Aging in The Brain
The impact of autophagy and specifically CMA, in physiological brain aging remains largely unknown, although some evidence is supporting the downregulation of relevant proteins involved in it (Table 1, Figure 3). Almost all of the studies published so far remain correlative, suggesting an impaired regulation of de novo LAMP2A synthesis. Among them, a study showed a significant decline of HSC70 (Loeffler et al., 2016) and little changes in LAMP2 (Loeffler et al., 2018) concentration in cerebrospinal fluid (CSF). Interestingly, neuronal nitric oxide synthase (nNOS) has been associated with neuronal aging in rodents, causing LAMP2A reduction and consequent accumulation of CMA substrates (Valek et al., 2019). In the same way, CMA impairment is detrimental for neuronal viability in vivo (Xilouri et al., 2016) and identified PARK7/DJ-1 and DPYSL2/CRMP-2 as the most significantly altered proteins upon LAMP2A downregulation in primary rat cortical neurons (Brekk et al., 2019). Interestingly, PARK7/DJ-1 contains overlapping canonical and acetylation-generated KFERQ-like motifs 10 residues upstream of 106C shown to be key for the activation of this protein by oxidative stress. It has been postulated that the oxidative status of 106C may change the exposure of the motif to HSC70 binding and subsequent CMA-mediated degradation, or prevent it by ubiquitylation (Kirchner et al., 2019). As oxidative stress has been associated with aging, this evidence makes it attractive to propose that post-translational modifications in KFERQ-like motifs of CMA substrates could have an impact on the aging phenotype.
Table 1.
Main studies revealing chaperone-mediated autophagy (CMA) downregulation in brain homeostasis and aging.
| Molecule | Role in CMA | Finding | Model of study | Reference |
|---|---|---|---|---|
| LAMP2A | Key effector | Its reduction induces progressive loss of nigral dopaminergic neurons, increased astro-microgliosis and motor deficits. | Rat substrantia nigra in vivo | Xilouri et al. (2016) |
| Alters the expression of PARK7/DJ-1 and DPYSL2/CRMP-2 proteins, with measurable effects in neuronal homeostasis and phenotype. | Primary rat cortical neurons | Brekk et al. (2019) | ||
| LAMP2 | Gene coding LAMP2A | Downregulation in the expression in aged patients. | Human CSF | Loeffler et al. (2018) |
| HSPA8 | Effector | Downregulation in the expression in aged patients. | Human CSF | Loeffler et al. (2016) |
| nNOS | Regulator | Associated to neuronal aging, causes an accumulation of CMA substrates and loss of LAMP2A. | Human neuroblastoma cell line | Valek et al. (2019) |
CFS, cerebrospinal fluid.
Figure 3.

Current knowledge regarding CMA regulators alteration in brain aging, neurodegenerative diseases, and glioblastoma. Comparison of the pieces of evidence among these three physiopathological conditions of the brain, highlighting the unexplored fields. CSF, cerebrospinal fluid; nNOS, neuronal nitric oxide synthase; PBMC, peripheral blood mononuclear cell.
Studies regarding altered regulation of LAMP2A stability in lysosomal membrane could also be associated with brain aging. In this line, p38MAPK, which phosphorylates LAMP2A causing its lysosomal membrane accumulation and active conformational change (Li et al., 2017a), has been recently identified as a regulator of neuronal and neural stem cell (NSC) activity during brain aging (Moreno-Cugnon et al., 2019, 2020), which support the idea that alterations in CMA regulation may be associated to brain aging.
Besides, a decline in the integrity of the blood-brain barrier (BBB) is a major feature of brain aging, and it has been speculated that downregulation of endothelial annexin A1, a CMA regulator (Liu et al., 2018), maybe partially responsible for the aged BBB phenotype (McArthur et al., 2016).
Considering all these ideas, it is reasonable to suggest that CMA activity could decrease in brain physiological aging, although still very preliminary (Figure 3). Thus, future studies should be directed to the elucidation of this hypothesis, focusing on different cell types among the brain and the crosstalk with UPS or macroautophagy, with a special interest in in vivo models.
Physiological Roles of Cma in The Brain
Protein degradation by CMA regulates diverse cellular processes so that its balanced activity is crucial, especially in the brain. Several studies have linked the onset of aging and age-associated neurodegenerative diseases to the downregulation of CMA (Cuervo and Dice, 2000a; Alfaro et al., 2018; Loeffler, 2019). Regarding malignant transformation, it has been described that CMA presents anti-oncogenic effects in untransformed healthy cells (Arias and Cuervo, 2020) suggesting that brain aging-associated CMA decline could be a risk factor for brain tumorigenesis. Once the damage is accumulated, tumor cells activate CMA constitutively, promoting brain cancer progression (Valdor et al., 2019).
It is widely accepted that both aging and cancer are two manifestations of the accumulation of cellular and DNA damage (López-Otín et al., 2013) so that CMA acts as a cell survival mechanism. In particular, CMA participates in the regulation of various common hallmarks of aging (López-Otín et al., 2013) and cancer (Hanahan and Weinberg, 2011), where, the existing data could suggest that, while brain cancer cells use CMA for survival, age-dependent CMA decline prevents recovery and restoration of damaged tissues (Figure 4).
Figure 4.
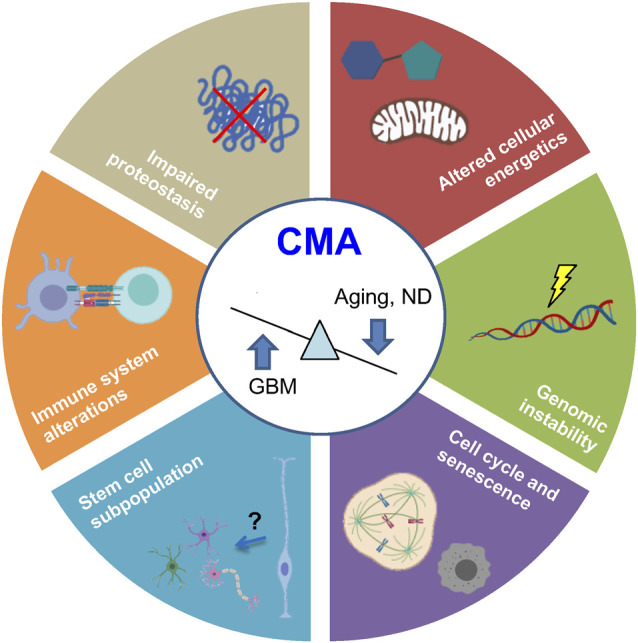
Common hallmarks of aging and cancer regulated by CMA. Recent evidence demonstrates that physiological aging and ND are associated with CMA downregulation, whereas glioblastoma (GBM) presents an upregulated activity. Processes regulated by CMA are represented in the figure. ND, neurodegenerative diseases.
Altered Proteostasis
The most characterized role of CMA is the maintenance of a correct proteostasis, which has been widely described to be impaired in aging and neurodegenerative diseases (López-Otín et al., 2013) and must be maintained in cancer cells to survive (Yang et al., 2019). In this sense, oxidative stress (Law et al., 2016; Issa et al., 2018) and hypoxic stress (Dohi et al., 2012) have been described to activate CMA to reduce protein damage in the brain.
Impaired Cellular Energetics and Metabolism
Besides quality control, there is currently evidence that supports that protein damage is not a requirement for CMA targeting (Kaushik and Cuervo, 2018). Cellular energetics, another common hallmark of aging and cancer, has been linked to CMA since its discovery when it was described that starvation-induced activation (Dice et al., 1986). This evidence was associated for a long time with a mechanism in which a pool of free amino acids was needed. However, recent studies show that the role of enzymes degraded by CMA in metabolism could be responsible for cellular energetic changes (Tasset and Cuervo, 2016). Interestingly, lipid and carbohydrate metabolic alterations caused by altered CMA have been described to be associated with physiological aging of the brain (Hallett et al., 2018) and neurodegenerative diseases (Alfaro et al., 2018). Not only that, but macrophage-specific LAMP2A deficient mice also exhibited significant intracellular lipid accumulation (Qiao et al., 2020), extending the impact of CMA to immune cells, which has been linked to brain homeostasis and disease (Li and Barres, 2018; Dulken et al., 2019). Regarding amino acid metabolism, the accumulation of cystine within the lysosome occurred in cystinosis, a disorder that affects also the brain (Jonas et al., 1987), which is associated with CMA activity (Zhang et al., 2017).
Augmented Genomic Instability
Genotoxic insults together with oxidative stress and starvation are the main stimulus which activates CMA (Park et al., 2015). In this regard, CHK1 participates in genome quality control, and it has been associated with physiological neuronal differentiation (Oshikawa et al., 2017) and resistance of glioblastoma cells to chemo/radiotherapy (Patties et al., 2019). Interestingly, CHK1 has been described as a CMA substrate, as it presents various KFERQ-like motifs in different domains (Kirchner et al., 2019). Indeed, phosphorylation of 345S, separated only by four amino acids from the canonical KFERQ-motif, produces conformational changes, altering HSC70 recognition, and subsequent degradation via CMA (Kirchner et al., 2019). The abnormal nuclear accumulation of phosphorylated CHK1 after a genotoxic insult in CMA-deficient condition alters proteins involved in cell cycle regulation and DNA repair, inducing levels of DNA damage marker p-γH2AX (Park et al., 2015). Indeed, ubiquitylation events in the putative motif of the catalytic region may also modulate the degradation of still inactive CHK1 (Kirchner et al., 2019), suggesting that many post-translational modifications within KFERQ-like motifs may modulate CHK1 degradation via CMA and consequently, regulate DNA repair response.
Cell Cycle Dysregulation and Senescence
Some studies describe the impact of CMA on cell cycle and senescence. On the one hand, tumor suppressor proteins such as p73 (Nguyen et al., 2020) or mutant p53 (Vakifahmetoglu-Norberg et al., 2013), and transcription factors MEF2A (Zhang et al., 2014) and MEF2D (Yang et al., 2009), previously associated to embryonic and adult neurogenesis processes, have been denominated as CMA substrates. Besides, it has been proposed that CMA dysfunction during aging promotes cellular senescence (Moreno-Blas et al., 2018). In contrast, CMA also participates in ferroptosis (Wu et al., 2019b), a specific necrotic cell death that has been linked to diverse age-associated brain diseases (Weiland et al., 2019).
Changes in Stem Cell Subpopulation
Alterations in stem cell subpopulations have been widely described in brain pathologies, with an age-dependent physiological decline of the NSC pool (Schultz and Sinclair, 2016) and the presence of malignant cancer stem cells in glioblastoma (Garros-Regulez et al., 2016; Arrizabalaga et al., 2017). Interestingly, old quiescent NSCs display defects in their lysosomal activity, with an increased accumulation of protein aggregates and reduced ability to be activated (Leeman et al., 2018). Many studies have focused on macroautophagy (Revuelta and Matheu, 2017; Sotthibundhu et al., 2018; Ryskalin et al., 2019), but the impact of CMA in NSC maintenance remains mostly elusive. To date, some studies could give insights about the possible link between stem cell regulation and the CMA process. On the one hand, retinoic acid has been associated with neural and astroglial differentiation during development (Hádinger et al., 2009; Tan et al., 2015), adulthood (Jacobs et al., 2006; Mishra et al., 2018), and glioblastoma pathogenesis (Ying et al., 2011) and this signaling pathway inhibits CMA (Anguiano et al., 2013). In the same way, hypoxia promotes NSC proliferation via Hypoxia-inducible factor 1-α (HIF1α; Hubbi and Semenza, 2015), which is targeted for lysosomal degradation by CMA (Ferreira et al., 2013; Hubbi et al., 2013). Moreover, mice with an extra copy of Arf/p53 display delayed exhaustion of NSCs and enhanced cognitive activity (Carrasco-Garcia et al., 2015, 2017). However, further research would be needed to test these associations and get direct evidence regarding CMA impact, if any, in stem cell maintenance.
Immune System Alterations
The first evidence that linked CMA to the immune system was that LAMP2A participates in the display of cytoplasmic epitopes via class II molecules in antigen-presenting cells (Zhou et al., 2005). Further research has shown a positive association between CMA and activation of T cells (Valdor et al., 2014) and tumor-associated macrophages in hepatocellular carcinoma (Guo et al., 2017) and breast cancer (Wang et al., 2019b). However, very little is known regarding the physiological role of CMA in this organ, although several studies have described the role of other types of autophagy in the brain’s immune system (Plaza-Zabala et al., 2017; Molina et al., 2019). Thus, CMA degrades IKKβ and thereby reduces TNFα expression in microglia (Liu et al., 2018) and CMA inhibition contributes to mesenchymal stem cell immunosuppressive function (Zhang et al., 2020). Further studies are needed to elucidate the role of CMA in the brain’s immune system and its crosstalk with other cell types.
Cma in Neurodegenerative Diseases
It was neurodegenerative Parkinson’s disease (PD) the first disorder associated with CMA (Cuervo et al., 2004). Since then, research has been extended to other prevalent neurodegenerative disorders, demonstrating a reduction in CMA activity in most of them (Table 2, Figure 3).
Table 2.
Main studies revealing CMA downregulation in neurodegenerative diseases.
| Disease type | Role in CMA | Molecule | Finding | Model of study | Reference |
|---|---|---|---|---|---|
| PD | Gene coding LAMP2A | LAMP2 | Study of LAMP2 promoter variants and its association with sporadic PD | Human peripheral leukocytes genomic DNA | Pang et al. (2012) |
| Expression diminishment in female LRRK2 PD | Human CFS | Klaver et al. (2018) | |||
| Effector | LAMP2A | Reduction in the expression of LAMP2A and Hsc70 in PD patients | Substantia nigra pars compacta and amygdala of PD and control brains | Alvarez-Erviti et al. (2010) | |
| Several miRNA regulate negatively LAMP2A in PD | Neuroblastoma cell line and human PD brains | Alvarez-Erviti et al. (2013) | |||
| Impaired endosome-to-golgi retrieval of LAMP2A accumulate α-syn | VPS35 mutant mice | Tang et al. (2015) | |||
| Overexpression of LAMP2A causes decreased α-syninduced dopaminergic neurodegeneration | Human neuroblastoma cells, rat primary cortical neurons in vitro, and nigral dopaminergic neurons in vivo | Xilouri et al. (2013) | |||
| Hsc70 | Reduction of the expresion in sporadic PD | Human PBMCs | Sala et al. (2014) | ||
| CMA substrate | α-syn | First evidence of α-syn degradation by CMA. Effect of mutant forms in CMA perforrmance | Rat ventral midbrain neuronal cultures | Cuervo et al. (2004) | |
| in vivo degradation of α-syn under normal and enhanced protein burden conditions | C57BL/6 and transgenic mice overexpressing wild-type mouse α-syn | Mak et al. (2010) | |||
| Dopamine-modified α-syn effect in CMA | Mouse ventral medial neuron culture, neuroblastoma cell line | Martinez-Vicente et al. (2008) | |||
| LRRK2 | First evidence of LRRK2 degradation by CMA. Effect of G2019S mutant form in CMA perforrmance | BAC transgenic for WT and mutant LRRK2, patient-derived iPSCs, human brain tissue (DMV) | Orenstein et al. (2013) | ||
| Regulator | UCH-L1 | UCH-L1 interaction with LAMP2A, Hsc70 and Hsp90. Effect of I93M UCH-L1 mutation | Human and murine fibroblasts | Kabuta et al. (2008) | |
| Nrf2 | Nrf2 in astrocytes delays CMA dysfunction observed in the hSYN(A53T) mice | GFAP-Nrf2/hSYN(A53T) mice strain | Gan et al. (2012) | ||
| hDJ-1 | Human DJ-1 in astrocytes protects from rotenone-induced neurodegeneration | hDJ-1 vector delivery to the substantia nigra and striatum of adult Lewis rats | De Miranda et al. (2018) | ||
| HDAC6 | Tubastatin A reduces astrocyte reactivity and neurodegeneration | Wilstar rat α-syn model | Francelle et al. (2020) | ||
| AD | CMA substrate | Tau | Tau degradation by CMA. Effect of mutant forms in CMA performance | Murine neuroblastoma cell line | Wang et al. (2009) |
| β-amyloid | Oligomers tagged with KFERQ motifs enter in the lysosomes | iPSC cortical neurons derived from AD patient fibroblasts | Dou et al. (2020) | ||
| Amyloid precursor protein (APP) contains KFERQ motif associated to its processing | Neuroblastoma cell line | Park et al. (2016) | |||
| HD | CMA inductor? | HTT | Mutant HTT induces the expression of LAMP2A and HSC70 | 18QHtt and 111QHtt knock-in mice. Striatum neuronal cultures from HD94 Human lymphoblast from HD patients | Koga et al. (2011a,b) |
| Attachment of KFERQ motifs to HTT induces it degradation by CMA | Murine neuroblastoma cell line, R6/2 HT mice | Bauer et al. (2010) | |||
| SCA7 | CMA substrate? | Ataxin-7 | May be degraded by CMA and poliQ expanded ataxin-7 blocks degradation of other CMA substrates | Mouse Purkinje cells | Duncan et al. (2010) |
| ALS/FTLD | CMA substrate | TDP-43 | TDP-43 degradation by CMA. Effect of mutant forms in CMA performance | Liver and brain of Sprague–Dawley rats | Ormeño et al. (2020) |
| Prion disease | CMA substrate | PrP | Downregulation of LAMP2A and HSC70. PLK3 induces degradation of PrP by CMA | 263K-infected hamster and SMB-S15 cell line | Wang et al. (2017) |
PD, Parkinson’s disease; AD, Alzheimer’s disease; HD, Huntington’s disease; SCA7, spinocerebellar ataxia 7; ALS, Amyotrophic lateral sclerosis; FTLD, Frontotemporal lobal degerenation; α-syn, α-synuclein; CFS, cerebrospinal fluid; DMV, Dorsal motor nucleus of the vagus nerve.
Parkinson’s Disease (PD)
PD is the second most common neurodegenerative disorder after Alzheimer’s disease. It is characterized by an extensive and progressive loss of dopaminergic neurons within the substantia nigra and aggregation of the protein α-synuclein in Lewy bodies. PD is mostly sporadic, except in a 5–10% familial, and mainly related to α-synuclein and leucine-rich repeat kinase 2 (LRRK2) mutations (Campbell et al., 2018; Tolosa et al., 2020). It has been shown that α-synuclein contains a KFERQ-like motif (Cuervo et al., 2004), whereas LRRK2 contains a total of 8 putative motifs (Orenstein et al., 2013). These pieces of evidence are accompanied by both in vitro and in vivo studies demonstrating that they can be degraded by CMA (Cuervo et al., 2004; Mak et al., 2010; Orenstein et al., 2013). Interestingly, this motif is masked in the structure of α-synuclein fibers, supporting the evidence that, once in an oligomeric state, the protein is no longer available for CMA degradation (Kirchner et al., 2019). Indeed, ubiquitylation in 96K and protein partners binding KFERQ-like motif may prevent degradation by CMA of free soluble α-synuclein (Kirchner et al., 2019). In the same line, pathogenic mutant or even dopamine-modified species of α-synuclein and LRRK2 are not able to be degraded by CMA but present a significantly higher affinity to bind to LAMP2A protein than other CMA substrates, blocking the degradation of the later ones (Cuervo et al., 2004; Martinez-Vicente et al., 2008; Orenstein et al., 2013). These data could suggest that changes in the KFERQ-like motif sequence or even any post-translational modification modifying it may be responsible for this augmented affinity of pathogenic forms to reach the lysosomal membrane. Currently, there is not any evidence regarding this idea, so future studies should be intended to determine its impact on CMA disruption in PD.
A familial PD-associated mutation in ubiquitin C-terminal hydrolase L1 (UCH-L1) has been also described as an inhibitor of CMA activity, by promoting an induced aberrant interaction with LAMP2A, HSC70, and HSP90 (Kabuta et al., 2008). Conversely, studies regarding alterations in de novo LAMP2A expression regulation have been also performed. In this line, mutations in LAMP2 gene promoter itself have been also associated with sporadic PD patients (Pang et al., 2012), and there is a decrease in LAMP2A and HSC70 expression in the substantia nigra and amygdala of PD brains compared to control samples (Alvarez-Erviti et al., 2010). Moreover, inductors of PD animal models such as 6-hydroxydopamine (6-OHDA) and rotenone, or even methamphetamine exposure, alter the expression of both Lamp2a and Hsc70, leading to neurotoxicity caused by α-synuclein accumulation (Gao et al., 2014; Sala et al., 2016; Wang et al., 2018b; Sun et al., 2019). Interestingly, several studies have also identified a set of miRNAs that regulate negatively LAMP2A and HSC70 in PD models (Alvarez-Erviti et al., 2013; Li et al., 2014).
In addition to expression alterations, impairment on LAMP2A trafficking has been also found in PD. In particular, it has been suggested that the impaired endosome-to-Golgi retrieval of Lamp2a in VPS35-deficient dopaminergic neurons may contribute to the accumulation of α-synuclein (Tang et al., 2015).
On the contrary, re-activation of CMA by upregulating lamp2a in neurons ameliorate α-synuclein-induced dopaminergic neurodegeneration in mice (Xilouri et al., 2013). Similarly, overexpression of CMA regulators such as nrf2 (Gan et al., 2012) or hDJ-1 in astrocytes (De Miranda et al., 2018) presented also a decreased α-synuclein accumulation. These studies reinforce the role of CMA in PD and highlight the benefits of CMA restoration.
The idea of using CMA effectors as biomarkers for the diagnosis of PD has been also investigated. Indeed, it has been shown that LAMP2 concentrations are decreased in female LRRK2 PD patients compared to healthy individuals (Klaver et al., 2018) in CSF samples. Besides, a significant reduction of HSC70 has been identified in peripheral blood mononuclear cells (PBMCs) of patients of sporadic PD, but no differences in LAMP2A were detected (Sala et al., 2014). Further research should be needed to explore in more detail the potential role of CMA as a PD trait biomarker.
Pharmacological activation of CMA has been postulated as a therapeutic strategy (Campbell et al., 2018). In support of this, co-culture of human pluripotent stem cell (iPSC)-derived astrocytes and neurons from control and PD patients, showed that chemical enhancement of CMA protected both from degeneration (di Domenico et al., 2019). In line with this, the treatment with the selective inhibitor of HDAC6 Tubastatin A reduces both astrocyte reactivity and neuron degeneration induced by α-synuclein in a rat PD model, increasing Lamp2a and Hsc70 expression and partially inducing α-synuclein acetylation (Francelle et al., 2020). Crosstalk between different protein degradation systems is also noteworthy in PD pathogenesis. It has been described that not only CMA but also UPS can participate in the degradation of soluble α-synuclein (Webb et al., 2003). As protein aggregation occurs, macroautophagy is activated as a secondary mechanism to compensate for CMA/UPS blockage and alleviate these conditions (Webb et al., 2003). Interestingly, the connection between autophagy and UPS is not limited to the removal of cytosolic proteins but also involves the removal of organelles. In this line, it has been described that ubiquitination of constituent proteins in the membrane of peroxisomes mediates their macroautophagy, whereas the familial form of PD-associated ubiquitin ligase parkin ubiquitinates mitochondrial proteins, inducing mitophagy (Wong and Cuervo, 2010).
Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common form of dementia in elderly people, characterized classically by the presence of β-amyloid plaques and neurofibrillary tangles of hyperphosphorylated and cleaved forms of the microtubule-associated protein tau (Weller and Budson, 2018). In AD, the translocated mutant tau products at the lysosomal membrane hamper the binding and translocation of other CMA substrates, decreasing their activity (Wang et al., 2009). Not only this, but reduced targeting to the lysosomes has been also observed in a mutant form of tau (Caballero et al., 2018), corroborating a general impaired CMA activity in AD. Interestingly, a regulator of calcineurin 1 (RCAN1) is elevated in AD and contains KFERQ motifs that habilitate it to be degraded by CMA (Liu et al., 2009). As RCAN1 is an inhibitor of calcineurin-dependent dephosphorylation of tau protein, it would be worthy to test the hypothesis that increased degradation of RCAN1 by activation of CMA could be mediating, at least in part, the reduction of the formation of tau aggregates. Together with this, AD-associated ubiquilin is degraded by CMA (Rothenberg et al., 2010), reinforcing the impact of this type of autophagy in AD pathogenesis. Interestingly, a recent study showed that tagging β-amyloid oligomers with multiple KFERQ motifs promoted their entering in endosomes and lysosomes, protecting human primary cultured cortical neurons from neurotoxicity (Dou et al., 2020). Indeed, amyloid precursor protein (APP) by itself, where β-amyloid is derived from, contains a KFERQ motif at its C-terminus which, intriguingly, has been associated with the correct APP processing, not to CMA-dependent degradation (Park et al., 2016). However, deletion of this motif increases the amount of APP cleaved products, suggesting that other KFERQ motif-dependent protein degradation system may be responsible for this effect.
Huntington’s Disease
Huntington’s disease (HD) is a dominantly inherited pathology caused by the accumulation of mutant huntingtin protein (HTT) that contains an expanded N-terminal polyglutamine (polyQ) tract (Bauer et al., 2010). HD is the only neurodegenerative disease with elevated LAMP2A and HSC70 expression (Koga et al., 2011a), as a result of both UPS and macroautophagy failure. The use of an adaptor that contains two copies of polyQ binding sequences and two different KFERQ motifs, directs specifically mutant HTT to the CMA machinery for its degradation, ameliorating symptoms, and extending the life span of an HD mouse model (Bauer et al., 2010). However, the efficiency of this compensatory mechanism may not be sufficient to control the evolution of the disease (Kon et al., 2011), due to the age-dependent CMA activity decline. Thus, the effect of high levels of CMA regulators and whether pharmacological activation of CMA could be beneficial in the evolution of the disease should be investigated.
Other Neurodegenerative Disorders
Associations between CMA alterations and other neurodegenerative disorders have been also described in the last few years. On the one hand, some studies showed that both wild-type and pathological forms of TAR DNA binding protein 43 kDa (TDP-43), whose accumulation in neurons is associated with amyotrophic lateral sclerosis and frontotemporal lobar degeneration, are substrates of CMA and can affect this degradative system (Ormeño et al., 2020). Interestingly, macroautophagy and endocytosis connection is also important in models of frontotemporal dementia. Impaired formation of multivesicular bodies due to ESCRT-III dysfunction in the membrane of the late endosomes reduces autophagy activity and induces autophagosome accumulation (Wong and Cuervo, 2010). This idea demonstrates the crosstalk between multiple types of lysosomal processes in this neurodegenerative disorder.
On the other hand, animal models for mutant prion protein (PrP) disease present downregulation of LAMP2A and HSC70 proteins, and that the overexpression of polo-like kinase 3 (PLK3) induces PrP degradation by CMA (Wang et al., 2017).
Overall, either by the negative effect of the mutant proteins or by the reduction on the expression of the machinery itself, a downregulation of CMA has been described in most neurodegenerative diseases. Further research directed to the study of the other regulatory levels of CMA discussed in previous sections in this work would contribute to the comprehensive view of the altered physiological mechanisms leading to CMA downregulation in ND (Figure 3). Together with this, the potential of CMA as a diagnosis/prognosis biomarker and its use as a therapeutic target remains to be elucidated.
Cma in Glioblastoma
Glioblastoma (GBM) represents the most common and aggressive primary brain tumor and it is classified as grade IV glioma, by the World Health Organization (WHO; Louis et al., 2016). Recent pieces of evidence support that CMA is hyper-activated in GBM (Table 3). Moreover, surgical samples from patients treated with temozolomide (TMZ) chemotherapy present induced expression of LC3B, LAMP1, and LAMP2A compared to the initial sample (Natsumeda et al., 2011). Curcumin treatment, which has shown anti-tumor effects in GBM, downregulates the expression of LAMP2A in GBM cell lines (Maiti et al., 2019) suggesting a cell-autonomous effect of CMA in GBM. In this direction, GBM-associated proteins such as TP53, EGFR, or HIF1α have been identified as CMA substrates (Vakifahmetoglu-Norberg et al., 2013). However, the implication of their altered CMA-mediated degradation in GBM remains to be elucidated. A quiescent subset of endogenous glioma cells named glioma stem cells (GSC) is responsible for sustaining long-term tumor growth and therapy resistance (Chen et al., 2012). The impact of macroautophagy in GSCs has been widely described (Nazio et al., 2019), but the role of CMA fully remains to be investigated. The fact that CMA substrates such as EGFR or HIF1α have been associated with GSCs population (Emlet et al., 2014), and that curcumin treatment potentially may target GSC subpopulation (Fong et al., 2010), indirectly links CMA to GSC activity. However, other studies claim that blocking CMA-mediated HIF1α degradation induces resistance to TMZ (Lo Dico et al., 2018) and that TMZ resistant cells are unable to increase cytoplasmic ROS levels and activate CMA, preventing thus GBM cell toxicity (Lo Dico et al., 2019). In this sense, it has been described that the inhibition of macroautophagy increases the susceptibility of GSCs to TMZ by augmenting ferroptosis, a CMA-associated necrotic cell death mechanism (Buccarelli et al., 2018). Thus, further studies are needed to elucidate the role of CMA in GSC subpopulation and therapy resistance.
Table 3.
Main studies revealing CMA upregulation in glioblastoma (GBM).
| Target | Role in CMA | Finding | Model of study | Reference |
|---|---|---|---|---|
| LAMP2A | Key effector | CMA defective pericytes reduce proliferation of GB. | Murine primary pericytes and U373/U87 GB cell lines. | Valdor et al. (2019) |
| Curcumin reduces LAMP2A expression. | U-87MG, GL261, F98, C6-glioma, and N2a cells. | Maiti et al. (2019) | ||
| TMZ treatment induce expression of LAMP2A | pre-/post-TMZ treatment human tumors. | Natsumeda et al. (2011) | ||
| Block of CMA-mediated HIF1α degradation induces resistance to TMZ. | U251, U87, T98, U138 GB cell lines. | Lo Dico et al. (2018) | ||
| TMZ resistant cells are unable to increase ROS levels and activate CMA, preventing GB cell toxicity | U251 and T98 cell lines. | Lo Dico et al. (2019) | ||
| Ferroptosis | Process in which CMA participate. | Macroautophagy inhibition increases susceptibility of GSCs to TMZ by inducing ferroptosis | Patient-derived GSCs and deparaffinized human tumor sections. | Buccarelli et al. (2018) |
Interestingly, a GBM mouse model grafted in vivo with CMA-defective pericytes shows reduced GB proliferation and effective immune response compared to the ones grafted with control pericytes (Valdor et al., 2019). This fact, together with other studies associating CMA to tumor-associated macrophages in other types of cancer (Guo et al., 2017; Wang et al., 2019b), highlights the potential role of CMA in the crosstalk between the tumor cells and the microenvironment.
Overall, studies regarding CMA in GBM have focused on the impact of the modulation of LAMP2A expression by itself (Figure 3). Nevertheless, the status of LAMP2A trafficking or stability in the lysosomal membrane or even post-translational modifications of LAMP2A, HSC70, or even KFERQ-like motifs of CMA substrates has not been explored.
Of note, it is known that GBM cells need to maintain balanced proteostasis for their survival and progression, by complex crosstalk between protein synthesis, unfolded protein response, stress response pathways, autophagy, and UPS (Yang et al., 2019). However, future studies will be necessary to assess the possible association of CMA with other protein degradation systems in GBM, as it is still a young field and no evidence has been published so far.
Therapeutic Targeting of Cma
The impact of autophagy as a therapeutic target in human brain pathologies has been studied mainly in terms of macroautophagy, where many preclinical studies in PD and GBM have been developed (Towers and Thorburn, 2016; Moors et al., 2017; Taylor et al., 2018). Indeed, few initial clinical trials in GBM with the lysosomal inhibitor chloroquine have been also performed, but with no significant overall survival improvement due to dose-limiting toxicity (Sotelo et al., 2006; Rosenfeld et al., 2014). Importantly, macroautophagy modulators such as 3-methyladenine, LY294002, vinblastine, or rapamycin do not alter CMA activity (Finn et al., 2005). Although to date, there are not specific modulators of CMA, several compounds have demonstrated to alter key effectors of this process. On the one hand, compounds such as geldanamycin (Pedrozo et al., 2013), 6-aminonicotinamide (Finn et al., 2005), glucose-6-phosphate dehydrogenase inhibitor (Finn et al., 2005), silymarin (Tripathi et al., 2020), chronic caffeine (Luan et al., 2018), manganese (Yan et al., 2019), trehalose (Rusmini et al., 2019), β-Asarone (Huang et al., 2016) and other compounds extracted from natural medicinal plants (Wu et al., 2019a), or even combination treatments with bortezomib and suberoylanilide hydroxamic acid (SAHA; Watanabe et al., 2010) increase LAMP2A levels and thus, CMA activity. This therapeutic strategy could be beneficial for neurodegenerative diseases, where, as mentioned before, CMA activity is downregulated.
On the other hand, treatment with compounds such as P140 (Macri et al., 2015), vitamin E (Cao et al., 2009), protein synthesis inhibitors anisomycin and cycloheximide (Finn et al., 2005) or p38MAPK inhibitor (Finn et al., 2005) downregulate CMA, a strategy that may be beneficial for cancer context. Overall, as mentioned before, the modulatory effects of all these compounds are not specific for CMA machinery, they have many other targets. Thus, novel strategies must be developed to get new insights regarding CMA-specific and thus, safer therapeutic approaches. For this, many advances have been done regarding both in vitro and in vivo models for the study of CMA (Juste and Cuervo, 2019). Among them, in vitro methods based on the use of a photoconvertible fluorescent reporter (Koga et al., 2011b) or a “GAPDH-HaloTag” fusion protein (Seki et al., 2012; Sato et al., 2016), and a recently described transgenic reporter mouse that allows dynamic measurement of CMA activity in vivo (Dong et al., 2020) will facilitate further advances in the field.
Concluding Remarks and Future Perspectives
CMA is a protein selective type of autophagy with increasing evidence revealing a role in homeostasis and pathology of a wide variety of organs, with special emphasis on aging and cancer. Regarding the brain, and in line with the protective role of CMA against protein damage and formation of toxic aggregates, much of the evidence gathered so far is focused on neurodegenerative diseases. Alterations in the expression of CMA regulators and the blocking effect of mutant CMA substrates show the reduced activity of this process in disorders such as PD or AD. The study of CMA in physiological brain aging is still a relatively young field, but recent pieces of evidence support the hypothesis that CMA could be involved in the decline in brain homeostasis and regeneration capacity. To consolidate this idea, future studies should be directed to understanding the impact of CMA in different cell types, with special interest on the neurogenic niches, whose impairment has been linked to age-associated cognitive and functional decline. Intriguingly, some evidence demonstrates that glioblastoma presents an upregulated activity of CMA, not only affecting tumor cells but also their crosstalk with pericytes. Similarly, recent studies in PD also show a CMA-mediated modulation of the crosstalk between astrocytes and neurons, highlighting its promising role in intercellular communication. Thus, further efforts should be done to elucidate the impact of CMA in the brain microenvironment during physiological and pathological aging.
Overall, CMA alterations in brain aging, neurodegenerative diseases, or glioblastoma could be interpreted in two directions. On the one hand, it could be postulated that CMA-mediated degradation of key disease-relevant proteins can be modulated, so that while aged cells are not able to degrade mutant species of physiological CMA substrates, cancer cells use CMA to eliminate proteins such as tumor suppressor. On the contrary, it may be suggested that it is the entire CMA process that is altered, being downregulated in aging and upregulated in glioblastoma progression. These ideas encourage further research to understand the specific mechanisms altering CMA activity in brain aging and glioblastoma, as well as to develop selective modulators of CMA as new strategies against age-related pathologies.
Author Contributions
JA-I wrote the draft and AM reviewed it and supervised the work.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. JA-I is the recipient of predoctoral fellowships from the Department of Education, University and Research of the Basque Government (PRE_2016_1_0375). AM lab is supported by grants from Instituto de Salud Carlos III and FEDER Funds (CP16/00039, DTS16/00184, PI16/01580, DTS18/00181, and PI19/01355), Diputacion Guipuzcoa and Industry and Health Departments of the Basque Country.
References
- Agarraberes F. A., Dice J. F. (2001). A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell. Sci. 114, 2491–2499. [DOI] [PubMed] [Google Scholar]
- Alfaro I. E., Albornoz A., Molina A., Moreno J., Cordero K., Criollo A., et al. (2018). Chaperone mediated autophagy in the crosstalk of neurodegenerative diseases and metabolic disorders. Front. Endocrinol. 9:778. 10.3389/fendo.2018.00778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Erviti L., Rodriguez-Oroz M. C., Cooper J. M., Caballero C., Ferrer I., Obeso J. A., et al. (2010). Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 67, 1464–1472. 10.1001/archneurol.2010.198 [DOI] [PubMed] [Google Scholar]
- Alvarez-Erviti L., Seow Y., Schapira A. H., Rodriguez-Oroz M. C., Obeso J. A., Cooper J. M. (2013). Influence of microRNA deregulation on chaperone-mediated autophagy and α-synuclein pathology in Parkinson’s disease. Cell Death Dis. 4:e545. 10.1038/cddis.2013.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anguiano J., Garner T. P., Mahalingam M., Das B. C., Gavathiotis E., Cuervo A. M. (2013). Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat. Chem. Biol. 9, 374–382. 10.1038/nchembio.1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias E., Cuervo A. M. (2020). Pros and cons of chaperone-mediated autophagy in cancer biology. Trends Endocrinol. Metab. 31, 53–66. 10.1016/j.tem.2019.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias E., Koga H., Diaz A., Mocholi E., Patel B., Cuervo A. M. (2015). Lysosomal mTORC2/PHLPP1/Akt regulate chaperone-mediated autophagy. Mol. Cell 59, 270–284. 10.1016/j.molcel.2015.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt V., Dick N., Tawo R., Dreiseidler M., Wenzel D., Hesse M., et al. (2010). Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr. Biol. 20, 143–148. 10.1016/j.cub.2009.11.022 [DOI] [PubMed] [Google Scholar]
- Arrizabalaga O., Moreno-Cugnon L., Auzmendi-Iriarte J., Aldaz P., Ibanez de Caceres I., Garros-Regulez L., et al. (2017). High expression of MKP1/DUSP1 counteracts glioma stem cell activity and mediates HDAC inhibitor response. Oncogenesis 6:401. 10.1038/s41389-017-0003-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydin Y., Stephens C. M., Chava S., Heidari Z., Panigrahi R., Williams D. D., et al. (2018). Chaperone-mediated autophagy promotes beclin1 degradation in persistently infected hepatitis C virus cell culture. Am. J. Pathol. 188, 2339–2355. 10.1016/j.ajpath.2018.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay U., Kaushik S., Varticovski L., Cuervo A. M. (2008). The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol. 28, 5747–5763. 10.1128/MCB.02070-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay U., Sridhar S., Kaushik S., Kiffin R., Cuervo A. M. (2010). Identification of regulators of chaperone-mediated autophagy. Mol. Cell 39, 535–547. 10.1016/j.molcel.2010.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer P. O., Goswami A., Wong H. K., Okuno M., Kurosawa M., Yamada M., et al. (2010). Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 28, 256–263. 10.1038/nbt.1608 [DOI] [PubMed] [Google Scholar]
- Boya P., Reggiori F., Codogno P. (2013). Emerging regulation and functions of autophagy. Nat. Cell Biol. 15, 713–720. 10.1038/ncb2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brekk O. R., Makridakis M., Mavroeidi P., Vlahou A., Xilouri M., Stefanis L. (2019). Impairment of chaperone-mediated autophagy affects neuronal homeostasis through altered expression of DJ-1 and CRMP-2 proteins. Mol. Cell. Neurosci. 95, 1–12. 10.1016/j.mcn.2018.12.006 [DOI] [PubMed] [Google Scholar]
- Buccarelli M., Marconi M., Pacioni S., De Pascalis I., D’Alessandris Q. G., Martini M., et al. (2018). Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 9:841. 10.1038/s41419-018-0864-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero B., Wang Y., Diaz A., Tasset I., Juste Y. R., Stiller B., et al. (2018). Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 17:e12692. 10.1111/acel.12692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell P., Morris H., Schapira A. (2018). Chaperone-mediated autophagy as a therapeutic target for Parkinson disease. Expert. Opin. Ther. Targets 22, 823–832. 10.1080/14728222.2018.1517156 [DOI] [PubMed] [Google Scholar]
- Cao L., Chen R., Xu J., Lin Y., Wang R., Chi Z. (2009). Vitamin E inhibits activated chaperone-mediated autophagy in rats with status epilepticus. Neuroscience 161, 73–77. 10.1016/j.neuroscience.2009.02.059 [DOI] [PubMed] [Google Scholar]
- Carrasco-Garcia E., Arrizabalaga O., Serrano M., Lovell-Badge R., Matheu A. (2015). Increased gene dosage of Ink4/Arf and p53 delays age-associated central nervous system functional decline. Aging Cell 14, 710–714. 10.1111/acel.12343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco-Garcia E., Moreno M., Moreno-Cugnon L., Matheu A. (2017). Increased Arf/p53 activity in stem cells, aging and cancer. Aging Cell 16, 219–225. 10.1111/acel.12574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Li Y., Yu T. S., McKay R. M., Burns D. K., Kernie S. G., et al. (2012). A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 488, 522–526. 10.1038/nature11287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi C., Leonard A., Knight W. E., Beussman K. M., Zhao Y., Cao Y., et al. (2019). LAMP-2B regulates human cardiomyocyte function by mediating autophagosome-lysosome fusion. Proc. Natl. Acad. Sci. U S A 116, 556–565. 10.1073/pnas.1808618116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo A. M., Dice J. F. (1996). A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273, 501–503. 10.1126/science.273.5274.501 [DOI] [PubMed] [Google Scholar]
- Cuervo A. M., Dice J. F. (2000a). Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 275, 31505–31513. 10.1074/jbc.M002102200 [DOI] [PubMed] [Google Scholar]
- Cuervo A. M., Dice J. F. (2000b). Unique properties of lamp2a compared to other lamp2 isoforms. J. Cell. Sci. 113, 4441–4450. [DOI] [PubMed] [Google Scholar]
- Cuervo A. M., Wong E. (2014). Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 24, 92–104. 10.1038/cr.2013.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo A. M., Mann L., Bonten E. J., d’Azzo A., Dice J. F. (2003). Cathepsin A regulates chaperone-mediated autophagy through cleavage of the lysosomal receptor. EMBO J. 22, 47–59. 10.1093/emboj/cdg002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo A. M., Palmer A., Rivett A. J., Knecht E. (1995). Degradation of proteasomes by lysosomes in rat liver. Eur. J. Biochem. 227, 792–800. 10.1111/j.1432-1033.1995.tb20203.x [DOI] [PubMed] [Google Scholar]
- Cuervo A. M., Stefanis L., Fredenburg R., Lansbury P. T., Sulzer D. (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. 10.1126/science.1101738 [DOI] [PubMed] [Google Scholar]
- De Duve C. (1963). The lysosome. Sci. Am. 208, 64–72. 10.1038/scientificamerican0563-64 [DOI] [PubMed] [Google Scholar]
- De Duve C., Wattiaux R. (1966). Functions of lysosomes. Annu. Rev. Physiol. 28, 435–492. 10.1146/annurev.ph.28.030166.002251 [DOI] [PubMed] [Google Scholar]
- De Miranda B. R., Rocha E. M., Bai Q., El Ayadi A., Hinkle D., Burton E. A., et al. (2018). Astrocyte-specific DJ-1 overexpression protects against rotenone-induced neurotoxicity in a rat model of Parkinson’s disease. Neurobiol. Dis. 115, 101–114. 10.1016/j.nbd.2018.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Domenico A., Carola G., Calatayud C., Pons-Espinal M., Muñoz J. P., Richaud-Patin Y., et al. (2019). Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson’s disease. Stem Cell Reports 12, 213–229. 10.1016/j.stemcr.2018.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dice J. F. (1990). Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 15, 305–309. 10.1016/0968-0004(90)90019-8 [DOI] [PubMed] [Google Scholar]
- Dice J. F., Chiang H. L., Spencer E. P., Backer J. M. (1986). Regulation of catabolism of microinjected ribonuclease A. Identification of residues 7-11 as the essential pentapeptide. J. Biol. Chem. 261, 6853–6859. [PubMed] [Google Scholar]
- Dohi E., Tanaka S., Seki T., Miyagi T., Hide I., Takahashi T., et al. (2012). Hypoxic stress activates chaperone-mediated autophagy and modulates neuronal cell survival. Neurochem. Int. 60, 431–442. 10.1016/j.neuint.2012.01.020 [DOI] [PubMed] [Google Scholar]
- Dong S., Aguirre-Hernandez C., Scrivo A., Eliscovich C., Arias E., Bravo-Cordero J. J., et al. (2020). Monitoring spatiotemporal changes in chaperone-mediated autophagy in vivo. Nat. Commun. 11:645. 10.1038/s41467-019-14164-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z., Cui H. (2018). The autophagy-lysosomal pathways and their emerging roles in modulating proteostasis in tumors. Cells 8:4. 10.3390/cells8010004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou J., Su P., Xu C., Wen Z., Mao Z., Li W. (2020). Targeting Hsc70-based autophagy to eliminate amyloid β oligomers. Biochem. Biophys. Res. Commun. 524, 923–928. 10.1016/j.bbrc.2020.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulken B. W., Buckley M. T., Navarro Negredo P., Saligrama N., Cayrol R., Leeman D. S., et al. (2019). Single-cell analysis reveals T cell infiltration in old neurogenic niches. Nature 571, 205–210. 10.1038/s41586-019-1362-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan C., Papanikolaou T., Ellerby L. M. (2010). Autophagy: polyQ toxic fragment turnover. Autophagy 6, 312–314. 10.4161/auto.6.2.11139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emlet D. R., Gupta P., Holgado-Madruga M., Del Vecchio C. A., Mitra S. S., Han S. Y., et al. (2014). Targeting a glioblastoma cancer stem-cell population defined by EGF receptor variant III. Cancer Res. 74, 1238–1249. 10.1158/0008-5472.CAN-13-1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endicott S. J., Boynton D. N., Jr., Beckmann L. J., Miller R. A. (2020). Long-lived mice with reduced growth hormone signaling have a constitutive upregulation of hepatic chaperone-mediated autophagy. Autophagy. 10.1080/15548627.2020.1725378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira J. V., Fôfo H., Bejarano E., Bento C. F., Ramalho J. S., Girão H., et al. (2013). STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 9, 1349–1366. 10.4161/auto.25190 [DOI] [PubMed] [Google Scholar]
- Ferreira J. V., Soares A. R., Ramalho J. S., Pereira P., Girao H. (2015). K63 linked ubiquitin chain formation is a signal for HIF1A degradation by chaperone-mediated autophagy. Sci. Rep. 5:10210. 10.1038/srep10210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn P. F., Mesires N. T., Vine M., Dice J. F. (2005). Effects of small molecules on chaperone-mediated autophagy. Autophagy 1, 141–145. 10.4161/auto.1.3.2000 [DOI] [PubMed] [Google Scholar]
- Fong D., Yeh A., Naftalovich R., Choi T. H., Chan M. M. (2010). Curcumin inhibits the side population (SP) phenotype of the rat C6 glioma cell line: towards targeting of cancer stem cells with phytochemicals. Cancer Lett. 293, 65–72. 10.1016/j.canlet.2009.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francelle L., Outeiro T. F., Rappold G. A. (2020). Inhibition of HDAC6 activity protects dopaminergic neurons from alpha-synuclein toxicity. Sci. Rep. 10:6064. 10.1038/s41598-020-62678-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara Y., Furuta A., Kikuchi H., Aizawa S., Hatanaka Y., Konya C., et al. (2013). Discovery of a novel type of autophagy targeting RNA. Autophagy 9, 403–409. 10.4161/auto.23002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L., Vargas M. R., Johnson D. A., Johnson J. A. (2012). Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J. Neurosci. 32, 17775–17787. 10.1523/JNEUROSCI.3049-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L., She H., Li W., Zeng J., Zhu J., Jones D. P., et al. (2014). Oxidation of survival factor MEF2D in neuronal death and Parkinson’s disease. Antioxid. Redox Signal. 20, 2936–2948. 10.1089/ars.2013.5399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garros-Regulez L., Aldaz P., Arrizabalaga O., Moncho-Amor V., Carrasco-Garcia E., Manterola L., et al. (2016). mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert. Opin. Ther. Targets 20, 393–405. 10.1517/14728222.2016.1151002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel T., Mausbach S., Tuermer A., Eltahir H., Winter D., Gieselmann V., et al. (2020). Proteaphagy in mammalian cells can function independent of ATG5/ATG7. Mol. Cell. Proteomics 19, 1120–1131. 10.1074/mcp.RA120.001983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z., Tasset I., Diaz A., Anguiano J., Tas E., Cui L., et al. (2018). Humanin is an endogenous activator of chaperone-mediated autophagy. J. Cell Biol. 217, 635–647. 10.1083/jcb.201606095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough N. R., Fambrough D. M. (1997). Different steady state subcellular distributions of the three splice variants of lysosome-associated membrane protein LAMP-2 are determined largely by the COOH-terminal amino acid residue. J. Cell Biol. 137, 1161–1169. 10.1083/jcb.137.5.1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B., Li L., Guo J., Liu A., Wu J., Wang H., et al. (2017). M2 tumor-associated macrophages produce interleukin-17 to suppress oxaliplatin-induced apoptosis in hepatocellular carcinoma. Oncotarget 8, 44465–44476. 10.18632/oncotarget.17973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hádinger N., Varga B. V., Berzsenyi S., Környei Z., Madarász E., Herberth B. (2009). Astroglia genesis in vitro: distinct effects of retinoic acid in different phases of neural stem cell differentiation. Int. J. Dev. Neurosci. 27, 365–375. 10.1055/s-0040-1721703 [DOI] [PubMed] [Google Scholar]
- Hallett P. J., Huebecker M., Brekk O. R., Moloney E. B., Rocha E. M., Priestman D. A., et al. (2018). Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol. Aging 67, 189–200. 10.1016/j.neurobiolaging.2018.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Heras-Sandoval D., Pérez-Rojas J. M., Hernández-Damián J., Pedraza-Chaverri J. (2014). The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 26, 2694–2701. 10.1016/j.cellsig.2014.08.019 [DOI] [PubMed] [Google Scholar]
- Huang L., Deng M., He Y., Lu S., Liu S., Fang Y. (2016). β-asarone increases MEF2D and TH levels and reduces α-synuclein level in 6-OHDA-induced rats via regulating the HSP70/MAPK/MEF2D/Beclin-1 pathway: Chaperone-mediated autophagy activation, macroautophagy inhibition and HSP70 up-expression. Behav. Brain Res. 313, 370–379. 10.1016/j.bbr.2016.07.028 [DOI] [PubMed] [Google Scholar]
- Hubbi M. E., Semenza G. L. (2015). Regulation of cell proliferation by hypoxia-inducible factors. Am. J. Physiol. Cell Physiol. 309, C775–C782. 10.1152/ajpcell.00279.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbi M. E., Hu H., Kshitiz, Ahmed I., Levchenko A., Semenza G. L. (2013). Chaperone-mediated autophagy targets hypoxia-inducible factor-1α (HIF-1α) for lysosomal degradation. J. Biol. Chem. 288, 10703–10714. 10.1074/jbc.M112.414771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa A. R., Sun J., Petitgas C., Mesquita A., Dulac A., Robin M., et al. (2018). The lysosomal membrane protein LAMP2A promotes autophagic flux and prevents SNCA-induced Parkinson disease-like symptoms in the Drosophila brain. Autophagy 14, 1898–1910. 10.1080/15548627.2018.1491489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs S., Lie D. C., DeCicco K. L., Shi Y., DeLuca L. M., Gage F. H., et al. (2006). Retinoic acid is required early during adult neurogenesis in the dentate gyrus. Proc. Natl. Acad. Sci. U S A 103, 3902–3907. 10.1073/pnas.0511294103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalali Z., Parvaz N. (2020). Molecular evolution of autophagy rate-limiting factor LAMP2 in placental mammals. Gene 727:144231. 10.1016/j.gene.2019.144231 [DOI] [PubMed] [Google Scholar]
- Jonas A. J., Conley S. B., Marshall R., Johnson R. A., Marks M., Rosenberg H. (1987). Nephropathic cystinosis with central nervous system involvement. Am. J. Med. 83, 966–970. 10.1016/0002-9343(87)90661-9 [DOI] [PubMed] [Google Scholar]
- Juste Y. R., Cuervo A. M. (2019). Analysis of chaperone-mediated autophagy. Methods Mol. Biol. 1880, 703–727. 10.1007/978-1-4939-8873-0_47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabuta T., Furuta A., Aoki S., Furuta K., Wada K. (2008). Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J. Biol. Chem. 283, 23731–23738. 10.1074/jbc.M801918200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S., Cuervo A. M. (2015). Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 17, 759–770. 10.1038/ncb3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S., Cuervo A. M. (2018). The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 19, 365–381. 10.1038/s41580-018-0001-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S., Massey A. C., Cuervo A. M. (2006). Lysosome membrane lipid microdomains: novel regulators of chaperone-mediated autophagy. EMBO J. 25, 3921–3933. 10.1038/sj.emboj.7601283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S., Massey A. C., Mizushima N., Cuervo A. M. (2008). Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol. Biol. Cell 19, 2179–2192. 10.1091/mbc.e07-11-1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiffin R., Kaushik S., Zeng M., Bandyopadhyay U., Zhang C., Massey A. C., et al. (2007). Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J. Cell. Sci. 120, 782–791. 10.1242/jcs.001073 [DOI] [PubMed] [Google Scholar]
- Kirchner P., Bourdenx M., Madrigal-Matute J., Tiano S., Diaz A., Bartholdy B. A., et al. (2019). Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 17:e3000301. 10.1371/journal.pbio.3000301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaver A. C., Coffey M. P., Aasly J. O., Loeffler D. A. (2018). CSF lamp2 concentrations are decreased in female Parkinson’s disease patients with LRRK2 mutations. Brain Res. 1683, 12–16. 10.1016/j.brainres.2018.01.016 [DOI] [PubMed] [Google Scholar]
- Klionsky D. J., Abdelmohsen K., Abe A., Abedin M. J., Abeliovich H., Acevedo Arozena A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. 10.1080/15548627.2015.1100356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga H., Martinez-Vicente M., Arias E., Kaushik S., Sulzer D., Cuervo A. M. (2011a). Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J. Neurosci. 31, 18492–18505. 10.1523/JNEUROSCI.3219-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga H., Martinez-Vicente M., Macian F., Verkhusha V. V., Cuervo A. M. (2011b). A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nat. Commun. 2:386. 10.1038/ncomms1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon M., Kiffin R., Koga H., Chapochnick J., Macian F., Varticovski L., et al. (2011). Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 3:109ra117. 10.1126/scitranslmed.3003182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law C.-Y., Siu C.-W., Fan K., Lai W.-H., Au K.-W., Lau Y.-M., et al. (2016). Lysosomal membrane permeabilization is involved in oxidative stress-induced apoptotic cell death in LAMP2-deficient iPSCs-derived cerebral cortical neurons. Biochem. Biophys. Rep. 5, 335–345. 10.1016/j.bbrep.2016.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Hong S. W., Kwon H., Park S. E., Rhee E. J., Park C. Y., et al. (2019). Resveratrol, an activator of SIRT1, improves ER stress by increasing clusterin expression in HepG2 cells. Cell Stress Chaperones 24, 825–833. 10.1007/s12192-019-01012-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeman D. S., Hebestreit K., Ruetz T., Webb A. E., McKay A., Pollina E. A., et al. (2018). Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 359, 1277–1283. 10.1126/science.aag3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Yang H., Zhu D., Huang H., Liu G., Lun P. (2014). Targeted suppression of chaperone-mediated autophagy by miR-320a promotes α-synuclein aggregation. Int. J. Mol. Sci. 15, 15845–15857. 10.3390/ijms150915845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Barres B. A. (2018). Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 18, 225–242. 10.1038/nri.2017.125 [DOI] [PubMed] [Google Scholar]
- Li W., Zhu J., Dou J., She H., Tao K., Xu H., et al. (2017a). Phosphorylation of LAMP2A by p38 MAPK couples ER stress to chaperone-mediated autophagy. Nat. Commun. 8:1763. 10.1038/s41467-017-01609-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Lu L., Luo N., Wang Y.-Q., Gao H. M. (2017b). Inhibition of PI3K/AKt/mTOR signaling pathway protects against d-galactosamine/lipopolysaccharide-induced acute liver failure by chaperone-mediated autophagy in rats. Biomed. Pharmacother. 92, 544–553. 10.1016/j.biopha.2017.05.037 [DOI] [PubMed] [Google Scholar]
- Liu H., Wang P., Song W., Sun X. (2009). Degradation of regulator of calcineurin 1 (RCAN1) is mediated by both chaperone-mediated autophagy and ubiquitin proteasome pathways. FASEB J. 23, 3383–3392. 10.1096/fj.09-134296 [DOI] [PubMed] [Google Scholar]
- Liu L., An D., Xu J., Shao B., Li X., Shi J. (2018). Ac2-26 Induces IKKβ degradation through chaperone-mediated autophagy via HSPB1 in NCM-treated microglia. Front. Mol. Neurosci. 11:76. 10.3389/fnmol.2018.00076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Dico A., Martelli C., Diceglie C., Lucignani G., Ottobrini L. (2018). Hypoxia-inducible factor-1α activity as a switch for glioblastoma responsiveness to temozolomide. Front. Oncol. 8:249. 10.3389/fonc.2018.00249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Dico A., Salvatore D., Martelli C., Ronchi D., Diceglie C., Lucignani G., et al. (2019). Intracellular redox-balance involvement in temozolomide resistance-related molecular mechanisms in glioblastoma. Cells 8:1315. 10.3390/cells8111315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler D. A. (2019). Influence of normal aging on brain autophagy: a complex scenario. Front. Aging Neurosci. 11:49. 10.3389/fnagi.2019.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler D. A., Klaver A. C., Coffey M. P., Aasly J. O., LeWitt P. A. (2016). Age-related decrease in heat shock 70-kDa protein 8 in cerebrospinal fluid is associated with increased oxidative stress. Front. Aging Neurosci. 8:178. 10.3389/fnagi.2016.00178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler D. A., Klaver A. C., Coffey M. P., Aasly J. O. (2018). Cerebrospinal fluid concentration of key autophagy protein lamp2 changes little during normal aging. Front. Aging Neurosci. 10:130. 10.3389/fnagi.2018.00130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C., Blasco M. A., Partridge L., Serrano M., Kroemer G. (2013). The hallmarks of aging. Cell 153, 1194–1217. 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis D. N., Perry A., Reifenberger G., von Deimling A., Figarella-Branger D., Cavenee W. K., et al. (2016). The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820. 10.1007/s00401-016-1545-1 [DOI] [PubMed] [Google Scholar]
- Luan Y., Ren X., Zheng W., Zeng Z., Guo Y., Hou Z., et al. (2018). Chronic caffeine treatment protects against α-synucleinopathy by reestablishing autophagy activity in the mouse striatum. Front. Neurosci. 12:301. 10.3389/fnins.2018.00301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macri C., Wang F., Tasset I., Schall N., Page N., Briand J. P., et al. (2015). Modulation of deregulated chaperone-mediated autophagy by a phosphopeptide. Autophagy 11, 472–486. 10.1080/15548627.2015.1017179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti P., Scott J., Sengupta D., Al-Gharaibeh A., Dunbar G. L. (2019). Curcumin and solid lipid curcumin particles induce autophagy, but inhibit mitophagy and the PI3K-Akt/mTOR pathway in cultured glioblastoma cells. Int. J. Mol. Sci. 20:399. 10.3390/ijms20020399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak S. K., McCormack A. L., Manning-Bog A. B., Cuervo A. M., Di Monte D. A. (2010). Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 285, 13621–13629. 10.1074/jbc.M109.074617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M., Talloczy Z., Kaushik S., Massey A. C., Mazzulli J., Mosharov E. V., et al. (2008). Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 118, 777–788. 10.1172/JCI32806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzella L., Ahlberg J., Glaumann H. (1981). Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 36, 219–234. 10.1007/BF02912068 [DOI] [PubMed] [Google Scholar]
- Massey A. C., Follenzi A., Kiffin R., Zhang C., Cuervo A. M. (2008). Early cellular changes after blockage of chaperone-mediated autophagy. Autophagy 4, 442–456. 10.4161/auto.5654 [DOI] [PubMed] [Google Scholar]
- Massey A. C., Kaushik S., Sovak G., Kiffin R., Cuervo A. M. (2006). Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. U S A 103, 5805–5810. 10.1073/pnas.0507436103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur S., Loiola R. A., Maggioli E., Errede M., Virgintino D., Solito E. (2016). The restorative role of annexin A1 at the blood-brain barrier. Fluids Barriers CNS 13:17. 10.1186/s12987-016-0043-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S., Kelly K. K., Rumian N. L., Siegenthaler J. A. (2018). Retinoic acid is required for neural stem and progenitor cell proliferation in the adult hippocampus. Stem Cell Reports 10, 1705–1720. 10.1016/j.stemcr.2018.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina M. L., García-Bernal D., Martinez S., Valdor R. (2019). Autophagy in the immunosuppressive perivascular microenvironment of glioblastoma. Cancers 12:102. 10.3390/cancers12010102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moors T. E., Hoozemans J. J., Ingrassia A., Beccari T., Parnetti L., Chartier-Harlin M. C., et al. (2017). Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 12:11. 10.1186/s13024-017-0154-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Blas D., Gorostieta-Salas E., Castro-Obregón S. (2018). Connecting chaperone-mediated autophagy dysfunction to cellular senescence. Ageing Res. Rev. 41, 34–41. 10.1016/j.arr.2017.11.001 [DOI] [PubMed] [Google Scholar]
- Moreno-Cugnon L., Arrizabalaga O., Llarena I., Matheu A. (2020). Elevated p38MAPK activity promotes neural stem cell aging. Aging 12, 6030–6036. 10.18632/aging.102994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Cugnon L., Revuelta M., Arrizabalaga O., Colie S., Moreno-Valladares M., Jimenez-Blasco D., et al. (2019). Neuronal p38α mediates age-associated neural stem cell exhaustion and cognitive decline. Aging Cell 18:e13044. 10.1111/acel.13044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsumeda M., Aoki H., Miyahara H., Yajima N., Uzuka T., Toyoshima Y., et al. (2011). Induction of autophagy in temozolomide treated malignant gliomas. Neuropathology 31, 486–493. 10.1111/j.1440-1789.2010.01197.x [DOI] [PubMed] [Google Scholar]
- Nazio F., Bordi M., Cianfanelli V., Locatelli F., Cecconi F. (2019). Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ. 26, 690–702. 10.1038/s41418-019-0292-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D., Yang K., Chao L., Deng Y., Zhou X., Zhang Z., et al. (2020). Inhibition of tumor suppressor p73 by nerve growth factor receptor via chaperone-mediated autophagy. J. Mol. Cell Biol. 12, 700–712. 10.1093/jmcb/mjaa017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino I., Fu J., Tanji K., Yamada T., Shimojo S., Koori T., et al. (2000). Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (danon disease). Nature 406, 906–910. 10.1038/35022604 [DOI] [PubMed] [Google Scholar]
- Obayashi H., Nagano Y., Takahashi T., Seki T., Tanaka S., Sakai N., et al. (2020). Histone deacetylase 10 knockout activates chaperone-mediated autophagy and accelerates the decomposition of its substrate. Biochem. Biophys. Res. Commun. 523, 246–252. 10.1016/j.bbrc.2019.12.048 [DOI] [PubMed] [Google Scholar]
- Oehme I., Linke J. P., Böck B. C., Milde T., Lodrini M., Hartenstein B., et al. (2013). Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl. Acad. Sci. U S A 110, E2592–E2601. 10.1073/pnas.1300113110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orenstein S. J., Kuo S. H., Tasset I., Arias E., Koga H., Fernandez-Carasa I., et al. (2013). Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 16, 394–406. 10.1038/nn.3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormeño F., Hormazabal J., Moreno J., Riquelme F., Rios J., Criollo A., et al. (2020). Chaperone mediated autophagy degrades TDP-43 protein and is affected by TDP-43 aggregation. Front. Mol. Neurosci. 13:19. 10.3389/fnmol.2020.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshikawa M., Okada K., Tabata H., Nagata K. I., Ajioka I. (2017). Dnmt1-dependent chk1 pathway suppression is protective against neuron division. Development 144, 3303–3314. 10.1242/dev.154013 [DOI] [PubMed] [Google Scholar]
- Pajares M., Rojo A. I., Arias E., Díaz-Carretero A., Cuervo A. M., Cuadrado A. (2018). Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 14, 1310–1322. 10.1080/15548627.2018.1474992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey U. B., Nie Z., Batlevi Y., McCray B. A., Ritson G. P., Nedelsky N. B., et al. (2007). HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863. 10.1038/nature05853 [DOI] [PubMed] [Google Scholar]
- Pang S., Chen D., Zhang A., Qin X., Yan B. (2012). Genetic analysis of the LAMP-2 gene promoter in patients with sporadic Parkinson’s disease. Neurosci. Lett. 526, 63–67. 10.1016/j.neulet.2012.07.044 [DOI] [PubMed] [Google Scholar]
- Park C., Cuervo A. M. (2013). Selective autophagy: talking with the UPS. Cell Biochem. Biophys. 67, 3–13. 10.1007/s12013-013-9623-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C., Suh Y., Cuervo A. M. (2015). Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 6:6823. 10.1038/ncomms7823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.-S., Kim D.-H., Yoon S.-Y. (2016). Regulation of amyloid precursor protein processing by its KFERQ motif. BMB Rep. 49, 337–342. 10.5483/bmbrep.2016.49.6.212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patties I., Kallendrusch S., Böhme L., Kendzia E., Oppermann H., Gaunitz F., et al. (2019). The Chk1 inhibitor SAR-020106 sensitizes human glioblastoma cells to irradiation, to temozolomide and to decitabine treatment. J. Exp. Clin. Cancer Res. 38:420. 10.1186/s13046-019-1434-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrozo Z., Torrealba N., Fernández C., Gatica D., Toro B., Quiroga C., et al. (2013). Cardiomyocyte ryanodine receptor degradation by chaperone-mediated autophagy. Cardiovasc. Res. 98, 277–285. 10.1093/cvr/cvt029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaza-Zabala A., Sierra-Torre V., Sierra A. (2017). Autophagy and microglia: novel partners in neurodegeneration and aging. Int. J. Mol. Sci. 18:598. 10.3390/ijms18030598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao L., Wang H. F., Xiang L., Ma J., Zhu Q., Xu D., et al. (2020). Deficient chaperone-mediated autophagy promotes lipid accumulation in macrophage. J. Cardiovasc. Transl. Res. 10.1007/s12265-020-09986-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revuelta M., Matheu A. (2017). Autophagy in stem cell aging. Aging Cell 16, 912–915. 10.1111/acel.12655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Navarro J. A., Kaushik S., Koga H., Dall’Armi C., Shui G., Wenk M. R., et al. (2012). Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc. Natl. Acad. Sci. U S A 109, E705–E714. 10.1073/pnas.1113036109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld M. R., Ye X., Supko J. G., Desideri S., Grossman S. A., Brem S., et al. (2014). A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 10, 1359–1368. 10.4161/auto.28984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg C., Srinivasan D., Mah L., Kaushik S., Peterhoff C. M., Ugolino J., et al. (2010). Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum. Mol. Genet. 19, 3219–3232. 10.1093/hmg/ddq231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout A. K., Strub M.-P., Piszczek G., Tjandra N. (2014). Structure of transmembrane domain of lysosome-associated membrane protein type 2a (LAMP-2A) reveals key features for substrate specificity in chaperone-mediated autophagy. J. Biol. Chem. 289, 35111–35123. 10.1074/jbc.M114.609446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusmini P., Cortese K., Crippa V., Cristofani R., Cicardi M. E., Ferrari V., et al. (2019). Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 15, 631–651. 10.1080/15548627.2018.1535292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryskalin L., Gaglione A., Limanaqi F., Biagioni F., Familiari P., Frati A., et al. (2019). The autophagy status of cancer stem cells in gliobastoma multiforme: from cancer promotion to therapeutic strategies. Int. J. Mol. Sci. 20:3824. 10.3390/ijms20153824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryzhikov M., Ehlers A., Steinberg D., Xie W., Oberlander E., Brown S., et al. (2019). Diurnal rhythms spatially and temporally organize autophagy. Cell Rep. 26, 1880.e6–1892.e6. 10.1016/j.celrep.2019.01.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu R., Kaushik S., Clement C. C., Cannizzo E. S., Scharf B., Follenzi A., et al. (2011). Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 20, 131–139. 10.1016/j.devcel.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala G., Marinig D., Riva C., Arosio A., Stefanoni G., Brighina L., et al. (2016). Rotenone down-regulates HSPA8/hsc70 chaperone protein in vitro: a new possible toxic mechanism contributing to Parkinson’s disease. Neurotoxicology 54, 161–169. 10.1016/j.neuro.2016.04.018 [DOI] [PubMed] [Google Scholar]
- Sala G., Stefanoni G., Arosio A., Riva C., Melchionda L., Saracchi E., et al. (2014). Reduced expression of the chaperone-mediated autophagy carrier hsc70 protein in lymphomonocytes of patients with Parkinson’s disease. Brain Res. 1546, 46–52. 10.1016/j.brainres.2013.12.017 [DOI] [PubMed] [Google Scholar]
- Salvador N., Aguado C., Horst M., Knecht E. (2000). Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J. Biol. Chem. 275, 27447–27456. 10.1074/jbc.M001394200 [DOI] [PubMed] [Google Scholar]
- Sato M., Seki T., Konno A., Hirai H., Kurauchi Y., Hisatsune A., et al. (2016). Fluorescent-based evaluation of chaperone-mediated autophagy and microautophagy activities in cultured cells. Genes Cells 21, 861–873. 10.1111/gtc.12390 [DOI] [PubMed] [Google Scholar]
- Schultz M. B., Sinclair D. A. (2016). When stem cells grow old: phenotypes and mechanisms of stem cell aging. Development 143, 3–14. 10.1242/dev.130633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki T., Yoshino K. I., Tanaka S., Dohi E., Onji T., Yamamoto K., et al. (2012). Establishment of a novel fluorescence-based method to evaluate chaperone-mediated autophagy in a single neuron. PLoS One 7:e31232. 10.1371/journal.pone.0031232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šentürk M., Lin G., Zuo Z., Mao D., Watson E., Mikos A. G., et al. (2019). Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat. Cell Biol. 21, 384–396. 10.1038/s41556-019-0281-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Czaja M. J. (2008). Compensatory mechanisms and the type of injury determine the fate of cells with impaired macroautophagy. Autophagy 4, 516–518. 10.4161/auto.5800 [DOI] [PubMed] [Google Scholar]
- Sotelo J., Briceño E., López-González M. A. (2006). Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 144, 337–343. 10.7326/0003-4819-144-5-200603070-00008 [DOI] [PubMed] [Google Scholar]
- Sotthibundhu A., Promjuntuek W., Liu M., Shen S., Noisa P. (2018). Roles of autophagy in controlling stem cell identity: a perspective of self-renewal and differentiation. Cell Tissue Res. 374, 205–216. 10.1007/s00441-018-2829-7 [DOI] [PubMed] [Google Scholar]
- Su M., Guan H., Zhang F., Gao Y., Teng X., Yang W. (2016). HDAC6 regulates the chaperone-mediated autophagy to prevent oxidative damage in injured neurons after experimental spinal cord injury. Oxid. Med. Cell. Longev. 2016:7263736. 10.1155/2016/7263736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L., Lian Y., Ding J., Meng Y., Li C., Chen L., et al. (2019). The role of chaperone-mediated autophagy in neurotoxicity induced by alpha-synuclein after methamphetamine exposure. Brain Behav. 9:e01352. 10.1002/brb3.1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan B. T., Wang L., Li S., Long Z. Y., Wu Y. M., Liu Y. (2015). Retinoic acid induced the differentiation of neural stem cells from embryonic spinal cord into functional neurons in vitro. Int. J. Clin. Exp. Pathol. 8, 8129–8135. [PMC free article] [PubMed] [Google Scholar]
- Tang F.-L., Erion J. R., Tian Y., Liu W., Yin D.-M., Ye J., et al. (2015). VPS35 in dopamine neurons is required for endosome-to-golgi retrieval of lamp2a, a receptor of chaperone-mediated autophagy that is critical for α-synuclein degradation and prevention of pathogenesis of Parkinson’s disease. J. Neurosci. 35, 10613–10628. 10.1523/JNEUROSCI.0042-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasset I., Cuervo A. M. (2016). Role of chaperone-mediated autophagy in metabolism. FEBS J. 283, 2403–2413. 10.1111/febs.13677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M. A., Das B. C., Ray S. K. (2018). Targeting autophagy for combating chemoresistance and radioresistance in glioblastoma. Apoptosis 23, 563–575. 10.1007/s10495-018-1480-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolosa E., Vila M., Klein C., Rascol O. (2020). LRRK2 in Parkinson disease: challenges of clinical trials. Nat. Rev. Neurol. 16, 97–107. 10.1038/s41582-019-0301-2 [DOI] [PubMed] [Google Scholar]
- Towers C. G., Thorburn A. (2016). Therapeutic targeting of autophagy. EBioMedicine 14, 15–23. 10.1016/j.ebiom.2016.10.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi M. K., Rasheed M. S. U., Mishra A. K., Patel D. K., Singh M. P. (2020). Silymarin protects against impaired autophagy associated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism. J. Mol. Neurosci. 70, 276–283. 10.1007/s12031-019-01431-8 [DOI] [PubMed] [Google Scholar]
- Vakifahmetoglu-Norberg H., Kim M., Xia H. G., Iwanicki M. P., Ofengeim D., Coloff J. L., et al. (2013). Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 27, 1718–1730. 10.1101/gad.220897.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdor R., García-Bernal D., Riquelme D., Martinez C. M., Moraleda J. M., Cuervo A. M., et al. (2019). Glioblastoma ablates pericytes antitumor immune function through aberrant up-regulation of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. U S A 116, 20655–20665. 10.1073/pnas.1903542116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdor R., Mocholi E., Botbol Y., Guerrero-Ros I., Chandra D., Koga H., et al. (2014). Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat. Immunol. 15, 1046–1054. 10.1038/ni.3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valek L., Heidler J., Scheving R., Wittig I., Tegeder I. (2019). Nitric oxide contributes to protein homeostasis by S-nitrosylations of the chaperone HSPA8 and the ubiquitin ligase UBE2D. Redox Biol. 20, 217–235. 10.1016/j.redox.2018.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Arrington J., Ratliff A. C., Chen J., Horton H. E., Nie Y., et al. (2019a). Methyltransferase-like 21c methylates and stabilizes the heat shock protein Hspa8 in type I myofibers in mice. J. Biol. Chem. 294, 13718–13728. 10.1074/jbc.RA119.008430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Wang H., Zhang D., Luo W., Liu R., Xu D., et al. (2018a). Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat. Commun. 9:3492. 10.1038/s41467-018-05449-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Tian C., Sun J., Chen L. N., Lv Y., Yang X. D., et al. (2017). Overexpression of PLK3 mediates the degradation of abnormal prion proteins dependent on chaperone-mediated autophagy. Mol. Neurobiol. 54, 4401–4413. 10.1007/s12035-016-9985-0 [DOI] [PubMed] [Google Scholar]
- Wang R., Liu Y., Liu L., Chen M., Wang X., Yang J., et al. (2019b). Tumor cells induce LAMP2a expression in tumor-associated macrophage for cancer progression. EBioMedicine 40, 118–134. 10.1016/j.ebiom.2019.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Zhai H., Wang F. (2018b). 6-OHDA induces oxidation of F-box protein fbw7β by chaperone-mediated autophagy in Parkinson’s model. Mol. Neurobiol. 55, 4825–4833. 10.1007/s12035-017-0686-0 [DOI] [PubMed] [Google Scholar]
- Wang Y., Martinez-Vicente M., Krüger U., Kaushik S., Wong E., Mandelkow E. M., et al. (2009). Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum. Mol. Genet. 18, 4153–4170. 10.1093/hmg/ddp367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T., Nagase K., Chosa M., Tobinai K. (2010). Schwann cell autophagy induced by SAHA, 17-AAG, or clonazepam can reduce bortezomib-induced peripheral neuropathy. Br. J. Cancer 103, 1580–1587. 10.1038/sj.bjc.6605954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb J. L., Ravikumar B., Atkins J., Skepper J. N., Rubinsztein D. C. (2003). Alpha-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 278, 25009–25013. 10.1074/jbc.M300227200 [DOI] [PubMed] [Google Scholar]
- Weiland A., Wang Y., Wu W., Lan X., Han X., Li Q., et al. (2019). Ferroptosis and its role in diverse brain diseases. Mol. Neurobiol. 56, 4880–4893. 10.1007/s12035-018-1403-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller J., Budson A. (2018). Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Res 7:F1000 Faculty Rev-1161. 10.12688/f1000research.14506.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong E., Cuervo A. M. (2010). Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 13, 805–811. 10.1038/nn.2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Chen S., Ammar A. B., Xu J., Wu Q., Pan K., et al. (2015). Crosstalk between macroautophagy and chaperone-mediated autophagy: implications for the treatment of neurological diseases. Mol. Neurobiol. 52, 1284–1296. 10.1007/s12035-014-8933-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J. Z., Ardah M., Haikal C., Svanbergsson A., Diepenbroek M., Vaikath N. N., et al. (2019a). Dihydromyricetin and salvianolic acid B inhibit alpha-synuclein aggregation and enhance chaperone-mediated autophagy. Transl. Neurodegener. 8:18. 10.1186/s40035-019-0159-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z., Geng Y., Lu X., Shi Y., Wu G., Zhang M., et al. (2019b). Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc. Natl. Acad. Sci. U S A 116, 2996–3005. 10.1073/pnas.1819728116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xilouri M., Brekk O. R., Landeck N., Pitychoutis P. M., Papasilekas T., Papadopoulou-Daifoti Z., et al. (2013). Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain 136, 2130–2146. 10.1093/brain/awt131 [DOI] [PubMed] [Google Scholar]
- Xilouri M., Brekk O. R., Polissidis A., Chrysanthou-Piterou M., Kloukina I., Stefanis L. (2016). Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 12, 2230–2247. 10.1080/15548627.2016.1214777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D.-Y., Liu C., Tan X., Ma Z., Wang C., Deng Y., et al. (2019). Mn-induced neurocytes injury and autophagy dysfunction in α-synuclein wild-type and knock-out mice: highlighting the role of alpha-synuclein. Neurotox Res 36, 66–80. 10.1007/s12640-019-00016-y [DOI] [PubMed] [Google Scholar]
- Yang Z., Klionsky D. J. (2010). Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12, 814–822. 10.1038/ncb0910-814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K., Niu L., Bai Y., Le W. (2019). Glioblastoma: targeting the autophagy in tumorigenesis. Brain Res. Bull. 153, 334–340. 10.1016/j.brainresbull.2019.09.012 [DOI] [PubMed] [Google Scholar]
- Yang Q., She H., Gearing M., Colla E., Lee M., Shacka J. J., et al. (2009). Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 323, 124–127. 10.1126/science.1166088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying M., Wang S., Sang Y., Sun P., Lal B., Goodwin C. R., et al. (2011). Regulation of glioblastoma stem cells by retinoic acid: role for notch pathway inhibition. Oncogene 30, 3454–3467. 10.1038/onc.2011.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y., Li W.-Z., Zhang S., Hu B., Li Y.-X., Li H.-D., et al. (2018). SNX10 mediates alcohol-induced liver injury and steatosis by regulating the activation of chaperone-mediated autophagy. J. Hepatol. 69, 129–141. 10.1016/j.jhep.2018.01.038 [DOI] [PubMed] [Google Scholar]
- Zhang C., Cuervo A. M. (2008). Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med. 14, 959–965. 10.1038/nm.1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Huang J., Gu Y., Xue M., Qian F., Wang B., et al. (2020). Inflammation-induced inhibition of chaperone-mediated autophagy maintains the immunosuppressive function of murine mesenchymal stromal cells. Cell. Mol. Immunol. 10.1038/s41423-019-0345-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Johnson J. L., He J., Napolitano G., Ramadass M., Rocca C., et al. (2017). Cystinosin, the small GTPase rab11 and the rab7 effector RILP regulate intracellular trafficking of the chaperone-mediated autophagy receptor LAMP2A. J. Biol. Chem. 292, 10328–10346. 10.1074/jbc.M116.764076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Sun Y., Fei M., Tan C., Wu J., Zheng J., et al. (2014). Disruption of chaperone-mediated autophagy-dependent degradation of MEF2A by oxidative stress-induced lysosome destabilization. Autophagy 10, 1015–1035. 10.4161/auto.28477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D., Li P., Lin Y., Lott J. M., Hislop A. D., Canaday D. H., et al. (2005). Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity 22, 571–581. 10.1016/j.immuni.2005.03.009 [DOI] [PubMed] [Google Scholar]
- Zhou J., Chong S. Y., Lim A., Singh B. K., Sinha R. A., Salmon A. B., et al. (2017). Changes in macroautophagy, chaperone-mediated autophagy and mitochondrial metabolism in murine skeletal and cardiac muscle during aging. Aging 9, 583–599. 10.18632/aging.101181 [DOI] [PMC free article] [PubMed] [Google Scholar]