

Abstract
De Novo metalloprotein design assesses the relationship between metal active site architecture and catalytic reactivity. Herein, we use an α-helical scaffold to control the iron coordination geometry when a heme cofactor is allowed to bind to either histidine or cysteine ligands, within a single artificial protein. Consequently, we uncovered a reversible pH-induced switch of the heme axial ligation within this simplified scaffold. Characterization of the specific heme coordination modes was done by using UV-Vis and Electron Paramagnetic Resonance spectroscopies. The penta- or hexa-coordinate thiolate heme (9 ≤ pH ≤ 11) and the penta-coordinate imidazole heme (6 ≤ pH ≤ 8.5) reproduces well the heme ligation in chloroperoxidases or cyt P450 monooxygenases and peroxidases, respectively. The stability of heme coordination upon ferric/ferrous redox cycling is a crucial property of the construct. At basic pHs, the thiolate mini-heme protein can catalyze O2 reduction when adsorbed onto a pyrolytic graphite electrode.
Keywords: De Novo Protein Design, cyt P450 monooxygenase, Heme Enzymes, thiolate ligands, EPR spectroscopy
Graphical Abstract
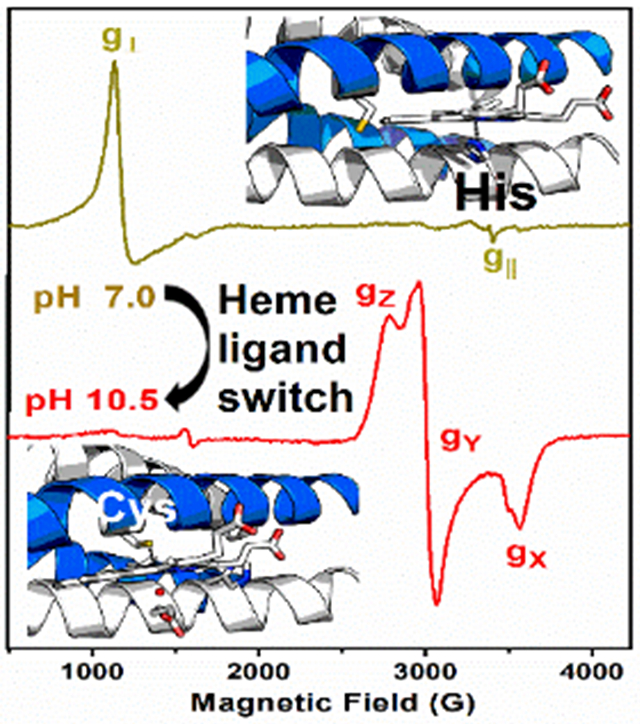
A pH-dependent preference for the heme ligation is achieved when using self-assembling α-helical coiled coils having Cys and His as binding sites. EPR and UV-Vis absorption spectroscopies uncovered a switch in heme axial ligand from His (6 ≤ pH ≤ 8.5) to Cys (9 ≤ pH ≤ 11), mimicking the heme sites in cyt P450 monooxygenase, chloroperoxidase and peroxidases, all three in a single artificial protein.
Protein engineering has developed into a powerful tool to interrogate biomolecular function using well-defined changes to enzyme active site structure. De Novo protein design has emerged as a subfield allowing assessment of fundamental features controlling protein structure and function. [1, 2] In particular, De Novo metalloprotein design has provided detailed insight into the relationship between the architecture of metal active sites and electron transfer or catalytic reactions. [3] In many cases, these synthetic systems utilize highly stable α-helical scaffolds capable of binding a broad range of metals and metal cofactors (reviewed in [4]). Among the most important and ubiquitous natural metalloenzymes are those containing the iron porphyrin cofactor, known as heme. Heme-containing proteins are involved in fundamental biological processes such as oxidative metabolism, oxygen storage and transport, signal transduction and drug metabolism.[5] The heme site is capable of catalyzing a variety of biologically-relevant chemical reactions, such as disproportionation of hydrogen peroxide (catalases[6]), oxidation and haloperoxidation of organic substrates (monofunctional heme peroxidases,[7] bi-functional catalase-peroxidases[8]) and oxygenation reactions (cyt P450 monooxygenases (Fig. S1) and nitric oxide synthases[7]). Such a functional versatility relies upon variations in the heme coordination (i.e. His, Tyr, Cys or Met axial ligand(s)), as well as being significantly modulated by the heme environment (2nd coordination sphere and H-bond networks). Studies of heme active sites using mutational analysis, structural biology and molecular spectroscopy provide essential information on the natural systems that delimit the catalytic processes. Yet, the complexity of the natural systems often masks the specific behavior of the metal cofactor. For this reason, small artificial proteins capable of binding heme at specified sites are being developed. [9,10] Our approach is to use α-helical scaffolds to provide ligation to the heme as cofactor to explore heme axial ligand selectivity, specifically hoping to define how one can control the iron coordination geometry when a heme is afforded an opportunity to bind either histidine (imidazole) or cysteine (thiolate) ligands. While many studies of de novo designed scaffolds with heme binding to nitrogenous ligands have been reported (reviewed in [11]), there have been limited studies examining either synthetic thiolate-bound FeIII-porphyrin complexes ([12] and references therein) or His-to-Cys mutations in natural heme enzymes [13,14] as structural models for the cyt P450 monooxygenases and chloroperoxidases (CPO) thiolate axial ligands. The putative heme preference between such ligands has not been addressed within a single artificial protein. In this communication, we assess the preference of heme binding to self-assembling α-helical coiled coils as a function of pH. We uncover a remarkable pH-driven conversion of the heme ligation, between the 5-coordinated (5-c) histidine structures (peroxidase-like) at neutral pH to thiolate bound structures that are 5-c at pH ≥ 9 or 6-coordinated (6-c) at pH > 10. In addition to providing rare examples of water-stable heme thiolate models, this work illustrates the ability to exploit proton concentration to switch heme coordination preferences in a protein through the simple expedient of pH. Thus, along with providing fundamental insight into metalloprotein structural preferences, our studies potentially allow for the pH-induced conversion from peroxidase-like catalytic activity (imidazole) to oxygenase (thiolate) reactivity.
The GRAND peptide family has been studied extensively for transition metal and heavy metalloid binding. [15] In the apo form, they generate highly stable three stranded coiled coils (3SCCs) at pH ≥ 5, or two stranded coiled coils (2SCCs) at more acidic pHs, illustrating the structural versatility in this system. [16] Furthermore, 2SCCs with oppositely charged residues at e and g heptade positions can associate to form antiparallel 4SCCs.[17] The coiled coils are stabilized through hydrophobic interactions (typically leucines) embedded at a and d positions of repeating heptads. These sites can be modified to include metal binding residues, such as cysteine or histidine, to incorporate a stabilizing metal within the hydrophobic interior. [18] Table 1 shows the specific peptide sequences used in this work. Each leucine mutation leads to a small destabilization of the system, although two or three changes are well tolerated. [19, 20]
Table 1.
Sequences of the GRAND Peptides Used in This Study.
| Peptide | 1 | 2 | 9 | 16 | 23 | 30 | 37 |
|---|---|---|---|---|---|---|---|
| GRAND L2W (GRW) [a] | Ac-G | WKALEEK | LKALEEK | LKALEEK | LKALEEK | LKALEEK | G-NH2 |
| GRW-L16CL30H | Ac-G | WKALEEK | LKALEEK | CKALEEK | LKALEEK | HKALEEK | G-NH2 |
| GRW-L16C | Ac-G | WKALEEK | LKALEEK | CKALEEK | LKALEEK | LKALEEK | G-HN2 |
| GRW-L30H | Ac-G | WKALEEK | LKALEEK | LKALEEK | LKALEEK | HKALEEK | G-NH2 |
Peptides are amidated and acetylated at the C- and N-terminus, respectively. The first residue of each heptad is labeled with its overall position within the peptide. Heme binding residues are in bold.
All the GRAND peptides (Table 1) showed CD spectra typical of well-folded coiled-coils across a wide pH range, both with and without heme (Fig. S2 and Fig. S3A). Also, once complexed with heme, peptides were thermodynamically more stable with TM increasing by 10 °C at basic pHs (Fig. S3B). Analytical Ultracentrifugation (AUC) studies confirmed that the apo peptides fold as 3SCCs (Fig. S4A); however, the estimated molecular mass indicated formation of tetramers upon heme complexation (Fig. S4B and S4C). We believe that the resulting GRAND mini-heme proteins are best described as dimers of antiparallel 2SCCs. Consequently, heme complexation to the 3SCCs apo GRAND peptides induces the conversion to 4SCCs, with a heme at the helical interface of each antiparallel dimer (Fig. 1 and Fig. S5). Conversion of a parallel 3SCC to an antiparallel 4SCC is not without precedent. [17] A recent report showed that two mutations in a model coiled coil peptide (HA2-DEL) were sufficient to convert the parallel 3SCC construct to an antiparallel 4SCC. [21]
Figure 1.
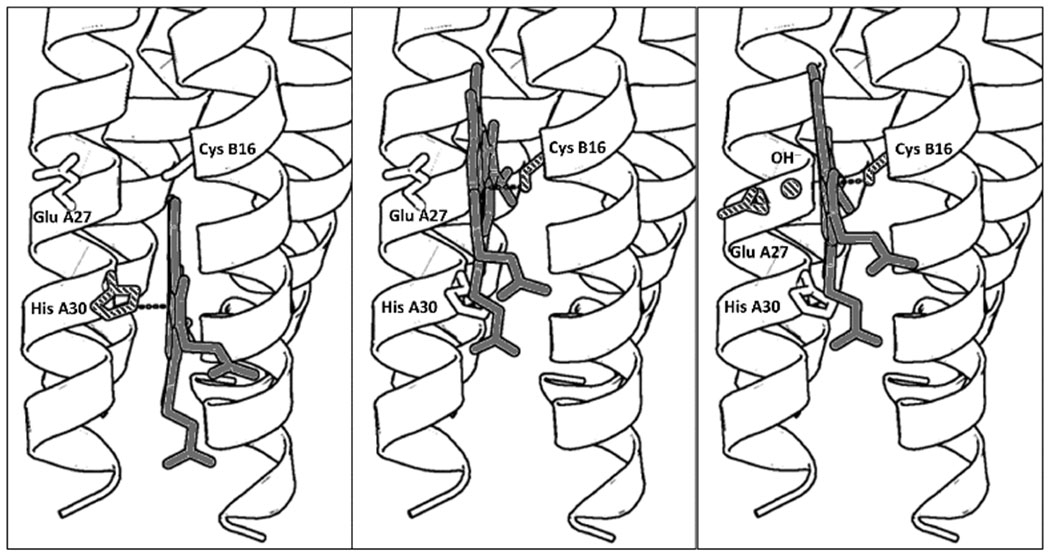
PyMol models illustrating the proposed folding of GRW-L16CL30H peptides complexed with heme (a dimer of antiparallel 2SCC) at selected pH values. The three coordination configurations concluded from our spectroscopic study, i.e. His-pentacoordinated heme (pH 7, left panel), Cys-pentacoordinated heme (pH 9.0, center panel) and Cys/hydroxide hexacoordinated heme (pH 10.5, right panel) are shown. Only the heme for one antiparallel 2SCC (A and B α-helices) is shown for clarity. The PyMol models are based on the crystal structure of a de novo designed antiparallel 4SCC (PDB code: 2B1F) as described in Supplementary Information. A color version of the models is shown in Fig. S5.
The heme to peptide stoichiometry for the mini-heme protein was assessed by monitoring the UV-Vis spectral changes of the heme. The absorption spectrum of hemin upon complexation with the GR2W-L16CL30H scaffold at pH 10.5 (Fig. 2 Inset, solid traces), a low-spin thiolate heme (see Fig. 3A), is clearly distinct to that of the hemin in buffer solution (Fig. 2 Inset, dashed traces). Hemin titration into the GRW-L16CL30H peptide showed the complexation of one heme per peptide dimer (Fig. 2). For higher hemin excess, the increasing spectral contribution of hemin in buffer solution was observed (Fig. 2 Inset, dashed trace).
Figure 2.
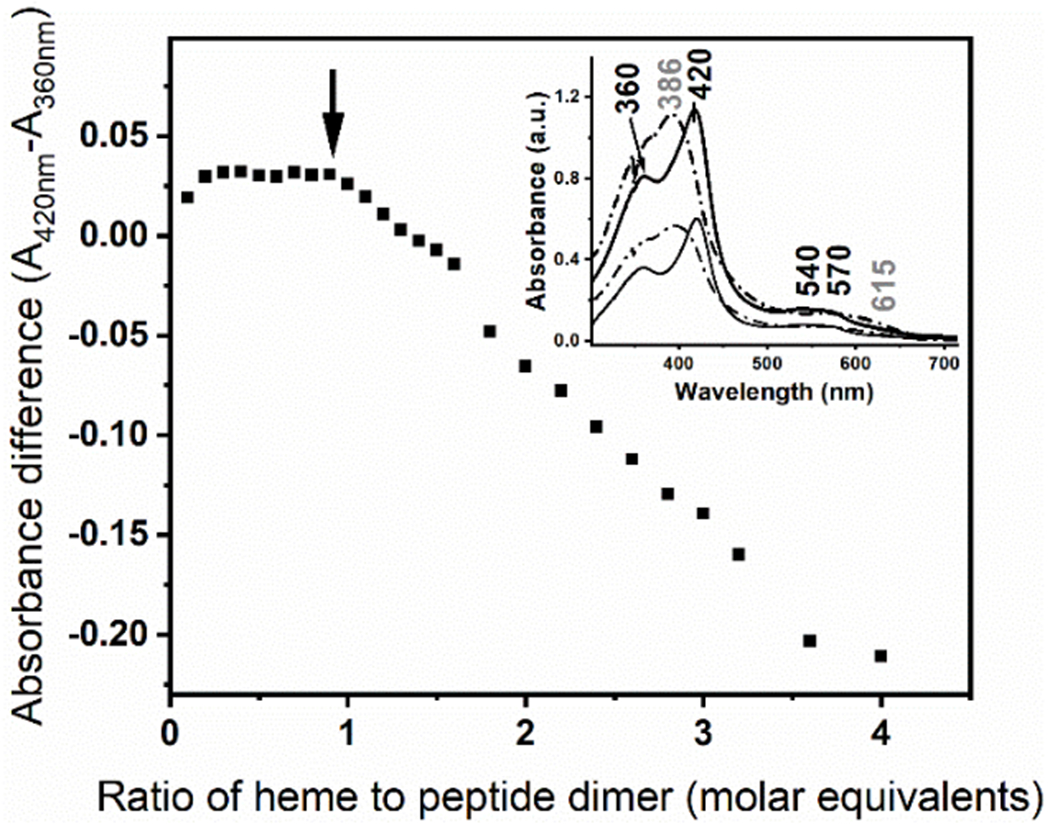
Hemin titration into the GRW-L16CL30H (20 μM monomeric peptide concentration) in 250 mM TRIS-maleate buffer at pH 10.3. The difference in absorbance of the split Soret bands (360 nm and 420 nm) of the GRW-L16CL30H mini-heme protein (Inset, solid thick trace) is plotted as a function of the added hemin to peptide-dimer ratio. It clearly shows that this ratio remains constant while there is no spectral contribution of the hemin in buffer solution. Inset: The absorption spectra of the mini-heme protein obtained with 0.5 (thin solid trace) and 1 (thick solid trace) molar equivalents of heme per peptide dimer are shown. Arithmetic spectral subtractions of the “1.5 minus 1.0” (thin dashed trace) and “2.0 minus 1.0” (thick dashed trace) experimental spectra shows the expected resulting spectrum of hemin in buffer solution in both cases.
Figure 3.
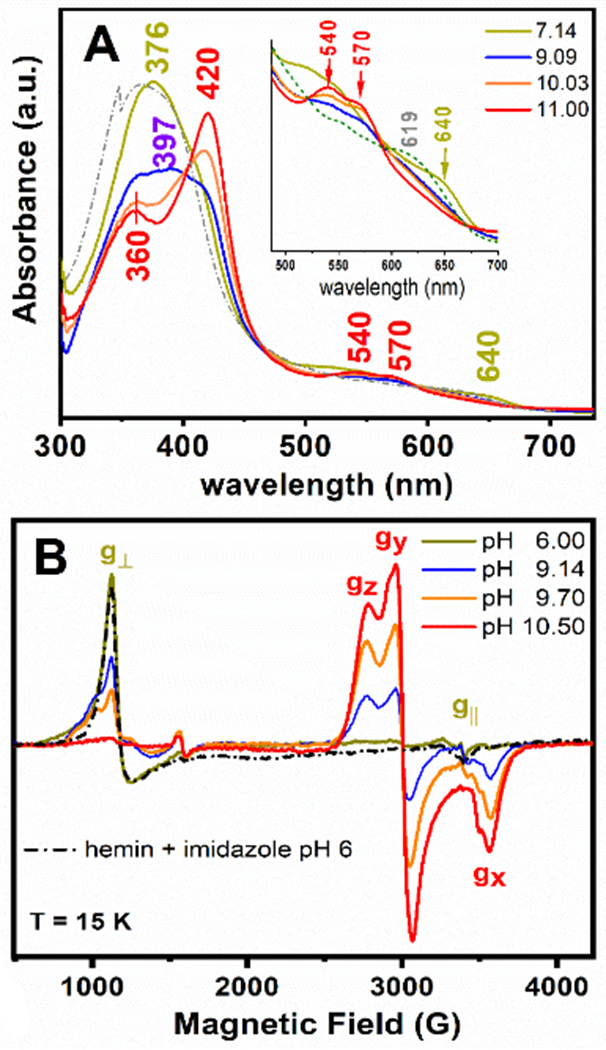
UV-Vis electronic absorption (Panel A) and 9-GHz EPR (Panel B) spectra of the GRW-L16CL30H mini-heme protein (15 μM and 300 μM heme concentration, respectively; one heme per peptide dimer). The absorption spectrum of hemin in buffer solution (pH 7, dash-dotted trace) and the EPR spectrum of hemin in imidazole buffer (pH 6, dash-dotted trace) are also shown. The pH-induced spectral changes were fully reversible. Freezing and thawing the EPR sample did not affect the ligation and the reversibility in heme ligation. EPR experimental conditions as in Fig. S6.
We combined UV-Vis and Electron Paramagnetic Resonance (EPR) spectroscopies to characterize the specific coordination mode of the heme to the GRW-L16CL30H peptide dimer in the pH range 6 to 11. Two clearly distinct absorption and EPR spectra (Fig. 3A and 3B) were obtained for neutral (olive green traces) and basic (red traces) pH values. The absorption spectrum at pH 7.14 (Fig. 3A, olive green trace) is characteristic of a 5-c heme, with a ligand-to-metal charge transfer (LMCT) band at 640 nm, reminiscent of peroxidases.[7] The corresponding axial EPR spectrum with g┴eff = 5.99 and g║eff = 1.98 resonances (Fig. 3B, olive green trace) is characteristic of heme iron in the ferric (FeIII) high-spin (HS) state, quite distinct to that of hemin in buffer solution (Fig. S5, dash-dotted gray trace) but virtually identical to that of hemin penta-coordinated to imidazole (Fig. 3B, black dash-dotted trace). Hence, considering the peptide sequence (Table 1), we can confidently conclude a 5-c heme with His30 as axial ligand at neutral pH, upon complexation with the GRW-L16CL30H dimer.
When raising the pH of the sample to 10.5 or higher, the absorption spectrum showed the total conversion to a 6-c heme (Fig. 3A, red trace), with the characteristic redshifted Soret band (to 420 nm) and the β and α bands at 540 and 570 nm, respectively. The observed split Soret band (360 nm and 420 nm) at pH ≥10 (Fig. 3A, orange and red traces) has been reported for natural proteins having a FeIII low-spin (LS) heme with thiolate ligand(s) as catalytic sites (reviewed in [22]). The new rhombic EPR signal observed for the pH 10.5 sample (Fig. 3B, red trace) is that of a ferric (S=1/2) heme iron in the LS state.[23] The g-anisotropy (Δg = gZ – gX = 0.56) of such rhombic EPR spectrum (effective g-values of gZ =2.45, gY = 2.25 and gX =1.89) is characteristic of ferric LS heme-thiolate proteins (reviewed in [22]), thus much lower than the expected value for a ferric LS His-coordinated hemes (Δg = 1.6). [23] Both the absorption and the EPR spectra of the mini-heme protein at pH ≥ 10.5 are very similar to those of cyt P450 monooxygenases, having a ferric LS heme with a Cys/thiolate and water as axial ligands. [7] Given the pH of the mini-heme protein, the ionized state of a water molecule should be the sixth ligand (Fig. 1).
The reversible change in the GRW-L16CL30H mini-heme protein from His30 axial ligation at neutral pH to Cys16/thiolate and water/hydroxyl at pH ≥ 10.5, reflects a relatively weak imidazole coordination favoring heme dissociation at basic pHs, as reported in 5-c heme maquettes, [24] and the concomitant deprotonation of Cys16 allowing a heme thiolate ligation. Accordingly, the main features of the absorption spectrum of GRW-L16CL30H mini-heme protein at pH 9.09 (Fig. 3A, blue trace), a Soret band with maximum at 397 nm and the broad LMCT band at ca. 625 nm, agree well with those of a 5-c heme with a thiolate axial ligand reported in CPO [25], the HS state of cyt P450s [26] and a 4α-helix bundle with a Cys axial ligand to a 5-c heme b [27]. The contribution of the LS 6-c species is indicative of a pH-dependent spin state equilibrium. Likewise, the 9-GHz EPR spectra (Fig. 3B) at pH 9.14 (blue trace) and 9.70 (orange trace) showed a new HS axial EPR signal (broad g┴ resonance) similar to that of hemin in cysteine buffer (Fig. S5, dashed gray trace), together with the LS-heme EPR signal observed at pH 10.5. The relative intensities of these two EPR signals for pH ≥ 9.0 were inversely proportional. These observations are best explained by heme being mostly 5-c to Cys16 at pH ≤ 9.5, and converting to a 6-c (with an OH trans to Cys16) at higher pHs. Similar spin state conversion and equilibrium of HS/LS species were reported for cyt P450 enzymes in vivo [28] and in vitro. [25]
The pH-dependent heme ligation pattern of the ferric mini-heme protein proved to be conserved upon reduction to the ferrous state (Fig. S7). In addition, the anaerobic reduction/re-oxidation cycle showed that the initial heme ligation is conserved for both His- and Cys-coordinated heme (Fig. S7). In cyt P450 and CPO enzymes, the thiolate axial ligand is crucial to exert the “push” effect on the heme iron during the catalytic reaction. [29] Cyclic voltammetry (CV) studies of the cyt P450 heme domain showed the catalytic reduction of O2, a crucial step prior to the substrate oxygenation reaction in the catalytic cycle (Fig. S1). Significantly, our CV studies on the GRW-L16CL30H mini-heme protein at pH 10.1 also proved that it can catalyze O2 reduction (Fig. 4).
Figure 4.
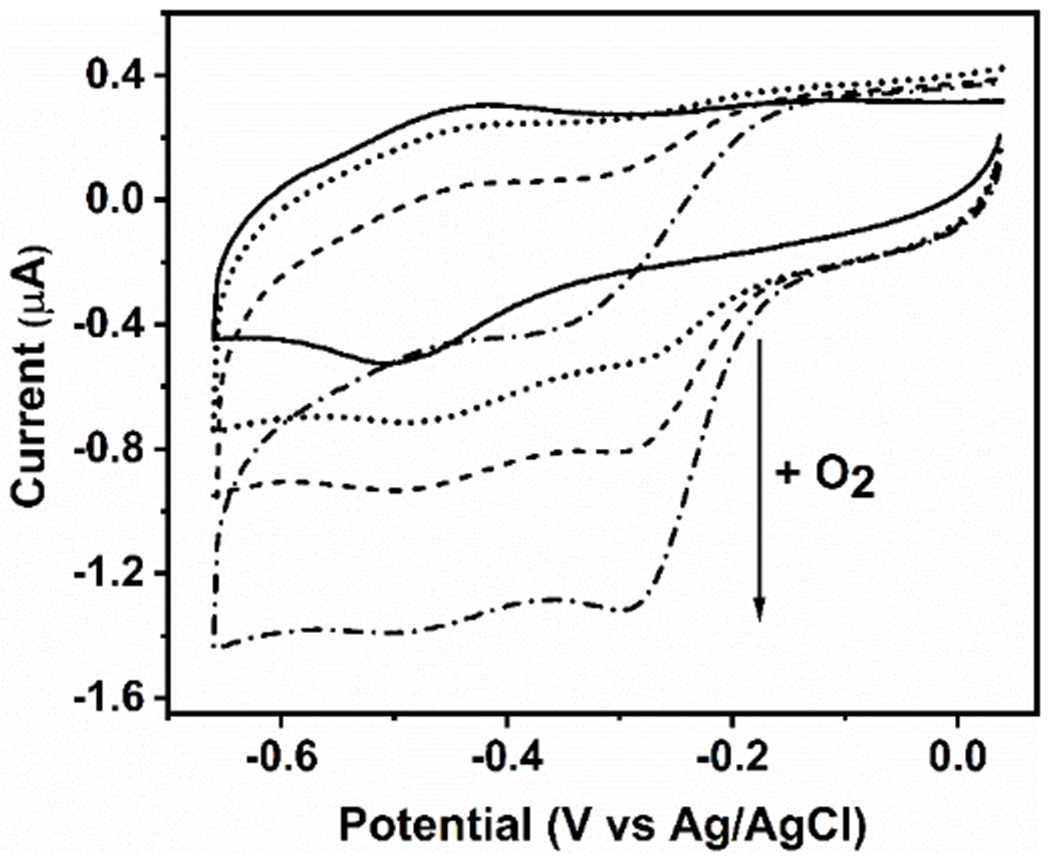
Cyclic voltammograms of the GRW-L16CL30H mini-heme protein (48 μM heme and 44 μM monomeric peptide concentrations) in 250 mM TRIS-maleate buffer at pH 10.1, entrapped in a thin layer at a pyrolytic graphite electrode, 20 mV/s scan rate. In strict anaerobic conditions, the well-defined redox peaks revealed a redox potential (E1/2) at -460 mV vs Ag/AgCl for the FeIII/FeII redox couple (solid trace). In the presence of O2, the amplitude of the additional cathodic wave, with no anodic counterpart (dotted trace), was proportional to the increase in O2 concentrations (see arrow), reflecting the catalytic reduction of dioxygen by the mini-heme protein. Experimental conditions as in Fig. S8.
We also characterized the GRAND scaffolds with single heme binding sites (Table 1). At pH 10.5, the UV-Vis absorption and EPR spectra of the GRW-L16C mini-heme protein (Fig. S9) were virtually identical to those of GRW-L16CL30H (Fig. 3). Moreover, the EPR spectra of GRW-L16C mini-heme protein at pH 7.0 (Fig. S9B) ruled out a 5-c heme with a Cys ligand [32] or hemin in buffer solution. Thus, at pH 7.0 and in the absence of coordinating amino acid, hydrophobic/non-covalent interactions accommodate the heme in the GRW-L16C scaffold. An equivalent situation was reported for 2α-helix bundle maquettes. [24] All in all, the GRW-L16C scaffold validates the coordination pattern concluded for the GRW-L16CL30H mini-heme protein at basic pHs and the prerequisite of Cys16 deprotonation at pH ≥ 9.0 to allow heme ligation.
The EPR spectra of the GRW-L30H mini-heme protein as a function of pH (Fig. S10) showed heme binding to His30 at 6.50 ≤ pH ≤ 8.50 and dissociating otherwise, most possibly due to a longer heme FeIII to NHis bond length than in natural peroxidases (see Fig. S7). This confirms the heme coordination behavior observed for GRW-L16CL30H mini-heme protein at neutral pHs, and irrespectively of the presence of a competing binding site. The GRW-L16C (at pH 10.5) and the GRW-L30H (at pH 7.0) mini-heme proteins showed stability upon their redox cycles (Fig. S11) as well as catalytic oxidation of ABTS substrate in solution, when using H2O2 as oxidant (Fig. S12).
In summary, we have established that a simple α-helical scaffold design opens exciting opportunities to explore heme enzyme chemistry. By using a short, repetitive amino acid sequence with two distinct iron binding residues (His and Cys) that folds into a dimer of antiparallel 2SCC upon heme complexation, we have developed a miniature protein with heme as redox cofactor and featuring a reversible pH-induced switch of the heme axial ligand. The resulting penta- or hexa-coordinate thiolate heme for 9 ≤ pH ≤ 11 and penta-coordinate imidazole heme for 6 ≤ pH ≤ 8.5, imitating the heme ligation in CPO, cyt P450s and peroxidases, all in a single synthetic protein, was not previously reported. Moreover, the minimal tertiary structure ensures on-demand heme coordination being stable upon redox cycling. This is a crucial property for catalysis as proved by the catalytic reduction of O2 by the GRW-L16CL30H and GRW-L16C mini-heme proteins at pH ≥ 9.0, adsorbed onto a pyrolytic graphite electrode. Further reactivity studies in solution are in progress to evaluate the extent of this very promising observation to develop versatile synthetic mini-heme catalysts.
Supplementary Material
Acknowledgements.
This work was funded by the US NIH (ES012236) (to V.L.P), the French National Centre for Scientific Research (CNRS/UMR 7281, Marseille), the French PACA Region (APR-EX ‘LIPCAT’) and the CNRS Program for International Collaborations (PICS 07624) (to A.I). The French EPR Federation/TGE RENARD (IR3443) is also acknowledged for partial support (to A.I.). UltraScan software’s development for AUC was funded by NIH grant GM120600 and data analysis by NSF/XSEDE allocation grant MCB070038-A13 (to B.D.). T.K. acknowledges a postdoctoral fellowship (APR-EX ‘LIPCAT’) and a foreign researcher allowance from the city of Marseilles. We thank Jill Harland (University of Michigan, Ann Arbor) for her assistance in preparing AUC samples and Valerie Monnier (Aix Marseille Univ, CNRS, Centrale Marseille, FSCM, Spectropole, Marseille) for detailed analysis of the ESI-MS data.
Footnotes
Experimental Section.
References
- [1].Huang P-S, Boyken SE, Baker D, Nature 2016, 537 (7620), 320–327. [DOI] [PubMed] [Google Scholar]
- [2].Korendovych I, DeGrado W, Q. Rev. Biophys 2020, 53, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yu F, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, Mocny CS, Ruckthong L, Qayyum H, Pecoraro VL, Chem. Rev 2014,114 (7), 3495–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pinter TBJ, Koebke KJ, Pecoraro VL, Angew. Chem. Int. Ed 2020, 59 (20), 7678–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chapman SK, Daft S, Munro AW, Springer-Verlag GmbH, BERLIN 33, vol. 88, 1997, pp. 39–70. [Google Scholar]
- [6].Nicholls P, Fita I, Loewen PC, Adv. Inorg. Chem 2000, 51, 51–106. [Google Scholar]
- [7].Poulos TL, Chem. Rev 2014, 114 (7), 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Njuma OJ, Ndontsa EN, C Goodwin D, Arch. Biochem. Biophys 2014, 544, 27–39. [DOI] [PubMed] [Google Scholar]
- [9].Lombardi A, Nastri F, Pavone V, Chem. Rev 2001, 101 (10), 3165–3189. [DOI] [PubMed] [Google Scholar]
- [10].Nastri F, Chino M, Maglio O, Bhagi-Damodaran A, Lu Y, Lombardi A, Chem. Soc. Rev 2016, 45 (18), 5020–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nastri F, D’Alonzo D, Leone L, Zambrano G, Pavone V, Lombardi A, Trends Biochem. Sci 2019, 44 (12), 1022–1040. [DOI] [PubMed] [Google Scholar]
- [12].Dasa PK, Samanta S, McQuarters AB, Lehnert N, Dey A, Proc. Natl. Acad. Sci. USA 2016, 113 (24), 6611–6616, and references therein [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Perera R, Sono M, Sigman JA, Pfister TD, Lu Y, Dawson JH, Proc. Natl. Acad. Sci. USA 2003, 100 (7), 3641–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vetter SW, Terentis AC, Osborne RL, Dawson JH, Goodin DB, J. Biol. Inorg. Chem 2008, 14, 179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zastrow ML, Pecoraro VL, Coord Chem Rev. 2013, 257(17-18), 2565–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dieckmann GR, McRorie DK, Lear JD, Sharp KA, DeGrado WF, Pecoraro VL, J. Molec. Biol 1998, 280 (5), 897–912. [DOI] [PubMed] [Google Scholar]
- [17].Apostolovic B, Danial M, Klok H-A, Chem. Soc. Rev 2010, 39, 3541–3575. [DOI] [PubMed] [Google Scholar]
- [18].Ghosh D, Pecoraro VL, Inorg. Chem 2004, 43 (25), 7902–7915. [DOI] [PubMed] [Google Scholar]
- [19].Wagschal K, Tripet B, Hodges RS, J. Mol. Biol 1999, 285, 785–803. [DOI] [PubMed] [Google Scholar]
- [20].Tolbert AE, Ervin CS, Ruckthong L, Paul TJ, Jayasinghe-Arachchige VM, Neupane KP, Stuckey JA, Prabhakar R, Pecoraro VL, Nat. Chem 2020, 12, 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Malashkevich VN, Higgins CD, Almo SC, Lai JR, J. Pept. Scie 104 (3), 178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Smith AT, Pazicni S, Marvin KA, Stevens DJ, Paulsen KM, Burstyn JN, Chem. Rev 2015, 115, 2532–2558. [DOI] [PubMed] [Google Scholar]
- [23].Walker AF, Coord. Chem. Rev 1999, 186, 471–534. [Google Scholar]
- [24].Choma CT, Lear JD, Nelson MJ, Dutton PL, Robertson DE, DeGrado WF, J. Am. Chem. Soc 1994, 116 (3), 856–865. [Google Scholar]
- [25].Blanke SR, Martinis SA, Sligar SG, Hager LP, Rux JJ, Dawson JH, Biochemistry 1996, 35 (46), 14537–14543. [DOI] [PubMed] [Google Scholar]
- [26].Conner KP, Schimpf AM, Cruce AA, McLean KJ, Munro AW, Frank DJ, Krzyaniak MD, Ortiz de Montellano PR, Bowman MK, Atkins WM, Biochemistry 2014, 53 (9), 1428–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Moser CC, Sheehan MM, Ennist NM, Kodali G, Bialas C, Englander MT, Discher BM, Dutton PL, Methods Enzymol. 2016, 580, 365–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Johnston WA, Hunter DJB, Noble CJ, Hanson GR, Stok JE, Hayes MA, De Voss JJ, Gillam EMJ, J. Biol Chem 2011, 286 (47), 40750–40759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Groves JT, Nat. Chem 2014, 6, 89–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schubert E, Florin N, Duthie F, Henning Brewitz H, Kühl T, Imhof D, Hagelueken G, Schiemann O, J. Inorg. Biochem 2015, 148, 49–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.