

Abstract
The emerging Coronavirus Disease 2019 (COVID-19) pandemic has posed a serious threat to the public health worldwide, demanding urgent vaccine provide. According to the virus feature as an RNA virus, a high rate of mutations imposes some vaccine design difficulties. Bioinformatics tools have been widely used to make advantage of conserved regions as well as immunogenicity. In this study, we aimed at immunoinformatic evaluation of SARS-CoV-2 proteins conservancy and immunogenicity to design a preventive vaccine candidate. Spike, Membrane and Nucleocapsid amino acid sequences were obtained, and four possible fusion proteins were assessed and compared in terms of structural features and immunogenicity, and population coverage. MHC-I and MHC-II T-cell epitopes, the linear and conformational B-cell epitopes were evaluated. Among the predicted models, the truncated form of Spike in fusion with M and N protein applying AAY linker has high rate of MHC-I and MCH-II epitopes with high antigenicity and acceptable population coverage of 82.95% in Iran and 92.51% in Europe. The in silico study provided truncated Spike-M-N SARS-CoV-2 as a potential preventive vaccine candidate for further in vivo evaluation.
Keywords: COVID-19, Vaccine design, In silico, SARS-CoV-2, Protein
Introduction
The COVID-19 pandemic caused by the SARS-CoV-2 virus has unexpectedly affected global health since emerging. As of 1 December 2020, more 68 million cases have been confirmed in 216 countries with more than 1.5 million deaths which clearly shows that a protective vaccine is urgently needed although several knowledge are still on this virus [1].
SARS-CoV-2 genome with ~ 30 kb size encodes multiple spike (S) protein, the envelope (E) protein, the membrane (M) protein, and the nucleocapsid (N) proteins and some non-structural ones. The spike (S) protein has pivotal roles in receptor binding, angiotensin-converting enzyme 2 (ACE2), and also membrane fusion. Therefore, it is widely investigated as an attractive antigen in vaccine designs aiming at virus binding/fusion blocking antibodies to neutralize virus infection. Since SARS-CoV is an RNA virus that imposes an error-prone genome and results in host immune response escape, targeting the full-length S protein in vaccine studies have not brought protective immunity against SARS outbreaks [2–5]. Although the spike protein is a promising protective immunogenic, antigen design optimization is critical to achieving optimal immune response. The S1 subunit includes the minimal receptor-binding domain (RBD, 318–510 aa), a conserved target for neutralizing antibody induction [6–9]. Therefore, this region could be more practical in comparison with full-length S protein.
The membrane glycoprotein)M) provides coronavirus assembly, is the most abundant envelope protein that facilitates viral components sortation and incorporation into virions coronavirus assembly [10–12]. M protein binding helps the virus to stabilize nucleocapsids and accelerates completion of viral assembly by N protein-RNA complex stabilization [13, 14]. The nucleocapsid protein (N) as a multifunctional RNA-binding protein is essential for viral RNA replication and transcription. It also has many vital roles in the viral RNA genome packaging, regulation of viral RNA synthesis in replication/transcription, and infected cell metabolism modulation. Some studies demonstrated that N protein regulates host–pathogen interactions, including actin reorganization, cell cycle progress, and apoptosis. This protein is also considered highly immunogenic based on abundantly expression during infection [15–17].
According to the critical demand on developing safe, effective approaches against SARS-CoV-2 have stepped on the way with some clinical evaluation worldwide [18–20]. There is no doubt that any approaches with generated vaccines could be highly valuable in possible outbreaks and probable seasonal re-emerging which is mainly depended on long-term protection evolution. MERS-CoV and SARS-CoV-1 vaccines progressions over the last years are crucial keys given their genetic similarity which provides vital awareness for SARS-CoV-2 vaccine development [21–25].
Therefore, multiple platforms have been under development since the emerging, including DNA- and RNA-based platforms and recombinant-subunit vaccines. Nevertheless, SARS-CoV-2 vaccine development poses some challenges even with novel platforms. For instance, preclinical studies on SARS and MERS vaccine candidates have brought concerns about exacerbating lung disease as an outcome of antibody-dependent enhancement or direct impact. Hence, testing in a suitable animal model and rigorous safety monitoring in clinical trials will be critical [26, 27].
Traditional approaches in vaccination based on laboratory experiments in the outbreak situation could not meet the urgent needs, and many therapeutic agents are being investigated [28–31]. Bioinformatics study is a strong tool specified in sortation, organization, and process large amounts of available data generated from other experiments to provide a large-scale immunological platform within a limited time. Since the virus genome and its protein sequences information are available, the presented epitopes and the virus characteristics could be predicted by in silico analysis, which significantly accelerates the progress of vaccine development [32–36].
In this study, we aimed at B-cell and T-cell epitope prediction of SARS-CoV-2s Spike SARS-CoV-2 Spike receptor-binding domain (RBD), M and N protein as fusion proteins and comparison in silico immunogenicity by applying bioinformatics methods to provide a subunit vaccine candidate against COVID-19.
Materials and Methods
Sequence Retrieval
Viral amino acid sequences of SARS-CoV-2 Spike (S), Membrane (M) and nucleocapsid (N) proteins (accession numbers S: YP_009724390.1, QIX12195.1, QJD47706.1, QJD47860.1, QJD25757.1, QIU78767.1, QIX12148.2, QIU80900.1, BCB97891.1, M: YP_009724393.1, QJD47709.1 QJD47863.1 QJD25760.1 QIU78770.1 QIX12151.1 QIX12198.1 QIU80903.1, BCB97894.1 and N: YP_009724397.2, QIU78775.1, QIX12156.1, QIX12203.1, MT186677.1, QIU80910.1, MT186677.1, BCB97898.1) were obtained from the GenBank [37]. The whole process is simply shown in Fig. 1.
Fig. 1.
Schematic view of the applied methods in the study
T-Cell Epitope and Antigenicity Prediction
The obtained sequences were submitted to MHCI- and MHCII-binding prediction tool http://tools.iedb.org/mhc/n in IEDB using different methods including Artificial Neural Network (ANN), Stabilized Matrix Method (SMM) or Scoring Matrices derived from Combinatorial Peptide Libraries (Comblib_Sidney2008) method. MHC-NP net CTLpan1.1server [38–40] and RankPEP server were also applied. The outcomes from all applied tools were in a similar range. Therefore, here, the IEDB outputs are reported.
T-cell epitopes lengths were defined as 9-mer for MHC class I and 15-mer for MHC class II for BALB/c and human separately. BALB/c MHC class I alleles included H2-Dd, H2-Kd and H2-Ld and MHC class II alleles were selected H2-IAd and H2-IEd. According to diversity of antigens and the recognition extent by the variable HLA molecules in a population and in considering the most popular HLA in the Persian population based on the available report [41–43], HLA-A*01, 02, 03, 11, 24, 26, 32, HLA-B*35, 51, 50, 27, 57 for MHC class I and HLA-DRB1*15, 11, 13, 03, 04, 07 for MHC class II were selected. The peptides which were predicted to bind to MHC class I and II molecules with percentile rank ≤ 1 were considered epitopic sequences.
The VaxiJen v2.0 online antigen prediction tool was applied to assess the antigenicity scores of predicted epitopes [44, 45], which provides antigen sorting according to the protein physicochemical qualities without the sequence alignment usage. Epitopes with antigenic score > 0.5 were considered antigenic.
Toxicity Analysis
We investigated the selected model of 4 for toxicity using ToxinPred [46]. This tool provides the confirmation of non-toxicity of epitopes for the host according to all physic-chemical parameters.
Population Coverage and Epitope Conservancy
MHC I and MHC II potential binders from the selected fusion form of model 4 were computed for population coverage analysis against the whole world population, especially the Persian population, with the selected human MHC I and MHC II interacting molecules using the IEDB population coverage calculation tool. Population coverage calculation is based on total HLA hits score which is achieved from the IEDB. These data are derived from a relative of an allele’s relative frequency at a particular locus in a population (Sequence identity threshold ≥ 100). In addition, we assessed the conservancy level of each potential epitope by searching identities in 10 amino acid sequences of S protein, 12 amino acid sequences of M protein and 12 amino acid sequences of N protein from different geographical area retrieved from the database.
B-Cell Epitope Prediction
BepiPred linear epitope prediction server [47] from the Immune Epitope Database was applied to predict linear B cell epitopes with threshold 0.35 and epitopes length is varied from 6 to higher residues.
For Recognition of other physicochemical properties of amino acids such as the antigenicity (Kolaskar and Tongaonkar) [48], surface accessibility [49], flexibility (Karplus and Schulz) [50], hydrophilicity [51] and beta-turns (Chou and Fasman) [52] methods were also assessed by the available tools at the platform of Immune Epitope Database (IEDB) Analysis Resource (http://tools.iedb.org/bcell). The protein sequence scanning window length for all methods was adjusted to seven residues. We applied ElliPro [53] at IEDB online tool for discontinuous B-cell epitope prediction with minimum score value set at 0.50. This method predicts epitopes by considering both the sequence- and structure-based information.
Structural Analysis
Physicochemical properties of fragments including weight, aliphatic index and Grand average of hydropathicity (GRAVY), theoretical pI and atomic composition were analyzed using Expasy’s ProtParam server [54]. Self-optimized prediction method with alignment (SOPMA) and Jpred tools were applied to generate and evaluate the secondary structure and assessment of a-helix, b sheets, random coils of the proteins [55, 56].
Homology Modeling and Validation
The 3D model were analyzed using the Threading ASSEmbly Refinement (I-TASSER) online server program [57] and IntFOLD Integrated Protein Structure and Function Prediction Server [58] that provides 3D models along with confidence score (C-Score) and model quality score. The further pattern evaluation was done by three indicators: Stereochemical qualities, C-score and DFIRE2 energy profile [59]. The Stereo chemical analysis of the 3D model was assessed by PROCHECK, ERRAT, VERIFY 3D and verified by structural Analysis and Verification server [60–62].
Results
The amino acid sequences of chain B, SARS-CoV-2 Spike receptor-binding domain (RBD), Spike, Membrane and Nucleocapsid proteins were obtained and four fusion forms as shown in Fig. 2 were predicted to be compared in term of immunogenicity. A proteasomal linker (AAY) was used to fuse the applied proteins.
Fig. 2.

Schematic view of predicted constructs with the flexible spacer (AAY)
MHC Class I and II Binding Prediction in BALB/c
We applied 9-mer and 15-mer lengths coverage of T-cell epitopes to design a vaccine model. Spike, M and N proteins were subjected to IEDB MHC I and MHCII binding prediction tool. The IEDB recommended, RANKPEP, net CTLpan1.1, MHC-NP, and netMHCpan3.0 server were used to predict the epitopes from selected proteins. High-affinity peptides with antigenic features are listed in Tables 1 and 2 (percentile rank ≤ 1).
Table 1.
BALB/c MHC class I epitopes in predicted models
| Antigenicity score | Percentile rank | Allele | Length | End | Start | Peptide |
|---|---|---|---|---|---|---|
| Model 1 | ||||||
| 1.426 | 0.3 | H-2-Kd | 9 | 69 | 61 | CYGVSPTKL |
| 0.596 | 0.3 | H-2-Dd | 9 | 195 | 187 | YQPYRVVVL |
| 0.578 | 0.64 | H-2-Kd | 9 | 178 | 170 | CYFPLQSYG |
| 0.5003 | 0.7 | H-2-Kd | 9 | 40 | 32 | VYAWNRKRI |
| 0.5107 | 0.8 | H-2-Dd | 9 | 179 | 171 | YFPLQSYGF |
| 0.5453 | 0.92 | H-2-Ld | 9 | 174 | 166 | EGFNCYFPL |
| Model 2 | ||||||
| 1.4263 | 0.3 | H-2-Kd | 9 | 69 | 61 | CYGVSPTKL |
| 0.5964 | 0.3 | H-2-Dd | 9 | 195 | 187 | YQPYRVVVL |
| 0.4821 | 0.4 | H-2-Kd | 9 | 314 | 306 | SYFIASFRL |
| 0.578 | 0.64 | H-2-Kd | 9 | 178 | 170 | CYFPLQSYG |
| 0.734 | 0.68 | H-2-Ld | 9 | 550 | 542 | SPRWYFYYL |
| 0.5003 | 0.7 | H-2-Kd | 9 | 40 | 32 | VYAWNRKRI |
| 0.8519 | 0.7 | H-2-Kd | 9 | 625 | 617 | SQASSRSSS |
| 0.611 | 0.75 | H-2-Ld | 9 | 746 | 738 | WPQIAQFAP |
| 0.5107 | 0.8 | H-2-Dd | 9 | 179 | 171 | YFPLQSYGF |
| 0.5453 | 0.92 | H-2-Ld | 9 | 174 | 166 | EGFNCYFPL |
| Model 3 | ||||||
| 1.4177 | 0.2 | H-2-Kd | 9 | 898 | 890 | QYIKWPWYI |
| 1.4263 | 0.3 | H-2-Kd | 9 | 69 | 61 | CYGVSPTKL |
| 0.596 | 0.3 | H-2-Dd | 9 | 195 | 187 | YQPYRVVVL |
| 0.8274 | 0.62 | H-2-Kd | 9 | 396 | 388 | AYSNNSIAI |
| 0.578 | 0.64 | H-2-Kd | 9 | 178 | 170 | CYFPLQSYG |
| 0.5003 | 0.7 | H-2-Kd | 9 | 40 | 32 | VYAWNRKRI |
| 0.853 | 0.7 | H-2-Kd | 9 | 408 | 400 | FTISVTTEI |
| 1.29 | 0.7 | H-2-Kd | 9 | 445 | 437 | QYGSFCTQL |
| 0.5107 | 0.8 | H-2-Dd | 9 | 179 | 171 | YFPLQSYGF |
| 0.5453 | 0.92 | H-2-Ld | 9 | 174 | 166 | EGFNCYFPL |
| Model 4 | ||||||
| 1.4177 | 0.2 | H-2-Kd | 9 | 898 | 890 | QYIKWPWYI |
| 1.4263 | 0.3 | H-2-Kd | 9 | 69 | 61 | CYGVSPTKL |
| 0.5964 | 0.3 | H-2-Dd | 9 | 195 | 187 | YQPYRVVVL |
| 0.4821 | 0.4 | H-2-Kd | 9 | 363 | 355 | SYQTQTNSP |
| 0.8274 | 0.62 | H-2-Kd | 9 | 396 | 388 | AYSNNSIAI |
| 0.578 | 0.64 | H-2-Kd | 9 | 178 | 170 | CYFPLQSYG |
| 0.734 | 0.68 | H-2-Ld | 9 | 1296 | 1288 | SPRWYFYYL |
| 0.5003 | 0.7 | H-2-Kd | 9 | 40 | 32 | VYAWNRKRI |
| 0.8535 | 0.7 | H-2-Kd | 9 | 408 | 400 | FTISVTTEI |
| 1.2906 | 0.7 | H-2-Kd | 9 | 445 | 437 | QYGSFCTQL |
| 0.8519 | 0.7 | H-2-Kd | 9 | 1371 | 1363 | SQASSRSSS |
| 0.611 | 0.75 | H-2-Ld | 9 | 1492 | 1484 | WPQIAQFAP |
| 0.5107 | 0.8 | H-2-Dd | 9 | 179 | 171 | YFPLQSYGF |
| 0.5453 | 0.92 | H-2-Ld | 9 | 174 | 166 | EGFNCYFPL |
Table 2.
BALB/c MHC class II epitopes in predicted models
| Antigenicity score | Percentile rank | Allele | Length | End | Start | Peptide |
|---|---|---|---|---|---|---|
| Model 1 | ||||||
| 0.3676 | 0.07 | H2-IEd | 15 | 29 | 43 | FASVYAWNRKRISNC |
| 0.4243 | 0.1 | H2-IEd | 15 | 28 | 42 | RFASVYAWNRKRISN |
| 0.3086 | 0.14 | H2-IEd | 15 | 30 | 44 | ASVYAWNRKRISNCV |
| 0.4963 | 0.14 | H2-IEd | 15 | 27 | 41 | TRFASVYAWNRKRIS |
| 0.1089 | 0.17 | H2-IEd | 15 | 130 | 144 | NYNYLYRLFRKSNLK |
| 0.2254 | 0.17 | H2-IEd | 15 | 131 | 145 | YNYLYRLFRKSNLKP |
| 0.3301 | 0.3 | H2-IEd | 15 | 31 | 45 | SVYAWNRKRISNCVA |
| 0.1801 | 0.36 | H2-IEd | 15 | 129 | 143 | GNYNYLYRLFRKSNL |
| 0.0415 | 0.38 | H2-IEd | 15 | 132 | 146 | NYLYRLFRKSNLKPF |
| 0.0814 | 0.71 | H2-IEd | 15 | 133 | 147 | YLYRLFRKSNLKPFE |
| 0.0207 | 0.74 | H2-IEd | 15 | 128 | 142 | GGNYNYLYRLFRKSN |
| Model 2 | ||||||
| 0.4243 | 0.1 | H2-IEd | 15 | 42 | 28 | RFASVYAWNRKRISN |
| 0.4614 | 0.14 | H2-Ied | 15 | 534 | 520 | QIGYYRRATRRIRGG |
| 0.4963 | 0.14 | H2-IEd | 15 | 41 | 27 | TRFASVYAWNRKRIS |
| 0.4072 | 0.27 | H2-Ied | 15 | 322 | 308 | FIASFRLFARTRSMW |
| 0.6649 | 0.27 | H2-IEd | 15 | 535 | 521 | IGYYRRATRRIRGGD |
| 0.4424 | 0.3 | H2-IEd | 15 | 323 | 309 | IASFRLFARTRSMWS |
| 0.7304 | 0.57 | H2-Ied | 15 | 324 | 310 | ASFRLFARTRSMWSF |
| 0.7955 | 0.73 | H2-IEd | 15 | 325 | 311 | SFRLFARTRSMWSFN |
| 0.7387 | 0.94 | H2-Ied | 15 | 260 | 246 | LLQFAYANRNRFLYI |
| 0.8634 | 0.98 | H2-IEd | 15 | 416 | 402 | DSGFAAYSRYRIGNY |
| Model 3 | ||||||
| 0.4243 | 0.1 | H2-IEd | 15 | 42 | 28 | RFASVYAWNRKRISN |
| 0.4963 | 0.14 | H2-Ied | 15 | 41 | 27 | TRFASVYAWNRKRIS |
| Model 4 | ||||||
| 0.4243 | 0.1 | H2-IEd | 15 | 42 | 28 | RFASVYAWNRKRISN |
| 0.4614 | 0.14 | H2-IEd | 15 | 1280 | 1266 | QIGYYRRATRRIRGG |
| 0.4963 | 0.14 | H2-IEd | 15 | 41 | 27 | TRFASVYAWNRKRIS |
| 0.4072 | 0.27 | H2-IEd | 15 | 1068 | 1054 | FIASFRLFARTRSMW |
| 0.6649 | 0.27 | H2-IEd | 15 | 1281 | 1267 | IGYYRRATRRIRGGD |
| 0.4424 | 0.3 | H2-IEd | 15 | 1069 | 1055 | IASFRLFARTRSMWS |
| 0.7304 | 0.57 | H2-IEd | 15 | 1070 | 1056 | ASFRLFARTRSMWSF |
| 0.7955 | 0.73 | H2-IEd | 15 | 1071 | 1057 | SFRLFARTRSMWSFN |
| 0.7387 | 0.94 | H2-IEd | 15 | 1006 | 992 | LLQFAYANRNRFLYI |
| 0.8741 | 0.98 | H2-IEd | 15 | 1162 | 1148 | DSGFAAYSRYRIGNY |
According to the generated data, in comparison between MCHI and MHCII in predicted models, the number of MHCI epitopes are clearly higher, meaning that the designed models could elicit cellular immunity responses in a mouse model. Moreover, among the 4 models, the last one, which is composed of truncated Spike, full M and full N proteins includes 14 MHCI epitopes with high antigenic scores rather than other models. In contrast with MHCI, analysis MHCII binders, only predicted model 2 and model 4 contain epitopic peptides with high antigenicity score (> 0.5).
Human T-Cell Epitope Prediction
According to the T-cell epitopes in mice, Models 2 and 4 had more antigenic epitopes. Therefore, we continued T-cell epitopes in human most prevalent HLA I and HLA II. The results are summarized in Tables 3 and 4. Model 2 and Model 4 contain at least 166 and 300 epitopes, respectively, from which we here only report the highly antigenic ones. Therefore, we continued the human epitope prediction study with truncated Spike + full M + full N as the best model. This fusion form has also 42 HLA class II epitopes (percentile rank < 1), from which 29 binders were assessed antigenic and are shown in Table 4.
Table 3.
Human class I epitopes in predicted model 4
| Antigenicity score | Percentile rank | Allele | Toxicity | End | Start | Peptide |
|---|---|---|---|---|---|---|
| 0.7571 | 0.01 | HLA-A*03:01 | Non-toxin | 1552 | 1544 | KTFPPTEPK |
| 0.7571 | 0.01 | HLA-A*11:01 | Non-toxin | 1552 | 1544 | KTFPPTEPK |
| 0.8132 | 0.01 | HLA-A*11:01 | Non-toxin | 755 | 747 | VTYVPAQEK |
| 0.7014 | 0.01 | HLA-A*03:01 | Non-toxin | 710 | 702 | ASANLAATK |
| 1.4278 | 0.01 | HLA-B*35:01 | Non-toxin | 586 | 578 | IPFAMQMAY |
| 0.5669 | 0.01 | HLA-B*57:01 | Non-toxin | 1457 | 1449 | KAYNVTQAF |
| 0.882 | 0.01 | HLA-B*51:01 | Non-toxin | 404 | 396 | IPTNFTISV |
| 0.5669 | 0.01 | HLA-B*57:01 | Non-toxin | 1457 | 1449 | KAYNVTQAF |
| 0.8132 | 0.01 | HLA-A*03:01 | Non-toxin | 755 | 747 | VTYVPAQEK |
| 1.4177 | 0.02 | HLA-A*24:02 | Non-toxin | 898 | 890 | QYIKWPWYI |
| 1.7462 | 0.02 | HLA-B*57:01 | Non-toxin | 1291 | 1283 | KMKDLSPRW |
| 0.53 | 0.02 | HLA-B*50:01 | Non-toxin | 871 | 863 | KEIDRLNEV |
| 0.7052 | 0.02 | HLA-B*51:01 | Non-toxin | 402 | 394 | IAIPTNFTI |
| 1.2394 | 0.02 | HLA-B*27:02 | Non-toxin | 1070 | 1062 | ARTRSMWSF |
| 1.7462 | 0.02 | HLA-B*57:01 | Non-toxin | 1291 | 1283 | KMKDLSPRW |
| 0.5107 | 0.03 | HLA-A*24:02 | Non-toxin | 179 | 171 | YFPLQSYGF |
| 1.1141 | 0.03 | HLA-B*27:02 | Non-toxin | 17 | 9 | VRFPNITNL |
| 1.6639 | 0.04 | HLA-A*02:01 | Non-toxin | 107 | 99 | KIADYNYKL |
| 0.8597 | 0.04 | HLA-B*50:01 | Non-toxin | 1100 | 1092 | LESELVIGA |
| 0.5781 | 0.04 | HLA-A*11:01 | Non-toxin | 517 | 509 | TLADAGFIK |
| 0.7785 | 0.05 | HLA-B*35:01 | Non-toxin | 1003 | 995 | FAYANRNRF |
| 0.9457 | 0.05 | HLA-B*35:01 | Non-toxin | 1136 | 1128 | VATSRTLSY |
| 0.6409 | 0.05 | HLA-B*50:01 | Non-toxin | 1102 | 1094 | SELVIGAVI |
| 0.7585 | 0.05 | HLA-B*51:01 | Non-toxin | 1257 | 1249 | FPRGQGVPI |
Table 4.
Human MHC class II epitopes in predicted Model 4
| Antigenicity score | Percentile rank | Allele | Toxicity | End | Start | Peptide |
|---|---|---|---|---|---|---|
| 0.6031 | 0.12 | HLA-DRB1*11:04 | Non-toxin | 1414 | 1400 | AALALLLLDRLNQLE |
| 0.5057 | 0.12 | HLA-DRB1*11:04 | Non-toxin | 1415 | 1401 | ALALLLLDRLNQLES |
| 0.5669 | 0.12 | HLA-DRB1*11:04 | Non-toxin | 1417 | 1403 | ALLLLDRLNQLESKM |
| 0.5531 | 0.12 | HLA-DRB1*11:04 | Non-toxin | 1413 | 1399 | DAALALLLLDRLNQL |
| 0.7357 | 0.12 | HLA-DRB1*11:04 | Non-toxin | 1416 | 1402 | LALLLLDRLNQLESK |
| 0.6286 | 0.12 | HLA-DRB1*11:04 | Non-toxin | 1418 | 1404 | LLLLDRLNQLESKMS |
| 0.6019 | 0.16 | HLA-DRB1*11:04 | Toxin | 918 | 904 | AGLIAIVMVTIMLCC |
| 0.6693 | 0.16 | HLA-DRB1*11:04 | Toxin | 919 | 905 | GLIAIVMVTIMLCCM |
| 0.7442 | 0.16 | HLA-DRB1*11:04 | Toxin | 921 | 907 | IAIVMVTIMLCCMTS |
| 0.6171 | 0.16 | HLA-DRB1*11:04 | Toxin | 920 | 906 | LIAIVMVTIMLCCMT |
| 0.6806 | 0.16 | HLA-DRB1*07:01 | Non-toxin | 409 | 395 | AIPTNFTISVTTEIL |
| 0.7719 | 0.4 | HLA-DRB1*07:01 | Non-toxin | 408 | 394 | IAIPTNFTISVTTEI |
| 1.1349 | 0.51 | HLA-DRB1*07:01 | Non-toxin | 411 | 397 | PTNFTISVTTEILPV |
| 0.8294 | 0.52 | HLA-DRB1*07:01 | Non-toxin | 410 | 396 | IPTNFTISVTTEILP |
| 1.1691 | 0.52 | HLA-DRB1*07:01 | Non-toxin | 412 | 398 | TNFTISVTTEILPVS |
| 0.6336 | 0.58 | HLA-DRB1*15:01 | Non-toxin | 445 | 431 | CSNLLLQYGSFCTQL |
| 0.9934 | 0.58 | HLA-DRB1*07:01 | Non-toxin | 1525 | 1511 | GTWLTYTGAIKLDDK |
| 0.6215 | 0.58 | HLA-DRB1*07:01 | Non-toxin | 1524 | 1510 | SGTWLTYTGAIKLDD |
| 1.2416 | 0.58 | HLA-DRB1*07:01 | Non-toxin | 1526 | 1512 | TWLTYTGAIKLDDKD |
| 0.8305 | 0.6 | HLA-DRB1*15:01 | Non-toxin | 446 | 432 | SNLLLQYGSFCTQLN |
| 0.6128 | 0.69 | HLA-DRB1*15:01 | Non-toxin | 19 | 5 | TESIVRFPNITNLCP |
| 1.2905 | 0.7 | HLA-DRB1*07:01 | Non-toxin | 1034 | 1020 | LACFVLAAVYRINWI |
| 0.7635 | 0.72 | HLA-DRB1*15:01 | Non-toxin | 444 | 430 | ECSNLLLQYGSFCTQ |
| 0.8668 | 0.75 | HLA-DRB1*15:01 | Non-toxin | 447 | 433 | NLLLQYGSFCTQLNR |
| 0.8548 | 0.76 | HLA-DRB1*07:01 | Non-toxin | 1031 | 1017 | PVTLACFVLAAVYRI |
| 1.0450 | 0.76 | HLA-DRB1*07:01 | Non-toxin | 1032 | 1018 | VTLACFVLAAVYRIN |
| 1.1115 | 0.88 | HLA-DRB1*07:01 | Non-toxin | 1035 | 1021 | ACFVLAAVYRINWIT |
| 1.3132 | 0.88 | HLA-DRB1*07:01 | Non-toxin | 1033 | 1019 | TLACFVLAAVYRINW |
| 0.6125 | 0.92 | HLA-DRB1*15:01 | Non-toxin | 20 | 6 | ESIVRFPNITNLCPF |
Toxicity Analysis
Model 4 at the final step was tested for toxicity using ToxinPred tool, as shown in Tables 3 and 4.
Population Coverage and Conservancy Analysis
Peptides predicted to interact with MHCI and II molecules in the selected Model 4 were tested for population coverage analysis using the IEDB population coverage tool to cover most HIV chronic infected individuals specifically the Persian population. Furthermore, we selected North America, Southwest Asia, South Asia, Europe, South America and Africa continent. The results of total population coverage in Persian and the other populations are listed in Table 5. The selected Model 4 has an acceptable coverage of 82.95% for MHC class I and II in the Persian population. To identify the Conservancy of predicted peptides of Model 4, we used the IEDB tool. Therefore, all peptides (with an antigenic score > 0.5) were submitted against related S, N, M sequences at a high threshold. Finally, we determined all epitopes were fully conserved (100%) epitopes.
Table 5.
Predicted epitopes of Model 4 interacting with combined of human MHC class I and II among different population worldwide
| Average of epitope hits | MHCI and MHCII Combined PPCa (%) | Population |
|---|---|---|
| 4.92 | 82.95 | Iran |
| 4.86 | 74.74 | Southwest Asia |
| 7.03 | 79.57 | South Asia |
| 7.87 | 92.51 | Europe |
| 6.83 | 89.20 | North America |
| 3.21 | 64.37 | South America |
| 3.04 | 48.97 | Africa |
aProjected population coverage
B-Cell Epitopes Recognition
The four predicted models were assessed by BepiPred server, and the antigenicity of predicted epitopes was evaluated by VaxiJen. The amino acid sequences, peptide lengths, and positions of these epitopes are shown in Table 5.
Among the predicted models, Model 2 (RBD + M + N) and Model 4 (Truncated Spike + M + N) have a high number of B-cell epitopes in comparison with the other models in agreement with T-cell prediction. Moreover, Model 4 includes 14 B-cell antigenic epitopes which shows to have the highest potency in the humoral response.
Surface accessibility, flexibility, hydrophilicity and antigenicity are essential features of B cell antigenic indexes in vaccine design. The selected Model 4 was assessed by different prediction at the BepiPred Sequential B-Cell Epitope Predictor, as shown in Fig. 3.
Fig. 3.
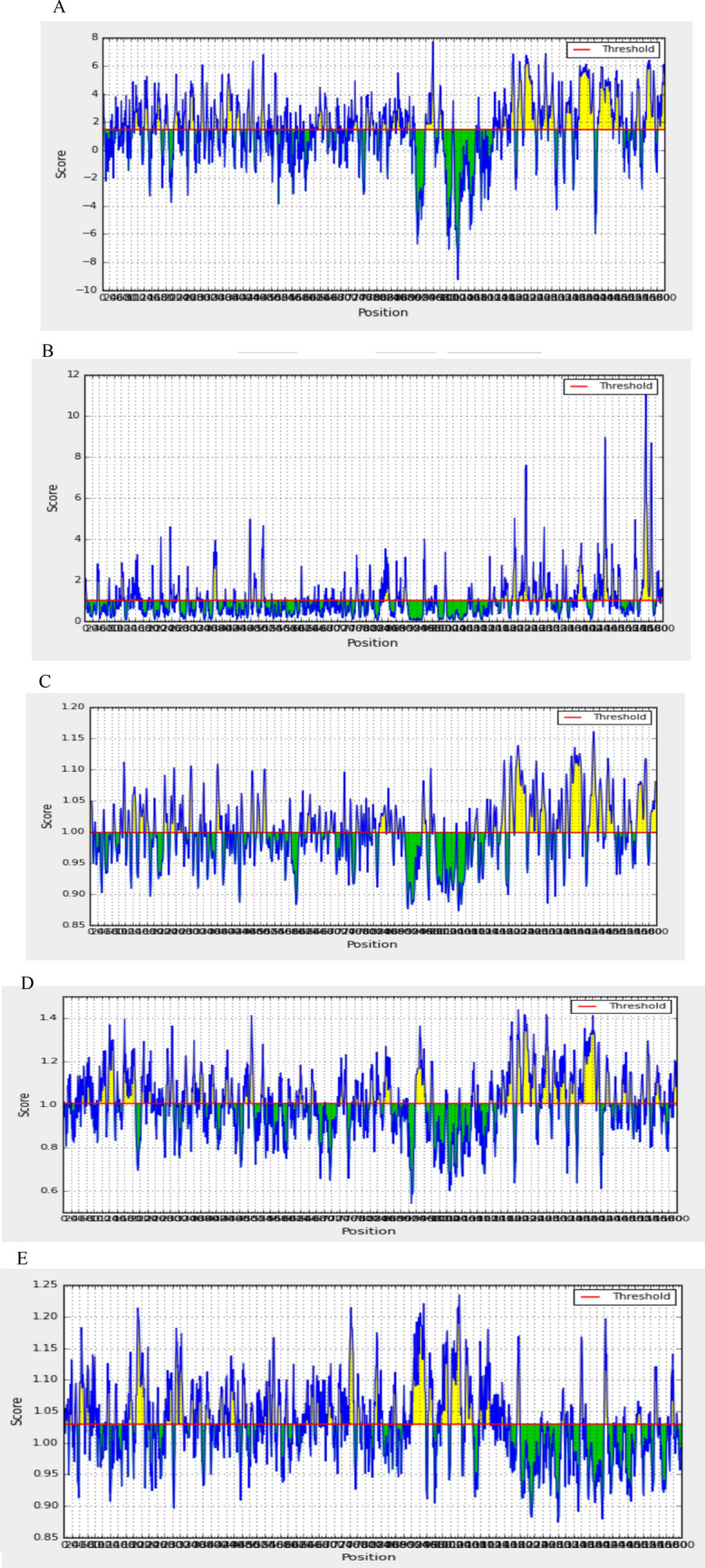
Graphical representation of B cell epitopes prediction by a Parker hydrophilicity prediction (threshold: 1.474), b Emini surface accessibility prediction (threshold: 1.000), c Karplus and Schulz flexibility prediction (threshold: 0.999), d Chou and Fasman beta turn prediction (threshold: 1.004) and e Kolaskar and Tongaonkar Antigenicity (threshold: 1.0). The yellow regions above the threshold (red line) are supposed to be a part of B cell epitope whereas the green areas are not (Color figure online)
In order to find conformational B-cell epitope in 3D structure, Ellipro was used. Ellipro predicted six discontinues epitopes for Model 1 with maximum score of 0.942 and minimum score of 0.542, eight epitopes for model 2 with maximum score of 0.802 and minimum score of 0.502 and nine epitopes for model 3 with maximum score of 0.816 and minimum score of 0.55 (data were not shown). Ellipro predicted a total of 61 discontinues epitopes for the chosen Model 4 with a maximum score of 0.994. Those scores greater than 0.8 were selected (Table 6).
Table 6.
B-cell linear epitopes for selected Model 4
| Antigenicity | Length | B cell (position) | Proteins |
|---|---|---|---|
| 1.2606 | 14 | VRQIAPGQTGKIAD (89) | Model 1 |
| 0.7136 | 12 | YGFQPTNGVGYQ (177) | |
| 0.8904 | 9 | NNLDSKVGG (121) | |
| 1.3668 | 6 | RVQPTE (1) | |
| 0.5455 | 53 | GTTLPKGFYAEGSRGGSQASSRSSSRSRNSSRNSTPGSSRGTSPARMAGNGGD (601) | Model 2 |
| 0.5302 | 38 | SKMSGKGQQQQGQTVTKKSAAEASKKPRQKRTATKAYN (669) | |
| 0.5605 | 30 | KTFPPTEPKKDKKKKADETQALPQRQKKQQ (798) | |
| 0.5570 | 28 | QHGKEDLKFPRGQGVPINTNSSPDDQIG (495) | |
| 1.1728 | 15 | AFGRRGPEQTQGNFG (710) | |
| 1.2606 | 14 | VRQIAPGQTGKIAD (89) | |
| 0.7136 | 12 | YGFQPTNGVGYQ (177) | |
| 0.8771 | 12 | RIRGGDGKMKDL (530) | |
| 2.1298 | 10 | KLDDKDPNFK (775) | |
| 0.8904 | 9 | NNLDSKVGG (121) | |
| 1.3668 | 6 | RVQPTE (1) | |
| 0.6838 | 6 | TDYKHW (733) | |
| 1.2606 | 14 | VRQIAPGQTGKIAD (89) | Model 3 |
| 0.7136 | 12 | YGFQPTNGVGYQ (177) | |
| 0.5322 | 12 | ILPDPSKPSKRS (487) | |
| 1.4039 | 11 | KNHTSPDVDLG (839) | |
| 0.8904 | 9 | NNLDSKVGG (121) | |
| 1.3668 | 6 | RVQPTE (1) | |
| 0.5455 | 53 | GTTLPKGFYAEGSRGGSQASSRSSSRSRNSSRNSTPGSSRGTSPARMAGNGGD (1347) | Model 4 |
| 0.5302 | 38 | SKMSGKGQQQQGQTVTKKSAAEASKKPRQKRTATKAYN (1415) | |
| 0.5605 | 30 | KTFPPTEPKKDKKKKADETQALPQRQKKQQ (1544) | |
| 1.1728 | 15 | AFGRRGPEQTQGNFG (1456) | |
| 1.2606 | 14 | VRQIAPGQTGKIAD (89) | |
| 0.7136 | 12 | YGFQPTNGVGYQ (177) | |
| 0.5322 | 12 | ILPDPSKPSKRS (487) | |
| 0.8771 | 12 | RIRGGDGKMKDL (1276) | |
| 1.4039 | 11 | KNHTSPDVDLG (839) | |
| 2.1298 | 10 | KLDDKDPNFK (1521) | |
| 0.8904 | 9 | NNLDSKVGG (121) | |
| 1.3668 | 6 | RVQPTE (1) | |
| 0.7417 | 6 | DSLSST (618) | |
| 0.6838 | 6 | TDYKHW (1479) |
Primary and Secondary Structure Analysis
Physiochemical characterization of selected Model 4 fusion protein was achieved using Expasy’s ProtParam server based on estimated molecular weight, theoretical isoelectric point, and average hydropathicity that indicates the solubility and hydrophobicity of protein. The fusion Model 4 with 1602 amino acids and 176.443 Da with pI: 8.77 and 157 positively charged residues (Arg + Lys) in the polypeptide and 134 negatively charged residues (Asp + Glu). This Model is also predicted to be soluble and hydrophilic (Grand average of hydropathicity (GRAVY): − 0.234).
SOPMA tool was used to predict secondary structure of Model 4 features, including alpha helixes, beta turns, random coils contribution, and C-score. Random coils and extended strands greater ratios are correlated with protein antigenic epitope formation enhancement. Subsequently, it is composed of 31.27% α-helix and 4.99% β-sheet, which beside the 42.88% of random coils, which is potential to form higher antigenic epitopes (Fig. 4a).
Fig.4.

Sequence and structural analysis of Model 4. a Secondary structure by SOPMA tool, b Three dimensional structure by PyMOL and c Ramachandran Plot generated to validate the modeled 3 structure of model 4 protein which indicates that 91.7% of residues are in the favored region
Homology Modeling Prediction and Validation
The three-dimensional structure of Model 4 was predicted using the IntFOLD Integrated Protein Structure and Function Prediction Server, which generates five top models with global model quality score. The one with the highest global model quality score represents the best model. This value of selected Model 4 was in the acceptable score. Chimera version v1.2 was applied to generate the protein image [62] (Fig. 4b). Moreover, the Ramachandran plot generated by the PROCHECK. Described the amino acid positions in the plot as well as the overall quality of the protein model. The plot showed that 91.7% amino acids were arranged in most favored core regions with 7% in allowed region, 1.1% generously allowed region, and 0.3% in disallowed region (Fig. 4c). Z-Score for 3D structure of model was − 5.62.
Discussion
Apart from the human coronaviruses, which are continuously circulating among human population, the originated viruses from animals have been shown to be lethal pathogens via crossing species barriers. Effective preventive approaches are urgent needs at the current situation. Potent epitopic vaccines predicted by bioinformatic analysis makes the vaccine design straightforward and fast compared to traditional vaccine approaches, which has been used in COVID-19 vaccine design recently [35, 63–65].
In this study, we evaluated four possible fusion forms of structural SARS-CoV-2 proteins in order to achieve the most immunogenic protein, which could elicit humoral and cellular immune responses as well. The amino acid sequences were applied to predict the probable antigenic epitopes of T-Cell, linear and conformational B-cell.
Among the four analyzed models, we found model 4 composed of truncated Spike, the full form of M and N proteins (S: 528–1293 + AAY + M + AAY + N) is the most immunogenic fusion form. The evaluation of Murine T-cell epitopes showed that it contains 14 MHCI binders which are all antigenic and also 10 MHCII peptides from which 5 are antigenic epitopes. Human investigation resulted in 24 highly immunogenic human MHC class I and 29 human MHC class II. Moreover, there are four epitopes, including KTFPPTEPK, VTYVPAQEK, KAYNVTQAF and KMKDLSPRW, which can bind to different HLAs.
B-cell evaluation also showed that this model contains 14 B-cell linear and 61 discontinues epitopes with maximum score of 0.994. Therefore, the in silico comparative analysis predicted this model to have a high potency in both immune arms induction. Structural analysis revealed that the selected model is a 176.443 Da protein composed of 1602 amino acids and 42.88% of random coils. In addition, the Ramachandran plot showed that 91.7% amino acids were arranged in most favored core regions. The predicted model is totally non-toxin with a great rate of population coverage especially in Iran and the Europe.
In a study by Joshi et al., SARS-COV-2 multiple virus proteins were assessed by in-silico methods. The obtained results showed that two epitopes ITLCFTLKR and VYQLRARSV are highly practical after docking and molecular dynamics simulation. Furthermore, these two epitopes were subjected to population coverage and toxicity analysis [66]. In our study, KTFPPTEPK from N protein was found highly potential to associate with two frequent HLA-A*03:01 and HLA-A*11:01. It is also a part of B-cell epitopes KTFPPTEPKKDKKKKADETQALPQRQKKQQ with a high score in the predicted method (Table 7).
Table 7.
Discontinuous B-cell epitopes predicted by Ellipro for model 4








Chen et al. investigated another in silico analysis [67]. They predicted 63 sequential B-cell epitopes of spike protein. They also showed that four peptides of Spike, including S 315–324, S 333–338, S 648–663 and S 1064–1079 are highly antigenic with optimum surface accessibility. In our study, one of the discontinuous B-cell predictions includes 38 residues (residues: L900, G901, F902, I903, A904, G905, L906, I907, A908, I909, V910, M911, V912, T913, I914, M915, L916, C917, C918, M919, T920, S921, C922, C923, S924, C925, L926, K927, G928, C929, C930, S931, C932, G933, S934, C935, C936, K937) with high score of 0.886. They also assessed HLA-binding peptides of nucleocapsid protein, which led to 81 and 64 peptides able to bind to MHC class I and MHC class II molecules. The HLA I and HLA II binders in our study were predicted lower due to the fact that we only considered the antigenic peptides at the high threshold (Tables 3 and 4).
The other bioinformatics-based assessment to achieve a vaccine against SARS-CoV-2 by Sahoo et al. focused on T-cell epitopes of similar targets including S, M and N [68]. Their study showed 36 T-cell potential epitopes that interacting with MHC-I alleles and also 25 T-cell epitopes interacting with MHC-II alleles. Among the predicted peptides, IGYYRRATR and YYRRATRRI from N protein and FRLFARTRS, FIASFRLFA and FARTRSMWS from M are predicted to interact by human alleles. These peptides are also supposed to be a BALB/c MHCII binder in our study (Table 2). FVLAAVYRI from M protein is predicted to interact with 31 HLA II and 3 HLA I. In our study, this peptide is also a part of seven HLA II-predicted epitopes (Table 4).
Therefore, immunoinformatics approaches have been already used identification of possible epitopes of novel human coronavirus, SARS-CoV-2. The outbreak of infection caused by this virus has brought great obstacles and challenges to public health. Thus, fast identification of immune epitopes and possible viral immunogenic products would be a superior way to monitor the candidates for vaccine development in comparison with other approaches at the impending pandemic era.
Conclusion
This study resulted in possible fusion forms prediction of SARS-CoV-2 structural proteins, which could be potential targets of neutralizing antibodies. The in silico evaluation of different fusion models have been effective in selecting the best fused model of S, M and N proteins. Truncated Spike + M + N is composed of 24 highly immunogenic human MHC class I and 29 MHC class II with 82.95% population coverage in Iran along with 14 B-cell linear and 61 discontinues epitopes.
The selected recombinant protein could highly elicit immune responses and will be evaluated in vitro and in vivo at the next step.
Acknowledgements
We would like to thank the health system staff worldwide who put all effort on controlling the emerging Coronavirus infection.
Abbreviations
- ACE2
Angiotensin-converting enzyme 2
- COVID-19
Coronavirus Disease 2019
- E
Envelope protein
- M
Membrane protein
- N
Nucleocapsid protein
- RBD
Receptor-binding domain
- S
Spike protein
Funding
The research was support by Pasteur Institute of Iran (Grant No. # 1159).
Compliance with Ethical Standards
Conflict of Interest
The authors declare no potential conflict of interest that could negatively influence the study.
Ethical Approval
The study was approved by Ethic committee of Pasteur Institute of Iran (No#IR.PII.REC.1399.016).
Research Involving Human and Animal Rights
This study was performed at in silico level.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mohammad Reza Aghadadeghi, Email: mrasadeghi@pasteur.ac.ir.
Golnaz Bahramali, Email: gbahramali@gmail.com.
References
- 1.World Health Organization . WHO Coronavirus Disease (COVID-19) Dashboard. Geneva: World Health Organization; 2020. [Google Scholar]
- 2.Huang Y, Yang C, Xu X-F, Xu W, Liu S-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacologica Sinica. 2020;41(9):1141–1149. doi: 10.1038/s41401-020-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Elslande J, Decru B, Jonckheere S, Van Wijngaerden E, Houben E, Vandecandelaere P, et al. Antibody response against SARS-CoV-2 spike protein and nucleoprotein evaluated by 4 automated immunoassays and 3 ELISAs. Clinical Microbiology and Infection. 2020;26(11):1557. doi: 10.1016/j.cmi.2020.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turoňová B, Sikora M, Schürmann C, Hagen WJH, Welsch S, Blanc FEC, von Bülow S, Gecht M, Bagola K, Hörner C, et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science. 2020;370(6513):203–208. doi: 10.1126/science.abd5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Littler DR, Gully BS, Colson RN, Rossjohn J. Crystal structure of the SARS-CoV-2 non-structural protein 9, Nsp9. Science. 2020;23:101258. doi: 10.1016/j.isci.2020.101258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alexpandi R, De Mesquita JF, Pandian SK, Ravi AV. Quinolines-based SARS-CoV-2 3CLpro and RdRp inhibitors and spike-RBD-ACE2 inhibitor for drug-repurposing against COVID-19: An in silico analysis. Frontiers in Microbiology. 2020;11:1796. doi: 10.3389/fmicb.2020.01796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao, W., Dong, C., Kim, S., Hou, D., Tai, W., Du, L., & Im, W. (2020). Biomechanical characterization of SARS-CoV-2 spike RBD and human ACE2 protein-protein interaction. bioRxiv, 2020.2007.2031.230730. [DOI] [PMC free article] [PubMed]
- 8.Trigueiro-Louro J, Correia V, Figueiredo-Nunes I, Gíria M, Rebelo-de-Andrade H. Unlocking COVID therapeutic targets: A structure-based rationale against SARS-CoV-2, SARS-CoV and MERS-CoV Spike. Computational and Structural Biotechnology Journal. 2020;18:2117–2131. doi: 10.1016/j.csbj.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chakraborty C, Sharma A, Bhattacharya M, Sharma G, Lee S-S. The 2019 novel coronavirus disease (COVID-19) pandemic: A zoonotic prospective. Asian Pacific Journal of Tropical Medicine. 2020;13:242–246. doi: 10.4103/1995-7645.281613. [DOI] [Google Scholar]
- 10.Siu YL, Teoh KT, Lo J, Chan CM, Kien F, Escriou N, et al. The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. Journal of Virology. 2008;82:11318–11330. doi: 10.1128/JVI.01052-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu DX, Yuan Q, Liao Y. Coronavirus envelope protein: A small membrane protein with multiple functions. Cellular and Molecular Life Sciences. 2007;64:2043–2048. doi: 10.1007/s00018-007-7103-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mukherjee S, Bhattacharyya D, Bhunia A. Host-membrane interacting interface of the SARS coronavirus envelope protein: Immense functional potential of C-terminal domain. Biophysical Chemistry. 2020;266:106452–106452. doi: 10.1016/j.bpc.2020.106452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Astuti I, Ysrafil, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes and Metabolic Syndrome. 2020;14:407–412. doi: 10.1016/j.dsx.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schoeman D, Fielding BC. Coronavirus envelope protein: current knowledge. Journal of Virology. 2019;16:69. doi: 10.1186/s12985-019-1182-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang S, Yang M, Hong Z, Zhang L, Huang Z, Chen X, et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharmaceutica Sinica B. 2020;10(7):1228–1238. doi: 10.1016/j.apsb.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Surjit M, Lal SK. The SARS-CoV nucleocapsid protein: a protein with multifarious activities. Infection, Genetics and Evolution. 2008;8:397–405. doi: 10.1016/j.meegid.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amrun SN, Lee CY-P, Lee B, Fong S-W, Young BE, Chee RS-L, et al. Linear B-cell epitopes in the spike and nucleocapsid proteins as markers of SARS-CoV-2 exposure and disease severity. EBioMedicine. 2020;58:102911. doi: 10.1016/j.ebiom.2020.102911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuttle KR. Impact of the COVID-19 pandemic on clinical research. Nature Reviews Nephrology. 2020;16(10):562–564. doi: 10.1038/s41581-020-00336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torreele E. The rush to create a covid-19 vaccine may do more harm than good. BMJ. 2020;370:m3209. doi: 10.1136/bmj.m3209. [DOI] [PubMed] [Google Scholar]
- 20.Prompetchara E, Ketloy C, Palaga T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pacific Journal of Allergy and Immunology. 2020;38:1–9. doi: 10.12932/AP-200220-0772. [DOI] [PubMed] [Google Scholar]
- 21.Moreno-Fierros L, García-Silva I, Rosales-Mendoza S. Development of SARS-CoV-2 vaccines: should we focus on mucosal immunity? Expert Opinion on Biological Therapy. 2020;20(8):831–836. doi: 10.1080/14712598.2020.1767062. [DOI] [PubMed] [Google Scholar]
- 22.Barnard D, Hu M, Jones T, Kenney R, Burt D, Lowell G. Intranasal protollin formulated recombinant SARS-CoV S protein elicits respiratory and serum neutralizing antibodies. Antiviral Research. 2007;74:A45–A45. doi: 10.1016/j.antiviral.2007.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, Xiao Z, Ye K, He X, Sun B, Qin Z, Yu J, Yao J, Wu Q, Bao Z, et al. SARS-CoV-2: Characteristics and current advances in research. Journal of Virology. 2020;17:117. doi: 10.1186/s12985-020-01369-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chakraborty C, Sharma AR, Sharma G, Bhattacharya M, Lee SS. SARS-CoV-2 causing pneumonia-associated respiratory disorder (COVID-19): Diagnostic and proposed therapeutic options. European Review for Medical and Pharmacological Sciences. 2020;24:4016–4026. doi: 10.26355/eurrev_202004_20871. [DOI] [PubMed] [Google Scholar]
- 25.Chakraborty C, Sharma AR, Sharma G, Bhattacharya M, Saha RP, Lee S-S. Extensive partnership, collaboration, and teamwork is required to stop the COVID-19 outbreak. Archives of Medical Research. 2020;51:728–730. doi: 10.1016/j.arcmed.2020.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amanat F, Krammer F. SARS-CoV-2 vaccines: Status Report. Immunity. 2020;52:583–589. doi: 10.1016/j.immuni.2020.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Zeng H, Gu J, Li H, Zheng L, Zou Q. Progress and prospects on vaccine development against SARS-CoV-2. Vaccines. 2020;8:153. doi: 10.3390/vaccines8020153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chakraborty C, Sharma AR, Bhattacharya M, Sharma G, Lee S-S, Agoramoorthy G. Consider TLR5 for new therapeutic development against COVID-19. Journal of Medical Virology. 2020;92:2314–2315. doi: 10.1002/jmv.25997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chakraborty C, Sharma AR, Bhattacharya M, Sharma G, Lee S-S, Agoramoorthy G. COVID-19: Consider IL-6 receptor antagonist for the therapy of cytokine storm syndrome in SARS-CoV-2 infected patients. Journal of Medical Virology. 2020;92:2260–2262. doi: 10.1002/jmv.26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saha A, Sharma AR, Bhattacharya M, Sharma G, Lee S-S, Chakraborty C. Tocilizumab: A therapeutic option for the treatment of cytokine storm syndrome in COVID-19. Archives of Medical Research. 2020;51:595–597. doi: 10.1016/j.arcmed.2020.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhattacharya M, Sharma AR, Patra P, Ghosh P, Sharma G, Patra BC, Lee S-S, Chakraborty C. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): Immunoinformatics approach. Journal of Medical Virology. 2020;92:618–631. doi: 10.1002/jmv.25736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen H, Tang L, Yu X, Zhou J, Chang Y, Wu X. Bioinformatics analysis of epitope-based vaccine design against the novel SARS-CoV-2. Infectious Diseases of Poverty. 2020;9(1):1–10. doi: 10.1186/s40249-020-00713-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiyotani K, Toyoshima Y, Nemoto K, Nakamura Y. Bioinformatic prediction of potential T cell epitopes for SARS-Cov-2. Journal of Human Genetics. 2020;65(7):569–575. doi: 10.1038/s10038-020-0771-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banerjee, S., Majumder, K., Gutierrez, G. J., Gupta, D., & Mittal, B. (2020). Immuno-informatics approach for multi-epitope vaccine designing against SARS-CoV-2. bioRxiv, 2020.2007.2023.218529.
- 35.Rakib A, Sami SA, Mimi NJ, Chowdhury MM, Eva TA, Nainu F, et al. Immunoinformatics-guided design of an epitope-based vaccine against severe acute respiratory syndrome coronavirus 2 spike glycoprotein. Computers in Biology and Medicine. 2020;124:103967. doi: 10.1016/j.compbiomed.2020.103967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saha A, Sharma AR, Bhattacharya M, Sharma G, Lee S-S, Chakraborty C. Probable molecular mechanism of remdesivir for the treatment of COVID-19: Need to know more. Archives of Medical Research. 2020;51:585–586. doi: 10.1016/j.arcmed.2020.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Research. 2008;37:D26–D31. doi: 10.1093/nar/gkn723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reche PA, Glutting JP, Zhang H, Reinherz EL. Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics. 2004;56:405–419. doi: 10.1007/s00251-004-0709-7. [DOI] [PubMed] [Google Scholar]
- 39.Stranzl T, Larsen MV, Lundegaard C, Nielsen M. NetCTLpan: Pan-specific MHC class I pathway epitope predictions. Immunogenetics. 2010;62:357–368. doi: 10.1007/s00251-010-0441-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giguere S, Drouin A, Lacoste A, Marchand M, Corbeil J, Laviolette F. MHC-NP: Predicting peptides naturally processed by the MHC. Journal of Immunological Methods. 2013;400–401:30–36. doi: 10.1016/j.jim.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 41.Abroun, S. (2010). Iran Royan Cord Blood Bank. (Royan Cord Blood Banking).
- 42.Shaiegan M, Yari F, Abolghasemi H, Bagheri N, Paridar M, Heidari A. Allele frequencies of HLA-A, B and DRB1 among People of Fars Ethnicity Living in Tehran. IJBC. 2011;3(4):55–59. [Google Scholar]
- 43.Esmaeili A, Rabe SZT, Mahmoudi M, Rastin M. Frequencies of HLA-A, B and DRB1 alleles in a large normal population living in the city of Mashhad, Northeastern Iran. The Iranian Journal of Basic Medical Sciences. 2017;20:940–943. doi: 10.22038/IJBMS.2017.9117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Doytchinova IA, Flower DR. Identifying candidate subunit vaccines using an alignment-independent method based on principal amino acid properties. Vaccine. 2007;25:856–866. doi: 10.1016/j.vaccine.2006.09.032. [DOI] [PubMed] [Google Scholar]
- 45.Doytchinova IA, Flower DR. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007;8(1):1–7. doi: 10.1186/1471-2105-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GP. In silico approach for predicting toxicity of peptides and proteins. PLoS ONE. 2013;8:e73957. doi: 10.1371/journal.pone.0073957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jespersen MC, Peters B, Nielsen M, Marcatili P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Research. 2017;45:W24–w29. doi: 10.1093/nar/gkx346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Letters. 1990;276:172–174. doi: 10.1016/0014-5793(90)80535-Q. [DOI] [PubMed] [Google Scholar]
- 49.Emini EA, Hughes JV, Perlow DS, Boger J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. Journal of Virology. 1985;55:836–839. doi: 10.1128/jvi.55.3.836-839.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karplus PA, Schulz GE. Prediction of chain flexibility in proteins. Naturwissenschaften. 1985;72:212–213. doi: 10.1007/BF01195768. [DOI] [Google Scholar]
- 51.Parker JM, Guo D, Hodges RS. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: Correlation of predicted surface residues with antigenicity and X-ray-derived accessible sites. Biochemistry. 1986;25:5425–5432. doi: 10.1021/bi00367a013. [DOI] [PubMed] [Google Scholar]
- 52.Chou PY, Fasman GD. Prediction of the secondary structure of proteins from their amino acid sequence. Advances in Enzymology and Related Areas of Molecular Biology. 1978;47:45–148. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]
- 53.Ponomarenko J, Bui H-H, Li W, Fusseder N, Bourne PE, Sette A, Peters B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinformatics. 2008;9:514. doi: 10.1186/1471-2105-9-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF. Protein identification and analysis tools in the ExPASy server. Methods MolBiol. 1999;112:531–552. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- 55.Geourjon C, Deleage G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Computer Applications in the Biosciences. 1995;11:681–684. doi: 10.1093/bioinformatics/11.6.681. [DOI] [PubMed] [Google Scholar]
- 56.Cuff JA, Clamp ME, Siddiqui AS, Finlay M, Barton GJ. JPred: A consensus secondary structure prediction server. Bioinformatics. 1998;14:892–893. doi: 10.1093/bioinformatics/14.10.892. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McGuffin LJ, Adiyaman R, Maghrabi AHA, Shuid AN, Brackenridge DA, Nealon JO, et al. IntFOLD: An integrated web resource for high performance protein structure and function prediction. Nucleic Acids Research. 2019;47:W408–w413. doi: 10.1093/nar/gkz322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang Y, Zhou Y. Specific interactions for ab initio folding of protein terminal regions with secondary structures. Proteins. 2008;72:793–803. doi: 10.1002/prot.21968. [DOI] [PubMed] [Google Scholar]
- 60.Laskowski RA, MacArthur M, Moss DS, Thornton JM. PROCHECK—A program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993;26(283–291):282. [Google Scholar]
- 61.Colovos C, Yeates T. Verification of protein structures: Patterns of non-bonded atomic interactions. Protein Science. 1993;9:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.BowieLuthy JUR, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 63.Poran A, Harjanto D, Malloy M, Arieta CM, Rothenberg DA, Lenkala D, et al. Sequence-based prediction of SARS-CoV-2 vaccine targets using a mass spectrometry-based bioinformatics predictor identifies immunogenic T cell epitopes. Genome Medicine. 2020;12:70. doi: 10.1186/s13073-020-00767-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Olvera A, Noguera-Julian M, Kilpelainen A, Romero-Martín L, Prado JG, Brander C. SARS-CoV-2 consensus-sequence and matching overlapping peptides design for COVID19 immune studies and vaccine development. Vaccines. 2020;8:444. doi: 10.3390/vaccines8030444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dong R, Chu Z, Yu F, Zha Y. Contriving multi-epitope subunit of vaccine for COVID-19: Immunoinformatics approaches. Frontiers in Immunology. 2020;11:1784. doi: 10.3389/fimmu.2020.01784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Joshi A, Joshi BC, Mannan MA, Kaushik V. Epitope based vaccine prediction for SARS-COV-2 by deploying immuno-informatics approach. Informatics in Medicine Unlocked. 2020;19:100338. doi: 10.1016/j.imu.2020.100338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen HZ, Tang LL, Yu XL, Zhou J, Chang YF, Wu X. Bioinformatics analysis of epitope-based vaccine design against the novel SARS-CoV-2. Infectious Diseases of Poverty. 2020;9(1):1–10. doi: 10.1186/s40249-020-00713-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Biswajit Sahoo KK, Rai NK, Chaudhary DK. Identification of T-cell epitopes in proteins of novel human coronavirus, SARS-Cov-2 for vaccine development. International Journal of Applied Biology and Pharmaceutical Technology. 2020;11:37–45. [Google Scholar]