

Abstract
ALFY is a large, multidomain protein involved in the degradation of protein aggregates by selective autophagy. The C-terminal FYVE domain of ALFY has been shown to bind phosphatidylinositol 3-phosphate (PI(3)P); however, ALFY only partially colocalizes with other FYVE domains in cells. Thus, we asked if the FYVE domain of ALFY has distinct membrane binding properties compared to other FYVE domains and whether these properties might affect its function in vivo. We found that the FYVE domain of ALFY binds weakly to PI(3)P containing membranes in vitro. This weak binding is the result of a highly conserved glutamic acid within the membrane insertion loop in the FYVE domain of ALFY that is not present in any other human FYVE domain. In addition, not only does this glutamic acid reduce binding to membranes in vitro and inhibits its targeting to membranes in vivo, but it is also important for the ability of ALFY to clear protein aggregates.
Keywords: ALFY, FYVE domain, autophagy, phosphatidylinositol 3-phosphate, nuclear magnetic resonance (NMR) spectroscopy, protein structure
1. Introduction
Maintaining protein homeostasis is critical for ensuring proper cellular functions. Protein homeostasis is disrupted by protein misfolding, which can lead to the accumulation of nonfunctional proteins and protein aggregates. To ensure that homeostasis is maintained, protein quality control mechanisms facilitate refolding, degradation, or sequestration of misfolded proteins.1-4 However, factors such as cellular stress, aging, and the accumulation of mutations can enhance the rate of misfolded proteins and overload protein quality control mechanisms resulting in the accumulation of protein aggregates.2,3 These aggregates are correlated with several neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s Disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS).5-7
One protein quality control mechanism that is employed to degrade protein aggregates is macroautophagy (hereafter, autophagy).2,3,8,9 Autophagy is an evolutionarily conserved process in which cytoplasmic cargos are sequestered into double membrane vesicles, termed autophagosomes, and delivered to lysosomes for degradation. The capture of cargos during this process can either be non-selective or selective. In non-selective autophagy, the core autophagy machinery facilitates the degradation of bulk cytosol in response to starvation.10,11 In selective autophagy, selective autophagy receptor (SAR) proteins recognize specific cargos, such as protein aggregates, organelles, or intracellular pathogens, and recruit the autophagy machinery to these cargos to facilitate their degradation.12,13 In addition to the SARs, some types of selective autophagy rely on autophagy adaptor proteins to bring the selected cargo and SARs in close contact with the core autophagy machinery and the autophagic membrane.13,14
Autophagy-linked FYVE protein (ALFY) is a 395 kDa multidomain protein that has been identified as an autophagy adaptor for the clearance of protein aggregates.15 Previous work has demonstrated that a loss of the homolog of ALFY in Drosophila melanogaster (bchs) resulted in the accumulation of ubiquitin positive aggregates, progressive deterioration in neural tissue, and premature death of adult flies.16 Furthermore, overexpression of a C-terminal fragment of ALFY led to a significant reduction in aggregation-prone huntingtin (Htt) inclusions in a neuronal model of HD.17 More recent work in an HD mouse model as well as in HD-patient-derived neurons has demonstrated that ALFY is required for efficient elimination of protein aggregates in the adult brain.18 All of these studies highlight the important role of ALFY in the degradation of protein aggregates.
The function of ALFY as an adaptor protein is to link cargo-bound SARs to the autophagic membrane. ALFY co-localizes with and interacts with p62/SQSTM1 and NBR1, both of which are SARs that bind to ubiquitinated protein aggregates.17,19 Furthermore, ALFY binds to ATG5, a core autophagy protein.17 ALFY has also been shown to interact indirectly with the autophagic membrane through its interaction with LC3C and the GABARAPs, which are ubiquitin like proteins that are conjugated to the headgroup of phosphatidylethanolamine (PE) in the autophagic membrane.20 Finally, ALFY also interacts with phosphatidylinositol 3-phosphate (PI(3)P) through its C-terminal FYVE domain allowing it to bind membranes directly.15 These interactions suggest that ALFY interacts both directly and indirectly with the autophagosomal membrane. However, the role of the FYVE domain of ALFY in this process has not been fully elucidated.
FYVE domains, named after the first four proteins to be identified to contain this domain (Fab1, YOTB, Vac 1, and EEA1), bind to PI(3)P found on the surface of early endosomes, on intraluminal vesicles of multivesicular endosomes, and on autophagosomes.21-23 Structurally, FYVE domains have two small double-stranded β-sheets and a C-terminal α-helix.24 The overall tertiary fold is stabilized by two Zn2+ ions that are each coordinated by four cysteine residues.24,25 FYVE domains contain three conserved motifs: a N-terminal WxxD, a central (R/K)(R/K)HHCR, and a R(V/I)C that mediate recognition and binding of PI(3)P.24 Upon recognition of PI(3)P, FYVE domains also contain a hydrophobic membrane insertion loop (MIL), also known as the “turret” loop, that penetrates into the lipid bilayer further anchoring the protein to the membrane.24,26-28
Full-length ALFY and the FYVE domain of ALFY (ALFY-FYVE) have been shown to bind to and have specificity for PI(3)P.15 ALFY has also been shown to colocalize with markers of autophagy and to colocalize in a limited manner with other FYVE domains. This suggests that the FYVE domain of ALFY may have only a minimal role in the localization of ALFY in vivo or that this domain may not bind strongly to PI(3)P.15 Therefore, we asked if ALFY-FYVE has distinct membrane binding preferences compared to other FYVE domains and whether these preferences might affect its function in vivo. To this end, we determined that ALFY-FYVE binds weakly to liposomes containing PI(3)P in comparison to the well-studied FYVE domain of EEA1 (EEA1-FYVE). We determined the structure of ALFY-FYVE using nuclear magnetic resonance (NMR) spectroscopy, and in combination with sequence analysis, we identified a glutamic acid residue that is highly conserved in ALFY-FYVE but is not present in any other human FYVE domains. Using mutagenesis, we further demonstrated that this residue is responsible for the weak liposome binding of ALFY-FYVE in vitro, affects the localization of ALFY-FYVE to membranes in cells, and is required for the efficient clearance of aggregates by ALFY.
2. Results
2.1. ALFY-FYVE is a weak binder of PI(3)P containing liposomes
GST-tagged ALFY-FYVE has been shown to bind to PI(3)P on a nitrocellulose membrane containing different lipids; however, in cells, ALFY only partially colocalizes with FYVE domains.15 This suggests that the FYVE domain of ALFY may contain distinct membrane binding properties compared to other FYVE domains. Therefore, we examined the lipid binding properties of ALFY-FYVE and compared them to the FYVE domain of EEA1 (EEA1-FYVE), as its binding to and specificity for PI(3)P has been studied extensively.15,26,29 We recombinantly expressed and purified ALFY-FYVE (residues 3428-3518 of ALFY) and EEA1-FYVE (residues 1347-1411 of EEA1) and performed liposome sedimentation assays with liposomes containing phosphatidylcholine (PC) and phosphatidylserine (PS) alone or containing PC and PS with 5% phosphatidylinositol (PI) or 5% PI(3)P. After incubation, liposomes were pelleted by ultracentrifugation, and the partitioning of protein between the supernatant (S) and the pellet (P) was probed by SDS-PAGE. Protein observed in the pellet fraction that was more than the background levels observed in the no liposome control is indicative of protein binding to liposomes. Only 21.8% of ALFY-FYVE was observed in the pellet fraction after incubation with PI(3)P containing liposomes. This is a small, but significant increase (p=0.02121) in ALFY-FYVE in the pellet fraction compared to the no liposome control (11.2% in the pellet fraction) (Figure 1A and 1B). In contrast, EEA1-FYVE was observed to be 50.9% in the pellet fraction after incubation with liposomes containing 5% PI(3)P, which is a highly significant increase (p<0.0001) over the no liposome control. Thus, ALFY-FYVE is a weaker binder of liposomes containing PI(3)P than EEA1-FYVE.
Figure 1. ALFY-FYVE weakly binds to liposomes containing PI(3)P.
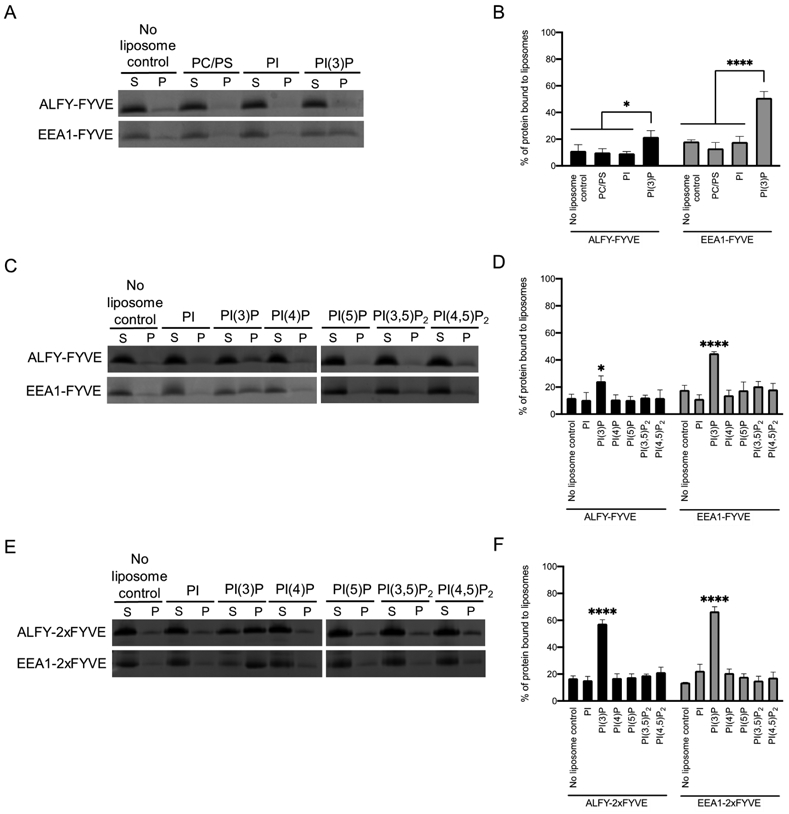
A) Liposome sedimentation assays were conducted with ALFY-FYVE or EEA1-FYVE and liposomes composed of PC, PS, and the indicated phosphoinositide at 5% molar ratio. Representative SDS-PAGE gels of the supernatant (S) and pellet (P) fractions are shown. B) The percent of protein in the pellet fraction was quantified by densitometry. Error bars represent the standard deviation from three independent experiments. Statistical significance was determined by ordinary two-way ANOVA with a Sidak’s multiple comparison test. *, p<0.0212; ****, p<0.0001 C) The specificity of ALFY-FYVE and EEA1-FYVE was probed using a liposome sedimentation assay. D) The percent of protein in the pellet fraction was quantified by densitometry. Error bars represent the standard deviation from three independent experiments. Statistical significance was determined by ordinary two-way ANOVA with Tukey’s multiple comparison test. *, p<0.0132; ****, p<0.0001. E) The specificity of ALFY-2xFYVE and EEA1-2xFYVE was probed using a liposome sedimentation assay. F) The percent of protein in the pellet fraction was quantified by densitometry. Error bars represent the standard deviation from three independent experiments. Statistical significance was determined by ordinary two-way ANOVA with Tukey’s multiple comparison test. ****, p<0.0001.
One possible explanation for the weak binding of PI(3)P containing liposomes by ALFY-FYVE is that ALFY-FYVE may have a more broad or different specificity for phosphoinositides than EEA1-FYVE.22 To test this, we performed liposomes sedimentation assays with liposomes containing different phosphoinositides (Figure 1C and 1D). Both ALFY-FYVE and EEA1-FYVE only bound to PI(3)P containing liposomes, with 24.3% and 44.9% in the pellet fraction, respectively. This demonstrates that ALFY-FYVE and EEA1-FYVE have similar specificities for PI(3)P despite the weaker binding of PI(3)P by ALFY-FYVE.
Since the binding of PI(3)P containing liposomes by ALFY-FYVE was weak, we also generated an ALFY-2xFYVE construct by fusing two copies of the ALFY-FYVE in tandem with a flexible linker in between them. This ALFY-2xFYVE construct should bind more strongly to liposomes and allow us to more easily probe the liposome binding properties of ALFY-FYVE. For comparison, we also generated an EEA1-2xFYVE domain construct in the same manner. We repeated liposome sedimentation with liposomes containing a variety of different phosphoinositides (Figure 1E and 1F). Both ALFY-2xFYVE and EEA1-2xFYVE only bound to PI(3)P with 57.5% and 66.7% in the pellet fraction for each of these proteins, respectively. This demonstrates that both constructs have similar specificities to their single copy counterparts with a preference for PI(3)P and no detectable binding for other phosphoinositides.
2.2. The structure of ALFY-FYVE
Since ALFY-FYVE bound to PI(3)P containing liposomes more weakly than EEA1-FYVE, we asked if there were structural differences between these domains that could account for the weaker binding of ALFY-FYVE. To probe this question, we determined the structure of ALFY-FYVE using nuclear magnetic resonance (NMR) spectroscopy. For the structure calculation of ALFY-FYVE, 1710 Nuclear Overhauser Effect (NOE) derived distance constraints and 327 dihedral angle constraints were used (Table 1). Excluding residues 3428-3444, which are disordered in the structure, this results in 29.5 distance constrains per amino acid. The 20 structures of the bundle aligned with an average pairwise backbone RMSD of 0.86 ± 0.10 Å for residues 3445-3518, indicating the good convergence of the calculated structures (Figure 2A). The overall structure of ALFY-FYVE is consistent with other FYVE domains, and it aligns well to EEA1-FYVE (PDB ID 1HYI) with an RMSD of 1.7 Å across Cα residues (Figure 2B and 2C).26
Table 1.
Structural statistics of AFLY-FYVE.
| ALFY-FYVE | |
|---|---|
| NMR distance and dihedral constraints | |
| Distance constraints | |
| Total NOE | 1710 |
| Intraresidue | 529 |
| Interresidue | 1181 |
| Sequential (∣i-j∣ = 1) | 440 |
| Medium range (1<∣i-j∣≤4) | 276 |
| Long range (∣i-j∣≥5) | 465 |
| Dihedral angle constraints | 327 |
| Structure statistics | |
| Violations (mean ± SD) | |
| Distance constraints (Å) | 0.264±0.005 |
| Dihedral angle constraints (°) | 0.484±0.105 |
| Ramachandran statistics | |
| Most favored | 64.3% |
| Additional allowed | 34.3% |
| Generously allowed | 1.4% |
| Disallowed | 0.0% |
| Average pairwise root mean square deviation (Å) | |
| Heavy (18-76) | 0.26±0.06 |
| Backbone (18-76) | 0.86±0.10 |
Figure 2. The NMR structure of ALFY-FYVE.

A) Ribbon diagram of ALFY-FYVE showing the overlay of the 20 structures within the bundle in two different orientations. B) Cartoon representation of ALFY-FYVE shown in the same two orientations as in A. The N and C termini are labeled, and the zinc atoms are shown as gray spheres. C) Overlay of the cartoon representation of ALFY-FYVE (cyan) with the cartoon representation of the EEA1-FYVE (magenta, PDB code 1HYI) shown in the same two orientations as in A. L3497 is shown as a stick representation to highlight the difference in the position of this loop between ALFY and EEA1. D) Two residues that coordinate the binding of the headgroup of PI(3)P are shown as stick representations from ALFY in cyan (R3472 and H3474) and EEA1 in magenta (R1370 and H1372).
The interaction of FYVE domains with the headgroup of PI(3)P occurs largely through an N-terminal aspartic acid, two histidine residues, and a cluster of four arginine/lysine residues.26,29 ALFY-FYVE contains all of the residues required for PI(3)P binding, and we compared the positions of each of these amino acids in ALFY-FYVE to those in EEA1-FYVE (Figure 2D). The side chains of these PI(3)P binding residues in ALFY-FYVE are in similar positions to the side chains of these residues in the structure of EEA1-FYVE. This suggests that the interaction with the headgroup of PI(3)P is unlikely to be different between these domains. A further comparison of the structure revealed that the biggest difference between ALFY-FYVE and EEA1-FYVE is the loop spanning residues 3493 to 3500 in ALFY. This loop is packed more tightly against the core of the FYVE domain in ALFY due to the presence of three hydrophobic amino acids (I3494, L3497, and I3499) that are not present in EEA1 (Figure 2C). However, this region is located away from the PI(3)P binding pocket and is therefore unlikely to affect membrane binding. Based on the structure of ALFY-FYVE, we did not find any features that were structurally distinct in ALFY-FYVE that could account for the weak binding to PI(3)P containing liposomes.
2.3. ALFY-FYVE contains a highly conserved glutamic acid that is not present in other FYVE domains
Without any obvious structural differences between ALFY-FYVE and EEA1-FYVE that could account for the weak membrane binding by ALFY-FYVE, we decided to compare the sequence of ALFY-FYVE to all other human FYVE domain sequences. To do this, we first aligned sequences of the FYVE domain from six ALFY homologues from Homo sapiens to Caenorhabditis elegans using Proviz to identify conserved residues in ALFY-FYVE (Supplemental Figure 1 and Figure 3A).30 Next, the sequences of all known human FYVE domain containing proteins was compiled using Blastp, Psi-Blastp, SMART, and Pfam searches and aligned (Figure 3B).31-33 Based on these alignments, we identified a series of residues that are evolutionarily conserved in ALFY-FYVE but that are not present in other human FYVE domains. One residue that stood out was a highly conserved glutamic acid in ALFY (E3471) that is the last residue in the MIL (Figure 3B and 3C). ALFY is the only human FYVE domain that contains a glutamic acid at this position out of 28 FYVE domains. DFCP1 is the only other protein that has a negatively charged aspartic acid residue (D615) at this position. Currently, DFCP1 is the only other FYVE domain containing protein that is thought to be involved in autophagy.34,35 The remaining 26 human FYVE domains contain a diversity of residues at this position. Eleven proteins contain a positively charged residue at this position, eight have a polar residue, and seven have a non-polar residue at this position (Figure 3B).
Figure 3. ALFY-FYVE contains a highly conserved glutamic acid not found in other human FYVE domains.

A) An alignment of the FYVE domain sequence of different ALFY homologues showing conservation of E3471 in red. B) An alignment of human FYVE domains sorted according to the identity of the last residue in the MIL. Negatively charged residues are red, positively charged residues are blue, polar residues are yellow, and nonpolar residues are green. C) An alignment of the structure of ALFY-FYVE to the structure of EEA1-FYVE bound to the headgroup of PI(3)P (PDB ID 1HYI) was performed. EEA1-FYVE was then was removed to visualize the expected position of PI(3)P binding to ALFY-FYVE.26 A cartoon representation of ALFY-FYVE is shown in cyan with E3471 and the headgroup of PI(3)P from PDB ID 1HYI shown as stick representations.
Since only ALFY and DFCP1 contain FYVE domains with negatively charged amino acids in the last position of the MIL, we wondered how evolutionarily conserved this position is in other FYVE domains. Proviz was used to align the available sequences of FYVE domain containing proteins from Homo sapiens, Mus musculus, Gallus gallus, Danio rerio, Xenopus tropicalis, Drosophila melanogaster, and Caenorhabditis elegans (Supplemental Figure 1). The amino acid at this position is evolutionarily conserved in most FYVE domains, which suggests that this residue is likely important for the function of these FYVE domains. In addition, this residue has been suggested to be important for membrane loop insertion and membrane binding.36,37 Taken together, our sequence analysis suggests that E3471 in ALFY may play an important role in defining the membrane binding properties of ALFY-FYVE.
2.4. E3471 in ALFY decreases FYVE domain binding to liposomes containing PI(3)P
To determine whether E3471 influences the liposome binding preferences of ALFY-FYVE, we mutated this residue to lysine, threonine, and valine as these are the most commonly occurring positively charged, polar, and non-polar residues found in this position in other human FYVE domains. We then performed liposome sedimentation assays with wild-type (WT) ALFY-FYVE and the different ALFY-FYVE mutants. Strikingly, there was an increase from 20.4% of WT ALFY-FYVE in the pellet fraction with liposomes containing PI(3)P to 85.3%, 73.1%, and 75.0% for the E3471K, E3471T, and E3471L mutants, respectively (Figure 4A and 4B). This indicates that E3471 in ALFY reduces the ability of ALFY-FYVE to bind liposomes containing PI(3)P.
Figure 4. E3471 affects liposome binding.
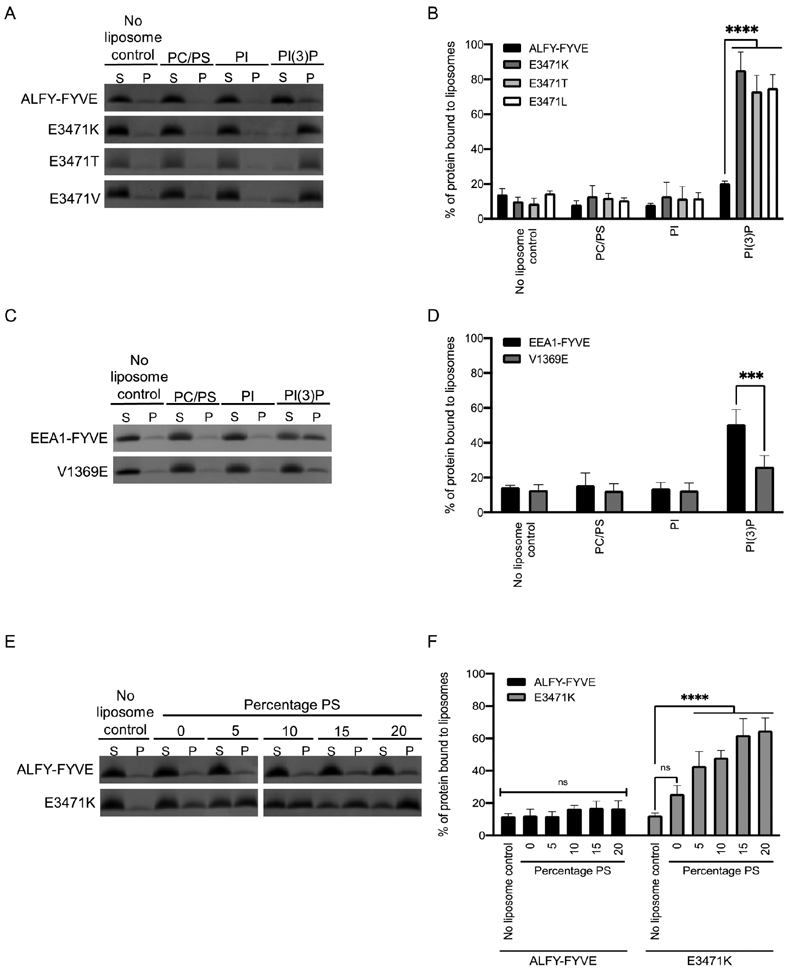
A) Liposome sedimentation assay was conducted with WT ALFY-FYVE and ALFY-FYVE E3471K, E3471T, or E3471V mutants. Liposomes were composed of PC, PS and the indicated phosphoinositide at 5% molar ratio. Representative SDS-PAGE gels of the supernatant (S) and pellet (P) fractions are shown. B) The percent of protein in the pellet fraction was quantified by densitometry. Error bars represent the standard deviation from four experiments. Statistical significance was determined by ordinary two-way ANOVA with Tukey’s multiple comparison test. ****, p<0.0001. C) Liposome sedimentation assay was conducted with WT EEA1-FYVE and EEA1-FYVE V1369E mutant. D) The percent of protein in the pellet fraction was quantified by densitometry. Error bars represent the standard deviation from three experiments. Statistical significance was determined by ordinary two-way ANOVA with Tukey’s multiple comparison test. ***, p=0.0006. E) Liposome sedimentation assay was conducted with ALFY-FYVE and ALFY-FYVE E3471K mutant in which the percentage of PS was varied from 0% to 20%. E) The percent of protein in the pellet fraction was quantified by densitometry. Error bars represent the standard deviation from three experiments. Statistical significance was determined by ordinary two-way ANOVA with Tukey’s multiple comparisons test. ****, p<0.0001.
EEA1-FYVE contains a valine (V1369) in the position corresponding to E3471 in ALFY-FYVE. To determine if this residue plays an important role in liposome binding for other FYVE domains, we mutated V1369 to a glutamic acid and performed liposome sedimentation with liposomes containing PI(3)P (Figure 4C and 4D). EEA1-FYVE V1369E had significantly lower binding (p=0.0001) to PI(3)P containing liposomes (29.4% in the pellet fraction) than the WT protein (59.8% in the pellet fraction). This finding further supports the importance of the last residue in the MIL in liposome binding and suggests that a negatively charged amino acid in the last position of the MIL may reduce liposome binding more broadly in other FYVE domains.
Based on its position in the MIL, E3471 in ALFY-FYVE is predicted to sit near the surface of the membrane when ALFY-FYVE binds to PI(3)P. This may create an electrostatic repulsion between E3471 in ALFY-FYVE and negatively charged lipids. Since the liposomes that we used in our previous experiments contained 19% of the negatively charged lipid PS, the high negative charge of these liposomes may be the cause of the weak binding that we observed by ALFY-FYVE. If this is the case, ALFY-FYVE may bind stronger to more neutral membranes. To test this, we generated liposomes containing PC and 5% PI(3)P with varying percentages of PS and probed liposome binding by ALFY-FYVE and ALFY-FYVE E3471K. We did not observe a statistically significant increase in binding of ALFY-FYVE to liposomes containing lower percentages of PS (Figure 4E and 4F). This suggests that E3471 does not aid in targeting ALFY to more neutral membranes. In contrast, ALFY-FYVE E3471K did have increased binding for liposomes with higher percentages of PS suggesting that the amino acid at this position may be able to provide some electrostatic interactions with the membrane (Figure 4E and 4F).
2.5. E3471 in ALFY-FYVE does not influence PI(3)P headgroup binding
Since E3471 is near the binding pocket for the PI(3)P headgroup, one additional possibility for the weak binding of PI(3)P liposomes by ALFY-FYVE is that E3471 may decrease the binding affinity for the PI(3)P headgroup. To test this, we wanted to determine the dissociation constant (Kd) of ALFY-FYVE and ALFY-FYVE E3471K for the headgroup of PI(3)P (inositol 1,3-bisphosphate (Ins(1,3)P2)) using NMR spectroscopy. However, the NMR assignment of ALFY-FYVE was completed using sodium phosphate buffer which could compete with Ins(1,3)P2 for binding to ALFY-FYVE. To avoid this competition, we decided to determine the Kd of ALFY-FYVE for Ins(1,3)P2 in Bis-Tris buffer. Therefore, we needed to first transfer the NMR assignment of ALFY-FYVE from the sodium phosphate buffer to Bis-Tris buffer. To do this, we recorded 1H-15N HSQC spectra of 15N-labeled ALFY-FYVE in buffer containing different ratios of sodium phosphate and Bis-Tris (Supplemental Figure 2). Next, we titrated Ins(1,3)P2 into 15N-labeled ALFY-FYVE or AFLY-FYVE E3471K and monitored chemical shift perturbations (CSPs) in a series of 1H-15N HSQC spectra. E3471 from AFLY-FYVE and E3471K from ALFY-FYVE E3471K were not monitored during this analysis as the peak for K3471 could not be assigned with confidence. R3472 was also excluded from this analysis due to overlap. With the exclusion of E3471 and R3472, the five residues that underwent the largest CSPs in ALFY-FYVE and ALFY-FYVE E3471K were H3450, K3453, G3455, R3473, and H3476 (Figure 5A). The Kd calculated for ALFY-FYVE binding to Ins(1,3)P2 ranged from 50.5 μM to 183.5 μM, while the Kd calculated for ALFY-FYVE E3471K binding to Ins(1,3)P2 ranged from 82.8 μM to 270.5 μM (Figure 5B-5E and Table 2). Comparing the Kd values determined for individual residues, there was roughly a 1.4-fold increase in the Kd of ALFY-FYVE for Ins(1,3)P2 in the E3471K mutant compared to WT ALFY-FYVE. This demonstrates that E3471 does not decrease the binding affinity to the headgroup of PI(3)P and instead may be involved in broadly repelling membranes.
Figure 5. E3471 does not decrease binding of ALFY-FYVE to Ins(1,3)P2.

A) CSP data for ALFY-FYVE in the presence of 250 μM Ins(1,3)P2. Peak assignments for A3448, R3473, H3474 could not be transferred from the initial ALFY-FYVE assignment and were given no CSP value. B) 1H-15N HSQC spectra of 50 μM ALFY-FYVE in the presence of increasing concentrations of Ins(1,3)P2 headgroup. The 1H-15N HSQC spectra of ALFY-FYVE in the absence of Ins(1,3)P2 headgroup is shown in blue. The inset shows the chemical shift perturbation for G3457 with the arrow pointing in the direction of the shift with increasing Ins(1,3)P2 headgroup concentration. C) CSP data for ALFY-FYVE H3450 (red), K3453 (orange), E3455 (green), R3473 (blue), and H3475 (purple) as a function of Ins(1,3)P2 headgroup concentration. All data points are shown as circles. The fits to the data are shown as lines of the same color as the data points. D) 1H-15N HSQC spectra of 50 μM ALFY-FYVE E3471K in the presence of increasing concentrations of Ins(1,3)P2 headgroup is shown as in B. E) CSP data for ALFY-FYVE E3471K H3450 (red), K3453 (orange), E3455 (green), R3473 (blue), and H3475 (purple) as a function of Ins(1,3)P2 concentration. All data points are shown as circles. The fits to the data are shown as lines of the same color as the data points.
Table 2.
ALFY-FYVE and ALFY-FYVE E3471K binding to Ins(1,3)P headgroup.
| ALFY-FYVE | ALFY-FYVE E3471K | |||
|---|---|---|---|---|
| Residue | Calculated Kd (μM) | R2 | Calculated Kd (μM) | R2 |
| H3450 | 183.5 | 0.99411 | 270.5 | 0.99275 |
| K3453 | 50.5 | 0.95089 | 82.8 | 0.94666 |
| E3455 | 81.3 | 0.97893 | 119.9 | 0.98325 |
| R3473 | 155.8 | 0.99040 | 192.0 | 0.99264 |
| H3476 | 135.9 | 0.99190 | 190.9 | 0.98936 |
2.6. E3471 in ALFY-FYVE prevents localization to PI(3)P-rich membranes
In order to understand if E3471 in ALFY impacts the ability of ALFY-FYVE to bind membranes in cells, we produced mCherry-ALFY-FYVE and mCherry-ALFY-FYVE E3471K fusion proteins and expressed these constructs in Saccharomyces cerevisiae. S. cerevisiae was selected to monitor the localization of ALFY-FYVE as its localization will not be influenced by potential binding partners in human cell lines and instead its localization should only be affected by its ability to bind membranes.15 Both mCherry-ALFY-FYVE and mCherry-ALFY-FYVE E3471K were dispersed in the cytosol with no distinctive punctate formation or localization to a particular membrane (Figure 6A). The diffuse localization of ALFY-FYVE or ALFY-FYVE E3471K is consistent with other FYVE domains, which in their monomeric states are often diffusely localized.38-40
Figure 6. E3471 modulates localization and aggregate clearance.

A) Cellular distribution of mCherry-ALFY-FYVE, mCherry-ALFY-2xFYVE, mCherry-ALFY-FYVE E3471K, and mCherry-ALFY-2xFYVE E3471K expressed in S. cerevisiae. Wide-field scale bar is 10 μm and narrow-field scale bar is 5 μm. B) Colocalization between mCherry-ALFY-2xFYVE or mCherry-ALFY-2xFYVE E3471K and GFP-organelle markers in S. cerevisiae. GFP-Sec7 was used as a marker for late Golgi/early endosomes, GFP-Vps4 was used as a marker for late endosomes, GFP-Vph1 was used as a marker for vacuoles, and GFP-Atg8 was used as a marker for autophagosomes. C) Exon1Htt103Q-HT cells were transiently transfected with Ctrl (tdTomato), tdTomato-ALFYC or tdTomato-ALFYC E3471K. A subset of Ctrl transfected cells was treated with Dox immediately post-transfection. Cell lysates were collected after 96 hrs, and then subject to filter trap analyses (FTA) to monitor the amount of insoluble Exon1Htt103Q-HT. The sample was slotted across a four-fold dilution from high to low as denoted by the triangle. D) Quantification of the FTA relative to Ctrl. The Mann-Whitney U non-parametric test reveals a significant difference between Ctrl and ALFYC (U=0; p=0.006); whereas no difference was detected between Ctrl and ALFYC E3471K (U=5; p=0.070). * indicates the significant difference observed between ALFYC and ALFYC E3471K (U=4; p=0.047). n-values are 3, 5, 5, 5 for +dox, Ctrl, ALFYC and ALFYC E3471K, respectively. E) Western blotting was performed on cells from B to monitor the amount of soluble halo tagged Exon1Htt103Q-HT protein. Vinculin was used as a loading control in these experiments.
A number of properties, including the strength of dimerization, have been cited to act synergistically to facilitate endosomal targeting by FYVE domains.29 Therefore, to monitor the localization of ALFY-FYVE in cells, we also produced mCherry-ALFY-2xFYVE and mCherry-ALFY-2xFYVE E3471K with both copies of the FYVE domain containing the E3471K mutation. mCherry-ALFY-2xFYVE was dispersed in the cytosol with no distinct localization, while mCherry-ALFY-2xFYVE E3471K contained a localization consistent with vacuolar membranes with some additional punctate structures appearing on the surface of the vacuolar membrane (Figure 6A).
To elucidate which membranes the ALFY-2xFYVE E3471K construct localized to, we co-expressed this construct with different organelle markers, including GFP-Sec7 for early endosomes, GFP-Vps4 for late endosomes, GFP-Vph1 for the vacuole, and GFP-Atg8 for autophagosomes (Figure 6B). ALFY-2xFYVE E3471K displayed partial colocalization with the late endosomal marker Vps4 and strong colocalization with the vacuole marker Vph1. This suggests that the mutant form of the protein is able to bind to late endosomes and the vacuole, while the WT protein is incapable of being targeted to any membranes even when expressed as two copies in tandem. These results indicate that E3471 in ALFY decreases the membrane binding of ALFY-FYVE in cells and inhibits the targeting of ALFY-FYVE to PI(3)P containing membranes.
2.7. E3471 in ALFY-FYVE aids in aggregate clearance in mammalian cells
Our colocalization experiments demonstrated that ALFY-FYVE E3471 prevents membrane targeting when ALFY-FYVE was expressed in yeast. However, ALFY is a mammalian protein whose main function is to clear protein aggregates. This ability to clear aggregates can be replicated using only an approximately 100 kDa fragment of ALFY, which includes the C-terminal WD40 repeat domain and the FYVE domain (ALFYC).17 If the weak binding of PI(3)P containing membranes by ALFY-FYVE is important for the function of ALFY in clearing protein aggregates, then we hypothesized that a mutation of E3471 should decrease aggregate clearance in mammalian cell culture. To test this, we expressed tdTomato-ALFYC and tdTomato-ALFYC E3471K in HeLa cells expressing aggregate-prone huntingtin protein (Exon1Htt103Q-HT). We then performed a filter trap assay to capture and quantitate the amount of Exon1Htt103Q-HT.41,42 Cells were lysed in buffer containing 8 M urea, 1% Triton x-100, and 0.1% SDS to aid in solubilization. Samples were then flowed through 200 nm pore cellulose acetate filters to separate solubilized Exon1Htt103Q-HT from insoluble aggregated Exon1Htt103Q-HT. The trapped aggregates (Figure 6C and 6D) and the soluble protein (Figure 6E) can then be monitored separately. tdTomato-ALFYC reduced the amount of SDS insoluble aggregated Exon1Htt103Q-HT by 23.0% compared to cells expressing tdTomato alone (Figure 6C and 6D). In contrast, tdTomato-ALFYC E3471K did not significantly reduce the levels of Exon1Htt103Q-HT compared to cells expressing tdTomato alone. Importantly, the difference in aggregate clearance between the ALFYC and ALFYC E3471K mutants was not due to differences in the transfection efficiencies of these plasmids or differences in the expression of these proteins as monitored by mean fluorescence intensity per cell (Supplemental Figure 3). Additionally, tdTomato-ALFYC and tdTomato-ALFYC E3471K had no effect on the amount of soluble Exon1Htt103Q-HT protein (Figure 6E). Taken together, these data demonstrate that the weak membrane binding of ALFY-FYVE is beneficial for its role in aggregate clearance.
3. Discussion
In this study, we have demonstrated that ALFY-FYVE binds weakly to liposomes containing PI(3)P even though its structure is similar to other FYVE domains and it has all of the residues required for PI(3)P binding. We also found that ALFY-FYVE has a highly conserved glutamic acid (E3471) located in its MIL. This residue is unique to ALFY and is not present in any other human FYVE domain. E3471 disrupts ALFY-FYVE binding to liposomes containing PI(3)P, but does not affect its ability to bind the headgroup of PI(3)P. Finally, despite the fact that E3471 reduces membrane binding and membrane targeting in vivo by ALFY-FYVE, this residue plays an important role in aggregate clearance by ALFY.
In FYVE domains, the MIL is typically referred to as the four residues preceding the PI(3)P headgroup binding (R/K)(R/K)HHCR motif. The residues within the MIL play an important role in defining the strength of membrane binding by the FYVE domain.29 Two independent studies have demonstrated that the middle two residues of the MIL in EEA1 (V1367 and T1368) penetrate farther into the membrane than the last residue in this loop (V1369).28,43 Since E3471 in ALFY is located at the same position as V1369 in EEA1, this suggests that the last residue in the MIL may interact more with the surface of the membrane rather than penetrating into it. Our results are in good agreement with this data as E3471 likely provides a general repulsion for all membranes, while ALFY-FYVE E3471K results in an increased binding to more negatively charged membranes. Taking this one step further, our work may indicate that FYVE domains with a negative charge in this position bind weakly to all membranes, while FYVE domains with positively charged residues in this position may bind stronger to more negatively charged membranes.
Five human FYVE domain containing proteins have a serine or threonine residue in the last position of the MIL. Since a glutamic acid in this position strongly inhibits membrane binding, it seems possible that phosphorylation at this position would do the same. Indeed, we demonstrated that mutating this position in ALFY to a threonine (ALFY-FYVE E3471T) allowed for significantly strong membrane binding, while the native sequence containing a glutamic acid had much weaker membrane binding (Figure 4A and 4B). Currently, there is no evidence that any of the five human FYVE domains with a threonine or serine in the last residue of the MIL are phosphorylated at this position. However, most FYVE domains contain at least one serine or threonine in their MIL (24 out of 28). Since these residues are highly conserved evolutionarily, this suggests that phosphorylation within the MIL may be a means to regulate membrane binding by FYVE domains. Indeed, FGD1, FGD3, FGD6, PIKFYVE, and PLEKHF2 all have documented phosphorylation events within their MILs, albeit not at the last MIL residue.44 It will be interesting to explore if these phosphorylation events reduce membrane binding.
We found that E3471 is highly conserved across ALFY homologues. In every organism that we checked from D. melanogaster to humans, this residue is always a glutamic acid. The only organism that did not have a glutamic acid in this position was C. elegans. Instead, C. elegans has an aspartic acid in this position, which conserves the negative charge. It is very intriguing that this residue is so highly conserved, despite the fact that it reduces the ability of ALFY-FYVE to bind membranes when compared to positively charged, polar, or nonpolar residues at this position (Figure 4A and 4B). This strongly suggests that the weak membrane binding of ALFY-FYVE is required for the proper function of ALFY in cells. Our work supports this idea as a mutation of E3471 to lysine (ALFY-FYVE E3471K) resulted in enhanced membrane binding when ALFY-FYVE mutant was expressed in yeast, but decreased aggregate clearance using mammalian cells containing aggregation-prone Htt. However, it is currently unclear why such weak membrane binding by ALFY-FYVE is advantageous for its ability to degrade protein aggregates.
Some FYVE domain containing proteins are capable of forming dimers or higher order oligomerization, and this property is a determinant of membrane binding ability and cellular localization.24,29,40 For example, EEA1 contains a coiled-coil region that dimerizes and SARA dimerizes via its FYVE domain and can also form higher order oligomers.39 Additionally, induced dimerization of Hrs results in endosomal targeting, which further supports the idea that multimerization of FYVE domains can regulate membrane targeting of FYVE domains in vivo.29 At this time, there is no evidence that ALFY is capable of self-associations to form a dimer or higher order oligomer. However, ALFY has been shown to bind to p62 aggregates, and p62 oligomerizes through its PB1 domain.19,45 As such, the weak binding of ALFY-FYVE to liposomes containing PI(3)P together with the ability of p62 to form higher order oligomers could serve as a regulatory mechanism to control the size of the protein aggregates that are targeted for degradation via autophagy. This may result in a system where ALFY is only able to target aggregates to membranes once the aggregate is large enough to accumulate sufficient copies of ALFY to overcome its weak PI(3)P binding. In this context, if the FYVE domain had a higher binding efficiency for membranes, this may result in targeting aggregates to autophagic membranes when the aggregates are too small to be efficiently packaged into autophagosomes.
Another possible reason for the weak membrane binding by ALFY-FYVE could be that the FYVE domain is not responsible for targeting ALFY to PI(3)P membranes, but instead strengthens the connection between the cargo and that autophagic membrane that are already in close proximity. During selective autophagy, once p62 binds to LC3, ALFY may reinforce the interaction between cargo and the membrane ensuring the selective encapsulation of the cargo during selective autophagy. In this context, a higher affinity for PI(3)P would not be required due to the close proximity of ALFY and the membrane. If the FYVE domain of ALFY did have a higher affinity for PI(3)P membranes, this could result in it erroneously targeting p62 bound aggregated protein complexes to other PI(3)P containing membranes in the cell. This, in turn, would decrease the efficiency of the clearance of aggregated proteins by selective autophagy.
4. Materials and Methods
4.1. Plasmid construction.
A gene encoding ALFY-FYVE, residues 3428-3518, was synthesized by Integrated DNA Technologies and cloned into the pET His6 GST TEV LIC cloning vector (1G), which was a gift from Scott Gradia (Addgene, plasmid #29655). ALFY-2xFYVE domain containing two copies of ALFY-FYVE, residues 3428-3518, with seven residue flexible linkers (GQSGES, GSGSGS, and GGGSGG) before, between, and after the first and second copy of ALFY-FYVE was synthesized by Integrated DNA Technologies. ALFY-2xFYVE was cloned into the pET His6 GST TEV LIC cloning vector (1G). ALFY-FYVE E3471K, E3471T, and E3471L mutants were produced via site-directed mutagenesis of ALFY-FYVE in pET His6 GST TEV LIC cloning vector (1G) using a Q5 site-directed mutagenesis kit (NEB, E0554S).
EEA1-FYVE was produced by subcloning residues 1347-1411 of EEA1 from GFP-EEA1 WT (Addgene, plasmid #42307) into pET His6 GST TEV LIC cloning vector (1G). EEA1-2xFYVE domain containing two copies of EEA1-FYVE, residues 1347-1411, with seven residue flexible linkers (GQSGES, GSGSGS, and GGGSGG) before, between, and after the first and second copy of EEA1-FYVE was synthesized with a His12 GST TEV sequence and cloned into pET-24a(+) using BamHI and XhoI by GenScript. EEA1-FYVE V1369E was synthesized with a His12 GST TEV sequence and cloned into pET-24a(+) using BamHI and XhoI by Twist Bioscience.
For localization studies in yeast, mCherry-pCu416Cup1 was produced by subcloning mCherry into pCu416Cup1 (ATCC, plasmid #87729) using SpeI and BamHI, without destroying the restriction sites. ALFY-FYVE and ALFY-2xFYVE were cloned into mCherry-pCu416Cup1 using BamHI and XhoI. ALFY-FYVE E3471K and ALFY-2xFYVE E3471K were produced using site-directed mutagenesis via a Q5 site-directed mutagenesis kit (NEB, E0554S). For ALFY-2xFYVE E3471K mutant, the mutation was produced in both copies of the FYVE domain. GFP-Atg8(414)/GFP-AUT7(414) was obtained from Addgene (Addgene, plasmid #49424).
For the filter trap assays, pDEST-Tomato-ALFY2285-3526 (tdTomato-ALFYC) was used as previously described.17 tdTomato-ALFYC E3471K was generated using site-directed mutagenesis via a Q5 site-direct mutagenesis kit (NEB, E0554S).
4.2. Protein expression and purification.
ALFY-FYVE, ALFY-2xFYVE, ALFY-FYVE mutants EEA1-FYVE, EEA1-2xFYVE, and EEA1-FYVE V1369V mutant were transformed into BL21-CodonPlus (DE3)-RILP competent cells (Agilent). For unlabeled protein expression, cultures were grown to LB Broth (Fisher Scientific, BP1426). To prepare uniformly 13C/15N-labeled and 15N-labeled proteins, bacterial cultures were grown in M9 minimal media containing selective antibiotics and 4 g/L [13C]-D-glucose and/or 1 g/L 15NH4Cl (Cambridge Isotope Laboratories, NLM-467 and DLM-2062) as the sole carbon and nitrogen sources, respectively. Cultures were grown to an optical density at 600 nm (OD600) of 0.6 at 37°C with shaking at 220 rpm. Protein expression was induced by the addition of 0.1 mM isopropylthio-β-D-galactose (IPTG) (IBI Scientific, IB02125) for all ALFY-1xFYVE and EEA1-1xFYVE constructs or by the addition of 0.5 mM IPTG for ALFY-2xFYVE and EEA1-2xFYVE constructs. All cultures were grown for an additional 3 hours at 37°C with shaking at 220 rpm. Cells were harvested and stored at −80°C. Cell pellets were thawed and resuspended in 50 mM Tris pH 8.0, 500 mM NaCl, 1% (v:v) Triton X-100, 5 mM MgCl2, 1 mM phenylmethanesulfonyl fluoride (PMSF), and complete Mini EDTA-free protease inhibitor tablets (Roche, 11836170001). Cells were lysed by passing the sample through a French Press (Thermo Electron, FA-032) four times at 4°C. Lysates were cleared by centrifugation at 40,000 x g for 50 minutes at 4°C. The supernatant was applied to TALON metal affinity resin (Clonetech, 635504), which was preequilibrated with 50 mM Tris pH 8.0 and 500 mM NaCl. The resin was washed with 50 mM Tris pH 8.0, 500 mM NaCl, and 2.5 mM imidazole, and the protein was eluted in 50 mM Tris pH 8.0, 500 mM NaCl, and 200 mM imidazole. TEV protease was added to fractions containing protein and incubated at 4°C rocking overnight to cleave off the His6/His12 GST tag. Fractions containing protein were pooled and applied to a HiLoad Superdex 75 PG column equilibrated in 20 mM Tris pH 7.4, 100 mM NaCl, and 0.2 mM tris(2-carbocyethyl)phosphine hydrochloride (TCEP) for liposome sedimentation assays, 20 mM sodium phosphate pH 6.5, 150 mM NaCl, and 0.2 mM TCEP for structural studies, or 20 mM Bis-Tris pH 6.5, 150 mM NaCl, and 0.2 TCEP for NMR titration studies. Fractions containing purified protein were pooled and concentrated.
4.3. Liposome sedimentation assay.
Synthetic liposomes made using L-α-phosphatidylcholine (PC) (Avanti, 840051) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (PS) (Avanti, 840034) with one of the following phosphoinositide when required L-α-phosphatidylinositol (PI) (Avanti 840042), 1,2-dioleoyl-sn-glycero-3-phospho-(1’-myo-inositol-3’-phosphate) (PI(3)P) (Avanti 850150), 1,2-dioleoyl-sn-glycero-3-phosph-(1’-myo-inositol-4’-phosphate) (PI(4)P) (Avanti 850151), 1,2-dioleoyl-sn-glycero-3-phospho-(1’-myo-inositol-5’-phosphate) (PI(5)P) (Avanti 850152), 1,2-dioleoyl-sn-glycero-3-phospho-(1’-myo-inositol-3’,5’-bisphosphate) (PI(3,5)P2) (Avanti 850154), 1,2-dioleoyl-sn-glycero-3-phospho-(1’-myo-insitol-4’,5’-bisphosphate) (PI(4,5)P2) (Avanti 850155). Liposomes were standardly composed of 80 mole percent PC and 20 mole percent PS or of 76 mole percent PC, 19 mole percent PS, and 5 mole percent of the required phosphoinositide. They were dried under a nitrogen stream for 30 min and dried for 18 hours in a vacuum oven. Dried liposomes were resuspended in 20 mM Tris pH 7.4, 100 mM NaCl, and 0.2 mM TCEP to a final concentration of 2.0 mg/mL. Liposomes were then extruded using an Avanti Mini Extruder using a 0.4 μm Nuclepore Track-Etched membrane (Whatman, 800282). Then 25 μL of 10 μM purified protein was mixed with 25 μL of 2.0 mg/mL liposomes. The mixture was incubated at 4°C for 60 min to afford complex formation, followed by centrifugation at 40,000 rpm for 40 min at 4°C using a TLA45 rotor. Supernatants were removed, and equal volumes of buffer were added to resuspend the pellets. Samples were run on Novex 10-20% tricine protein gels (Invitrogen, EC6625BOX).
Densitometry was performed on both the pellet (P) and supernatant (S) fractions using Image Lab v 5.1 (Bio-Rad). The supernatant and pellet band intensity were summed together to determine the total intensity of protein in each sample. The percentage of protein bound to liposomes was determined by taking the ratio of the pellet band intensity over the total intensity. Each experiment was performed in triplicate or quadruplicate as indicated. The results were averaged and plotted with error bars representing the standard deviations of the three or four independent experiments. Statistical analysis was performed using Prism software v 8.2.1 (GraphPad). Data was compared using an ordinary two-way ANOVA with Sidak’s multiple comparison tests (Figure 1B) or with Tukey’s multiple comparison test (Figure 1D, 1F, 4B, 4D, and 4F). *, p<0.0212; ***, p=0.0006; ****, p<0.0001.
4.4. Dynamic Light Scattering.
To determine the liposome size and homogeneity, dynamic light scattering was performed on liposomes at 2.0 mg/mL at 20°C using DynaPro NanoStar (Wyatt Technology). Light scattering data were analyzed using Dynamics v7.1.8 (Wyatt Technology).
4.5. NMR spectroscopy.
All NMR experiments were performed at 298 K on a Bruker Avance 700 MHz. NMR spectra were processed using Topspin 3.5 (Bruker). 1H chemical shifts were referenced to 0 ppm methyl resonance of 2,2-dimethyl-2-silapentane-5-sulfonate (DSS), whereas 13C and 15N chemical shifts were indirectly references according to the IUPAC recommendations.46 All peaks were assigned using CARA (http://cara.nmr.ch/). The sequence-specific backbone assignment was determined using 2D 1H-15N HSQC, 3D HNCA, 3D HN(CO)CA, 3D HNCO, 3D HN(CA)CO, 3D CBCA(CO)NH, 3D HNCACB, and 3D HBHA(CBCACO)NH experiments. Aliphatic side chain assignments were determined using 2D 1H-13C HSQC, 3D (H)CCH-TOCSY, 3D HC(C)H-COSY, 3D HC(C)H-TOCSY, 3D H(CCCO)NH, and 3D (H)CC(CO)NH experiments. For aromatic side chain assignments, purified protein was dialyzed into 20 mM sodium phosphate pH 6.5, 150 mM NaCl, and 0.2 mM TCEP made in 100% D2O (Cambridge Isotope Laboratories, DLM-4) and 2D COSY, 2D TOCSY and 2D NOESY spectra were recorded. Backbone and side chain assignments were deposited in the BMRB under accession 30736. For structure calculation, a 2D NOESY, 3D 15N-resolved NOESY, and 3D 13C-resolved NOESY were recorded and processed using Topspin 3.5 (Bruker). UNIO was used for iterative automated NOE peak picking and NOE assignment by ATNOS/CANDID with CYANA was used for structure calculation.47,48 The quality of the final ensemble of 20 structures was validated using NMR-Procheck.49 The final structure was deposited in the PDB under ID 6W9N. All structures figures were generated using the MacPyMOL: PyMOL Molecular Graphics System, Version 1.7.6.2 (Shrodinger, LLC).
4.6. Sequence Alignments.
The sequences of all known human FYVE domain containing proteins that met the criteria discussed subsequently were compiled using Blastp, Psi-Blastp, SMART, and Pfam searches.31-33 The FYVE domain must contain four conserved CXXC motif for zinc binding and contain a RRHH, RQHH, RKHH, KKHH, or RYHH sequencing in the basic PI(3)P coordination binding site. ProVis was use to obtain the amino acid sequences of the homologue of the human FYVE domain containing proteins found in Mus musculus, Gallus gallus, Danio rerio, Xenopus tropicalis, Caenorhabditis elegans, and Drosophila melanogaster. 16-18 A sequence alignment was performed and a consensus sequence as well as a conservation plot produced for each individual human FYVE domains using CLC Sequence Viewer, Version 7.5 (QIAGEN).
4.7. Ins(1,3)P2 binding.
In order to transfer the peak assignment of 1H-15N HSQC spectra collected in sodium phosphate buffer for structural calculations to the Bis-Tris buffer for the binding experiments, 1H-15N HSQC spectra of 15N-labeled ALFY-FYVE were collected in the following buffers: 6 mM Bis-Tris, 14 mM sodium phosphate pH 6.5, 150 mM NaCl, and 0.2 mM TCEP; 10 mM Bis-Tris, 10 mM sodium phosphate pH 6.5, 150 mM NaCl, and 0.2 mM TCEP; and 15 mM Bis-Tris, 5 mM sodium phosphate pH 6.5, 150 mM NaCl, and 0.2 mM TCEP. The inositol 1,3-bisphosphate (Ins(1,3)P2) (Echelon, Q-0013) was suspended in 20 mM Bis-Tris, pH 6.5, 150 mM NaCl, and 0.2 mM TCEP. The Ins(1,3)P2 concentration was varied from 0 μM to 250 μM. ALFY-FYVE and ALFY-FYVE E3471K were uniformly 15N-labeled and purified as described above. The final purification buffer of 20 mM Bis-Tris pH 6.5, 150 mM NaCl, and 0.2 mM TCEP. 15N-labeled ALFY-FYVE or 15N-labeled ALFY-FYVE E3471K were mixed with Ins(1,3)P2 to a final protein concentration of 50 μM. 1H15N HSQC spectra were recorded on a Bruker Avance 700 MHz spectrometer using a 1.7 mm cryoprobe. CSP values were determined using δΔ= √((δ1H)2+0.14(δ1N)2). CSP data were fit using MATLAB (MathWorks) to f(x) = CSPmax*((Kd + x + Ptot) – sqrt(Kd + x + Ptot)2– 4*(x* Ptot)))/(2* Ptot)) where Ptot is the total concentration of protein (50 μM).
4.8. Cellular localization.
To determine the cellular localization, SEY6210 yeast cells were transformed with ALFY-FYVE, ALFY-2xFYVE, ALFY-FYVE E3471K, or ALFY-2xFYVE E3471K in mCherry pCu416Cup1 vector (Figure 6A).50 YDR170C (GFP-Sec7), YPR173C (GFP-Vps4), or YOR270C (GFP-Vph1) yeast cells were transformed with mCherry-ALFY-2xFYVE or mCherry-ALFY-2xFYVE E3471K in pCu416Cup1 vector (Figure 6B).51 SEY6210 yeast cells were also co-transformed with mCherry-ALFY-2xFYVE or mCherry-ALFY-2xFYVE E3471K in pCu416Cup1 vector and GFP-Atg8(414)/GFP-AUT7(414) (Figure 6B). Freshly transformed cells were grown at 30°C in synthetic minimal medium (SMD; 0.67% [wt:vol] yeast nitrogen base with ammonium sulfate, 2% [wt:vol] glucose, supplemented with the appropriate amino acids) to an OD600 of approximately 1. No additional copper was added to drive the expression of mCherry-ALFY-FYVE or mCherry-ALFY-FYVE E3471K; instead, expression was driven as a result of the copper in the media. Cells were then plated in SMD on 35 mm MatTek glass bottom dishes and allowed to settle for 5 minutes before imaging. Images were acquired on a Nikon Eclipse Ti microscope fitted with a Yokogawa CSU-W1 confocal scanner unit. Images were acquired using a Nikon alpha Plan-Apochromatic 100x oil lens with NA=1.45 using Nikon immersion oil type F at room temperature. Emission was detected using a Photometrics PRIM BSI detector. The central plane of the cells was imaged in z-stacks to a final height of 2 μm. Acquisition was performed using the NIS elements software and analyzed using ImageJ software. No gamma correction was applied. Single plane images are shown.
4.9. Filter Trap Analysis.
The FTA was performed as described previously.41,42 Briefly, HeLa cells stably expressing Exon1Htt103Q-HT were maintained without doxycycline for several passages to stabilize aggregate number. Cells were then plated and transfected with Lipofectamine 2000 per manufacturer’s instructions with tdTomato, tdTomato-ALFYC or tdTomato-ALFYC E3471K. 24 hours after transfection, a subset of wells transfected with tdTomato were treated with dox to inhibit Exon1Htt103Q-HT expression. All samples were collected then lysed in PBS + 1% Triton-X100 + 8M Urea 96 hours post-transfection. Lysates were immediately pelleted at 14000 rpm. Lysates were quantified using a DC protein assay kit (Bio-Rad). 120 μg and 30 μg of protein were then slotted onto a cellulose acetate membrane (20 μM) using a Biodot (Biorad) and probed with anti-Halotag antibody (Promega). 20 μg of protein was also run on the 4-12% Bis Tris gels (ThermoFisher) and probed with anti-Halotag and anti-Vinculin (ThermoFisher). Densitometry was performed on ImageJ.
Supplementary Material
Synopsis:
ALFY is a FYVE domain containing protein involved in the clearance of misfolded aggregates, including huntingtin, by autophagy. We have demonstrated that the FYVE domain of ALFY contains a highly conserved glutamic acid within its membrane insertion loop that is not present in any other human FYVE domain. This glutamic acid dramatically reduces the ability of ALFY to bind membranes in vitro and in vivo and is required for efficient aggregate clearance.
Acknowledgments:
This research was supported by start-up funds from Dartmouth College and the National Institute of Health awards P20GM113132 and R35GM128662 to M.J.R, National Institute of Health awards R01NS101663, R01NS077111, and R01NS063973 to A.Y, and by a National Science Foundation Graduate Research Fellowship to E.F.R. DNA sequencing was performed using the Genomics and Molecular Biology Shared Resources (GMBSR) at the Norris Cotton Cancer Center at Dartmouth with NCI Cancer Center Support Grant P30CA023108.
Footnotes
Conflict of Interest
The authors declare no potential conflict of interest.
References
- 1.Amm I, Sommer T & Wolf DH Protein quality control and elimination of protein waste: The role of the ubiquitin–proteasome system. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1843, 182–196 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Chen B, Retzlaff M, Roos T & Frydman J Cellular Strategies of Protein Quality Control. Cold Spring Harb Perspect Biol 3, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gregersen N, Bross P, Vang S & Christensen JH Protein Misfolding and Human Disease. Annual Review of Genomics and Human Genetics 7, 103–124 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Ciechanover A & Kwon YT Protein Quality Control by Molecular Chaperones in Neurodegeneration. Front. Neurosci 11, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor JP, Hardy J & Fischbeck KH Toxic proteins in neurodegenerative disease. Science 296, 1991–1995 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Ross CA & Poirier MA Protein aggregation and neurodegenerative disease. Nature Medicine 10, S10–S17 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Aguzzi A & O’Connor T Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nature Reviews Drug Discovery 9, 237–248 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Yamamoto A & Simonsen A The elimination of accumulated and aggregated proteins: A role for aggrephagy in neurodegeneration. Neurobiol Dis 43, 17–28 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamark T & Johansen T Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol 2012, 736905 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaart AVD, Mari M & Reggiori F A Picky Eater: Exploring the Mechanisms of Selective Autophagy in Human Pathologies. Traffic 9, 281–289 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Kissová I et al. Selective and non-selective autophagic degradation of mitochondria in yeast. Autophagy 3, 329–336 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Anding AL & Baehrecke EH Cleaning House: Selective Autophagy of Organelles. Dev. Cell 41, 10–22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reggiori F, Komatsu M, Finley K & Simonsen A Autophagy: more than a nonselective pathway. Int J Cell Biol 2012, 219625 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isakson P, Holland P & Simonsen A The role of ALFY in selective autophagy. Cell Death Differ 20, 12–20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simonsen A et al. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. Journal of Cell Science 117, 4239–4251 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Finley KD et al. blue cheese Mutations Define a Novel, Conserved Gene Involved in Progressive Neural Degeneration. The Journal of Neuroscience 23, 1254–1264 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Filimonenko M et al. The Selective Macroautophagic Degradation of Aggregated Proteins Requires the PI3P-Binding Protein Alfy. Molecular Cell 38, 265–279 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fox LM et al. Huntington’s Disease Pathogenesis Is Modified In Vivo by Alfy/Wdfy3 and Selective Macroautophagy. Neuron (2019) doi: 10.1016/j.neuron.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clausen TH et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 6, 330–344 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Lystad AH et al. Structural determinants in GABARAP required for the selective binding and recruitment of ALFY to LC3B-positive structures. EMBO Rep. 15, 557–565 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gillooly DJ, Simonsen A & Stenmark H Cellular functions of phosphatidylinositol 3-phosphate and FYVE domain proteins. Biochem J 355, 249–258 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stenmark H, Aasland R & Driscoll PC The phosphatidylinositol 3-phosphate-binding FYVE finger. FEBS Letters 513, 77–84 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Wallroth A & Haucke V Phosphoinositide conversion in endocytosis and the endolysosomal system. J. Biol. Chem 293, 1526–1535 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Misra S & Hurley JH Crystal Structure of a Phosphatidylinositol 3-Phosphate-Specific Membrane-Targeting Motif, the FYVE Domain of Vps27p. Cell 97, 657–666 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Stenmark H, Aasland R, Toh B-H & D’Arrigo A Endosomal Localization of the Autoantigen EEA1 Is Mediated by a Zinc-binding FYVE Finger. J. Biol. Chem 271,24048–24054 (1996). [DOI] [PubMed] [Google Scholar]
- 26.Kutateladze T & Overduin M Structural Mechanism of Endosome Docking by the FYVE Domain. Science 291, 1793–1796 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Stahelin RV et al. Phosphatidylinositol 3-Phosphate Induces the Membrane Penetration of the FYVE Domains of Vps27p and Hrs. J. Biol. Chem 277, 26379–26388 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Kutateladze TG et al. Multivalent Mechanism of Membrane Insertion by the FYVE Domain. J. Biol. Chem 279, 3050–3057 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Hayakawa A et al. Structural Basis for Endosomal Targeting by FYVE Domains. J. Biol. Chem 279, 5958–5966 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Jehl P, Manguy J, Shields DC, Higgins DG & Davey NE ProViz—a web-based visualization tool to investigate the functional and evolutionary features of protein sequences. Nucleic Acids Res 44, W11–W15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul SF et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schultz J, Copley RR, Doerks T, Ponting CP & Bork P SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res. 28, 231–234 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bateman A et al. Pfam 3.1: 1313 multiple alignments and profile HMMs match the majority of proteins. Nucleic Acids Res 27, 260–262 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Axe EL et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. The Journal of Cell Biology 182, 685–701 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Itakura E & Mizushima N Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6, 764–776 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.He J et al. Membrane insertion of the FYVE domain is modulated by pH. Proteins 76, 852–860 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blatner NR et al. The Molecular Basis of the Differential Subcellular Localization of FYVE Domains. J. Biol. Chem 279, 53818–53827 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Mari M, Macia E, Marchand-Brustel YL & Cormont M Role of the FYVE Finger and the RUN Domain for the Subcellular Localization of Rabip4. J. Biol. Chem 276, 42501–42508 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Ridley SH et al. FENS-1 and DFCP1 are FYVE domain-containing proteins with distinct functions in the endosomal and Golgi compartments. J. Cell. Sci 114, 3991–4000 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Lawe DC, Patki V, Heller-Harrison R, Lambright D & Corvera S The FYVE domain of early endosome antigen 1 is required for both phosphatidylinositol 3-phosphate and Rab5 binding. Critical role of this dual interaction for endosomal localization. J. Biol. Chem 275, 3699–3705 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Eenjes E, Dragich JM, Kampinga HH & Yamamoto A Distinguishing aggregate formation and aggregate clearance using cell-based assays. J. Cell. Sci 129, 1260–1270 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eenjes E, Yang-Klingler YJ & Yamamoto A Monitoring Aggregate Clearance and Formation in Cell-Based Assays in Protein Misfolding Diseases: Methods and Protocols (ed. Gomes CM) 157–169 (Springer, 2019). doi: 10.1007/978-1-4939-8820-4_9. [DOI] [PubMed] [Google Scholar]
- 43.Yokogawa M et al. NMR Analyses of the Interaction between the FYVE Domain of Early Endosome Antigen 1 (EEA1) and Phosphoinositide Embedded in a Lipid Bilayer. J Biol Chem 287, 34936–34945 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hornbeck PV et al. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43, D512–D520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jakobi AJ et al. Structural basis of p62/SQSTM1 helical filaments and their role in cellular cargo uptake. Nat Commun 11, 1–15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Markley JL et al. Recommendations for the presentation of NMR structures of proteins and nucleic acids--IUPAC-IUBMB-IUPAB Inter-Union Task Group on the standardization of data bases of protein and nucleic acid structures determined by NMR spectroscopy. Eur. J. Biochem 256, 1–15 (1998). [DOI] [PubMed] [Google Scholar]
- 47.Herrmann T, Güntert P & Wüthrich K Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J. Biomol. NMR 24, 171–189 (2002). [DOI] [PubMed] [Google Scholar]
- 48.Herrmann T, Güntert P & Wüthrich K Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol 319, 209–227 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R & Thornton JM AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8, 477–486 (1996). [DOI] [PubMed] [Google Scholar]
- 50.Robinson JS, Klionsky DJ, Banta LM & Emr SD Protein sorting in Saccharomyces cerevisiae: isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol Cell Biol 8, 4936–4948 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu J et al. A Validated Set of Fluorescent-Protein-Based Markers for Major Organelles in Yeast (Saccharomyces cerevisiae). mBio 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.