

Abstract
Alcoholic liver disease (ALD), due to the multifactorial damage associated with alcohol (ethanol/EtOH) consumption and metabolism, is one of the most prevalent liver diseases in the United States. The liver is the primary site of EtOH metabolism and is subsequently injured due to the production of reactive oxygen species (ROS), acetaldehyde, and metabolic stress. Building evidence suggests that dihydromyricetin (DHM), a bioactive flavonoid isolated from Hovenia dulcis, provides hepatoprotection by enhancing EtOH metabolism in the liver by maintaining hepatocellular bioenergetics, reductions of oxidative stress, and activating lipid oxidation pathways. The present study investigates the utility of DHM on hepatic mitochondrial biogenesis via activation of the AMP-activated protein kinase (AMPK)/Sirtuin (Sirt)-1/PPARG coactivator 1 (PGC)-1α signaling pathway. We utilized a forced drinking ad libitum study that chronically fed 30% EtOH to male C57BL/6J mice over 8 weeks and induced ALD pathology. We found that chronic EtOH feeding resulted in the suppression of AMPK activation and cytoplasmic Sirt1 and mitochondrial Sirt3 expression, effects that were reversed with daily DHM administration (5 mg/kg; i.p.). Chronic EtOH feeding also resulted in hepatic hyperacetylation of PGC-1α, which was improved with DHM administration and its mediated increase of Sirt-1 activity. Furthermore, EtOH-fed mice were found to have increased expression of mitochondrial transcription factor A (TFAM), reduced mitochondrial biogenesis as assessed by mitochondrial DNA to nuclear DNA ratios, and significantly lower levels of hepatic ATP. In contrast, DHM administration significantly increased TFAM expression relative to water and EtOH-fed mice, increased hepatic ATP concentrations, and induced mitochondrial biogenesis. In total, this work demonstrates a novel mechanism of DHM that improves hepatic bioenergetics, metabolic signaling, and mitochondrial biogenesis that supports its utility for treatment of ALD and other metabolic disorders.
Keywords: Ethanol, Dihydromyricetin, Alcoholic Liver Disease (ALD), Mitochondria, Sirtuin, PGC-1α
Introduction
Alcoholic liver disease (ALD) is one of the most prevalent liver diseases in the United States, and it includes a spectrum of diseases ranging from reversible fatty liver to alcoholic hepatitis, and cirrhosis (Mellinger et al., 2018). The majority of patients diagnosed with ALD are also suffering from alcohol use disorder (AUD), where this latter condition affects over 15 million people in the United States. A concerning trend is the increased incidences of younger adults being diagnosed with ALD due to increasing rates of alcohol (ethanol/EtOH) abuse. The mechanisms involved in the development of ALD are multifaceted, and it is becoming evident that the disease spectrum and subsequent progress to late-stage ALD with prolonged EtOH abuse results from the multifactorial injurious responses that occur throughout the body (Rehm et al., 2010, 2013; Seitz et al., 2018). Of the multiple mechanisms of injury, one major contributing factor resulting in the pathology of ALD, and subsequent development of late-stage ALD, is the metabolism of EtOH in the liver that relies on the central role of mitochondria (Han et al., 2012).
EtOH is primarily metabolized in the liver by cytosolic alcohol dehydrogenase (ADH) and the inducible cytochrome P450 2E1 (CYP2E1) in mitochondria and endoplasmic reticulum (Lieber et al., 1970; Seitz et al., 2018). The metabolism of EtOH by ADH1 and CYP2E1 results in the oxidation of EtOH to acetaldehyde (ACH), a highly reactive and toxic metabolite, which is then further metabolized to acetate by aldehyde dehydrogenase (ALDH2) in the liver mitochondria. Throughout this metabolic process, ROS formation occurs, with CYP2E1 oxidation contributing to much of the ROS stress in the liver (Lieber, 1997; Neve and Ingelman-Sundberg, 2000; Leung and Nieto, 2013). The combination of EtOH metabolism, increased reactive oxygen species (ROS), and production of ACH results in multiple responses in the liver that dysregulate energy signaling pathways, lipid metabolism, and lead to the induction of inflammatory responses (Ceni et al., 2014; Seitz et al., 2018). Additionally, ACH and ROS alter mitochondrial structure and activity, thereby leading to functional impairment, including decreased oxidative phosphorylation (ATP generation), ROS exacerbation, and a decrease in ALDH2 activity resulting in elevated ACH (O’Shea et al., 2010). Furthermore, the nicotinamide adenine dinucleotide (NAD)-dependent metabolism of EtOH/ACH, which results in the depletion of hepatic NAD+, contributes to additional metabolic stressors (French, 2016; Wang et al., 2018). In an effort to adapt to the ongoing hepatic injury from high levels of EtOH exposure, mitochondria undergo various responses, such as biogenesis, to maintain mitochondrial integrity and prevent further injury resulting from dysfunctional metabolic responses (Serviddio et al., 2010; Degli Esposti et al., 2012; Pessayre et al., 2012). Collectively this work suggests that pharmacological agents that target these aspects of EtOH-mediated stress on mitochondria could be critical in protecting hepatocytes from energy impairments that result from mitochondrial-related stress in the liver.
One key regulator of mitochondrial biogenesis and cellular energy metabolism is the peroxisome proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1α), which belongs to the family of PGC-1 transcription coactivators (Handschin, 2009; Scarpulla, 2011). PGC-1α is present at low but inducible levels in the liver, where it also regulates most metabolic pathways, such as fatty acid β-oxidation, gluconeogenesis, and ketogenesis (Yoon et al., 2001; Puigserver et al., 2003; Rhee et al., 2003). Under normal physiological stress conditions (e.g., energy deprivation, fasting, and/or low temperatures), PGC-1α is activated via cyclic AMP response element-binding protein (CREB) and post-translationally via adenosine monophosphate-activated protein kinase (AMPK) phosphorylation and NAD+-dependent sirtuin (SIRT)-1 deacetylation (Scarpulla, 2011). However, EtOH metabolism inhibits the AMPK-dependent phosphorylation of PGC-1α necessary for activation, and depletes NAD+ concentrations critical for Sirt-1-driven deacetylation, thereby inhibiting PGC-1α activation via ROS stress and energy depletion (Chaung et al., 2008; You et al., 2015; French, 2016). This inhibition of mitochondrial biogenesis in addition to mitochondrial stress and damage results in the accumulation of dysfunctional mitochondrial and reduced hepatic ability to selectively remove mitochondria by mitophagy (Eid et al., 2013; Williams et al., 2015).
Dihydromyricetin (DHM), an active bioflavonoid isolated from Hovenia dulcis, has been used in Chinese traditional medicines for centuries and has been shown to protect the liver against chemically-induced liver damage via increased Sirt-1 signaling (Ma et al., 2019). Related to this work, we and others have shown that DHM, can have multiple medicinal benefits, including anti-inflammatory, antioxidant, hepatoprotective, and anti-alcohol properties (Qi et al., 2012; Shen et al., 2012; Chen et al., 2015; Martínez-Coria et al., 2019; Silva et al., 2020). As part of this later effort, we found that DHM enhances EtOH metabolism in the liver, in part due to increased NAD+ concentrations, and can reduce hepatic lipid accumulation via AMPK activation in EtOH-fed male C57BL/6J mice (Silva et al., 2020). However, much remains to be elucidated regarding the mechanism by which these metabolic changes in response to EtOH occur. The current study addresses this issue by investigating the hepatoprotective role of DHM in the AMPK/Sirt-1/PGC-1α signaling axis and subsequent mitochondrial biogenesis that is otherwise dysregulated with chronic EtOH feeding.
Methods
Animals and Experimental Design
A forced drinking ad libitum study was conducted as previously described using 8-week old male C57BL/6J mice and the provision of a single bottle EtOH [EtOH-fed], starting at 5% and gradually increasing to 30% EtOH, or tap water [water-fed controls] for a total of 8 weeks (Keegan et al., 1995; Brandon-Warner et al., 2012; Silva et al., 2020). Mice were grouped as follows: 1) Water-fed + saline intraperitoneal (i.p). injections (n=6), 2) EtOH-fed + saline i.p. injections (n=6), and 3) EtOH-fed + DHM i.p. injections (5 mg/kg; n=10). Mice were administered DHM or saline 5 days a week via i.p. injection. All experimental procedures were approved by the USC IACUC committee, and all methods were carried out in accordance with relevant guidelines and regulations. At the end of the experimental period, the mice were euthanized via CO2 and cervical dislocation. Fresh liver tissue was snap-frozen in liquid nitrogen, followed by preservation at −80°C until utilized.
Immunoprecipitation
Protein concentration of hepatic extracts was determined by Bradford Assay. Sufficient amount of PGC-1α (Cell Signaling) was added into 200 μg of protein and gently rotated overnight at 4°C. The immunocomplex was captured by adding 120 μl protein Dynabeads Protein G (ThermoFisher Scientific, San Jose, CA, USA) and gently rotating at 4°C for 3 h and centrifuged at 1500 × g for 5 min. The precipitate was washed three times with ice-cold RIPA buffer, resuspended in 3X sample buffer, eluted with Pierce IgE Elution Buffer (Thermo Fisher Scientific), and subjected Western blot.
Mitochondrial isolation, protein extraction, and Western blot analysis
Mitochondrial isolation, protein extraction, and Western blot analyses were conducted as previously published (Silva et al., 2020). All primary and secondary antibodies were purchased from Cell Signaling (Beverly, MA). All trials were repeated in triplicates to confirm changes in protein expression. Densitometry analysis was performed using the ImageJ Gel Analysis Tool and normalized against untreated controls.
NovaQuant mtDNA/nDNA Ratio Protocol:
Real-time PCR analysis was performed with NovaQUANT Mouse Mitochondrial to Nuclear DNA Ratio Kit (EMD Millipore, Billerica, MA) according to the manufacturer’s instructions. DNA extractions were performed on frozen mouse liver tissue using a Qiagen Dneasy Blood & Tissue Kit (QIAGEN, Chatsworth, VA). A set of four optimized PCR primer pairs targeting two mitochondrial genes (trLEV and 12s RNA) and two nuclear genes (BECN1 and NEB) were pre-aliquoted in an Applies Biosystems MicroAmp Fast Optical 96-well Reaction Plate. A fast real-time qPCR system (Applied Biosystems, Foster City, CA) was used to measure the ratio of mtDNA to nuclear DNA, the relative mtDNA copy number, reflecting the relative mtDNA per cell. The results of the qPCR reactions were analyzed with 2-ΔCT method and normalized to WT control.
Hepatic ATP Measurements
Livers were homogenized in pre-cooled Tris-EDTA extractant (0.1 M Tris-acetate buffer + 2 mM EDTA, pH 7.75) using a Branson Digital Sonifier 150 ultrasonic tissue disruptor-homogenizer (Emerson., St. Louis, MO). The homogenate was centrifuged at 10,000 × g for 10 minutes in a refrigerated centrifuge at 4°C and supernatant was collected. Aliquots of supernatant were re-adjusted to pH 7.8 (according to required pH for assay guidelines) with 320 μL of 2.5 M KOH and precipitate removed by a second centrifugation (10,000 g for 10 min). Aliquots of supernatant were transferred to a fresh tube, on ice, and 240 μL of Tris-HCL/EDTA (pH 7.75) was added. For supernatant ATP levels to be accurately assayed using the Sigma ATP Bioluminescent Kit, final supernatant pH levels were re-adjusted to pH 7.8 (Sigma-Aldrich, St. Louis, MO). ATP levels of isolated and grouped mouse brain extracts (n= 4 for each group) were measured by using 100 μL of supernatant with an ATP luciferin bioluminescence assay kit according to the manufacturer’s guidelines. ATP assay mix was diluted with 5 mL of sterile water and remained on ice and protected from light for one hour to assure complete dissolution. 100 μL of ATP Assay Mix was added to each well and incubated at room temperature for 3 minutes to allow for hydrolysis of endogenous ATP. Immediately after adding 100 μL of tissue homogenates, standards, or water controls with ATP Assay Mix, the sample was measured for luciferase light production. Luminescence was measured using the BioTek Synergy H1 Hybrid Multi-Mode Reader plate reader (BioTek, Winooski, VT). Relative luminescent units from 4 measurements were averaged for calculations.
Data Analysis
Animal biochemical analyses were conducted using 6 separate samples from mice groups, and 3 separate samples for Western blot analysis. The data are presented as mean ± standard error mean (SEM). Statistical analysis included one-way analysis of variance followed by Bonferroni multiple comparison tests using Prism (GraphPad Software, Inc., La Jolla, CA). Differences among groups were stated to be statistically significant when p ≤ 0.05.
Results
DHM Reverses EtOH-Mediated Inhibition of Hepatic AMPK and Increased Sirt-1 and Mitochondrial Sirt-3
We evaluated the expression of cytoplasmic Sirt-1, activated pAMPK (Thr172) relative to total AMPK, and mitochondrial Sirt-3 in the liver of chronic EtOH-fed and water-fed mice. EtOH-feeding significantly reduced Sirt-1 and Sirt-3 expression by 37% and 31%, respectively (Fig. 1). Additionally, chronic EtOH-feeding inhibited the activation of AMPK (phosphorylation at Thr172) by 43% compared to water-fed controls (†p<0.05 vs. water-fed; Fig 1B). DHM administration (5 mg/kg; i.p.) significantly reversed these EtOH-mediated responses in the liver (*p<0.05; Fig 1B).
Fig 1.
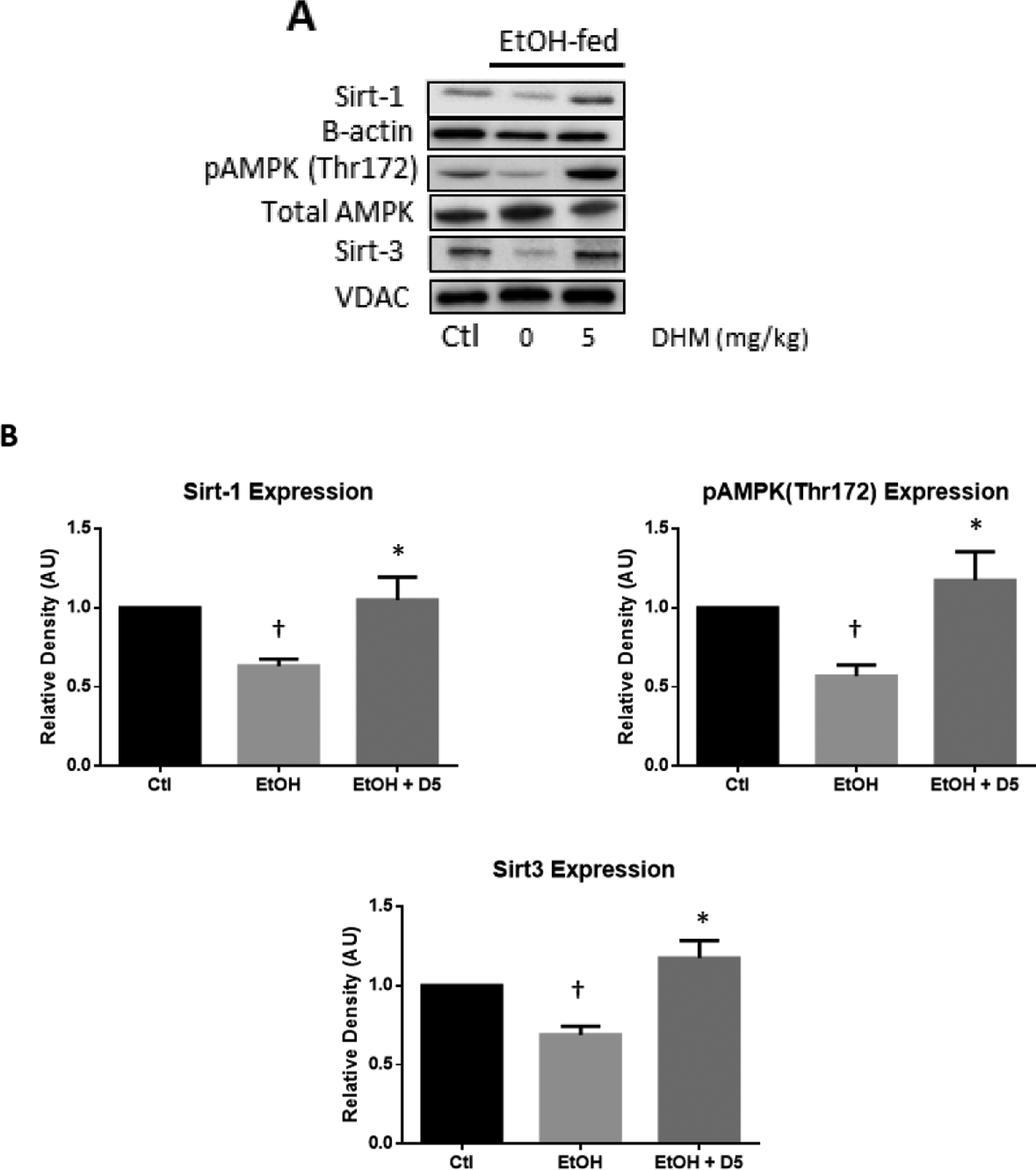
DHM administration significantly increased the expression of hepatic Sirt-1, pAMPK (Thr172), and Sirt-3 compared to EtOH fed mice. A) Representative Western blot images of Sirt-1, β-actin loading control, pAMPK (Thr172), total AMPK, Sirt-3, and VDAC loading control. B) ImageJ quantification of designated triplicate blots. The Western blot images are representative of Western blots obtained from 3 different biological experiments; n=3/group.
DHM Increases Sirt-1-mediated Deacetylation of PGC-1α in EtOH-fed Mouse Livers
Recently, we found that DHM administration can significantly increase NAD+ concentrations in the liver of EtOH-fed mice (Silva et al., 2020). To assess DHM modifications of Sirt-1 activity in response to increased NAD+ concentrations (Silva et al., 2020), we measured the deacetylation extent of hepatic PGC-1α in EtOH-fed mice (Fig 2). Chronic EtOH-feeding significantly increased PGC-1α acetylation (Fig 2; ††p<0.01 vs. water-fed control), suggesting reduced Sirt1-driven deacetylation. DHM administration reversed this outcome and resulted in enhanced deacetylation of PGC-1α (Fig 2; **p<0.01 vs. EtOH-fed control), correlating with the elevated Sirt-1 expression (Fig 1) and increased NAD+ concentrations (Silva et al., 2020).
Fig 2.
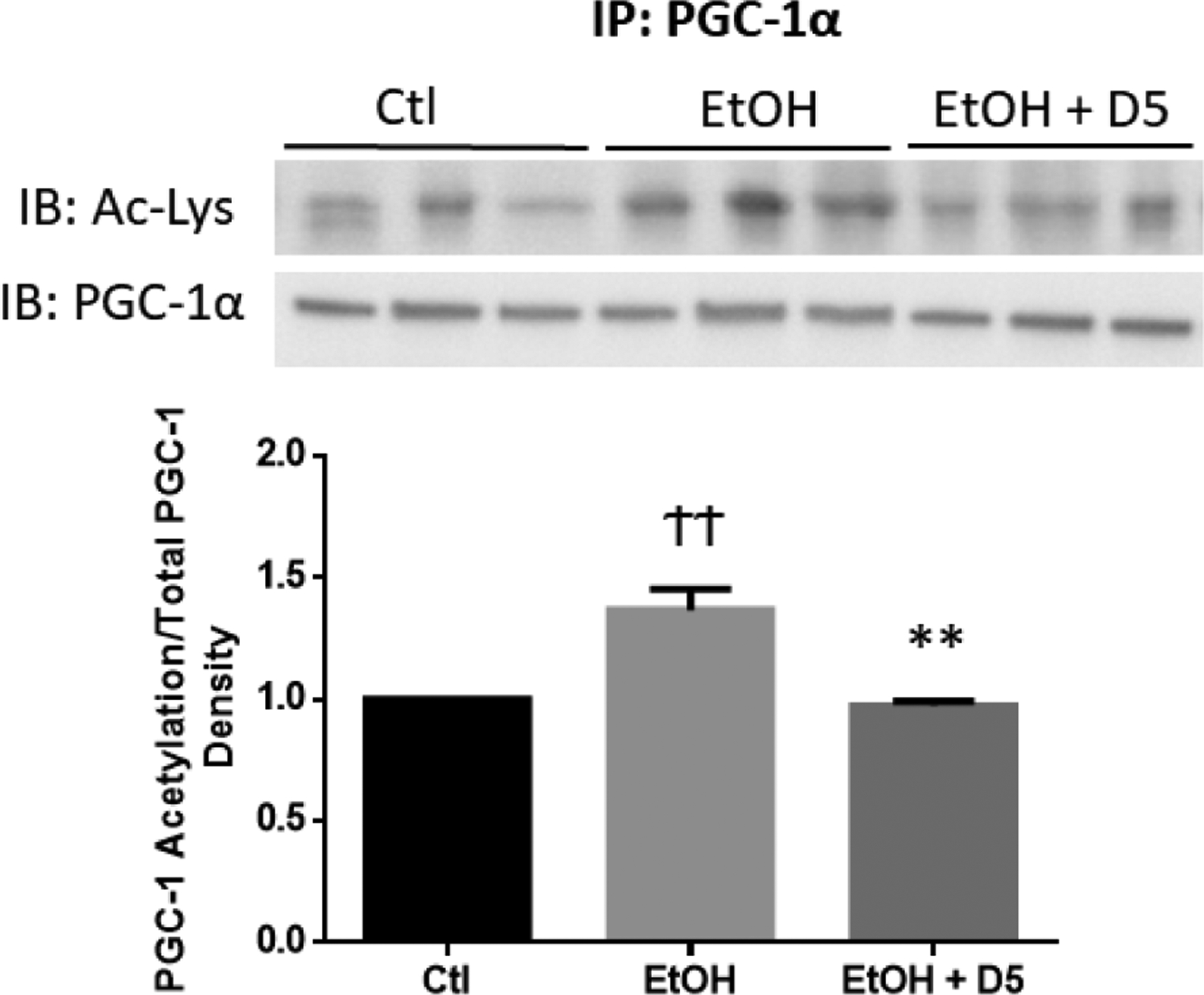
DHM administration significantly increased Sirt-1 deacetylation of PGC-1α in the livers of EtOH-fed mice. Liver extracts were first immunoprecipitated with anti-PGC1 antibody, and the immunoprecipitates were analyzed by Western blot with acetyl-lysine. ††p<0.01 vs. water-fed control (Ctl) and **p<0.01 vs. EtOH-fed control (EtOH).
DHM Increases Mitochondrial Biogenesis and Hepatic ATP Levels in EtOH-Fed Mice
Due to the activation of AMPK (pAMPK Thr172, Fig 1) and increased PGC-1α deacetylation (Fig 2), we next evaluated the downstream response of PGC-1α on mitochondrial biogenesis and ATP responses. Upon activation of PGC-1α and Nrf transcription factors, TFAM is produced and transported to the mitochondria to induce mitochondrial transcription and biogenesis (Picca and Lezza, 2015; Kunkel et al., 2016). Therefore, we expected to observe a reduction of mitochondrial TFAM expression in the livers of EtOH-fed mice due to our previous findings of reduced hepatic Nrf2 (Silva et al., 2020) and reduced PGC-1α activation shown in Fig 2. Interestingly, hepatic TFAM was found to be elevated by 30% in EtOH-fed mice relative to water-fed controls (†p<0.05), suggesting a compensatory mechanism inherent in the ability of TFAM to bind or activate mitochondrial genes. Administration of DHM alongside EtOH-feeding resulted in a much larger (86%) increase of TFAM expression compared to water-fed controls and a 54% increases compared to EtOH-fed controls (**p<0.01; Fig 3A). Although elevated TFAM expression was observed in EtOH-fed liver, hepatic ATP concentrations were significantly reduced to 2.43 nmol/mg (†p<0.05 vs. water-fed; Fig 3B). This effect on ATP concentrations was significantly reversed (3.898 nmol/mg) with daily DHM administration (*p<0.05 vs. EtOH-fed; Fig 3B). Furthermore, chronic EtOH feeding resulted in a 40% reduction of mtDNA/nDNA, suggesting reduced mitochondrial content (†p<0.05 vs. water-fed; Fig 3C). Notably, DHM administration significantly reversed this effect, resulting in a 20% increase compared to EtOH-fed controls (*p<0.05; Fig 3C).
Fig 3.
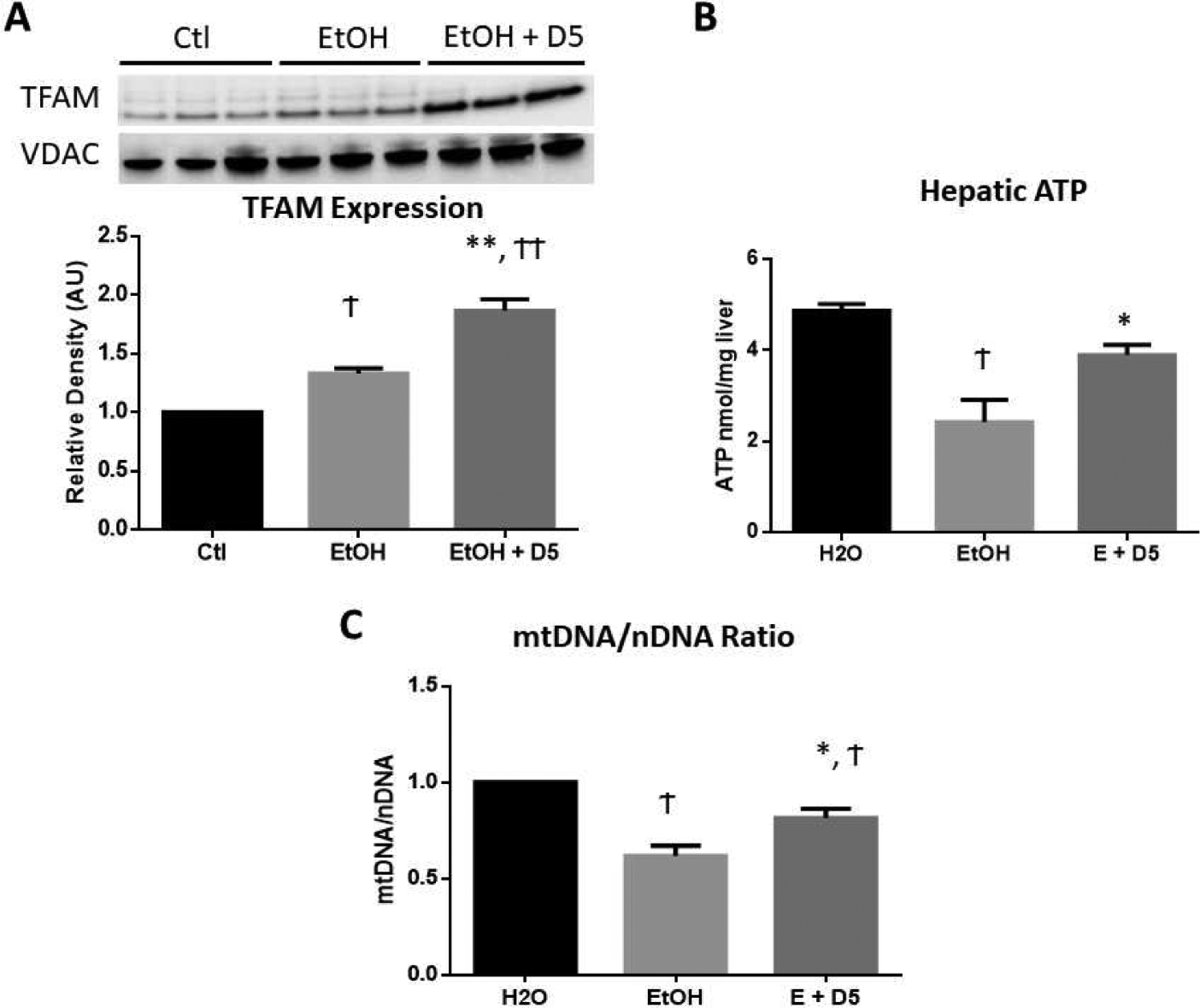
DHM significantly increased hepatic mitochondrial biogenesis and preserved ATP concentrations. A) Western blot image and ImageJ quantification of hepatic TFAM protein expression normalized to VDAC loading control (n=3/group). B) Hepatic ATP concentrations (nmol/mg; n=6/group). C) mtDNA copy number in the liver, measured as the ratio of mtDNA to nDNA (mtDNA/nDNA), and normalized to water-fed mice (n=6/group). †p<0.05 vs. water-fed control (Ctl), ††p<0.01 vs. Ctl, *p<0.05 vs. EtOH-fed control (EtOH), and **p<0.01 vs. EtOH.
Discussion
The outcomes from the present work represent the first demonstration that DHM pharmacologically induces mitochondrial biogenesis under EtOH conditions that suppress biogenesis and mitochondrial viability. These data support the hypothesis that DHM pharmacological effects on NAD bioenergetics result in the activation of the AMPK/Sirt-1/PGC-1α axis and subsequent modification of mitochondrial biogenesis and ATP activity. Beyond the hepatoprotective effects identified in our forced drinking ad libitum model (Silva et al., 2020), we report herein that DHM also protects the liver against EtOH-mediated injury via modification of hepatocellular bioenergetics and activation of signaling pathways resulting in mitochondrial biogenesis. These mechanisms, in combination with those found to be engaged in anti-inflammatory responses, lipid oxidation, and mitochondrial lipid transport (Silva et al., 2020), further illustrate the multifactorial benefits of DHM pharmacological responses in the liver (Fig 4).
Fig 4.
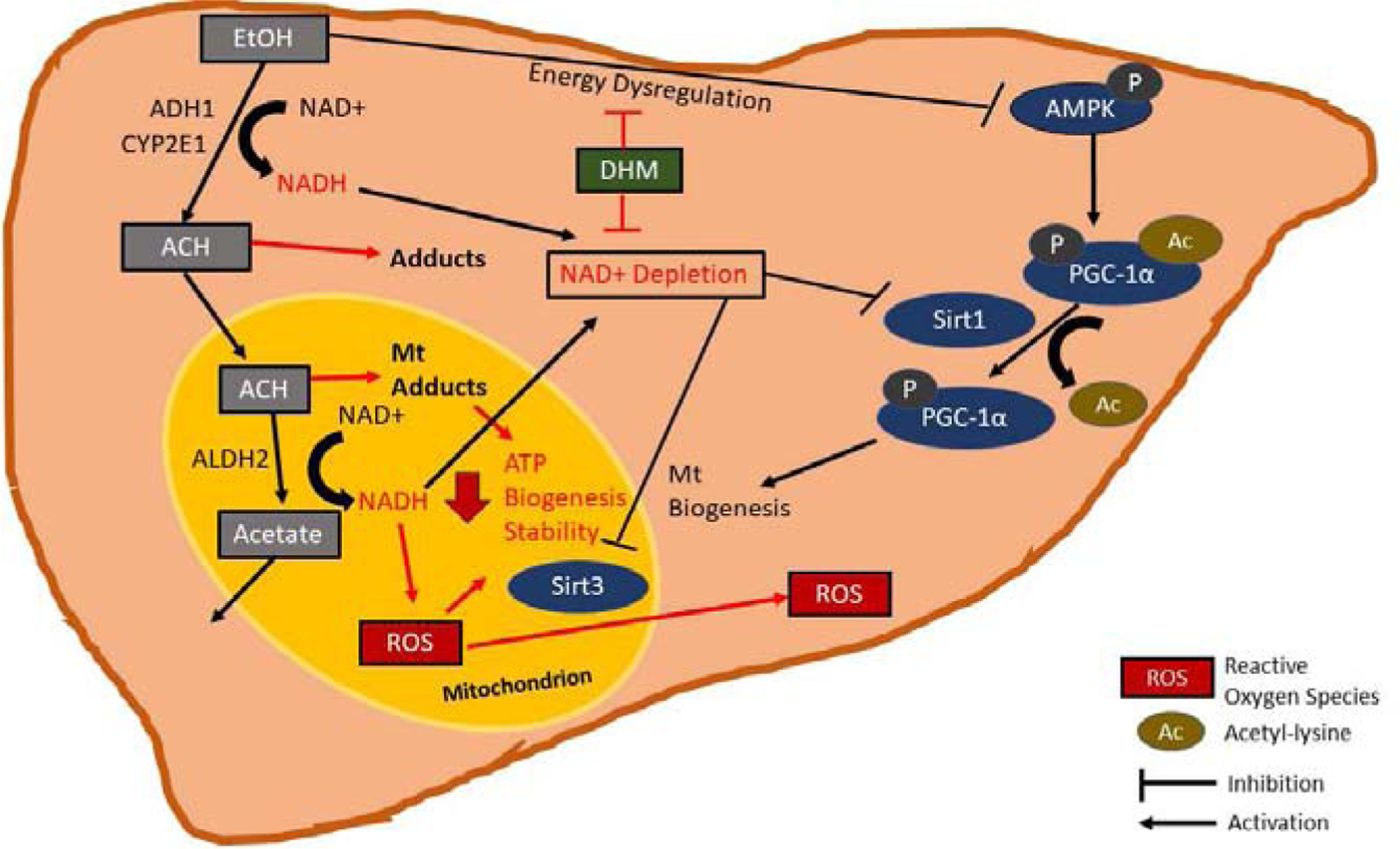
EtOH-mediated suppression of AMPK/Sirt1/PGC-1α via NAD depletion is reversed with DHM administration. EtOH is primarily metabolized in the liver by alcohol dehydrogenase (ADH) and cytochrome P450 2E1 (CYP2E1), resulting in the production of acetaldehyde (ACH). Toxic ACH is metabolized in the mitochondria by ALDH2, thereby producing a non-toxic metabolite, acetate, that is then utilized in the liver or transported to the blood. Due to the process of EtOH and ACH metabolism, nicotinic adenine dinucleotide (NAD)+ is reduced to NADH, contributing to the overall hepatic NAD depletion. Furthermore, reactive oxygen species (ROS) formed in the mitochondria are further exacerbated by the increased NADH levels. The presence of ACH and ROS in the mitochondria then contributes to mitochondrial injury and instability due to adduct formation and oxidative stress, thereby reducing ATP output. The increase in NADH relative to NAD+ leads to NAD depletion that inhibits the activity of NAD-dependent mitochondrial Sirtuin (Sirt)3 and cytoplasmic Sirt1. Furthermore, the effects of EtOH metabolism inhibit the activity of AMP Kinase (AMPK) and subsequent priming for peroxisome proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1α) activation by Sirt1 deacetylation, thereby resulting in reduced PGC-1α activity on mitochondrial biogenesis. We hypothesize that dihydromyricetin (DHM) reverses the EtOH-mediated inhibition of AMPK and NAD depletion leading to maintained Sirt1 and Sirt3 activity and PGC-1α activation that results in mitochondrial biogenesis and preserved ATP output.
To investigate potential pharmacological mechanisms of DHM that benefit critical metabolic signaling pathways in the liver, we assessed the expression of AMPK, Sirt1, and Sirt3 in the liver following chronic EtOH feeding or EtOH feeding with daily DHM administration. As expected with chronic EtOH metabolism and injury, a significant reduction of AMPK activation and expression of both cytoplasmic Sirt1 and mitochondrial Sirt3 was observed (Fig 1). The reduction of these key enzymes is likely contributed to by NAD depletion resulting from NAD-dependent EtOH metabolism, production of toxic ACH, and additional stressors in the liver (Fig 4). Consequently, the decrease of these Sirt enzymes in the liver exacerbates EtOH-mediated damage by inhibiting pathways that favor oxidation and energy expenditure, thereby contributing to mechanisms that promote hepatic lipogenesis and inflammation (Fritz et al., 2012; Yin et al., 2012, 2014; Choi et al., 2013). Notably, these effects are not limited to lipid metabolism, as the resulting NAD+-depletion following EtOH metabolism can also contribute to several signaling dysregulations involved in critical metabolic processes, such as glucose intolerance (Fritz et al., 2012; Choi et al., 2013; Yin et al., 2014). Furthermore, hyperacetylation of mitochondrial ALDH2, and other antioxidant enzymes, due to reduced Sirt3 activity likely reduces its detoxification of ACH and ROS and contributes to mitochondrial instability (Fritz et al., 2012; Ding et al., 2017). Therefore, the multifactorial responses in the liver result in injury due to shifts in bioenergetics responses and reduced enzymatic metabolism/clearance of EtOH, its toxic metabolite, ACH, and associated increases in ROS. However, the DHM-mediated modification of NAD+ in the liver of EtOH-fed mice (Silva et al., 2020), likely increased by the activity of AMPK, partly explains the preserved expression of Sirt1/3 (Cantó et al., 2009) and supports our earlier findings of DHM treatment inhibiting lipid synthesis, steatosis, and oxidative stress in the liver. Further evaluation is necessary to better understand the DHM-mediated effects on the transcriptional activity of Sirt1 and Sirt3 in the liver and other tissues affected by shifts in NAD/NADH and reduced Sirt expression.
As illustrated in Fig 4, PGC-1α is a downstream target of both AMPK and Sirt1 that requires phosphorylation by AMPK and the subsequent deacetylation by Sirt1 for activation of ROS suppressing pathways, lipid oxidation, and mitochondrial biogenesis (Chaung et al., 2008; Scarpulla, 2011). Consistent with previous findings, we identified that ad libitum chronic EtOH feeding resulted in the hyperacetylation of PGC-1α that is due to the inhibition of Sirt deacetylase activity and expression (You et al., 2008; Shepard and Tuma, 2009; Choi et al., 2013). DHM administration significantly reversed the hyperacetylation of PGC-1α (Fig 2), supporting earlier findings of increased lipid oxidation signaling and suppression of oxidative stress (Silva et al., 2020). In total, this activity helps explain that DHM administration increases the activity of cytoplasmic Sirt1 on PGC-1α. This increase of PGC-1α deacetylation, in addition to AMPK activation, is likely to result in the activation of PGC-1α, thereby contributing to multiple benefits, including oxidative suppression, mitochondrial biogenesis, and hepatic glucose homeostasis (Yoon et al., 2001; Wu et al., 2016). Moreover, these findings align with previous investigations that report DHM administration improving nonalcoholic fatty liver disease (NAFLD) outcomes (Chen et al., 2015), which is linked to preserved Sirt-3 deacetylation in the mitochondria (Zeng et al., 2019). Results from the present study are in agreement with this evidence in that we found that DHM administration increased the expression of mitochondrial Sirt3 after EtOH feeding. We further expanded these results by illustrating the beneficial effects of DHM to increase cytoplasmic deacetylation of PGC-1α linked to Sirt1 activity while maintaining NAD+ levels (Silva et al., 2020).
Notably, as illustrated in Fig 3, we found that EtOH-fed mice using a forced drinking ad libitum model resulted in the inhibition of mitochondrial biogenesis and reduced mitochondrial ATP output. The copy number of mtDNA is reflective of mtDNA transcription and ATP production (Kunkel et al., 2016), and was assessed to validate mitochondrial biogenesis and the restorative conditions of the liver following chronic EtOH injury (Picca and Lezza, 2015). TFAM is a promoter-specific enhancer of mtDNA and a major mitochondrial gene regulator that is produced in the nucleus and transported to the mitochondria (Picca and Lezza, 2015). Interestingly, TFAM was found to be elevated in the mitochondria of EtOH-fed mice, similar to what has been reported with intragastric EtOH feeding (Han et al., 2012), but the overall mtDNA content and ATP concentration were significantly reduced. The elevation of TFAM expression and localization in the mitochondria can likely be attributed to hepatic compensation and adaptations to chronic EtOH feeding. However, the significant reduction of mtDNA number with increased TFAM localization requires further elucidation of the potential dysregulation of TFAM binding to mtDNA or changes in post-translational modifications that may contribute to decreased mtDNA transcription. Similar to other investigations (Lamlé et al., 2008; Shin et al., 2013), we previously identified a decrease in hepatic nuclear Nrf2 expression using a forced drinking ad libitum model (Silva et al., 2020), which is involved in mitochondrial biogenesis through co-activation with PGC-1α (Athale et al., 2012; MacGarvey et al., 2012). In the present study, we measured an increase in TFAM expression alongside our previous findings of reduced Nrf2 (Silva et al., 2020). This outcome needs further investigation to determine the activity of other potential factors and their involvement in mitochondrial biogenesis and composition, such as Nrf1 or hypoxia-inducible factor 1 (HIF-1) signaling, following EtOH feeding. None the less, we found that DHM administration over the 8-week drinking period improved hepatic mitochondrial biogenesis as assessed by the increases in TFAM expression and mtDNA number, and is potentially linked to the anti-inflammatory benefits and increased expression/activity of Nrf2 in the liver (Silva et al., 2020). Furthermore, DHM preserved hepatic ATP concentrations with chronic EtOH-feeding, thereby suggesting improved mitochondrial viability. Overall, these findings illustrate the potential for DHM to improve mitochondrial outcomes in response to injury, and provide a novel pharmacological mechanism of DHM that supports its utility for preventing/reducing ALD and other mitochondrial-related disorders.
As discussed above, the present work represents the first demonstration that DHM reverses EtOH-mediated suppression of mitochondrial biogenesis and adds to the evidence that DHM can be used to improve hepatic bioenergetics and mitochondrial outcomes that are otherwise dysregulated with chronic EtOH feeding. Our findings suggest a therapeutic value for DHM as a Sirt activating compound that can modulate many aspects of oxidative stress and mitochondrial function via preservation of hepatic bioenergetics. Importantly, building evidence demonstrates that these factors contribute to many of the EtOH induced pathologies and that DHM supplementation can be used to benefit the management of various metabolic disorders beyond ALD.
Dihydromyricetin (DHM), a bioactive flavonoid, improved mitochondrial outcomes in the liver of male C57BL/6J mice after chronic alcohol feeding
Alcohol inhibition of key metabolic enzymes, AMPK and Sirtuins, and mitochondrial injury are reversed with DHM administration
These findings support the utility of DHM, a dietary supplement, as a novel therapeutic for the reduction/prevention of alcohol-related mitochondrial injury in the liver
This dietary flavonoid provides key metabolic responses that support its use for other mitochondria-related disorders
Acknowledgments
This work was supported by funding and grants from the NIH NIAA R01AA022448, USC GoodNeighbors Campaign, USC School of Pharmacy, and American Foundation for Pharmaceutical Education (AFPE).
Abbreviations:
- EtOH
ethanol
- DHM
dihydromyricetin
- ALD
alcoholic liver disease
- Sirt
sirtuin
- NAD
nicotinamide adenine dinucleotide
- AMPK
AMP-activated protein kinase
- PGC-1α
PPARG coactivator-1α
- ROS
reactive oxygen species
- Nrf
nuclear respiratory factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Athale J, Ulrich A, Chou MacGarvey N, Bartz RR, Welty-Wolf KE, Suliman HB, et al. (2012). Nrf2 promotes alveolar mitochondrial biogenesis and resolution of lung injury in Staphylococcus aureus pneumonia in mice. Free Radic. Biol. Med 53, 1584–1594. doi: 10.1016/j.freeradbiomed.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon-Warner E, Schrum LW, Schmidt CM, and McKillop IH (2012). Rodent models of alcoholic liver disease: Of mice and men. Alcohol 46, 715–725. doi: 10.1016/j.alcohol.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. (2009). AMPK regulates energy expenditure by modulating NAD + metabolism and SIRT1 activity. Nature 458, 1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceni E, Mello T, and Galli A (2014). Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol 20, 17756–17772. doi: 10.3748/wjg.v20.i47.17756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaung WW, Jacob A, Ji Y, and Wang P (2008). Suppression of PGC-1alpha by Ethanol: Implications of Its Role in Alcohol Induced Liver Injury. Int. J. Clin. Exp. Med 1, 161–70. Available at: http://www.ncbi.nlm.nih.gov/pubmed/19079670%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC2596315. [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhao X, Wan J, Ran L, Qin Y, Wang X, et al. (2015). Dihydromyricetin improves glucose and lipid metabolism and exerts anti-inflammatory effects in nonalcoholic fatty liver disease: A randomized controlled trial. Pharmacol. Res 99, 74–81. doi: 10.1016/j.phrs.2015.05.009. [DOI] [PubMed] [Google Scholar]
- Choi SE, Fu T, Seok S, Kim DH, Yu E, Lee KW, et al. (2013). Elevated microRNA-34a in obesity reduces NAD+ levels and SIRT1 activity by directly targeting NAMPT. Aging Cell 12, 1062–1072. doi: 10.1111/acel.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degli Esposti D, Hamelin J, Bosselut N, Saffroy R, Sebagh M, Pommier A, et al. (2012). Mitochondrial roles and cytoprotection in chronic liver injury. Biochem. Res. Int 2012. doi: 10.1155/2012/387626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding RB, Bao JL, and Deng CX (2017). Emerging roles of SIRT1 in fatty liver diseases. Int. J. Biol. Sci 13, 852–867. doi: 10.7150/ijbs.19370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid N, Ito Y, Maemura K, and Otsuki Y (2013). Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: An immunohistochemical and electron microscopic study. J. Mol. Histol 44, 311–326. doi: 10.1007/s10735-013-9483-x. [DOI] [PubMed] [Google Scholar]
- French SW (2016). Chronic alcohol binging injures the liver and other organs by reducing NAD+ levels required for sirtuin’s deacetylase activity. Exp. Mol. Pathol 100, 303–306. doi: 10.1016/j.yexmp.2016.02.004. [DOI] [PubMed] [Google Scholar]
- Fritz KS, Galligan JJ, Hirschey MD, Verdin E, and Petersen DR (2012). Mitochondrial acetylome analysis in a mouse model of alcohol-induced liver injury utilizing SIRT3 knockout mice. J. Proteome Res 11, 1633–1643. doi: 10.1021/pr2008384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Ybanez MD, Johnson HS, McDonald JN, Mesropyan L, Sancheti H, et al. (2012). Dynamic adaptation of liver mitochondria to chronic alcohol feeding in mice: Biogenesis, remodeling, and functional alterations. J. Biol. Chem 287, 42165–42179. doi: 10.1074/jbc.M112.377374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handschin C (2009). The biology of PGC-1α and its therapeutic potential. Trends Pharmacol. Sci 30, 322–329. doi: 10.1016/j.tips.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Keegan A, Martini R, and Batey R (1995). Ethanol-related liver injury in the rat: a model of steatosis, inflammation and pericentral fibrosis. J. Hepatol 23, 591–600. doi: 10.1016/0168-8278(95)80067-0. [DOI] [PubMed] [Google Scholar]
- Kunkel GH, Chaturvedi P, and Tyagi SC (2016). Mitochondrial pathways to cardiac recovery: TFAM. Heart Fail. Rev 21, 499–517. doi: 10.1007/s10741-016-9561-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamlé J, Marhenke S, Borlak J, von Wasielewski R, Eriksson CJP, Geffers R, et al. (2008). Nuclear Factor-Eythroid 2-Related Factor 2 Prevents Alcohol-Induced Fulminant Liver Injury. Gastroenterology 134, 1159–1168. doi: 10.1053/j.gastro.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Leung TM, and Nieto N (2013). CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J. Hepatol 58, 395–398. doi: 10.1016/j.jhep.2012.08.018. [DOI] [PubMed] [Google Scholar]
- Lieber CS (1997). Cytochrome P-4502E1: Its physiological and pathological role. Physiol. Rev 77, 517–544. doi: 10.1152/physrev.1997.77.2.517. [DOI] [PubMed] [Google Scholar]
- Lieber CS, Rubin E, and DeCarli LM (1970). Hepatic microsomal ethanol oxidizing system (MEOS): Differentiation from alcohol dehydrogenase and NADPH oxidase. Biochem. Biophys. Res. Commun 40, 858–865. doi: 10.1016/0006-291X(70)90982-4. [DOI] [PubMed] [Google Scholar]
- Ma JQ, Sun YZ, Ming QL, Tian ZK, Yang HX, and Liu CM (2019). Ampelopsin attenuates carbon tetrachloride-induced mouse liver fibrosis and hepatic stellate cell activation associated with the SIRT1/TGF-β1/Smad3 and autophagy pathway. Int. Immunopharmacol 77, 105984. doi: 10.1016/j.intimp.2019.105984. [DOI] [PubMed] [Google Scholar]
- MacGarvey NC, Suliman HB, Bartz RR, Fu P, Withers CM, Welty-Wolf KE, et al. (2012). Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am. J. Respir. Crit. Care Med 185, 851–861. doi: 10.1164/rccm.201106-1152OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Coria H, Mendoza-Rojas MX, Arrieta-Cruz I, and López-Valdés HE (2019). Preclinical research of dihydromyricetin for brain aging and neurodegenerative diseases. Front. Pharmacol 10, 1–6. doi: 10.3389/fphar.2019.01334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellinger JL, Shedden K, Winder GS, Tapper E, Adams M, Fontana RJ, et al. (2018). The high burden of alcoholic cirrhosis in privately insured persons in the United States. Hepatology 68, 872–882. doi: 10.1002/hep.29887. [DOI] [PubMed] [Google Scholar]
- Neve EPA, and Ingelman-Sundberg M (2000). Molecular basis for the transport of cytochrome P450 2E1 to the plasma membrane. J. Biol. Chem 275, 17130–17135. doi: 10.1074/jbc.M000957200. [DOI] [PubMed] [Google Scholar]
- O’Shea RS, Dasarathy S, McCullough AJ, Shuhart MC, Davis GL, Franco J, et al. (2010). Alcoholic liver disease. Hepatology 51, 307–328. doi: 10.1002/hep.23258. [DOI] [PubMed] [Google Scholar]
- Pessayre D, Fromenty B, Berson A, Robin MA, Lettéron P, Moreau R, et al. (2012). Central role of mitochondria in drug-induced liver injury. Drug Metab. Rev 44, 34–87. doi: 10.3109/03602532.2011.604086. [DOI] [PubMed] [Google Scholar]
- Picca A, and Lezza AMS (2015). Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions. Useful insights from aging and calorie restriction studies. Mitochondrion 25, 67–75. doi: 10.1016/j.mito.2015.10.001. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, et al. (2003). Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature 423, 550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- Qi S, Xin Y, Guo Y, Diao Y, Kou X, Luo L, et al. (2012). Ampelopsin reduces endotoxic inflammation via repressing ROS-mediated activation of PI3K/Akt/NF-κB signaling pathways. Int. Immunopharmacol 12, 278–287. doi: 10.1016/j.intimp.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Rehm J, Samokhvalov AV, and Shield KD (2013). Global burden of alcoholic liver diseases. J. Hepatol 59, 160–168. doi: 10.1016/j.jhep.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Rehm J, Taylor B, Mohapatra S, Irving H, Baliunas D, Patra J, et al. (2010). Alcohol as a risk factor for liver cirrhosis: A systematic review and meta-analysis. Drug Alcohol Rev. 29, 437–445. doi: 10.1111/j.1465-3362.2009.00153.x. [DOI] [PubMed] [Google Scholar]
- Rhee J, Inoue Y, Yoon JC, Puigserver P, Fan M, Gonzalez FJ, et al. (2003). Regulation of hepatic fasting response by PPARγ coactivator-1α (PGC-1): Requirement for hepatocyte nuclear factor 4α in gluconeogenesis. Proc. Natl. Acad. Sci. U. S. A 100, 4012–4017. doi: 10.1073/pnas.0730870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC (2011). Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta - Mol. Cell Res 1813, 1269–1278. doi: 10.1016/j.bbamcr.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz HK, Bataller R, Cortez-Pinto H, Gao B, Gual A, Lackner C, et al. (2018). Alcoholic liver disease. Nat. Rev. Dis. Prim 4. doi: 10.1038/s41572-018-0014-7. [DOI] [PubMed] [Google Scholar]
- Serviddio G, Bellanti F, Sastre J, Vendemiale G, and Altomare E (2010). Targeting Mitochondria: A New Promising Approach for the Treatment of Liver Diseases. Curr. Med. Chem 17, 2325–2337. doi: 10.2174/092986710791698530. [DOI] [PubMed] [Google Scholar]
- Shen Y, Lindemeyer K, Gonzalez C, Shao XM, Spigelman I, Olsen RW, et al. (2012). Dihydromyricetin as a novel anti-alcohol intoxication medication. J. Neurosci 32, 390–401. doi: 10.1523/JNEUROSCI.4639-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard BD, and Tuma PL (2009). Alcohol-induced protein hyperacetylation: Mechanisms and consequences. World J. Gastroenterol 15, 1219–1230. doi: 10.3748/wjg.15.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SM, Yang JH, and Ki SH (2013). Role of the Nrf2-are pathway in liver diseases. Oxid. Med. Cell. Longev 2013. doi: 10.1155/2013/763257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J, Yu X, Moradian R, Folk C, Spatz MH, Kim P, et al. (2020). Dihydromyricetin Protects the Liver via Changes in Lipid Metabolism and Enhanced Ethanol Metabolism. Alcohol. Clin. Exp. Res 44, 1046–1060. doi: 10.1111/acer.14326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wan T, Ye M, Qiu Y, Pei L, Jiang R, et al. (2018). Nicotinamide riboside attenuates alcohol induced liver injuries via activation of SirT1/PGC-1α/mitochondrial biosynthesis pathway. Redox Biol. 17, 89–98. doi: 10.1016/j.redox.2018.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Ni HM, Ding Y, and Ding WX (2015). Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am. J. Physiol. - Gastrointest. Liver Physiol 309, G324–G340. doi: 10.1152/ajpgi.00108.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Deng X, Shi Y, Su Y, Wei J, and Duan H (2016). PGC-1α, glucose metabolism and type 2 diabetes mellitus. J. Endocrinol 229, R99–R115. doi: 10.1530/JOE-16-0021. [DOI] [PubMed] [Google Scholar]
- Yin H, Hu M, Liang X, Ajmo JM, Li X, Bataller R, et al. (2014). Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 146, 801–811. doi: 10.1053/j.gastro.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Hu M, Zhang R, Shen Z, Flatow L, and You M (2012). MicroRNA-217 promotes ethanol-induced fat accumulation in hepatocytes by down-regulating SIRT1. J. Biol. Chem 287, 9817–9826. doi: 10.1074/jbc.M111.333534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, et al. (2001). Control of hepatic gluconeogenesis through the transcriptional coaotivator PGC-1. Nature 413, 131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- You M, Jogasuria A, Taylor C, and Wu J (2015). Sirtuin 1 signaling and alcoholic fatty liver disease. Hepatobiliary Surg. Nutr 4, 88–100. doi: 10.3978/j.issn.2304-3881.2014.12.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M, Liang X, Ajmo JM, and Ness GC (2008). Involvement of mammalian sirtuin 1 in the action of ethanol in the liver. Am. J. Physiol. - Gastrointest. Liver Physiol 294, 892–898. doi: 10.1152/ajpgi.00575.2007. [DOI] [PubMed] [Google Scholar]
- Zeng X, Yang J, Hu O, Huang J, Ran L, Chen M, et al. (2019). Dihydromyricetin Ameliorates Nonalcoholic Fatty Liver Disease by Improving Mitochondrial Respiratory Capacity and Redox Homeostasis Through Modulation of SIRT3 Signaling. Antioxidants Redox Signal. 30, 163–183. doi: 10.1089/ars.2017.7172. [DOI] [PubMed] [Google Scholar]