

Abstract
CRISPR is a revolutionary genome-editing tool that has been broadly used and integrated within novel biotechnologies. A major component of existing CRISPR design tools is the search engines that find the off-targets up to a predefined number of mismatches. Many CRISPR design tools adapted sequence alignment tools as the search engines to speed up the process. These commonly used alignment tools include BLAST, BLAT, Bowtie, Bowtie2 and BWA. Alignment tools use heuristic algorithm to align large amount of sequences with high performance. However, due to the seed-and-extend algorithms implemented in the sequence alignment tools, these methods are likely to provide incomplete off-targets information for ultra-short sequences, such as 20-bp guide RNAs (gRNA). An incomplete list of off-targets sites may lead to erroneous CRISPR design. To address this problem, we derived four sets of gRNAs to evaluate the accuracy of existing search engines; further, we introduce a search engine, namely CRISPR-SE. CRISPR-SE is an accurate and fast search engine using a brute force approach. In CRISPR-SE, all gRNAs are virtually compared with query gRNA, therefore, the accuracies are guaranteed. We performed the accuracy benchmark with multiple search engines. The results show that as expected, alignment tools reported an incomplete and varied list of off-target sites. CRISPR-SE performs well in both accuracy and speed. CRISPR-SE will improve the quality of CRISPR design as an accurate high-performance search engine.
INTRODUCTION
Clustered regularly interspaced short palindromic repeats (CRISPR)/ CRISPR-associated (Cas) system was first discovered in the prokaryotic genome and used to cleavage foreign DNA sequence during phage infection in various bacteria (1–3). Recently, CRISPR-Cas9 based technologies were adapted for genome-editing in mouse and human genomes (4,5). The CRISPR-Cas9 system could modify or delete genomic regions through the designed 20-mer gRNA sequences with the upstream regions of photospacer adjacent motif (PAM) (2,6–8). Various studies have shown that CRISPR-Cas9 system could lead to off-target effect, deletion or modification occurs at nontargeting genomic regions; the binding of gRNA to target genomic region tolerates few mismatches located nearby PAM motif (9–11). To minimize the potential off-target effects, a search engine for list of off-target sites is desired for CRISPR design.
The off-target sites for a query gRNA are list of gRNAs with less or equal to a predefined maximum number of mismatches found in the reference genome. For instance, there are over 200 million unique gRNAs in human or mouse genome. It is straightforward to perform a linear scan followed with sorting to find of all unique gRNAs; however, it would be very time-consuming to calculate off-target sites in a large scale without optimized data structure and algorithm. For example, the estimated processing time for GuideScan (12) is at least three months for genome-wide gRNA design. GuideScan uses the ‘trie’ data structure with a brute-force algorithm that guarantees the search accuracy. To speed up this process, many CRISPR design methods use existing sequence alignment tools as a search engine to identify potential off-target sites. We listed 27 CRISPR design methods found with detailed method descriptions and active online-tools (Table 1). Note that not all CRISPR design methods require list of off-target sets, and CRISPR design methods differ in post processing, on-target scoring function, off-target scoring functions and many other research focuses.
Table 1.
Many CRISPR design methods require a search engine to retrieve list of off-target sites. Newer methods are likely to use accurate brute force approaches
Common sequence alignment tools include BLAST (13), BLAT (14), Bowtie (15), Bowtie2 (16), BWA (17) and customized search engines (18–20). The BLAST tool was developed in 1990 by Samuel Karlin and Stephen Altschul; as an early version of the sequence alignment tool, the BLAST tool can align a query against the whole RefSeq database in a few minutes, where the RefSeq database includes >1.9 trillion nucleotides. BLAST is commonly used with a small amount of input up to a few thousands of bases.
BLAT is the BLAST-like alignment tool. It was developed by Jim Kent at UCSC in early 2000, and it is well-known as a sequence alignment tool integrated within UCSC genome browser. Bowtie use developed in 2009 by Ben Langmead et al. at the University of Maryland. In 2011, the Bowtie 2 was released. Bowtie 2 is suitable to find longer, gapped alignment; it also runs faster with longer reads, supports gapped alignment and has no upper limit on read length. BWA was developed by Heng Li in 2009. BWA is another sequence alignment tool that was commonly used in standard data processing pipelines. Bowtie, Bowtie 2 and BWA are used to map millions of next-generation sequencing (NGS) reads to the user-specified genome.
Alignment tools first create indices from the reference genome using a K-mer (seed) hash table; the K-mer hash table store both K-mer sequences and the locations of the sequence. For each query sequence, the K-mer table is used to trace the locations of all K-mer sub-sequence within the query sequences; then the sequence alignment tools merge these locations as potential alignment sites; next, the top candidates are selected base on the extended alignment to the complete query sequence (Supplementary Figure S1). The seed-and-extend approaches are efficient for sequence alignment with exact match. For instance, many sequence alignment tools use a hash-table with approximately 20-mer, and the query sequences are normally longer (≥50 bp); the locations of the query sequences can be traced-back quickly when a single K-mer sub-sequence in query sequence matches entries in K-mer table; next, the local extensions are performed to match the complete query sequence; the extension processes allow multiple mismatches in the extended regions which make it appealing to be a fast off-targets search engine.
Sequence alignment tools rely on minimum one K-mer exact match, the algorithm is likely to miss off-targets of high number of mismatches for the ultra-short gRNAs (20-mer). Incomplete off-target information will lead to unexpected off-target effects and generate false-positive results for downstream analysis. To the best of our knowledge, these problems have not been solved.
Recently, numerous methods have been developed with brute-force approaches (12-21). GuideScan uses a ‘trie’ data structure with a brute-force algorithm that guarantees the search accuracy. Cas-OfFinder uses GPU to speed up the search. FlashFry uses a block-compressed binary format to keep potential gRNA information. FlashFry is written in Scala language and run with Java virtual machine. Crisflash used an N-ary tree structure, which search up to four mismatches. CRISPRitz used a four-bit-based encoding to represent each nucleotide to allow for efficient bitwise operations. CRISPRitz supports off-targets with both mismatches and indels. Crackling focused on off-targeting scoring. Crackling use the Inverted Signature Slice Lists (ISSL) for the off-target search.
In order to test the accuracy and performance of the K-mer based alignment methods and the brute force approaches, we created four clusters of gRNAs based on the minimum numbers of mismatches to the gRNAs in the reference genome. We then evaluated the accuracy of K-mer based alignment methods and the speed of different search engines. We show that using optimized data structure and algorithm Figure 1. CRISPR-SE identifies list of off-target sets quickly and accurately.
Figure 1.
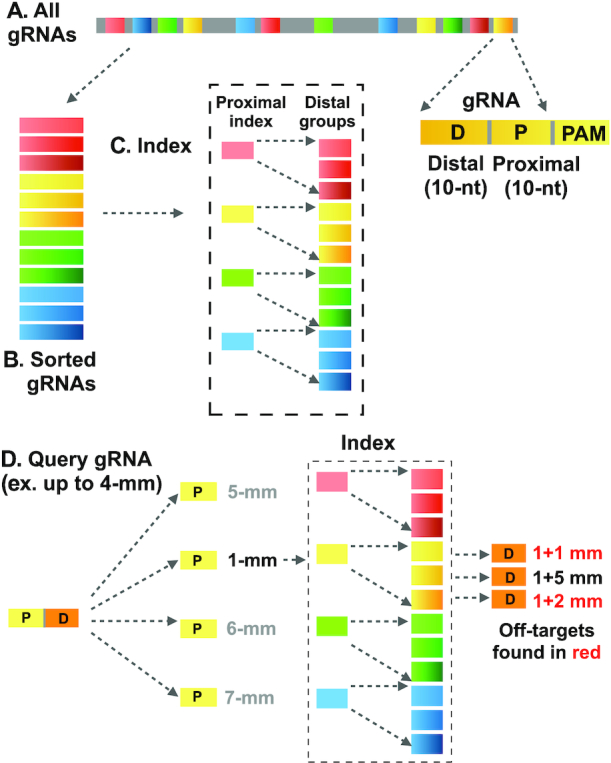
Schematic of CRISPR-SE: (A) CRISPR-SE scans all of the 20-nucleotides (20-mer) gRNAs; (B) sort all gRNAs by the proximal region (10-nt close to PAM sequence); (C) Group gRNAs with the same proximal regions. (D) Calculate the mismatches in the proximal region; add the mismatches in the distal region only with similar proximal region. Using this approach, a query gRNA are virtually compared with all gRNAs.
MATERIALS AND METHODS
To evaluate the accuracy and performance of existing search engines, we constructed four gRNA clusters derived from the hg38 and mm10 reference genome (Supplementary Table S1). The gRNAs were clustered based on the minimum number of mismatches compared to all other gRNAs in the reference genome. A gRNA is named as N-mm gRNA when it has exactly N mismatches with at least one gRNA. To identify the N-mm gRNAs clusters (N = 1–4), we first used CRISPR-SE to search gRNAs with minimum N or more mismatches (n = 1–5) to any other gRNAs. The gRNA datasets found with 1 or more mismatches are named as 1+mm dataset. Next, we constructed gRNA sets from 2+mm to 5+mm dataset. We further construct the 1-mm gRNAs cluster by excluding all 2+mm gRNAs from 1+mm; therefore, the gRNAs in 1-mm cluster have exactly 1 mismatch with at least another one gRNA. Similarly, we derived 2-mm to 4-mm clusters for the benchmark of gRNA search engines by excluding all [N+1]+mm dataset from N+mm dataset. As an example, the 4-mm gRNA TGGTGTACGATCTACTCTCG locates at chr1:858163-858182 on hg38; it has four mismatches with the gRNA TGGTGTACAATCTAGTCACA at chr18:63700374-63700393; the 4-mm gRNA have four or more gRNAs compared with any other gRNAs found in hg38 reference genome. The repeated gRNAs with exact matches are 0-mm cluster since there are at least two such gRNAs having the same gRNA sequence. In the benchmark, we also validate the gRNA clusters base on the off-targets searching results of each search engine to ensure the correctness of the cluster construction.
Benchmark
We perform the off-target search using the five common K-mer based alignment methods: BLAST, BLAT, Bowtie, Bowtie 2 and BWA; for the brute force approaches, we included FlashFry, Crisflash and CRISPR-SE. GuideScan (12) computes the genome wide gRNAs, the estimated processing time for GuideScan is at least three months for the genome-wide gRNA design. We also excluded Cas-OffFinder because the software requires the presence of GPU hardware. Also, we excluded the CRISPRitz method because CRISPRitz has been reported slower than FlashFry. The Crackling method was also excluded because Crackling focuses on the scoring function and the method does not report the alignment information. For each of the search engine, we performed the off-targets search using 1-mm to 4-mm gRNA datasets. Due to the time limit, only the first 10 000 gRNAs from each cluster are used. All programs were provided with the same computational resource (8 x 2300 MHz AMD Opteron 6276 processors, up to 384 GB memory).
A gRNA has a fixed off-target sets searched against a reference genome, we evaluate the accuracy of a search engine by checking if the search engine can report an off-target with the minimum number of mismatches, as to classify an N-mm gRNA correctly. If the classification is incorrect, it is sufficient to show that the off-targets searching results are incomplete. For each of the five K-mer based alignment method, we evaluate both the accuracy and speed. For the brute force approaches, we only perform the speed test because the methods would report the same results using the same parameters as long as the method is implemented correctly.
Parameters
We used the search parameters found in the publications as well as from the source code (Supplementary Table S2). For the K-mer based alignment methods, the most important parameters are ‘-a’ for Bowtie that reports all alignments (22–28); ‘-k 100’ for Bowtie2 to report up to 100 alignments (29,30); ‘-N’ for BWA to search all hits (31–34); ‘-task blastn’ for BLAST for short sequences (35–37); and ‘-oneOff=1’ for BLAT to triggers all alignments (38). Note that these parameters are required for the alignment methods to perform off-target search; the alignment tools will run in a ‘slow mode’ that enforce the alignment tools to report more alignments. We also extended the query gRNAs from 20-mer to 23-mer by adding each of the four nucleotides followed by GG. The extensions were applied to overcome the error of ‘query sequence too short’ and not all search engines accept the wild nucleotide ‘N’ in the input such as Bowtie. For the brute force approaches, we used the default parameters for each method.
RESULTS
Validation
Alignment validation
A search engine also acts as a sequence alignment tool that reports the original positions of the gRNAs on the reference genome. For each of the methods, we compared the positions of the alignments with the original location of the gRNAs. We confirmed that the search engines align the query gRNAs to their original positions. The comparisons include all of the five alignment tools and the methods using brute force approaches.
Cluster validation
For each of the searching result, we verified that none of the search engines report gRNAs with less than expected mismatches. For instance, none of the off-targets identified for a 3-mm query gRNA have two mismatches or less compared to the query gRNA. This confirmed that the clusters were constructed correctly. A search engine may classify a 2-mm gRNA as a 3-mm cluster when the off-targets search is incomplete. To the best of our knowledge, similar datasets have not been reported elsewhere.
Accuracy comparison
We performed the accuracy comparison of the five alignment tools and the CRISPR-SE. The alignment tools use the seed-and-extend algorithm (Figure 1): in the processing of tracing back K-mer positions using indices (step B), the off-targets may not be found when all K-mer subsequences contain one or more mismatches; for instance, to search four mismatches within 20-mer gRNAs, there could be one mismatch in every 5-mer sub-sequence; or perfect match in a 16-mer sub-sequence. Therefore, the algorithm is more likely to miss potential off-targets as the number of matches increase. As shown in Table 2A, all alignment tools partially classified one or more of the four gRNA clusters. The percentages of the correctly classified gRNAs drop as the required number of mismatches increases.
Table 2.
(A) The ratio of correctly classified off-targets using the first 10 000 1-mm to 4-mm gRNA datasets. (B) Processing time with the same 1-mm to 4-mm datasets (seconds)
| 1-mm(%) | 2-mm(%) | 3-mm(%) | 4-mm(%) | |||||
|---|---|---|---|---|---|---|---|---|
| hg38 | mm10 | hg38 | mm10 | hg38 | mm10 | hg38 | mm10 | |
| (A) Accuracy comparisons: | ||||||||
| Alignment tools: | ||||||||
| BLAST | 99 | 99 | 46 | 43 | 26 | 31 | 7 | 8 |
| BLAT | 88 | 94 | 51 | 50 | 48 | 57 | 36 | 39 |
| Bowtie | 100 | 100 | 100 | 100 | 100 | 100 | 0 | 0 |
| Bowtie2 | 100 | 100 | 24 | 15 | 0 | 0 | 0 | 0 |
| BWA | 25 | 36 | 0 | 0 | 0 | 0 | 0 | 0 |
| Brute force: | ||||||||
| CRISPR-SE | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| (B) Speed comparisons: | ||||||||
| 1-mm(s) | 2-mm(s) | 3-mm(s) | 4-mm(s) | |||||
| hg38 | mm10 | hg38 | mm10 | hg38 | mm10 | hg38 | mm10 | |
| Alignment tools: | ||||||||
| BLAST | 23 595 | 14 333 | 19 344 | 14 718 | 14 523 | 15 083 | 8869 | 8897 |
| BLAT | 2874 | 4325 | 2144 | 3100 | 1286 | 2161 | 622 | 853 |
| Bowtie | 58 | 122 | 483 | 565 | 384 | 482 | 271 | 234 |
| Bowtie2 | 949 | 936 | 876 | 943 | 920 | 926 | 981 | 743 |
| BWA | 4092 | 5827 | 5144 | 6316 | 4469 | 6171 | 3409 | 3554 |
| Brute force: | ||||||||
| FlashFry | 122 | 151 | 194 | 282 | 531 | 563 | 1828 | 1851 |
| Crisflash | 155 | 182 | 1,641 | 1799 | 10 376 | 11 619 | 53 359 | 55 863 |
| CRISPR-SE | 235 | 260 | 229 | 232 | 270 | 235 | 282 | 274 |
| Top | Bowtie | Bowtie | FlashFry | SE | SE | SE | SE | SE |
Partial classification
All alignment tools misclassified one or more of the four gRNA clusters. We evaluated the accuracy of a search engine using the percentage of correct classifications. This is a simplified condition to check if a classification is correct. There are more stringent conditions such as checking if all off-targets match the expected. If a search classified a gRNA incorrectly, it implies that the off-targets do not match the expected; and when the off-targets match the expected, the search must classify the gRNA correctly. The search results show that the ratio of correct classifications varies from 100% (mostly with 1-mm) to 0% (mostly with 4-mm). The results show that the simplified condition is sufficient to demonstrate that the off-target search using alignment tools are incomplete.
We further compared the off-targets identified using Bowtie with CRISPR-SE, we found that Bowtie not only classified the gRNAs correctly for 1-mm to 3-mm, the off-targets reported by Bowtie also match 100% with CRISPR-SE. Bowtie search off-targets up to three mismatches; for all gRNAs in 4-mm clusters, Bowtie only reports the source of the gRNAs as expected. CRISPR-SE reports 100% off-targets for all four clusters.
Number of mismatches
The percentages of the correctly classified gRNAs mostly stay the same or drop as the required number of mismatches increases. For 1-mm clusters, four alignment tools perform well: Bowtie and Bowtie 2 score 100% of accuracy, BLAST and BLAT reach 99%/99% and 94%/88% for hg38/mm10. For 2-mm clusters, there are >50% of gRNAs are 2-mm clusters (Supplementary Table S1), the accuracies stay 100% for Bowtie, drop to 43%–51% for BLAST and BLAT, drop much quicker for Bowtie 2 (15% and 24% for hg38 and mm10); and BWA drop to 0% for both reference genome. For the 3-mm clusters, the accuracies for BLAT increased to 57% from 49.7% for hg38, and drop to 48% from 51.2% for mm10; Bowtie remains 100% high accuracy; and the accuracies for BLAST keep dropping to 31% and 26% for hg38 and mm10; and Bowtie 2 and BWA drop to 0%. For the 4-mm clusters, BLAST and BLAT drop to 7–8%, and Bowtie, Bowtie 2, and BWA drop to 0%. Comparing to the accuracy changes by different number of clusters, the accuracies between hg38 and mm10 are much less different. Bowtie has the best performance(100%) up to three mismatches; BLAT reach higher accuracies than BLAST for all four clusters. Bowtie 2 reach 100% accuracy only for 1-mm cluster; and the BWA performed the lowest accuracies with all four clusters.
Implementation details
The accuracy comparison imply that (i) the K-mer alignment algorithm partially reports the off-targets and (ii) different alignment tools implement off-targets search details differently. For instance, the accuracy comparison shows that BLAT, a newer version of the BLAST-like alignment tool, performs better than BLAST; BLAST and BLAT identifies 7.9% and 39.1% of 4-mm clusters. For the 3-mm clusters, Bowtie 2 report 0% of off-targets as Bowtie report 100% accurately; whereas Bowtie and Bowtie 2 are developed by same group. (iii) Alignment tools are top candidates for sequence alignment. Bowtie and Bowtie 2 are actively developed in 2020, and BWA is commonly used in standard sequence alignment pipeline for large consortiums like ENCODE(https://www.encodeproject.org/) and 4DN(https://www.4dnucleome.org/).
Speed comparison
We performed the speed comparison for both alignment and brute force methods (Table 2B). It is mandatory for the alignment tools run in the ‘slow mode’ to search the off-targets for the ultra-short 20-mer gRNAs. Alignment tools using default parameters will run much faster for alignment; however, it will yield to very low off-targets information. Bowtie reported that it ‘aligns short DNA sequences (reads) to the human genome at a rate of over 25 million 35-bp reads per hour’, equivalent to ∼7000 reads per second, which yield to <2 s for each 10 000 cluster. The alignment tools are mostly used to align millions of reads (50–300 bp). As NGS reads getting longer with higher sequencing qualities, the alignment tools focus on mapping high quality read with less mismatches and speed. For example, Bowtie 2 runs faster with longer reads, supports gapped alignment, and has no upper limit on read length.
Alignment tools
The processing time varies between alignment tools. BLAST took the longest time as 23 595 seconds, almost 6.5 h for the 1-mm cluster (99.4%); where bowtie finished in 58 s for the same 1-mm cluster(100%), with about 400 times different. The differences drop as the number of mismatches increases: BLAST took about 15 000 s for 3-mm clusters with 26.0–30.6% accuracy as bowtie took 384–482 seconds, about 30 times different. Similar trends are observed between other alignment tools.
The processing time is likely dependent on individual methods. In most cases, the speed are ordered by Bowtie > Bowtie 2 > BLAT > BWA > BLAST. The processing time of BLAST and BLAT drop as the number of maximum mismatches increase. For Bowtie 2 and BWA, the processing time are less affected by the number of mismatches. Bowtie run 6–8 times faster for 1-mm than 2-mm and 3-mm clusters.
A longer processing time does not necessarily lead to higher accuracy. Bowtie performs 100% up to 3-mm clusters, meanwhile, the processing time is the least among all five alignment methods. The processing time of Bowtie 2 drops from 100% (1-mm) to 14.8% (2-mm) where the processing time only drops 8% (from 949 to 876 s) with the hg38 dataset.
Brute force approaches
The speed differences in brute force are related to number of mismatches. Brute force approaches use tree structures to speed up the processes. The tree structures are preferably used for exact match, where a search identifies a target without traveling the tree structure back and forth. The off-targets search with multiple mismatches requires visiting the tree multiple times to search for multiple mismatches. Thus, the numbers of visiting increase exponentially as the number of mismatches increase. This is the primary reason that FlashFry slows down rapidly as the required number of mismatches increase. CRISPR-SE applied multiple methods to optimization the process: (i) Minimize the number of tree depth to two, such that the searching speed would not be affected much by the number of mismatches. (ii) Use 2-bit representation to minimize the memory usage; as the guide RNA candidate pool is large, a computer runs faster with less memory by introducing less page faults. (iii) Multi-threading: CRISPR-SE uses an array data structure that shared by multiple processors to calculate the off-targets in parallel.
Speed comparison
For all the search engine, Bowtie uses the least time for 1-mm cluster; FlashFry runs faster for the 2-mm cluster in hg38; CRISPR-SE runs faster for the other clusters. With the 4-mm cluster, CRISPR-SE runs six times faster than FlashFry, and about 200 times faster than Crisflash.
CRISPR-SE
CRISPR-SE support off-targets search in query mode and batch mode; in the query mode, user provides query sequences, such as the four clusters used in the benchmark. In the batch mode, CRISPR-SE search all genome-wide gRNAs against itself without query sequence. On average, CRISPR-SE processes ∼40 gRNAs per second in query mode, and the processing time was less dependent on the number of mismatches. In the batch mode, CRISPR-SE processes ∼193 gRNAs/s, about 6 h with 48 CPUs, to design genome-wide gRNAs with genome size similar to human or mouse genome.
Scoring function
Scoring function is another important component for CRISPR design. Numerous research have been conducted for gRNAs on-target (efficiency) and off-target (specificity) scores functions as also summarized in the review of (39). The on-target scores evaluate the cleavage efficiency and the off-target scores assess the risks of genome modifications at the nonintended cutting sites. Despite of much progress made by many methods, both computational design for on-target and off-target prediction remain challenging due to experimental limitations, affects of multiple on-target features and computational complexities for off-target effects prediction.
On-target cleavage efficiency
CRISPR cleavage efficiency is affected by various sets of features including sequence compositions, nucleotide positions, GC contents, chromatin accessibility, gene coding, RNA secondary structures, melting temperatures and free energies. Using various sets of features, multiple computation models, algorithm and machine learning methods have been developed to predict the cleavage efficiency and many web-tools are available with integrated scoring functions (28,40–53). For instance, as a comprehensive web-tool, CRISPOR provides 10 different on-target scoring functions and FlashFry outputs two on-target scoring functions.
Off-target cutting specificity
Many CRISPR off-target scoring function rely on list of potential off-target sites identified by sequence alignment tools (54–57,58). The off-target score can also be evaluated using a weight matrix where the weight matrix are derived from large scale experimental tests (25,46,53,59–63). Two commonly used off-target scoring functions are MIT score (59) and CFD score (46). Both CRISPOR and FlashFry reports MIT and CFD scores. Similar to on-target scoring function, collective off-target scoring functions are developed using machine learning, linear regression and deep learning methods. These methods utilize the off-target sites identified using alignment tools as well as the features utilized in on-target scoring function (61–63).
CRISPR-SE
CRISPR-SE was initially developed to overcome the accuracy problem exists in heuristic algorithm (Table 2A) and the computational challenges in large scale CRISPR design (64). In the study, we designed >10 000 pairs of gRNA within two million base POU5F1 locus in human embryonic stem cells; we chose gRNAs with at least four mismatch-counts with any other gRNA found in the reference genome, where the mismatches in the proximal region where counted twice.
CRISPR-SE can effortlessly be used to replace existing alignment tools for complete lists of off-target sites. As for demonstration, we provided the instructions of how to replace BWA with CRISPR-SE in CRISPOR web-site, a live-demo of CRISPOR integrated with CRISPR-SE and a simple input. We also provided a script that converts the output of CRISPR-SE into FlashFry format to use additional scoring functions.
CONCLUSION
CRISPR-Cas9 based technology has been broadly applied in many biotechnologies and we showed that the gRNAs selected using heuristic approaches are incomplete, which would lead to higher off-target effects. CRISPR-SE serves as an accurate and high-performance search engine for CRISPR design and it can be utilized for precise genome-editing applications and novel biotechnology studies.
DATA AVAILABILITY
We built a web interface with pre-computed gRNAs for human and mouse genomes. All scripts and results were available online at http://renlab.sdsc.edu/CRISPR-SE/. The source code is available at https://github.com/bil022/CRISPR-SE.
Supplementary Material
ACKNOWLEDGEMENTS
B.L. developed the CRISPR-SE algorithm. B.L., P.B.C. and Y.D. conceived the development of CRISPR-SE. B.L. and P.B.C wrote the paper.
Contributor Information
Bin Li, Department of Cellular and Molecular Medicine, University of California, San Diego School of Medicine, La Jolla, CA 92093, USA.
Poshen B Chen, Department of Cellular and Molecular Medicine, University of California, San Diego School of Medicine, La Jolla, CA 92093, USA.
Yarui Diao, Department of Cell Biology, Department of Orthopaedic Surgery, and Regeneration Next Initiative, Duke University Medical Center, Durham, NC 27710, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
NIH [1UM1HG009402].
Conflict of interest statement. None declared.
REFERENCES
- 1. Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science (New York, N.Y.). 2007; 315:1709–1712. [DOI] [PubMed] [Google Scholar]
- 2. Garneau J.E., Dupuis M.-È., Villion M., Romero D.A., Barrangou R., Boyaval P., Fremaux C., Horvath P., Magadán A.H., Moineau S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010; 468:67–71. [DOI] [PubMed] [Google Scholar]
- 3. Brouns S.J.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R. J.H., Snijders A.P.L., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science (New York, N.Y.). 2008; 321:960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A. et al. Multiplex genome engineering using CRISPR/Cas systems. Science (New York, N.Y.). 2013; 339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mali P., Yang L., Esvelt K.M., Aach J., Güell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science (New York, N.Y.). 2013; 339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mojica F. J.M., Díez-Villaseñor C., García-Martínez J., Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology (Reading, England). 2009; 155:733–740. [DOI] [PubMed] [Google Scholar]
- 7. Shah S.A., Erdmann S., Mojica F. J.M., Garrett R.A. Protospacer recognition motifs: mixed identities and functional diversity. RNA Biol. 2013; 10:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (New York, N.Y.). 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013; 31:827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013; 31:822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pattanayak V., Lin S., Guilinger J.P., Ma E., Doudna J.A., Liu D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013; 31:839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perez A.R., Pritykin Y., Vidigal J.A., Chhangawala S., Zamparo L., Leslie C.S., Ventura A. GuideScan software for improved single and paired CRISPR guide RNA design. Nat. Biotechnol. 2017; 35:347–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mount D.W. Using the Basic Local Alignment Search Tool (BLAST). CSH Protoc. 2007; 2007:doi:10.1101/pdb.top17. [DOI] [PubMed] [Google Scholar]
- 14. Kent W.J. BLAT–the BLAST-like alignment tool. Genome Res. 2002; 12:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England). 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Blin K., Pedersen L.E., Weber T., Lee S.Y. CRISPy-web: An online resource to design sgRNAs for CRISPR applications. Synth. Syst. Biotechnol. 2016; 1:118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pliatsika V., Rigoutsos I. ‘Off-Spotter’: very fast and exhaustive enumeration of genomic lookalikes for designing CRISPR/Cas guide RNAs. Biol. Direct. 2015; 10:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Naito Y., Hino K., Bono H., Ui-Tei K. CRISPRdirect: software for designing CRISPR/Cas guide RNA with reduced off-target sites. Bioinformatics (Oxford, England). 2015; 31:1120–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cancellieri S., Canver M.C., Bombieri N., Giugno R., Pinello L. CRISPRitz: rapid, high-throughput and variant-aware in silico off-target site identification for CRISPR genome editing. Bioinformatics. 2019; 36:2001–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu H., Wei Z., Dominguez A., Li Y., Wang X., Qi L.S. CRISPR-ERA: a comprehensive design tool for CRISPR-mediated gene editing, repression and activation. Bioinformatics (Oxford, England). 2015; 31:3676–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Montague T.G., Cruz J.M., Gagnon J.A., Church G.M., Valen E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014; 42:W401–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Couvin D., Bernheim A., Toffano-Nioche C., Touchon M., Michalik J., Néron B., P C Rocha E., Vergnaud G., Gautheret D., Pourcel C. CasFinder:CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018; 46:W246–W251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stemmer M., Thumberger T., del Sol Keyer M., Wittbrodt J., Mateo J.L. CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PLOS ONE. 2015; 10:e0124633-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O’Brien A., Bailey T.L. GT-Scan: identifying unique genomic targets. Bioinformatics (Oxford, England). 2014; 30:2673–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heigwer F., Zhan T., Breinig M., Winter J., Brügemann D., Leible S., Boutros M. CRISPR library designer (CLD): software for multispecies design of single guide RNA libraries. Genome Biol. 2016; 17:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Heigwer F., Kerr G., Boutros M. E-CRISP: fast CRISPR target site identification. Nat. Methods. 2014; 11:122–123. [DOI] [PubMed] [Google Scholar]
- 29. Zhu H., Richmond E., Liang C. CRISPR-RT: a web application for designing CRISPR-C2c2 crRNA with improved target specificity. Bioinformatics (Oxford, England). 2018; 34:117–119. [DOI] [PubMed] [Google Scholar]
- 30. Zhu H., Misel L., Graham M., Robinson M.L., Liang C. CT-Finder: A web service for CRISPR optimal target prediction and visualization. Scientific Reports. 2016; 6:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oliveros J.C., Franch M., Tabas-Madrid D., San-León D., Montoliu L., Cubas P., Pazos F. Breaking-Cas-interactive design of guide RNAs for CRISPR-Cas experiments for ENSEMBL genomes. Nucleic Acids Res. 2016; 44:W267–W271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma J., Köster J., Qin Q., Hu S., Li W., Chen C., Cao Q., Wang J., Mei S., Liu Q. et al. CRISPR-DO for genome-wide CRISPR design and optimization. Bioinformatics (Oxford, England). 2016; 32:3336–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Concordet J.-P., Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018; 46:W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pulido-Quetglas C., Aparicio-Prat E., Arnan C., Polidori T., Hermoso T., Palumbo E., Ponomarenko J., Guigó R., Johnson R. Scalable Design of Paired CRISPR Guide RNAs for Genomic Deletion. PLoS Comput. Biol. 2017; 13:e1005341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rastogi A., Murik O., Bowler C., Tirichine L. PhytoCRISP-Ex: a web-based and stand-alone application to find specific target sequences for CRISPR/CAS editing. BMC Bioinformatics. 2016; 17:261–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Biswas A., Gagnon J.N., Brouns S. J.J., Fineran P.C., Brown C.M. CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol. 2013; 10:817–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu H., Ding Y., Zhou Y., Jin W., Xie K., Chen L.-L. CRISPR-P 2.0: An Improved CRISPR-Cas9 Tool for Genome Editing in Plants. Mol. Plant. 2017; 10:530–532. [DOI] [PubMed] [Google Scholar]
- 38. Güell M., Yang L., Church G.M. Genome editing assessment using CRISPR Genome Analyzer (CRISPR-GA). Bioinformatics (Oxford, England). 2014; 30:2968–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu G., Zhang Y., Zhang T. Computational approaches for effective CRISPR guide RNA design and evaluation. Comput. Struct. Biotechnol. J. 2020; 18:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Moreno-Mateos M.A., Vejnar C.E., Beaudoin J.-D., Fernandez J.P., Mis E.K., Khokha M.K., Giraldez A.J. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods. 2015; 12:982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Housden B.E., Valvezan A.J., Kelley C., Sopko R., Hu Y., Roesel C., Lin S., Buckner M., Tao R., Yilmazel B. et al. Identification of potential drug targets for tuberous sclerosis complex by synthetic screens combining CRISPR-based knockouts with RNAi. Sci. Signal. 2015; 8:rs9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chari R., Mali P., Moosburner M., Church G.M. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat. Methods. 2015; 12:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chari R., Yeo N.C., Chavez A., Church G.M. sgRNA Scorer 2. 0: a species-independent model to predict CRISPR/Cas9 activity. ACS Synth. Biol. 2017; 6:902–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xu H., Xiao T., Chen C.-H., Li W., Meyer C.A., Wu Q., Wu D., Cong L., Zhang F., Liu J.S. et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015; 25:1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wong N., Liu W., Wang X. WU-CRISPR: characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol. 2015; 16:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Doench J., Fusi N., Sullender M., Hegde M., Vaimberg E., Donovan K., Smith I., Tothova Z., Wilen C., Orchard R. et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016; 34:181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Labuhn M., Adams F.F., Ng M., Knoess S., Schambach A., Charpentier E.M., Schwarzer A., Mateo J.L., Klusmann J.-H., Heckl D. Refined sgRNA efficacy prediction improves large-and small-scale CRISPR–Cas9 applications. Nucleic Acids Res. 2018; 46:1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rahman M.K., Rahman M.S. CRISPRpred: a flexible and efficient tool for sgRNAs on-target activity prediction in CRISPR/Cas9 systems. PLoS one. 2017; 12:e0181943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mendoza B.J., Trinh C.T. Enhanced guide-RNA design and targeting analysis for precise CRISPR genome editing of single and consortia of industrially relevant and non-model organisms. Bioinformatics. 2018; 34:16–23. [DOI] [PubMed] [Google Scholar]
- 50. Kim H.K., Min S., Song M., Jung S., Choi J.W., Kim Y., Lee S., Yoon S., Kim H.H. Deep learning improves prediction of CRISPR–Cpf1 guide RNA activity. Nat. Biotechnol. 2018; 36:239. [DOI] [PubMed] [Google Scholar]
- 51. Peng H., Zheng Y., Blumenstein M., Tao D., Li J. CRISPR/Cas9 cleavage efficiency regression through boosting algorithms and Markov sequence profiling. Bioinformatics. 2018; 34:3069–3077. [DOI] [PubMed] [Google Scholar]
- 52. Wilson L.O., Reti D., O’Brien A.R., Dunne R.A., Bauer D.C. High activity target-site identification using phenotypic independent CRISPR-Cas9 core functionality. CRISPR J. 2018; 1:182–190. [DOI] [PubMed] [Google Scholar]
- 53. Zhang D., Hurst T., Duan D., Chen S.-J. Unified energetics analysis unravels SpCas9 cleavage activity for optimal gRNA design. Proc. Natl. Acad. Sci. 2019; 116:8693–8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics (Oxford, England). 2014; 30:1473–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. McKenna A., Shendure J. FlashFry: a fast and flexible tool for large-scale CRISPR target design. BMC Biol. 2018; 16:74–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jacquin A. L.S., Odom D.T., Lukk M. Crisflash: open-source software to generate CRISPR guide RNAs against genomes annotated with individual variation. Bioinformatics. 2019; 35:3146–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xiao A., Cheng Z., Kong L., Zhu Z., Lin S., Gao G., Zhang B. CasOT: a genome-wide Cas9/gRNA off-target searching tool. Bioinformatics. 2014; 30:1180–1182. [DOI] [PubMed] [Google Scholar]
- 58. Xie S., Shen B., Zhang C., Huang X., Zhang Y. sgRNAcas9: a software package for designing CRISPR sgRNA and evaluating potential off-target cleavage sites. PLoS one. 2014; 9:e100448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013; 31:827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Alkan F., Wenzel A., Anthon C., Havgaard J.H., Gorodkin J. CRISPR-Cas9 off-targeting assessment with nucleic acid duplex energy parameters. Genome Biol. 2018; 19:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Abadi S., Yan W.X., Amar D., Mayrose I. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput. Biol. 2017; 13:e1005807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Listgarten J., Weinstein M., Kleinstiver B.P., Sousa A.A., Joung J.K., Crawford J., Gao K., Hoang L., Elibol M., Doench J.G. et al. Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat. Biomed. Eng. 2018; 2:38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chuai G., Ma H., Yan J., Chen M., Hong N., Xue D., Zhou C., Zhu C., Chen K., Duan B. et al. DeepCRISPR: optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018; 19:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Diao Y., Fang R., Li B., Meng Z., Yu J., Qiu Y., Lin K.C., Huang H., Liu T., Marina R.J., Jung I., Shen Y., Guan K.-L., Ren B. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat. Methods. 2017; 14:629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We built a web interface with pre-computed gRNAs for human and mouse genomes. All scripts and results were available online at http://renlab.sdsc.edu/CRISPR-SE/. The source code is available at https://github.com/bil022/CRISPR-SE.