

Abstract
Nonalcoholic steatohepatitis (NASH), the progressive form of nonalcoholic fatty liver disease, is increasing in prevalence. NASH-related alterations in hepatic protein expression (e.g., transporters) and in overall physiology may affect drug exposure by altering drug disposition and elimination. The aim of this study was to build a physiologically-based pharmacokinetic (PBPK) model to predict drug exposure in NASH by incorporating NASH-related changes in hepatic transporters. Morphine and morphine-3-glucuronide (M3G) were used as model compounds. A PBPK model of morphine with permeability-limited hepatic disposition was extended to include M3G disposition and enterohepatic recycling (EHR). The model captured the area under the plasma concentration-time curve (AUC) of morphine and M3G after intravenous morphine administration within 0.82- and 1.94-fold of observed values from three independent clinical studies for healthy adult subjects (n= 6, 10, and 14 individuals). When NASH-related changes in multidrug resistance-associated protein 2 (MRP2) and MRP3 were incorporated into the model, the predicted M3G mean AUC in NASH was 1.34-fold higher compared to healthy subjects, which is slightly lower than the observed value (1.63-fold). Exploratory simulations on other physiological changes occurring in NASH (e.g., moderate decreases in glomerular filtration rate and portal vein blood flow) revealed that the effect of transporter changes was most prominent. Additionally, NASH-related transporter changes resulted in decreased morphine EHR, which could be important for drugs with extensive EHR. This study is an important first step to predict drug disposition in complex diseases such as NASH using PBPK modeling.
Keywords: transporters, drug transport, metabolites, physiologically-based pharmacokinetics, liver
Introduction
The liver is a major organ of drug elimination. Specific metabolic enzymes and transport proteins in the liver transform and excrete endogenous compounds and xenobiotics, including drugs. Diseases affecting the liver can change the expression and function of these proteins and, therefore, alter drug exposure.1, 2 Predicting drug exposure in patients with liver disease is important, especially when developing drugs that are specifically targeted to the liver.
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease globally.3 NAFLD is characterized by lipid accumulation in hepatocytes and is linked to obesity, insulin resistance and the metabolic syndrome.4 It is estimated that up to 25% of the global population suffers from NAFLD.3 The high prevalence of NAFLD is of concern, because its progressive form, nonalcoholic steatohepatitis (NASH), can lead to cirrhosis and hepatocellular carcinoma. Recently, NAFLD has been linked to COVID-19 progression (p<0.001, n=202 patients)5, and attention has been called to whether NAFLD patients are more vulnerable to the widespread SARS-CoV-2 infection or if COVID-19 might increase NAFLD progression to NASH.6
In NASH, steatosis is accompanied by inflammation, hepatocyte ballooning and fibrosis. Importantly from a pharmacokinetics (PK) and drug safety viewpoint, there is growing evidence that proteins involved in drug absorption, distribution, metabolism and excretion (ADME) are altered in patients with NASH.7–12 There is, however, limited information about the functional consequences of NASH-related changes in ADME proteins due to the small number of compounds that have been studied in clinical trials. To date, transporter changes in NASH have been linked to altered PK of acetaminophen glucuronide, morphine glucuronides and the nuclear imaging agent 99mTc-mebrofenin.13–16 Conversely, NAFLD had no effect on apixaban, rosuvastatin or metformin PK17, 18; however, this may not be the case in patients with NASH.
Morphine is an opioid analgesic that is used to treat moderate and severe pain. It undergoes high extraction in the liver and has low oral bioavailability (0.2 – 0.47) due to the extensive first-pass metabolism.19–21 Morphine can permeate membranes passively22 and is metabolized by several uridine 5’-diphospho-glucuronosyltransferase (UGT) enzymes. Although it can be metabolized in the intestine,23 morphine is taken up primarily into the liver by organic cation transporter (OCT) 1, where it is rapidly metabolized mainly by UGT2B7 to its major metabolites morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G).20, 24, 25 OCT1 activity and UGT2B7 mRNA levels do not appear to be altered in NASH,8, 11, 18 which is consistent with similar morphine PK profiles in healthy subjects and patients with NASH.14 M3G formed in the liver undergoes excretion into bile (canalicular efflux) via multidrug-resistance associated protein (MRP) 2 and basolateral efflux to the systemic circulation via MRP3.26, 27 M3G excreted in bile can undergo enterohepatic recycling (EHR).20, 21, 27–29 The reported increase in hepatic MRP3 abundance and MRP2 mislocalization in liver tissue from patients with NASH could explain the altered exposure of morphine glucuronides in NASH.7, 11, 30
Physiologically-based pharmacokinetic (PBPK) modeling is a mathematical method that combines drug properties, physiological data and clinical trial information. Clinical observations, e.g. plasma concentration data, can be used to inform model building in a middle-out approach of PBPK modeling. By altering specific physiological parameters (e.g. metabolic enzyme or transporter abundance), PBPK modeling can be utilized to simulate exposure in disease states. It also is useful to examine the contribution of specific changes in system (and drug) parameters on overall PK. This can be beneficial when studying multifactorial diseases such as NASH, where alterations occur not only in ADME proteins, but also in physiological factors, such as body weight.4
The aim of this study was to develop a PBPK model to describe the disposition of morphine and M3G in healthy subjects, and extend the model to predict the disposition in patients with NASH based on alterations in transporter abundance and activity. To the best of our knowledge, this is the first PBPK model to include EHR of the metabolite, and luminal metabolism of the metabolite back to parent compound, to evaluate the effect of NASH-related transporter changes on EHR. In addition, simulations were performed to evaluate the contribution of different physiological factors relevant to NASH on morphine and M3G PK.
Methods
PBPK model development for M3G in healthy subjects
An overview of the modeling workflow and a schematic depicting the disposition and enterohepatic recycling of morphine are presented in Figure 1. All simulations were performed using Simcyp simulator software v 18.1 (Certara UK Ltd, Sheffield, UK). A previously published PBPK model of morphine31 was used as the basis for model development. A compound file was developed for M3G, using compound specific and measured physico-chemical and blood-binding data (Table 1). The maximum rate of transporter-mediated efflux (Vmax) for MRP2 and MRP3, as well as the passive permeability clearance in the liver, were optimized using the parameter estimation module with a Maximum Likelihood population-based fitting with M3G serum and urine data from individual healthy subjects.14 The passive permeability of M3G in the liver was constrained to less than the corresponding permeability of morphine (0.0034 ml/min/million hepatocytes) because glucuronides are expected to exhibit lower passive diffusion compared to the parent.32 A Kp scalar of 0.2 was applied to enable the model to capture the observed peak concentration (Cmax) of M3G. Only M3G was included in the model because of the large number of data points that were below the lower limit of quantification for M6G, the less abundant but active metabolite of morphine.
Figure 1.
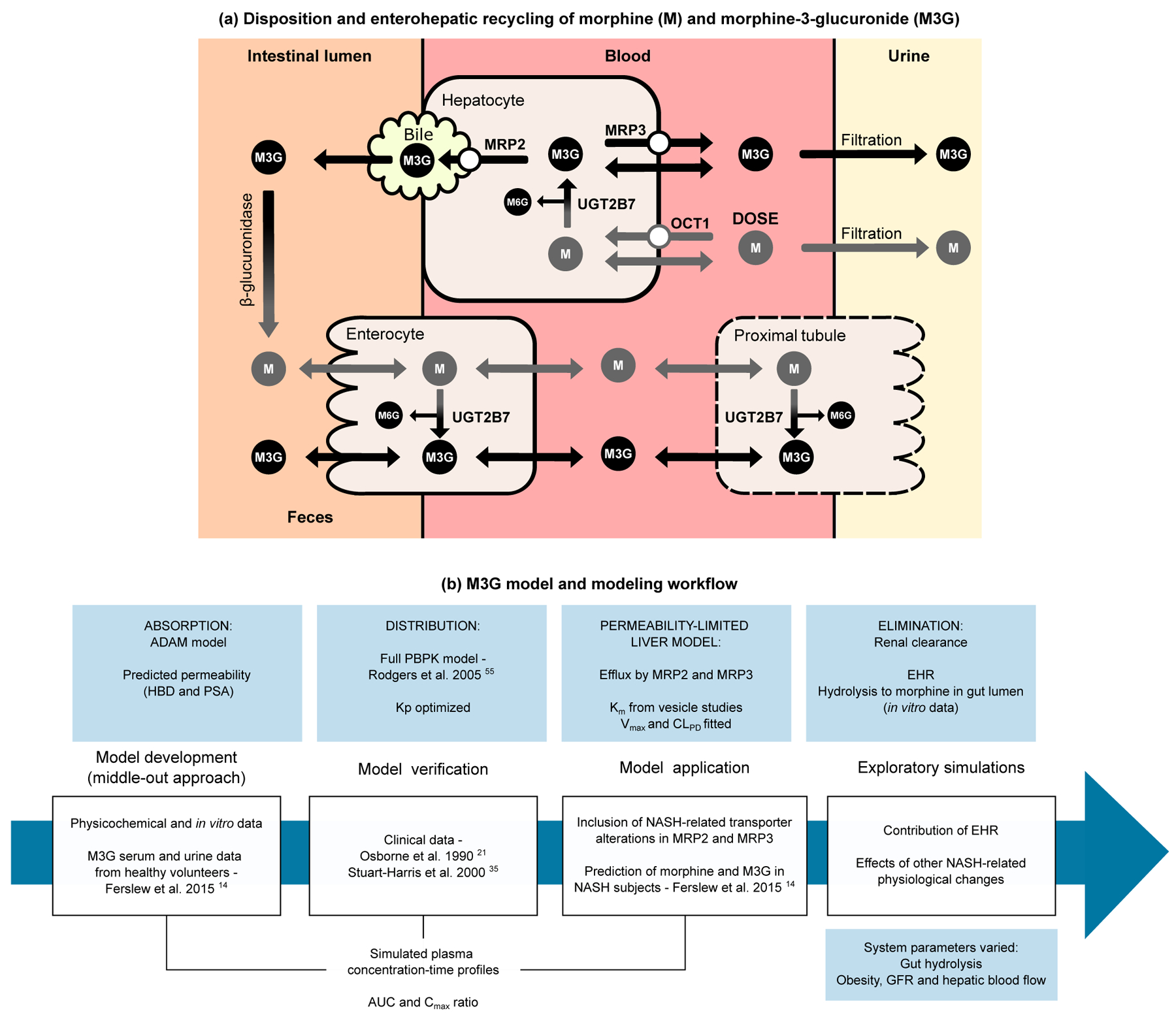
Schematic depicting the disposition and enterohepatic recycling of morphine (M) and morphine-3-glucuronide (M3G) (a) and the workflow for the development of the M3G physiologically-based pharmacokinetic (PBPK) model and its application to subjects with nonalcoholic steatohepatitis (NASH) (b). Only the disposition pathways considered in the PBPK model are included in the schematic (a). Note that metabolism of morphine in the enterocyte is low, and that the kidney was modeled as a perfusion-limited compartment as part of the full PBPK model. Morphine-6-glucuronide (M6G) disposition is similar to M3G, but was not included in the model due to limited clinical data. ADAM, advanced dissolution, absorption, and metabolism model; AUC, area under concentration-time curve; CLPD, passive diffusion clearance; Cmax, peak concentration in plasma; EHR, enterohepatic recycling; GFR, glomerular filtration rate; HBD, hydrogen bond donor; Km, Michaelis-Menten constant; Kp, equilibrium distribution ratio; MRP, multidrug-resistance associated protein; OCT1, organic cation transporter 1; PSA, polar surface area; UGT2B7, uridine 5’-diphospho-glucuronosyltransferase 2B7; Vmax, maximum rate of transporter-mediated efflux.
Table 1.
Final input parameters used in the morphine-3-glucuronide model.
| Parameter | Value | Reference |
|---|---|---|
| Molecular weight | 461.46 g/mol | |
| LogP | −1.1 | 52 |
| Compound type | Ampholyte | |
| pKa1 | 2.86 (acid) | 52 |
| pKa2 | 7.93 (base) | 53 |
| Blood-to-plasma ratio | 0.61 | 54 |
| Fraction unbound in plasma | 0.85 | 41 |
| Main plasma binding component | Albumin | |
| Distribution | Full PBPK model | |
| Vss | Predicted | Method 255 |
| Kp scalar | 0.2 | Optimized |
| Renal clearance | 8.4 l/h (for 70 kg subject) | 20 |
| Permeability limited liver | ||
| Passive permeability | 0.0006 ml/min/106 hepatocytes | Optimized |
| MRP3 | ||
| Vmax | 7300 pmol/min/106 hepatocytes | Optimized |
| Km | 500 μM | 26 |
| MRP2 | ||
| Vmax | 590 pmol/min/106 hepatocytes | Optimized |
| Km | 50 μM | 27 |
| Polar surface area | 149 Å2 | Pubchem |
| Number of hydrogen bond donors | 5 | Pubchem |
| Peff,man | 0.0266*10−4 cm/s | Predicted |
| fugut | 1 | Assumed |
| Gut luminal hydrolysis CLint | 6000 μl/h/g luminal contents | 33, 34 |
CLint, intrinsic clearance; fugut, fraction of drug unbound in the enterocyte; LogP, log of the octanol-water partition coefficient for the neutral compound; Km, Michaelis-Menten constant; Kp, equilibrium distribution ratio; MRP, multidrug-resistance associated protein; Peff,man, effective human jejunum permeability; Vmax, maximum rate of transporter-mediated efflux; Vss, volume of distribution at steady-state.
An EHR component was included in the model that incorporated MRP2-mediated transport of M3G into bile and subsequent release into the gastrointestinal tract, cleavage of the glucuronide moiety by intestinal (bacterial) β-glucuronidases to release morphine, and subsequent reabsorption of morphine. The effective intestinal permeabilities (Peff,man) of morphine (2.07 *10−4 cm/s) and M3G (0.0266 *10−4 cm/s) were predicted based on the polar surface area and number of hydrogen bond donors using the prediction model within the simulator. The relative abundance of hydrolyzing enzymes in the gastrointestinal lumen was set to increase towards the colon, because M3G cleavage/hydrolysis has been shown to be much higher in the colon than small intestine in laboratory animals.28, 33, 34 For example, the initial hydrolysis rate of morphine glucuronide in rat intestines in vitro was up to 178-fold higher in the large intestine compared to the small intestine.34 Therefore, the colon was set as the reference segment and the relative activities were set to 0.0001 in the stomach, 0.001 in the duodenum and jejunum I, 0.1 in the jejunum II and ileum I, and 1.0 in the ileum II – IV compartments and colon to describe this increase. The hydrolysis rate was estimated to be 6000 μl/h/g luminal content, acting on the total concentration in the lumen.33, 34 Simulations were performed in the fasted state.
The observed area under the concentration time curve (AUC) values were calculated from the original Ferslew et al.14 serum data in Phoenix® version 8.2. All AUC0−∞ values from the simulations also were calculated in Phoenix®.
Model verification with clinical data from healthy volunteers
Model performance was verified using two independent sets of clinical data describing morphine and M3G disposition after intravenous administration of morphine sulfate.21, 35 Plasma concentration-time data were extracted from the publications using the online tool WebPlot Digitizer (https://automeris.io/WebPlotDigitizer/). The goodness of fit of the data were evaluated by visual inspection of the simulated plasma concentration time curves with 90% confidence intervals and by comparing simulated plasma AUC values for morphine and M3G, and Cmax values for M3G, with the observed data. The absolute average fold error (AAFE) was calculated across both healthy and NASH simulations according to equation 1:
| (Equation 1) |
where Pred and Obs are the predicted and observed values, respectively, of AUC and Cmax. Predictions of AUC and Cmax within the two-fold range, i.e. predicted/observed ratios between 0.5- and 2-fold, were regarded as acceptable.
PBPK modeling in NASH incorporating changes in transporters
Simulations were performed using virtual individuals matched for age, weight, height and sex with biopsy-proven noncirrhotic NASH patients.14 Matching was used because the weight distribution of individuals in the clinical trial was not consistent with either the “Sim-Healthy Volunteers” or the “Sim-Obese” population libraries within the Simcyp Simulator. Previously reported data on MRP2 and MRP3 changes in NASH livers were used to inform the activity changes incorporated into the NASH M3G model. A relative activity factor of 2.5 was used for MRP3 based on immunoblot data.7 For MRP2, a relative activity factor of 0.5 was approximated based on information about the mislocalization of MRP2/Mrp2 in NASH,7, 30 and the observed ≈ 50% decrease in (1) biliary clearance of the MRP2 substrate 99mTc-mebrofenin in patients with NASH,15 and (2) biliary excretion of M3G in rats with methionine-choline diet induced NASH.30 These scaling factors are also in line with recently reported proteomic data showing increased MRP3 and decreased MRP2 abundance.11
Exploratory simulations
The contribution of different physiological factors that are affected by NASH on morphine and M3G PK was studied by simulation. First, the effect of obesity was investigated by running simulations with the “Sim-Healthy Volunteers”, and the two obesity populations in the default library: “Sim-Obese” (BMI 30–40 kg/m2) and “Sim-Morbidly obese” (BMI >40 kg/m2).36
Further simulations were performed using the “Sim-Obese” population library, but the base glomerular filtration rate (GFR) in the population was set to equal the GFR in the “Sim-Healthy volunteers” population based on published data suggesting that estimated GFR (eGFR) may be comparable in healthy individuals and patients with steatotic livers.14, 37 First, the contribution of EHR to morphine and M3G exposure was evaluated by performing simulations where the hydrolysis of M3G to morphine in the gut lumen was disabled in both the control population and the population with NASH-related transporter changes. Next, the effects of NASH-related alterations in renal function and portal vein blood flow were examined. In contrast to data reported for obese patients,36 patients with NASH may have slightly decreased renal clearance (14% lower eGFR than healthy controls) and are at a higher risk of chronic kidney disease.38 Impaired GFR was incorporated into the model by increasing the serum creatinine values of the obese population to obtain approximately 14% lower mean GFR in the virtual obese subjects than in the virtual healthy population. GFR was calculated using the Modification of Diet in Renal Disease (MDRD) method. The effects of portal vein blood flow were studied in simulations where portal vein blood flow (described as % of cardiac blood flow) was reduced by 15% and 30% to mimic observations in patients with NASH and different stages of fibrosis.39 Finally, the effect of altered MRP2 and MRP3 transport on morphine and M3G was simulated, along with simulating the combined effects of the other physiological changes. Ten trials of ten subjects, 50% female, age 33–63 years were simulated in each of the exploratory simulations.
Results
Model development and verification
Model development was performed using the M3G serum and urine data from individual healthy volunteers (n = 14).14 The parameter values used in the final model for M3G are compiled in Table 1. Based on visual inspection of the plasma concentration-time profiles, the model captured the M3G data adequately for the dataset used for model development as well as the two datasets used for model verification21, 35 (Figure 2). The ratios of the predicted to observed mean AUC and Cmax values were within the two-fold acceptance criterion and ranged from 0.82–1.49, except the AUC0−∞ ratio for morphine in the model development set which was 1.94 (Table 2).
Figure 2.
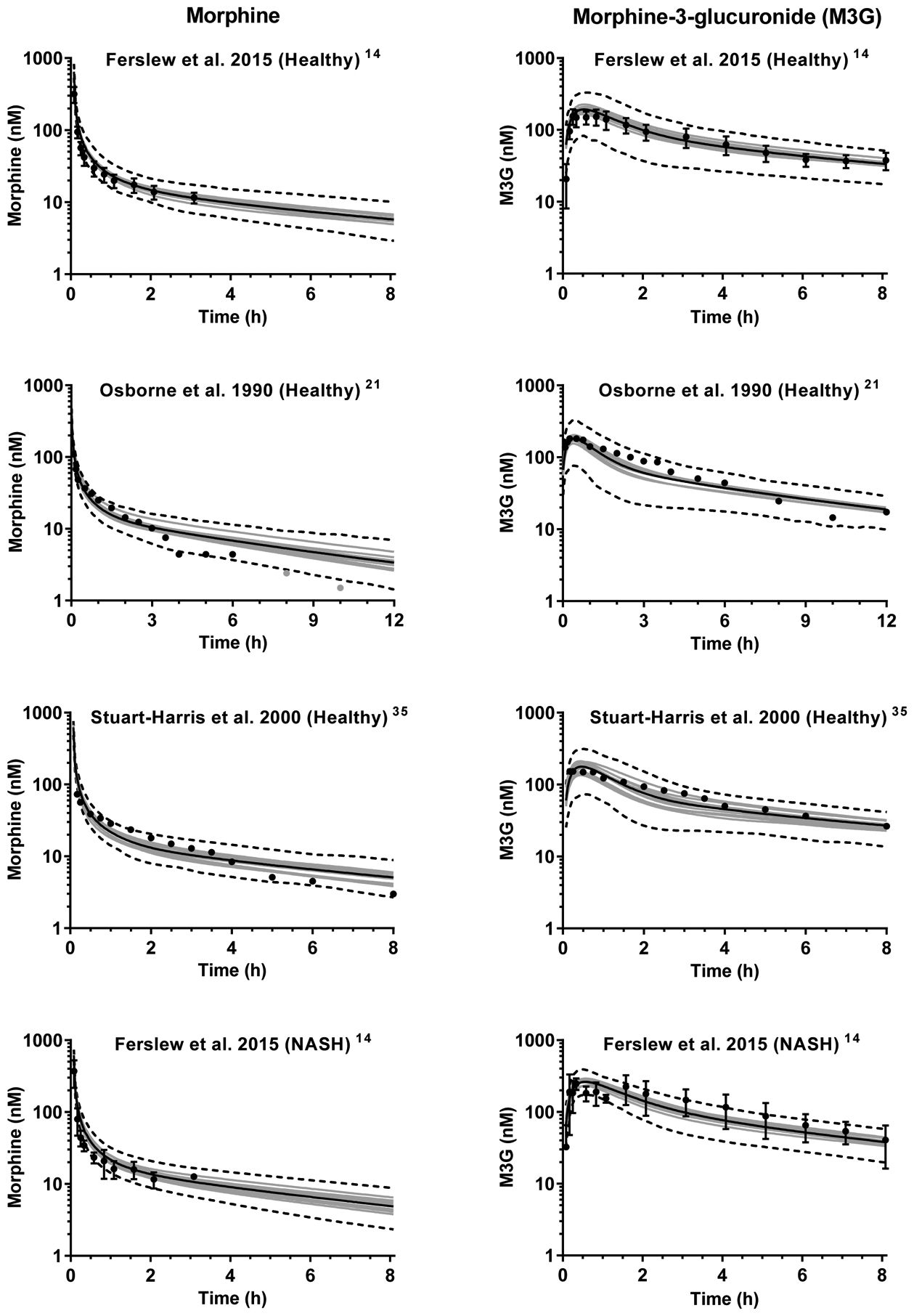
Simulated plasma concentration-time profiles of morphine (left panel) and morphine-3-glucuronide (M3G; right panel) after intravenous administration of 3.76 mg morphine (corresponding to 5 mg morphine sulfate) to virtual subjects in ten trials. The number of subjects in each virtual trial was matched with the corresponding clinical study. Gray lines represent the mean of one trial and the black solid line represents the mean of all trials. Dashed lines show the simulated 5th and 95th percentiles. Simulations are overlaid with mean plasma/serum data for fourteen14, ten21 and six35 healthy volunteers and seven patients with nonalcoholic steatohepatitis (NASH).14 The error bars in the observed data from Ferslew et al.14 show the 95% confidence interval. The light gray points in the Osborne et al.21 morphine panel indicate observed values close to the analytical lower limit of quantification.
Table 2.
Predicted and observed AUC of morphine and AUC and Cmax of M3G in plasma/serum.
| Predicted | Observed | Ratio predicted/observed | |
|---|---|---|---|
| Model development | |||
| Ferslew et al. 2015 (n=14)14 | |||
| Morphine AUC0−∞ (nM*h) | 211 ± 15 [190 – 235] |
109 ± 45 | 1.94 [1.74 – 2.16] |
| Morphine AUC0-last (nM*h)a | 115 ± 4 [109 – 121] |
88 ± 37 | 1.31 [1.24 – 1.38] |
| M3G AUC0–8h (nM*h) | 613 ± 59 [541 – 737] |
588 ± 175 | 1.04 [0.92 – 1.25] |
| M3G Cmax (nM) | 193 ± 18 [165 – 226] |
211 ± 62 | 0.91 [0.78 – 1.07] |
| Model verification | |||
| Osborne et al. 1990b (n=10)21 | |||
| Morphine AUC0−∞ (nM*h) | 170 ± 24 [144 – 223] |
114 ± 18 | 1.49 [1.26 – 1.96] |
| M3G AUC0–12h (nM*h) | 639 ± 55 [542 – 712] |
784 ± 110 | 0.82 [0.69 – 0.91] |
| M3G Cmax (nM) | 182 ± 15 [155 – 204] |
204 ± 50 | 0.89 [0.76 – 1.00] |
| Stuart-Harris et al. 2000b (n=6)34 | |||
| Morphine AUC0–8h (nM*h) | 149 ± 12 [129 – 165] |
135 ± 31 | 1.10 [0.96 – 1.22] |
| M3G AUC0–8h (nM*h) | 499 ± 84 [378 – 638] |
511 ± 128 | 0.98 [0.74 – 1.25] |
| M3G Cmax (nM) | 178 ± 28 [135 – 211] |
167 ± 38 | 1.07 [0.81 – 1.26] |
| NASH simulation | |||
| Ferslew et al. 2015 (n=7)14 | |||
| Morphine AUC0−∞ (nM*h) | 183 ± 20 [160 – 219] |
98 ± 36 | 1.87 [1.63 – 2.23] |
| Morphine AUC0-last (nM*h)a | 104 ± 6 [96 – 115] |
77 ± 30 | 1.35 [1.25 – 1.49] |
| M3G AUC0–8h (nM*h) | 823 ± 67 [699 – 920] |
961 ± 331 | 0.86 [0.73 – 0.96] |
| M3G Cmax (nM) | 263 ± 23 [231 – 292] |
325 ± 72 | 0.81 [0.71 – 0.90] |
| NASH effectc | |||
| Morphine AUC0-last | 0.90 | 0.88 | 1.02 |
| M3G AUC0–8h | 1.34 | 1.63 | 0.82 |
| M3G Cmax | 1.36 | 1.54 | 0.88 |
Data are presented as the mean ± SD of ten simulated trials with the corresponding number of subjects (Sim-Healthy Volunteers) as the clinical trial in each simulated trial. For the NASH simulations, NASH-related transporter changes were incorporated into the model. The range of means from individual simulated trials are shown in brackets.
The area under plasma concentration-time curve (AUC) range from simulations was matched to the available data from the matching clinical study subjects.
Reported values were transformed to correspond to a dose of 5 mg morphine sulfate.
The NASH effect was calculated as the ratio of the parameter in the NASH simulation and the model development simulation. Cmax, peak concentration in plasma.
PBPK modeling in NASH incorporating changes in transporters
The plasma PK of morphine and M3G were simulated by incorporating NASH-related transporter changes into the developed model. The performance of the model was similar to the simulations for healthy subjects, with the ratio of predicted to observed values ranging from 0.81–1.87 (Table 2, Figure 2), which met the two-fold acceptance criterion. Across all simulations, the AAFE of morphine AUC was 1.48 and the AAFEs of M3G AUC and Cmax were 1.12 and 1.13, respectively. M3G exposure (AUC0–8h) was predicted to be 1.34-fold higher in patients with NASH compared to healthy controls whereas the observed value was 1.63-fold. Similarly, the predicted M3G Cmax change in patients with NASH (1.36-fold) was lower than the observed value (1.54-fold).
Evaluation of individual simulations
In addition to evaluating the mean fits, observed data for each subject from Ferslew et al.14 were overlaid with the plasma concentration-time profiles from the ten matched virtual individuals for both healthy subjects and patients with NASH. Characteristics of virtual individuals with aberrant simulated profiles were investigated to uncover possible reasons for these abnormalities. Examples of cases are presented in Figure 3. High OCT1 values (> 2.8-fold of average) resulted in lower morphine concentrations in some simulated subjects (Subject A2 and B2), whereas a low OCT1 value (0.11-fold of control) increased morphine concentrations (Subject B1). In these examples, individuals with a low MRP3/MRP2 ratio (≤ 0.15) and high OCT1 activity (> 2.1-fold of average) showed relatively low and sustained concentrations of M3G over the simulation period (Subject A1 and B2). M3G concentrations were also lower with a combination of lower than average UGT2B7 activity (0.78-fold of average) and a low MRP3/MRP2 ratio (0.12) (Subject B3). In this individual, OCT1 activity was slightly below average (0.75-fold of average) and the slope of the M3G terminal phase was similar to other curves despite the low MRP3/MRP2 ratio. For Subject A2, a combination of high OCT1 (3.05-fold of average), high metabolism to M3G (1.40-fold of average) and a high MRP3/MRP2 ratio (7.30) resulted in high M3G exposure.
Figure 3.
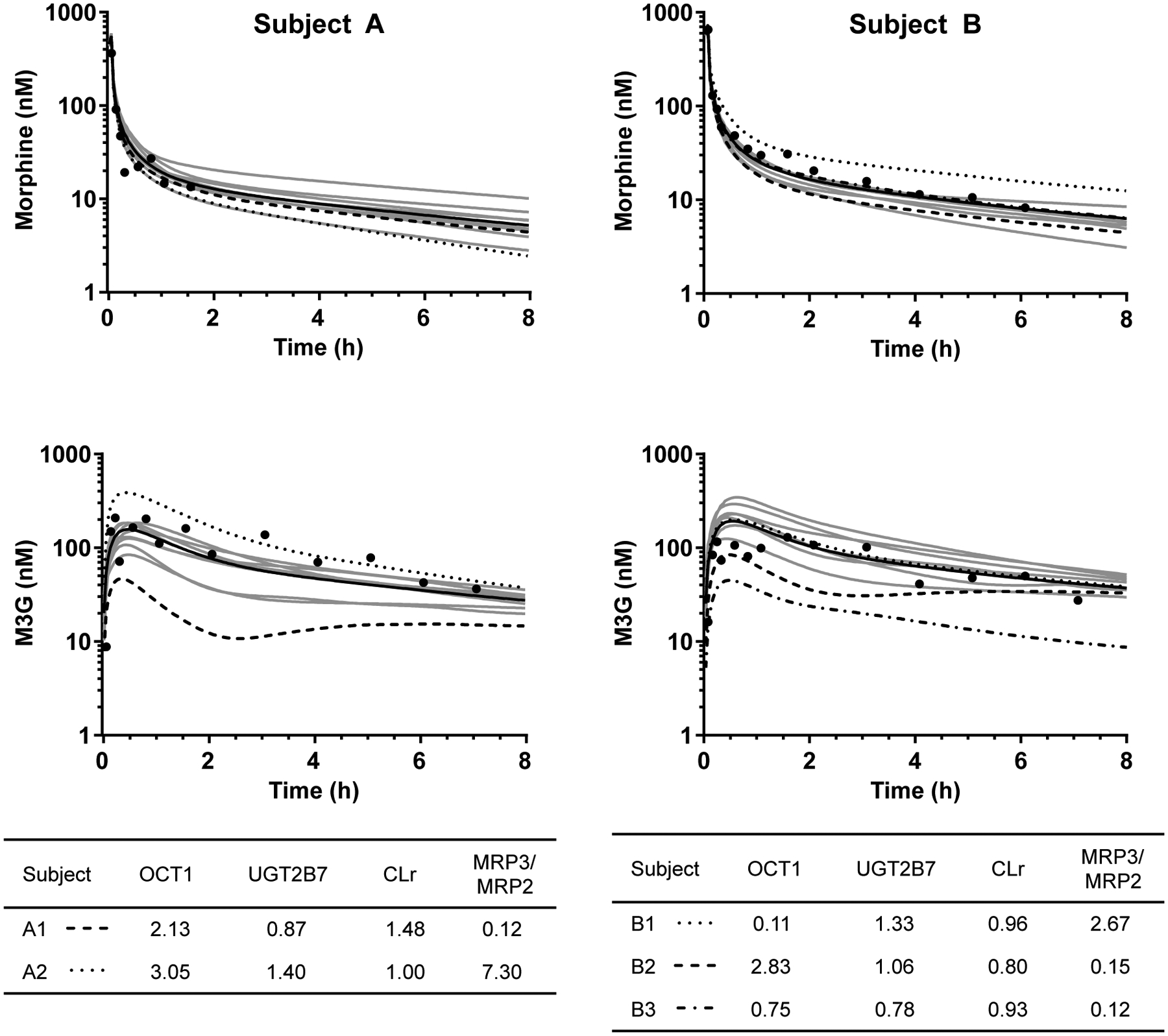
Two examples of simulated individual plasma concentration-time profiles of morphine and morphine-3-glucuronide (M3G) in healthy subjects. Shown are observed concentrations (black circles), the mean of simulated profiles of ten virtual subjects matched for age, weight, height and sex with the corresponding study subject (black line) and individual profiles of the virtual subjects. With this matching, virtual individuals share characteristics linked to body surface area and age (e.g., liver weight), but have individual enzyme and transporter phenotypes. Specific features of highlighted individuals are given in tables. Values in the OCT1, UGT2B7 and CLr columns are normalized to the population mean. OCT1, maximal hepatic uptake clearance of morphine; UGT2B7, maximal hepatic clearance of morphine to M3G; CLr, renal clearance of morphine; MRP3/MRP2, ratio of maximal basolateral and canalicular efflux clearance.
Exploratory simulations
There are differences in the demographics of the populations simulated using the Simcyp library populations for obese and morbidly obese subjects (Supplementary data Table S1). Body and liver weight were increased as was the volume of distribution at steady-state (Vss). Notably, the GFR in simulated obese and morbidly obese individuals was 30% and 65% higher, respectively, than healthy volunteers. Hepatic blood flow was not different between the populations. Morphine AUC0–12h was not markedly affected by obesity (<20% decrease), but mean M3G AUC0–12h decreased by 25% in obese and 45% in morbidly obese populations compared to the healthy population (Figure 4A and B). In subsequent simulations, the obese population was used, but the GFR was set equal to that of the healthy population.
Figure 4.
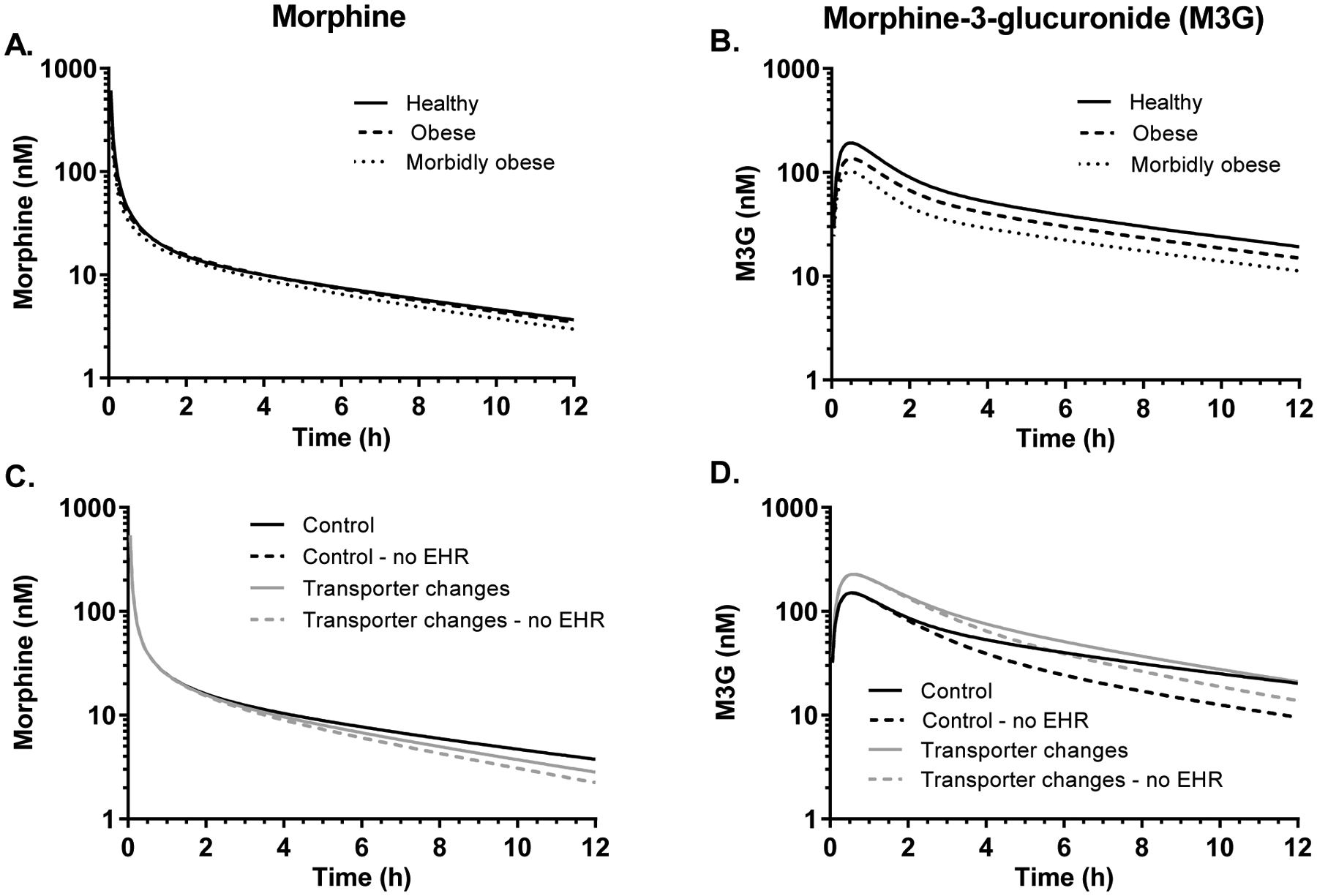
Simulated plasma concentration-time profiles of morphine (left panel) and morphine-3-glucuronide (M3G; right panel) from exploratory simulations. One hundred virtual individuals were simulated using the Simcyp library healthy volunteers, obese and morbidly obese populations (A and B). In panels C and D, simulations were performed using the “Sim-obese” population where glomerular filtration rate (GFR) was set to match the healthy volunteers (Control) and the same populations with NASH-related transporter changes with and without enterohepatic recycling (EHR). In the simulations with no EHR, the luminal hydrolysis of M3G to morphine was disabled. All results are shown as the mean of all simulations. In C, the “Control – no EHR” and the “Transporter changes – no EHR” profiles are overlapping.
The contribution of EHR was simulated by disabling the hydrolysis of M3G to morphine in the gastrointestinal lumen, which is the major component of morphine EHR.28, 29 The M3G AUC0–12h was 21% lower when the hydrolysis of M3G to morphine was disabled in the model; the morphine AUC0–12h was decreased by 9% (Figure 4 C and D, Table 3). When NASH-related transporter changes were included in the model, the difference in M3G AUC0–12h with and without EHR was approximately 13%.
Table 3.
Results from the simulations investigating the effect of enterohepatic recycling (EHR) and different nonalcoholic steatohepatitis (NASH)–related physiological changes on morphine and morphine-3-glucuronide (M3G) AUC.
| Condition | Morphine | M3G | |||
|---|---|---|---|---|---|
| AUC0–12h (nM*h) | Ratio (condition/ control)a | AUC0–12h (nM*h) | Ratio (condition/ control)a |
||
| Control (Obese with healthy GFR) | 175 ± 7 | 628 ± 88 | |||
| No EHR | 160 ± 5 | 0.91 ± 0.05 | 494 ± 76 | 0.79 ± 0.16 | |
| Transporter changes with no EHR | 160 ± 5 | 0.91 ± 0.05 (1 ± 0.05b) |
781 ± 84 | 1.24 ± 0.22 (1.58 ± 0.30b) |
|
| Reduced GFR by 14% | 178 ± 7 | 1.02 ± 0.06 | 722 ± 102 | 1.15 ± 0.23 | |
| Reduced portal vein blood flow by 15% | 188 ± 6 | 1.07 ± 0.06 | 612 ± 85 | 0.97 ± 0.19 | |
| Reduced portal vein blood flow by 30% | 205 ± 6 | 1.17 ± 0.06 | 591 ± 82 | 0.94 ± 0.19 | |
| Transporter changes | 166 ± 6 | 0.95 ± 0.05 (1.04 ± 0.05c) |
879 ± 86 | 1.40 ± 0.24 (1.13 ± 0.16c) |
|
| Transporter changes and reduced GFR by 14% | 169 ± 7 | 0.97 ± 0.05 | 1022 ± 100 | 1.63 ± 0.28 | |
| Transporter changes, reduced GFR by 14% and portal vein blood flow by 30% | 195 ± 6 | 1.11 ± 0.06 | 970 ± 95 | 1.54 ± 0.26 | |
One hundred obese fasted subjects, 50% female, age 33–63 years were simulated for each condition. The control represents the virtual obese population with the glomerular filtration rate (GFR) equal to healthy volunteers. Transporter changes include simultaneous scaling of multidrug-resistance associated protein (MRP) 2 and MRP3 relative activity by 0.5- and 2.5-fold of control, respectively. In the simulations with no EHR, the hydrolysis of M3G in the gut lumen to morphine was disabled. Data are reported as mean ± SD area under the plasma concentration-time curve from 0 to 12 h (AUC0–12h).
The ratio (condition/control) represents the simulated condition to the control value (Obese with healthy GFR), except where otherwise noted.
represents a comparison between the ‘Transporter changes with no EHR’ and ‘No EHR’;
represents a comparison between ‘Transporter changes” and the ‘Transporter changes with no EHR’.
To examine the effects of reduced GFR, the baseline healthy GFR was modified to simulate the 14% decrease observed in patients with NASH by Targher et al.38. The reduction in GFR had little effect on morphine exposure, but increased M3G exposure by 15% (Table 3, Supplementary Figure S1 A and B). Reducing portal vein blood flow by 15% and 30% in simulations led, in both cases, to ≤ 6% decrease in M3G exposure, but the 30% reduction in portal vein flow increased morphine AUC0–12h by 17% (Table 3, Supplementary Figure S1 C and D).
Of the tested NASH-related physiological changes, NASH-mediated transporter alterations (i.e., simultaneous scaling of MRP2 and MRP3 relative activity by 0.5- and 2.5-fold of control, respectively) had the highest effect on M3G exposure (Table 3, Supplementary Figure S1 E and F). M3G AUC was increased 40%, but morphine exposure was not markedly altered. The combination of transporter changes, reduced GFR and a 30% decrease in portal vein blood flow increased the exposure of M3G by 54% compared to the control group (Table 3, Supplementary Figure S1 G and H).
Discussion
Alongside the obesity epidemic, the prevalence of NAFLD and NASH is increasing rapidly.3 Recently, the worldwide COVID-19 pandemic has raised concerns that NAFLD may predispose patients to SARS-CoV-2 infection and COVID-19 complications, and that COVID-19 could expedite NAFLD progression.6 However, this remains to be studied. At present, NASH is one of the top three causes of hepatic transplants in the U.S. and there is no direct treatment for NASH beyond lifestyle changes.3 NASH patients are often burdened by other diseases such as cardiovascular disease and type 2 diabetes that require drug treatment.3, 4 Therefore, understanding NASH-associated alterations in drug PK is not only important for the development of drugs to treat NASH, but also for ensuring safe treatment of other comorbidities in patients with NASH using currently-marketed drugs.
One of the challenges of predicting exposure in this patient population is that NAFLD is progressive. Liver size and composition can vary and it has been shown that there are differences in transporter abundance not only between normal and NAFLD livers, but also between simple steatosis and NASH, as well as NASH with fatty liver and NASH without fatty liver.7, 11 However, it is difficult to determine the stage of liver disease of a patient or a study subject, because the most reliable way to evaluate steatosis and fibrosis in NAFLD and to diagnose NASH is liver biopsy, which is invasive.4 Typically, the diagnosis is determined by the presence of hepatic steatosis based on imaging and histology, no evidence of significant alcohol consumption and no competing causes of steatosis or chronic liver disease.
Due to the challenges of diagnosing NASH stage and evaluating the associated degree of changes in transporters and metabolic enzymes, it may be difficult to make PK predictions on an individual level. However, PBPK modeling can be used to gauge interindividual variability and the risk of altered exposure and potential adverse effects. Furthermore, it can be utilized to study the effects of specific factors that cannot be distinguished from in vivo data either due to restrictive inclusion criteria in clinical studies or the complexity of the in vivo system.
In this study, a PBPK model of morphine and M3G was constructed and verified using data from healthy volunteers. By incorporating NASH-related changes in transporter abundance and activity into the model, M3G exposure was predicted to increase 1.34-fold compared to healthy subjects, consistent with increased M3G exposure in patients with NASH.14 The increase was driven by the combined increased MRP3 (2.5-fold) and decreased MRP2 function (0.5-fold). The changes in MRP2 and MRP3 function had the greatest effect on M3G exposure in our exploratory simulations. Although there is some evidence for increased total protein abundance of MRP2 in NASH, changes in glycosylation and localization in hepatocytes suggest that decreased abundance at the canalicular membrane contribute to decreased function.7, 9 A decrease in MRP2 function is supported by a 48% decrease in biliary AUC of M3G observed in a rat model of NASH, which has similar histologic hepatic hallmarks as human NASH.30 This decrease also could explain the observed 1.6-fold increase in hepatic 99mTc-mebrofenin exposure and the ~2-fold decrease in biliary clearance observed in patients with NASH.15
Obesity is highly associated with NAFLD and NASH and it is estimated that the majority of severely obese patients undergoing bariatric surgery have NAFLD.40 Therefore, simulations were performed using obese and morbidly obese virtual populations.36 A major difference in these virtual populations compared to healthy volunteers was the increase in GFR (Supplementary Table S1), which is propagated as increased renal clearance. This increase in renal clearance explains the simulated decrease in morphine and M3G exposure in the obese and morbidly obese populations, because renal excretion is the primary route of elimination for morphine glucuronides and excretion is related to renal function.21, 41 The simulated decrease in M3G exposure in the obese populations is contradictory to a study by de Hoogd et al.42, where the plasma concentration of M3G was higher in morbidly obese patients than in healthy subjects. However, NAFLD/NASH status was not evaluated by these authors, so the effect of obesity cannot be distinguished from possible NASH-related transporter alterations.
NASH is associated with an increased risk of chronic kidney disease (CKD) and eGFR is reported to be slightly decreased (8–14%) in overweight subjects with NASH (n=80) compared with nonsteatotic matched controls (n=80), and in morbidly obese NASH subjects (n=37) compared with subjects with simple steatosis (n=111).37, 38 Although CKD risk also was reported to be increased in a Japanese overweight population with NASH, statistical testing for differences in eGFR were inconclusive.43 For the simulations corresponding to the patients with NASH in the Ferslew et al.14 study, GFR was not adjusted because there was no significant difference in eGFR between the study cohorts. However, in the present study, the effect of decreased GFR was simulated in the exploratory simulations and a 14% decrease in GFR resulted in a 15% increase in M3G exposure. The combination of reduced GFR with NASH-related transporter changes increased M3G exposure to similar levels as observed in the clinical study by Ferslew et al.14. Decreases in portal vein blood flow primarily affected morphine exposure, which is consistent with previous work showing that morphine clearance is sensitive to changes in cardiac output in both adults and children.31
One of the novel features of this PBPK model is the incorporation of morphine EHR. In the exploratory simulations, EHR was shown to account for 21% of M3G exposure (M3G AUC0–12h decreased from 628 to 494 nM*h in the absence of M3G hydrolysis). Due to the extensive first-pass metabolism of morphine to M3G, the effect on morphine exposure was slightly less (9%). The effect of EHR was most pronounced when the ratio of MRP3 to MRP2 transport was low (i.e., canalicular efflux was dominant) and OCT1 transport was higher than average, resulting in sustained M3G concentration profiles (Figure 3). In line with this, EHR had less of an impact in the NASH population, because M3G excretion into bile via MRP2 is diminished in NASH (Table 3, Figure 4.). This difference in EHR between healthy subjects and patients with NASH could be important for some drugs (e.g., ezetimibe) that have an intestinal target and undergo extensive glucuronidation and EHR.44 Another factor to consider regarding EHR is possible alterations in the intestinal microbiome in NASH that could affect glucuronide cleavage by gut bacteria. These alterations are supported by observed increases in secondary bile acids that are formed in the gut by the gut microflora in patients with NASH.45
A limitation of this study is that it is focused on M3G, which is the inactive glucuronide metabolite of morphine. Only M3G data were incorporated in the model due to the large number of M6G concentrations that were below the limit of quantification and the resulting low confidence in estimating the clearance values for MRP2 and MRP3 efflux. Based on the clinical data, however, NASH effects appear similar for M6G and M3G.14
To avoid overparameterization, the model focused on the major transporters for morphine (OCT1) and M3G (MRP2 and MRP3). However, in vitro data indicate that morphine also is a substrate of OATP2B1,46 and clinical data suggest that P-glycoprotein may affect morphine absorption.47 Therefore, inclusion of these transporters could be important for modeling oral dosing data, which was not the focus in this study. In addition, studies in HEK293-OCT2 cells suggest that morphine is a substrate of the renal transporter OCT2.48 However, the influence of OCT2 on morphine disposition is expected to be low since only < 10% of morphine is recovered unchanged in urine.20, 21. Although M3G is not a substrate of OATP2B1,48 data from uptake studies in hepatocytes suggest that OATP1B transporters are involved in M3G uptake into the liver.11 The significance of OATP1B transporters, which are altered in NASH,9, 11, 12, 15 on M3G disposition in healthy subjects and in NASH remains to be further evaluated to address identifiability issues between OATP1B uptake and MRP3 efflux at the hepatic basolateral membrane. Even a clinical study focusing on the effects of OATP1B1 genotypes on M3G disposition could only help to answer this question if the OATP1B contribution is significantly affecting the PK of M3G.
Another point to consider is that although NAFLD and NASH affect the liver directly, the disease could result in compensatory changes elsewhere in the body. Unfortunately, there is a lack of information on transporter alterations in the intestine and kidneys of NASH patients, but changes might be expected based on findings from rodent models of NASH.49, 50 Active secretion in the kidney does not appear to be significant for M3G, but renal metabolism of morphine does contribute to M3G formation.20, 41, 51 More clinical data are needed to incorporate NASH-related changes in tissues other than the liver, for example the intestine, into the PBPK model. Another limitation is that in the model, body surface area is used to scale organ size, which results in larger organs for obese subjects. However, it is known that in addition to increased liver size with obesity, the composition of the liver is altered in NASH by definition, and the functional mass might be decreased with disease progression. Further work and clinical data are required to study the effects of these changes on drug disposition.
Conclusions
The PBPK model constructed here for morphine and M3G highlighted the importance of NASH-related transporter changes for M3G disposition, and was able to capture some of the changes in M3G exposure observed in patients with NASH. Other physiological factors that are altered in NASH had less pronounced effects on M3G simulations. They should, however, be accounted for within a PBPK model for NASH since these minor effects can become more relevant in combination with each other. The contribution of EHR to M3G exposure was decreased in NASH due to altered M3G transport. More clinical data are needed to better understand NASH-related changes in the liver and other tissues in order to incorporate these into PBPK modeling. This work is an important first step, because modeling is an indispensable tool to study and predict drug disposition in complex diseases such as NASH. However, since the physiological changes in NASH are so complex, we focused on the evaluation and relevance of a limited number of transporters for the present study. More clinical studies that consider the fate of the metabolite are needed to extend our understanding of NASH, so that future models can be used to draw conclusions about the effects of NASH and its progression on other drugs/metabolites.
Supplementary Material
Table S1. Demographics of virtual populations.
Figure S1. Simulated plasma concentration-time profiles of morphine and morphine-3-glucuronide from exploratory simulations.
Supplementary Materials. Simcyp workspace file of the morphine and morphine-3-glucuronide model corresponding to the Stuart-Harris et al. (2000) verification simulation.
Study Highlights.
-
What is the current knowledge on the topic?
Nonalcoholic steatohepatitis (NASH) is increasing in prevalence. NASH-related alterations in physiology and protein expression (e.g., transporters) may affect drug exposure by altering drug disposition and elimination.
-
What question did this study address?
This study investigated the use of physiologically-based pharmacokinetic (PBPK) modeling to predict drug exposure in NASH. NASH-related changes in hepatic transporters were incorporated and the effects of other NASH-related physiological changes were investigated using morphine and morphine-3-glucuronide (M3G) as model compounds.
-
What this study adds to our knowledge?
Simulations revealed that NASH-related changes in hepatic transporters can explain a majority of the increased M3G exposure in NASH patients. The effect of transporters was more prominent than moderate decreases in GFR and portal blood flow. In addition, transporter changes resulted in decreased morphine enterohepatic recycling.
-
How might this change drug discovery, development, and/or therapeutics?
PBPK models incorporating NASH-related changes may aid in identifying drugs with increased risk of altered exposure, understanding sources of interindividual variability and helping ensure safe and efficacious drug therapy in this patient population.
Acknowledgements
The authors would like to acknowledge Certara UK (Simcyp Division) that granted free access to the Simcyp Simulator through an academic license (subject to conditions). Certara is also acknowledged for providing Phoenix® software to the UNC Eshelman School of Pharmacy, Division of Pharmacotherapy and Experimental Therapeutics, as part of the Pharsight Academic Center of Excellence Program. This work was presented, in part, at the 2019 American Society of Clinical Pharmacology and Therapeutics Annual Meeting.
Funding information
This study was supported by the Sigrid Jusélius Foundation (N.S.) and the National Institute of General Medical Sciences of the National Institutes of Health (NIH) under award number R35 GM122576 (K.L.R.B). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Conflict of Interest statement
S.N. is an employee of Certara UK Ltd.. All other authors declared no competing interests for this work.
References
- 1.Dietrich CG, Gotze O, Geier A. Molecular changes in hepatic metabolism and transport in cirrhosis and their functional importance. World J Gastroenterol 22 72–88. (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thakkar N, Slizgi JR, Brouwer KLR. Effect of Liver Disease on Hepatic Transporter Expression and Function. J Pharm Sci 106 2282–2294. (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Younossi Z, et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 69 2672–2682. (2019) [DOI] [PubMed] [Google Scholar]
- 4.Chalasani N, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67 328–357. (2018) [DOI] [PubMed] [Google Scholar]
- 5.Ji D, et al. Non-alcoholic fatty liver diseases in patients with COVID-19: A retrospective study. J Hepatol 73 449–473. (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prins GH, Olinga P. Potential implications of COVID-19 in non-alcoholic fatty liver disease. Liver Int. doi. 10.1111/liv.14484 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardwick RN, Fisher CD, Canet MJ, Scheffer GL, Cherrington NJ. Variations in ATP-binding cassette transporter regulation during the progression of human nonalcoholic fatty liver disease. Drug Metab Dispos 39 2395–2402. (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hardwick RN, et al. Altered UDP-glucuronosyltransferase and sulfotransferase expression and function during progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos 41 554–561. (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clarke JD, Novak P, Lake AD, Hardwick RN, Cherrington NJ. Impaired N-linked glycosylation of uptake and efflux transporters in human non-alcoholic fatty liver disease. Liver Int 37 1074–1081. (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fisher CD, et al. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab Dispos 37 2087–2094. (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vildhede A, Kimoto E, Pelis RM, Rodrigues AD, Varma MVS. Quantitative Proteomics and Mechanistic Modeling of Transporter-Mediated Disposition in Nonalcoholic Fatty Liver Disease. Clin Pharmacol Ther 107 1128–1137. (2020) [DOI] [PubMed] [Google Scholar]
- 12.Clarke JD, et al. Synergistic interaction between genetics and disease on pravastatin disposition. J Hepatol 61 139–147. (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canet MJ, et al. Altered regulation of hepatic efflux transporters disrupts acetaminophen disposition in pediatric nonalcoholic steatohepatitis. Drug Metab Dispos 43 829–835. (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferslew BC, et al. Altered morphine glucuronide and bile acid disposition in patients with nonalcoholic steatohepatitis. Clin Pharmacol Ther 97 419–427. (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ali I, et al. Transporter-Mediated Alterations in Patients With NASH Increase Systemic and Hepatic Exposure to an OATP and MRP2 Substrate. Clin Pharmacol Ther 104 749–756. (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barshop NJ, Capparelli EV, Sirlin CB, Schwimmer JB, Lavine JE. Acetaminophen pharmacokinetics in children with nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr 52 198–202. (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tirona RG, et al. Apixaban and Rosuvastatin Pharmacokinetics in Nonalcoholic Fatty Liver Disease. Drug Metab Dispos 46 485–492. (2018) [DOI] [PubMed] [Google Scholar]
- 18.Sundelin EIO, et al. Hepatic exposure of metformin in patients with non-alcoholic fatty liver disease. Br J Clin Pharmacol 85 1761–1770. (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sawe J, Kager L, Svensson Eng JO, Rane A. Oral morphine in cancer patients: in vivo kinetics and in vitro hepatic glucuronidation. Br J Clin Pharmacol 19 495–501. (1985) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hasselstrom J, Sawe J. Morphine pharmacokinetics and metabolism in humans. Enterohepatic cycling and relative contribution of metabolites to active opioid concentrations. Clin Pharmacokinet 24 344–354. (1993) [DOI] [PubMed] [Google Scholar]
- 21.Osborne R, Joel S, Trew D, Slevin M. Morphine and metabolite behavior after different routes of morphine administration: demonstration of the importance of the active metabolite morphine-6-glucuronide. Clin Pharmacol Ther 47 12–19. (1990) [DOI] [PubMed] [Google Scholar]
- 22.Wandel C, Kim R, Wood M, Wood A. Interaction of morphine, fentanyl, sufentanil, alfentanil, and loperamide with the efflux drug transporter P-glycoprotein. Anesthesiology 96 913–920. (2002) [DOI] [PubMed] [Google Scholar]
- 23.Stone AN, Mackenzie PI, Galetin A, Houston JB, Miners JO. Isoform selectivity and kinetics of morphine 3- and 6-glucuronidation by human udp-glucuronosyltransferases: evidence for atypical glucuronidation kinetics by UGT2B7. Drug Metab Dispos 31 1086–1089. (2003) [DOI] [PubMed] [Google Scholar]
- 24.Tzvetkov MV, et al. Morphine is a substrate of the organic cation transporter OCT1 and polymorphisms in OCT1 gene affect morphine pharmacokinetics after codeine administration. Biochem Pharmacol 86 666–678. (2013) [DOI] [PubMed] [Google Scholar]
- 25.Morrish GA, Foster DJ, Somogyi AA. Differential in vitro inhibition of M3G and M6G formation from morphine by (R)- and (S)-methadone and structurally related opioids. Br J Clin Pharmacol 61 326–335. (2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zelcer N, et al. Mice lacking multidrug resistance protein 3 show altered morphine pharmacokinetics and morphine-6-glucuronide antinociception. Proc Natl Acad Sci U S A 102 7274–7279. (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van de Wetering K, et al. Multidrug resistance proteins 2 and 3 provide alternative routes for hepatic excretion of morphine-glucuronides. Mol Pharmacol 72 387–394. (2007) [DOI] [PubMed] [Google Scholar]
- 28.Walsh CT, Levine RR. Studies of the enterohepatic circulation of morphine in the rat. J Pharmacol Exp Ther 195 303–310. (1975) [PubMed] [Google Scholar]
- 29.Parker RJ, Hirom PC, Millburn P. Enterohepatic recycling of phenolphthalein, morphine, lysergic acid diethylamide (LSD) and diphenylacetic acid in the rat. Hydrolysis of glucuronic acid conjugates in the gut lumen. Xenobiotica 10 689–703. (1980) [DOI] [PubMed] [Google Scholar]
- 30.Dzierlenga AL, et al. Mechanistic basis of altered morphine disposition in nonalcoholic steatohepatitis. J Pharmacol Exp Ther 352 462–470. (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emoto C, et al. Characterization of Contributing Factors to Variability in Morphine Clearance Through PBPK Modeling Implemented With OCT1 Transporter. CPT Pharmacometrics Syst Pharmacol 6 110–119. (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petri N, et al. Absorption/metabolism of sulforaphane and quercetin, and regulation of phase II enzymes, in human jejunum in vivo. Drug Metab Dispos 31 805–813. (2003) [DOI] [PubMed] [Google Scholar]
- 33.Kenyon EM, Calabrese EJ. Extent and implications of interspecies differences in the intestinal hydrolysis of certain glucuronide conjugates. Xenobiotica 23 373–381. (1993) [DOI] [PubMed] [Google Scholar]
- 34.Pollack GM, Spencer AP, Horton TL, Brouwer KL. Site-dependent intestinal hydrolysis of valproate and morphine glucuronide in the developing rat. Drug Metab Dispos 22 120–123. (1994) [PubMed] [Google Scholar]
- 35.Stuart-Harris R, Joel SP, McDonald P, Currow D, Slevin ML. The pharmacokinetics of morphine and morphine glucuronide metabolites after subcutaneous bolus injection and subcutaneous infusion of morphine. Br J Clin Pharmacol 49 207–214. (2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghobadi C, et al. Application of a systems approach to the bottom-up assessment of pharmacokinetics in obese patients: expected variations in clearance. Clin Pharmacokinet 50 809–822. (2011) [DOI] [PubMed] [Google Scholar]
- 37.Machado MV, et al. Impaired renal function in morbid obese patients with nonalcoholic fatty liver disease. Liver Int 32 241–248. (2012) [DOI] [PubMed] [Google Scholar]
- 38.Targher G, et al. Relationship between kidney function and liver histology in subjects with nonalcoholic steatohepatitis. Clin J Am Soc Nephrol 5 2166–2171. (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shigefuku R, et al. Correlations of Hepatic Hemodynamics, Liver Function, and Fibrosis Markers in Nonalcoholic Fatty Liver Disease: Comparison with Chronic Hepatitis Related to Hepatitis C Virus. Int J Mol Sci 17 doi. 10.3390/ijms17091545. (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Subichin M, et al. Liver disease in the morbidly obese: a review of 1000 consecutive patients undergoing weight loss surgery. Surg Obes Relat Dis 11 137–141. (2015) [DOI] [PubMed] [Google Scholar]
- 41.Milne RW, Nation RL, Somogyi AA, Bochner F, Griggs WM. The influence of renal function on the renal clearance of morphine and its glucuronide metabolites in intensive-care patients. Br J Clin Pharmacol 34 53–59. (1992) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Hoogd S, et al. Influence of Morbid Obesity on the Pharmacokinetics of Morphine, Morphine-3-Glucuronide, and Morphine-6-Glucuronide. Clin Pharmacokinet 56 1577–1587. (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yasui K, et al. Nonalcoholic steatohepatitis and increased risk of chronic kidney disease. Metabolism 60 735–739. (2011) [DOI] [PubMed] [Google Scholar]
- 44.Patrick JE, et al. Disposition of the selective cholesterol absorption inhibitor ezetimibe in healthy male subjects. Drug Metab Dispos 30 430–437. (2002) [DOI] [PubMed] [Google Scholar]
- 45.Ferslew BC, et al. Altered Bile Acid Metabolome in Patients with Nonalcoholic Steatohepatitis. Dig Dis Sci 60 3318–3328. (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang ZZ, et al. siRNA capsulated brain-targeted nanoparticles specifically knock down OATP2B1 in mice: a mechanism for acute morphine tolerance suppression. Sci Rep 6 33338 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kharasch ED, Hoffer C, Whittington D, Sheffels P. Role of P-glycoprotein in the intestinal absorption and clinical effects of morphine. Clin Pharmacol Ther 74 543–554. (2003) [DOI] [PubMed] [Google Scholar]
- 48.Zhu P, et al. Irinotecan Alters the Disposition of Morphine Via Inhibition of Organic Cation Transporter 1 (OCT1) and 2 (OCT2). Pharm Res 35 243 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Canet MJ, et al. Renal xenobiotic transporter expression is altered in multiple experimental models of nonalcoholic steatohepatitis. Drug Metab Dispos 43 266–272. (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laho T, et al. Effect of nonalcoholic steatohepatitis on renal filtration and secretion of adefovir. Biochem Pharmacol 115 144–151. (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mazoit JX, Sandouk P, Scherrmann JM, Roche A. Extrahepatic metabolism of morphine occurs in humans. Clin Pharmacol Ther 48 613–618. (1990) [DOI] [PubMed] [Google Scholar]
- 52.Avdeef A, Barrett DA, Shaw PN, Knaggs RD, Davis SS. Octanol-, chloroform-, and propylene glycol dipelargonat-water partitioning of morphine-6-glucuronide and other related opiates. J Med Chem 39 4377–4381. (1996) [DOI] [PubMed] [Google Scholar]
- 53.Kaufman JJ, Semo NM, Koski WS. Microelectrometric titration measurement of the pKa’s and partition and drug distribution coefficients of narcotics and narcotic antagonists and their pH and temperature dependence. J Med Chem 18 647–655. (1975) [DOI] [PubMed] [Google Scholar]
- 54.Skopp G, Potsch L, Ganssmann B, Aderjan R, Mattern R. A preliminary study on the distribution of morphine and its glucuronides in the subcompartments of blood. J Anal Toxicol 22 261–264. (1998) [DOI] [PubMed] [Google Scholar]
- 55.Rodgers T, Leahy D, Rowland M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci 94 1259–1276. (2005) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Demographics of virtual populations.
Figure S1. Simulated plasma concentration-time profiles of morphine and morphine-3-glucuronide from exploratory simulations.
Supplementary Materials. Simcyp workspace file of the morphine and morphine-3-glucuronide model corresponding to the Stuart-Harris et al. (2000) verification simulation.