

Abstract
Electrochemical aptamer-based (E-AB) sensors offer a powerful and general means for analyte detection in complex samples for various applications. Paper-based E-AB sensors could enable portable, low-cost, and rapid detection of a broad range of targets, but it has proven challenging to fabricate suitable three-electrode systems on paper. Here, we demonstrate a simple, economic, and environmentally-friendly strategy for fabricating aptamer-modified paper electrochemical devices (PEDs) via ambient vacuum filtration. The electrode material, shape, size, and thickness of the three-electrode PED system can be fully customized. We developed aptamer-modified PEDs that enable sensitive and specific detection of small molecules in minimally processed biosamples. The sensitivity and stability of the PEDs is comparable to E-AB sensors based on commercial gold electrodes. We believe our fabrication strategy can lead to the development of high performance PEDs that enable on-site detection of a variety of analytes, simplifying the development of portable sensors for real-world applications.
Keywords: Biosensors, paper electrochemical devices, small molecules, nanomaterials, aptamers
Graphical Abstract
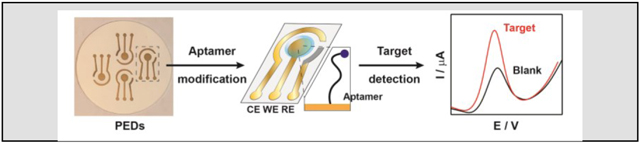
A simple, economic, and environmentally-friendly strategy is developed to fabricate paper electrochemical devices (PEDs) with three-electrode systems that can be readily modified with aptamers to achieve a sensitive and specific detection of small molecules in minimally processed biosamples. Aptamer-modified PEDs serve as a general sensing platform for the point-of-care detection of analytes like biomolecules and pharmaceuticals.
Introduction
Electrochemical sensors can potentially provide a facile means for achieving rapid and sensitive detection in applications such as diagnostics, environmental monitoring, forensics, and biomedical research.[1,2] For example, ion-selective electrodes[3] and the personal glucose meter[4] are widely used in the context of on-site or point-of-care detection. However, the number of commercially available portable electrochemical sensors remains relatively low overall. This is partly because only a handful of analytes can be electrochemically detected directly or via enzymatic transformation. Electrochemical aptamer-based (E-AB) sensors[5] offer a promising platform for extending the utility of electrochemical detection.[6] The E-AB sensor architecture consists of a redox tag-labeled aptamer immobilized onto a gold electrode surface via thiol-gold chemistry.[7,8] Target binding to the aptamer induces a conformational change in the aptamer that changes the distance of the redox tag relative to the electrode surface. This produces a change in peak current at the reduction potential of the tag, which can be detected via square wave or alternating current voltammetry.[5] Compared with other electrochemical sensors, the E-AB sensor is a general platform for the detection of almost any analyte as aptamers can be isolated for essentially any target with high affinity and specificity through an in vitro process known as SELEX (systematic evolution of ligands by exponential enrichment).[9,10] Another advantage of E-AB sensors is their suitability for detection in complex matrices such as foodstuffs,[11] environmental samples,[12] and even biological specimens.[13] The modularity and robustness of E-AB sensors has led to the development of more advanced architectures, such as microfluidic[14] or miniaturized sensors for the real-time detection of drug molecules in undiluted blood—including in the circulation of live animals.[15] However, there has been little effort in transitioning the E-AB sensing platform from laboratory settings to portable devices that can be readily employed for field-testing.
Paper electrochemical devices (PEDs) are particularly attractive for on-site target detection in many applications because they are cost-effective, lightweight, disposable, and environmentally friendly.[16] The first PED, developed by Dungchai et al., contained a carbon working electrode and was used to detect small molecules in biological samples via enzymatic reactions.[17] Subsequent works established methods for the fabrication of PEDs with metallic-particle three-electrode systems. Hu et al. used an inkjet printer along with a particle enhancement process to fabricate an all-gold three-electrode system on paper.[18] However, such device is not suitable for E-AB sensing because reference electrodes based on metals other than silver cannot provide reproducible and drift-free voltammetric measurements.[19] An ideal PED for E-AB sensing should have a gold working electrode for aptamer immobilization and a silver reference electrode for controlling and stabilizing the potential used during measurements.[8] Recently, Yang et al. employed an electron-beam evaporator, stencil, and parylene C to fabricate PEDs on newspaper containing a three-electrode system made of different metals.[20] However, the fabrication process is complicated and time consuming, requiring the use of multiple expensive masks, a standard cleanroom, and sophisticated instrumentation. It would be challenging for developing countries to produce such devices for on-site testing.
We describe here a new strategy for the reproducible fabrication of customized aptamer-modified PEDs for electrochemical sensing using a basic filtration setup with inexpensive plastic stencils and nanomaterials. The process is simple, rapid (~20 min), economical, and does not require time-consuming deposition processes or sophisticated instrumentation. To make the device, single-walled carbon nanotubes (SWCNTs) are first filtered onto a filter paper substrate under vacuum along with a customized stencil to form a conductive underlay with a three-electrode pattern. Metal nanoparticles are then added onto the SWCNT-patterned paper under vacuum, which results in the formation of metallic films that serve as electrodes and electrical connections on the PED. With the use of stencils, different metal nanoparticles can be deposited onto the same PED to form different electrodes. These stencils enable customization of electrodes in terms of material, thickness, and shape. We assessed the electrochemical performance of the resulting PEDs by first testing the electrochemical response of electroactive molecules in solution. We then modified these PEDs with aptamers and demonstrated rapid and specific detection of small molecule drugs in both buffer and biosamples. Importantly, the performance of our PEDs is comparable to conventional gold disk electrodes in traditional three-electrode electrochemical cells. We therefore believe that our fabrication strategy represents a practical and effective solution for preparing compact, inexpensive, and robust PEDs that are suitable for on-site detection of a diverse range of molecular targets and can even be adapted with hybrid materials with combinations of different size or shape to make custom PEDs for specific applications.
Results and Discussion
Fabrication of PEDs with all-AuNP electrodes.
We previously reported an ambient filtration method for rapid fabrication of highly conductive gold thin films on mixed cellulose ester filter papers.[21] This paper-based gold film consisted of two layers: a conductive SWCNT underlayer and a nanometer-thin AuNP-based metal film directly on top. Based on this initial success, we posited that this filtration method could be used to prepare different thin metal film electrodes on a single paper substrate. To demonstrate this, we developed a new fabrication strategy which utilizes this vacuum filtration method alongside homemade stencils and mask (Supporting Information (SI), Fig. S1) to fabricate PEDs comprising multiple nanometer-thick electrodes of unique size, shape, and material. Each PED consists of a working electrode (WE), a counter electrode (CE), and a reference electrode (RE). We first determined if it was possible to make pre-designed patterned electrodes on a paper substrate by preparing PEDs with only SWCNTs using a stencil permanently laminated onto the paper. The stencil has two purposes: it aids in electrode patterning by restricting the filtration of SWCNTs to the engraved parts, and it serves as a hydrophobic surface on the non-engraved areas and makes the device sturdier. We first used a water-impermeable polyethylene terephthalate film to prepare a top stencil which contained a pattern for four PEDs (Fig. 1A). Such stencils can be made with virtually any shape with micrometer resolution using a desktop cutting machine that costs less than $200. The engraved parts of the stencil were carefully removed using tweezers, leaving openings with a total area of 2.6 cm2. The resulting top stencil was then carefully overlapped with a cellulose filter paper with the same diameter and fed into a desktop thermal laminating machine to make the stencil adhere to the paper substrate (Fig. 1B). After this process, only the engraved parts of the laminated filter paper were permeable to water (Fig. 1C). The filter paper was then placed onto a Buchner funnel under vacuum and 10 mL of 5 μg/mL SWCNT solution dispersed in 1% Triton-X 100 was filtered through the paper at ambient conditions. We found that only the exposed area on the paper was covered with a uniform layer of SWCNTs, and there was no bleeding of SWCNTs into the masked areas (Fig. 1D). The SWCNT-patterned paper was then washed with a copious amount of deionized water to remove excessive surfactant, followed by vacuum drying. Finally, the four PEDs - each with dimensions of 1.2 cm × 0.9 cm (SI, Fig. S2) - were cut from the paper. The fabricated PED comprised a three-electrode-patterned filter paper between PET-parafilm films (SI, Fig. S3). These results demonstrate that the vacuum filtration method and stencil enable facile high-resolution custom patterning on paper.
Figure 1.
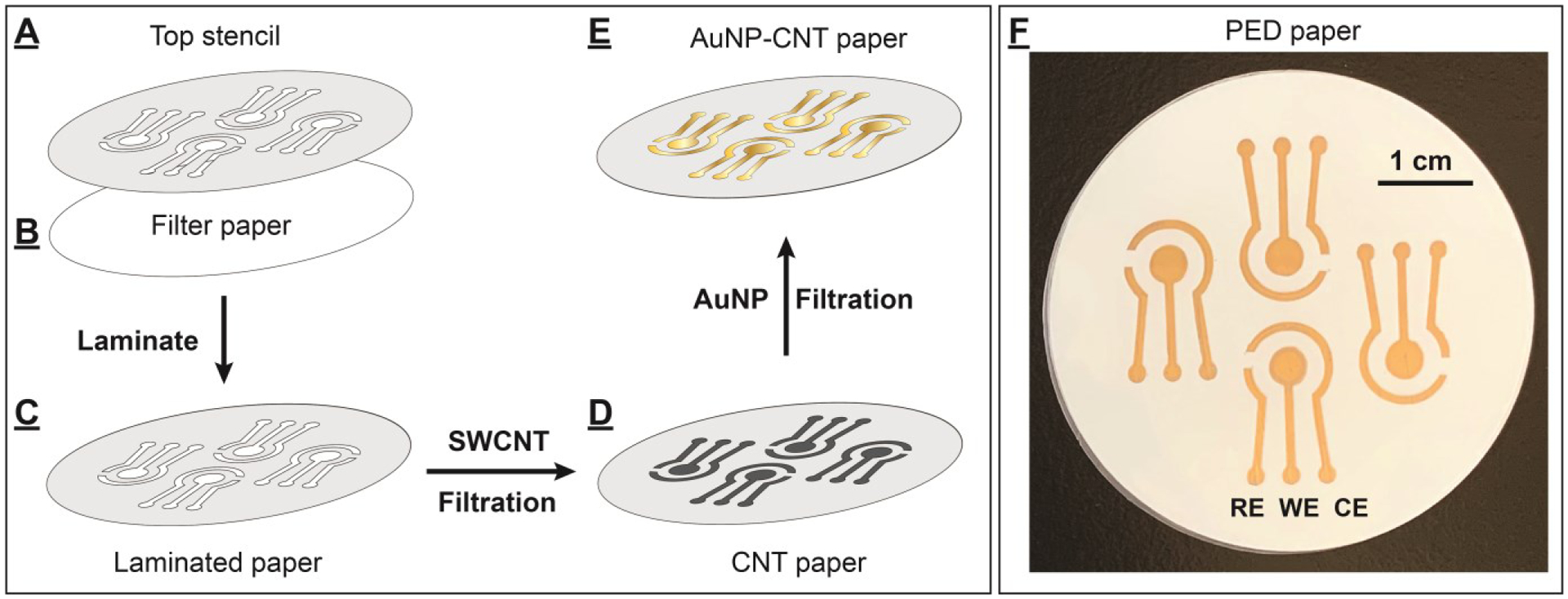
Fabrication of PEDs using an ambient vacuum filtration technique. (A) The top stencil was thermally laminated to (B) a filter paper to create (C) a laminated and patterned paper. (D) SWCNTs were layered onto the designated area to form SWCNT-fabricated PEDs. (E) Gold film was further formed on the SWCNT surface by vacuum-filtration of an AuNP solution to produce (F) four PEDs on a single piece of paper, each with AuNP-fabricated RE, WE, and CE electrodes.
We then determined whether PEDs with metallic electrodes could be accurately patterned on pre-designated areas of filter papers by preparing PEDs with all-AuNP electrodes. We also chose to prepare the electrical contact trails from AuNPs because they have higher conductivity and lower capacitance current than pure SWCNTs.[21] We synthesized citrate-capped AuNPs using a modified Turkevich method,[22] yielding AuNPs with an average diameter of 15±1 nm based on transmission electron microscopy (SI, Fig. S4) with a concentration of 3.5 nM. After preparing SWCNT-patterned paper using the process described above, we filtered 10 mL of 0.35 nM AuNPs (3.5 picomoles, 0.47 mg) through this paper. A reflective and uniform gold film immediately formed on the SWCNT-coated area (Fig. 1E), without contamination on any areas protected by the top stencil. We observed that the filtrate byproduct was colorless (SI, Fig. S5A), and the UV-vis spectra of filtrates did not show any absorption peaks from AuNPs (λmax = 520 nm) (SI, Fig. S5B). Four AuNP-fabricated PEDs were prepared from each paper after drying (Fig. 1F), and no peeling or stripping of the gold films was observed.
To evaluate the performance of the resulting PEDs with all-AuNP electrodes, we tested their electrochemical response to ferro/ferricyanide, a well-known redox pair, in a solution of 1 mM K3[Fe(CN)6] in 0.1 M KCl. We perform cyclic voltammetry (CV) using these PEDs and observed oxidation of [Fe(CN)6]4- and reduction of [Fe(CN)6]3-, and obtained a peak-to-peak separation of 99 ± 2 mV at a scan rate of 100 mV/s, which rivaled the performance of a commercial gold slide electrode (a peak-to-peak separation of 95 ± 2 mV at the same scan rate) (SI, Fig. S6). In contrast, we observed a peak-to-peak separation of 600 mV under the same scan rate with SWCNT-fabricated PEDs, which demonstrated completely irreversible electrochemical behavior (SI, Fig. S6). This indicated that the addition of AuNPs greatly improved the electrochemical performance of the PEDs. Clearly, our method allows for the fabrication of PEDs with high-performance metallic electrodes on paper substrates.
Preparation of PEDs with AgNP pseudo-REs.
Despite the excellent performance of the working electrode on the all-gold PED, the peak reduction and oxidation potentials were much more negative (Epc = −0.16 V and Epa = −0.075 V) compared to those obtained using the gold slide with a standard Ag/AgCl RE (Epc = 0.18 V and Epa = 0.28 V) (SI, Fig. S6). Additionally, the oxidation/reduction peaks for each fabricated PEDs appeared at seemingly arbitrary potentials (data not shown). This clearly indicated that the AuNP-fabricated RE on the PEDs was not suitable for electrochemical detection. We therefore developed a method to fabricate PEDs with gold WE and CE and a silver pseudo-RE for more stable and reproducible electrochemical measurements (Fig. 2). Two different bottom stencils were prepared with polyethylene terephthalate film to allow two-step deposition of different metallic nanoparticles at pre-designated locations. Both bottom stencils had a diameter of 55 mm, slightly larger than the diameter of the filter paper (47 mm). Bottom stencil 1 was engraved with the outline of the WE, CE and connection circuits and bottom stencil 2 contained the outline of the RE. To fabricate the PEDs, the top stencil was laminated onto a filter paper (Fig. 2A) and the filtration procedure described above was used to form a uniform SWCNT pattern on the paper (Fig. 2B). Then, bottom stencil 1 (Fig. 2C) was placed beneath the filter paper and carefully aligned with the SWCNT-laden paper, and 9 mL of 0.35 nM AuNP solution (3.15 picomoles, 0.42 mg) were filtered through the paper. This stencil only allowed the AuNP solution to flow through the engraved area, forming a conductive gold layer for the WE, CE, and connection circuit. (Fig. 2C), with no AuNP deposition on the un-engraved parts of the bottom stencil. Bottom stencil 1 was then removed and bottom stencil 2 was placed beneath the filter paper. The silver RE was prepared by flowing 6.9 nM AgNPs (0.2 mL total, 0.12 picmoles, 12.7 μg) (30 ± 2 nm diameter, SI, Fig. S7) through the filter paper to form an AgNP layer at the defined area without contamination of the gold electrodes (Fig. 2D). Bottom stencil 2 was then removed, and the paper was dried under vacuum. A single filter paper yielded four PEDs (Fig. 2E), each featuring an AgNP pseudo-RE, and an AuNP WE, and an AuNP CE (Fig. 2F). The devices are cut from the paper using scissors; a schematic depicting this is shown in Figure S8.
Figure 2.
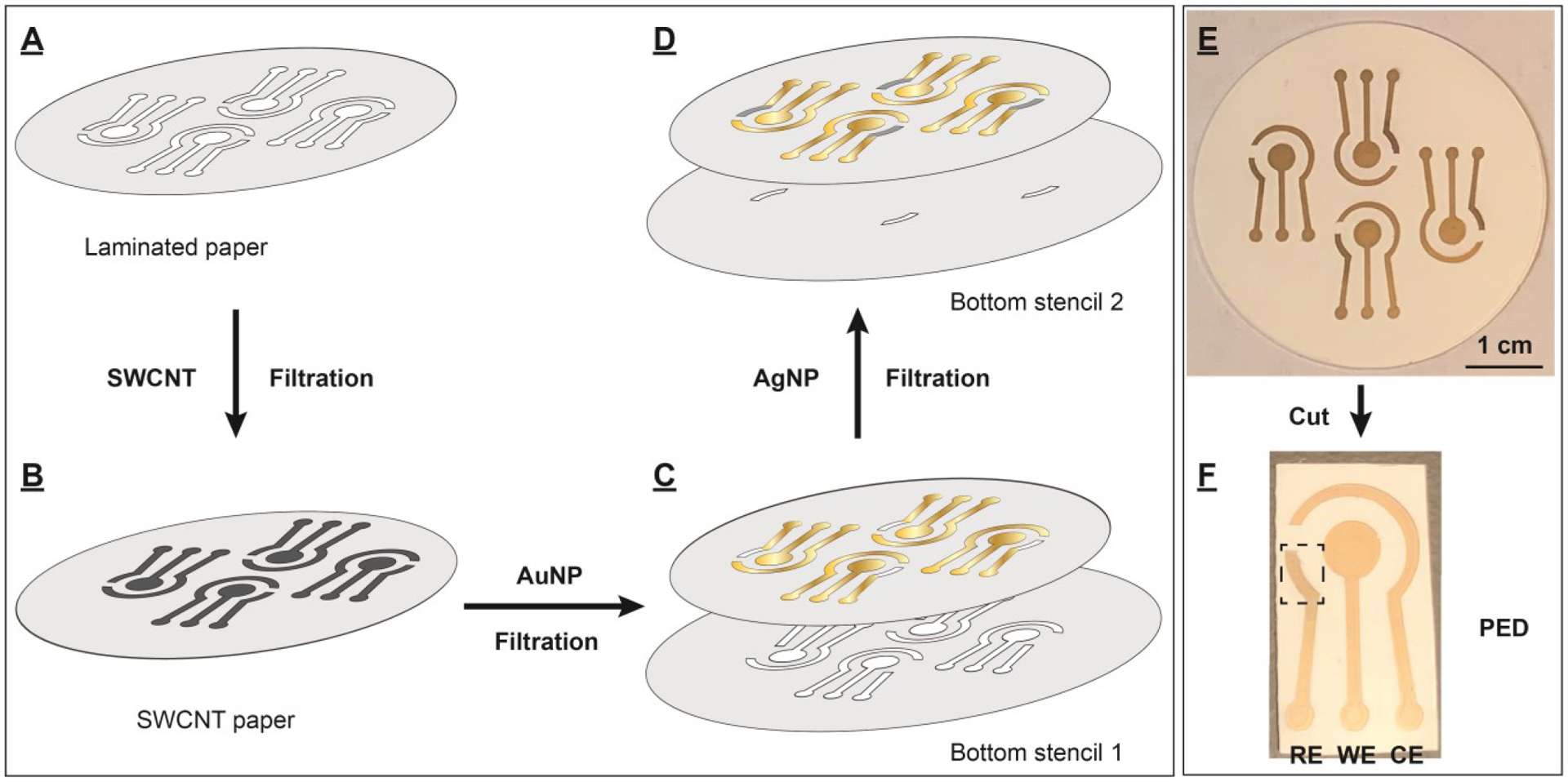
Fabrication of PEDs using various nanomaterials, stencils, and ambient filtration. (A) The top stencil was laminated to a filter paper to create a patterned paper. (B) SWCNTs were first deposited onto the patterned area. Different bottom stencils were then placed beneath the SWCNT-laden paper, followed by filtration of different metal nanoparticles to generate layers of (C) AuNPs (bottom stencil 1) and (D) AgNPs (bottom stencil 2) on designated areas on the paper. (E) Each paper contains four PEDs, which each contain (F) an AgNP pseudo-RE, an AuNP WE, and a AuNP CE. The RE is indicated by a box.
Electrochemical performance and stability of PEDs with AgNP pseudo-REs.
We first evaluated the quality of the fabricated PEDs by performing CV scans in 1 mM K3[Fe(CN)6] solution with 0.1 M KCl and determining peak-to-peak separation (ΔEp). This metric provides an assessment of the stability of the AgNP pseudo-RE. We found that this PED achieved an Epc of 0.08 V and an Epa of 0.18 V for [Fe(CN)6]3-/4-. Both peak potentials were more positive relative to the all-gold PED, but the stability of this reference electrode was still unsatisfactory because both peak potentials exhibited negative shifts after multiple CV scans. For example, Epa shifted from 0.18 V to 0.06 V after four cycles (Fig. 3A). This could be because the AgNP layer was too thin to stabilize the applied potentials during the measurements. To remedy this, we fabricated a new batch of PEDs using the same amount of SWCNTs and AuNPs but with twice the amount of AgNPs. When measuring the electrochemical response of 1 mM K3[Fe(CN)6], these PEDs had much more positive peak potentials (Epc = 0.12 V and Epa = 0.20 V), which are close to those of the commercial gold slide working electrode with a standard Ag/AgCl RE (Epc = 0.18 V and Epa = 0.28 V) (SI, Fig. S9). Importantly, the stability of this thicker AgNP pseudo-RE was greatly improved, and both peak potentials remained constant after ten cyclic voltammetry scans with a scan-rate of 100 mV/s (Fig. 3B). We found that the AgNP pseudo-RE provided stable reference potentials throughout all scans over a total duration of 120 s with a ΔEp of 98 ± 2 mV. No potential shift of anodic (Epa) or cathodic (Epc) peak currents was observed (Fig 3B). This level of stability is more than sufficient for a disposable PED, which would typically employ single-scan measurements lasting no more than 10 s.
Figure 3.
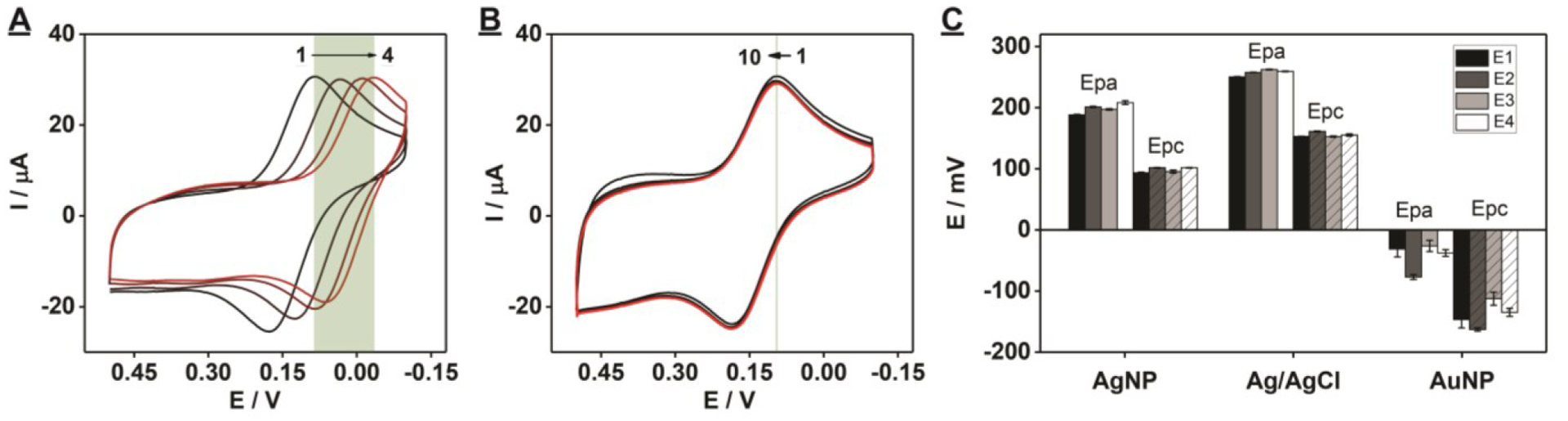
Stability test of the AgNP pseudo-REs of our PED. Cyclic voltammetry scans recorded with 1 mM K3[Fe(CN)6] solution in 0.1 M KCl with a scan rate of 100 mV/s using PEDs prepared with 50 μg SWCNTs, 3.15 picomoles AuNPs, and (A) 0.12 pmole AgNPs or (B) 0.24 picomoles AgNPs. The highlighted area indicates the shift of reduction potential over CV scans. The black to red gradient represents scans 1 through 4, respectively. (C) Potential drift in Epa and Epc produced by the PEDs with an AuNP pseudo-RE, AgNP pseudo-RE, or standard Ag/AgCl RE. Four PEDs (E1–E4) were tested for each reference electrode, and 10 CV scans were performed for each PED with a scan-rate of 100 mV/s. Error bars represent standard deviation from four PEDs.
We then tested the potential stability of PEDs with a AuNP pseudo-RE, AgNP pseudo-RE, or standard Ag/AgCl RE by performing 10 cycles of CV in 1 mM K3[Fe(CN)6] with 0.1 M KCl at a scan-rate of 100 mV/s. The AgNP pseudo-RE and standard Ag/AgCl RE provided highly stable and reproducible measurements with minimal drift in voltage of Epa and Epc (Fig. 3C). Epa and Epc were similar for four different PEDs with AgNP pseudo-REs (SI, Fig. S10A) or a standard Ag/AgCl RE (SI, Fig. S10B). In contrast, PEDs with an AuNP pseudo-RE demonstrated high levels of potential drift (SI, Fig. S10C). On average, the AuNP pseudo-RE had 25- and 6-fold greater deviations in Epa and Epc, respectively, over several scans relative to devices with AgNP pseudo-Res (Fig. 3C). In addition, there was a large deviation in Epa and Epc among PEDs containing AuNP pseudo-RE (Fig. 3C). These results demonstrate that PEDs containing AgNP pseudo-REs can provide stable reference potentials that are superior to AuNP pseudo-REs and similar to standard Ag/AgCl REs during electrochemical measurements.
We further characterized the electrochemical performance of the PEDs by assessing the electron transfer rates of the electroactive molecules ferrocyanide and methylene blue. First, we performed cyclic voltammetry measurements of 1 mM K3[Fe(CN)6] in 0.1 M KCl at various scan rates. We observed a linear relationship between peak current and scan-rate, which indicates that mass transfer in this system is diffusion-limited (Fig. 4A). Using the Nicolson method,[23] we calculated an electron transfer rate (ks) of 3.36 × 10−3 cm/s, which is slightly less than that of the commercial gold slide (ks = 5.46 × 10−3 cm/s) (SI, Fig. S11A). We then tested the electrochemical characteristics of methylene blue on the PEDs and observed a quasi-reversible electrochemical response with a peak-to-peak separation of 98 mV at a scan rate of 100 mV/s (Fig. 4B). The similar linear relationship between peak current and scan rate clearly demonstrated that the process of oxidation and reduction is also diffusion limited. The electron transfer rate of methylene blue on PEDs is slightly lower (ks = 2.25 × 10−3 cm/s) compared to the commercial gold slide (ks = 3.77 × 10−3 cm/s) (SI, Fig. S11B). Nevertheless, these results demonstrated that these PEDs have reliable electrochemical behavior with rapid electron transfer rates. To test the long-term stability of our PEDs, we prepared eight PEDs and measured their electron transfer rates upon fabrication and after 1, 2, 4, and 6 weeks. We observed reliable and reproducible electrochemical behavior with rapid electron transfer rates even after six weeks of storage at room temperature (SI, Fig. S12).
Figure 4.
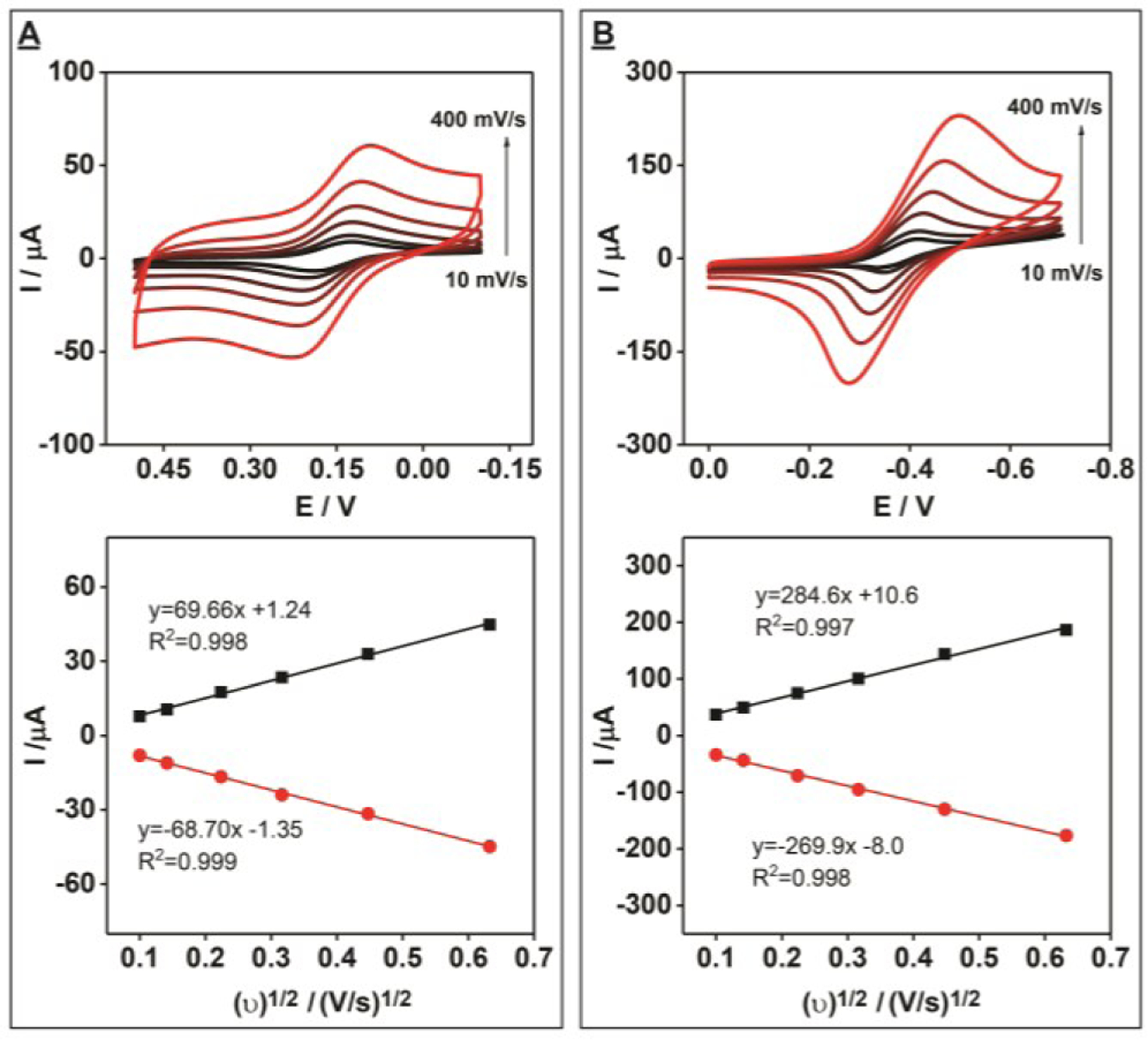
Electrochemical performance of unmodified PEDs with electroactive small molecules. Cyclic voltammetry scans at different scan rates (top) (increasing scan rate is indicated by the black to red color gradient) and the relationship between the square root of the scan rate and the observed current (bottom) for 1 mM (A) K3[Fe(CN)6] or (B) methylene blue in 0.1 M KCl. Black and red lines indicate oxidation and reduction peak currents, respectively. Scan rates: 10, 20, 50, 100, 200 and 400 mV/s.
Finally, to determine the reproducibility of our fabrication method, we prepared eight different batches of PEDs, totaling 32 devices in total, and tested the consistency of their electrochemical performance by determining their electron transfer rates. We observed that all 32 PEDs have very similar electron transfer rates (average ks = 3.32 × 10−3 cm/s), with a relative standard deviation of 4% (SI, Fig. S13). This result demonstrated that our device fabrication method has excellent reproducibility.
Specific detection of small molecules using aptamer-modified PEDs.
We next modified the PEDs with small-molecule-binding aptamers in order to assess their performance as E-AB sensors for the detection of small-molecule analytes. We first electrochemically cleaned the AuNP working electrode surface (Fig. 5A) by performing 10 cycles of cyclic voltammetry over the potential range of 0.2 to 1.5 V in 0.05 M H2SO4 (Fig. 5B and SI, Fig. S14). Afterwards, the PED was rinsed with distilled water and dried with nitrogen gas. We then modified the cleaned working electrode surface with a thiolated methylene-blue-modified aptamer (Fig. 5C) which has micromolar binding affinity for cocaine.[24,25] After backfilling the aptamer-modified PEDs with 6-mercapto-1-hexanol (Fig. 5D), alternating current voltammetry was used to perform cocaine detection in the range of 0–600 μM (Fig. 5E). The reduction peak current of methylene blue increased with increasing cocaine concentrations, with a measurable limit of detection of 1 μM in buffer (Fig. 5F). To demonstrate the generality of the aptamer-based PED design, we next modified PEDs with a recently-isolated synthetic cathinone-binding aptamer with nanomolar affinity for this family of designer drugs,[26] and tested its analytical performance for detecting one such drug, methylenedioxypyrovalerone (MDPV), in the concentration range of 0–100 μM. We obtained a measurable limit of detection of 0.1 μM for MDPV in buffer (Fig. 5G), Importantly, our aptamer-modified PEDs also demonstrated the capability to detect both cocaine and MDPV in complex samples, demonstrating clear target concentration-dependent signals (Figs. 5H–I) with detection limits of 5 μM cocaine and 1 μM MDPV in 50% calf serum (SI, Fig. S15). We then demonstrated the on-site applicability of our aptamer-based PEDs by performing detection in minimally-processed biological samples. Specifically, we detected cocaine in 50% saliva and MDPV in 50% urine using these aptamer-modified PEDs, achieving limits of detection of 5 μM and 1 μM, respectively (Figs. 5H–I and SI, Fig. S15).
Figure 5.
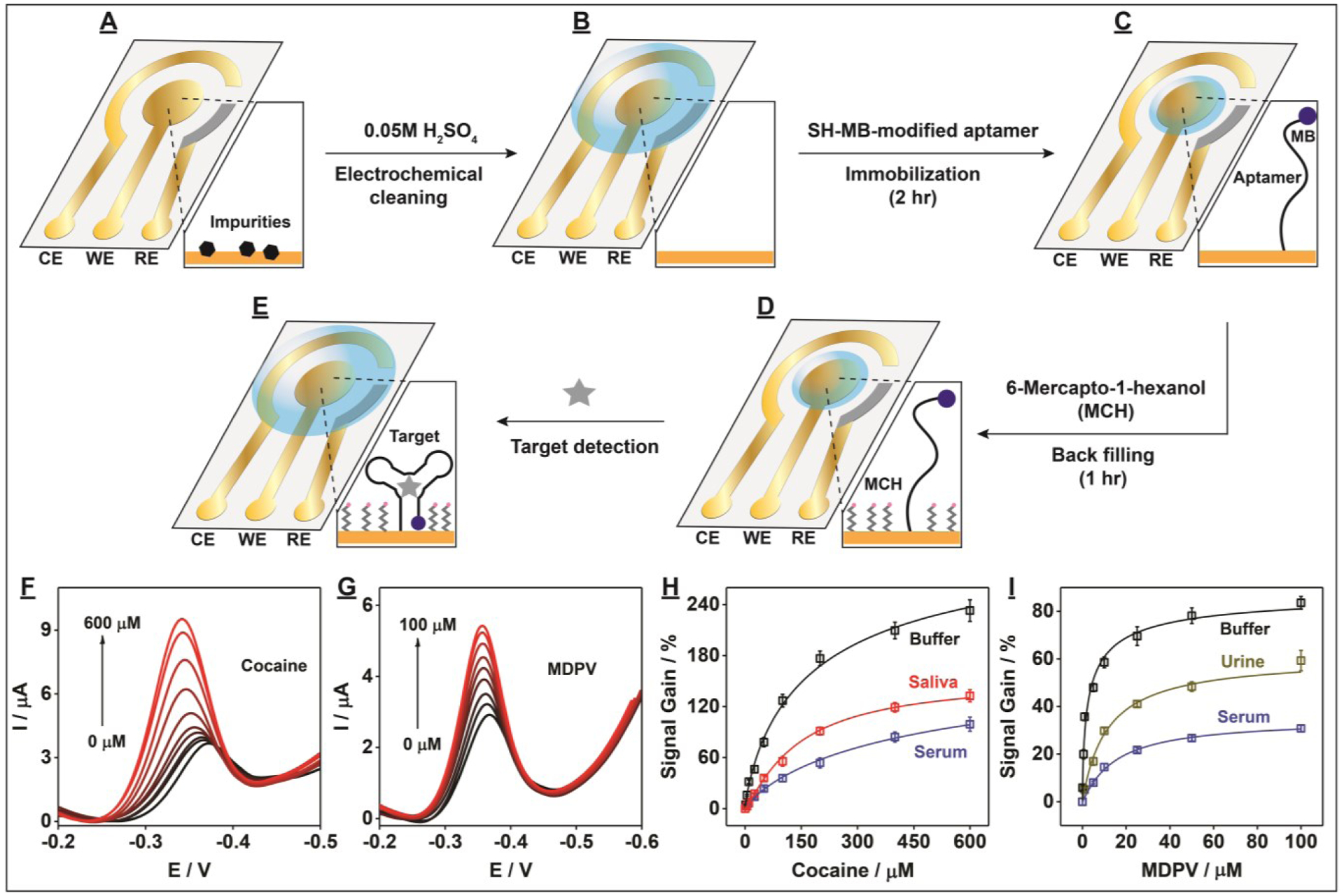
Sensing performance of aptamer-based PEDs for the specific detection of small-molecule targets. (A-E) Schematic of the fabrication process of aptamer-modified PEDs and their employment to detect small molecules. Alternating current voltammograms for detection of (F) cocaine and (G) MDPV in buffer (10 mM Tris (pH 7.4), 20 mM NaCl, 0.5 mM MgCl2)and calibration curves of (H) cocaine and (I) MDPV in buffer (black lines), 50% calf serum (blue lines), 50% saliva (red line), or 50% urine (gold line). Error bars represent standard deviation from three experiments.
To validate the performance of our PEDs, we fabricated cocaine- and MDPV-aptamer-modified E-AB sensors using conventional gold disk working electrodes and detected their respective analytes in buffer and biological samples (SI, Fig. S16). In all tested matrices, the conventional E-AB sensors had similar detection limits as the aptamer-based PEDs (SI, Table S1). Additionally, the signal gains produced by the conventional sensors were reduced by 5–10-fold in biological samples relative to that in buffer, similar to the PED E-ABs. This is probably due to the aptamers have reduced affinity in these matrices and/or protein fouling on the electrode surface, which has been reported previously for E-AB sensors.[5,13,27] Finally, to determine if the aptamer-modified PEDs produced by our method had good reproducibility and low batch-to-batch variation, we developed eight PEDs modified with the cocaine-binding aptamer and tested their detection performance. All yielded similar signal gain values (163.5 ± 2.9%) in the presence of 250 μM cocaine (SI, Fig. S17). Additionally, we found that these PEDs produced reliable and reproducible sensing behavior even after one week of storage at room temperature (SI, Fig. S18).
Conclusion
Aptamer-based PEDs could offer an ideal solution for general on-site testing of a variety of analytes, even in complex sample matrices. However, it has been challenging to develop such PEDs due to the lack of methods for reproducibly fabricating electrodes made of different materials and of different size or shape. Here, we introduce a novel, simple, and cost-effective strategy for fabricating aptamer-modified PEDs that are well-suited for on-site biosensing of small-molecule targets in biological samples. The resulting devices can be used to directly detect electroactive molecules, and can be extended for the bioreceptor-based detection (e.g. aptamer, antibodies, enzymes) of any analyte. Based on our previously established concept of metallic thin-film formation through vacuum filtration,[21] we introduce a new methodology using a combination of stencils and vacuum filtration for the precise deposition of different metal nanoparticles onto the desired areas of a single paper device to form a highly customizable three-electrode system. Specifically, we demonstrated successful fabrication of PEDs with a three-electrode system comprising AuNP WE and CE and an AgNP pseudo-RE. In terms of their sturdiness, our PEDs have similar durability to standard electrochemical glucose test strips. We then modified the PED WE with aptamers to develop aptamer-modified PEDs that can robustly detect small molecules even in complex samples with equally high sensitivity and stability as E-AB sensors employing conventional gold disk electrodes and Ag/AgCl reference electrodes. We believe that our PEDs should be fully compatible with portable potentiostats produced by manufacturers such as PalmSens and Metrohm for on-site applications. Notably, this fabrication method is very environmentally friendly because no precious metals are wasted (unlike etching processes), there are no high-energy requirements (e.g. laser, E-beam), and is chemically ‘green’ as no hazardous solvents or reagents are involved.
Screen printing is the most frequently used procedure to fabricate electrochemical sensors on substrates such as ceramic, plastic, and paper. In this method, ink is deposited on the substrate in a patterned manner using a stencil with the aid of a squeegee. Although both screen-printing and our vacuum filtration method are comparably simple, screen printing has certain disadvantages related to cost, accessibility, and quality. First, the screen-printing process generates printed electrodes with thicknesses of 20–100 μm,[28] and thus requires large quantities of ink material. When precious electrode materials such as gold are used, fabrication becomes expensive. In contrast, our fabrication method requires only a small quantity of materials (5 μg SWCNTs, 470 μg AuNPs, 12.7 μg AgNPs), as our electrodes have thickness < 100 nm,[21] and does not depend on sophisticated instruments or lithography. Second, screen-printed electrodes have high batch-to-batch variability due to the aggregation of nanoparticles in the ink.[29] In contrast, our device is fabricated using nanoparticles that are well-dispersed in aqueous solution. It is very simple and straightforward to prepare such nanoparticles, and uniform metal films can be formed on paper substrates by filtering the particles under vacuum force. As such, our devices have minimal inter-device variability. Third, our fabrication method also does not require sophisticated instruments or energy-intensive processes like high temperature heating (e.g., 120–900 °C required for screen printing).[30,31] Our process utilizes equipment readily available in any scientific laboratory: standard vacuum filtration glassware and inexpensive desktop thermal laminator and a paper cutting machine. Finally, it is difficult to quickly develop a new screen-printing stencil with a different pattern.[32] In contrast, the stencils used in our method can be readily customized. Electrode patterns can be designed via software like Adobe Illustrator, and new stencils can be prepared from plastic film with a benchtop cutting machine in just a few minutes. We believe this feature of method will greatly accelerate the prototyping process for PED design and sensor development.
There are a few reports on the E-AB sensing platform being adapted to screen-printed electrodes.[33] For example, the Crooks group demonstrated the successful fabrication of paper E-AB devices for the detection of thrombin.[34] However, our method is simpler, faster, less energy-intensive, has low batch-to-batch variation and offers greater customization of device design. First, our method is only two steps and yields four devices, while the Crooks method requires three steps and was demonstrated to produce one electrode at a time. Second, our method greatly simplifies and accelerates the custom fabrication of new stencil designs relative to screen-printing procedures, thereby enabling faster prototyping of electrochemical devices in a low-cost and user-friendly manner. To demonstrate this, we evaluated the architectural resolution and size limitation of our fabrication method. Specifically, we designed a stencil containing multiple rectangular shapes with lengths of 8 mm but different widths (100, 200, 300, 400, 500, 700, 900, and 1,000 μm) (SI, Fig. S19A) and utilized our vacuum filtration technique to deposit a SWCNT (SI, Fig. S19B) or AuNP-SWCNT (SI, Fig. S19C) film on the paper. We observed that a uniform SWCNT and shiny AuNP-SWCNT film formed on the exposed rectangular areas with width as low as 200 μm. We also measured the conductivity of the AuNP-SWCNT films, and observed a linear relationship between film width and resistance (SI, Fig. S19D), implying that the thickness of the film remains the same regardless of the width. This result therefore suggests that at least twice the number of PEDs can be fabricated on the same sized paper substrate at a time.
Our device fabrication method is versatile, allowing for deposition of different metals on the same paper substrate. This is an important consideration, because alternative electrode compositions (e.g. Pt-Au hybrid working electrodes) may be required for particular analytical applications. Although we demonstrated the detection of analytes in minimally-processed biological samples using aptamer-modified PEDs, we believe that the sensitivity of our aptamer-modified PEDs can be further improved by fabricating electrodes from nanomaterials of different sizes or shapes. For example, Soleymani et al. demonstrated highly sensitive detection of analytes using spiky nanoparticle-modified gold electrodes.[35] Creating PEDs with existing functionalized or forthcoming advanced nanomaterials may also prove beneficial for mitigating other aspects of the PED such as greater surface anti-fouling ability to improve detection in complex samples.[36] Finally, we envision that our filtration strategy can be used to fabricate PEDs based bioreceptors (e.g., with aptamers, enzymes, or antibodies) with multiple working electrodes for multiplexed analyte detection in a single sample. This would prove especially useful for analytical applications in the areas of medical diagnostics, environmental monitoring, and food safety.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health - National Institute on Drug Abuse R21DA045334-01A1 and the National Science Foundation (1905143). O.A. acknowledges the support from the Presidential Fellowship, which is awarded by the University Graduate School of Florida International University.
Footnotes
Publisher's Disclaimer: This manuscript has been accepted after peer review and appears as an Accepted Article online prior to editing, proofing, and formal publication of the final Version of Record (VoR). This work is currently citable by using the Digital Object Identifier (DOI) given below. The VoR will be published online in Early View as soon as possible and may be different to this Accepted Article as a result of editing. Readers should obtain the VoR from the journal website shown below when it is published to ensure accuracy of information. The authors are responsible for the content of this Accepted Article.
Supporting information for this article is given via a link at the end of the document.
References
- [1].Labib M, Sargent EH, Kelley SO, Chem. Rev 2016, 116, 9001–9090. [DOI] [PubMed] [Google Scholar]
- [2].Ronkainen NJ, Halsall HB, Heineman WR, Chem. Soc. Rev 2010, 39, 1747–1763. [DOI] [PubMed] [Google Scholar]
- [3].Gupta VK, Ganjali MR, Norouzi P, Khani H, Nayak A, Agarwal S, Crit. Rev. Anal. Chem 2011, 41, 282–313. [DOI] [PubMed] [Google Scholar]
- [4].Clarke SF, Foster JR, Br. J. Biomed. Sci 2012, 69, 83–93. [PubMed] [Google Scholar]
- [5].Xiao Y, Lubin AA, Heeger AJ, Plaxco KW, Angew. Chemie Int. Ed 2005, 44, 5456–5459. [DOI] [PubMed] [Google Scholar]
- [6].Schoukroun-Barnes LR, Macazo FC, Gutierrez B, Lottermoser J, Liu J, White RJ, Annu. Rev. Anal. Chem 2016, 9, 163–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fan C, Plaxco KW, Heeger AJ, Proc. Natl. Acad. Sci. U. S. A 2003, 100, 9134–9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xiao Y, Lai RY, Plaxco KW, Nat. Protoc 2007, 2, 2875–2880. [DOI] [PubMed] [Google Scholar]
- [9].Ellington AD, Szostak JW, Nature 1990, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- [10].Tuerk C, Gold L, Science 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- [11].Li H, Somerson J, Xia F, Plaxco KW, Anal. Chem 2018, 90, 10641–10645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Xiao Y, Rowe AA, Plaxco KW, J. Am. Chem. Soc 2007, 129, 262–263. [DOI] [PubMed] [Google Scholar]
- [13].Baker BR, Lai RY, Wood MS, Doctor EH, Heeger AJ, Plaxco KW, J. Am. Chem. Soc 2006, 128, 3138–3139. [DOI] [PubMed] [Google Scholar]
- [14].Swensen JS, Xiao Y, Ferguson BS, Lubin AA, Lai RY, Heeger AJ, Plaxco KW, Soh HT, J. Am. Chem. Soc 2009, 131, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Arroyo-Currás N, Somerson J, Vieira PA, Ploense KL, Kippin TE, Plaxco KW, Proc. Natl. Acad. Sci 2017, 114, 645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mettakoonpitak J, Boehle K, Nantaphol S, Teengam P, Adkins JA, Srisa-Art M, Henry CS, Electroanalysis 2016, 28, 1420–1436. [Google Scholar]
- [17].Dungchai W, Chailapakul O, Henry CS, Anal. Chem 2009, 81, 5821–5826. [DOI] [PubMed] [Google Scholar]
- [18].Hu C, Bai X, Wang Y, Jin W, Zhang X, Hu S, Anal. Chem 2012, 84, 3745–3750. [DOI] [PubMed] [Google Scholar]
- [19].Pavlovic E, Lai RY, Wu TT, Ferguson BS, Sun R, Plaxco KW, Soh HT, Langmuir 2008, 24, 1102–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yang MH, Jeong SW, Chang SJ, Kim KH, Jang M, Kim CH, Bae NH, Sim GS, Kang T, Lee SJ, et al. , ACS Appl. Mater. Interfaces 2016, 8, 34978–34984. [DOI] [PubMed] [Google Scholar]
- [21].Guntupalli B, Liang P, Lee J-H, Yang Y, Yu H, Canoura J, He J, Li W, Weizmann Y, Xiao Y, ACS Appl. Mater. Interfaces 2015, 7, 27049–27058. [DOI] [PubMed] [Google Scholar]
- [22].Turkevich J, Stevenson PC, Hillier J, Discuss. Faraday Soc 1951, 11, 55–75. [Google Scholar]
- [23].Nicholson RS, Anal. Chem 1965, 37, 1351–1355. [Google Scholar]
- [24].Slavkovic S, Altunisik M, Reinstein O, Johnson PE, Bioorg. Med. Chem 2015, 23, 2593–7. [DOI] [PubMed] [Google Scholar]
- [25].Stojanovic MN, de Prada P, Landry DW, J. Am. Chem. Soc 2001, 123, 4928–4931. [DOI] [PubMed] [Google Scholar]
- [26].Yang W, Yu H, Alkhamis O, Liu Y, Canoura J, Fu F, Xiao Y, Nucleic Acids Res. 2019, 47, e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Campuzano S, Pedrero M, Yáñez-Sedeño P, Pingarrón JM, Int. J. Mol. Sci 2019, 20, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Metters JP, Kadara RO, Banks CE, Analyst 2011, 136, 1067–1076. [DOI] [PubMed] [Google Scholar]
- [29].Antuña-Jiménez D, González-García MB, Hernández-Santos D, Fanjul-Bolado P, Biosensors 2020, 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Susmel S, Guilbault GG, O’Sullivan CK, Biosens. Bioelectron 2003, 18, 881–889. [DOI] [PubMed] [Google Scholar]
- [31].García-González R, Fernández-Abedul MT, Pernía A, Costa-García A, Electrochim. Acta 2008, 53, 3242–3249. [Google Scholar]
- [32].Tortorich RP, Shamkhalichenar H, Choi JW, Appl. Sci 2018, 8, 288. [Google Scholar]
- [33].Grabowska I, Sharma N, Vasilescu A, Iancu M, Badea G, Boukherroub R, Ogale S, Szunerits S, ACS Omega 2018, 3, 12010–12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cunningham JC, Brenes NJ, Crooks RM, Anal. Chem 2014, 86, 6166–6170. [DOI] [PubMed] [Google Scholar]
- [35].Soleymani L, Fang Z, Sargent EH, Kelley SO, Nat. Nanotechnol 2009, 4, 844–848. [DOI] [PubMed] [Google Scholar]
- [36].Li H, Dauphin-Ducharme P, Arroyo-Currás N, Tran CH, Vieira PA, Li SG, Shin C, Somerson J, Kippin TE, Plaxco KW, Angew. Chemie - Int. Ed 2017, 56, 7492–7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.