

Abstract
Hydrogen mass repartitioning (HMR) that permits time steps of all-atom molecular dynamics simulation up to 4 fs by increasing the mass of hydrogen atoms has been used in protein and phospholipid bilayers simulations to improve conformational sampling. Molecular simulation input generation via CHARMM-GUI now supports HMR for diverse simulation programs. In addition, considering ambiguous pH at the bacterial outer membrane surface, different protonation states, either −2e or −1e, of phosphate groups in lipopolysaccharides (LPS) are also supported in CHARMM-GUI LPS Modeler. To examine the robustness of HMR and the influence of protonation states of phosphate groups on LPS bilayer properties, eight different LPS-type all-atom systems with two phosphate protonation states are modeled and simulated utilizing both OpenMM 2-fs (standard) and 4-fs (HMR) schemes. Consistence in the conformational space sampled by standard and HMR simulations shows the reliability of HMR even in LPS, one of the most complex biomolecules. For systems with different protonation states, similar conformations are sampled with a PO41− or PO42− group, but different phosphate protonation states make slight impacts on lipid packing and conformational properties of LPS acyl chains. Systems with PO41− have slightly smaller area per lipid and thus slightly more ordered lipid A acyl chains compared to those with PO42−, due to more electrostatic repulsion between PO42− even with neutralizing Ca2+ ions. HMR and different protonation states of phosphates of LPS available in CHARMM-GUI are expected to be useful for further investigations of biological systems of diverse origin.
Graphical Abstract

INTRODUCTION
Conventional all-atom molecular dynamics (MD) simulations suffer from slow and incomplete coverage of phase space, which can limit their applicability to slowly evolving systems. Thus, there is a great need to identify and implement methods to improve conformational sampling.1–4 A major contributing factor to the slow coverage of phase space is the small integration time-steps (1 or 2 fs) that are required to conserve the total energy.5 Hydrogen mass repartitioning (HMR) is a simulation technique that enables all-atom MD to employ a 4-fs time-step by distributing a heavy atom mass to hydrogen atom(s) attached to the heavy atom. It was originally proposed by Feenstra et al. in 19995 and was shown to be practically useful for protein simulations by Hopkins et al. in 2015.6 Among different HMR schemes, the most popular one is to increase each hydrogen atom mass by a factor of 3 and subtract the total increased mass from the heavy atom bearing hydrogen atom(s). HMR has become popular in the MD simulation community with its recent application to phospholipid bilayers.7–9 CHARMM-GUI (http://www.charmm-gui.org)10–11 now supports this HMR scheme for NAMD12, GROMACS13, AMBER14, GENESIS15, LAMMPS16, Desmond17, and OpenMM18. In this work, we have tested the HMR scheme by performing MD simulations of various lipopolysaccharide (LPS) bilayer systems using OpenMM.
LPS molecules, which are found in the outer leaflet of the outer membranes (OM) of Gram-negative bacteria, are composed of lipid A, core oligosaccharide, and O-antigen polysaccharide, representing one of the most complex biological molecules.19–20 In LPS, phosphate groups are often attached to the glucosamine dimer of lipid A and L-glycero-D-manno-heptose (Hep) of a core oligosaccharide. At 25 °C, two pKa values of glucose-1-phosphate and glucose-4-phosphate are 1.10/6.13 and 0.84/5.67, respectively.21–22 Under physiological conditions, however, the protonation states, either −2e or −1e, of phosphate groups in LPS are difficult to be determined unambiguously due to unknown pH at the bacterial OM surface. Therefore, LPS Modeler23–24 and Membrane Builder25–27 in CHARMM-GUI now supports different protonation states of LPS phosphate groups. In this work, the influences of different protonation states on LPS bilayer properties have been extensively tested by performing MD simulations of various LPS bilayer systems. Note that this work was partially motivated by a recent simulation study by Amy et al. that explored the influence of the protonation states of lipid A glucosamine phosphate groups on the properties of bilayers composed of Salmonella enterica lipid A and Rc core (two Kdo (2-keto-3-deoxyoctulosonate), three Hep, and one Glc (D-glucose)).28 Among many recent progresses in MD simulations of various LPS systems29–31, their study is the first work (to the best of our knowledge) to investigate the different phosphate protonation states and LPS-ion interactions.
In this work, as shown in Table 1, eight different LPS-type systems were modeled and simulated: three from Pseudomonas aeruginosa (P. aeruginosa or Pa), two from Escherichia coli (E. coli or Ec), and three from Burkholderia cepacia (B. cepacia or Bc). These LPS types differ by lipid A, core, and O-antigen regions, such that extensive simulations of these complex LPS bilayers are valuable in order to examine the usage of HMR, as well as the influences of phosphate protonation states on LPS bilayer properties.
Table 1.
LPS system information in this study.†
| System name | LPS type |
|---|---|
| Pa-Kdo | P. aeruginosa lipid A + 2 Kdo sugars |
| Pa-G2 | P. aeruginosa lipid A + core 2 |
| Pa-O10 | P. aeruginosa lipid A + core 2 + 2 repeating units of O10 O-antigen |
| Ec-Kdo | E. coli lipid A + 2 Kdo sugars |
| Ec-K12 | E. coli lipid A + K12 core |
| Bc-T1 | B. cepacia lipid A (Type 1) + core A |
| Bc-T2 | B. cepacia lipid A (Type 2) + core A |
| Bc-T3 | B. cepacia lipid A (Type 3) + core A |
Unless specified explicitly, lipid A is type 1 defined in CHARMM-GUI LPS Modeler. LPS structures are shown in Figure S1.
METHODS
System details
To investigate the effects of LPS phosphate protonation states on the outer leaflet of bacterial OMs, symmetric bilayers with different LPS types (Table 1) were modeled and simulated. For P. aeruginosa systems, three different systems were built: Pa-Kdo with P. aeruginosa lipid A and two Kdo residues, Pa-G2 with lipid A and G2 core oligosaccharides, and Pa-O10 with lipid A, G2 core, and two repeating units of O-10 O-antigen (hereinafter, O10-antigen). All symmetric bilayers with 49 LPS molecules in each leaflet were constructed using Membrane Builder in CHARMM- GUI. Ca2+ ions were added to the LPS lipid A and core regions to neutralize each system and 150 mM KCl was added to the bulk region to mimic the bulk ionic solution. An initial area per lipid (APL) of 180 Å2 was used in the modeling of Pa-Kdo and Pa-G2 systems, while both 180 Å2 and 200 Å2 APL were used for the Pa-O10 system (Pa-O10180 and Pa-O10200) to check the simulation convergence depending on the initial membrane area (Figure 1). For each Pa system, both protonation states, either −2e or −1e, of phosphate groups in the glucosamine dimer of lipid A (Figure S2) were considered.
Figure 1.

Molecular graphics snapshots of systems Pa-Kdo, Pa-G2, and Pa-O10. Lipid A in each LPS is colored by atom: cyan for carbon, red for oxygen, blue for nitrogen, and tan for phosphorus. Kdo residues are colored yellow. The G2 core is colored yellow and orange, and two O10 polysaccharide repeating units are colored red.
E. coli and B. cepacia systems were prepared with the same procedure used for P. aeruginosa systems. A total of 49 LPS molecules were added in each leaflet to model symmetric systems. For the E. coli systems, two different systems were built: Ec-Kdo with E. coli lipid A and two Kdo residues and Ec-K12 with lipid A and K-12 core oligosaccharides. Like in the Pa system, two protonation states (−2e or −1e) of phosphate groups in the glucosamine dimer of lipid A were considered. For Ec-K12 systems, in addition to lipid A, different phosphate protonation states were also considered for Hep residues in the K12 core (Ec-K12a and Ec-K12b for Hep with −2e or −1e phosphate group, respectively). For B. cepacia systems, the same core oligosaccharide sequence was used with a variation in lipid A structures (Figure S3). Type 1 lipid A includes penta-acylated tails and two phosphate groups on each end of the glucosamine dimer. Both phosphate groups were assigned to two different protonation states, −2e or −1e, in different simulations. Type 2 lipid A has an additional 4-amino-4-deoxy-L-arabinose (L-Ara4N) connected to the first phosphate group. The second phosphate group was assigned to −2e or −1e. Type 3 lipid A has an additional L-Ara4N at both phosphate groups, making the net charge neutral. An initial APL of 200 Å was used for both E. coli and B. cepacia systems.
Simulation details
During simulations, together with a TIP3P water model,32–33 the CHARMM36 force field for LPS24, 34, lipids35, and carbohydrates36–40 was used to describe the system energetics. For each system, following the CHARMM-GUI standard membrane equilibration protocol, equilibrations were first conducted in NVT (constant particle number, volume, and temperature) ensemble with gradually decreasing restraints applied to the lipids and water molecules to ensure the gradual equilibration of the assembled system. After equilibration, 2-μs (Pa systems), 1.5-μs (Ec systems), or 2.5-μs (Bc systems) NPT (constant particle number, pressure, and temperature) production simulation with a time-step of 2 fs was conducted for each system. During simulations, all bonds containing hydrogen atoms were fixed using SHAKE.41 The van der Waals interactions were smoothly switched off over 10–12 Å by a force-switching function42 and the long-range electrostatic interactions were calculated using the particle-mesh Ewald method.43 For OpenMM simulations, Langevin dynamics was used for the temperature coupling with a collision frequency of 1 ps−1. A semi-isotropic Monte Carlo barostat method with a pressure coupling frequency of 100 steps was used to maintain the pressure.44–45 Unless specified explicitly, for all simulations, the temperature was maintained at 310.15 K and the pressure was set to 1 bar. For HMR simulations, all simulation details were the same as the standard ones except for 4-fs time-step used in production runs. Three independent replicas with different random seed numbers were simulated to improve sampling and to check the simulation convergence.
RESULTS AND DISCUSSION
The simulation results are presented in the following order: (1) results of Pa, Ec, and Bc systems with different phosphate protonation states using OpenMM 2-fs time-step; (2) results of Pa systems using OpenMM 4-fs time-step with HMR.
Influences of different phosphate protonation states on conformational preference
For each Pa system, comparisons of pairwise root-mean-square deviation (RMSD of the entire LPS including the acyl chain after superposition) calculated between 1.0 – 1.5 μs and 1.5 – 2.0 μs are shown in Figure S4. The RMSD distributions are almost identical between the two time periods, indicating simulations reach convergence and similar conformational ensembles are sampled during last 1-μs simulations with either PO41− or PO42− groups. As expected, the Pa-O10 systems are more dynamic and flexible with broader RMSD distributions compared to Pa-G2 and Pa-Kdo systems due to its large size. In the following, unless specified explicitly, the last 500-ns trajectories are used for analyses.
To investigate the influence of phosphate protonation states on the conformational variations, pairwise RMSD distributions with PO41− and PO42− groups are shown in Figure 2A. The well overlapped distributions for each system indicate that similar conformational ensembles are sampled and no significant structural variations are observed between the two protonation states. The averaged Z-lengths between phosphate groups of lipid A and the first residue of each O10-antigen repeating unit are shown in Figure 2C; there are two O10 repeating units in Pa-O10 systems. In both protonation states, conformations with similar Z-lengths (about 26 ± 2 Å and 34 ± 4 Å, respectively) are sampled during simulations, which is consistent with the result of pairwise RMSD distributions. Thus, although different protonation states are used for lipid A, each Pa system adopts similar conformation with PO41− or PO42−. For Ec and Bc systems, similar pairwise RMSD distributions are also observed between two protonation states (Figure S5), consistent with the conclusion for Pa systems that similar conformations are sampled with PO41− or PO42− groups.
Figure 2.
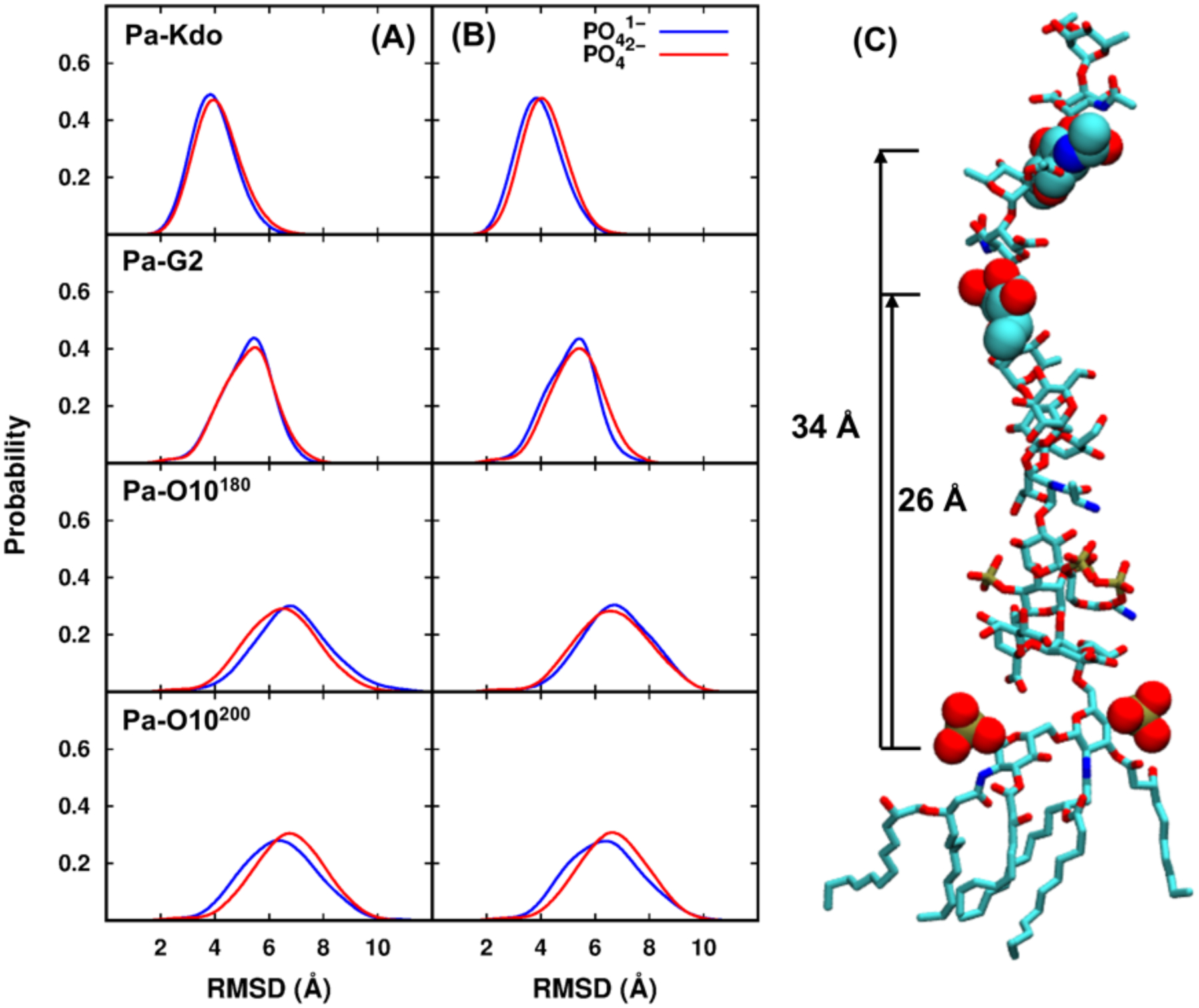
Comparisons of pairwise RMSD distributions between two phosphate protonation states in (A) standard and (B) HMR simulations for each Pa system. Pa-O10180 and Pa-O10200 denote the systems with the initial areas per lipid of 180 Å2 and 200 Å2, respectively. (C) Averaged Z-lengths between the centers of mass of phosphate groups of lipid A and the first residue of each O10-antigen repeating unit. The structure is a snapshot from Pa-O10 with PO41−.
Different protonation states of lipid A phosphate groups could also alter the distributions of divalent cations that maintain the stability and integrity of LPS membrane systems. Figure S6A and Figure S7 show the density distributions of Ca2+ ions along the Z axis (i.e., the membrane normal with the bilayer center at Z = 0) for Pa, Ec, and Bc systems. Ca2+ ions are dominantly occupied in the lipid headgroup and core regions. For Pa-O10 systems, some Ca2+ ions moved slightly toward the O10-antigen region indicated by the small peak at Z = 46 Å. As expected, when phosphate group charges are −2e, more Ca2+ ions are bound to the lipid A phosphate regions (around Z = 17 Å) for each Pa system simply because there are more neutralizing Ca2+ ions. Comparison between Ec-K12a and Ec-K12b systems also indicates more Ca2+ ions bound in the core region due to the −2e phosphate charge for HEP residues in Ec-K12a.
Influences of different phosphate protonation states on bilayer properties
Although similar conformations are sampled with PO41− and PO42− groups for each system, different phosphate protonation states could make impacts on lipid packing and conformational properties of LPS acyl chains. Figure S8 shows the time series of averaged area per lipid (APL) every 100 ns and its corresponding standard errors for each Pa system in both protonation states. Both averaged APL and its standard errors (Table S1) generally reach a plateau after about 1-μs simulation. It is noticeable that systems with PO41− have slightly smaller APL compared to those with PO42−, indicating the lipid A acyl chains are slightly more ordered and straight with PO41−. In addition, for systems Pa-O10180 and Pa-O10200 started with two different initial APL values, there still exists slight differences for the averaged APL (even after 2-μs simulation), which results from the slow lateral relaxation of LPS. The hydrophobic thicknesses calculated using carbons in the purple circles in Figure S2 are shown in Figure 3A and Table S1. Expected from the APL results, the hydrophobic thickness of each Pa system with PO42− is slightly thinner than the corresponding Pa system with PO41−.
Figure 3.
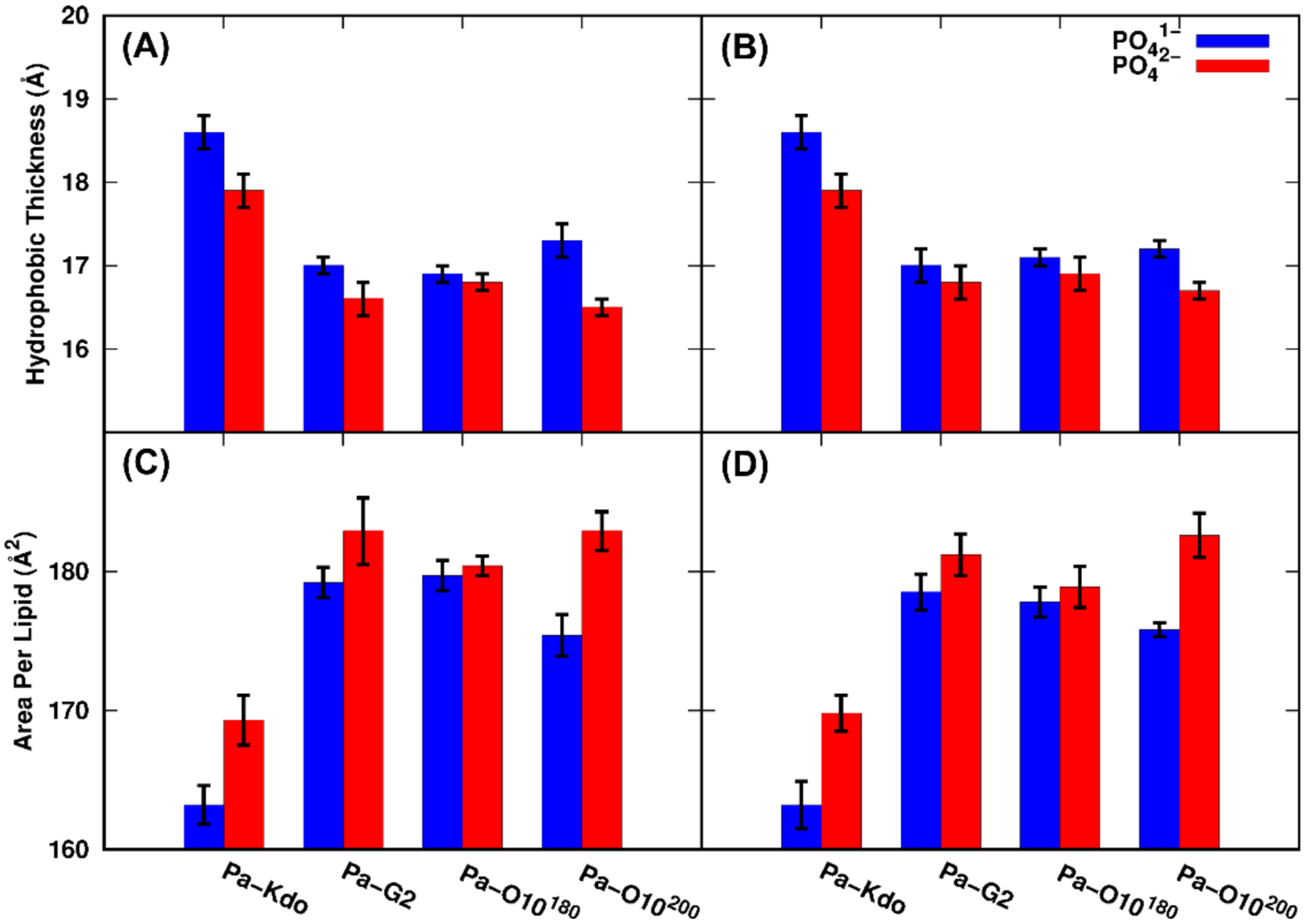
Averaged hydrophobic thickness and area per lipid for each Pa system with two phosphate protonation states: (A and C) standard and (B and D) HMR simulations.
For Ec systems, averaged APL along with simulation time generally reach convergence after 1-μs as shown in Figure S9. However, there still exists significant differences for simulations of Bc systems among the three replicas, which clearly manifests the slow lateral relaxation of these LPS types. In this work, for Bc systems, temperature was increased to 400 K at 500 ns and then cooled down to 310 K at 1.5 μs to obtain better convergence as shown in Figure S9. The last 500-ns trajectories at 310 K were used for analyzes. Averaged APL and its standard error are shown in Table S2. Except for Bc-T2 systems, averaged APL with PO41− groups are all smaller than those with PO42−, and hydrophobic thickness is correspondingly larger, which is consistent with the Pa results. For Bc-T2 systems, there is only one phosphate group having −2e or −1e (Figure S3), and the APL difference is within the errors. For Ec-K12 systems, it is also found that the APL becomes larger when the phosphate group of Hep is −2e, regardless of the lipid A protonation state. Results from analysis of APL and hydrophobic thickness indicate that the protonation state of phosphate groups in lipid A could affect the lipid packing and conformations of acyl chains of lipid A.
The order parameters (SCD) of lipid acyl chains are also used to provide information regarding the overall order of the membrane. SCD= |<3 × cos2(θCH) − 1>⁄2|, where θCH is the angle between a C-H bond vector and the Z axis, and the bracket represents the time and ensemble average. Figure 4A shows the calculated SCD of lipid A acyl chain 3 (in Figure S2) in each Pa system. Overall, the acyl chain is more ordered with PO41− compared to that with PO42−, indicated by larger SCD values. The lipid A analysis again agrees well with the hydrophobic thickness and APL results, indicating that LPS bilayers with PO42− in lipid A are slightly more dynamic and flexible. SCD of acyl chain 2 for Ec and Bc systems are also calculated and plotted in Figure S10. The more ordered acyl chains with PO41− groups are detected for Ec and Bc systems, which is consistent with the conclusion from the Pa systems.
Figure 4.
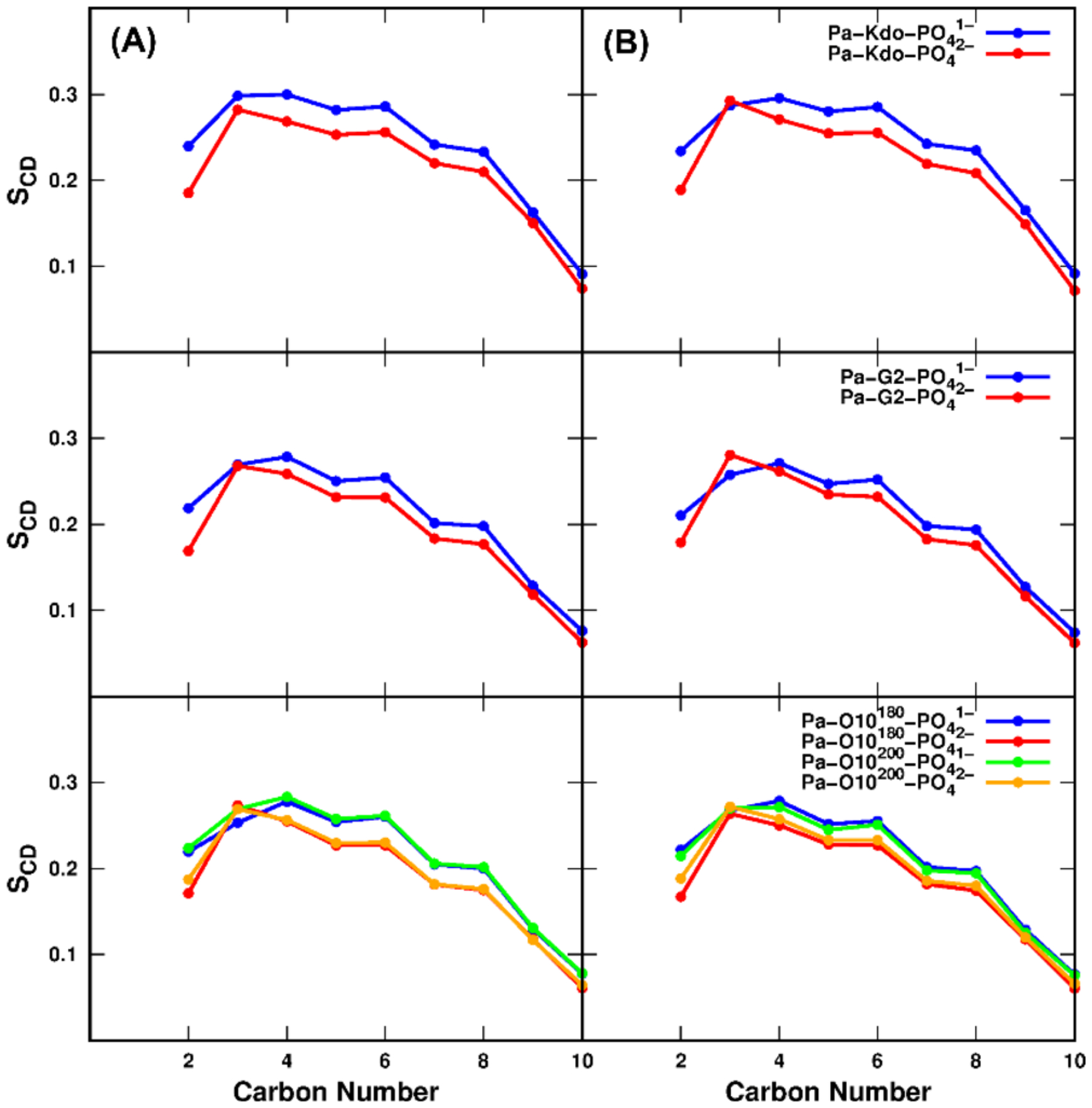
Calculated chain order parameters for acyl chain 3 of lipid A for each Pa system with two phosphate protonation states: (A) standard and (B) HMR simulations.
The protonation state of lipid A phosphate groups also affects the electrostatic interactions among LPS as well as between LPS and divalent cations. Figure 5A shows the sum of the per-LPS average number of inter-lipid A hydrogen bonds and the per-LPS average salt bridges between Ca2+ and phosphate groups for Pa systems (see Figure S11 for Ec and Bc systems). For Pa systems with PO41−, more inter-lipid A hydrogen bonds are detected than those with PO42− due to more hydrogen bond donors in PO41− (Figure S12A). However, the overall stabilization of the leaflet integrity is maintained by the electrostatic interactions formed by salt bridges between Ca2+ ions and phosphate groups of lipid A, indicated by much larger number of salt bridges than that of inter-lipid A hydrogen bonds (Figure S12C). While there are more salt bridges for the PO42− cases due to more neutralizing Ca2+ ions, stronger electrostatic repulsion between PO42− groups appears to make the APL of LPS with PO42− larger than that with PO41−.
Figure 5.
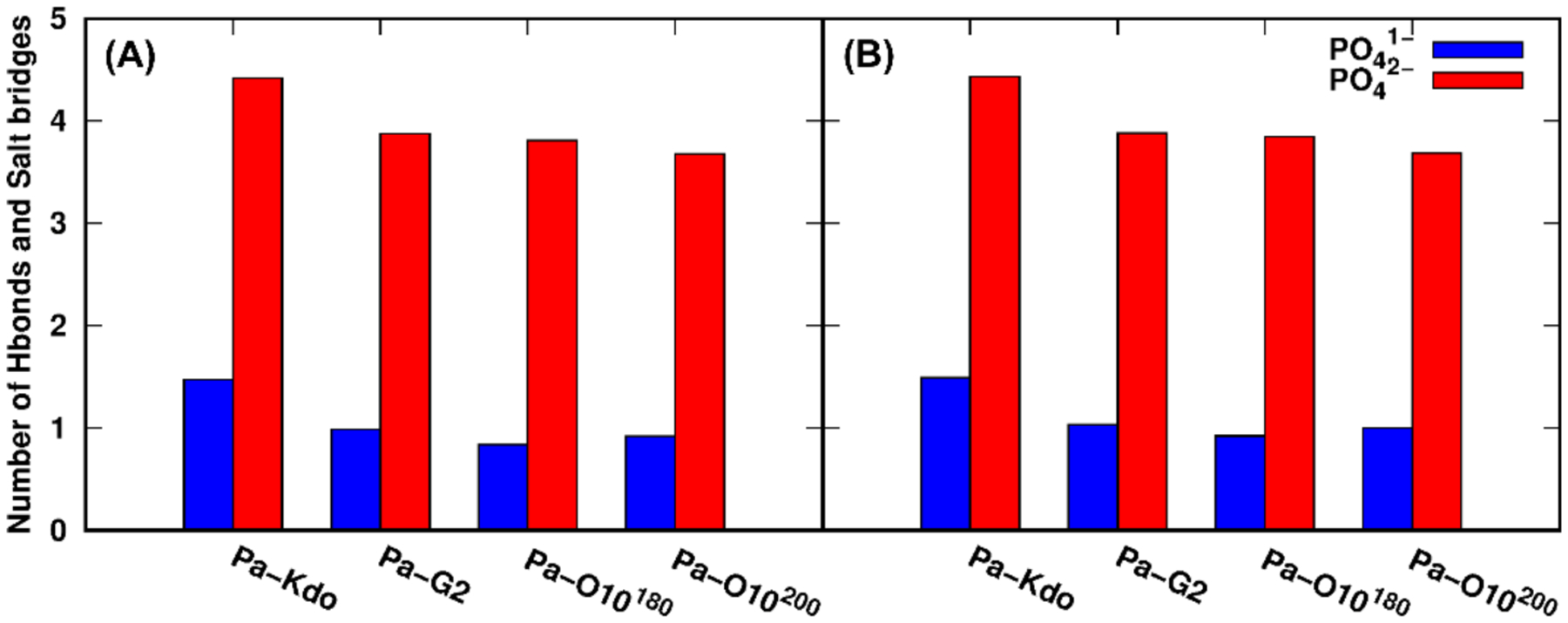
Sum of the per-LPS average number of inter-lipid A hydrogen bonds and the per-LPS average salt bridges between Ca2+ and phosphate groups for two phosphate protonation states: (A) standard and (B) HMR simulations. A hydrogen bond is counted when the distance between the donor and acceptor is less than 3 Å and the angle is larger than 120°. A salt bridge is counted when the distance between Ca2+ and any atom on phosphate groups of lipid A is less than 4 Å.
Axial relaxation time constants, which are related to both lipid structure and the membrane environment, are measured by the second-rank reorientational correlation function, C2(t), for specific atoms.
where P2 is the second Legendre polynomial and is a heavy atom vector. In this work, the vectors between the last carbons of each acyl chain were selected to estimate the slow relaxation time: C1.12 (12th carbon from the carbonyl carbon in chain 1 in Figure S2) – C2.9, C3.10–C4.10, C4.12– C5.9, and C1.10–C6.10. Three exponential fits were used to determine time constants for each cross-acyl chain:
The time constants represent the fast and slow relaxation times associated with the lipid’s fast isomerization and slow wobbling motions, respectively.46–48 As shown in Table 2, for LPS membrane systems, the relaxation time is much longer than bilayers comprised of dipalmitoylphosphatidylcholine (DPPC) lipids, for which the slow relaxation time is ~10 ns and the full rotational motion is an order of magnitude longer.48 In this study, the slow relaxation time of adjacent chains is ~300 ns, and for the long-range vector (C1.10–C6.10), which represents the overall lipid rotational-like motion, the correlation is still very high during last 1-μs simulations (Figure S13), i.e., longer relaxation time than adjacent chain vectors.
Table 2.
Average of three-exponential fitted relaxation time (in ns) of the cross-chain correlation function in standard and HMR simulations.
| cross chain | OpenMM 2-fs | OpenMM HMR | ||||
|---|---|---|---|---|---|---|
| τ1 | τ2 | τ3 | τ1 | τ2 | τ3 | |
| C1.12-C2.9 | 0.36 ± 0.04 | 11.60 ± 2.23 | 328.7 ± 116.3 | 0.37 ± 0.02 | 11.23 ± 1.28 | 322.9 ± 50.36 |
| C3.10-C4.10 | 0.38 ± 0.03 | 13.43 ± 1.01 | 294.0 ± 36.0 | 0.39 ± 0.03 | 14.15 ± 1.75 | 362.5 ± 36.78 |
| C4.12-C5.9 | 0.38 ± 0.06 | 11.41 ± 2.38 | 335.2 ± 123.9 | 0.37 ± 0.06 | 10.56 ± 2.43 | 271.5 ± 64.92 |
| C1.10-C6.10 | 0.59 ± 0.05 | 15.69 ± 1.54 | 398.0 ± 127.0 | 0.65 ± 0.07 | 18.07 ± 3.32 | 479.5 ± 194.2 |
Comparison between standard and HMR simulations for Pa systems
The HMR technique permits all-atom MD simulation time steps of up to 4 fs and thus improves simulation efficiency with reasonable stability. Since HMR is now available in CHARMM-GUI and can be used for various systems, the methodology was herein employed to perform simulations and examine its robustness by investigating the conformational properties for Pa systems. Pairwise RMSD distributions for two protonation states are shown in Figure 2B. Compared to standard 2-fs simulations, similar RMSD distributions with comparable probabilities are observed for each system. The averaged Z-lengths between phosphate groups in lipid A and the first residue of each O10-antigen repeating unit are also close to that in the standard simulations (~26 ± 2 Å and 34 ± 4 Å, respectively) for both protonation states, indicating that similar conformations are sampled under HMR and standard OpenMM simulations. In addition, consistent with standard simulations, the well overlapped pairwise-RMSD distributions with PO41− or PO42− indicate similar conformational space of LPS sampled under two protonation states. Density distributions of Ca2+ ions are shown in Figure S6B. Similar pairwise RMSD and Ca2+ ions distributions between HMR and standard simulations prove the usefulness of HMR.
Averaged APL and its corresponding standard errors under two protonation states from HMR simulations are summarized in Table S1. The APL under HMR is very close to those under standard simulations, and the difference between two simulations falls within its standard errors, indicating that a similar conformational ensemble is sampled under both HMR and standard simulations. For systems Pa-O10180 and Pa-O10200 there also exists slight differences for the averaged APL. Hydrophobic thickness, number of hydrogen bonds and salt bridges (Figure 3B and Figure 5B) are also consistent with those from the standard simulations. Relaxation times were also determined for HMR simulations to examine if the HMR method changes the lipid dynamics compared to standard simulations. Table 2 indicates that HMR results are comparable with standard ones, indicating the reliability of HMR simulations.
CONCLUSIONS
The HMR and different protonation states of phosphates in LPS are now supported in CHARMM-GUI for various biomolecular MD simulations. In this work, eight different LPS-type systems were modeled and simulated utilizing standard OpenMM 2-fs and HMR 4-fs schemes to examine the usage of HMR, as well as the influences of phosphate protonation states on LPS bilayer properties. Comparisons of pairwise RMSD distributions, area per lipid, hydrophobic thickness, chain order parameters, and number of inter-lipid hydrogen bonds between standard and HMR simulations reveal the robustness of HMR, supporting that HMR can be used for complex biomolecular simulations including LPS. For systems with two phosphate protonation states, similar conformations are sampled with either a PO41− or PO42− group, indicated by pairwise RMSD distributions. However, slight impacts on lipid packing and conformational properties of LPS acyl chains are also observed with Ca2+ neutralizing ions. Systems with PO41− group(s) have slightly smaller area per lipid and thus slightly more ordered lipid A acyl chains compared to those with PO42−. More hydrogen bonds between Lipid A molecules are detected in the PO41− case than the PO42− case due to more hydrogen bond donors in PO41– groups. However, the stabilization of the leaflet integrity is dominated by the electrostatic interactions between Ca2+ ions and phosphate groups of lipid A, supported by the much larger number of salt bridges than of inter-lipid A hydrogen bonds. Axial relaxation time constants were determined by fitting the second-rank reorientational correlation functions of cross acyl chains; for LPS systems, the slow relaxation time (~300 ns) is much longer than bilayers comprised of phospholipids (~10 ns), indicating the slow lateral relaxation of LPS systems that is also manifested by slight APL differences even after 2-μs simulations of the systems with two different initial APL values. The availability of HMR and different protonation states of phosphates of LPS in CHARMM-GUI are expected to be useful for MD studies of different biomolecular systems.
Supplementary Material
Figure S1: Schematic structures of Pa-O10, Ec-K12, and Bc-T1/T2/T3 LPS. Figure S2–S3: Chemical structures of P.aeruginosa lipid A and three types of B.cepacia lipid A. Figure S4: Comparisons of pairwise RMSD distributions (of the entire LPS) with two phosphate protonation states between 1.0 – 1.5 μs and 1.5 – 2.0 μs standard simulations for each Pa system. Figure S5: Comparisons of pairwise RMSD distributions (of the entire LPS) between two phosphate protonation states for each Ec and Bc systems. Figure S6–7: Comparisons of distributions of Ca2+ ions along the Z-axis (i.e., the membrane normal) between two phosphate protonation states for each Pa systems (in standard and HMR simulations) and for each Ec and Bc systems. Figure S8–9: Time series of averaged APL every 100 ns for each Pa, Ec, and Bc systems in both protonation states and its corresponding standard errors. Figure S10: Calculated chain order parameters for acyl chain 2 of lipid A for each Ec and Bc system with two phosphate protonation states. Figure S11: Sum of the per-LPS average number of inter-lipid A hydrogen bonds and the per-LPS average salt bridges between Ca2+ and phosphate groups for each Ec and Bc system with two phosphate protonation states. Figure S12: Per-LPS average number of inter-lipid A hydrogen bonds and salt bridges between Ca2+ and any atom on phosphate groups of lipid A for each Pa system (standard and HMR simulations). Figure S13: Correlation function C2(t) for the cross acyl chains in Pa-O10 system. Table S1: Averaged area per lipid and hydrophobic thickness with standard errors for each Pa system in standard and HMR simulations with two phosphate protonation states. Table S2: Averaged area per lipid and hydrophobic thickness with standard errors for each Ec/Bc system with two phosphate protonation states.
ACKNOWLEDGEMENT
This work was supported in part by grants from the NSF MCB-1810695, NSF DBI-1660380, NIH GM138472, XSEDE MCB070009, Friedrich Wilhelm Bessel Research Award from the Humboldt foundation (to WI), the Swedish Research Council 2017-03703 (GW), KIAS individual grant (CG080501) (SK), the National Natural Science Foundation of China (62072296) (YG), NSF CHE-2003912 and MCB-1951425 (to JBK), and Engineering and Physical Sciences Research Council, EP/R029407/1 (to SK).
References
- 1.Saiz L; Klein ML, Computer Simulation Studies of Model Biological Membranes. Acc. Chem. Res 2002, 35, 482–489. [DOI] [PubMed] [Google Scholar]
- 2.Marrink SJ; Risselada HJ; Yefimov S; Tieleman DP; de Vries AH, The Martini Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y; Markwick PRL; de Oliveira CAF; McCammon JA, Enhanced Lipid Diffusion and Mixing in Accelerated Molecular Dynamics. Biophys. J 2012, 102, 3199–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shearer J; Marzinek JK; Bond PJ; Khalid S, Molecular Dynamics Simulations of Bacterial Outer Membrane Lipid Extraction: Adequate Sampling? J. Chem. Phys 2020, 153, 044122. [DOI] [PubMed] [Google Scholar]
- 5.Feenstra KA; Hess B; Berendsen HJC, Improving Efficiency of Large Time-Scale Molecular Dynamics Simulations of Hydrogen-Rich Systems. J. Comput. Chem 1999, 20, 786–798. [DOI] [PubMed] [Google Scholar]
- 6.Hopkins CW; Le Grand S; Walker RC; Roitberg AE, Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J. Chem. Theory Comput 2015, 11, 1864–1874. [DOI] [PubMed] [Google Scholar]
- 7.Olesen K; Awasthi N; Bruhn DS; Pezeshkian W; Khandelia H, Faster Simulations with a 5 Fs Time Step for Lipids in the Charmm Force Field. J. Chem. Theory Comput 2018, 14, 3342–3350. [DOI] [PubMed] [Google Scholar]
- 8.Balusek C; Hwang H; Lau CH; Lundquist K; Hazel A; Pavlova A; Lynch DL; Reggio PH; Wang Y; Gumbart JC, Accelerating Membrane Simulations with Hydrogen Mass Repartitioning. J. Chem. Theory Comput 2019, 15, 4673–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee J; Hitzenberger M; Rieger M; Kern NR; Zacharias M; Im W, Charmm-Gui Supports the Amber Force Fields. J. Chem. Phys 2020, 153. [DOI] [PubMed] [Google Scholar]
- 10.Jo S; Kim T; Iyer VG; Im W, Charmm-Gui: A Web-Based Graphical User Interface for Charmm. 2008, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- 11.Lee J; Cheng X; Swails JM; Yeom MS; Eastman PK; Lemkul JA; Wei S; Buckner J; Jeong JC; Qi YF; Jo S; Pande VS; Case DA; Brooks CL; MacKerell AD; Klauda JB; Im W, Charmm-Gui Input Generator for Namd, Gromacs, Amber, Openmm, and Charmm/Openmm Simulations Using the Charmm36 Additive Force Field. J. Chem. Theory Comput 2016, 12, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K, Scalable Molecular Dynamics with Namd. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abraham MJ; Murtola T; Schulz R; Páll S; Smith JC; Hess B; Lindahl E, Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar]
- 14.Case DA; Cheatham TE; Darden T; Gohlke H; Luo R; Merz KM; Onufriev A; Simmerling C; Wang B; Woods RJ, The Amber Biomolecular Simulation Programs. J. Comput. Chem 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung J; Mori T; Kobayashi C; Matsunaga Y; Yoda T; Feig M; Sugita Y, Genesis: A Hybrid-Parallel and Multi-Scale Molecular Dynamics Simulator with Enhanced Sampling Algorithms for Biomolecular and Cellular Simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci 2015, 5, 310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plimpton S, Fast Parallel Algorithms for Short-Range Molecular-Dynamics. J. Comput. Phys 1995, 117, 1–19. [Google Scholar]
- 17.Bowers KJ; Chow DE; Xu H; Dror RO; Eastwood MP; Gregersen BA; Klepeis JL; Kolossvary I; Moraes MA; Sacerdoti FD; Salmon JK; Shan Y; Shaw DE In Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters, SC ‘06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, 11–17 Nov.; 2006; pp 43–43. [Google Scholar]
- 18.Eastman P; Swails J; Chodera JD; McGibbon RT; Zhao YT; Beauchamp KA; Wang LP; Simmonett AC; Harrigan MP; Stern CD; Wiewiora RP; Brooks BR; Pande VS, Openmm 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. Plos Comput. Biol 2017, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simpson BW; Trent MS, Pushing the Envelope: Lps Modifications and Their Consequences. Nat. Rev. Microbiol 2019, 17, 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stahle J; Widmalm G, Lipopolysaccharides of Gram-Negative Bacteria: Biosynthesis and Structural Aspects. Trends Glycosci. Glyc 2019, 31, E159–E171. [Google Scholar]
- 21.Cori CF; Colowick SP; Cori GT, The Isolation and Synthesis of Glucose-1-Phosphoric Acid. J. Biol. Chem 1937, 121, 465–477. [Google Scholar]
- 22.Pontis HG, Preface In Methods for Analysis of Carbohydrate Metabolism in Photosynthetic Organisms, Pontis HG, Ed. Academic Press: Boston, 2017. [Google Scholar]
- 23.Lee J; Patel DS; Stahle J; Park SJ; Kern NR; Kim S; Lee J; Cheng X; Valvano MA; Holst O; Knirel YA; Qi YF; Jo S; Klauda JB; Widmalm G; Im W, Charmm-Gui Membrane Builder for Complex Biological Membrane Simulations with Glycolipids and Lipoglycans. J. Chem. Theory Comput 2019, 15, 775–786. [DOI] [PubMed] [Google Scholar]
- 24.Jo S; Wu EL; Stuhlsatz D; Klauda JB; MacKerell AD; Widmalm G; Im W, Lipopolysaccharide Membrane Building and Simulation In Glycoinformatics, Lütteke T; Frank M, Eds. Springer New York: New York, NY, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jo S; Kim T; Im W, Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations. Plos One 2007, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jo S; Lim JB; Klauda JB; Im W, Charmm-Gui Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes. Biophys. J 2009, 97, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu EL; Cheng X; Jo S; Rui H; Song KC; Davila-Contreras EM; Qi YF; Lee JM; Monje-Galvan V; Venable RM; Klauda JB; Im W, Charmm-Gui Membrane Builder toward Realistic Biological Membrane Simulations. J. Comput. Chem 2014, 35, 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rice A; Rooney MT; Greenwood AI; Cotten ML; Wereszczynski J,Lipopolysaccharide Simulations Are Sensitive to Phosphate Charge and Ion Parameterization. J. Chem. Theory Comput 2020, 16, 1806–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Im W; Khalid S, Molecular Simulations of Gram-Negative Bacterial Membranes Come of Age. In Annual Review of Physical Chemistry, Vol 71, Johnson MA; Martinez TJ, Eds. 2020; Vol. 71, pp 171–188. [DOI] [PubMed] [Google Scholar]
- 30.Messias A; Santos DES; Pontes FJS; Lima FS; Soares TA, Out of Sight, out of Mind: The Effect of the Equilibration Protocol on the Structural Ensembles of Charged Glycolipid Bilayers. Molecules 2020, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel DS; Qi Y; Im W, Modeling and Simulation of Bacterial Outer Membranes and Interactions with Membrane Proteins. Curr. Opin. Struc. Biol 2017, 43, 131–140. [DOI] [PubMed] [Google Scholar]
- 32.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- 33.Neria E; Fischer S; Karplus M, Simulation of Activation Free Energies in Molecular Systems. J. Chem. Phys 1996, 105, 1902–1921. [Google Scholar]
- 34.Kim S; Patel DS; Park S; Slusky J; Klauda JB; Widmalm G; Im W, Bilayer Properties of Lipid a from Various Gram-Negative Bacteria. Biophys. J 2016, 111, 1750–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klauda JB; Venable RM; Freites JA; O’Connor JW; Tobias DJ; Mondragon-Ramirez C; Vorobyov I; MacKerell AD; Pastor RW, Update of the Charmm All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guvench O; Greene SN; Kamath G; Brady JW; Venable RM; Pastor RW; Mackerell AD, Additive Empirical Force Field for Hexopyranose Monosaccharides. J. Comput. Chem 2008, 29, 2543–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guvench O; Hatcher E; Venable RM; Pastor RW; MacKerell AD, Charmm Additive All-Atom Force Field for Glycosidic Linkages between Hexopyranoses. J. Chem. Theory Comput 2009, 5, 2353–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hatcher E; Guvench O; MacKerell AD, Charmm Additive All-Atom Force Field for Aldopentofuranoses, Methyl-Aldopentofuranosides, and Fructofuranose. J. Phys. Chem. B 2009, 113, 12466–12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guvench O; Mallajosyula SS; Raman EP; Hatcher E; Vanommeslaeghe K; Foster TJ; Jamison FW; MacKerell AD, Charmm Additive All-Atom Force Field for Carbohydrate Derivatives and Its Utility in Polysaccharide and Carbohydrate-Protein Modeling. J. Chem. Theory Comput 2011, 7, 3162–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park S-J; Lee J; Patel DS; Ma H; Lee HS; Jo S; Im W, Glycan Reader Is Improved to Recognize Most Sugar Types and Chemical Modifications in the Protein Data Bank. Bioinformatics 2017, 33, 3051–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryckaert J-P; Ciccotti G; Berendsen HJC, Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of N-Alkanes. J. Comput. Phys 1977, 23, 327–341. [Google Scholar]
- 42.Steinbach PJ; Brooks BR, New Spherical-Cutoff Methods for Long-Range Forces in Macromolecular Simulation. J. Comput. Chem 1994, 15, 667–683. [Google Scholar]
- 43.Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG, A Smooth Particle Mesh Ewald Method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- 44.Chow KH; Ferguson DM, Isothermal Isobaric Molecular-Dynamics Simulations with Monte-Carlo Volume Sampling. Comput. Phys. Commun 1995, 91, 283–289. [Google Scholar]
- 45.Aqvist J; Wennerstrom P; Nervall M; Bjelic S; Brandsdal BO, Molecular Dynamics Simulations of Water and Biomolecules with a Monte Carlo Constant Pressure Algorithm. Chem. Phys. Lett 2004, 384, 288–294. [Google Scholar]
- 46.Klauda JB; Eldho NV; Gawrisch K; Brooks BR; Pastor RW, Collective and Noncollective Models of Nmr Relaxation in Lipid Vesicles and Multilayers. J. Phys. Chem. B 2008, 112, 5924–5929. [DOI] [PubMed] [Google Scholar]
- 47.Klauda JB; Roberts MF; Redfield AG; Brooks BR; Pastor RW, Rotation of Lipids in Membranes: Molecular Dynamics Simulation, 31P Spin-Lattice Relaxation, and Rigid-Body Dynamics. Biophys. J 2008, 94, 3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Monje-Galvan V; Klauda JB, Modeling Yeast Organelle Membranes and How Lipid Diversity Influences Bilayer Properties. Biochemistry 2015, 54, 6852–6861. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Schematic structures of Pa-O10, Ec-K12, and Bc-T1/T2/T3 LPS. Figure S2–S3: Chemical structures of P.aeruginosa lipid A and three types of B.cepacia lipid A. Figure S4: Comparisons of pairwise RMSD distributions (of the entire LPS) with two phosphate protonation states between 1.0 – 1.5 μs and 1.5 – 2.0 μs standard simulations for each Pa system. Figure S5: Comparisons of pairwise RMSD distributions (of the entire LPS) between two phosphate protonation states for each Ec and Bc systems. Figure S6–7: Comparisons of distributions of Ca2+ ions along the Z-axis (i.e., the membrane normal) between two phosphate protonation states for each Pa systems (in standard and HMR simulations) and for each Ec and Bc systems. Figure S8–9: Time series of averaged APL every 100 ns for each Pa, Ec, and Bc systems in both protonation states and its corresponding standard errors. Figure S10: Calculated chain order parameters for acyl chain 2 of lipid A for each Ec and Bc system with two phosphate protonation states. Figure S11: Sum of the per-LPS average number of inter-lipid A hydrogen bonds and the per-LPS average salt bridges between Ca2+ and phosphate groups for each Ec and Bc system with two phosphate protonation states. Figure S12: Per-LPS average number of inter-lipid A hydrogen bonds and salt bridges between Ca2+ and any atom on phosphate groups of lipid A for each Pa system (standard and HMR simulations). Figure S13: Correlation function C2(t) for the cross acyl chains in Pa-O10 system. Table S1: Averaged area per lipid and hydrophobic thickness with standard errors for each Pa system in standard and HMR simulations with two phosphate protonation states. Table S2: Averaged area per lipid and hydrophobic thickness with standard errors for each Ec/Bc system with two phosphate protonation states.