

Abstract
While vital to platelet and leukocyte adhesion, the role of integrin affinity modulation in adherent cells remains controversial. In endothelial cells, atheroprone hemodynamics and oxidized lipoproteins drive an increase in the high affinity conformation of α5β1 integrins in endothelial cells in vitro, and α5β1 integrin inhibitors reduce proinflammatory endothelial activation to these stimuli in vitro and in vivo. However, the importance of α5β1 integrin affinity modulation to endothelial phenotype remains unknown. We now show that endothelial cells (talin1 L325R) unable to induce high affinity integrins initially adhere and spread but show significant defects in nascent adhesion formation. In contrast, overall focal adhesion number, area, and composition in stably adherent cells are similar between talin1 wildtype and talin1 L325R endothelial cells. However, talin1 L325R endothelial cells fail to induce high affinity α5β1 integrins, fibronectin deposition, and proinflammatory responses to atheroprone hemodynamics and oxidized lipoproteins. Inducing the high affinity conformation of α5β1 integrins in talin1 L325R endothelial cells suggest that NF-κB activation and maximal fibronectin deposition require both integrin activation and other integrin-independent signaling. In endothelial-specific talin1 L325R mice, atheroprone hemodynamics fail to promote inflammation and macrophage recruitment, demonstrating a vital role for integrin activation in regulating endothelial phenotype.
Keywords: Talin1, Inflammation, Oxidized LDL, Shear Stress, Integrin
Graphical Abstract
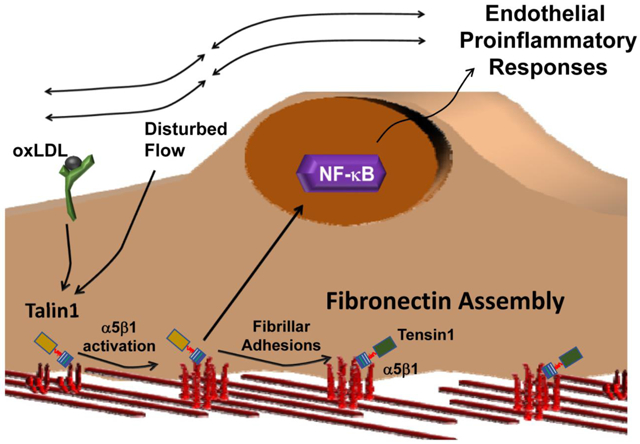
Introduction
In the arterial microenvironment at atherosclerosis prone regions, local hemodynamics and matrix composition critically regulate endothelial activation and early atherosclerosis [1–3]. Disturbed flow primes endothelial cells to activation by systemic factors, in part through nuclear factor-κB (NF-κB) activation [4, 5]. However, the presence of a fibronectin-rich matrix significantly enhances disturbed flow-induced endothelial activation, whereas basement membrane proteins (collagen IV, laminin) support minimal endothelial activation [6–9]. Similarly, matrix content plays a vital role in oxidized low density lipoprotein (oxLDL)-mediated proinflammatory responses, where a fibronectin-rich matrix enhances endothelial activation in response to oxLDL [10, 11]. Fibronectin supports atherogenic endothelial activation through specific integrin signaling responses. Integrins, the largest family of receptors for extracellular matrix proteins, are heterodimers of α and β subunits that mediate both cell-dependent extracellular matrix remodeling and matrix-dependent changes in cell phenotype [12]. Blocking fibronectin-binding integrins (α5β1, αvβ3) suppresses endothelial activation by oxLDL and by disturbed flow both in vitro and in vivo [10, 11, 13, 14], suggesting that cell-matrix interactions serve as an essential component of early atherogenic endothelial activation.
Integrins exist in multiple activation states, including a bent, closed state (low affinity), an intermediate extended state with a closed headpiece (intermediate affinity), and a fully extended, open conformation (high affinity) [15, 16]. The intermediate affinity of extended, closed integrins mediate weak interactions, such as during integrin-mediated leukocyte slow rolling [17], whereas the extended, open conformation is required for high affinity interactions, such as leukocyte firm adhesion. The common final step in integrin affinity modulation involves interactions between the integrin β subunit’s cytoplasmic tail and talin1 [18], a cytoskeletal adaptor protein with a head domain that binds the integrin β tail and a rod domain that binds the actin cytoskeleton [19]. The talin1 head domain binds to an NPxY motif on the β integrin subunit driving a transition from the bent, closed to the extended, closed (intermediate affinity) conformation. A second interaction between the talin1 head domain and the β tail membrane-proximal region drives the final transition to an extended, open (high affinity) conformation [20]. A talin1 mutant (W359A) that prevents talin1 binding to the β integrin NPxY motif prevents integrin extension and integrin–mediated slow rolling [21, 22]. A mutation in the talin1 head domain that prevents talin1 interactions with the integrin β subunit membrane-proximal region (L325R) allows talin1 interaction with integrins and actin but prevents the transition to the extended, open (high affinity) conformation [21, 23, 24]. Mice expressing only talin1 L325R in platelets show defects in αIIbβ3 and β1 integrin activation and thrombus formation [23, 25]. Similarly, mice expressing only talin1 L325R in neutrophils show defects in neutrophil firm adhesion, although integrin-mediated neutrophil slow rolling remains intact presumably through integrins in the intermediate affinity conformation [21].
Endothelial integrins mediate distinct responses based on the integrin-ligand pair involved [26]. The fibronectin-binding integrin α5β1 plays a clear role in atherogenic endothelial activation. Atheroprone areas show elevated α5 mRNA and protein expression [27, 28], and both disturbed flow and oxLDL induce α5β1-dependent fibronectin matrix assembly and endothelial proinflammatory responses including NF-κB activation and intercellular adhesion molecule-1 (ICAM-1)/vascular cell adhesion molecule-1 (VCAM-1) expression [10, 11, 29, 30]. Mice deficient for endothelial α5 integrin signaling show reduced atherogenic endothelial activation and diminished plaque formation [10, 31]. In unstimulated cells, a majority of α5β1 exists in the bent, closed (low affinity) conformation [16], and both oxLDL and shear stress enhance α5β1 affinity for ligand [10, 32]. However, the role of talin1-dependent integrin affinity modulation in endothelial cell function remains poorly defined. Global knockout of talin1 is embryonic lethal at 8.5–9.5 days and associated with vascular defects [33], and inducible talin1 deletion in endothelial cells results in an unstable intestinal vasculature, resulting in hemorrhage and premature death [34]. However, these effects may be due to talin1’s role in linking integrins to the cytoskeleton rather than integrin affinity modulation. Therefore, we sought to utilize endothelial cells expressing only talin1 L325R to characterize the importance of talin1-dependent integrin affinity modulation in endothelial phenotype in the context of atherogenic endothelial activation.
Results
Talin1 L325R mutant endothelial cells lack oxLDL and shear stress-induced α5β1 integrin affinity modulation.
To characterize the role of integrin affinity modulation in endothelial cell function, we isolated mouse lung endothelial cells (MLECs) from talin1fl/L325R mice that express one floxed talin1 allele and one L325R mutant allele. Talin1fl/L325R endothelial cells were then treated with adenovirus expressing either green fluorescent protein (GFP; talin1 wildtype (WT)) or expressing GFP-Cre recombinase (talin1 L325R) to delete the floxed allele. While the talin1Δ/L325R (hereafter talin1 L325R) endothelial cells contain only one copy of the talin1 gene, talin1fl/L325R (hereafter talin1 WT) and talin1 L325R endothelial cells show equivalent protein levels of talin1 (Figure 1A). Consistent with previous reports in talin deficient cells [35], talin1 L325R MLECs show slowed adhesion and spreading compared to talin1 WT MLECs (Figure 1B/C). While talin1/2 deficient cells eventually lose the spread cell phenotype [35], talin1 L325R MLECs remain stably adherent, suggesting that the retained ability of talin1 L325R to link integrins to the cytoskeleton is sufficient to support stable cell adhesion. In contrast, talin1 L325R endothelial cells show significantly reduced formation of vinculin-positive (Figure 1D/E) or active β1 integrin-positive (Supplemental Figure Ia–c) nascent adhesions at the leading edge of protrusions, consistent with unstable protrusions and poor cell spreading. In isolated focal adhesion fractions, we observed that talin1 WT and talin1 L325R endothelial cells show similar recruitment of talin1, α5, and β1 integrins (Figure 1F, Supplemental Figure IIa–c), as well as the talin1-binding proteins paxillin and vinculin (Figure 1F, Supplemental Figure IId–e). Positive (integrin-linked kinase (ILK)) and negative (β-tubulin) controls demonstrate purity of the focal adhesion fraction [36]. Similar to the focal adhesion fractions, immunocytochemistry demonstrates similar staining for talin1, active β1 integrins (9EG7 antibody), vinculin, and paxillin in talin1 WT and talin1 L325R endothelial cells (Supplemental Figure IIIa/b). Quantification of focal adhesions after staining for active β1 integrins (9EG7 antibody) and vinculin shows that talin1 WT and talin1 L325R endothelial cells form similar number, size, and area of focal adhesions (Figure 1G–J, Supplemental Figure IV), suggesting that stable integrin adhesions are unaffected by preventing high affinity endothelial integrins.
Figure 1. Talin1 L325R Endothelial Cells Show Deficient Spreading and Nascent Adhesions but Normal Focal Adhesion Structure.
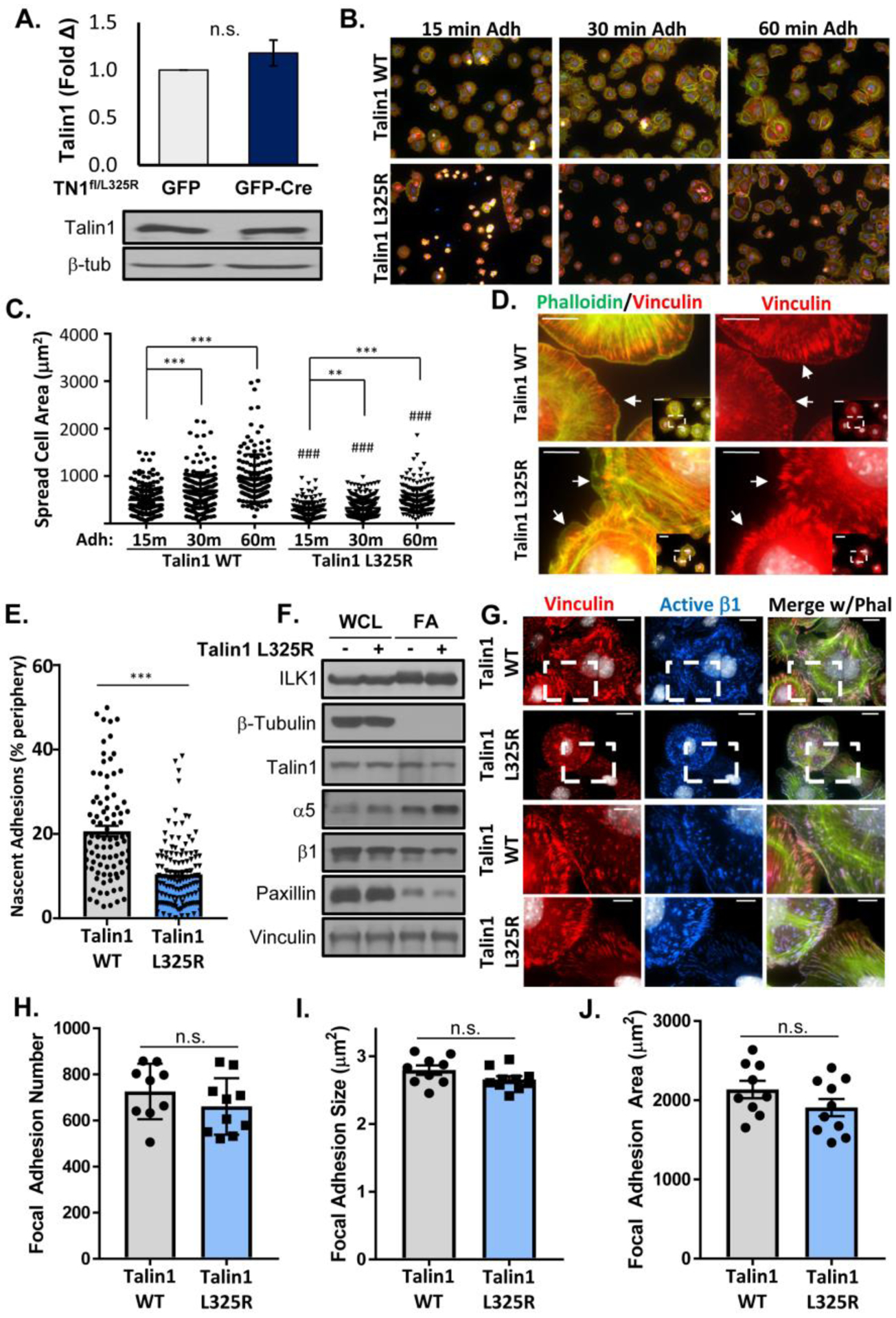
A) Representative Western blots of MLEC from Talin1fl/L325R after expression of GFP or GFP-Cre to delete the floxed allele. B/C) Talin1 WT and talin1 L325R MLECs plated on fibronectin for 15, 30, and 60 minutes were fixed and immunostained for active β1 integrin (9EG7, red) and actin (phalloidin, green). Spread cell area was quantified for each cell across three independent experiments. Representative 20x micrographs are shown. D-E). Cells were stained as in (B) and nascent adhesions forming within 1 μm of the cell edge at 30 minutes adhesion were visualized and quantified. Representative micrographs are shown with 60x inset. Scale bar = 10 μm. F) Focal adhesion fraction in talin1 WT and talin1 L325R MLECs were blotted for various focal adhesion proteins. ILK serves a positive control for the focal adhesion fraction, whereas β-tubulin serves as a negative control for the focal adhesion fraction. n=4 (quantified in Supplemental Figure II). G-J) Talin1 WT and talin1 L325R MLECs were plated on fibronectin for 2 hours and stained for active β1 (9EG7, blue), vinculin (red), and actin (phalloidin, green). H) Focal adhesion number, (I) average size, and (J) area per high powered field were quantified for each of three separate experiments. Representative micrographs are shown with 60x inset. Scale bar = 20 μm and 10 μm for image within boxed area. Values are means ±SE. Statistical analysis was performed using Student t test. ** p<0.01, *** p<0.001. n.s. indicates not significant.
The presence of only the talin1 L325R mutant impairs the induction of high affinity αIIbβ3 in platelets [23, 25] and high affinity αLβ2 in leukocytes [21]. Therefore, we assessed whether talin1 L325R endothelial cells show a similar reduction in α5β1 integrin affinity modulation in response to acute onset of shear stress and to oxLDL [10, 32]. Talin1 WT and L325R endothelial cells were exposed to shear stress or oxLDL, and α5β1 affinity modulation was measured using a glutathione S-transferase (GST) tagged portion of the fibronectin (FN) protein (FN type III repeats 9–11, GST-FNIII9–11), an α5β1 ligand mimetic that binds high affinity, unligated integrins [32]. Retention of GST-FNIII9–11 is specifically mediated by α5β1 and is sensitive the both positive and negative regulators of integrin affinity modulation [32]. While basal levels of GST-FNIII9–11 retention were similar between talin1 WT and talin1 L325R endothelial cells (Figure 2A/B), the enhanced levels of high affinity α5β1 observed in response to acute onset of laminar shear stress (Figure 2A), and to oxLDL (Figure 2B) was absent in talin1 L325R endothelial cells. To validate this result, talin1 WT and L325R endothelial cells were immunostained for active β1 (9EG7), which demonstrates both high affinity, unligated and high affinity, ligated integrins. Following treatment with oxLDL or shear stress, active β1 staining increased in talin1 WT endothelial cells but not talin1 L325R endothelial cells, consistent with an impaired integrin affinity modulation response (Figure 2C/D). These results suggest that talin1 is dispensable for the formation of stable adhesions but is required for the induction of high affinity integrins in endothelial cells that contribute to nascent adhesion formation and the endothelial response to oxLDL and shear stress.
Figure 2. Talin1 Mediates Shear Stress- and OxLDL-Induced Integrin Activation.
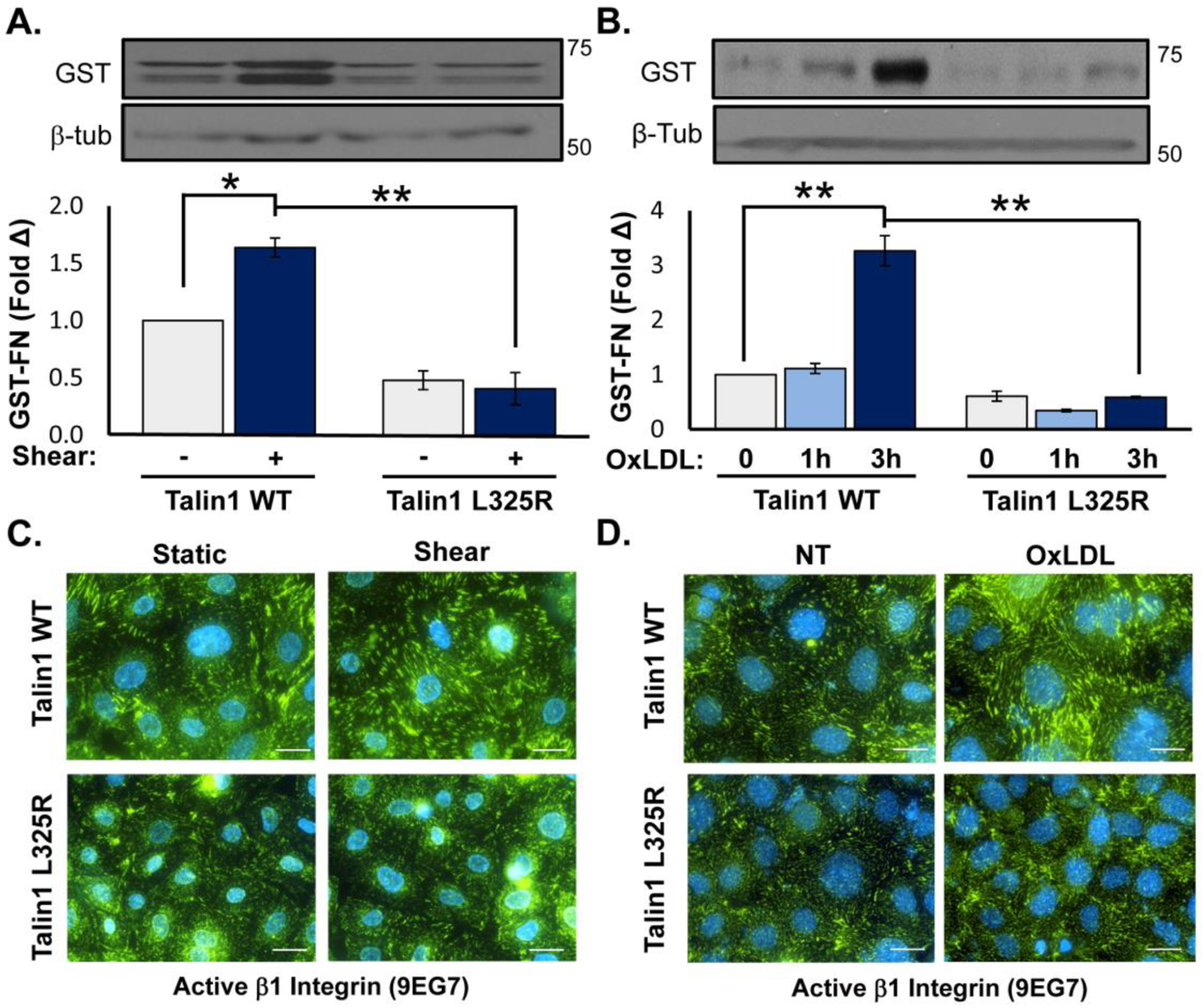
A/B) Talin1 WT and talin1 L325R MLECs were plated on fibronectin and exposed to acute onset of laminar flow (“Shear”, 5 minutes) or treated with oxLDL (100 μg/ml) for 1 or 3 hours. α5β1 activation was assessed by measuring retention of the α5β1 ligand mimetic GST-FNIII9–11 by Western blotting. n=4. C/D) Talin1 WT and talin1 L325R MLECs were plated on fibronectin and either (C) exposed to shear stress or (D) treated with oxLDL for 1 hour. β1 integrin activation was assessed by staining with the 9EG7 antibody that binds β1 only when it is in the active conformation. Representative images are shown. Scale bar = 20 μm. n=4–5. Values are means ±SE. *p<0.05, **p<0.01. Statistical analysis was performed using 2-way ANOVA with Bonferroni posttest.
Talin1 L325R mutation blocks endothelial inflammation on fibronectin.
At atherosclerosis prone regions, NF-κB signaling plays a major role in driving proinflammatory endothelial activation [5]. Signaling through α5β1 mediates NF-κB activation and enhanced proinflammatory gene expression in response to both oxLDL [10, 11] and shear stress [29, 31, 37–39]. Since integrin inhibitors (small molecule inhibitors, blocking antibodies) blunt shear stress and oxLDL-induced NF-κB activation, we and others have postulated that new integrin-matrix interactions following integrin affinity modulation likely drive endothelial responses to both stimuli. However, these inhibitors may also affect stable integrin-matrix interactions or promote certain integrin signaling responses. Therefore, we utilized talin1 WT and L325R endothelial cells to elucidate the specific role of integrin affinity modulation in response to shear stress and oxLDL. Whereas both oxLDL and acute onset of laminar shear stress induce phosphorylation of the NF-κB p65 subunit (hereafter NF-κB), neither oxLDL nor shear stress induced significant NF-κB phosphorylation in talin1 L325R endothelial cells (Figure 3A/B). Similarly, NF-κB nuclear translocation in response to oxLDL and shear stress was observed in talin1 WT but not talin1 L325R endothelial cells (Figure 3C/D). In contrast, other known shear-responsive signaling events, such as extracellular signal-regulated kinase 1/2, AKT, and endothelial nitric oxide synthase (Supplemental Figure V) were activated similarly in talin1 WT and talin1 L325R endothelial cells, indicating these signaling responses are independent of shear stress-induced integrin affinity modulation [40]. Since α5β1 mediates oxLDL and oscillatory shear stress (OSS)-induced proinflammatory gene expression, we examined the effect of blocking talin1-dependent integrin affinity modulation in this response. Consistent with the suppressed NF-κB activation, our results show a marked inhibition of VCAM-1 and ICAM-1 in response to chronic OSS (Figure 4A/B) and oxLDL (Figure 4C/D) in talin1 L325R endothelial cells. However, talin1-dependent integrin affinity modulation is not required for endothelial VCAM-1 expression in response to tumor necrosis factor α (TNFα), interleukin-1β (IL-1β), and lipopolysaccharide (LPS) (Figure 4E), consistent with a specific role for integrin signaling in oxLDL and OSS-induced proinflammatory endothelial activation.
Figure 3. Blocking Talin1-Dependent Integrin Activation Blunts Shear- and OxLDL-Induced NF-κB Activation.
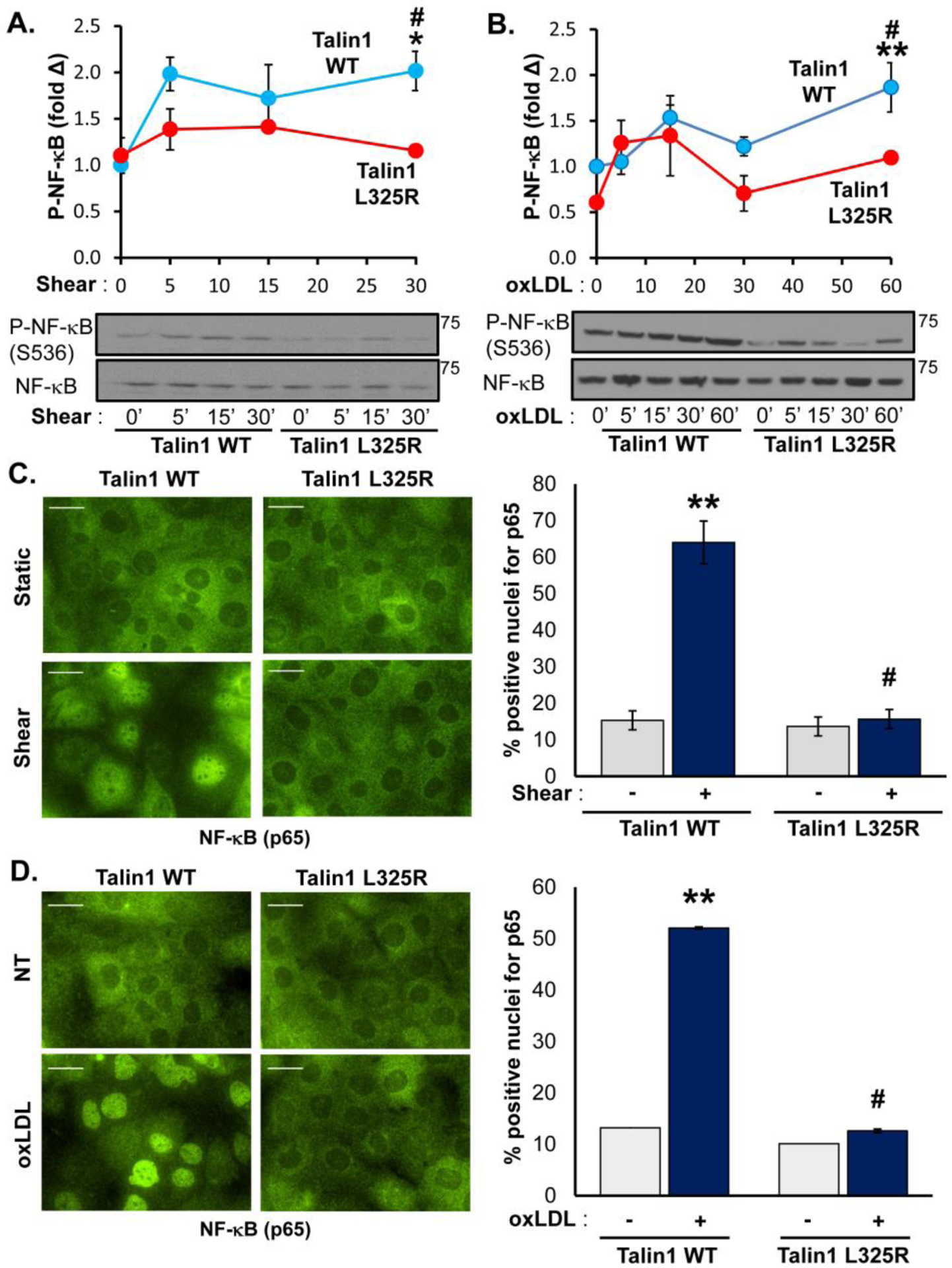
A/B) Talin1 WT and talin1 L325R MLECs were plated on fibronectin and either (A) exposed to onset of laminar flow (shear) or (B) treated with oxLDL (100 μg/ml) for the indicated time points. NF-κB activation was assessed by Western blotting for NF-κB phosphorylation (P-NF-κB (p65, Ser536)). Representative Western blots are shown. n=4–5. C/D) Talin1 WT and talin1 L325R MLECs were treated with either (C) shear for 30 minutes or (D) oxLDL for an hour, and NF-κB activation was assessed by immunocytochemistry for NF-κB nuclear translocation. At least 100 cells were assessed per experiment. Representative images are shown. n=4. Scale bar = 25 μm. Values are means ±SE. *p<0.05, **p<0.001 indicates significance compared to untreated controls, and #p<0.05 indicates significance between talin1 WT and talin1 L325R MLECs. Statistical analysis was performed using 2-way ANOVA with Bonferroni posttest.
Figure 4. Talin1-Mediated Integrin Activation Is Required for OSS- and OxLDL-Induced Inflammation.
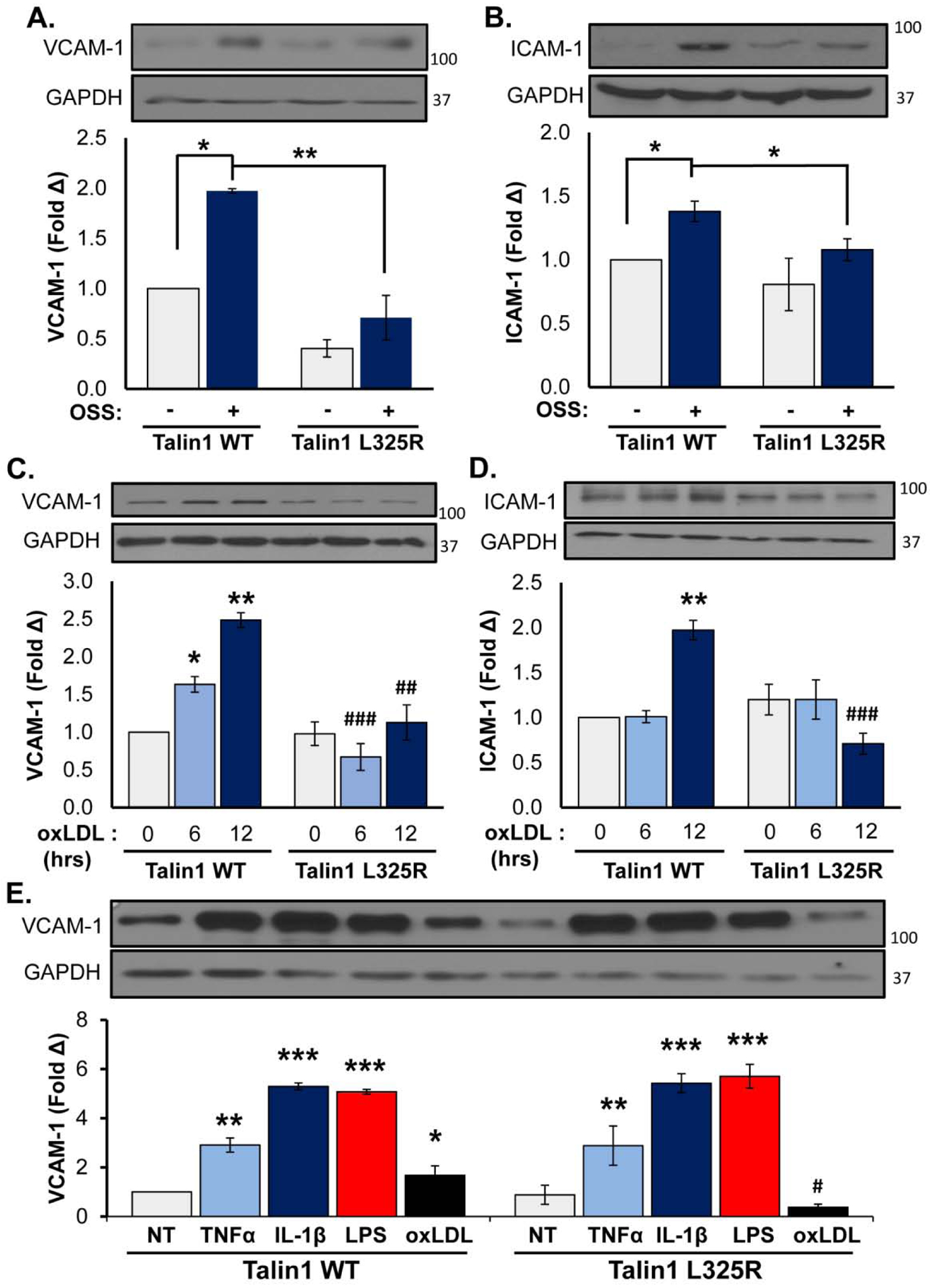
A/B) Talin1 WT and talin1 L325R MLECs were plated on fibronectin and exposed to oscillatory shear stress (OSS) for 18 hours. VCAM-1 and ICAM-1 protein expression was measured using Western blot and normalized to GAPDH. Representative images are shown. n=4–5. C/D) Talin1 WT and talin1 L325R MLECs were treated with oxLDL (100 μg/ml) for the indicated time points, and expression of (C) VCAM-1 and (D) ICAM-1 was assessed by Western blotting and normalized to GAPDH. Representative images are shown. n=4. E) Talin1 WT and talin1 L325R MLECs were treated with TNFα (1 μg/ml), IL-1β (5 ug/ml), LPS (10 μg/ml), or oxLDL for 6 hours, and VCAM-1 expression was assessed by Western blotting. Representative images are shown (n=3). Values are means ±SE. *p<0.05, **p<0.01, ***p<0.001 indicates statistical significance compared with untreated samples, whereas #p<0.05, ##p<0.01, and ###p<0.001 indicates statistical significance comparing talin1 WT and talin1 L325R MLECs. Statistical analysis was performed using 2-way ANOVA with Bonferroni posttest.
Aberrant fibronectin fibrillogenesis in endothelial cells expressing talin1 L325R mutation.
Fibronectin deposition occurs early during atherogenesis and contributes to early endothelial activation in part through inducing α5β1 proinflammatory signaling [6, 13, 29, 31, 41]. However, α5β1 integrins also regulate fibronectin deposition in response to oxLDL and shear stress [11, 39], and it is unclear whether integrin affinity modulation is required to mediate fibronectin matrix deposition. Therefore, we sought to examine fibronectin deposition in talin1 L325R mutant endothelial cells. Talin1 WT and talin1 L325R endothelial cells were plated on basement membrane proteins (diluted Matrigel) for 4 hours and then were exposed to either OSS or oxLDL for 24 hours. Fibronectin deposition into the deoxycholate (DOC)-insoluble matrix fraction was compared using Western blotting and immunocytochemistry as previously described [11]. Talin1 WT endothelial cells showed a low level of fibronectin deposition under unstimulated conditions that increased significantly in response to both OSS (Figure 5A) and oxLDL (Figure 5B). While fibronectin deposition did not differ between talin1 WT and talin1 L325R endothelial cells under unstimulated conditions, talin1 L325R endothelial cells failed to show enhanced fibronectin deposition in response to either stimulus. To gain further insight into this response, the fibronectin matrix was visualized by immunocytochemistry. As previously shown, both OSS and oxLDL increased fibronectin staining in the subendothelial matrix (Figure 5C/D), associated with both increased fluorescence intensity for fibronectin (Figure 5E/F) and increased fibronectin fibril length (Figure 5G/H). While fibronectin staining and fibril length were similar between talin1 WT and talin1 L325R endothelial cells in unstimulated conditions, both OSS and oxLDL failed to enhance fibronectin staining intensity or fibronectin fibril length in talin1 L325R endothelial cells.
Figure 5. Talin1-Mediated Integrin Activation Promotes Fibronectin Fibrillogenesis.
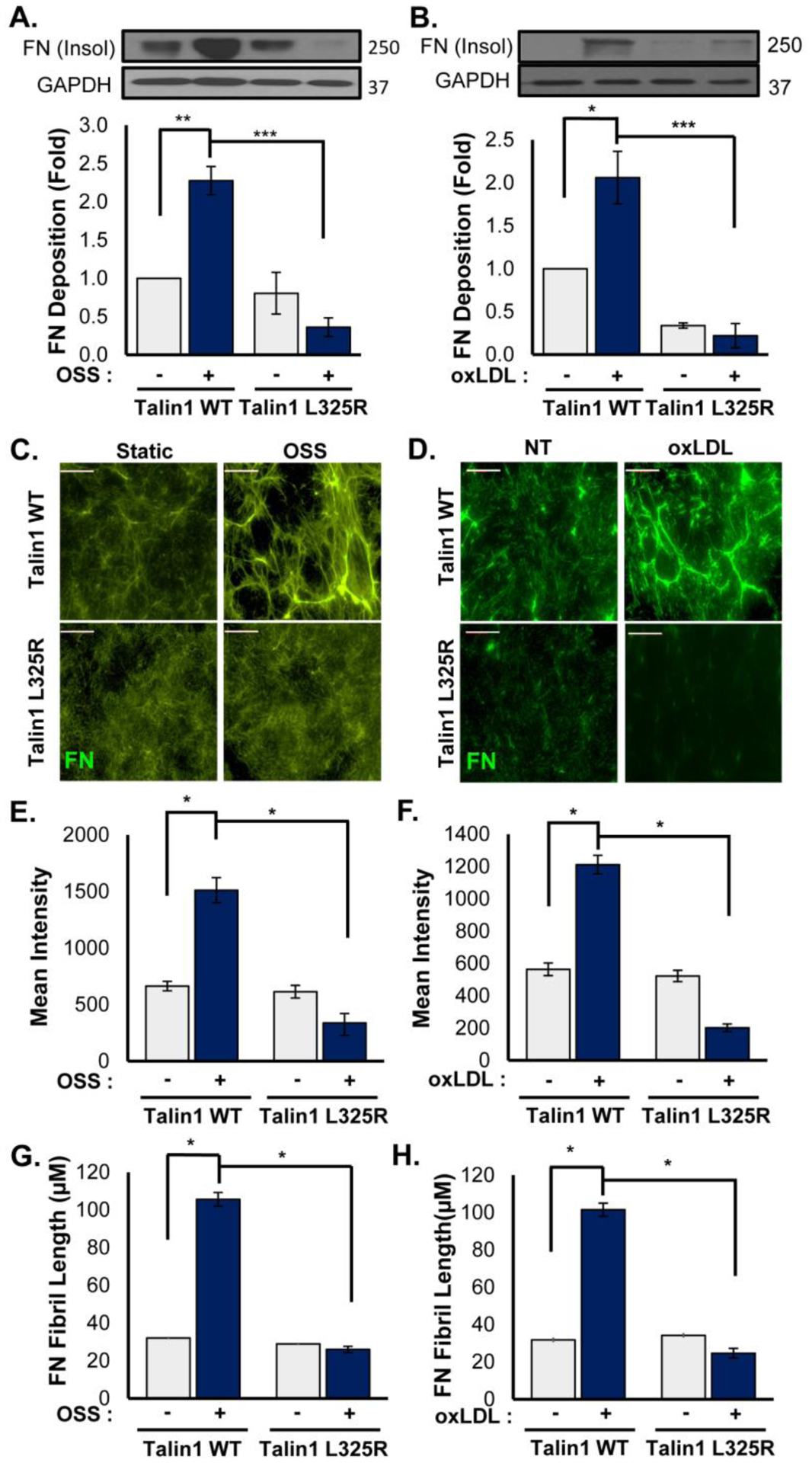
A/B) Talin1 WT and talin1 L325R MLECs were plated on basement membrane proteins (diluted Matrigel) for 4 hours and either exposed to OSS (18 hrs) or treated with oxLDL (100 μg/ml, 24 hrs). The DOC-insoluble matrix fraction was collected, and Western blotting for fibronectin was performed to assess fibronectin matrix assembly. n=4. C-H) Talin1 WT and talin1 L325R MLECs were treated as in A/B, and fibronectin matrix deposition was assessed by immunocytochemistry. C/D) Represented images are shown, and E/F) Mean fluorescent intensity and (G/H) fibronectin fibril length were assessed. n=4–5. Scale bar = 25 μm. Values are means ±SE. *p<0.05, **p<0.01, ***p<0.001. Statistical analysis was performed using 2-way ANOVA with Bonferroni posttest.
Although the talin1 L325R endothelial cells were deficient for inducible fibronectin deposition, fibronectin expression was similar between the talin1 WT and talin1 L325R endothelial cells (Supplemental Figure VIa–d), suggesting the defect may be in matrix deposition. Fibronectin fibril assembly involves α5β1 translocation from peripheral focal adhesions into tensin1-rich fibrillar adhesions that organize the fibronectin dimers into higher order fibrils [11, 42]. To assess whether talin1-dependent integrin affinity modulation affects the formation of fibrillar adhesions in response to oxLDL or OSS, talin1 WT and talin1 L325R endothelial cells were treated with oxLDL or OSS, and the composition of endothelial adhesions was assessed by immunocytochemistry and Western blotting analysis of the isolated focal adhesion fraction. Both oxLDL and OSS stimulated enhanced recruitment of α5 integrins into the focal adhesion fraction in talin1 WT endothelial cells (Figure 6A/B), consistent with an increase in fibrillar adhesion formation. However, levels of α5 in the focal adhesion fraction were unchanged in talin1 L325R endothelial cells. Consistent with this result, immunocytochemistry showed enhanced α5 staining in endothelial adhesions in response to both oxLDL and OSS (Figure 6C/D). To determine if the enhanced α5 recruitment was associated with fibrillar adhesions, we assayed tensin recruitment into these adhesions following oxLDL and OSS treatment. Like α5 integrins, tensin showed enhanced recruitment into the focal adhesion fraction in response to both oxLDL and OSS in talin1 WT endothelial cells but not talin1 L325R endothelial cells (Figure 6E/F). This defective recruitment of tensin into fibrillar adhesions in response to oxLDL and OSS in the talin1 L325R endothelial cells was verified by immunocytochemistry (Figure 6G/H). Together, these data suggest that inducible high affinity integrins and the formation of new cell-matrix interactions are critical for fibrillar adhesion development and endothelial fibronectin deposition in response to oxLDL and OSS.
Figure 6. Talin1 L325R Endothelial Cells Show Reduced Fibrillar Adhesions in Response to OSS and OxLDL.
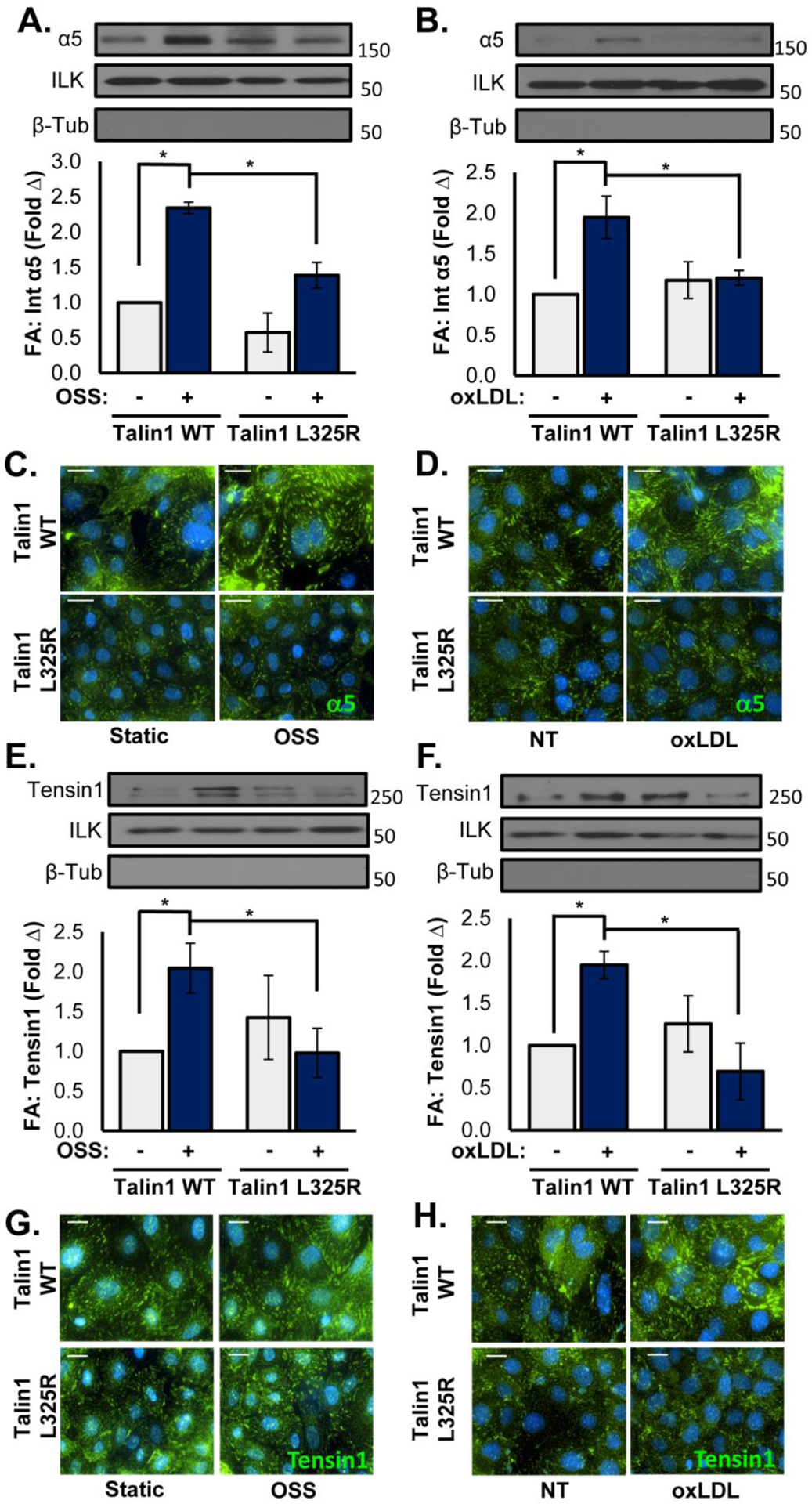
A/B) Talin1 WT and talin1 L325R MLECs were either exposed to OSS (18 hrs) or treated with oxLDL (100 μg/ml, 24 hrs). Focal adhesions were extracted using hydrodynamic shock, and α5 levels in focal adhesions was assessed by Western blotting. Representative Western blots for α5, ILK (focal adhesion positive control), and β-tubulin (focal adhesion negative control) are shown. n=4. Scale bar = 25 μm. C/D) Talin1 WT and talin1 L325R MLECs treated as in A/B were stained for α5 integrins by immunocytochemistry. Representative images are shown. n=4. E/F) Talin1 WT and talin1 L325R MLECs were treated as in A/B, and tensin1 recruitment into the focal adhesion fraction was assessed by Western blotting. Representative Western blots for tensin1, ILK, and β-tubulin are shown. n=4–5. G/H) MLECs treated as in A/B were stained for tensin1 by immunocytochemistry. Representative images are shown. n=4. Scale bar = 20 μm. Values are means ±SE. *p<0.05. Statistical analysis was performed using 2-way ANOVA with Bonferroni posttest.
Requirement of integrin signaling for endothelial activation and matrix remodeling.
While these data indicate that a subset of endothelial responses to oxLDL require inducible high affinity integrins, it remains unclear whether integrin affinity modulation and signaling are sufficient for these responses. To test the role of integrin affinity modulation in endothelial proinflammatory gene expression and fibronectin deposition, we employed computationally designed transmembrane α-helical peptides (CHAMP) targeted to the α5 integrin transmembrane region to selectively induce high affinity α5β1 integrins in the talin1 L325R MLECs [43]. Treatment with the α5 CHAMP peptide enhanced fibronectin deposition in talin1 L325R endothelial cells (Figure 7A/B), but treatment with a combination of α5 CHAMP peptides and oxLDL resulted in a significantly increased fibronectin deposition (Figure 7A/B). While α5 is clearly required for oxLDL-induced NF-κB activation [10], activation of α5β1 with the α5 CHAMP peptide was not sufficient to induce NF-κB activation (Figure 7C). However, simultaneous treatment with both oxLDL and the α5 CHAMP peptide restored NF-κB activation in talin1 L325R endothelial cells (Figure 7C). Taken together, these data suggest that NF-κB activation and maximal fibronectin assembly requires co-stimulatory integrin-dependent and integrin-independent signaling pathways.
Figure 7. α5 Integrin Activation Requirement is Different for Matrix Remodeling Versus Inflammation.
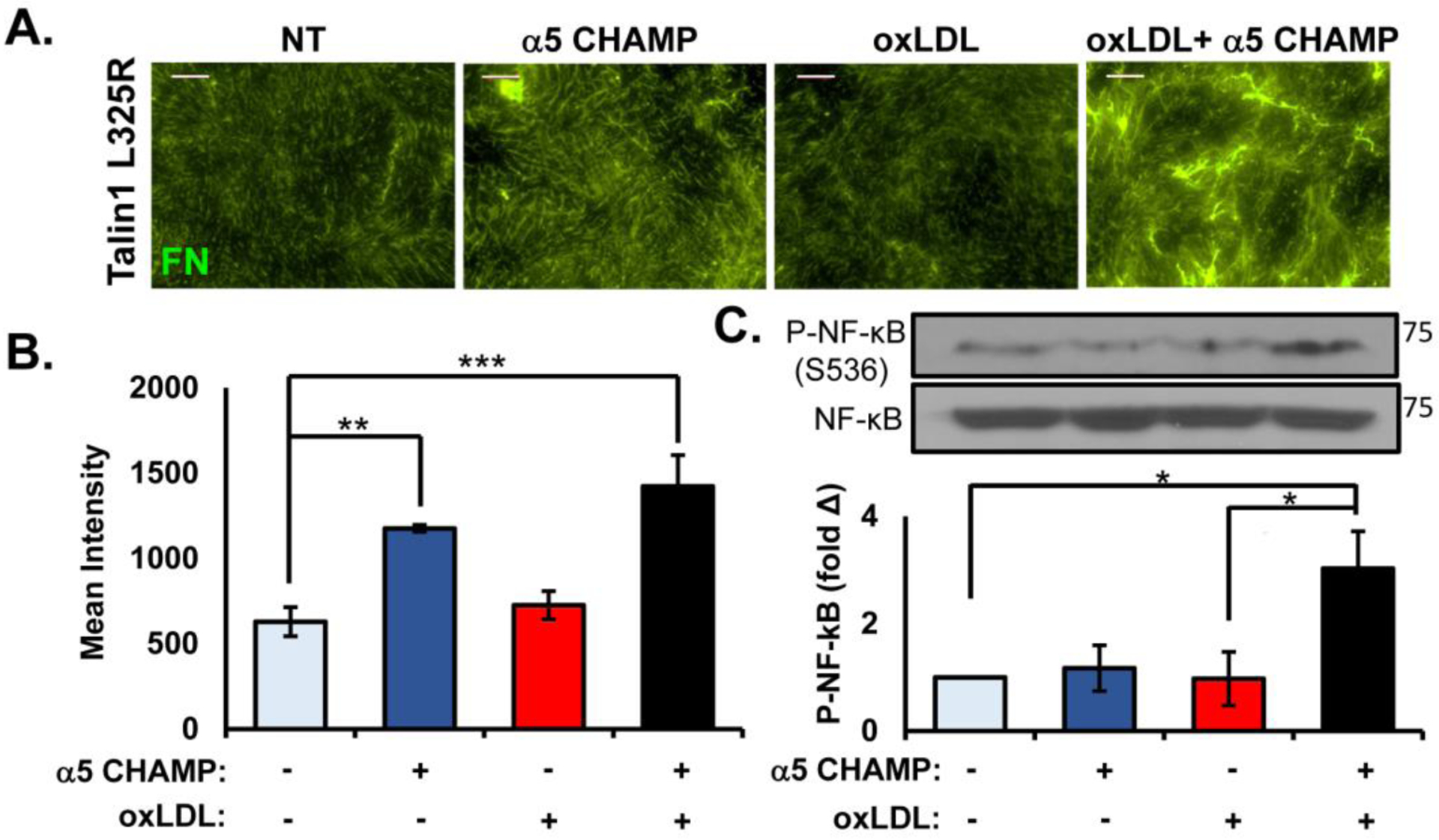
A/B) Talin1 L325R MLECs were plated on basement membrane proteins (diluted Matrigel) for 4 hours and treated with either oxLDL (100 μg/ml), α5 CHAMP (4 μM), or both for 24 hours. Fibronectin in the DOC-insoluble matrix was visualized by immunocytochemistry, and (B) quantified for mean fluorescent intensity. Representative images are shown. n=4–7. Scale bar = 25 μm. C. Talin1 L325R MLECs were plated on fibronectin and treated with either oxLDL, α5 CHAMP, or both for 1 hour. NF-κB activation was assessed by Western blotting for NF-κB phosphorylation (P-NF-κB (p65, Ser536)). Representative blots are shown. n=4. Values are means ±SE. *p<0.05, **p<0.01, ***p<0.001. Two-way ANOVA with Bonferroni posttest was used for statistical analyses.
Mice expressing endothelial talin1 L325R are protected from atherogenic inflammation.
Since our in vitro studies show a clear role for talin1-dependent integrin affinity modulation in atherogenic endothelial activation, we next examined whether mice expressing only talin1 WT or talin1 L325R in their endothelial cells show differential susceptibility to disturbed flow-induced endothelial activation in vivo. Tamoxifen-inducible, endothelial-specific talin1 WT (iEC-Talin1 WT; Talin1WT/flox, VE-Cadherin-CreERTtg/?) and talin1 L325R (iEC-Talin1 L325R; Talin1flox/L325R, VE-Cadherin-CreERTtg/?) mice underwent partial ligation of the left carotid artery to induce disturbed flow. The right carotid artery remained exposed to laminar flow and was used as an internal control. After 48 hours, intimal mRNA was isolated to analyze endothelial proinflammatory gene expression using quantitative real time PCR (qRT-PCR). Intimal mRNA preparations were shown to be enriched for CD31 and deficient in smooth muscle actin compared to medial mRNA (Figure 8A). Both iEC-Talin1 WT and iEC-Talin1 L325R mice show reduced Kruppel-like factor 2 (KLF2) expression in the left carotid artery following partial ligation, consistent with a loss of laminar flow (Figure 8B). However, only iEC-Talin1 WT mice showed a significant increase in proinflammatory gene expression (ICAM-1, VCAM-1) in the left carotid (compared to the nonligated right carotid), whereas iEC-Talin1 L325R mice showed no upregulation of proinflammatory gene expression (Figure 8B). In addition to mRNA analysis, we harvested carotid arteries from iEC-Talin1 WT and iEC-Talin1 L325R mice 7 days after partial carotid ligation for immunohistochemical analysis. While the unligated right carotid showed no signs of inflammation, as assessed by staining for NF-κB, VCAM-1, and macrophages (Mac2), all of these inflammatory markers were enhanced in the ligated left carotids in iEC-Talin1 WT mice (Supplemental Figure VII). Consistent with our in vitro data, iEC-Talin1 L325R mice show a reduction of endothelial NF-κB nuclear translocation (Figure 8C/D, Supplemental Figure VIII), VCAM-1 expression (Figure 8E/F), and macrophage (Mac2-positive) recruitment (Figure 8G/H). Diminished macrophage recruitment in the iEC-Talin1 L325 mice was also verified by staining for CD68 (Supplemental Figure IX). In contrast, carotid endothelial (CD31-positive) and smooth muscle (smooth muscle actin-positive) content was similar between iEC-Talin1 WT and iEC-Talin1 L325R mice (Supplemental Figure X). Altogether, these data show that modulating integrin affinity in endothelial cells contributes to proinflammatory endothelial activation both in vitro and in vivo.
Figure 8. Mice Expressing Endothelial Talin1 L325R Mutation Are Protected from Atherogenic Inflammation.
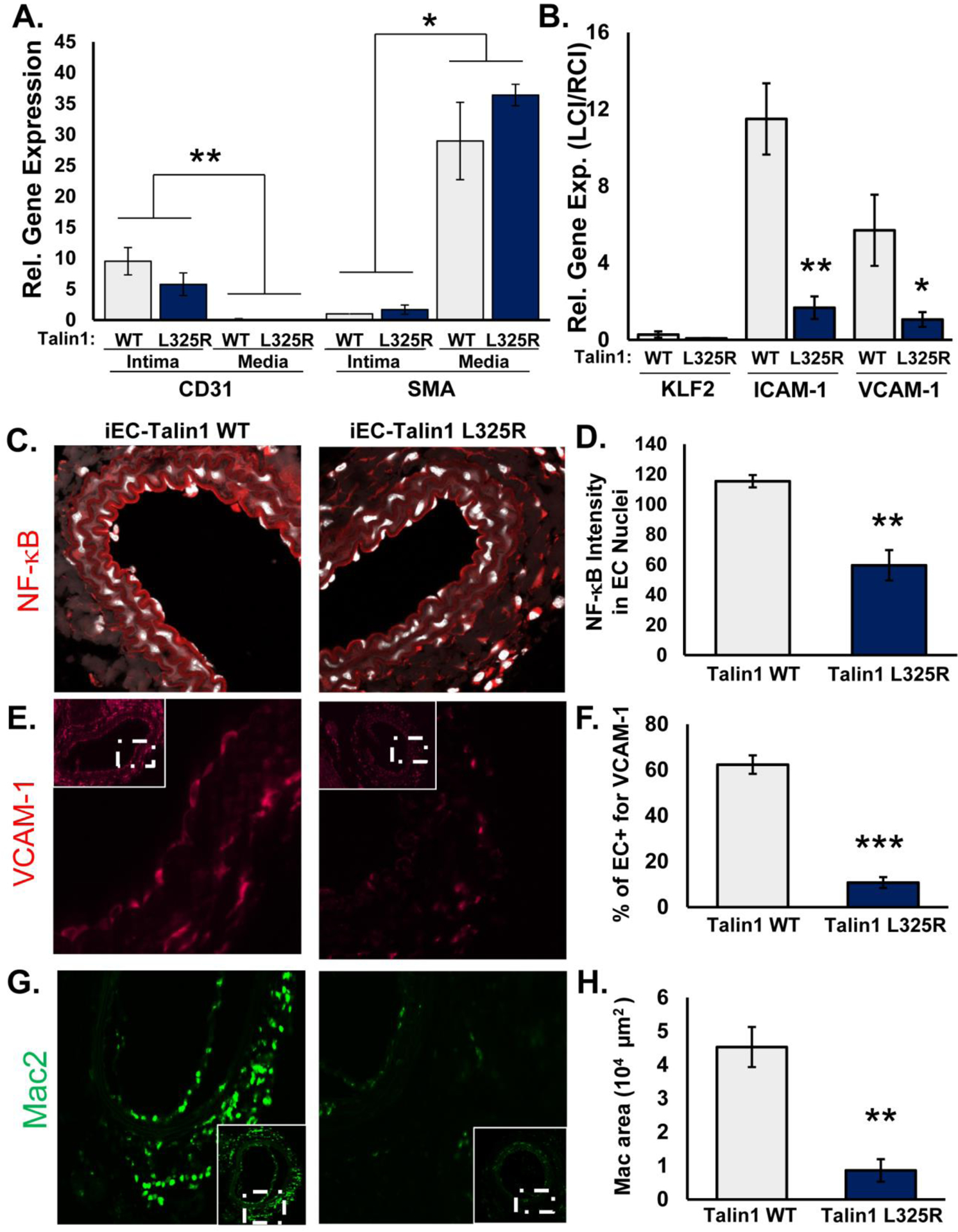
A/B) iEC-Talin1 WT (Talin1+/flox, VE-cadherin-CreERT2tg/?) and iEC-Talin1 L325R (Talin1flox/L325R, VE-Cadherin-CreERT2tg/?) mice were treated with tamoxifen for 5 consecutive days. Four weeks later, mice underwent partial carotid ligation of their left carotid artery, and mRNA was isolated from the intimal (endothelial) and medial (smooth muscle) layers after 48 hours. A) Expression of CD31 and smooth muscle actin (SMA) was assessed by qRT-PCR to ensure fraction purity. B) Endothelial expression of Klf2, ICAM-1, and VCAM-1 was determined by qRT-PCR. Expression was normalized to β2 microglobulin and compared between the ligated left carotid and the non-ligated right carotid artery for each mouse. n=5–6. C-H) iEC-Talin1 WT and iEC-Talin1 L325R underwent partial carotid ligation, and vessel remodeling was assessed by immunohistochemistry after 7 days. C/D) NF-κB activation (red, visualized by confocal microscopy) was assessed by examining nuclear (DAPI, white) staining intensity in the endothelium. E/F) VCAM1 expression in the endothelium was determined and G/H) macrophage (Mac2-positive staining) levels were assessed. Representative images are shown (n=5–6). Values are means ±SE. *p<0.05, **p<0.01, and ***p<0.001. Statistical analysis for (A) utilized Two-Way ANOVA with Bonferroni posttest, whereas analysis in (B-H) utilized Student t test.
Discussion
Cell-matrix interactions play a critical role in endothelial activation and early atherosclerosis [2]. However, the role of integrin affinity modulation in this context remains enigmatic. Previously, we showed that fibronectin enhances proinflammatory responses to OSS and oxLDL and deleting fibronectin-binding integrins in the endothelium blunts atherogenic inflammation [10, 11]. To characterize the role of integrin activation in endothelial cell phenotype, we isolated MLECs from mice expressing the L325R talin1 mutation that selectively inhibits integrin affinity modulation without affecting talin1’s ability to link the integrin β tail with actin. Consistent with cells lacking talin1 [35], talin1 L325R endothelial cells adhere and form focal adhesions but fail to form nascent adhesions at the edge of membrane protrusions resulting in a reduction in endothelial cell spreading. However, talin1 L325R endothelial cells form stable adhesions that do not differ significantly in size or number from talin1 WT endothelial cells under unstimulated conditions. Additionally, we showed that talin1-dependent α5β1 affinity modulation in response to oxLDL and OSS is critical for fibronectin deposition, NF-κB activation, and proinflammatory gene expression. One caveat to these studies is the lack of data in human endothelial cells and the sole reliance on MLECs from talin1 WT and talin1 L325R mice due to the difficulties inherent in mutating endogenous genes in human cells. Lastly, mice harboring a talin1 L325R mutation in endothelial cells show a significant reduction in proinflammatory gene expression and macrophage accumulation in the partial carotid ligation model of disturbed flow, suggesting that induction of high affinity α5β1 integrins in endothelial cells critically regulates atherogenic endothelial activation.
Deletion and rescue models suggest that talin1 and talin2 likely play non-redundant roles in cell adhesion. Like our results in endothelial cells, talin1-deficient fibroblasts adhere and spread normally [35], and siRNA-mediated knockdown of talin2 in these cells does not blunt initial cell adhesion and spreading. However, in the absence of talin2, the cells fail to form stable adhesions and ultimately lose the spread cell phenotype [35]. Expression of the integrin-activating talin1 head domain in these cells restores nascent adhesion formation, but only rescue with full length talin1 rescued focal adhesion formation, suggesting that tension-independent integrin activation mediates nascent adhesion formation whereas mechanical coupling through talin1-actin interactions critically regulates focal adhesion formation. Furthermore, talin1, but not talin2, is preferentially recruited to protrusions and localizes to peripheral adhesions, suggesting that talin2 likely plays a minimal role in nascent adhesion formation during cell motility and spreading [44]. Compared to fibroblasts, endothelial cells express minimal talin2 [24, 45], and deletion of endothelial talin1, but not talin2, results in defective cardiovascular development [33, 46]. While talin1 knockdown in endothelial cells reduces cell adhesion and focal adhesion formation, rescue with either talin1 or talin2 is sufficient to rescue both adhesion and focal adhesion formation, whereas expression of W359A and L325R talin1 did not [24]. In contrast, our data suggest that talin1 L325R mutant endothelial cells show normal focal adhesion formation but impaired nascent adhesion formation and impaired α5β1 affinity modulation in response to oxLDL or OSS.
While talin1 critically regulates cell adhesion in leukocytes and platelets, the residual cell adhesion and spreading in talin1 L325R endothelial cells may involve multiple mechanisms. Like neutrophils [21], the presence of talin1 L325R in endothelial cells may allow the endothelial cell integrins to attain the extended, closed (intermediate affinity) conformation, which could mediate weak integrin-matrix interactions to support initial cell adhesion and spreading in the mild mechanical environment of static cell culture. However, the reduced nascent adhesion formation in talin1 L325R MLECs suggest that these intermediate affinity integrins may not be sufficient to support nascent adhesion formation to stabilize membrane protrusions, thereby resulting in delayed spreading. Consistent with this hypothesis, low levels of force application to extended, closed integrins may promote integrins to attain the extended, open conformation and stabilize this high affinity conformation through a process termed outside-in integrin activation [47]. This method of integrin activation may be insensitive to talin1-dependent transition to a high affinity state in the context of mechanical coupling. Nascent adhesions form in a tension-independent manner and may be insensitive to outside-in affinity modulation through mechanical coupling [48], whereas outside-in integrin activation may support focal adhesion remodeling to force in the absence of inducible inside-out affinity modulation.
To dissect the role of integrin signaling in endothelial cell phenotype, several groups have used a variety of integrin inhibitors and blocking antibodies to implicate integrin affinity modulation and new integrin-matrix interactions in endothelial proinflammatory responses, particularly in response to OSS and oxLDL [6, 10, 14, 29, 31, 32, 37, 41, 49]. However, integrin inhibitors and blocking antibodies have the potential to affect pre-existing integrin-matrix interactions as well, making it difficult to determine the specific role of integrin affinity modulation and new integrin-matrix interactions. In addition, the use of ligand-mimetic inhibitors may stimulate ligation-dependent signaling, and anti-integrin antibodies may induce a subset of integrin signaling due to clustering. Our model of talin1 L325R mutant endothelial cells eliminates these barriers by specifically eliminating talin1-dependent integrin affinity modulation without affecting talin1-dependent focal adhesion formation. The talin1 L325R mutant endothelial cells provide the first confirmation of an important role for endothelial integrin affinity modulation in fibronectin deposition, NF-κB activation, and proinflammatory adhesion molecule expression (VCAM-1, ICAM-1). Furthermore, we utilize α5β1 activating CHAMP peptides to show that α5β1 affinity modulation is partially sufficient to induce fibronectin deposition, similar to previous studies by Wu et al. using constitutively active integrins and integrin activating antibodies [50]. However, CHAMP peptide-induced high affinity α5β1 integrins were not sufficient to induce maximal fibronectin deposition and NF-κB activation, suggesting that both integrin-dependent and integrin-independent signaling pathways contribute to these responses.
Much of our current understanding of integrin affinity modulation derives from studies in platelets and leukocytes, leading to the development of a multiple therapeutics for hematological and cardiovascular diseases. However, far less is known concerning integrin affinity modulation in adherent cell types, such as the endothelium. Previous studies in mouse models clearly implicate cell-matrix interactions in atherogenic endothelial activation but fail to connect endothelial integrin affinity modulation to these responses. Deletion of fibronectin or fibronectin-binding integrins (α5β1, αvβ3) limits endothelial NF-κB activation and proinflammatory gene expression in atherosclerosis models [11, 13], whereas β1 integrin inhibition reduces vascular permeability in response to proinflammatory mediators (LPS, IL-1β) [51]. In contrast, endothelial talin1 knockout mice show destabilized endothelial cell-cell junctions, mostly due to disruption of vascular endothelial-cadherin organization [34]. While talin1 reexpression restores endothelial adherens junction structure, rescuing integrin activation with the talin1 head domain (but not a L325R head domain mutant) only partially reverts this phenotype, suggesting an important role for both talin1-dependent integrin affinity modulation and talin1-mediated mechanical coupling in maintaining vascular barrier function [34]. In this manuscript, we showed that selectively preventing talin1-dependent integrin activation in endothelial cells (iEC-Talin1 L325R mice) blunts early atherogenic endothelial activation, demonstrating for the first time a vital role for endothelial integrin affinity modulation in the regulation of endothelial phenotype and proinflammatory gene expression in vitro and in vivo.
Experimental procedures
Cell culture-
MLECs were isolated from Talin1fl/L325R mice (gift of Brian Petrich, Emory, Atlanta, GA) as previously described [34]. Briefly, lung tissue was harvested, minced, and pushed through a 16G needle and subjected to enzymatic digestion to obtain a single cell suspension. After sorting with magnetic beads coupled to intercellular adhesion molecule-2 antibodies (eBiosource), cells were transformed using a retrovirus expressing temperature-sensitive large T-antigen. The floxed allele of talin1 was deleted using adenovirus harboring either GFP-Cre or GFP alone and then sorted for GFP positivity. Lung endothelial cells were grown in DMEM with 10% fetal bovine serum, 1% penicillin/streptomycin, and 1% Glutamax. For endothelial shear stress treatments, cells were plated on 38 × 75 mm glass slides (Corning) at confluency, and slides were assembled into a parallel plate flow chamber as previously described [49]. For acute shear stress, cells were exposed to acute onset of laminar flow at 12 dynes/cm2 for up to 1 hour. For chronic OSS (model of disturbed flow), cells were exposed to ±5 dynes/cm2 with 1 dyne/cm2 forward flow for media exchange. Human low density lipoprotein (Intracell) was oxidized by dialysis in PBS containing 13.8 μM Cu2SO4 followed by 50 μM EDTA as previously described [10, 32]. CHAMP peptides were a gift of Dr. William DeGrado (University of California – San Francisco) [43].
Western blot-
Cell lysis and western blot was done as previously described [32]. Briefly, cells were lysed in 2X laemmli buffer, separated by SDS-PAGE gels and then transferred to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA). Consequently, membranes were blocked in 5% dry milk in TBST for an hour, then incubated with primary antibodies overnight (Supplemental Table I). The following day after rinsing with TBST, horseradish peroxidaseconjugated secondary antibodies (Jackson ImmunoResearch) in blocking buffer were applied for two hours. Antibodies were detected using Pierce ECL solution (ThermoFisher) and x-ray film (Phenix Research Products).
Immunocytochemistry-
Cells were plated on coverslips or slides and fixed in formaldehyde and permeabilized using 0.2% Triton X-100 solution. Coverslips-containing cells were blocked with 1% heat denatured bovine serum albumin for an hour. Subsequently, cells were washed and incubated with primary antibodies overnight (Supplemental Table I). Next, fluorochrome-tagged secondary antibodies (Life Technologies) were added to coverslips for 2 hours. After rinsing, cells were counterstained with DAPI. We used Nikon Eclipse Ti inverted epifluorescence microscope equipped with a Photometrics CoolSNAP120 ES2 camera and the NIS Elements 3.00, SP5 imaging software. Nascent adhesions were defined as vinculin or β1 integrin positive adhesions less than 1μm2 in size and within 1 μm of the cell edge. Focal adhesions were defined as integrin adhesions between 1–10μm2 [52, 53].
Adhesion/spreading assay-
Cells were plated sparsely on fibronectin-coated coverslips for 15, 30, or 60 minutes in serum-free DMEM. Next, cells were fixed with PBS-buffered, 4% formaldehyde and co-stained with 546-conjugated phalloidin and 9EG7 (active β1 integrin).
Integrin Activation Assay-
Activation of α5β1 integrins was assessed using a specific α5β1 ligand mimetic as previously described [10]. Briefly, after cells were stimulated, GST-FNIII9–11 was added to the cell culture medium for 30 minutes. Cell were rinsed with fresh media and then lysed with 2X laemmli buffer, subjected to Western blotting, and probed for GST-FNIII9–11 (~70kDa).
Focal Adhesion Isolation-
Endothelial cells were plated on glass slides and treated. After experiments, cells were exposed to hypertonic shock using triethanolamine (2.5 mmol/L at pH 7.0) for 3 minutes. Next, cell bodies were removed by pulsed hydrodynamic force (Conair WaterPIK) at ≈0.5 cms from and ≈90° to the surface of the slide scanning the entire length 3 times [36]. Slides were visualized under a microscope to ensure complete removal of cell bodies before lysis in 2X Laemmli buffer.
DOC Solubility Assay-
Fibronectin integrated into extracellular matrix is insoluble in a DOC detergent. Discerning between the soluble fibronectin and matrix-bound fibronectin was done using a DOC solubility assay, as previously described [11]. Briefly, after stimulation, cells were rinsed with ice cold PBS, incubated with DOC buffer (2% Sodium Deoxycholate, 20mM Tris-HCL, pH 8.8, 2 mM PMSF, 2mM Iodoacetic Acid, 2mM N-ethylmaleimide, 10mM EDTA, pH 8), lysed, and then passed through a 25G needle. The DOC-insoluble matrix fraction was pelleted by centrifugation, and the DOC-soluble supernatant fraction was collected. After rinsing the pellet with additional DOC buffer, the DOC-insoluble faction was lysed in a solubilization buffer (2% SDS, 25mM Tris-HCL, pH 8, 2mM PMSF, 2mM Iodoacetic Acid, 2mM N-ethylmaleimide, 10mM EDTA, pH 8). Samples were subjected to western blotting and probed with rabbit anti-fibronectin.
qRT-PCR-
qRT-PCR was done as previously described[14]. Briefly, mRNA was extracted from tissues using TRIzol (Life Technologies, Inc., Carlsbad, CA). Next, iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) was used to synthesize Complimentary DNA. qRT-PCR was performed using Bio-Rad iCycler with the use of SYBR Green Master mix (Bio-Rad). We used the online Primer3 software and verified by sequencing the PCR products (Supplemental Table I). Results were expressed as fold change by using the 2-ΔΔCT method.
Animals and Tissue Harvesting-
The Louisiana State University Health Sciences Center-Shreveport Animal Care and Use Committee approved all animal protocols, and all animals were cared for according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. C57Bl/6J mice with a tamoxifen inducible, VE-cadherin-CreERT2 transgene (from Ralf Adams, Max Planck, Germany) with either wildtype talin1, floxed alleles for talin1, or with an L325R mutation in talin1 [25]. All mice were backcrossed to C57BL/6J mice for at least 7 generations. At 8 weeks of age, mice were treated with 1 mg/kg tamoxifen (Sigma-Aldrich, St Louis, MO) via intraperitoneal injection every day for 5 total injections to induce Cre expression and gene excision. Four weeks after tamoxifen injection, mice underwent partial carotid ligation as previously described [54]. Briefly, a superficial midline incision on the neck of mice under isoflurane anesthesia was done to expose the left carotid artery. Two 7–0 silk sutures were used to tie-off (occlude flow) to the internal, external, and occipital branches of the left carotid artery, whereas flow remained patent through the superior thyroid artery branch. The incision was closed with surgical glue and 6–0 silk sutures. Carprofen (5mg/kg) was administered as a post-surgical analgesic and 800uL of saline solution was given subdurally to prevent dehydration stress. Surgical success was confirmed via echocardiography 1 to 2 days prior to endpoint for confirmation of ligation. Mice were euthanized by pneumothorax under isoflurane anesthesia for tissue collection either 2- or 7-days post-surgery. After 2 days, carotid arteries were collected for RNA isolation performed by a TRIzol flush as previously described [54]. Briefly, carotids were cleaned of perivascular adipose tissue and flushed with 150 mL TRIzol from an insulin syringe. The remaining media/adventitia were then placed in 150 mL TRIzol and sonicated to lyse the tissue. Samples were then frozen until analysis by quantitative PCR. After 7 days, the carotids were excised, placed in 4% PBS buffered formaldehyde, and processed for immunohistochemistry.
Immunohistochemistry-
Tissue was fixed in PBS-buffered 4% formaldehyde, processed for paraffin embedding, and cut into 5 μm thick sections onto Superfrost plus glass sides. After heated and subjected sodium citrate antigen retrieval (Vector Labs, H-3300), tissue was blocked using 10% horse serum, 1% BSA in PBS. Primary antibodies were applied overnight, followed by Alexa Fluor conjugated secondary antibodies (Thermo Fisher). Stains are imaged on a Nikon Eclipse Ti inverted fluorescent microscope. Images are captured at either a 10X, 20X, or 60X (oil objective) using the Photometrics Coolsnap120 ES2 camera and the NIS Elements BR 3.00, SP5 imaging software. NF-κB nuclear translocation was visualized by Leica TCS SP5 confocal microscope using 63X (oil objective) and the Nikon NIS-Elements C software.
Statistical analysis-
Statistical analysis was performed using GraphPad Prism software. All of the data were tested for normality using Kolmogorov-Smirnov test. Data passing the normality tests were analyzed using either Student’s t-test, 1-way ANOVA with Newman-Keuls post-test, or 2-way ANOVA with Bonferroni post-tests depending upon the number of independent variables and groups. Data failing the normality test were analyzed using the nonparametric Mann-Whitney U test and the Kruskal-Wallis test with post hoc analysis.
Supplementary Material
Highlights:
Endothelial cells expressing only the talin1 L325R mutant deficient in integrin activation are able to adhere and spread normally, however, show aberrant nascent adhesion formation.
Talin1-dependent integrin activation is required for endothelial proinflammatory responses and fibronectin deposition in response to both shear stress and oxidized LDL.
Oxidized LDL-induced endothelial NF-κB activation and maximal fibronectin deposition require both integrin activation-dependent and integrin activation-independent signaling pathways.
Mice expressing only the talin1 L325R mutant in endothelial cells show reduced inflammation to disturbed flow in vivo.
Acknowledgements:
CHAMP peptides were a generous gift of Dr. William DeGrado (University of California - San Francisco).
Source of Funding
This work was supported by National Heart, Lung, and Blood Institute R01 HL098435, HL133497, HL141155, and GM121307 (to A.W.O.), R01 HL117061 (to B.G.P.), by an American Heart Association Pre-doctoral Fellowship (19PRE34380751) and Malcolm Feist Cardiovascular Research Endowment Pre-doctoral Fellowship (to Z.A.Y.).
Abbreviations:
- CHAMP
computationally designed transmembrane α-helical peptides
- DOC
deoxycholate
- FN
fibronectin
- GFP
green fluorescent protein
- ILK
integrin-linked kinase
- ICAM-1
intercellular adhesion molecule-1
- IL-1β
interleukin-1β
- KLF2
Kruppel-like factor 2
- LPS
lipopolysaccharide
- MLEC
mouse lung endothelial cell
- NF-κB
nuclear factor-κB
- OSS
oscillatory shear stress
- oxLDL
oxidized low density lipoprotein
- qRT-PCR
quantitative real time PCR
- TNFα
tumor necrosis factor α
- VCAM-1
vascular cell adhesion molecule-1
- WT
wildtype
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
The authors declare no conflicts.
References:
- [1].Pober JS, Sessa WC, Evolving functions of endothelial cells in inflammation, Nat Rev Immunol 7(10) (2007) 803–15. [DOI] [PubMed] [Google Scholar]
- [2].Yurdagul A Jr., Orr AW, Blood Brothers: Hemodynamics and Cell-Matrix Interactions in Endothelial Function, Antioxidants & redox signaling 25(7) (2016) 415–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhang X, Sun D, Song JW, Zullo J, Lipphardt M, Coneh-Gould L, Goligorsky MS, Endothelial cell dysfunction and glycocalyx - A vicious circle, Matrix biology: journal of the International Society for Matrix Biology 71–72 (2018) 421–431. [DOI] [PubMed] [Google Scholar]
- [4].Collins T, Cybulsky MI, NF-kappaB: pivotal mediator or innocent bystander in atherogenesis?, Journal of Clinical Investigation 107(3) (2001) 255–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI, The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation, Proceedings of the National Academy of Sciences of the United States of America 97(16) (2000) 9052–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA, The subendothelial extracellular matrix modulates NF-kappaB activation by flow: a potential role in atherosclerosis, The Journal of cell biology 169(1) (2005) 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Feaver RE, Gelfand BD, Wang C, Schwartz MA, Blackman BR, Atheroprone hemodynamics regulate fibronectin deposition to create positive feedback that sustains endothelial inflammation, Circulation research 106(11) (2010) 1703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Green J, Yurdagul A Jr., McInnis MC, Albert P, Orr AW, Flow patterns regulate hyperglycemia-induced subendothelial matrix remodeling during early atherogenesis, Atherosclerosis 232(2) (2014) 277–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pozzi A, Yurchenco PD, Iozzo RV, The nature and biology of basement membranes, Matrix biology: journal of the International Society for Matrix Biology 57–58 (2017) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yurdagul A Jr., Green J, Albert P, McInnis MC, Mazar AP, Orr AW, alpha5beta1 integrin signaling mediates oxidized low-density lipoprotein-induced inflammation and early atherosclerosis, Arteriosclerosis, thrombosis, and vascular biology 34(7) (2014) 1362–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Al-Yafeai Z, Yurdagul A Jr., Peretik JM, Alfaidi M, Murphy PA, Orr AW, Endothelial FN (Fibronectin) Deposition by alpha5beta1 Integrins Drives Atherogenic Inflammation, Arteriosclerosis, thrombosis, and vascular biology 38(11) (2018) 2601–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hynes RO, Integrins: bidirectional, allosteric signaling machines, Cell 110(6) (2002) 673–87. [DOI] [PubMed] [Google Scholar]
- [13].Rohwedder I, Montanez E, Beckmann K, Bengtsson E, Duner P, Nilsson J, Soehnlein O, Fassler R, Plasma fibronectin deficiency impedes atherosclerosis progression and fibrous cap formation, EMBO Mol Med 4(7) (2012) 564–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen J, Green J, Yurdagul A Jr., Albert P, McInnis MC, Orr AW, alphavbeta3 Integrins Mediate Flow-Induced NF-kappaB Activation, Proinflammatory Gene Expression, and Early Atherogenic Inflammation, The American journal of pathology 185(9) (2015) 2575–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shattil SJ, Kim C, Ginsberg MH, The final steps of integrin activation: the end game, Nature reviews. Molecular cell biology 11(4) (2010) 288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li J, Su Y, Xia W, Qin Y, Humphries MJ, Vestweber D, Cabanas C, Conformational equilibria and intrinsic affinities define integrin activation, 36(5) (2017) 629–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Salas A, Shimaoka M, Kogan AN, Harwood C, von Andrian UH, Springer TA, Rolling adhesion through an extended conformation of integrin alphaLbeta2 and relation to alpha I and beta I-like domain interaction, Immunity 20(4) (2004) 393–406. [DOI] [PubMed] [Google Scholar]
- [18].Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, Calderwood DA, Talin binding to integrin beta tails: a final common step in integrin activation, Science (New York, N.Y.) 302(5642) (2003) 103–6. [DOI] [PubMed] [Google Scholar]
- [19].Calderwood DA, Campbell ID, Critchley DR, Talins and kindlins: partners in integrin-mediated adhesion, Nature reviews. Molecular cell biology 14(8) (2013) 503–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wegener KL, Partridge AW, Han J, Pickford AR, Liddington RC, Ginsberg MH, Campbell ID, Structural basis of integrin activation by talin, Cell 128(1) (2007) 171–82. [DOI] [PubMed] [Google Scholar]
- [21].Yago T, Petrich BG, Zhang N, Liu Z, Shao B, Ginsberg MH, McEver RP, Blocking neutrophil integrin activation prevents ischemia-reperfusion injury, The Journal of experimental medicine 212(8) (2015) 1267–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Han J, Lim CJ, Watanabe N, Soriani A, Ratnikov B, Calderwood DA, Puzon-McLaughlin W, Lafuente EM, Boussiotis VA, Shattil SJ, Ginsberg MH, Reconstructing and deconstructing agonist-induced activation of integrin alphaIIbbeta3, Curr Biol 16(18) (2006) 1796–806. [DOI] [PubMed] [Google Scholar]
- [23].Stefanini L, Ye F, Snider AK, Sarabakhsh K, Piatt R, Paul DS, Bergmeier W, Petrich BG, A talin mutant that impairs talin-integrin binding in platelets decelerates alphaIIbbeta3 activation without pathological bleeding, Blood 123(17) (2014) 2722–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kopp PM, Bate N, Hansen TM, Brindle NP, Praekelt U, Debrand E, Coleman S, Mazzeo D, Goult BT, Gingras AR, Pritchard CA, Critchley DR, Monkley SJ, Studies on the morphology and spreading of human endothelial cells define key inter- and intramolecular interactions for talin1, European journal of cell biology 89(9) (2010) 661–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Haling JR, Monkley SJ, Critchley DR, Petrich BG, Talin-dependent integrin activation is required for fibrin clot retraction by platelets, Blood 117(5) (2011) 1719–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Finney AC, Stokes KY, Pattillo CB, Orr AW, Integrin signaling in atherosclerosis, Cellular and molecular life sciences: CMLS 74(12) (2017) 2263–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pickering JG, Chow LH, Li S, Rogers KA, Rocnik EF, Zhong R, Chan BM, alpha5beta1 integrin expression and luminal edge fibronectin matrix assembly by smooth muscle cells after arterial injury, The American journal of pathology 156(2) (2000) 453–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cai WJ, Li MB, Wu X, Wu S, Zhu W, Chen D, Luo M, Eitenmuller I, Kampmann A, Schaper J, Schaper W, Activation of the integrins alpha 5beta 1 and alpha v beta 3 and focal adhesion kinase (FAK) during arteriogenesis, Mol Cell Biochem 322(1–2) (2009) 161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yun S, Budatha M, Dahlman JE, Coon BG, Cameron RT, Langer R, Anderson DG, Baillie G, Schwartz MA, Interaction between integrin alpha5 and PDE4D regulates endothelial inflammatory signalling, Nat Cell Biol 18(10) (2016) 1043–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lv H, Wang H, Quan M, Zhang C, Fu Y, Zhang L, Lin C, Liu X, Yi X, Chen J, Wang X, Cheng T, Ai D, Kong W, Zhu Y, Cartilage Oligomeric Matrix Protein Fine-tunes Disturbed Flow-induced Endothelial Activation and Atherogenesis, Matrix biology: journal of the International Society for Matrix Biology (2020). [DOI] [PubMed] [Google Scholar]
- [31].Sun X, Fu Y, Gu M, Zhang L, Li D, Li H, Chien S, Shyy JY, Zhu Y, Activation of integrin alpha5 mediated by flow requires its translocation to membrane lipid rafts in vascular endothelial cells, Proceedings of the National Academy of Sciences of the United States of America 113(3) (2016) 769–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Orr AW, Ginsberg MH, Shattil SJ, Deckmyn H, Schwartz MA, Matrix-specific suppression of integrin activation in shear stress signaling, Molecular biology of the cell 17(11) (2006) 4686–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Monkley SJ, Kostourou V, Spence L, Petrich B, Coleman S, Ginsberg MH, Pritchard CA, Critchley DR, Endothelial cell talin1 is essential for embryonic angiogenesis, Dev Biol 349(2) (2011) 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pulous FE, Grimsley-Myers CM, Kansal S, Kowalczyk AP, Petrich BG, Talin-Dependent Integrin Activation Regulates VE-Cadherin Localization and Endothelial Cell Barrier Function, Circulation research 124(6) (2019) 891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhang X, Jiang G, Cai Y, Monkley SJ, Critchley DR, Sheetz MP, Talin depletion reveals independence of initial cell spreading from integrin activation and traction, Nat Cell Biol 10(9) (2008) 1062–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kuo JC, Han X, Yates JR 3rd, Waterman CM, Isolation of focal adhesion proteins for biochemical and proteomic analysis, Methods Mol Biol 757 (2012) 297–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yun S, Hu R, Schwaemmle ME, Scherer AN, Zhuang Z, Koleske AJ, Pallas DC, Schwartz MA, Integrin alpha5beta1 regulates PP2A complex assembly through PDE4D in atherosclerosis, The Journal of clinical investigation 130 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li B, He J, Lv H, Liu Y, Lv X, Zhang C, Zhu Y, Ai D, c-Abl regulates YAPY357 phosphorylation to activate endothelial atherogenic responses to disturbed flow, The Journal of clinical investigation 129(3) (2019) 1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Budatha M, Zhang J, Zhuang ZW, Yun S, Dahlman JE, Anderson DG, Schwartz MA, Inhibiting Integrin alpha5 Cytoplasmic Domain Signaling Reduces Atherosclerosis and Promotes Arteriogenesis, Journal of the American Heart Association 7(3) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA, A mechanosensory complex that mediates the endothelial cell response to fluid shear stress, Nature 437(7057) (2005) 426–31. [DOI] [PubMed] [Google Scholar]
- [41].Hahn C, Orr AW, Sanders JM, Jhaveri KA, Schwartz MA, The subendothelial extracellular matrix modulates JNK activation by flow, Circulation research 104(8) (2009) 995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Georgiadou M, Lilja J, Jacquemet G, AMPK negatively regulates tensin-dependent integrin activity, 216(4) (2017) 1107–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mravic M, Hu H, Lu Z, Bennett JS, Sanders CR, Orr AW, DeGrado WF, De novo designed transmembrane peptides activating the alpha5beta1 integrin, Protein engineering, design & selection: PEDS 31(5) (2018) 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Praekelt U, Kopp PM, Rehm K, Linder S, Bate N, Patel B, Debrand E, Manso AM, Ross RS, Conti F, Zhang MZ, Harris RC, Zent R, Critchley DR, Monkley SJ, New isoform-specific monoclonal antibodies reveal different sub-cellular localisations for talin1 and talin2, European journal of cell biology 91(3) (2012) 180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Liu J, Wang Y, Goh WI, Goh H, Baird MA, Ruehland S, Teo S, Bate N, Critchley DR, Davidson MW, Kanchanawong P, Talin determines the nanoscale architecture of focal adhesions, 112(35) (2015) E4864–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Debrand E, Conti FJ, Bate N, Spence L, Mazzeo D, Pritchard CA, Monkley SJ, Critchley DR, Mice carrying a complete deletion of the talin2 coding sequence are viable and fertile, Biochemical and Biophysical Research Communications 426(2–3) (2012) 190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sun Z, Costell M, Integrin activation by talin, kindlin and mechanical forces, 21(1) (2019) 25–31. [DOI] [PubMed] [Google Scholar]
- [48].Choi CK, Vicente-Manzanares M, Zareno J, Whitmore LA, Mogilner A, Horwitz AR, Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner, Nature cell biology 10(9) (2008) 1039–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Orr AW, Hahn C, Blackman BR, Schwartz MA, p21-activated kinase signaling regulates oxidant-dependent NF-kappa B activation by flow, Circulation research 103(6) (2008) 671–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wu C, Keivens VM, O’Toole TE, McDonald JA, Ginsberg MH, Integrin activation and cytoskeletal interaction are essential for the assembly of a fibronectin matrix, Cell 83(5) (1995) 715–24. [DOI] [PubMed] [Google Scholar]
- [51].Hakanpaa L, Kiss EA, Jacquemet G, Miinalainen I, Lerche M, Guzman C, Mervaala E, Eklund L, Ivaska J, Saharinen P, Targeting beta1-integrin inhibits vascular leakage in endotoxemia, Proceedings of the National Academy of Sciences of the United States of America 115(28) (2018) E6467–e6476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Owen GR, Meredith DO, ap Gwynn I, Richards RG, Focal adhesion quantification - a new assay of material biocompatibility? Review, Eur Cell Mater 9 (2005) 85–96; discussion 85–96. [DOI] [PubMed] [Google Scholar]
- [53].Balaban NQ, Schwartz US, Riveline D, Goichberg P, Tzur G, Sabanay I, Mahula D, Safran S, Beshadsky A, Addadi L, Geiger B, Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates, Nat. Cell Biol 3 (2001) 466–472. [DOI] [PubMed] [Google Scholar]
- [54].Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison D, Giddens D, Jo H, Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis, Am J Physiol Heart Circ Physiol 297(4) (2009) H1535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.