

Abstract
RTK-RAS-MAPK systems are major signaling pathways for cell fate decisions. Among the several RTK species, it is known that the transient activation of ERK (MAPK) stimulates cell proliferation, whereas its sustained activation induces cell differentiation. In both instances however, RAS activation is transient, suggesting that the strict temporal regulation of its activity is critical in normal cells. RAS on the cytoplasmic side of the plasma membrane is activated by SOS through the recruitment of GRB2/SOS complex to the RTKs that are phosphorylated after stimulation with growth factors. The adaptor protein GRB2 recognizes phospho-RTKs both directly and indirectly via another adaptor protein, SHC. We here studied the regulation of GRB2 recruitment under the SHC pathway using single-molecule imaging and fluorescence correlation spectroscopy in living cells. We stimulated MCF7 cells with a differentiation factor, heregulin, and observed the translocation, complex formation, and phosphorylation of cell signaling molecules including GRB2 and SHC. Our results suggest a biphasic regulation of the GRB2/SOS-RAS pathway by SHC: At the early stage (<10 min) of stimulation, SHC increased the amplitude of RAS activity by increasing the association sites for the GRB2/SOS complex on the plasma membrane. At the later stage however, SHC suppressed RAS activation and sequestered GRB2 molecules from the membrane through the complex formation in the cytoplasm. The latter mechanism functions additively to other mechanisms of negative feedback regulation of RAS from MEK and/or ERK to complete the transient activation dynamics of RAS.
Keywords: cell signaling, FCCS, single-molecule imaging, RTK-RAS-MAPK, temporal regulation
Significance
The temporal regulation of cell signaling pathways is emerging as an indispensable process for normal cell activity but many of the underlying mechanisms remain unknown. We report in our current study that an apparently redundant pathway, RTK-SHC-GRB2, operates in parallel to the direct RTK-GRB2 pathway to regulate the dynamics of the GRB2 signaling activation of RAS, which is a key mechanism for cell fate changes. Our present results suggest that SHC changes the intracellular location of its major functions over time to enable the positive and negative regulation of GRB2 function and thereby realize the transient activation of RAS.
Introduction
Receptor tyrosine kinases (RTKs) play a multifaceted role in different biological processes by connecting the extracellular and intracellular environments. The human genome contains 58 RTK genes, and although each RTK protein executes distinct roles and receives specific sets of extracellular signals, the intracellular signaling pathways downstream of all RTKs are very similar and involve the same RAS-MAPK system. Hence, one of the central questions in relation to the biology of the RTKs is how reaction networks that are mostly same can code distinct signals from the extracellular environment. In terms of distinguishing between cell proliferation and differentiation signals, temporal coding is known to be used by some types of RTK [1–3], whereby RTK signaling induces a transient (i.e. within ten minutes) or a sustained (more than several tens of minutes) activation of MAPK (extracellular signal regulated kinase [ERK] in the RAS-MAPK system) in accordance with the stimulation received from a proliferation factor or a differentiation factor, respectively.
Surprisingly however, RAS activation, a key event during RTK-RAS-MAPK signaling, is transient in many cases and is independent of ERK signaling dynamics. It is possible that this is because an uncontrolled elevation in RAS signaling can lead to cancer progression [4], and cells thus need to strictly regulate the duration of RAS activation. Although the involvement of negative feedback loops from ERK to the factors that operate upstream of RAS has been suggested [5–7], the molecular mechanisms underlying the temporal regulation of RAS activation remain to be fully understood. The roles of the signaling pathways that operate between the RTKs and RAS thus require investigation.
The association of extracellular signaling molecules with RTKs typically causes multiple tyrosine phosphorylation events in its cytoplasmic domain, which induce the recruitment of various adapter proteins including GRB2 and p52SHC (hereafter referred to as SHC) from the cytoplasm to the plasma membrane that contains embedded RTK molecules [8,9]. The adapter proteins transmit the signals from RTKs to the RAS/MAPK pathway. Despite their lack of enzymatic activity, these adapter proteins play important roles in both normal and oncogenic cell signaling [10–12]. Upon their membrane translocation, GRB2 and SHC recognize the tyrosine phosphorylation of RTKs using a Src homology 2 (SH2) domain [13] and phosphotyrosine binding (PTB) domain [14], respectively.
Once it has associated with activated RTKs, SHC is phosphorylated by the kinase activity of the RTKs at its Tyr239/240 and Tyr317 residues in its central region containing a collagen homology 1 (CH1) domain [15,16]. These phosphotyrosine residues serve as specific binding sites for PTB and/or SH2 domain-containing proteins including GRB2 [17]. This indicates that GRB2 is involved in two molecular interactions for its translocation to active RTKs; one is the direct binding to phosphorylated RTKs (pRTKs) and the other is indirect binding via phosphorylated SHC (pSHC).
GRB2 possesses two Src homology 3 (SH3) domains that can associate with the proline-rich motifs in other proteins, including Son of Sevenless (SOS), to form protein complexes [18,19]. The membrane translocation of GRB2/SOS complexes recognizing pRTKs or pSHC results in the conversion of neighboring molecules of the inactive GDP-bound form of RAS protein into their active GTP-bound form [20–22]. This RAS activation event in turn activates the MAPK cascade, a three-tiered kinase cascade consisting of RAF, MEK, and ERK [23]. The direct and indirect (pSHC-mediated) recruitment of GRB2 to the pRTKs have been considered to be comparable in their ability to activate the downstream RAS/MAPK signaling pathway [24–26]. In contrast, it has also been reported that the existence of SHC is significant; it plays important roles in the full activation of RAS/MAPK signaling while stimulating cells with low concentrations of growth factors [27], and for a stronger activation of DNA synthesis [24]. These studies suggested that SHC regulates the amplitude of RAS/MAPK signaling.
On the other hand, the roles of SHC in the temporal regulation of RAS/MAPK signaling pathway have not yet been elucidated or even discussed in any detail. We recently found that in a human mammalian cancer derived cell line, MCF7, stimulation with a differentiation factor, heregulin (HRG), induces the prolonged phosphorylation of the ERBB receptors (ERBBs), the RTKs for HRG, and the sustained translocation of SHC, but the translocation of GRB2 to the plasma membrane was transient. This difference in the signaling dynamics between GRB2 and SHC suggested specific roles of SHC in the temporal regulation of the RAS-MAPK system. In our current study, we further investigated how SHC regulates GRB2/SOS signaling in MCF7 cells under HRG stimulation.
Methods
Plasmid construction
CMV-p52SHC and GFP-p52SHC constructs were kindly provided by Kenichi Sato at Kyoto Sangyo University. The pmEGFP-C2 transfer vector (BD Biosciences, Franklin lakes, NJ) harboring a monomeric mutation of GFP was constructed through a direct point mutation of A206K in the pmEGFP-C2 vector, as described previously [28]. To construct the mEGFP-GRB2 transfer vectors (GFP-GRB2), CMV-GRB2 [29] fragments was subcloned into the BglII-XhoI site of the pmEGFP-C2 vector. The Halo7-C1 vector was constructed by exchanging mEGFP for Halo7 (pFN19 HaloTag® T7 SP6 Flexi vector; Promega, Madison, WI) in the EGFP-C1 vector (Takara Bio, Kusatsu, Japan). To construct Halo7-p52SHC transfer vectors (Halo-SHC), CMV-p52SHC inserts were subcloned into the SacI-KpnI sites of the Halo7-C1 vector, respectively. The cDNA of Halo-SHC3F (Y239F/Y240F/Y313F) was generated by exchanging tyrosines 239, 240, and 313 of Halo-SHC with phenylalanine using the SLiCE method. To construct the mEGFP-SOS1 vector (GFP-SOS), the pCGN-HA-hSOS1 fragment [30] was subcloned into the Hind III-SalI sites of the pmEGFP-C2 plasmid. The construction of mEGFP-RAF1 (GFP-RAF) has been described previously [31]. The construction of the cDNA of Halo7-SOS (Halo-SOS) has been described previously [32]. The cDNA of Halo-SOS (R1131K) was constructed through a direct point mutation of R1131K in Halo-SOS as described previously [33].
Cell culture and transfection
MCF7 cells were cultured in Dulbecco’s modified Eagle’s medium (Wako Pure Chemical, Osaka, Japan) supplemented with 10% fetal bovine serum in a 5% CO2 incubator at 37°C. These cultures were transfected with different expression vectors using previously described methods [32]. For the inducible knockdown of SHC, we used ON-TARGETplus Human SHC1 siRNA (Dharmacon, Lafayette, CO). After incubation at 37°C for 20 h, the cells were serum-starved in minimal essential medium (MEM; Nissui, Tokyo, Japan) containing 1.5 mg/mL NaHCO3, 0.3 mg/mL L-glutamine, 15 mM HEPES (pH 7.4, Nacalai Tesque, Kyoto, Japan) and 0.1% fatty acid free BSA (Wako Pure Chemical). Cells were cultures in a 5% CO2 incubator at 37°C for 24 h. To detect Halo-p52SHC and Halo-SHC3F expression, the cells were labeled with 100 nM HaloTag® tetramethylrhodamine (TMR; Promega) in culture medium at 37°C for 15 min and washed repeatedly with HBSS (Sigma-Aldrich, St. Louis, MO). The medium was then replaced with MEM containing 5 mM HEPES (pH 7.4) and 0.1% BSA.
Fluorescence microscopy
GFP-GRB2 and fluorescently labeled Halo-SHC and Halo-SHC3F expression in living cells were observed under a total internal reflection fluorescence (TIRF) microscope to detect translocation to the basal cell surface. This microscope system is based on an inverted microscope (IX83; Olympus, Tokyo, Japan) equipped with a 60×, NA 1.49 oil immersion objective (Apo N; Olympus), and was used as described previously [34] with some modifications. Briefly, for the selective fluorescent excitation of GFP and TMR, OPSL lasers (Sapphire 488 LP and Sapphire 561 LP; Coherent, Santa Clara, CA) and a ZT 488/561 rpc dichroic mirror (Chroma Technology, Bellows Falls, VT) were used. To separate emission signals at the 505–530 nm (GFP) and 560–650 nm (TMR) wavelengths, an image splitting system, WVIEW GEMINI-2C (Hamamatsu Photonics, Hamamatsu, Japan) was used, equipped with a T560lpxr dichroic mirror (Chroma Technology) and two emission filters, i.e., FF01-514/30-25 514 nm (Semrock, Rochester, NY) for GFP and FF01-605/64-25 605/64 nm BrightLine® single-band bandpass filter (Laser 2000, Cambridgeshire, UK) for TMR. Fluorescence images were acquired using two ORCA-Flash 4.0 V3 Digital CMOS cameras (Hamamatsu Photonics) at 20 fps.
Under the microscope at 25°C, the cells were stimulated with recombinant human NRG1-β1/HRG1-β1 EGF domain (HRG; R&D Systems, Minneapolis, MN) at a final concentration of 10 nM. For inhibition of the kinase activities of MEK, cells were pretreated with 10 μM U0126 (Promega) for 15 min prior to stimulation. The dynamics of the cell signaling protein translocation to the plasma membrane from the cytoplasm were observed on the basal plasma membranes of the cells for 60 min after HRG stimulation via 1.5-min time-lapse imaging [34]. Fluorescent particles of GFP and TMR on the basal plasma membrane in each frame were detected with G-Count Software (G-Angstrom, Sendai, Japan). The relative numbers of these molecules on the plasma membrane were calculated by multiplying the number of fluorescent particles by their fluorescence intensity. The amplitude dynamics of these protein translocation events were normalized with the values obtained before stimulation in each cell. The effects of photobleaching were normalized using the cytoplasmic fluorescence intensities detected by epi-illumination.
Kinetics analysis
Kinetics and statistical analyses of GRB2 on the plasma membrane were performed with Igor Pro 8.0 (WaveMetrics) as described previously [34].
FCCS measurement
Fluorescence cross correlation spectroscopy (FCCS) measurements were performed using a ConfoCor2 instrument (Carl Zeiss, Oberkochen, Germany) equipped with a 40×, NA 1.2 water-immersion objective (C-Apochromat, Carl Zeiss) at 25°C, as described previously [35]. For GFP and TMR excitation, a 488-nm Ar+ laser and a 543-nm He-Ne laser were used, respectively. Emission wavelengths of 505–530 nm for GFP and 600–650 nm for TMR were detected using two avalanche photodiodes after the emission signal was split by a 570-nm beam splitter.
Data analysis was performed with ConfoCor2 software, as described previously [35,36]. The fluorescence autocorrelation and cross-correlation functions were calculated from the following equation:
| (1) |
where, τ is the time delay, I is the fluorescence intensity in the GFP (i) and TMR (j) channels, and the angle brackets denote the time average. The calculated G (τ) values were fitted with the following equation described as previously [37]:
| (2) |
where, Fi and τi are the fraction and diffusion time of the component i, respectively, N is the average number of fluorescent particles in the excitation-detection volume, and s is the structure parameter calibrated using Rhodamine-6G standard solution at room temperature. The relative concentration of GFP ([GFPtotal]), Halotag-TMR ([TMRtotal]), and GFP-TMR complex ([Complex]) were calculated with the following equations:
| (3) |
| (4) |
| (5) |
where, GG (τ) and GR (τ) are the fluorescence autocorrelation functions from the GFP and TMR channels, respectively, Gcross (τ) is the fluorescence cross-correlation function, NA is the Avogadro number, and V is the detection volume. Total (probe plus endogenous) concentrations of SHC [SHCtotal] and GRB2 [GRB2total], and corrected complex [cComplex] were calculated from the following equations:
| (6) |
| (7) |
and
| (8) |
The expression ratio of the endogenous and exogenous GRB2 and SHC in cells simultaneously expressing GRB2 and SHC probes were estimated to be 0.70 and 0.30, respectively. These values were determined from the expression ratios (endogenous/probe) in all cell population detected by western blotting analysis (1.47 and 1.26 for GRB2 and SHC, respectively) and the percentages of cells expressing the probe molecules (0.48 and 0.24 for GFP-GRB2 and Halo-SHC, respectively) (Fig. 1). From the definition of the dissociation constant (Kd), the fraction ratios of the complex to the total [GRB2total] or [SHCtotal] were derived from equations (9) and (10), as described previously [38]:
Figure 1 .
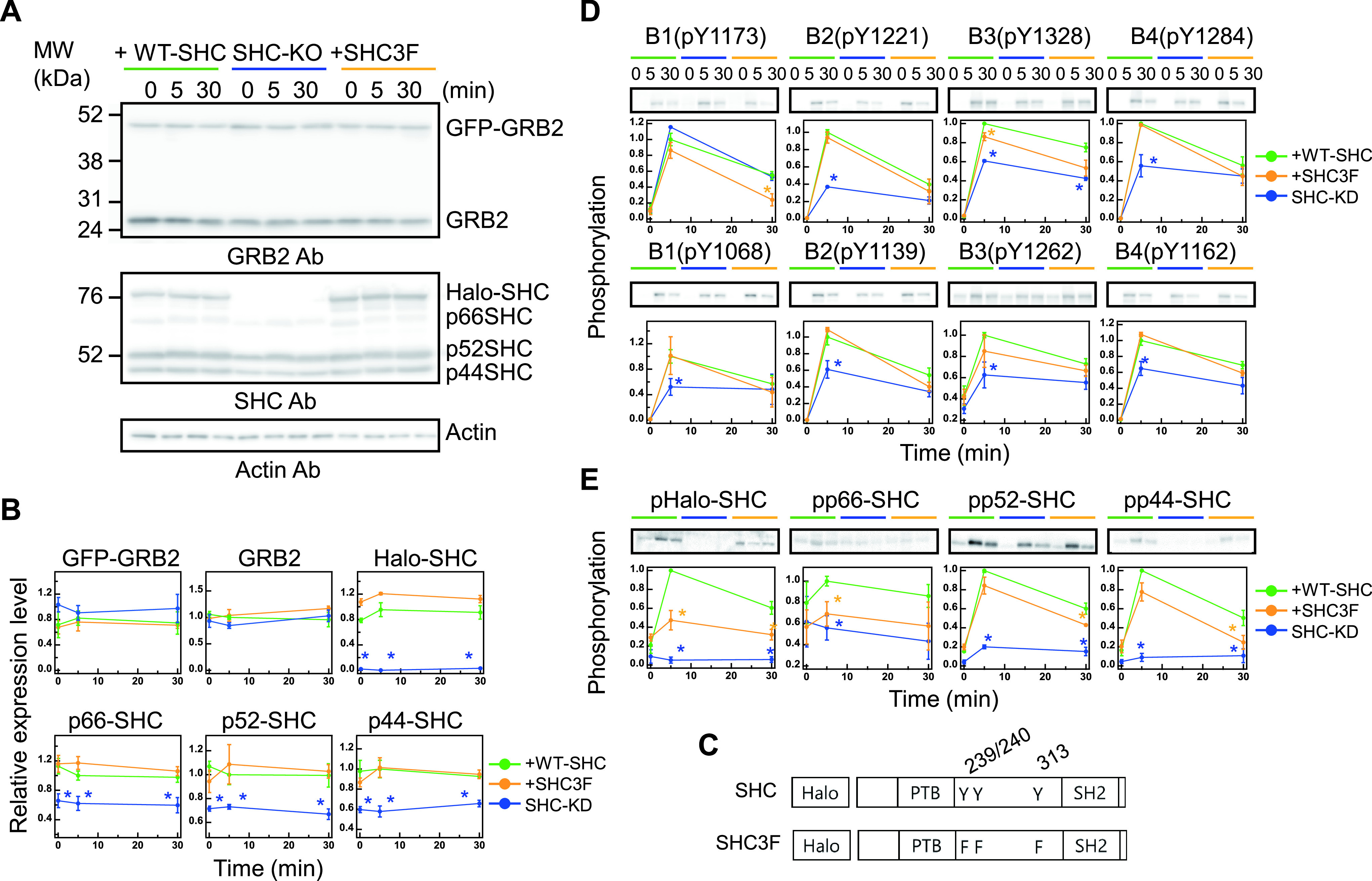
Protein expression and phosphorylation dynamics. (A) The expression profiles of GFP-GRB2 (upper panel), Halo-SHC or Halo-SHC3F (middle panel), and the corresponding endogenous proteins under the indicated cell conditions were assessed by western blotting analysis of cell lysates with the indicated antibodies (Ab). MW, molecular weight markers. (B) Changes in GRB2 and SHC expression after HRG stimulation (0, 5, and 30 min). Values were normalized as described in the Methods section. The expression levels of GFP-GRB2 and Halo-SHC were normalized using the corresponding endogenous protein levels without stimulation, respectively. (C) Schematic structures of the SHC constructs. The HaloTag was fused to the N-termini of the constructs. Numbers represent the amino acid positions. (D, E) Time course analysis of the ERBB receptor (D) and SHC (E) phosphorylations after HRG stimulation (0, 5, and 30 min). Phosphorylations shown in the upper and lower panels in (D) are those of the major SHC and GRB2 binding sites, respectively. Values were normalized as described in the Methods section. In (B, D, E), the mean values for three independent experiments were plotted along with the standard error. Asterisks denote statistical significance against the values obtained in control cells expressing WT-SHC (p<0.05 by t test).
| (9) |
| (10) |
Western blotting
Primary antibodies against the following proteins were used to quantify their expression levels: GRB2 (610111; BD biosciences), SHC (06-203; Millipore, Burlington, MA), and β-actin (A5441; Sigma-Aldrich). The following primary antibodies were used to quantify the indicated phosphorylated protein levels: anti-pERBB1 (pTyr1068; 3777S; CST), anti-pERBB1 (pTyr1173; 4407; CST), anti-pERBB2 (pTyr1139; ab53290; Abcam, Cambridge, UK), anti-pERBB2 (pTyr1221/2; 2243; CST), anti-pERBB3 (pTyr1262; AF5817-sp; R&D Systems), anti-pERBB3 (pTyr1328; ab131444; Abcam), anti-pERBB4 (pTyr1162; ab68478; Abcam), anti-pERBB4 (pTyr1284; 4757; CST), and anti-pSHC (pTyr317; 2431; CST).
Three normalization steps were taken to compare the phosphorylation levels from the immunoblotting data. First, all experimental data were normalized to the staining intensities of β-actin. Second, time series data obtained from the same experiment were normalized using the maximum intensities. Last, these normalized data were further normalized by the values obtained in cells expressing wild type SHC after 5 min of HRG stimulation.
Results and Discussion
The phosphorylation of the ERBBs and SHC is maintained after HRG stimulation
To investigate the spatiotemporal changes of GRB2 and SHC in response to RTK signals, these adaptor proteins were fused with GFP and Halo protein at the N-terminus, respectively (hereafter referred to as GFP-GRB2 and Halo-SHC), and expressed in MCF7 cells. The relative expression levels of GFP-GRB2 and Halo-SHC were measured by western blotting analysis (the expected mobilities on SDS-PAGE for these proteins are 52 and 86 kDa, respectively (Fig. 1A). The fold-increases in the expression levels of GFP-GRB2 and Halo-SHC in comparison with their endogenous counterparts were determined to be 1.4 and 3.3, respectively, after corrections using cell fractions expressing probe molecules. These values were not affected by HRG stimulation (Fig. 1B). In addition to the wild-type (WT) SHC probe, we constructed a dominant-negative mutant SHC probe (SHC3F), in which all three known tyrosine phosphorylation sites (Y239/240 and Y313) were substituted for phenylalanine (Fig. 1C). The phosphorylation of these three tyrosine residues has been reported to generate specific binding sites for GRB2, and the phenylalanine substitutions of these residues do not transmit phosphotyrosine-dependent signals downstream of RTK [15,39,40]. To directly assess the roles of SHC in cell signaling dynamics, we used SHC-knockdown cells. Following the knockdown, expression levels of all three endogenous SHC isoforms (p46, p52, and, p66) were decreased to about 60–70% of the corresponding levels in MCF7 cells (Fig. 1B). The expression of the GRB2 and SHC probes was not affected by the expression of endogenous molecules, and the knockdown of SHC did not affect the expression of GRB2 (Fig. 1A, B).
The ERBB family, which is one of the RTKs, consists of four members (ERBB1–B4), and HRG is a ligand for ERBB3 and B4. The association of HRG with ERBB3 and B4 is known to induce multiple tyrosine phosphorylations among all ERBB family members through a mutual phosphorylation network. We measured the phosphorylation time courses of the major GRB2 and SHC binding sites of all four ERBBs following stimulation with a saturation level concentration of HRG (Fig. 1D). The phosphorylation levels were increased at 5 min then decreased but maintained at significantly high levels (40–70% of 5-min peak) until 30 min after stimulation. The phosphorylation time course of SHC at the major GRB2 binding site (pTyr317 in p52 and its corresponding sites in p46 and p66) was also detected (Fig. 1E). The time courses for WT Halo-SHC and two endogenous isoforms of SHC (p52 and p46) were similar to those of the ERBBs. The p66 isoform showed a higher basal phosphorylation level and lower response to HRG. We used the p52 probe of SHC because it is the isoform mainly responsible for GRB2 signaling [17]. The co-expression of Halo-SHC3F slightly decreased the phosphorylation time course of the ERBBs and endogenous SHC isoforms (Fig. 1D, E).
As shown by our current data, increases in the phosphorylation of ERBBs at the GRB2 and SHC binding sites, and of SHC at the GRB2 binding site, exhibited similar time courses and were maintained for long as 30 min after HRG stimulation. In the conventional model, assuming the parallel recruitment of GRB2 and SHC to the pERBBs, and tandem association of pERBB, pSHC, and GRB2, we expected similar translocation dynamics of GRB2 and SHC to the plasma membrane from the cytoplasm. Upon ligand binding and activation, RTKs are often internalized within the cytoplasm through clathrin-mediated endocytosis [41,42]. However, since the endocytosis of HRG-bound receptors is reported to be slow [43], most ERBB molecules probably remained on the cell surface under our experimental conditions. The maintained phosphorylation status of the binding sites on the ERBBs and SHC suggested a sustained translocation of both GRB2 and SHC to the plasma membrane.
The HRG-induced phosphorylations of the ERBBs, excluding the SHC-binding site (pY1173) of ERBB1, were found to be significantly decreased at 5 min in the SHC-knockdown cells (Fig. 1D). These decreases in ERBB phosphorylation should be a reason of decreased phosphorylation in the endogenous SHC isoforms (Fig. 1E). Mechanism of the decrease in pERBBs after SHC knockdown is unknown. SHC binding may have a function in protecting the dephosphorylation of pERBBs.
HRG induces the transient and sustained membrane localization of SHC and GRB2
Using the fluorescence probe molecules, we observed the translocation dynamics of GRB2 and SHC to the plasma membrane in living cells after stimulation with HRG. In the experiments, a Halo protein fused with SHC was conjugated with tetramethylrhodamine ligand (TMR). Both GFP-GRB2 and Halo-SHC were found to be mainly distributed in the cytoplasm. In addition, occasional associations of the protein particles were observed on the cytoplasmic surface of the plasma membrane by TIRF microscopy even before HRG stimulation, suggesting circulation between the cytoplasm and the plasma membrane (Fig. 2A). Membrane associations prior to stimulation could be caused by basal phosphorylation of the tyrosine residues in ERBBs and other membrane proteins.
Figure 2 .
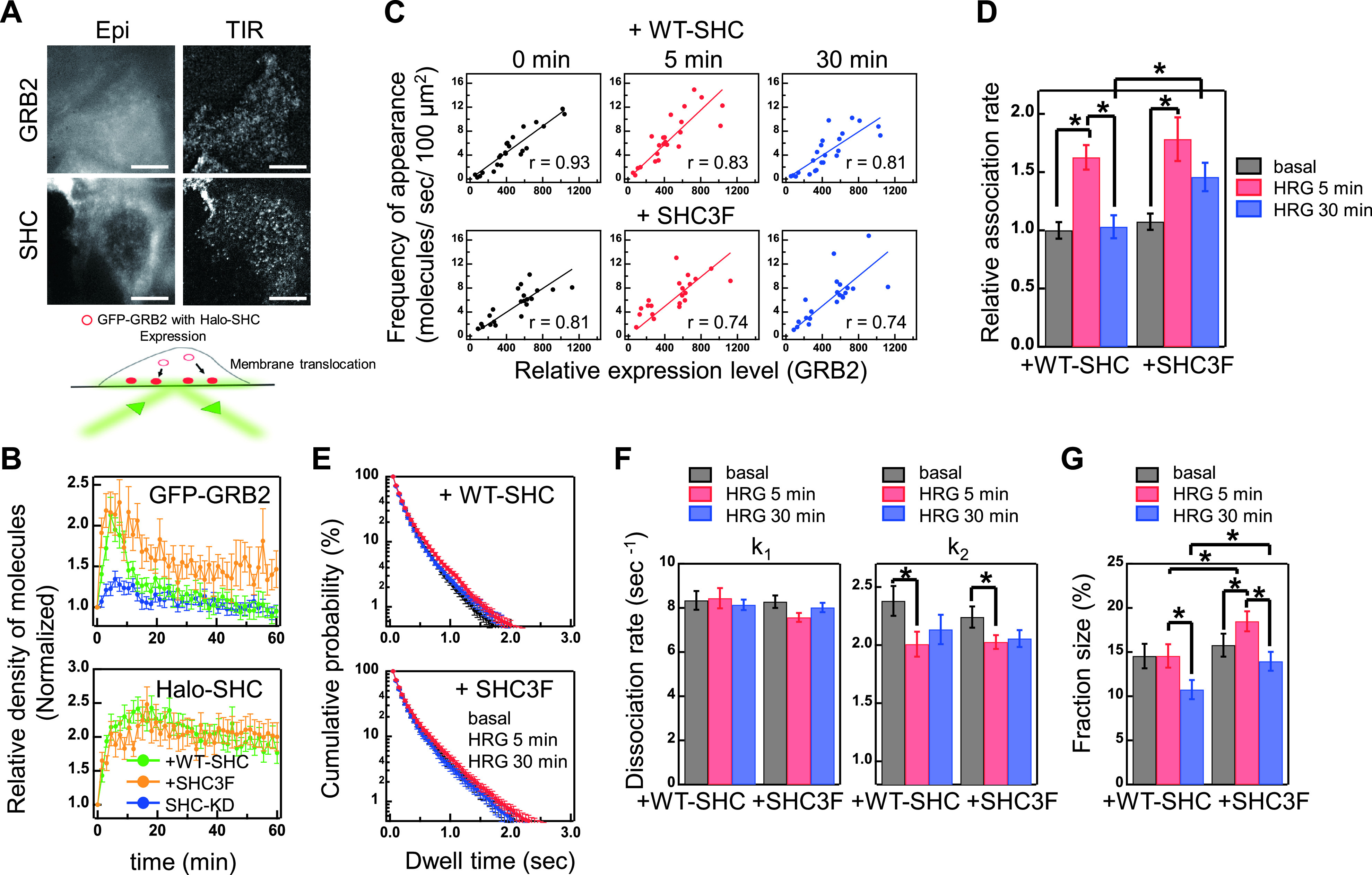
Membrane translocation of GRB2 and SHC. (A) Epi-fluorescence (Epi) and TIRF (TIR) images of GFP-GRB2 and Halo-SHC in MCF7 cells. Scale bar,10 μm. (B) Time course of GFP-GRB2 (left) and Halo-SHC (right) translocations to the plasma membrane. Cells were stimulated with 10 nM HRG at time 0. Cells co-expressing GFP-GRB2 and WT (green) or 3F mutant (orange) of Halo-SHC and expressing GFP-GRB2 with an SHC-knockdown (blue) were examined. Vertical axes were normalized to the values obtained before stimulation. The mean values for 16–32 cells were plotted along with the standard errors. (C) Frequencies of appearance of GFP-GRB2 molecules (relative numbers/sec/100 μm2) on the plasma membrane were plotted against the relative expression level of GFP-GRB2 observed by epi-illumination. Results of linear fitting (lines) are shown with the correlation coefficient, r. (D) Relative association rate constants for GRB2. (E) Dwelling time distributions of single GFP-GRB2 particles on the basal plasma membrane. (F) Dissociation rate constants for the fast (k1, left) and slow (k2, right) dissociation components of GRB2. (G) Fraction sizes for the slow dissociation component of GRB2. Cells in (C-G) were co-expressed with GFP-GRB2 and either WT SHC or its 3F mutant. In (D-G), the mean values before (black), and at 5 min (red), and 30 min (blue) after HRG stimulation are shown with standard errors. Number of cells examined were 21–24 (D), and 27–32 (E-G). Asterisks denote statistical significance (p<0.05 by t test).
The addition of HRG to the culture medium increased the densities of both adaptor proteins on the plasma membrane. The increased membrane localization of GFP-GRB2 was transient for 10 min in cells co-expressing WT Halo-SHC, while the translocation of Halo-SHC in the same population of cells was sustained for at least 60 min after HRG stimulation (Fig. 2B). Because the expression levels of these proteins were not affected by HRG (Fig. 1B), the increases in the densities of these proteins were due to increased density and/or affinity of the binding sites on the plasma membrane after HRG stimulation. This observation was unexpected and clearly indicated that GRB2 and SHC have distinct membrane localization dynamics in MCF7 cells stimulated with HRG, for which the underlying mechanisms are not known.
SHC regulates the amplitude and sustainability of GRB2 translocation
We next examined the membrane localization of GFP-GRB2 when co-expressed with Halo-SHC3F (Fig. 2B). Both the amplitude and sustainability of Halo-SHC3F were comparable to WT Halo-SHC, i.e. the increased membrane localization of Halo-SHC3F was sustained for as long as that of Halo-SHC after HRG stimulation. We anticipated a dominant-negative effect of SHC3F on the membrane translocation of GRB2 but found in contrast that Halo-SHC expression sustained GRB2 translocation for longer (Fig. 2B). On the other hand, the membrane translocation of GFP-GRB2 after HRG stimulation was considerably suppressed in the SHC-knockdown cells at the early stage (<10 min) of signal transduction. This suppression may have been caused by the reduction of indirect pSHC-mediated membrane association sites on GRB2 under the conditions of reduced SHC expression. A decrease in the phosphorylation levels of ERBBs in cells with a SHC-knockdown (Fig. 1D) is likely to be an additional cause of the suppression of the membrane translocation of GRB2.
The effect of SHC on the GRB2 membrane localization was therefore biphasic i.e. at the early stage, SHC increased the amplitude of GRB2 translocation, while at the later stage, SHC3F positively regulated GRB2 translocation, suggesting negative roles of pSHC. Taken together, it appears from our current observations that WT-SHC regulates GRB2 dynamics to be transient after HRG stimulation, even when the upstream ERBB phosphorylation events are sustained.
SHC affects the kinetics of the GRB2 interaction with the plasma membrane
The association kinetics of GFP-GRB2 in relation to the binding sites on the plasma membrane were examined by using single molecule observations. We first measured the appearance frequency of GFP-GRB2 particles per unit of time and unit of area on the plasma membrane [34]. Under our experimental conditions, the frequencies were almost linearly correlated with the relative expression level of GFP-GRB2 detected from the fluorescence intensity in the cytoplasm observed under epi-illumination (Fig. 2C). The relative values of the second-order association rate constants (kon) were determined by normalizing the appearance frequency with the relative expression level (Fig. 2D and Table 1). The time courses of the changes in kon values were similar to those in the membrane translocation of GRB2. Under WT Halo-SHC expression, the kon increased at 5 min after HRG stimulation, then decreased to the basal value at 30 min. Halo-SHC3F expression also increased the kon values at 5 min, which again decreased but were sustained at a higher level than that before stimulation for at least 30 min. At the late stage of HRG stimulation, the kon value in cells expressing Halo-SHC3F was higher than that in the cells with WT Halo-SHC. Increases in the kon values under Halo-SHC3F expression are not readily explained if we expect a dominant-negative effect of SHC3F that will prevent increases in pSHC as the binding site for GRB2 on the plasma membrane.
Table 1 .
The relative association rate constants of GRB2 with the plasma membrane
| HRG | 0 min | 5 min | 30 min |
|---|---|---|---|
| GRB2+WT-SHC | 1.00±0.07a
(4.90±0.7)b |
1.63±0.10 (7.33±0.9) |
1.03±0.10 (5.03±0.8) |
| GRB2+SHC3F | 1.07±0.07 (5.14±0.6) |
1.78±0.19 (7.28±0.7) |
1.46±0.12 (7.16±0.9) |
Times after HRG stimulation are indicated. Averages in 21–23 cells are shown with SE. These are relative values to that obtained before (0 min) HRG stimulation in cells expressing wild type SHC.
a Mean values (+/– SE) of the relative association rate constants.
b Frequency of appearance (molecules/sec/100 μm2).
We next measured the dwelling times of individual GFP-GRB2 particles on the plasma membrane to determine their dissociation kinetics. Distributions of the dwell times can be fitted with a two-component exponential decay function both in cells expressed WT Halo-SHC or Halo-SHC3F (Fig. 2E). Values for the two dissociation rate constants (koff) and fraction sizes of the slow component were calculated from the fitting results (Fig. 2F, G and Table 2). The values of koff1 (for the fast components) showed no significant difference in cells expressed with WT Halo-SHC and Halo-SHC3F and constant with times for cell stimulation. The values of koff2 (for slow components) were also not significantly different between cells with WT Halo-SHC and Halo-SHC3F but decreased after HRG stimulation in both conditions. Fraction sizes of the slow components were different between under expressions of WT Halo-SHC and Halo-SHC3F: The slow component in cells with WT Halo-SHC expression was decreased after 30 min of stimulation, while it was increased in cells with Halo-SHC3F expression after 5 min of stimulation and back to the basal level after 30 min (Fig. 2G).
Table 2 .
The dissociation rate constants of GRB2 with the plasma membrane and its fraction size
| HRG | 0 min | 5 min | 30 min | |||
|---|---|---|---|---|---|---|
| fraction | k1 | k2 | k1 | k2 | k1 | k2 |
| GRB2+WT-SHC | 8.34±0.42a
(85.4±1.4)b |
2.38±0.13 (14.6±1.4) |
8.43±0.46 (85.4±1.3) |
2.01±0.11 (14.6±1.3) |
8.14±0.24 (89.3±1.1) |
2.14±0.13 (10.7±1.1) |
| GRB2+SHC3F | 8.28±0.28 (84.2±1.3) |
2.24±0.09 (15.8±1.3) |
7.57±0.20 (81.5±1.1) |
2.03±0.06 (18.5±1.1) |
7.57±0.20 (81.5±1.1) |
2.06±0.07 (14.0±1.1) |
Times after HRG stimulation are indicated. Averages in 27–32 cells are shown with SE.
a Mean values (+/– SE) of the dissociation rate constant (s–1).
b fraction size (%).
These kinetic analyses revealed that the larger kon value and fraction of slow dissociation component at the later stage of stimulation (30 min) produced the more sustained the membrane translocation of GRB2 in cells with Halo-SHC3F expression. Since SHC3F is a dominant negative mutant which cannot serve all three GRB2 binding sites, the increase in the ratio of slow dissociation fraction with Halo-SHC3F expression likely means that slower fraction represents the direct complex with pERBBs but not the indirect one via pSHC. In other words, in cells with WT-SHC, accumulation of pSHC (faster dissociation site of GRB2) on the pERBBs, which occurs later than the production of pERBBs (slower dissociation site of GRB2) resulted in the decreased density of GRB2 on the plasma membrane at the later stage, and have a role to induce transient translocation of GRB2. In addition, we expect that changes in the kon values (Fig. 2D) contributed to the transient GRB2 dynamics in cells with WT-SHC.
Cytoplasm SHC inhibits the translocation of GRB2 to the plasma membrane
One possibility is that pSHC dissociated into the cytoplasm regulates GRB2 translocation to the plasma membrane. To examine this, we measured interaction between GFP-GRB2 and Halo-SHC in the cytoplasm by using fluorescence cross correlation spectroscopy (FCCS) (Fig. 3A) and detected in addition to the autocorrelation (FCS) signals, significant levels of cross-correlation signals (Fig. 3B). The cytoplasmic concentrations of GFP-GRB2 and TMR-SHC (WT and 3F) determined from the FCS measurements were not changed significantly with time (Fig. 3C), i.e., the membrane bound fractions were small to the cytoplasmic fractions. Values of the dissociation constant (Kd) between GFP-GRB2 and Halo-SHC (or Halo-SHC3F) in the cytoplasm were determined in the same conditions of TIRF imaging before and after HRG stimulation (Fig. 3D). The apparent Kd values between GFP and TMR probes were translated into the values between GRB2 and SHC, including interactions with non-fluorescent endogenous molecules, normalized with the relative expression amounts of the endogenous and probe molecules (Fig. 1; METHODS). In this calculation, total amount of GRB2 and SHC were assumed to be constant during the measurements (Fig. 1).
Figure 3 .

Cytoplasm interaction of GRB2 and SHC. (A) Schematic representation of FCCS measurement (left) and fluorescence micrographs of a cell co-expressing GFP-GRB2 and Halo-SHC (middle). Fluctuations in the fluorescence intensities (count rate) in a detection area (arrows in the middle) were detected for FCCS analysis (right). Scale bar, 10 μm. (B) Typical autocorrelation (left) for GFP-GRB2 (green) and Halo-SHC (red), or cross-correlation (right) curves in the same detection volumes. Curves were fitted (blue lines) with functions described in the Methods section. (C) The relative concentrations of GFP-GRB2 (upper) and Halo-SHC (WT or 3F, lower) were determined from the autocorrelation curves. (D, E) The dissociation constant (Kd) of GRB2/SHC complex (D) and fraction ratios of the complex to the total GRB2 or SHC levels (E) were calculated from the FCCS analysis. The mean values for 14–16 cells are plotted along with the standard errors. Asterisks denote statistical significance (p<0.05 using the t test).
Application of HRG to the culture medium resulted in about two-fold decrease in the Kd value between GRB2 and WT-SHC in the cytoplasm, and this decrease continued for at least 30 min after HRG stimulation (Fig. 3D). On the other hand, no decrease in the Kd values were observed between GRB2 and SHC3F. The decrease in Kd between GRB2 and WT SHC suggests that SHC phosphorylated by the ERBBs on the plasma membrane dissociated into the cytoplasm remaining in the phosphorylated state and formed a complex with cytoplasmic GRB2. We have estimated fraction of the cytoplasmic GRB2/SHC complex within the total amounts of these proteins including endogenous ones (Fig. 3E). The fraction of SHC-GRB2 complex was increased after cell stimulation to about 20% of the total GRB2 and 30% of the total SHC molecules. Because it is highly likely that the cytoplasmic association depends on the phosphotyrosine residues of pSHC similarly to the interaction on the plasma membrane, it must be inhibitory to the recruitment of GRB2 to the plasma membrane.
FCCS analysis suggested that the effective concentration of GRB2 in the cytoplasm was reduced by the complex formation with pSHC produced after HRG stimulation. Reduction of kon must be caused by the reduction of pERBBs on the plasma membrane with time as well (Fig. 1D). However, the observed level of pERBB reduction was insufficient to explain the transient GRB2 dynamics in cells with WT Halo-SHC (Fig. 2B). In cells with Halo-SHC3F expression, increase in the cytoplasmic GRB2/SHC complex was not observed (Fig. 3E) and the change in kon with time (Fig. 2D) was similar to those of pERBB amounts (Fig. 1D). We can conclude that sequestration of GRB2 into the cytoplasm by pSHC is one reason of the transient dynamics of GRB2 translocation to the plasma membrane.
Multiple mechanisms regulate GRB2 translocation dynamics
The GRB2/SOS complex recruited to the plasma membrane activates RAS, which in turn recruits RAF from the cytoplasm for its activation. RAF is the MAPKKK of ERK. We measured the plasma membrane translocation of GFP-SOS and GFP-RAF in cells after HRG stimulation (Fig. 4A). The membrane translocation dynamics of GFP-SOS was transient in all three experimental conditions of SHC examined in this study. The initial peak amplitude was considerably suppressed in cells with SHC3F expression and SHC-knockdown. The RAF dynamics seems to be reflecting the SOS dynamics. Since RAF is a downstream effector molecule that recognizes SOS-activated RAS on the plasma membrane [20,31,44], we anticipated that these dynamics are reflecting the RAS activation.
Figure 4 .

SOS and RAF translocation and effects of a MEK inhibitor. (A) Membrane translocation time courses of GFP-SOS (left) and GFP-RAF (right) in cells co-expressing Halo-SHC (green) or Halo-SHC3F (orange), or with an SHC-knockdown (blue). (B, C) Membrane translocation time courses of GFP-GRB2, Halo-SHC, GFP-SOS, and GFP-RAF in cells pretreated with 10 μM U0126. GRB2, SOS, and RAF were co-expressed with Halo-SHC (green) or Halo-SHC3F (orange). (D) Membrane translocation time courses of SOS R1131K-Halo in cells with co-expression of WT-SHC (green) and SHC3F (orange). Vertical axes were normalized to the values obtained before stimulation. The mean values for 8–40 (A) or 13–30 (B, C) or 8–16 (D) cells were plotted along with the standard errors.
Although we observed an increase in the sustainability of GRB2 translocation in cells with Halo-SHC3F expression (Fig. 2B), SOS translocation was not extended in these cells. It is possible under normal conditions in cells that SHC principally functions as an amplifier of initial GRB2 signaling to RAF, and that its function in the later stage to induce transient GRB2 dynamics is hidden by other mechanisms operating simultaneously on SOS signaling. Negative feedback pathways known to operate for ERK also possibly influence the membrane translocation dynamics of SOS and RAF. For example, ERK induces the expression and phosphorylation of Sprouty protein that binds to GRB2 and inhibits the recruitment of the GRB2/SOS complex to RTK receptors [45,46]. Also, ERK phosphorylates SOS directly and thereby disrupts its association with GRB2 and inhibits SOS recruitment to the plasma membrane [6,47]. These mechanisms may induce the transient localization dynamics of SOS even under prolonged GRB2 translocation conditions.
We measured the membrane translocation dynamics of GFP-GRB2, Halo-SHC, and Halo-SHC3F in cells after pretreatment with a MEK (MAPKK of ERK) inhibitor, U0126, which inhibits ERK activation (Fig. 4B). In cells with WT-SHC expression, GRB2 translocation was prolonged in the presence of this MEK inhibitor, and the expression of SHC3F sustained this even further. The membrane translocations of WT-SHC and SHC3F were not affected by exposure to the MEK inhibitor. These results suggest that the phosphorylation of SHC and a negative feedback signal from MEK and/or ERK are additively suppressing sustained GRB2 translocation. MEK inhibition also increased the temporal widths of the SOS and RAF translocations to the plasma membrane (Table 3). However, the SOS dynamics were still significantly more transient than that of GRB2, and SHC3F had no further effect on the membrane translocation of SOS or RAF after MEK inhibition (Fig. 4C), indicating that SOS regulation independent of GRB2 translocation is not disrupted by this MEK inhibitor.
Table 3 .
Temporal widths of the protein translocation dynamics to the plasma membrane
| Inhibitor | U1026 | ||||
|---|---|---|---|---|---|
| condition | +WT-SHC | +SHC3F | SHC-KD | +WT-SHC | +SHC3F |
| GFP-SOS | 5.6±0.5 (8) | 6.6±0.8 (21) | 6.0±1.4 (8) | *7.2±0.4 (21) | *8.2±0.4 (20) |
| HaloSOS-R1131K | *9.2±1.0 (8) | *9.8±1.2 (16) | |||
| GFP-RAF | 11.8±0.6 (40) | *8.5±0.6 (27) | 9.6±1.3 (19) | *20.9±1.9 (30) | *18.8±2.3 (12) |
a Means values (+/– SE) of the full-width at half maximum (min). Numbers in parentheses are the number of measured cells. Asterisks indicate statistical significances against WT-SHC condition (p<0.05 in t-test).
To separate negative feedback effects on GRB2/SOS complex formation from other steps of RAS signaling, we next examined the membrane translocation of SOS in cells with expression of R1131K mutant of SOS. This mutation has been suggested to increase GRB2/SOS association and reduce the negative effect of SOS phospholyations in its GRB2 association domain [33]. As expectedly, the duration of SOS translocation became more sustained after co-expression of SHC-3F (Fig. 4D). These results suggested SOS phosphorylation by a negative feedback loop is another mechanism to produce transient RAS activation.
In this study, we analyzed the temporal regulation of GRB2 translocation. GRB2 is an adaptor protein connecting activated RTKs, including the ERBBs, and SOS, an activator of RAS. We found that another adaptor protein, SHC regulates GRB2 dynamics. Our results suggest a biphasic model of SHC function (Fig. 5) i.e. in the early stages of signaling, SHC increases the amplitude of GRB2 translocation by increasing the phosphorylation levels of the ERBBs and also providing an additional recruitment pathway from GRB2 to the ERBBs. This function is executed on the plasma membrane. At the later stage, the role of SHC in relation to the GRB2 pathway is inhibitory. This latter role occurs in the cytoplasm through the generation of a complex with GRB2 that reduces its effective cytoplasmic concentration. Thus, SHC changes its location in accordance with the timing of cell signaling processes.
Figure 5 .
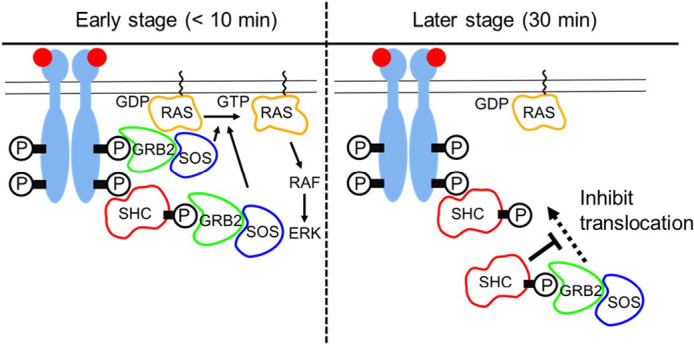
Biphasic spatiotemporal regulation of GRB2 by SHC. Recognizing the phosphorylation sites (P) on the activated EGFR molecules, GRB2 and SHC are translocated to the plasma membrane. At the early stage (<10 min), SHC increases the density of GRB2/SOS complex on the plasma membrane to increase the amplitude of RAS activity. Whereas, at the later stage (30 min), SHC inhibits the membrane translocation of GRB2 through the complex formation in the cytoplasm and suppresses RAS activation.
In conclusion, cells have multiple mechanisms that operate simultaneously to achieve the temporal regulation of RAS activation. SHC functions in a GRB2 translocation step. Other mechanisms, including negative feedback loops downstream of MEK, regulate GRB2 translocation and GRB2/SOS interaction. Hence, the amplitude and timing of RAS activation is controlled in living cells.
Acknowledgments
RY was supported by a Grant-in Aid for JSPS Fellows. YS was supported by MEXT Japan with Grants-in-Aid for Scientific Research (17H06021, 19H05647) and by JST CREST (JPMJCR1912). We thank Hiromi Sato for technical assistance and Kenichi Sato for providing expression vectors.
Conflicts of Interest
The authors declare no conflicts of interest in relation to this study.
Author Contributions
Conceived and designed the experiments: RY, NU, MM, and YS. Performed the experiments: RY and AY. Analyzed the data: RY. Wrote the paper: RY and YS.
References
- [1].Marshall, C. J. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80, 179–185 (1995). DOI: 10.1016/0092-8674(95)90401-8 [DOI] [PubMed] [Google Scholar]
- [2].Kao, S., Jaiswal, R. K., Kolch, W. & Landreth, G. E.. Identification of the mechanisms regulating the differential activation of the mapk cascade by epidermal growth factor and nerve growth factor in PC12 cells. J. Biol. Chem. 276, 18169–18177 (2001). DOI: 10.1074/jbc.M008870200 [DOI] [PubMed] [Google Scholar]
- [3].Nagashima, T., Shimodaira, H., Ide, K., Nakakuki, T., Tani, Y., Takahashi, K., et al. Quantitative transcriptional control of ErbB receptor signaling undergoes graded to biphasic response for cell differentiation. J. Biol. Chem. 282, 4045–4056 (2007). DOI: 10.1074/jbc.M608653200 [DOI] [PubMed] [Google Scholar]
- [4].Sharma, S. V., Bell, D. W., Settleman, J. & Haber, D. A.. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 7, 169–181 (2007). DOI: 10.1038/nrc2088 [DOI] [PubMed] [Google Scholar]
- [5].Lake, D., Correa, S. A. & Muller, J.. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell. Mol. Life Sci. 73, 4397–4413 (2016). DOI: 10.1007/s00018-016-2297-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Langlois, W. J., Sasaoka, T., Saltiel, A. R. & Olefsky, J. M.. Negative feedback regulation and desensitization of insulin- and epidermal growth factor-stimulated p21ras activation. J. Biol. Chem. 270, 25320–25323 (1995). DOI: 10.1074/jbc.270.43.25320 [DOI] [PubMed] [Google Scholar]
- [7].Porfiri, E. & McCormick, F.. Regulation of epidermal growth factor receptor signaling by phosphorylation of the ras exchange factor hSOS1. J. Biol. Chem. 271, 5871–5877 (1996). DOI: 10.1074/jbc.271.10.5871 [DOI] [PubMed] [Google Scholar]
- [8].Mendoza, M. C., Er, E. E. & Blenis, J.. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem. Sci. 36, 320–328 (2011). DOI: 10.1016/j.tibs.2011.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nepstad, I., Hatfield, K. J., Grønningsæter, I. S. & Reikvam, H.. The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 21, 2907 (2020). DOI: 10.3390/ijms21082907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Davol, P. A., Bagdasaryan, R., Elfenbein, G. J., Maizel, A. L.& Frackelton, A. R., Jr.. Shc proteins are strong, independent prognostic markers for both node-negative and node-positive primary breast cancer. Cancer Res. 63, 6772–6783 (2003). [PubMed] [Google Scholar]
- [11].Gu, H., Maeda, H., Moon, J. J., Lord, J. D., Yoakim, M., Nelson, B. H., et al. New role for Shc in activation of the phosphatidylinositol 3-kinase/Akt pathway. Mol. Cell. Biol. 20, 7109–7120 (2000). DOI: 10.1128/mcb.20.19.7109-7120.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ursini-Siegel, J., Hardy, W. R., Zuo, D., Lam, S. H., Sanguin-Gendreau, V., Cardiff, R. D., et al. ShcA signalling is essential for tumour progression in mouse models of human breast cancer. EMBO J. 27, 910–920 (2008). DOI: 10.1038/emboj.2008.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pelicci, G., Lanfrancone, L., Grignani, F., McGlade, J., Cavallo, F., Forni, G., et al. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 70, 93–104 (1992). DOI: 10.1016/0092-8674(92)90536-l [DOI] [PubMed] [Google Scholar]
- [14].Pawson, T. & Scott, J. D.. Signaling through scaffold, anchoring, and adaptor proteins. Science 278, 2075–2080 (1997). DOI: 10.1126/science.278.5346.2075 [DOI] [PubMed] [Google Scholar]
- [15].van der Geer, P., Wiley, S., Gish, G. D. & Pawson, T.. The Shc adaptor protein is highly phosphorylated at conserved, twin tyrosine residues (Y239/240) that mediate protein-protein interactions. Curr. Biol. 6, 1435–1444 (1996). DOI: 10.1016/s0960-9822(96)00748-8 [DOI] [PubMed] [Google Scholar]
- [16].Rozakis-Adcock, M., McGlade, J., Mbamalu, G., Pelicci, G., Daly, R., Li, W., et al. Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature 360, 689–692 (1992). DOI: 10.1038/360689a0 [DOI] [PubMed] [Google Scholar]
- [17].Ravichandran, K. S. Signaling via Shc family adapter proteins. Oncogene 20, 6322–6330 (2001). DOI: 10.1038/sj.onc.1204776 [DOI] [PubMed] [Google Scholar]
- [18].Songyang, Z., Shoelson, S. E., McGlade, J., Olivier, P., Pawson, T., Bustelo, X. R., et al. Specific motifs recognized by the SH2 domains of Csk, 3BP2, fps/fes, GRB-2, HCP, SHC, Syk, and Vav. Mol. Cell. Biol. 14, 2777–2785 (1994). DOI: 10.1128/mcb.14.4.2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sparks, A. B., Rider, J. E., Hoffman, N. G., Fowlkes, D. M., Quillam, L. A. & Kay, B. K.. Distinct ligand preferences of Src homology 3 domains from Src, Yes, Abl, Cortactin, p53bp2, PLCgamma, Crk, and Grb2. Proc. Natl. Acad. Sci. USA 93, 1540–1544 (1996). DOI: 10.1073/pnas.93.4.1540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rozakis-Adcock, M., Fernley, R., Wade, J., Pawson, T. & Bowtell, D.. The SH2 and SH3 domains of mammalian Grb2 couple the EGF receptor to the Ras activator mSos1. Nature 363, 83–85 (1993). DOI: 10.1038/363083a0 [DOI] [PubMed] [Google Scholar]
- [21].Bisson, N., James, D. A., Ivosev, G., Tate, S. A., Bonner, R., Taylor, L., et al. Selected reaction monitoring mass spectrometry reveals the dynamics of signaling through the GRB2 adaptor. Nat. Biotechnol. 29, 653–658 (2011). DOI: 10.1038/nbt.1905 [DOI] [PubMed] [Google Scholar]
- [22].Pawson, T. Dynamic control of signaling by modular adaptor proteins. Curr. Opin. Cell Biol. 19, 112–116 (2007). DOI: 10.1016/j.ceb.2007.02.013 [DOI] [PubMed] [Google Scholar]
- [23].Widmann, C., Gibson, S., Jarpe, M. B. & Johnson, G. L.. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol. Rev. 79, 143–180 (1999). DOI: 10.1152/physrev.1999.79.1.143 [DOI] [PubMed] [Google Scholar]
- [24].Oku, S., van der Meulen, T., Copp, J., Glenn, G. & van der Geer, P.. Engineering NGF receptors to bind Grb2 directly uncovers differences in signaling ability between Grb2- and ShcA-binding sites. FEBS Lett. 586, 3658–3664 (2012). DOI: 10.1016/j.febslet.2012.08.021 [DOI] [PubMed] [Google Scholar]
- [25].Saucier, C., Khoury, H., Lai, K. M., Peschard, P., Dankort, D., Naujokas, M. A., et al. The Shc adaptor protein is critical for VEGF induction by Met/HGF and ErbB2 receptors and for early onset of tumor angiogenesis. Proc. Natl. Acad. Sci. USA 101, 2345–2350 (2004). DOI: 10.1073/pnas.0308065101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Saucier, C., Papavasiliou, V., Palazzo, A., Naujokas, M. A., Kremer, R. & Park, M.. Use of signal specific receptor tyrosine kinase oncoproteins reveals that pathways downstream from Grb2 or Shc are sufficient for cell transformation and metastasis. Oncogene 21, 1800–1811 (2002). DOI: 10.1038/sj.onc.1205261 [DOI] [PubMed] [Google Scholar]
- [27].Lai, K. M. & Pawson, T.. The ShcA phosphotyrosine docking protein sensitizes cardiovascular signaling in the mouse embryo. Genes Dev. 14, 1132–1145 (2000). [PMC free article] [PubMed] [Google Scholar]
- [28].Hibino, K., Shibata, T., Yanagida, T. & Sako, Y.. Activation kinetics of RAF protein in the ternary complex of RAF, RAS-GTP, and kinase on the plasma membrane of living cells: single-molecule imaging analysis, J. Biol. Chem. 286, 36460–36468 (2011). DOI: 10.1074/jbc.M111.262675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Morimatsu, M., Takagi, H., Ota, K. G., Iwamoto, R., Yanagida, T. & Sako, Y.. Multiple-state reactions between the epidermal growth factor receptor and Grb2 as observed by using single-molecule analysis. Proc. Natl. Acad. Sci. USA 104, 18013–18018 (2007). DOI: 10.1073/pnas.0701330104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chardin, P., Camonis, J. H., Gale, N. W., van Aelst, L., Schlessinger, J., Wigler, M. H., et al. Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science 260, 1338–1343 (1993). DOI: 10.1126/science.8493579 [DOI] [PubMed] [Google Scholar]
- [31].Hibino, K., Watanabe, T. M., Kozuka, J., Iwane, A. H., Okada, T., Kataoka, T., et al. Single- and multiple-molecule dynamics of the signaling from H-Ras to cRaf-1 visualized on the plasma membrane of living cells. Chemphyschem 4, 748–753 (2003). DOI: 10.1002/cphc.200300731 [DOI] [PubMed] [Google Scholar]
- [32].Nakamura, Y., Hibino, K., Yanagida, T. & Sako, Y.. Switching of the positive feedback for RAS activation by a concerted function of SOS membrane association domains. Biophys. Physicobiol. 13, 1–11 (2016). DOI: 10.2142/biophysico.13.0_1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nakamura, Y., Umeki, N., Abe, M. & Sako, Y.. Mutation-Specific Mechanisms of Hyperactivation of Noonan Syndrome SOS Molecules Detected with Single-molecule Imaging in Living Cells. Sci. Rep. 7, 14153 (2017). DOI: 10.1038/s41598-017-14190-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yoshizawa, R., Umeki, N., Yanagawa, M., Murata, M. & Sako, Y.. Single-molecule fluorescence imaging of RalGDS on cell surfaces during signal transduction from Ras to Ral. Biophys. Physicobiol. 14, 75–84 (2017). DOI: 10.2142/biophysico.14.0_75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pack, C. G., Yukii, H., Toh-e, A., Kudo, T., Tsuchiya, H., Kaiho, A., et al. Quantitative live-cell imaging reveals spatio-temporal dynamics and cytoplasmic assembly of the 26S proteasome. Nat. Commun. 5, 3396 (2014). DOI: 10.1038/ncomms4396 [DOI] [PubMed] [Google Scholar]
- [36].Pack, C., Saito, K., Tamura, M. & Kinjo, M.. Microenvironment and effect of energy depletion in the nucleus analyzed by mobility of multiple oligomeric EGFPs. Biophys. J. 91, 3921–3936 (2006). DOI: 10.1529/biophysj.105.079467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Park, H., Han, S. S., Sako, Y. & Pack, C. G.. Dynamic and unique nucleolar microenvironment revealed by fluorescence correlation spectroscopy. FASEB J. 29, 837–848 (2015). DOI: 10.1096/fj.14-254110 [DOI] [PubMed] [Google Scholar]
- [38].Sadaie, W., Harada, Y., Matsuda, M. & Aoki, K.. Quantitative in vivo fluorescence cross-correlation analyses highlight the importance of competitive effects in the regulation of protein-protein interactions. Mol. Cell. Biol. 34, 3272–3290 (2014). DOI: 10.1128/MCB.00087-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kim, B., Cheng, H. L., Margolis, B. & Feldman, E. L.. Insulin receptor substrate 2 and Shc play different roles in insulin-like growth factor I signaling. J. Biol. Chem. 273, 34543–34550 (1998). DOI: 10.1074/jbc.273.51.34543 [DOI] [PubMed] [Google Scholar]
- [40].Ishihara, H., Sasaoka, T., Wada, T., Ishiki, M., Haruta, T., Usui, I., et al. Relative involvement of Shc tyrosine 239/240 and tyrosine 317 on insulin induced mitogenic signaling in rat1 fibroblasts expressing insulin receptors. Biochem. Biophys. Res. Commun. 252, 139–144 (1998). DOI: 10.1006/bbrc.1998.9621 [DOI] [PubMed] [Google Scholar]
- [41].Di Guglielmo, G. M., Baass, P. C., Ou, W. J., Posner, B. I. & Bergeron, J. J.. Compartmentalization of SHC, GRB2 and mSOS, and hyperphosphorylation of Raf-1 by EGF but not insulin in liver parenchyma. EMBO J. 13, 4269–4277 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kholodenko, B. N. Four-dimensional organization of protein kinase signaling cascades: the roles of diffusion, endocytosis and molecular motors. J. Exp. Biol. 206, 2073–2082 (2003). DOI: 10.1242/jeb.00298 [DOI] [PubMed] [Google Scholar]
- [43].Wang, Q., Chen, X. & Wang, Z.. Dimerization drives EGFR endocytosis through two sets of compatible endocytic codes. J. Cell Sci. 128, 935–950 (2015). DOI: 10.1242/jcs.160374 [DOI] [PubMed] [Google Scholar]
- [44].Katz, M., Amit, I. & Yarden, Y.. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim. Biophys. Acta 1773, 1161–1176 (2007). DOI: 10.1016/j.bbamcr.2007.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hanafusa, H., Torii, S., Yasunaga, T. & Nishida, E.. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat. Cell Biol. 4, 850–858 (2002). DOI: 10.1038/ncb867 [DOI] [PubMed] [Google Scholar]
- [46].Guy, G. R., Jackson, R. A., Yusoff, P. & Chow, S. Y.. Sprouty proteins: modified modulators, matchmakers or missing links? J. Endocrinol. 203, 191–202 (2009). DOI: 10.1677/JOE-09-0110 [DOI] [PubMed] [Google Scholar]
- [47].Corbalan-Garcia, S., Yang, S. S., Degenhardt, K. R. & Bar-Sagi, D.. Identification of the mitogen-activated protein kinase phosphorylation sites on human Sos1 that regulate interaction with Grb2. Mol. Cell. Biol. 16, 5674–5682 (1996). DOI: 10.1128/mcb.16.10.5674 [DOI] [PMC free article] [PubMed] [Google Scholar]