

Summary:
In this issue of Cancer Discovery , Sodir and colleagues employ a pancreatic ductal adenocarcinoma mouse model with mutant KRAS and inducible MYC to demonstrate that MYC acts as a reversible driver of malignant tumor progression. Abrogation of MYC triggers rapid regression and disassembly of the ensemble tumor through both cancer cell–intrinsic and cancer cell–extrinsic mechanisms, providing a compelling rationale for therapeutic targeting of MYC.
In this issue of Cancer Discovery , Sodir and colleagues present a mouse model with pancreatic expression of mutant KRAS and switchable expression of MYC to demonstrate that MYC acts as a reversible driver of malignant pancreatic ductal adenocarcinoma (PDAC; ref. 1 ). MYC activation in KRAS G12D -expressing epithelium instructs malignant progression from the precursor disease in a cell-intrinsic manner, concurrent with induction of cell-extrinsic signals that drive the accompanying desmoplastic stromal expansion and tumor-suppressive immune microenvironment characteristic of human PDAC. Importantly, MYC deactivation in vivo triggers rapid regression and disassembly of the ensemble tumor through both cancer cell–intrinsic and cancer cell–extrinsic mechanisms to restore tissue architecture, as depicted in Fig. 1 . This, together with recent literature in other cancers, provides a strong rationale for therapeutic targeting of MYC.
Figure 1.
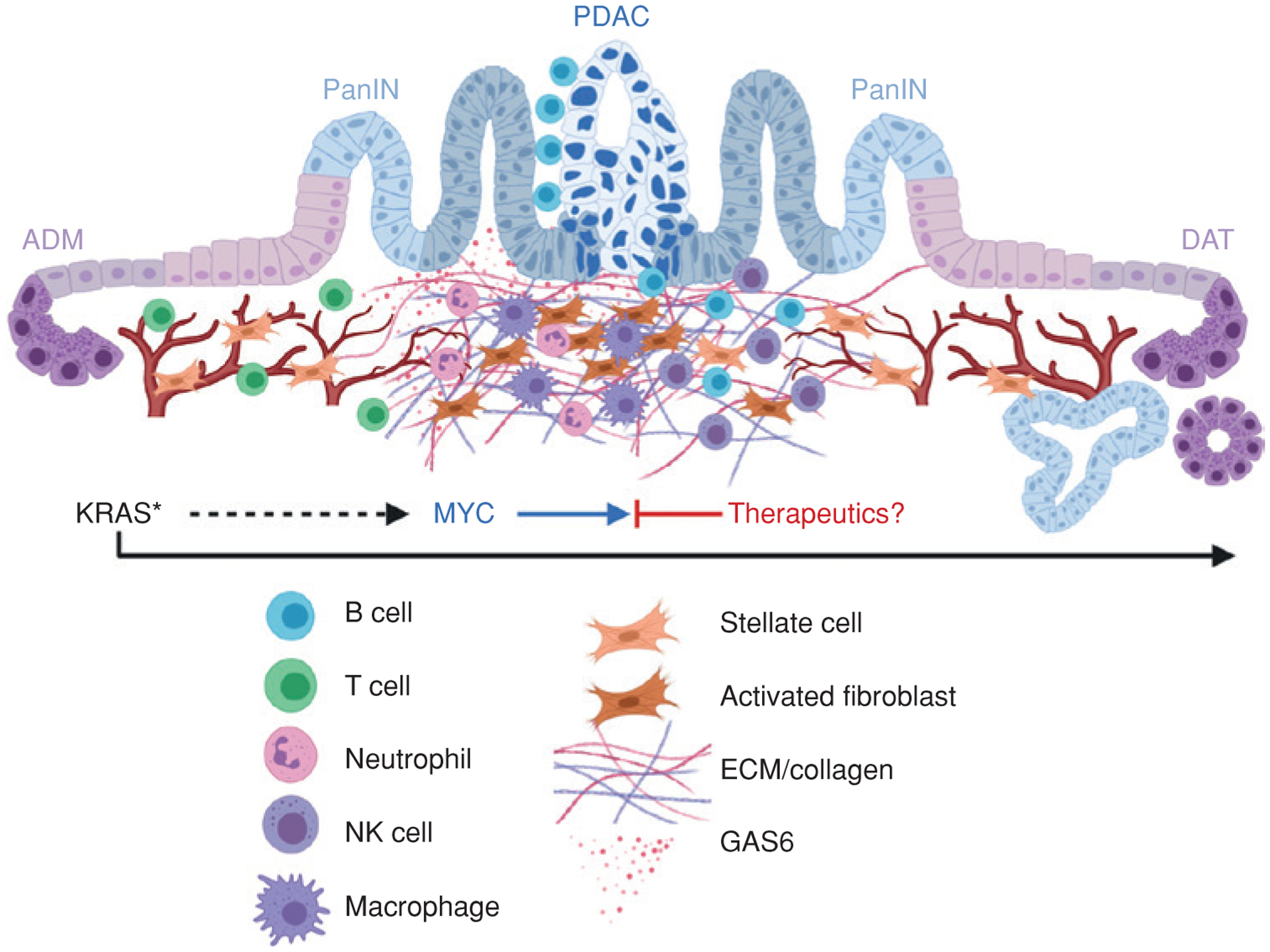
Schematic representation of the role of MYC as a reversible driver of cell-intrinsic and cell-extrinsic PDAC progression. Epithelial acinar-to-ductal metaplasia (ADM), pancreatic intraepithelial neoplasia (PanIN), progression to pancreatic ductal adenocarcinoma (PDAC), and reversion to PanIN and ductal-to-acinar transdifferentiation (DAT) are indicated. Desmoplastic stromal and vascular changes and immune cell infiltrates are depicted. ECM, extracellular matrix; KRAS*, mutationally activated KRAS.
PDAC ranks among the top three most aggressive cancers in the United States and is projected to become the second leading cause of cancer-related deaths by 2021, with a 5-year survival rate that the American Cancer Society only just reported has reached 10%. Although specific targeted inhibitors and immune-based therapeutic agents have improved patient outcomes for some cancer types, PDAC is generally found to be refractory to these therapies, and standard of care relies on combination chemotherapy regimens that provide only a modest (months) increase in median survival. The aggressiveness and therapy-resistant nature of this deadly cancer are thought to arise from its signature dense desmoplastic and immune-suppressive tumor microenvironment that comprises the majority of the tumor. In recent years, efforts have focused on strategies to disrupt the dense desmoplasia and diminish immune suppression. Unfortunately, a clinical trial with the hyaluronidase PEGPH20, aimed at reducing stromal density, showed no improvement in overall survival, and mouse models with similar goals have resulted in the unleashing of a more aggressive tumor phenotype, suggesting a tumor-suppressive role for the desmoplasia ( 2 ). Immune checkpoint inhibitors have likewise been unsuccessful in treating this recalcitrant cancer. Currently, the only FDA-approved immunotherapy in PDAC, the PD-1 checkpoint inhibitor pembrolizumab, is exclusively indicated for patients with unresectable or metastatic disease that is also microsatellite instability-high or mismatch repair-deficient, both rare in PDAC. Thus, there is an urgent need to understand the complexity of stromal expansion and dysfunctional tumor immunity to realize meaningful therapies for this aggressive disease. This study by Sodir and colleagues demonstrates that the tumor cell–intrinsic master transcription factor MYC instructs the PDAC microenvironment, and targeting MYC represents an alternative strategy to induce PDAC regression.
MYC is a master transcription factor needed for cell proliferation, and many other hallmark cancer phenotypes. When deregulated, MYC can promote tumor aggressiveness and therapeutic resistance in cancers. RAS is an upstream activator of MYC, increasing its stability and DNA binding ( 3 ), and KRAS mutations occur in approximately 95% of PDAC. These oncogenes cooperate in driving PDAC—genetic loss of endogenous Myc in mutant KRAS models inhibits pancreatic intraepithelial neoplasia (PanIN) progression to PDAC ( 4 ), and the Der lab has shown MYC to be the primary resistance mechanism to KRAS signaling inhibition ( 5 ). Recent work to explore the impact of MYC on the tumor microenvironment of multiple cancers, including PDAC, supports a role in suppression of immune surveillance and limiting T-cell infiltration ( 6 ). To this end, the Stanger lab showed a positive correlation between MYC overexpression and chemokine (C-X-C motif) ligand 1 (CXCL1) expression in T cell–low PDAC tumor clones, suggesting that MYC may allow some PDAC tumors to suppress tumor immunity ( 7 ).
Sodir and colleagues crossed their previously developed inducible Rosa-LSL-MycERT2 mouse model with mice carrying LSL-KrasG12D and a pancreas-specific promoter (p48) driving CRE recombinase (KMp48). In this model, mutant KRAS expression induces PanIN formation, and tamoxifen treatment in adult mice activates sustained physiologic levels of MYC in pancreatic epithelial cells. Signature features of aggressive and spontaneous human PDAC were observed in these mice after 3 weeks of tamoxifen-induced MYC expression. The authors not only observed more proliferating tumor cells, but also identified stromal alterations, including an influx of immunosuppressive macrophages and neutrophils, accumulation of tumor peripheral B lymphocytes, eradication of CD3+ T cells, and activation of stellate cells to cancer-associated fibroblasts along with subsequent desmoplasia, hypoxia, and loss of vascularity. The remodeling of the tumor microenvironment began after only 24 hours of tamoxifen exposure, suggesting a rapid instruction of these changes by MYC. Molecular analyses revealed that the remodeling involved known pathways of desmoplastic stroma including Sonic Hedgehog signaling, with neutrophil and macrophage recruitment by CXCL5 and CCL9, respectively. However, the authors also uncovered a novel component of what may be a parallel MYC-driven neutrophil recruitment pathway in PDAC: the ligand/receptor GAS6/AXL pathway, which has previously been implicated in diverse neutrophil dynamics.
Follow-up studies showed that inhibition of GAS6 had little to no effect on MYC-induced T-cell depletion and only modest effects on macrophage influx. However, GAS6 inhibition blocked the recruitment of neutrophils and accumulation of peritumoral B cells induced by MYC, slowed stellate cell activation and proliferation, and impeded tumor growth. Having previously established that MYC induces PD-L1- expressing macrophage-mediated immune evasion in lung adenocarcinoma (8), the authors also tested PD-L1 blockade in the KMp48 tumors, which exhibit PD-L1 expression in the epithelial tumor cells. With PD-L1 blockade, T cells were no longer expelled from the tumors, but no change was observed in tumor growth or PanIN-to-PDAC progression, pointing away from a prominent role for T-cell exclusion and further emphasizing the importance of GAS6 on neutrophil recruitment in their KRAS/MYC-driven PDAC model.
Strikingly, deactivation of ectopic MYC in the KMp48 mice through tamoxifen withdrawal caused complete regression of PDAC, including collapse of the desmoplastic, inflamed stroma, immune remodeling, including T-cell influx and death or transdifferentiation of neoplastic duct cells. MYC loss restored a state of normal acinar cells and early PanINs, along with recovery of blood-vessel patency and local tissue oxygenation. The authors showed this phenomenon to occur even after long-term MYC activation, although extensive scarring and cystic phenotypes endured in some cases. To test the importance of endogenous MYC, the authors used expression of OmoMYC (9), a peptide inhibitor of MYC, within a KrasG12D;Trp53null-driven PDAC mouse model, and observed a similar reversal in PDAC progression and stromal desmoplasia.
In an effort to further define key immune effectors of MYC deactivation–induced regression of PDAC, Sodir and colleagues depleted the repopulated CD4+ and CD8+ T cells separately, but this had no effect on the observed tumor collapse. MYC inactivation also shifted CD20+ B cells from the periphery toward the center of the tumor, and triggered recruitment of NKp46+ natural killer (NK-like) cells; and depletion studies of either of these cell types showed both to be essential for cancer cell death caused by MYC withdrawal.
Altogether, Sodir and colleagues demonstrated the importance of MYC activation in combination with mutant KRAS for PDAC progression and elucidated a novel neutrophil recruitment and fibroblast activation pathway (GAS6/AXL) that could be an important target in the treatment of PDAC. In addition, the authors compared tissue-specific effects of MYC activation in pancreatic adenocarcinoma to their lung adenocarcinoma model (8). Both models progressed to adenocarcinoma characterized by influx of immunosuppressive macrophages, expulsion of T cells, and tumor cell proliferation and invasion. However, influx of neutrophils and peritumoral B cells was observed only in the PDAC model, and induction of PD-L1 was observed on tumor epithelium in PDAC, whereas it occurred on incoming macrophages in the lung. Furthermore, desmoplasia caused by fibroblast activation was observed only in the PDAC model. The authors posit that this profound tissue specificity is indicative of MYC “hacking” an endogenous tissue regenerative program during tumor expansion and that regression of the tumor after deactivation of MYC is enabled by co-opting a tissue-specific injury resolution program.
Systemic therapies for cancer including immune-targeted treatments are plagued by cancer site–agnostic and often debilitating side effects. By highlighting the importance of tumor cell–intrinsic factors in initiating and sustaining immune suppression and stromal remodeling, the work by Sodir and colleagues may foster alternative approaches to deconstructing tumors and reengaging the immune system by targeting cancer cell–intrinsic drivers that can take advantage of endogenous tissue repair programs. Various strategies to inhibit MYC are in development, although most are not cancer cell–specific. However, studies by the Evan lab suggest that there is little toxicity with inhibiting MYC in adult mice. Alternatively, targeting tumor-specific upstream effectors of MYC activity, such as mutant KRAS, would be cancer cell- intrinsic. Similar to MYC, targeting mutant RAS has been a long-sought goal. Exciting progress has recently been made with mutant KRASG12C inhibitors. One such inhibitor, AMG 510, has shown single-agent efficacy in immune-competent mice (10). Treatment with this specific inhibitor improved CD8+ T-cell infiltration and upregulation of proinflammatory cytokines. Synergy with immune checkpoint blockade therapies with lower doses of AMG 510 was also demonstrated, providing an exciting path in the clinic. The insights from the study by Sodir and colleagues encourage the field to closely examine cross-talk between the tumor cell and its microenvironment, which will be essential for effective therapeutic strategies in the treatment of cancers.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
REFERENCES
- 1.Sodir NM, Kortlever RM, Barthet VJA, Campos T, Pellegrinet L, Kupczak S, et al. MYC instructs and maintains pancreatic adenocarcinoma phenotype. Cancer Discov 2020;10:588–607. [DOI] [PubMed] [Google Scholar]
- 2.Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014;25:719–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farrell AS, Sears RC. MYC degradation. Cold Spring Harb Perspect Med 2014;4:pii:a014365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014;511:483–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, et al. Long-Term ERK inhibition in KRAS-mutant pancreatic cancer is associated with MYC degradation and senescence-like growth suppression. Cancer Cell 2016;29:75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Casey SC, Baylot V, Felsher DW. MYC: Master regulator of immune privilege. Trends Immunol 2017;38:298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity 2018;49:178–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Swigart LB, et al. Myc cooperates with Ras by programming inflammation and immune suppression. Cell 2017;171:1301–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, et al. Modelling Myc inhibition as a cancer therapy. Nature 2008; 455:679–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575:217–23. [DOI] [PubMed] [Google Scholar]