

1. Introduction
1.1. Background
In 2008, several synthetic cannabinoid receptor agonists (referred to as “synthetic cannabinoids” throughout the rest of this document) were detected in herbal smoking blends, which were sold on the Internet and in specialized shops under a variety of brand names such as “Spice Silver”, “Spice Gold”, “Spice Diamond”, “Yucatan Fire” and “Smoke” [[1], [2]]. These colourful and professionally designed packages of herbal products typically contain about 0.5–3 g of finely cut plant material to which one or more synthetic cannabinoids have been added [3,4]. Generally, they do not contain cannabis, but may produce cannabis-like effects. Furthermore, they are usually administered by smoking either as a joint or in a water-pipe.
Before 2008, the use of these herbal products seemed to be restricted to a small number of experimental users. However, in 2008, these products achieved immense popularity in Germany and other European countries through the Internet and in subsequent media reports, where they were referred to as “legal alternatives” to cannabis, thus unintentionally promoting their use. Since then, hundreds of new herbal products with different brand names have been marketed. The synthetic additives in these products could vary significantly in terms of quantity as well as the types of synthetic cannabinoids used [2,3,[5], [6], [7], [8], [9], [10], [11], [12], [13], [14], [15], [16], [17], [18], [19]].
One of the first synthetic cannabinoids detected in the market that persisted over a number of years was JWH-018 a representative of the napthoyl indole group of substances. In the intervening years, synthetic cannabinoids have evolved to be quite structurally diverse, although some trends have been observed (Fig. 1). These include simple halide substitution in a number of positions in the alkyl chain or varying the length of the alkyl chain. Subsequently, changes to the linked group from naphthalene to different groups such as adamantyl and methoxyphenyl were observed. One evolution in the structure of synthetic cannabinoids that led to a dramatic increase in substances in 2013 was the introduction of indazole heterocycles and the use of amide and ester linking groups. Substances of this type continue to emerge to this day, although to a lesser extent in recent years.
Fig. 1.

Examples of the evolution of synthetic cannabinoids structures.
Currently, relatively little is known about the pharmacology and toxicology of the various (frequently changing) synthetic cannabinoids that are added to the herbal products. A number of these substances may have a higher addictive potential compared to cannabis due to quicker development of tolerance, which could lead to a tendency towards higher acute and long-term toxicity [[20], [21], [22]]. Between 2015 and 2019 fourteen synthetic cannabinoids were placed under International control, in schedule II of the 1971 Convention on Psychotropic Substances. These are JWH- 018, AM-2201, MDMB-CHMICA, 5F-APINACA (5F-AKB-48), MDMB-CHMICA, XLR-11, AB-PINACA, AB-CHMINACA, 5F-PB-22, UR-144, 5F-MDMB-PIN- ACA, ADB-FUBINACA, FUB-AMB (MMB-FUBINACA, AMB-FUBINACA), CUMYL-4CN-BINACA and ADB-CHMINACA (MAB-CHMINACA). A further four substances were placed in schedule II of the same convention following scheduling decisions of the 63rd Session of the Commission on Narcotic Drugs, 2–6 March 2020. These are AB-FUBINACA, 5F-AMB-PINACA (5F-AMB, 5F-MMB-PINACA), 5F-MDMB-PICA (5F-MDMB-2201) and 4F-MDMB-BINACA.
At the national level, countries have used a variety of different types of legislation to deal with the emergence of synthetic cannabinoids and other groups of NPS. These include individual listing or scheduling substances through generic controls, analogue legislation, through temporary bans and rapid procedures or by other leg-islative approaches. Moreover, some countries have developed legislation based on the purported psychoactive effect of synthetic cannabinoids, that of binding to can-nabinoid CB1 receptors. For further information and details of these and other types of legislative responses by Member States to address NPS, the reader is directed to the UNODC Early Warning Advisory on NPS (www.unodc.org/nps) and the United Nations toolkit on Synthetic Drugs (www.unodc.org/unodc/en/opioid-crisis/un-toolkit-on-synthetic-drugs.html).
1.2. Purpose and use of the manual
The present manual is one in a series of similar publications dealing with the identification and analysis of various types of drugs under control. These manuals are the outcome of a programme pursued by UNODC since the early 1980s, aimed at the harmonization and establishment of recommended methods of analysis for national drug analysis laboratories.
The present Manual is a revision of the manual on Recommended Methods for the Identification and Analysis of Synthetic Cannabinoids in Seized Materials (ST/NAR/48), which was published in 2013. It has been prepared taking into account the continued emergence of new synthetic cannabinoids, the inclusion of a number of substances in the International Drug Conventions and the latest developments in analytical technology with a view to providing the basis for reliable forensic scientific evidence on seized materials containing synthetic cannabinoids.
In line with the overall objective of the series, the present manual suggests approaches that may assist drug analysts in the selection of methods appropriate to the sample under examination and provide data suitable for the purpose at hand, leaving room also for adaptation to the level of sophistication of different laboratories and the various legal needs. The majority of methods included in the present manual are validated methods, which have been used in reputable laboratories. The reader should be aware, however, that there are a number of other methods, including those published in the forensic science literature, which may also produce acceptable results. Any new method that is about to be used in the reader’s laboratory must be validated and/or verified prior to casework use.
In addition, there are a number of more sophisticated approaches, but they may not be necessary for routine operational applications. Therefore, the methods described here should be understood as guidance, and when needed, minor modifications can be made to suit local circumstances, and validation should be performed on the modified methods before use on casework. The choice of the methodology and approach to analysis as well as the decision whether or not additional methods are required remain with the analyst and may also be dependent on the availability of appropriate instrumentation and the level of legally acceptable proof in the jurisdic-tion within which the analyst works.
Attention is also drawn to the vital importance of the availability to drug analysts of reference materials and literature on drugs of abuse and analytical techniques. Moreover, the analyst must of necessity keep abreast of current trends in drug analysis, consistently following current analytical and forensic science literature.
2. General aspects
2.1. Definition of synthetic cannabinoids
Synthetic cannabinoids are referred to as substances with structural features which allow binding to one of the known cannabinoid receptors, that is, CB1 or CB2, present in human cells. The CB1 receptor is located mainly in the brain and spinal cord and is responsible for the typical physiological and particularly the psychotropic effects of cannabis, whereas the CB2 receptor is located mainly in the spleen and cells of the immune system and may mediate immune-modulatory effects.
With the exception of endocannabinoids, naturally occurring cannabinoids are limited to chemical constituents of cannabis such as Δ9-tetrahydrocannabinol and cannabidiol. In contrast, synthetic cannabinoids as defined above, could encompass a great variety of structurally dissimilar compounds with the possibility for further structural changes, that is, analogues and derivatives, which could potentially show affinity to either one of the cannabinoid receptors as well.
The binding of synthetic cannabinoids to cannabinoid receptors may result in (par-tial) agonistic, inverse agonistic or antagonistic effects. Synthetic cannabinoids of interest in forensic science contexts are mainly compounds showing “sufficient” affinity to the CB1 receptor and show agonistic or partial agonistic activity, as the typical psychotropic cannabis-like effects are mediated typically via agonistic stimu-lation of this receptor type.
2.2. Chemical classification
Cannabinoid receptor agonists can be classified to a large degree based on their chemical structures into the following main groups [23,24]. Examples of each group are given in section 3.
-
i.Classical cannabinoids
-
–tetrahydrocannabinol, other chemical constituents of cannabis and their structurally related synthetic analogues
-
–
-
iiNon-classical cannabinoids
-
–cyclohexylphenols or 3-arylcyclohexanols
-
–
-
iiiHybrid cannabinoids
-
–combinations of structural features of classical and non-classical cannabinoids
-
–
-
iv.Aminoalkylindoles, which can be further divided into the following groups:
-
a.Naphthoylindoles
-
b.Phenylacetylindoles
-
c.Benzoylindoles
-
d.Naphthylmethylindoles
-
e.Cyclopropoylindoles
-
f.Adamantoylindoles
-
g.Indole carboxamides
-
h.Indole carboxylates
-
a.
-
v.Aminoalkylindazoles, which can be further divided into the following groups:
-
a.Naphthoylindazoles
-
b.Indazole carboxamides
-
a.
-
vi.Eicosanoids
-
–endocannabinoids and their synthetic analogues
-
–
- vii.
Many derivatives and analogues in the above classes of compounds could be syn-thesized by the addition of a halogen, alkyl, alkoxy or other substituents to one of the aromatic ring systems. Other small changes such as variation of the length and configuration of the alkyl chain can also be made.
2.3. Products and modes of administration
Before the appearance of the wide variety of synthetic cannabinoids in “ready-to- smoke” products, a few synthetic cannabinoids such as CP-55,940 or WIN-55,212–2 were commercially available in small quantities used in research institutes for pharmacological research.
Around 2004, the first products containing synthetic cannabinoids emerged. They were added to plant material, for example, crushed leaves or strips of leaves, by soaking or spraying a solution of one or more synthetic cannabinoids in an organic solvent which was later evaporated. In some cases, synthetic cannabinoids in solid form (crystalline powder) were used, leading to an inhomogeneous distribution of the active compound in the plant material. A minority of these products were found to resemble hashish in colour and texture and are used in a similar manner, that is, mixed with tobacco in a joint or smoked pure in pipe.
2.3.1. General aspects 7
In recent years, a growing number of online shops and traders started to offer synthetic cannabinoids as “research chemicals” in variable amounts from milligramme to kilogramme quantities. These substances are not only procured by mass-producers of these herbal products but also by end-users who would concoct their own blend of herbal mixtures. Some of these substances were of high purity [27], while others were contaminated with synthetic by-products or artefacts due to insufficient clean-up [18]. Products containing synthetic cannabinoids in e-cigarettes have also been observed in recent years [28]. In both Europe and Brazil, paper and other types of materials have been found to be used as supports for impregnated synthetic cannabinoids intended to be smuggled into prison facilities [[29], [30], [31]]. In Brazil, synthetic cannabinoids have also appeared on LSD-type blotters [32]. Other means of administration such as intravenous injection or snorting have not been reported to play a significant role.
3. Description of the pure compounds
The pure compounds are mostly in the form of fine crystalline powders with colours ranging from white to a grey, brownish or yellowish hue. Most of the compounds are highly lipophilic and show good solubility in non-polar or medium-polarity solvents such as methanol, ethanol, acetonitrile, ethyl acetate, acetone or isooctane. Generally, water solubility of synthetic cannabinoids is low.
The following table includes examples of synthetic cannabinoids in the respective classes as defined in section 2.2. Note that the primary name used below is the most common name/abbreviation in the UNODC early warning advisory and the reader is directed there for more comprehensive list of alternate names and abbreviations. Also note that substances can have different acceptable chemical or IUPAC names.
-
i)
Classical cannabinoids
| Name | Structure | Chemical name | CAS No | Molecular formula |
|---|---|---|---|---|
| THC (Δ9-Tetrahydrocannabinol) | 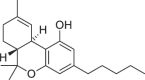 |
(6aR,10aR)-6a,7,8,10a-tetrahydro-6,6,9-trimethyl-3-pentyl-6H-dibenzo[b,d]pyran-1-ol | 1972-08-3 | C21H30O2 |
|
HU-210 (11-Hydroxy-Δ8-THC) |
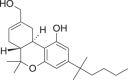 |
6aR,10aR)-6a,7,10,10a-tetrahydro-6,6-dimethyl-9-(hydroxymethyl)-3-(2-methyloctan-2-yl)-6H-dibenzo[b,d]pyran-1-ol | 112,830-95-2 | C25H38O3 |
-
ii)
Non-classical cannabinoids
| Name | Structure | Chemical name | CAS No | Molecular formula |
|---|---|---|---|---|
| CP-47,497 |  |
rel-2[(1S, 3R)- 3- hydroxycyclohexyl]- 5- (2- methyloctan- 2- yl)phenol | 70,434-82-1 | C21H34O2 |
| CP-47,497-C6 | 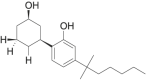 |
rel- 2[(1S, 3R)- 3- hydroxycyclohexyl]- 5- (2- methylheptan- 2- yl)phenol | 70,435-06-2 | C20H32O2 |
| CP-47,497-C8 |  |
rel- 2- [(1S, 3R)- 3- hydroxycyclohexyl]- 5- (2- methylnonan- 2- yl)phenol | 70,434-92-3 | C22H36O2 |
| CP-47,497-C9 | 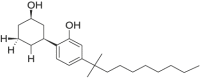 |
rel- 2[(1S, 3R)- 3- hydroxycyclohexyl]- 5- (2- methyldecan- 2- yl)phenol | 70,435–084 | C23H38O2 |
| CP-55,940 | 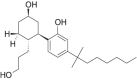 |
rel- 2- [(1R, 2R, 5R)- 5- hydroxy- 2- (3- hydroxypropyl)cyclohexyl]- 5- (2- methyloctan- 2- yl)phenol | 83,003-12-7 | C24H40O3 |
| Dimethyl CP-47,497-C8 | 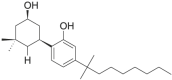 |
rel- 2- [(1S, 3R)- 3- hydroxy-5,5-dimethylcyclohexyl]- 5- (2- methylnonan- 2- yl)phenol | not available | C24H40O2 |
-
iii)
Hybrid cannabinoids
| Name | Structure | Chemical name | CAS No | Molecular formula |
|---|---|---|---|---|
| AM-4030 |  |
(6S,6aR,9R,10aR)-9-(hydroxymethyl)-6-[(E)-3-hydroxyprop-1-enyl]-6-methyl-3-(2-methyloctan-2-yl)-6a,7,8,9,10,10a-hexahydrobenzo[c]chromen-1-ol | 587,023-54-9 | C27H42O4 |
-
iv)
Aminoalkylindoles
a) Naphthoylindoles
| Name | Structure | Chemical name | CAS No | Molecular formula |
|---|---|---|---|---|
| AM-1220 | 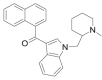 |
(naphthalen-1-yl) [1- [(1- methyl piperidin-2-yl)methyl]- 1H- indol- 3- yl]methanone | 137,642-54-7 | C26H26N2O |
| AM-1220 azepane isomer |  |
(naphthalen-1- yl)[1- (1- methylazepan- 3- yl)- 1H- indol- 3- yl]methanone | 1,348,081-04-8 | C26H26N2O |
| AM-2201 |  |
(naphthalen-1-yl) [1- (5- fluoropentyl)- 1H- indol- 3- yl]methanone | 335,161-24-5 | C24H22FNO |
| AM-2232 |  |
5-(3-(1-naphthoyl)-1H-indol-1-yl)pentanenitrile | 335,161-19-8 | C24H20N2O |
| JWH-007 | 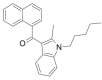 |
(naphthalen-1-yl) (2- methyl- 1- pentyl- 1H- indol- 3- yl)methanone | 155,471-10-6 | C25H25NO |
| JWH-015 |  |
(naphthalen-1-yl) (2- methyl- 1- propyl- 1H- indol- 3- yl)methanone | 155,471-08-2 | C23H21NO |
| JWH-018 |  |
(naphthalen-1-yl) (1- pentyl- 1H- indol- 3- yl)methanone | 209,414-07-3 | C24H23NO |
| JWH-019 | 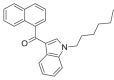 |
(naphthalen-1-yl) (1- hexyl- 1H- indol- 3- yl)methanone | 209,414-08-4 | C25H25NO |
| JWH-020 | 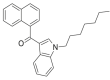 |
(naphthalen-1-yl) (1-heptyl-1H-indol-3-yl)methanone | 209,414-09-5 | C26H27NO |
| JWH-022 |  |
(naphthalen-1-yl)[1- (pent-4-en- 1- yl)- 1H- indol- 3- yl]methanone | 209,414-16-4 | C24H21NO |
| JWH-072 | 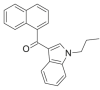 |
(naphthalen-1-yl) (1- propyl- 1H- indol- 3- yl)methanone | 209,414-06-2 | C22H19NO |
| JWH-073 | 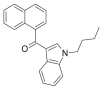 |
(naphthalen-1-yl) (1- butyl- 1H- indol- 3- yl)methanone | 208,987-48-8 | C23H21NO |
| JWH-073 (4-methylnaphthyl) |  |
(4- methylnaphthalen- 1- yl) (1- butyl- 1H- indol- 3- yl)methanone | 1,354,631-21-2 | C24H23NO |
| JWH-081 |  |
(4- methoxynaphthalen-1-yl) (1- pentyl- 1H- indol- 3- yl)methanone | 210,179-46-7 | C25H25NO2 |
| JWH-122 | 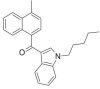 |
(4- methylnaphthalen-1-yl) (1- pentyl- 1H- indol- 3- yl)methanone | 619,294-47-2 | C25H25NO |
| JWH-200 | 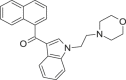 |
(naphthalen-1-yl) [1- [2- (morpholin-4-yl)ethyl]- 1H- indol- 3- yl]methanone | 103,610-04-4 | C25H24N2O2 |
| JWH-210 |  |
(4- ethyl naphthalen-1-yl) (1- pentyl- 1H- indol- 3- yl) methanone | 824,959-81-1 | C26H27NO |
| JWH-387 | 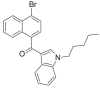 |
(4-bromonaphthalen-1-yl) (1-pentyl-1H-indol-3-yl)methanone | 1,366,067-59-5 | C24H22BrNO |
| JWH-398 |  |
(4-chloronaphthalen-1-yl) (1-pentyl-1H-indol-3-yl)methanone | 1,292,765-18-4 | C24H22ClNO |
| JWH-412 | 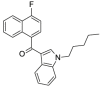 |
(4-fluoronaphthalen-1-yl) (1-pentyl-1H-indol-3-yl)methanone | 1,364,933-59-4 | C24H22FNO |
| MAM-2201 | 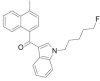 |
(4- methylnaphthalen-1-yl)[1- (5- fluoropentyl)- 1H- indol- 3- yl]methanone | 1,354,631-24-5 | C25H24FNO |
b) Phenylacetylindoles
| Name | Structure | Chemical name | CAS No | Molecular formula |
|---|---|---|---|---|
| Cannabipiperidi-ethanone | 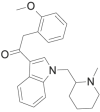 |
2- (2- methoxyphenyl)- 1- [1- [(1- methylpiperidin-2-yl)methyl]- 1H- indol- 3- yl]ethanone | 1,345,970-43-5 | C24H28N2O2 |
| JWH-201 |  |
2- (4- methoxyphenyl)- 1- (1- pentyl- 1H- indol- 3- yl)ethanone | 864,445-47-6 | C22H25NO2 |
| JWH-203 | 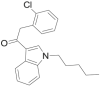 |
2- (2- chlorophenyl)- 1- (1- pentyl- 1H- indol- 3- yl)ethanone | 864,445-54-5 | C21H22ClNO |
| JWH-250 |  |
2- (2- methoxyphenyl)-1- (1- pentyl- 1H- indol- 3- yl) ethanone | 864,445-43-2 | C22H25NO2 |
| JWH-251 |  |
2- (2- methylphenyl)- 1- (1- pentyl- 1H- indol- 3- yl)ethanone | 864,445-39-6 | C22H25NO |
| JWH-302 | 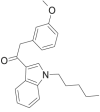 |
2- (3- methoxyphenyl)- 1- (1- pentyl- 1H- indol- 3- yl) ethanone | 864,445-45-4 | C22H25NO2 |
| RCS-8 | 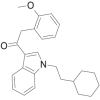 |
2- (2- methoxyphenyl)-1- (1- (2- cyclohexylethyl)- 1H- indol- 3- yl)ethanone | 1,345,970-42-4 | C25H29NO2 |
c) Benzoylindoles
| Name | Structure | Chemical name | CAS No | Molecular Formula |
|---|---|---|---|---|
| AM-694 | 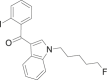 |
(2- iodophenyl)[1- (5- fluoropentyl)- 1H- indol- 3- yl]methanone | 335,161-03-0 | C20H19FINO |
| AM-694 chloro derivative |  |
(2- iodophenyl)[1- (5- chloropentyl)- 1H- indol- 3- yl]methanone | not available | C20H19ClINO |
| AM-2233 | 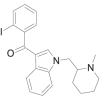 |
(2- iodophenyl)[1- [(1- methylpiperidin-2-yl)methyl]- 1H- indol- 3- yl]methanone | 444,912-75-8 | C22H23IN2O |
| RCS-4 |  |
(4- methoxyphenyl) (1- pentyl- 1H- indol- 3- yl)methanone | 1,345,966-78-0 | C21H23NO2 |
| RCS-4 ortho isomer (or 2-methoxy isomer) | 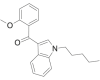 |
(2- methoxyphenyl) (1- pentyl- 1H- indol- 3- yl)methanone | 1,345,966-76-8 | C21H23NO2 |
| RCS-4 butyl homologue (C4 homologue) | 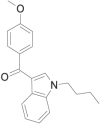 |
(4- methoxyphenyl) (1- butyl- 1H- indol- 3- yl)methanone | 1,345,966-77-9 | C20H21NO2 |
| WIN 48,098 | 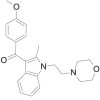 |
(4- methoxyphenyl)[(2- methyl)- 1- [2- (morpholin-4-yl)ethyl]- 1H- indol- 3- yl]methanone | 92,623-83-1 | C23H26N2O3 |
d) Naphthylmethylindoles
| Name | Structure | Chemical name | CAS No | Molecular Formula |
|---|---|---|---|---|
| JWH-184 | 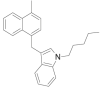 |
(3-[(4-methyl-1-naphthalenyl)methyl]-1-pentyl-1H-indole | 619,294-37-0 | C25H27N |
e) Cyclopropoylindoles
| Name | Structure | Chemical name | CAS No | Molecular Formula |
|---|---|---|---|---|
| FUB-144 | 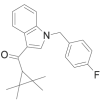 |
(1-(4-fluorobenzyl)-1H-indol-3-yl)-(2,2,3,3-tetramethylcyclopropyl)methanone | 2,185,863-15-2 | C23H24FNO |
| UR-144 |  |
(2, 2, 3, 3- tetramethylcyclopropyl) (1- pentyl- 1H- indol- 3- yl)methanone | 1,199,943-44-6 | C21H29NO |
| XLR-11 | 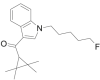 |
(2, 2, 3, 3- tetramethylcyclopropyl) (1- (5- fluoropentyl)- 1H- indol- 3- yl)methanone | 1,364,933-54-9 | C21H28FNO |
f) Adamantoylindoles
| Name | Structure | Chemical name | CAS No | Molecular Formula |
|---|---|---|---|---|
| AB-001 |  |
(1-adamantyl) (1- pentyl- 1H- indol- 3- yl)methanone | 1,345,973-49-0 | C24H31NO |
| AM-1248 | 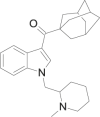 |
(1-adamantyl)[1- [(1- methylpiperidin-2-yl)methyl]- 1H- indol- 3- yl] methanone | 335,160-66-2 | C26H34N2O |
g) Indole carboxamides
| Name | Structure | Chemical name | CAS No | Molecular Formula |
|---|---|---|---|---|
| AB-CHMICA |  |
N-[1-amino-3-methyl-1-oxobutan-2-yl]-1-(cyclohexylmethyl)-1H-indole-3-carboxamide | 2,219,330-90-0 | C21H29N3O2 |
| ADBICA |  |
N-[1-amino-3,3-dimethyl-1-oxobutan-2-yl]-1-pentyl-1H-indole-3-carboxamide | 1,445,583-48-1 | C20H29N3O2 |
| APICA |  |
N-(1-adamantyl)-1-pentyl-1H-indol-3-carboxamide | 1,345,973-50-3 | C24H32N2O |
| APP-CHMICA | 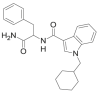 |
N-[1-amino-1-oxo-3-phenylpropan-2-yl]-1-(cyclohexylmethyl)-1H-indole-3-carboxamide | Not available | C24H28N3O2 |
| 5F-MN-24 | 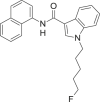 |
1-(5-fluoropentyl)-N-(naphthalen-1-yl)-1H-indole-3-carboxamide | 1,445,580-60-8 | C24H23FN2O |
| 5-Fluoro APP-PICA (PX 1) |  |
N-[1-amino-1-oxo-3-phenylpropan-2-yl]-1-(5-fluoropentyl)-1H-indole-3-carboxamide | 2,221,100-71-4 | C23H26FN3O2 |
| CUMYL-5F-PICA | 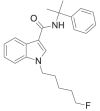 |
1-(5-fluoropentyl)-N- (2-phenylpropan-2yl)-1H-indole-3-carboxamide | 1,400,742-18-8 | C23H27FN2O |
| 5F-MDMB-PICA | 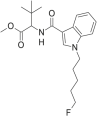 |
methyl 2-[1-(5-fluoropentyl)-1H-indole-3-carboxamido]-3, 3-dimethylbutanoate; | 1,971,007-88-1 | C21H29FN2O3 |
| MDMB-CHMICA |  |
methyl 2-(1-(cyclohexylmethyl)-1H-indole-3-carboxamido)-3,3-dimethylbutanoate | 1,971,007-95-0 | C23H32N2O3 |
| MMB-CHMICA | 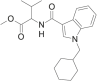 |
methyl 2-[1-(cyclohexylmethyl)-1H-indole-3-carboxamido]-3-methylbutanoate | 1,971,007-94-9 | C22H30N2O3 |
| MDMB-FUBICA | 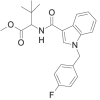 |
methyl 2-[1-(4-fluorobenzyl)-1H-indole-3-carboxamido]-3,3-dimethylbutanoate | 1,971,007-91-6 | C23H25FN2O3 |
| MMB-FUBICA |  |
methyl 2-[1-(4-fluorobenzyl)-1H-indole-3-carboxamido]-3-methylbutanoate | 1,971,007-90-5 | C22H23FN2O3 |
| 5F-APICA |  |
N-(1-adamantyl)-1-(5-fluoropentyl)-1H-indol-3-carboxamide | 1,354,631-26-7 | C24H31FN2O |
h) Indole carboxylates
| Name | Structure | Chemical name | CAS No | Molecular formula |
|---|---|---|---|---|
| BB-22 |  |
quinolin-8-yl 1-(cyclohexylmethyl)-1H-indole-3-carboxylate | 1,400,742-42-8 | C25H24N2O2 |
| FDU-PB-22 | 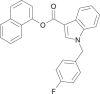 |
napthalen-1-yl 1-(4-fluorobenzyl)-1H-indole-3-carboxylate | 1,883,284-94-3 | C26H18FNO2 |
| 5F-PB-22 |  |
quinolin-8-yl 1-(5-fluoropentyl)-1H-indole-3-carboxylate | 1,400,742-41-7 | C23H21FN2O2 |
| FUB-PB-22 | 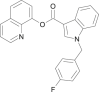 |
quinolin-8-yl 1-(4-fluorobenzyl)-1H-indole-3-carboxylate | 1,800,098-36-5 | C25H17FN2O2 |
| NM-2201 | 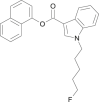 |
naphthalen-1-yl 1-(5-fluoropentyl)-1H-indole-3-carboxylate | 2,042,201-16-9 | C24H22FNO2 |
| PB-22 | 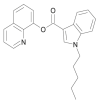 |
quinolin-8-yl 1-pentyl-1H-indole-3-carboxylate | 1,400,742-17-7 | C23H22N2O2 |
-
v)
Aminoalkylindazoles
a) Naphthoylindazoles
| Name | Structure | Chemical name | CAS No | Molecular formula |
|---|---|---|---|---|
| THJ-018 |  |
Naphthalen-1-yl[1-(pent-1-yl)-1H-indazol-3-yl]methanone | 1,364,933-55-0 | C23H22N2O |
| THJ-2201 | 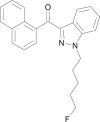 |
[1-(5-Fluoropent-1-yl)-1H-indazol-3-yl](naphthalen-1-yl)methanone | 1,801,552-01-1 | C23H21FN2O |
b) Indazole carboxamides
| Name | Structure | Chemical name | CAS No | Molecular formula |
|---|---|---|---|---|
| AB-CHMINACA |  |
N-(1-amino-3-methyl-1-oxobutan-2-yl)-1-(cyclohexylmethyl)-1H-indazole-3-carboxamide | 1,185,887-21-1 | C20H28N4O2 |
| AB-FUBINACA | 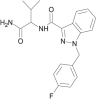 |
N-(1-amino-3-methyl-1-oxobutan-2-yl)-1-(4-fluorobenzyl)-1H-indazole-3-carboxamide | 1,185,282-01-2 | C20H21FN4O2 |
| AB-PINACA | 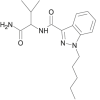 |
N-(1-amino-3-methyl-1-oxobutan-2-yl)-1-pentyl-1H-indazole-3-carboxamide | 1,445,752-09-9 | C18H25N4O2 |
| ADB-FUBINACA |  |
N-(1-amino-3,3-dimethyl-1-oxobutan-2-yl)-1-(4-fluorobenzyl)-1H-indazole-3-carboxamide | 1,445,583-51-6 | C21H23FN4O2 |
| ADB-PINACA | 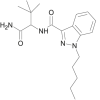 |
N-(1-amino-3,3-dimethyl-1-oxobutan-2-yl)-1-pentyl-1H-indazole-3-carboxamide | 1,633,766-73-0 | C19H27N4O2 |
| APINACA |  |
N-(1-adamantyl)-1-pentyl-1H-indazole-3-carboxamide | 1,345,973-53-6 | C23H31N3O |
| APP-CHMINACA | 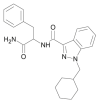 |
N-(1-amino-1-oxo-3-phenylpropan-2-yl)-1-(cyclohexylmethyl)-1H-indazole-3-carboxamide | 1,185,887-14-2 | C24H28N4O2 |
| 5Cl-AB-PINACA | 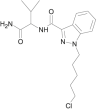 |
N-(1-amino-3-methyl-1-oxobutan-2-yl)-1-(5-chloropentyl)-1H-indazole-3-carboxamide | 1,801,552-02-2 | C18H25ClN4O2 |
| CUMYL-4CN-BINACA | 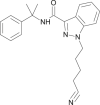 |
N-(2-phenylpropan-2-yl)-1-(4-cyanobutyl)-1H-indazole-3-carboxamide | 1,631,074-54-8 | C22H24N4O |
| 5F-AB-PINACA | 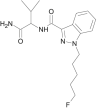 |
N-(1-amino-3-methyl-1-oxobutan-2-yl)-1-(5-fluoropentyl)-1H-indazole-3-carboxamide | 1,800,101-60-3 | C18H25FN4O2 |
| 5F-ADB-PINACA | 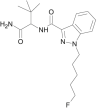 |
N-[1-(amino-3,3-dimethyl-1-oxo-butan-2-yl)-1-(5-fluoropentyl)-1H-indazole-3-carboxamide | 1,863,065-90-0 | C19H27FN4O2 |
| 5F-AMB-PINACA |  |
methyl 2-[1-(5-fluoropentyl)-1H-indazole-3-carboxamido]-3-methylbutanoate | 1,801,552-03-3 | C19H26FN3O3 |
| 5F-APINACA | 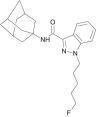 |
N-(1-adamantyl)-5-fluoropentyl-1H-indazole-3-carboxamide | 1,400,742-13-3 | C23H30FN3O |
| 5F- APP-PINACA |  |
N-(1-amino-1-oxo-3-phenylpropan-2-yl)-1-(5-fluoropentyl)-1H-indazole-3-carboxamide | Not available | C22H25FN4O2 |
| 5F- EDMB-PINACA |  |
ethyl 2-(1-(5-fluoropentyl)-1H-indazole-3-carboxamido-3,3-dimethylbutanoate | Not available | C21H31FN3O3 |
| 5F- MDMB-PINACA | 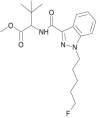 |
methyl 2-[1-(5-fluoropentyl)-1H-indazole-3-carboxamido]-3,3-dimethylbutanoate | 1,838,134-16-9 | C20H28FN3O3 |
| FUB-APINACA | 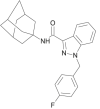 |
N-(1-adamantyl)-1-(4-fluorobenzyl)-1H-indazole-3-carboxamide | Not available | C25H26FN3O |
| AMB-CHMINACA |  |
methyl 2-[1-(cyclohexylmethyl)-1H-indazole-3-carboxamido]-3-methylbutanoate | Not available | C21H29N3O3 |
| MAB-CHMINACA |  |
N-(1-amino-3,3-dimethyl-1-oxobutan-2-yl)-1-(cyclohexylmethyl)-1H-indazole-3-carboxamide | 1,863,065-92-2 | C21H30N4O2 |
| MDMB-CHMINACA |  |
methyl 2-[1-(cyclohexylmethyl)-1H-indazole-3-carboxamido]-3,3-dimethylbutanoate | 1,185,888-32-7 | C22H31N3O3 |
| MDMB-FUBINACA | 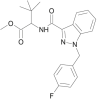 |
methyl 2-[1-(4-fluorobenzyl)-1H-indazole-3-carboxamido]-3,3-dimethylbutanoate | 1,971,007-93-8 | C22H24FN3O3 |
| FUB-AMB (MMB-FUBINACA, AMB-FUBINACA) | 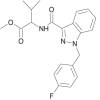 |
methyl 2-[1-(4-fluorobenzyl)-1H-indazole-3-carboxamido]-3-methylbutanoate | 1,971,007-92-7 | C21H22FN3O3 |
-
vi)
Eicosanoids
| Name | Structure | Chemical name | CAS No | Molecular Formula |
|---|---|---|---|---|
| AM-356 | 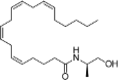 |
N- (2- hydroxy- 1R- methylethyl)- 5Z, 8Z, 11Z, 14Z- eicosatetraenamide | 157,182-49-5 | C23H39NO2 |
-
vii)
Selected other synthetic cannabinoids
| Name | Structure | Chemical name | CAS No | Molecular Formula |
|---|---|---|---|---|
| CRA-13 | 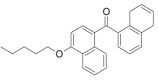 |
(naphthalen-1-yl) (4-pentyloxynaphthalen-1-yl)methanone | 432,047-72-8 | C26H24O2 |
| CUMYL-PeGaClone | 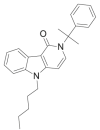 |
2,5-dihydro-2-(2-phenylpropan-2-yl)-5-pentyl-1H-pyrido[4,3-b]indol-1-one | 2,160,555-55-3 | C25H28N2O |
| 5-Fluoro-CUMYL-PeGaClone | 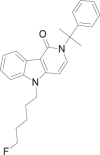 |
5-(5-fluoropentyl)-2-(2-phenylpropan-2-yl)-2,5-dihydro-1H-pyrido[4,3-b]indol-1-one | Not available | C25H27FN2O |
| FUBIMINA |  |
(1-(5-fluoropentyl)-1H-benzimidazol-2-yl) (naphthalen-1-yl)methanone | 1,984,789-90-3 | C23H21FN2O |
| JWH-030 | 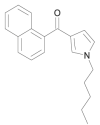 |
naphthalen-1-yl(1-pentyl-1H-pyrrol-3-yl)methanone | 162,934-73-8 | C20H21NO |
| JWH-031 | 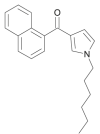 |
(1-hexyl-1H-pyrrol-3-yl) (naphthalen-1-yl)methanone | 162,934-74-9 | C21H23NO |
| JWH-145 |  |
naphthalen-1-yl(1-pentyl-5-phenyl-1H-pyrrol-3-yl)methanone | 914,458-19-8 | C26H25NO |
| JWH-147 | 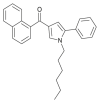 |
(1-hexyl-5-phenyl-1H-pyrrol-3-yl) (naphthalen-1-yl)methanone | 914,458-20-1 | C27H27NO |
| JWH-176 |  |
E−1-[1-(1-Naphthalenylmethylene)-1H-inden-3-yl]pentane | 619,294-62-1 | C25H24 |
| JWH-307 | 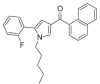 |
[5-(2-fluorophenyl)-1-pentyl-1H-pyrrol-3-yl] (naphthalen-1-yl) methanone | 914,458-26-7 | C26H24FNO |
| JWH-309 |  |
naphthalen-1-yl[5-(naphthalen-1-yl)-1-pentyl-1H-pyrrol-3-yl]methanone | 914,458-42-7 | C30H27NO |
| JWH-368 | 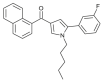 |
[5-(3-fluorophenyl)-1-pentyl-1H-pyrrol-3-yl](naphthalen-1-yl)methanone | 914,458-31-4 | C26H24FNO |
| JWH-369 | 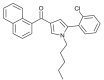 |
[5-(2-chlorophenyl)-1-pentyl-1H-pyrrol-3-yl](naphthalen-1-yl)methanone | 914,458-27-8 | C26H24ClNO |
| JWH-370 | 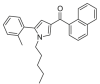 |
[5- (2- methylphenyl)- 1- pentyl- 1H- pyrrol- 3- yl] naphthalen-1-yl)methanone | 914,458-22-3 | C27H27NO |
| Org 27,569 |  |
5-chloro-3-ethyl-1H-indol-2-carboxylic acid [2-(4-piperidin-1-ylphenyl)ethyl]amide | 868,273-06-7 | C24H28ClN3O |
| Org 27,759 | 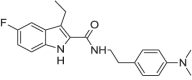 |
5-fluoro-3-ethyl-1H-indol-2-carboxylic acid [2-(4-dimethylaminophenyl)ethyl]amide | 868,273-09-0 | C21H24FN3O |
| Org 29,647 | 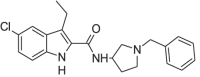 |
5-chloro-3-ethyl-1H-indol-2-carboxylic acid (1-benzylpyrrolidin-3-yl)amide | not available | C22H24ClN3O |
| WIN-55,212–2 | 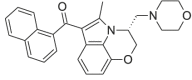 |
(naphthalen-1-yl)[(3R)- 2, 3- dihydro- 5- methyl- 3- (4- morpholinylmethyl)pyrrolo[1, 2, 3- de]- 1, 4- benzoxazin- 6- yl]methanone | 131,543-23-2 | C27H26N2O3 |
4. Production and diversion
4.1. Synthesis of pure compounds
In the years following the initial emergence of synthetic cannabinoids, amino-alkylindoles were among the most prevalent compounds found in herbal products. However, more recently as the structures of synthetic cannabinoids have continued to evolve and substances with indole/indazole carboxamide functional groups have become more common. This may be due to the fact that syntheses of both classes of synthetic cannabinoids are less elaborate and less complicated than syntheses of classical, non-classical or hybrid cannabinoids. In general, they can be synthesized without sophisticated laboratory equipment using inexpensive reagents and chemicals. The synthetic route of some of the synthetic cannabinoids from these classes are described in this manual, as adapted from procedures published in the literature.
4.1.1. Synthesis of aminoalkylindoles – the naphthoylindoles
The naphthoylindoles are synthesized by Friedel-Crafts acylation at C3 followed by N-alkylation of a (substituted) indole or vice versa (Scheme 1). The common precursors used are:
-
i.
1-Alkylindoles and 1-alkyl-2-methylindoles (alkyl: butyl, pentyl, hexyl or others, halogenated if applicable)
-
ii.
1-Naphthoyl chlorides (e.g., substituted at C4)
Scheme 1.

Syntheses of naphthoylindolesa.
Potassium tert
-butoxide, butyl iodide or pentyl iodide, tetrahydrofuran, room temperature, AlCl3, dichloromethane, 0 °C.
One example of a synthetic route for naphthoylindoles such as JWH-073, JWH-073 (4-methylnaphthyl), JWH-018 and JWH-122 [33] is shown in Scheme 1.
4.1.2. Synthesis of indole/indazole carboxamides
Indole/indazole carboxamide can be readily synthesized by reacting the respective indole/indazole carboxylic acids with the amino acid amides (e.g. tert-leucinamide or valinate) [34]. The acid amides may be purchased commercially or synthesized through a threestep synthesis (refer to Scheme 2).
Scheme 2.

Synthesis of L-tert-leucinamide.
Reagents and conditions: (a) NaOH, benzyl chloroformate, 0 °C to room temperature, 2h, 99%; (b) NH4Cl, Et3N, hydroxybenzotriazole, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, dimethylformamide, room temperature, 16h, 84%; (c) 10% Pd/C, H2, THF, 12h, 48%.
The syntheses of indazole carboxamides such as ADB-FUBINACA (R1 = 4- fluorobenzyl), ADB-PINACA (R1 = C5H11) and 5F-ADB-PINACA (R1 = C5H10F) are shown in Scheme 3.
Scheme 3.

Syntheses of indazole carboxamides.
Reagents and conditions: (a) conc. H2SO4, MeOH, reflux, 4h, 76%, (b) BrR1, potassium tert-butoxide, tetrahydrofuran, 0 °C to room temperature, 48 h, 67–77%, (c) NaOH, MeOH, room temperature, 24h, 76–96%; (d) 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride, hydroxybenzotriazole, diiso-propylethylamine, L-tert-leucinamide, dimethylformamide, room temperature, 24 h, 31–63%.
The syntheses of indole carboxamides such as ADB-FUBICA (R1 = 4-fluorobenzyl), ADBICA (R1 = C5H11) and 5F-ADBICA (R1 = C5H10F) are shown in Scheme 4.
Scheme 4.

Syntheses of indole carboxamides.
Reagents and conditions: (a) (i) NaH, BrR1, dimethylformamide, 0 °C to room temperature, 1 h, (ii) (CF3CO)2O, dimethylformamide, 0 °C to room temperature 1h; (b) KOH, MeOH, toluene, reflux, 2 h, 54–68% (over two steps); (c) 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride, hydroxybenzo-triazole, diisopropylethylamine, L-tert-leucinamide, room temperature, 24 h, 65–86%.
4.2. Production of herbal preparations
Although synthetic cannabinoids can generally be administered as pure substances, end products are usually designed for smoking. Most of these end products are made of herbal material laced with one or more synthetic cannabinoids and natural/arti-ficial flavourings.
The mixing of the plant material with synthetic cannabinoids could be performed by filling the plant material in a cement mixer and adding a solution of synthetic cannabinoids in an organic solvent (e.g., acetone) to soak the material. After drying, the cannabinoids are distributed more or less homogeneously on the plant material. In many cases, traces of other synthetic cannabinoids in addition to the main com-pounds could be detected in the end products. This could be a consequence of the mixing vessel not being cleaned thoroughly after each production cycle and hence leading to cross contamination. Sometimes crystalline powder is visible at the bot-tom of the packets or on the surface of the vegetable matter when viewed under the microscope. This is likely due to a simple mixing of the plant material with the drugs in powder form, and resulting in an inhomogeneous mixture of the active compounds and the plant material (see Fig. 2).
Fig. 2.

White crystalline substance on the surface of vegetable matter (viewed under microscope).
4.3. Precursors and sources
Synthetic cannabinoids can be obtained from commercial chemical companies as certified reference materials (CRMs), which are of high purity, and fulfil quality requirements in line with international standards. However, CRMs are typically expensive and usually supplied in small quantities, hence they are not generally used for preparation of herbal mixtures. Alternatively, synthetic cannabinoids have been available to purchase online from companies often located in Asia, although sources in Europe have also been reported. In some cases, the substances purchased may be of high purity [27], whereas in other cases, the quality of these compounds does not meet pharmaceutical standards and can often be contaminated with synthetic by-products and derivatives originating from inefficient synthetic processes [35]. To mislead customs authorities, these products are usually shipped using wrong declarations, for example, “polyphosphate”, “maleic acid”, “fluorescent whitening agent”, “ethyl vanillin”, “cotton”, “paper sample”, “TiO2” (titanium dioxide) or “fish tank cleaner”. Chemicals that are common precursors to the manufacture of carboxamide synthetic cannabinoids include indole, indazole and amino acids such as tert-leucine.
4.4. Typical seized materials
The most prevalent forms of seized products are ready-to-smoke mixtures of plant material laced with synthetic cannabinoids. They often contain more than one active compound, rising to about six in the same product. Seized materials containing the pure substances in powder form are also common and are often used for large scale production of herbal preparations or by the end users who would concoct their own blend of herbal mixtures. Products resembling hashish in their appearance are not so commonly encountered. Other forms of synthetic cannabinoids reported include in liquid for vaping and impregnated on paper often to facilitate trafficking.
4.5. Adulterants/masking agents
In the first herbal products containing synthetic cannabinoids that emerged, adulterants such as tocopherols or oleamide were frequently added [1]. It remains unclear if the purpose was to mask the active ingredients or if they were added as preservatives. Tocopherol acts as an antioxidant and was mainly found in products containing CP-47,497-C8. Oleamide on the other hand exhibits cannabis-like behavioural responses when ingested and may have been added to modify the psychotropic effects. These additives are no longer commonly found in herbal products. However, many products have been known to contain natural/artificial flavourings such as ethylvanillin, eugenol or other terpenoids [36]. It is unlikely that these compounds have any significant impact on the pharmacological activity of the products.
5. Qualitative and quantitative analysis of materials containing synthetic cannabinoids
In attempting to establish the identity of a controlled drug in suspect material, the general analytical approach must entail the determination of at least two uncorrelated parameters. One of these parameters should provide information on the chemical structure of the analyte, for example: infrared (IR) spectroscopy, mass spectrometry (MS), or nuclear magnetic resonance (NMR) spectroscopy.
It is recognized that the selection of these parameters in any particular case would take into account the drug involved and the laboratory resources available to the analyst. It is also accepted that unique requirements in different jurisdictions may dictate the actual practices followed by a particular laboratory.

5.1. General aspects
As synthetic cannabinoids are often found in herbal mixtures, the strategy for analysis can differ to some extent from the analysis of classical herbal drugs such as cannabis or drugs in other forms such as heroin, cocaine and amphetamine-type stimulants. Some important aspects of analysis that should be considered are summarized in Table 1.
Table 1.
Analytical aspects and considerations for analysis.
| Analytical aspects | Considerations |
|---|---|
| i. Sampling |
|
| ii. Homogeneity |
|
| iii. Extraction |
|
| iv. Sensitivity |
|
| iv. Other important aspects |
|
Qualitative analysis may be performed by thin layer chromatography (TLC), ion mobility mass spectrometry (IMS), infra-red spectroscopy (IR), gas chromatography-flame ionization detector (GC-FID), gas chromatography-infra-red detection (GC- IRD), gas chromatography-mass spectrometry (GC-MS), liquid chromatography (LC) or liquid-chromatography-mass spectrometry (LC-MS). GC-MS can be regarded as the gold standard, as it provides not only excellent chromatographic resolution but can also allow for the identification of active ingredients by their electron impact-mass spectra (EI-MS. However, GC-MS may have limitations in analysing closely related isomers. To distinguish these, additional measurements using other analytical techniques (e.g., IR or GC-IRD, NMR) are necessary for unambiguous identification of the correct isomer.
TLC is an inexpensive and rapid technique which allows processing of high numbers of samples and thus can serve to significantly reduce the number of required GC-MS analyses. Coupling TLC with ambient mass spectrometric techniques, such as desorption electrospray ionization mass spectrometry (DESI-MS) can enable the identification of a broad range of analytes. With regard to IMS, it can be regarded as a sensitive screening method similar to other presumptive tests, such as colour tests, however microcrystal tests are not suitable to analyse herbal products.
For solid material containing pure substances, IR or Raman spectroscopy techniques may be applied. Mobile systems are also useful in the field for rapid screening of seized materials suspected to contain pure synthetic cannabinoids in powder form. If there is only a single synthetic cannabinoid in a seized sample, identification of the compound by IR is also possible with extracts of herbal mixtures after evapora-tion of the solvent on the attenuated total reflectance (ATR) diamond cell.
The recommended minimum requirements for qualitative analysis have been formu-lated by the Scientific Working Group for the Analysis of Seized Drugs (SWG- DRUG) and are available online at the website: www.swgdrug.org/. Generally, at least two techniques should be used for the identification of a drug, with one tech-nique of sufficient selectivity to provide structural information (such as the use of IR, MS or NMR) and a second technique with an intermediate level of selectivity through physical or chemical characteristics (such as the use of GC, LC, or TLC).
For quantitative analyses, GC-FID, LC and LC-MS (or LC-MS/MS) methods can be used. Liquid chromatography methods may be superior to gas chromatography methods in cases of the presence of high amounts of fatty acid derivatives, which might cause interferences in gas chromatographic signals.
5.2. Sampling
The principal reason for a sampling procedure is to permit an accurate and meaningful chemical analysis. Because most methods (both qualitative and quantitative) used in forensic drug analysis laboratories require very small aliquots of material, it is vital that these small aliquots be representative of the bulk from which they have been drawn. Sampling should conform to the principles of analytical chemistry, as laid down, for example, in national pharmacopoeias or by regional or international organizations. For general aspects of representative drug sampling of multi-unit samples, refer to the Guidelines on Representative Drug Sampling (www.unodc.org/unodc/en/scientists/guidelines-on-representative-drug-sampling_new.html). For seized material with obvious external characteristics, a sampling method based on the Bayes’ model may be preferred over the hypergeometric approach.
The use of an approved sampling system also helps to preserve valuable resources and time by reducing the number of determinations needed. It is recognized that there may be situations where, for legal reasons, the normal rules of sampling and homogenization cannot be followed. With herbal mixtures, modified sampling strate-gies may be required, particularly in cases whereby a large variety of different brands are encountered in the same seizure. It should be noted that the content of a particular brand of product could change over time as well. If a large number of identical products or bulk material is seized, commonly used sampling strategies may be applied.
Sampling procedures for determination of purity have specific aspects that need to be considered and, the reader is directed to the Guidelines on Sampling of Illicit Drugs for Quantitative Analysis published by the European Network of Forensic Science Institutes (ENFSI) (http://enfsi.eu/wp-content/uploads/2016/09/guidelines_quant_sampling_dwg_printing_vf4.pdf).
5.3. Extraction and sample preparation
5.3.1. Qualitative analysis
Add 1 ml of medium-polar or non-polar solvents such as methanol, ethanol, acetonitrile, ethyl acetate, acetone or isooctane to a small portion of sample (e.g., 100 mg of plant material or 1–2 mg of solid material). Sonicate the extract and filter or centrifuge, if necessary, before analysis.
5.3.2. Quantitative analysis
Pulverize and homogenize the plant/solid materials before taking samples for analy-sis. Homogenization can also be performed in an electric grinder or frozen with liquid nitrogen in a mortar. Homogenization of only an aliquot of the sample should be avoided, as the cannabinoids tend to settle down at the bottom of a sample. At least two individual samples should be generated from the homogenate; however, one sub-sample may only be possible in cases where the mass or homogeneity of the original material does not permit more than one sample to be taken.
Extract the samples using medium-polar or non-polar solvents such as methanol, ethanol, acetonitrile, ethyl acetate, acetone or isooctane. Sonicate the mixture for more effective extraction and filter before analysis. For better recovery efficiency, the number of extractions performed could be increased. Soxhlet extraction may also be used although this may be too elaborate for routine use in forensic labora-tories. Note: the use of alcohol solvents such as methanol or ethanol for extraction of cannabimimetic quinolinyl carboxylates, such as PB-22, 5F-PB-22 and FUB- PB-22, may cause transesterification to occur. 8-Quinolinol has been observed as a degradation product during GC-MS analysis [37].
5.4. Analysis of synthetic cannabinoids
5.4.1. Presumptive tests
Presumptive tests such as colour tests and microcrystal tests would not be appropri-ate due to low concentrations of the analytes in the herbal mixtures, possible inter-ferences by the sample matrix and the limited selectivity of presumptive tests with regard to synthetic cannabinoids. Although there are some commercially available presumptive tests for a few specific synthetic cannabinoids, there are currently no presumptive tests which cover the whole range of synthetic cannabinoids.
5.4.2. Thin layer chromatography
Thin layer chromatography (TLC) is a commonly used technique for the separation and detection of drugs. It is inexpensive, rapid and flexible in the selection of both the stationary and mobile phase and amenable to a wide variety of substances, in base and salt form, ranging from most polar to non-polar materials. Unlike HPLC columns, which are susceptible to contamination (of the stationary phase) by matrix compounds (such as fatty acid derivatives), TLC plates are single-use products and hence immune to these problems.
Certain classical and non-classical cannabinoids (e.g., HU-210) can be selectively and sensitively detected with UV light, Fast Blue RR reagent, iodine as well as iodoplatinate, whereas aminoalkylindoles (e.g. JWH-018, JWH-081 and JWH-210) and indole/indazole carboxamides (e.g., FUB-AMB and 5F-MDMB-PINACA) can be detected with UV light, iodine or iodoplatinate.
5.4.2.1. TLC plates (stationary phases)
Coating: Silica gel G with layer thickness of 0.25 mm and containing an inert indicator, which fluoresces under UV light wavelength 254 nm (Silica gel GF254).
Typical plate sizes: 20 × 20 cm; 20 × 10 cm; 10 × 5 cm (the latter should be used with the 10 cm side vertical with the TLC tank).
Plates that are prepared by the analyst must be activated before use by placing them into an oven at 120 °C for at least 10–30 min. Plates are then stored in a grease-free desiccator over orange silica gel∗. Heat activation is not required for commer-cially available coated plates.
5.4.2.2. Methods
5.4.2.2.1. Developing solvent systems (Table 2)
Table 2.
TLC developing systems.
Prepare developing solvent system (System A, B or C as shown in Table 2) as accurately as possible by using pipettes, dispensers and measuring cylinders. Leave the solvent system in the TLC tank for a sufficient time to allow vapour phase saturation to be achieved prior to analysis (with adsorbent paper-lined tanks, this takes approximately 5 min).
5.4.2.2.2. Preparation of sample solutions
As the purpose of the TLC assay of herbal products is qualitative analysis, homog-enization of the herbal material is not necessary. To a suitable amount of herbal mixture, for example, 100 mg, extract with approximately 10-fold amount of solvent under ultrasonication for at least 10 min and subsequently centrifuge the mixture. Suitable solvents are acetonitrile (well defined sample spots observed) or methanol (better solvent for synthetic cannabinoids but less well defined sample spots observed).
∗Blue silica gel can also be used. However, due care should be taken as blue silica gel contains cobalt(II) chloride which is possibly carcinogenic to humans.
5.4.2.2.3. Preparation of standard solutions
Standard solutions are prepared at a concentration of 0.5 mg per mL in a suitable solvent.
5.4.2.2.4. Spotting and developing
Apply, as separate spots, 1 μL and 5 μL aliquots of sample solution, 2 μL of the standard solutions and 2 μL of solvent (as a negative control) on the TLC plate. Spotting must be done carefully to avoid damaging the surface of the plate.

5.4.2.2.5. Visualization/detection
The plates must be dried prior to visualization. This can be done at room temperature or by use of a drying box, oven or hot air. In the latter cases, care must be taken that no component of interest is subject to thermal decomposition. Use of a fume hood is recommended for ii–iv below.
5.4.2.2.6. Visualization/detection methods
i. UV light at 254 nm
Dark spots against a green background are observed. The spots are marked and if necessary, a digital photograph recorded.
ii. Freshly prepared Fast Blue RR reagent
Dissolve 0.10 g of Fast Blue RR in 10 ml of distilled water and add 4 mL of 20% (w/v) sodium hydroxide solution. The classical or non-classical cannabinoids appear as orange-reddish spots when the plate is sprayed with the reagent. If necessary, the plate is photographed after drying for documentation.
iii Iodine
Place the dried plate in a TLC chamber containing solid iodine crystals. The synthetic cannabinoids appear as yellow to brown spots. If necessary, the plate is photographed for documentation.
iv. Iodoplatinate
Dissolve 5 g of chloroplatinic acid hexahydrate and 35 g of potassium iodide in 1650 ml of distilled water. Then, add 49.5 mL of concentrated hydrochloric acid. The synthetic cannabiniods appear as green/yellow, white/pink or purple spots. If necessary, the plate is photographed after drying for documentation.
5.4.2.2.7. Interpretation
After visualization, mark spots (e.g., by pencil) and calculate retardation factor (Rf) values.
5.4.2.2.8. Results
While TLC is a useful and practical separation technique, it will not be suitable to differentiate certain closely related compounds such as 5F-MDMB-PINACA and 5F-EDMB-PINACA due to their similar chromatographic properties. It is essential that other more selective methods (e.g., GC-MS, GC-IRD) be used to confirm the presence of these substances. see (Table 3)

Table 3.
Rf values for selected synthetic cannabinoids using the above methods.
|
Rfvalues |
System C | |||
|---|---|---|---|---|
| Compound | System A | System B | ||
| JWH-200 | 0.02 | 0.60 | 0.85 | |
| HU-210 | 0.05 | 0.34 | 0.78 | |
| RCS-4 ortho isomer | 0.16 | – | – | |
| RCS-4 | 0.18 | 0.67 | 0.87 | |
| AM-2201 | 0.18 | 0.75 | 0.82 | |
| AM-694 | 0.18 | – | – | |
| JWH-015 | 0.22 | 0.73 | 0.91 | |
| JWH-018 | 0.25 | 0.76 | 0.91 | |
| JWH-250 | 0.26 | 0.74 | 0.91 | |
| JWH-072 | 0.31 | – | – | |
| JWH-007 | 0.31 | – | – | |
| JWH-307 | 0.35 | – | – | |
| JWH-073 | 0.36 | 0.75 | 0.91 | |
| JWH-251 | 0.36 | 0.71 | 0.88 | |
| JWH-203 | 0.40 | – | – | |
| JWH-081 | 0.41 | 0.71 | 0.88 | |
| JWH-122 | 0.41 | – | – | |
| JWH-019 | 0.42 | 0.76 | 0.91 | |
| JWH-020 | 0.44 | – | – | |
| JWH-412 | 0.44 | – | – | |
| JWH-210 | 0.45 | 0.75 | 0.85 | |
| JWH-398 | – | 0.71 | 0.88 | |
| CP-47,497 | – | 0.31 | 0.77 | |
| CP-47,497-C8 | – | 0.31 | 0.77 | |
| CP-55,940 | – | 0.14 | 0.52 | |
| RCS-8 | – | 0.70 | 0.88 | |
| WIN-55,212-2 | – | 0.58 | 0.86 | |
5.4.3. Infrared spectroscopy
In general, infrared spectroscopy (IR) is a straightforward and useful tool for iden-tification of closely related synthetic cannabinoids due to unique IR fingerprint bands for each compound. It can also be a useful tool for identification of new substances [39] with the use of reference databases or literature.
Qualitative analysis of synthetic cannabinoids in herbal mixtures by infrared spec-troscopy can be more challenging due to the complex matrix and the comparatively low concentration of the synthetic cannabinoids present in the herbal products. Sometimes, with an extraction step, it is possible to obtain a good IR spectrum after evaporating the extract directly on the attenuated total reflection (ATR) diamond cell, depending on the complexity of the sample matrix. Nevertheless, the correlation factors that are calculated by the software of the IR spectrometer for synthetic cannabinoids in extracts of herbal mixtures will be slightly lower than for pure substances due to interferences from the sample matrix. Hence, it is important to perform a visual comparison of the reference spectrum of the pure cannabinoid vs. the spectrum of the analysed sample extract.
In such situations, the use of gas chromatography with infrared detection (GC-IRD) can be a more suitable technique for the identification of certain synthetic cannabi-noids in herbal mixtures. The GC separates the different drug components in the sample matrix and the IRD can identify them based on their individual IR spectra. Two examples are shown in this manual to illustrate its use in differentiating both positional and structural isomers [40].
Solid deposition GC-IRD operating conditions GC conditions: GC oven conditions: 80°C for 5 min, increased to 300°C at a rate of 20°C/min, and then held isothermal at 300°C for 20 min. Total run time: 36 min Column: 5% phenyl/95% methyl silicone column (HP-5MS), 30 m length x 0.25 mm i.d., 0.25 μm film thickness Injection parameters: 2 μl aliquot of sample injected with a split ratio of 5:1 Injector temp: 280°C Carrier gas: IRD conditions: Helium, 1.2 mL /min constant flow Transfer line temperature: 300°C Oven temperature: 300°C Restrictor temperature: 300°C Disk temperature: -40°C Dewar cap temperature: 30°C Disk speed: 12 mm per minute Pressure: Data processing: Approx. 3 x 10-4 Torr or less Software: Thermo galactic GRAMS/AI spectroscopy and chromatography software IR algorithm matching: First derivative correlation.
Example 1
Differentiation of fluoropentyl positional isomers of AM-2201 using GC-IRD (see Fig. 3, Fig. 4 and Table 4)
Fig. 3.

Full scan IR spectra of AM-2201 and its fluoro positional isomers.
Fig. 4.

IR spectra of AM-2201 and its fluoro positional isomers at the fingerprint region of 2000–800 cm-1.
Table 4.
Quality match factor data.
| Drug standard | Quality match factor (QMF) to the respective drug |
|||
|---|---|---|---|---|
| AM-2201 | AM-2201 2-isomer |
AM-2201 3-isomer |
AM-2201 4-isomer |
|
| AM-2201 | 0.0407 | 0.3334 | 0.2601 | 0.1729 |
| AM-2201 2-isomer | 0.3181 | 0.0386 | 0.2990 | 0.2933 |
| AM-2201 3-isomer | 0.2595 | 0.3121 | 0.0478 | 0.1456 |
| AM-2201 4-isomer | 0.1542 | 0.2953 | 0.1323 | 0.0392 |
∗With the laboratory validated method, a quality match factor of 0 indicates a perfect match while a number less than 0.1 is considered a positive identification.
From the fingerprint region 1000–1300 cm-1 (boxed in red), the four isomers showed different IR bands of different intensities, allowing differentiation among them. Particularly, for AM-2201 3-isomer and AM-2201 4-isomer, although their IR looks similar visually, they can be differentiated based on the instrument-generated IR quality match factor. For instance, the infra-red quality match factor (IRQMF) of 0.1323 and 0.1456 obtained from 3-isomer and 4-isomer when matched against each other, did not pass the laboratory validated threshold criteria of 0.1 for a positive identification.
Example 2
Differentiation of JWH-018 and its structural isomers at the pentyl chain with GC-IRD (see Fig. 5 and Table 5)
Fig. 5.

Full scan IR spectra of JWH-018 and its structural isomers at the pentyl chain.
Table 5.
Quality match factor to the respective cannabinoid.
| Drug Standard | Quality match factor to the respective drug |
|||||||
|---|---|---|---|---|---|---|---|---|
| JWH-018 | Isomer 1 | Isomer 2 | Isomer 3 | Isomer 4 | Isomer 5 | Isomer 6 | Isomer 7 | |
JWH-018
|
0.0012 | 0.5703 | 0.5721 | 0.5373 | 0.5745 | 0.5305 | 0.2209 | 0.1639 |
| JWH-018 N-(1,1-dimethylpropyl) 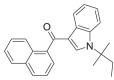 ISOMER 1 |
0.5707 | 0.0013 | 0.5913 | 0.7732 | 0.5312 | 0.5738 | 0.6059 | 0.5248 |
JWH-018 N-(1,2-dimethylpropyl) ISOMER 2 |
0.5724 | 0.5916 | 0.0014 | 0.7432 | 0.2087 | 0.2423 | 0.5243 | 0.5141 |
JWH-018 N-(2,2-dimethylpropyl)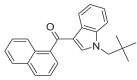 ISOMER 3 |
0.5347 | 0.7728 | 0.7438 | 0.0016 | 0.7906 | 0.7745 | 0.5377 | 0.5402 |
JWH-018 N-(1-ethylpropyl)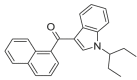 Isomer 4 |
0.5732 | 0.5312 | 0.2103 | 0.7890 | 0.0013 | 0.2161 | 0.5683 | 0.5359 |
JWH-018 N-(1-methylbutyl) Isomer 5 |
0.5300 | 0.5730 | 0.2418 | 0.7744 | 0.2151 | 0.0014 | 0.5004 | 0.4720 |
JWH-018 N-(2-methylbutyl) Isomer 6 |
0.2199 | 0.6039 | 0.5227 | 0.5374 | 0.5684 | 0.4989 | 0.0013 | 0.2087 |
JWH-018 N-(3-methylbutyl)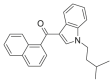 Isomer 7 |
0.1618 | 0.5236 | 0.5139 | 0.5414 | 0.5376 | 0.4715 | 0.2087 | 0.0015 |
∗With the laboratory validated method, a quality match factor of 0 indicates a perfect match while a number less than 0.1 is considered a positive identification.
Similarly, the GC-IR analyses of JWH-018 and its structural isomers demonstrated that they can be differentiated by their IR spectra. JWH-018 and all 7 isomers are correctly identified with IRQMF ranging from 0.0012 to 0.0016. In contrast, JWH- 018 and all isomers gave IRQMF above 0.1 (ranging from 0.1618 to 0.7906) when they were matched to the wrong isomers. This is a factor of about 130–500 times higher than the correct match, demonstrating the high selectivity of IR in this type of analysis.

5.4.4. Gas chromatography-mass spectrometry
Gas chromatography-mass spectrometry (GC-MS) is one of the most commonly used techniques for the identification of drug samples in forensics. As a hyphenated technique, it combines the separation power and sensitivity of a GC with the analyte specificity of a spectrometric technique. It can provide highly specific spectral data on individual compounds in a complex mixture without prior isolation.
5.4.4.1. Sample preparation and extraction procedure
Add 1 mL of a medium-polar or non-polar solvent such as methanol, ethanol, ace-tonitrile, ethyl acetate, acetone or isooctane to a small portion of sample (e.g., 100 mg of plant material or 1–2 mg of solid material). Sonicate the extract and filter before analysis.
5.4.4.2. Preparation of internal standard solution (for retention time locking if required)
Dissolve N,N-dibenzyl-2-chlorobenzamide in methanol to give a concentration of 20 μg/ml. Add an aliquot of the internal standard to the sample/standard solution if retention time locking of the analysis is required.
5.4.4.3. Preparation of standard solutions
Prepare a standard solution of the synthetic cannabinoid of interest at a concentra-tion of 1 mg/mL with an appropriate solvent (e.g., methanol, ethanol, acetonitrile, ethyl acetate, acetone or isooctane).
5.4.4.4. Results
GC retention times (RT) for selected synthetic cannabinoids using the above operat-ing conditions are as follows (Table 6):
Table 6.
GC retention times.
| Compound | GC RT (min) |
Characteristic GC-MS ions (m/z) | |
|---|---|---|---|
| Method 1 | Method 2 | ||
| ADB-FUBINACA | 10.7 | 109, 145, 253, 338, 382 M+ | |
| AM-1220 | 16.3 | 16.7 | 98, 127, 155, 382 M+ |
| AM-1248 | 15.6 | 15.5 | 70, 98, 99, 390 M+ |
| AM-2201 | 13.7 | 13.0 | 127, 144, 232, 284, 342, 359 M+ |
| AM-2232 | 16.2 | 16.7 | 127, 225, 284, 352 M+ |
| AM-694 | 11.8 | 220, 232, 360, 435 M+ | |
| APINACA | 11.9 | 11.7 | 145, 215, 294, 365 M+ |
| BB-22 | 17.4 | 116, 144, 240, 384 M+ | |
| CBM-2201 | 17.5 | 144, 232, 374 M+ | |
| CP-47,497 | 6.8 | 8.9 | 215, 233, 300, 318 M+ |
| CP-47,497-C8 (1S/3R or 1R/3S) | 7.7 | 215, 233, 314, 332 M+ | |
| CP-47,497-C8 (1S/3S or 1R/3R) | 7.4 | 215, 233, 314, 332 M+ | |
| CUMYL-4CN-BINACA | 11.5 | 145, 226, 345, 360 M+ | |
| FDU-PB-22 | 17.7 | 109, 143, 252, 395 M+ | |
| 5F-ADB-PINACA | 11.4 | 131, 145, 233, 289, 318, 362 M+ | |
| 5F-APINACA | 12.5 | 145, 233, 294, 326, 338, 355, 383 M+ | |
| 5F-CUMYL-PeGaClone | 14.2 | 91, 197, 272, 390 M+ | |
| 5F-CUMYL-PICA | 11.3 | 144, 173, 232, 248, 366 M+ | |
| 5F-EDMB-PINACA | 9.5 | 131, 145, 233, 289, 318, 335, 391 M+ | |
| 5F-MDMB-PICA | 10.1 | 144, 232, 260, 288, 320, 376 M+ | |
| 5F-MDMB-PINACA | 9.4 | 131, 145, 233, 289, 321, 377 M+ | |
| 5F-PB-22 | 14.3 | 116, 144, 232, 376 M+ | |
| FUB-144 | 9.6 | 109, 252, 334, 349 M+ | |
| FUB-APINACA | 14.5 | 109, 150, 253, 294, 375, 403 M+ | |
| FUB-PB-22 | 18.7 | 109, 252, 396 M+ | |
| Internal standard | 8.1 | 139, 141, 244, 335 M+ | |
| JWH-007 | 13.0 | 12.3 | 298, 340, 354, 355 M+ |
| JWH-015 | 11.4 | 11.2 | 270, 310, 326, 327 M+ |
| JWH-018 | 12.6 | 11.6 | 214, 284, 324, 341 M+ |
| JWH-019 | 13.5 | 12.9 | 228, 284, 338, 355 M+ |
| JWH-073 | 11.8 | 11.1 | 200, 284, 310, 327 M+ |
| JWH-081 | 15.3 | 214, 314, 354, 371 M+ | |
| JWH-122 | 13.9 | 13.2 | 214, 298, 338, 355 M+ |
| JWH-200 | 16.8 | 17.2 | 100, 127, 155, 384 M+ |
| JWH-203 | 10.0 | 144, 116, 214, 339 M+ | |
| JWH-210 | 14.5 | 14.0 | 214, 312, 352, 369 M+ |
| JWH-250 | 10.2 | 10.5 | 144, 116, 214, 335 M+ |
| JWH-251 | 9.2 | 10.1 | 144, 116, 214, 319 M+ |
| JWH-307 | 13.2 | 155, 188, 314, 385 M+ | |
| JWH-412 | 12.2 | 145, 173, 302, 359 M+ | |
| MAM-2201 | 14.8 | 14.6 | 232, 298, 356, 373 M+ |
| MDMB-CHMICA | 11.0 | 144, 240, 268, 296, 328, 384 M+ | |
| MDMB-CHMINACA | 10.0 | 131, 145, 241, 297, 329, 385 M+ | |
| MDMB-FUBINACA | 10.0 | 109, 145, 253, 341, 397 M+ | |
| MMB-CHMICA | 10.8 | 144, 240, 256, 370 M+ | |
| MMB-FUBICA | 10.8 | 109, 252, 268, 382 M+ | |
| FUB-AMB | 9.9 | 109, 253, 269, 324, 383 M+ | |
| NM-2201 | 13.8 | 115, 144, 232, 375 M+ | |
| Org 27,569 | 19.3 | 174, 187, 253, 409 M+ | |
| Org 27,759 | 12.5 | 118, 134, 147, 353 M+ | |
| Org 29,647 | 15.1 | 91, 143, 159, 381 M+ | |
| PB-22 | 13.8 | 116, 144, 214, 358 M+ | |
| RCS-4 | 10.7 | 10.7 | 135, 214, 264, 321 M+ |
| RCS-4 ortho isomer | 8.8 | 144, 264, 304, 321 M+ | |
| UR-144 | 6.1 | 8.8 | 144, 214, 296, 311 M+ |
| XLR-11 | 6.7 | 9.1 | 144, 232, 314, 329 M+ |
Note: M+ refers to molecular ion.
Identification is accomplished by comparing the retention time and mass spectrum of the analyte with that of a reference standard. All compounds identified by GC-MS ideally should be compared to a current mass spectrum of the appropriate reference standard, preferably obtained from the same instrument, operated under the same conditions. When a reference standard is not analysed concurrently with the sample, one can make use of various chromatographic techniques such as retention time locking [41], relative retention time [42] or retention index [43] to minimize any variation in the retention time of the analyte.
For the correct identification of regioisomers, additional techniques such as IR might be necessary. One case study is shown below where IR is required to differentiate JWH-018 from its N-(3-methylbutyl) isomer as their mass spectra are very similar (see Fig. 6, Fig. 7 and Table 7).
Fig. 6.

Mass spectra of JWH-018 (top) and JWH-018 N-(3-methylbutyl) isomer (bottom).
Fig. 7.

IR spectra of JWH-018 (top) and JWH-018 N-(3-methylbutyl) isomer (bottom).
Table 7.
Instrument-generated mass spectrum and infrared spectrum quality match factor (MSQMF and IRQMF).
| Drug standard | MSQMF of the respective isomer |
IRQMF of the respective isomer |
||
|---|---|---|---|---|
| JWH-018 | JWH 018 N-(3-methylbutyl) |
JWH-018 | JWH 018 N-(3-methylbutyl) |
|
| JWH-018 | 99 | 91 | 0.0012 | 0.1639 |
|
JWH 018 N-(3-methylbutyl) |
99 | 99 | 0.1618 | 0.0015 |
| Note: The MS quality match factors are generated using probability-based matching (PBM) algorithm using the Agilent Technologies ChemStation software, version E.02.02.143. With this algorithm, the closer the number is to 100, the closer is the match with the reference spectrum in the library. | Note: The IR quality match factors are generated using first derivative algorithm using thermo galactic GRAMS/AI spectroscopy and chromatography software, version 9.1. With this algorithm, the lower the quality match factor, the closer the match. With the laboratory validated method, IRQMF of <0.1 indicates a positive identification. | |||
5.4.4.5. Case study: MS and IR analyses of JWH-018 and its N-(3-methylbutyl) isomer [40]
In this example, the mass spectra of both JWH-018 and JWH-018 N-(3-methylbutyl) are very similar visually and the instrument software also computes a high correlation (quality match) between the two spectra. Hence both compounds cannot be differentiated based on the MS data. On the other hand, the IR data as shown in Fig. 3 show obvious differences in the IR bands in the region of 2800–3100 cm-1 and the IR quality match factor when matched against the wrong isomer is greater than 0.1, which is the threshold for positive identification. This shows that the two synthetic cannabinoids that have very similar chemical structure can be differentiated based on the IR data and their quality match factors.
5.4.4.6. Co-eluting synthetic cannabinoids
Some synthetic cannabinoids that have emerged in recent years have been observed to co-elute (e.g. MMB-CHMICA with MDMB-CHMICA) during GC-MS analysis using the HP-5 column. The co-elution may be resolved by using GC column pack-ing of different polarities, such as the more polar DB-35 column.
5.4.4.6.1. An example of GC-MS run using both HP-5MS and DB-35 columns
Retention time of selected synthetic cannabinoids using methods 1 and 2 (Table 8):
Table 8.
Comparison of methods 1 and 2.
| Compound | Retention time (min) |
|
|---|---|---|
| Method 1 | Method 2 | |
| MMB-CHMICA | 19.41 | 20.69 |
| MDMB-CHMICA | 21.01 | |
| ADB-FUBINACA | 22.06 | |
5.4.5. Gas chromatography with flame ionization detection
Gas chromatography with flame ionization detection (GC-FID) could be employed for both qualitative and quantitative determinations of synthetic cannabinoids, GC- FID alone will not be sufficient for confirmation of drug identity based on its reten-tion time as many synthetic cannabinoids have similar retention times. The method for the quantitative GC-FID analysis of selected synthetic cannabinoids is described here to be used as a guide for adaptation and modification, which would be required for other synthetic cannabinoids of interest [33]. It should be noted that for samples with very low concentrations, it would be more advisable to employ a more sensi-tive technique, for example, LC-MS or LC-MS/MS for quantitative determinations.
5.4.5.1. Preparation of internal standard (IS) solution (for quantitative analysis)
Dissolve methyl oleate in methanol to give a concentration of 0.8 mg/mL.
5.4.5.2. Preparation of synthetic cannabinoid standard solutions (for quantitative analysis)
Prepare standard solutions of targeted synthetic cannabinoids in an appropriate work-ing concentration range. This method could be validated for the concentration range of 0.02–2.00 mg/mL in methanol. Usually at least five standard solutions should be prepared for a good linear calibration curve. Then, add 500 μL of the internal standard solution to 500 μL of each standard solution and vortex the mixture. Inject 1 μL of the mixture into the gas chromatograph.
5.4.5.3. Preparation of sample solutions (unknown “herbal mixture“)
For qualitative analysis, the herbal material can be soaked in a suitable solvent and an aliquot removed for analysis. For quantitative analysis, obtain a representative sample from the seized material. Homogenize and accurately weigh 50 mg of seized material into a centrifugation tube and add 5 mL of methanol. Sonicate and centri-fuge the mixture for 5 min at 2500 rpm. Then, add 500 μL of the internal standard solution to 500 μl of the supernatant solution and vortex the mixture. Inject 1 μL of the mixture into the gas chromatograph. At least a duplicate analysis should be carried out.
| GC operating conditions | |
|---|---|
| Detector | FID |
| Column | Factor Four VF-5MS containing 5% phenyl methyl polysiloxane or equivalent, 30 m × 0.25 mm i.d., 0.25 μm film thickness |
| Carrier gas | Helium 1.2 mL/min |
| Detector gas | Hydrogen 35 mL/min, air 350 mL/min |
| Inlet temp | 250 °C |
| Detector temp | 280 °C |
| Oven temp | Column temp. Initially set at 70 °C and ramped to 180 °C at a rate of 40 °C/min and then ramped to 300 °C at a rate of 10 °C/min |
| Injection volume | 1 μL |
| Split ratio | 30:1 |
Note: The above conditions may be altered as long as appropriate validation is carried out.
5.4.5.4. Results
Elution order and the corresponding retention time are shown in Table 9:
Table 9.
Elution order.
| Compound | Retention time (min) |
|---|---|
| Internal standard | 9.3 |
| JWH-073 | 18.3 |
| JWH-018 | 19.4 |
| JWH-073 (4-methylnaphthyl) | 20.1 |
| JWH-122 | 22.8 |
5.4.5.5. Calculations for quantitative analysis
The general calculation of the amount of synthetic cannabinoid in a sample is shown below. This calculation can apply to both GC and LC analyses. The percentage of targeted synthetic cannabinoid in the sample is calculated by first plotting a linear calibration curve of the response ratio observed from the calibration standards (i.e., peak area of cannabinoid standard/peak area of IS) against concentration of can-nabinoid standard used (mg/mL). From the response of the unknown sample solution and the corresponding value from the calibration curve, the percentage of synthetic cannabinoid in the sample could be obtained using the formula below:
Where;
V: Volume of extraction solvent used (mL)
RS: Response ratio observed for the sample (i.e., peak area of cannabinoid/peak area of IS)
a: Gradient/slope of the calibration curve
b: Intercept of the calibration curve
WS: Weight of the sample (mg).
Generally, with modern instrumentation and software, manual calculation of purity would not be required. Usually after input by the operator of the concentrations of the different calibration standards and the unknown sample solution, the calibration curve will be established and calculations will be performed automatically for any single point along the curve upon completion of the analytical run. Typically, the result will then be expressed as the percentage content of the unknown drug in the original sample material, that is, as the sample purity (weight of the analyte relative to the sample weight).
5.4.6. Liquid chromatography
Liquid chromatography (LC) is a technique used in many laboratories to separate, components in a mixture. It is typically coupled to an ultraviolet (UV) detector and the analytes may be identified by their UV/VIS spectra by comparison to the spectra of suitable reference standards although it should be noted that the UV spectra of many compounds are too similar to allow for identification. Separation occurs based on the interaction of the sample with the mobile phase and the stationary phase of the column. Due to differing affinities of the respective analytes with the mobile and stationary phases, separation is achieved and the analytes elute with different retention times.
Ultra-high performance liquid chromatography (UHPLC) systems are instruments with enhanced chromatographic capabilities compared to traditional high-performance liquid chromatography (HPLC). UHPLC instruments are designed to withstand higher operating back pressures, which allows the use of columns with sub-2 μm particles giving rise to higher separation efficiencies. The separation speed of the UHPLC is also significantly greater which allows for faster sample throughput. Furthermore, it is more environmentally friendly with lower solvent consumption and reduced waste disposal.
Since there are a large variety of stationary and mobile phases available to the analyst, one method each for quantitative and qualitative LC analysis is described below and can be modified for improved performance. This method has been field-tested within forensic casework and is considered fit-for-purpose. With adequate verification and validation, the same method can also be extended to other synthetic cannabinoids.
5.4.6.1. Standard/sample preparation
5.4.6.1.1. Preparation of internal standard (IS) solution
Weigh 20 mg of 1-pyrenebutyric acid into a 10 mL volumetric flask and dilute to volume with methanol to give a concentration of 2.0 mg/mL.
5.4.6.1.2. Preparation of synthetic cannabinoid standard solutions (for quantitative analysis)
Accurately weigh 5 mg of analyte into a 5 mL volumetric flask and dilute to volume with methanol to give a stock solution with a concentration of 1.0 mg/mL. For some analytes (e.g., JWH-018, JWH-019 and JWH-073) solutions with 1.0 mg/mL con-centrations are commercially available. The stock solution can be stored for at least one year with appropriate refrigeration. Prepare accurately an appropriate working concentration range. Usually at least five standard solutions should be prepared for a good linear calibration curve. An example of the preparation of a 6-point calibra-tion curve is given below (Table 10):
Table 10.
Calibration data.
| Calibration |
Volume of standard stock solution |
Volume of IS solution |
Total volume |
Final concentration |
Final |
|---|---|---|---|---|---|
| after dilution with methanol |
concentration of cannabinoids |
||||
| level | added (μL) | added (μL) | (mL) | of IS (μg/mL) | (μg/mL) |
| Level 1 | 10 | 40 | 10 | 8 | 1 |
| Level 2 | 10 | 8 | 2 | 8 | 5 |
| Level 3 | 25 | 4 | 1 | 8 | 25 |
| Level 4 | 50 | 4 | 1 | 8 | 50 |
| Level 5 | 37.5 | 2 | 0.5 | 8 | 75 |
| Level 6 | 50 | 2 | 0.5 | 8 | 100 |
5.4.6.1.3. Preparation of sample solutions (unknown “herbal mixture“)
For qualitative analysis, the herbal material can be soaked in a suitable solvent and an aliquot removed for analysis. For quantitative analysis, obtain a representative sample from the seized material and carefully homogenize. Accurately weigh 200 mg of sample into a flask and add quantitatively 2 mL of methanol. Extract under sonication for 15 min, invert flask at least 10 times, and centrifuge for 2 min at 5000 rpm, or allow to settle. Then, transfer the liquid to another flask and repeat extraction step twice with portions of 2 mL of methanol. Take an aliquot of approximately 2 mL of the combined extracts and filter using a syringe filter (≤0.45 μm). Then, accurately pipette 50 μL of the filtrate and 8 μl of IS solution into a 2 ml volumetric flask and dilute to volume with mobile phase A. Inject 5 μL of the sample solution into the UHPLC. At least a duplicate analysis should be carried out.
5.4.6.1.4. Qualitative analysis
Identification is accomplished by comparing the retention time of the analyte with the retention time of a reference standard. The internal standard, if used, allows the use of retention index as an additional identification criterion. Furthermore, the UV spectrum of the analyte should be compared with that of a reference material (Table 11, Table 12).
| HPLC operating conditions (Method A) | |
|---|---|
| Column | Hypersil-BDS column, 150 × 2.1 mm i.d., 5 μm particle size Isothermal at 25 °C |
| Mobile phase | Solvent A: 10 mM triethylammonium phosphate (TEAP) buffer solution (pH = 3.0) |
| Solvent B: Acetonitrile | |
| Flow rate | 0.4 mL/min |
| Detection | UV-photodiode array detector (DAD), detection wavelength at 210 nm |
| Total run time | 29 min |
| Elution programme | 1–50% B in first 5 min, ramp up to 90% B for the next 21 min, then ramp down to 1% B |
| UHPLC operating conditions (Method B) | |
| Column | Acquity UHPLC BEH Phenyl, 100 mm × 2.1 mm i.d., 1.7 μm particle size |
| Mobile phase | A: 95% acetonitrile, 4.9% water, 0.1% formic acid |
| B: 95% water, 4.9% acetonitrile, 0.1% formic acid | |
| Gradient | 0.0–12.5 min 41% A |
| 12.5–20.0 min 50% A | |
| 20.0–23.0 min 60% A | |
| 23.0–27.5 min 41% A | |
| Flow rate | 0.4 mL/min |
| Pressure | 512 bar |
| Temp | 30 °C |
| Detection | UV-DAD detection wavelengths (see below) |
| Injection volume: | 5 μL |
Note: The above conditions may be altered as long as appropriate validation is carried out.
Table 11.
HPLC retention times for Method A and UV spectra λmax for selected synthetic cannabinoids.
| Drug | Retention time (min) | λmax (nm) |
|---|---|---|
| FUB-144 | 17.3 | 214, 302, 244 |
| FUB-AMB | 13.2 | 208, 302 |
| 5F-MDMB-PINACA | 13.5 | 208, 302 |
| 5F-MDMB-PICA | 12.4 | 218, 290 |
| MMB-CHMICA | 14.3 | 218, 292 |
| 5F-PB-22 | – | 216, 294 |
| 5F-CUMYL-PICA | 12.5 | 218, 290 |
| JWH-018 | 18.2 | 218, 314 |
| AM-2201 | 14.7 | 218, 314 |
| THJ-2201 | 15.5 | 220, 320 |
| CUMYL-4CN-BINACA | 11.2 | 208, 302 |
| 5F-EDMB-PINACA | 14.9 | 208, 302 |
| BB-22 | 17.2 | 216, 294 |
| PB-22 | – | 216, 294 |
| 5F-UR-144 | 16.1 | 218, 304, 248 |
| MMB-FUBICA | – | 216, 290 |
Table 12.
UHPLC retention times for Method B and the detection wavelength used for selected synthetic cannabinoids.
| Compounds | Retention time (min) | Detection wavelength (nm) |
|---|---|---|
| JWH-200 | 1.9 | 217 |
| AM-1220 | 2.3 | 217 |
| Internal Standard | 5.7 | 198/242 |
| AM-694 | 11.8 | 209 |
| RCS-4 | 12.8 | 209 |
| CP-47,497 | 13.7 | 198 |
| JWH-250 | 15.5 | 209 |
| JWH-073 | 16.3 | 217 |
| CP-47,497-C8 | 16.6 | 198 |
| JWH-251 | 17.0 | 209 |
| JWH-203 | 17.6 | 209 |
| JWH-018 | 19.2 | 217 |
| JWH-007 | 20.0 | 217 |
| JWH-081 | 20.6 | 209 |
| JWH-122 | 21.9 | 217 |
| JWH-019 | 22.5 | 217 |
| JWH-210 | 24.0 | 217 |
5.4.6.1.5. Quantitative analysis
Due to possible matrix interactions, internal standard calibration is strongly advised. The use of peak area for quantitation is recommended because negative effects from peak broadening can be minimized. Previously characterized “herbal mixtures” or blends can be employed as precision controls. The calculation on the percentage of targeted synthetic cannabinoid in a sample is shown in section 5.4.5.

5.4.7. Liquid chromatography-tandem mass spectrometry
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) is a powerful tech-nique which combines the separation features of conventional HPLC or UHPLC with the detection capabilities of a tandem mass spectrometer, resulting in signifi-cantly increased selectivity and reduced interference between active ingredients and matrix. Its low limits of detection allow for trace analysis and the analysis of bio-logical specimens such as urine, blood and hair. With high sensitivity and selectivity, LC-MS/MS is suitable for both qualitative and quantitative analysis of synthetic cannabinoids at low concentrations in complex herbal mixtures.
One method for both qualitative and quantitative LC-MS/MS analysis is described below and can be modified for improved performance. This method has been field-tested within forensic casework and is considered fit-for-purpose. With adequate verification and validation, the same method can also be extended to other synthetic cannabinoids.
5.4.7.1. Standard/sample preparation
5.4.7.1.1. Preparation of internal standard (IS) solution
Prepare a solution of diphenylamine (DPA) in a suitable volume of ethanol to give a final concentration of 100 mg/L.
5.4.7.1.2. Preparation of synthetic cannabinoid standard stock solution (for quantitative analysis)
Prepare a standard stock solution containing all analytes to be quantified (e.g., JWH- 018, JWH-019 and JWH-073) in concentrations of 1.0 mg/L and the internal standard diphenylamine at a concentration of 100 μg/L as follows:
Accurately pipette 100 μL IS solution of 100 mg/L and 100 μL of 1 mg/mL solu-tions of each analyte (1 mg/mL concentrations are commercially available) into a 100 mL volumetric flask and dilute to volume with ethanol. The stock solution can be stored for at least one year with refrigeration.
5.4.7.1.3. Preparation of synthetic cannabinoid standard working solution (for quantitative analysis)
For making up the working standard solutions, the IS solution of 100 mg/L should be first diluted 1000 times to give a concentration of 100 μg/L (Drug internal standard (DIS) solution). This solution is used to dilute the synthetic cannabinoid standard stock solution to the desired concentration.
Prepare accurately an appropriate working concentration range. Usually, at least five standard solutions should be prepared for a good linear calibration curve. An example of the preparation of a 5-point calibration curve is given below (Table 13):
Table 13.
Example of calibration data.
| Volume of |
||||
|---|---|---|---|---|
| volumetric flask | ||||
| Volume of | used to dilute | Final | ||
| standard stock | to volume with | Final | concentration of | |
| Calibration | solution added | DIS solution | concentration of | cannabinoids |
| level | (μL) | (mL) | IS (μg/L) | (μg/L) |
| Level 1 | 30 | 10 | 100 | 3 |
| Level 2 | 100 | 10 | 100 | 10 |
| Level 3 | 300 | 10 | 100 | 30 |
| Level 4 | 1000 | 10 | 100 | 100 |
| Level 5 | 2000 | 10 | 100 | 200 |
5.4.7.1.4. Preparation of sample solutions (unknown “herbal mixture“)
For qualitative analysis, the herbal material can be soaked in a suitable solvent and an aliquot removed for analysis. For quantitative analysis, obtain a representative sample from the seized material and carefully homogenize. Accurately weigh 100 mg of sample into a 50 mL volumetric flask and make up to the mark with IS solution (100 mg/L). Extract under sonication for 5 min, invert flask for at least 10 times, and centrifuge for 2 min at 5000 rpm, or allow to settle. Take an aliquot of approximately 2 mL and filter using a syringe filter (≤0.45 μm). Then, accurately pipette 50 μL of the filtrate into a 50 mL volumetric flask and dilute to volume with ethanol. Inject 5 μL of the sample solution into the LC-MS/MS. At least a duplicate analysis should be carried out.
| LC-MS/MS operating conditions | ||
|---|---|---|
| LC: | Method 1 | Method 2 |
| Column: | C18 analytical column (e.g., 100 mm × 2.1 mm i.d.,3.5 μm), C18 guard column(10 mm × 2.1 mm i.d.,3.5 μm) | CORTECS UHPLC C18 column, (100 mm × 2.1 mm id 1.6 μm) |
| Mobile phase: | 0.1% formic acid (A): water (B): methanol (C) | 10 mM ammonium formate with 0.1% formic acid (A):acetonitrile with 0.1% formic acid (B) |
| Gradient: | Initial A:B:C = 10:70:20, linear to 10:5:85 within 10 min | Initial A:B = 70:30, linear to 50:50 within 3 min |
| 10 min isocratic | 1.5 min isocratic Linear to 5:95 within 4.5 min 0.5 min isocratic | |
| back to initial conditions within 1 min | back to initial conditions within 0.1 min | |
| 4 min equilibration (total run time 25 min) | 2.4 min equilibration time (total run time 12 min) | |
| Flow rate: | 0.2 mL/min | 0.4 mL/min |
| Column temperature: | 30 °C | 30 °C |
| Injection volume: | 5 μL | 2 μL |
| MS/MS: | ||
| Detection mode: | Multiple reaction monitoring (MRM) | |
| Ionization mode: | Simultaneous positive and negative electrospray ionization (ESI+ and ESI-) | ESI+ |
| Capillary voltage: | 3.5kV | 0.5 kV |
| Drying gas temperature | 350 °C at 650 L/h | 400 °C at 800 L/hr |
Note: The above conditions may be altered as long as appropriate validation is carried out.
Table 14 shows mass spectrometric data and parameters for some selected synthetic cannabinoids and the diphenylamine (DPA) internal standard (see Table 15).
Table 14.
Mass spectrometric data.
| Analyte | Ionization mode | Precursor ion (m/z) | Product ions (m/z) | Cone voltage (V) | Collision energy (eV) | Retention times (min) (method) |
|---|---|---|---|---|---|---|
| AM-2201 | ESI+ | 360 | 155 | 30 | 25 | 16.2 (1) |
| 145 | 40 | |||||
| BB-22 | ESI+ | 385 | 240 | 30 | 19 | 7.9 (2) |
| 144 | 36 | |||||
| 116 | 55 | |||||
| CP-47,497 | ESI- | 317 | 299 | 45 | 26 | 19.2 (1) |
| 160 | 55 | |||||
| CUMYL-4CN- BINACA | ESI+ | 361 | 226 | 30 | 20 | 4.9 (2) |
| 243 | 10 | |||||
| 119 | 25 | |||||
| DPA (IS) | ESI+ | 170 | 93 | 31 | 28 | 15.0 (1) |
| 5F-APINACA | ESI+ | 384 | 135 | 30 | 23 | 8.2 (2) |
| 93 | 47 | |||||
| 107 | 47 | |||||
| 5F-MDMB- PINACA | ESI+ | 378 | 233 | 30 | 22 | 6.5 (2) |
| 318 | 14 | |||||
| 213 | 30 | |||||
| 5F-PB-22 | ESI+ | 377 | 232 | 30 | 18 | 6.2 (2) |
| 144 | 38 | |||||
| 116 | 54 | |||||
| 5F-UR-144 | ESI+ | 330 | 125 | 30 | 23 | 7.6 (2) |
| 232 | 25 | |||||
| 144 | 39 | |||||
| JWH-018 | ESI+ | 342 | 155 | 30 | 25 | 18.1 (1) |
| 145 | 42 | |||||
| JWH-019 | ESI+ | 356 | 1545 | 34 | 25 | 18.9 (1) |
| 127 | 44 | |||||
| JWH-073 | ESI+ | 328 | 155 | 33 | 22 | 17.2 (1) |
| 1267 | 50 | |||||
| JWH-081 | ESI+ | 372 | 185 | 33 | 25 | 18.5 (1) |
| 214 | 25 | |||||
| JWH-122 | ESI+ | 356 | 169 | 29 | 25 | 19.0 (1) |
| 214 | 25 | |||||
| JWH-200 | ESI+ | 385 | 1545 | 25 | 20 | 11.7 (1) |
| 114 | 25 | |||||
| JWH-210 | ESI+ | 370 | 183 | 33 | 26 | 19.9 (1) |
| 214 | 26 | |||||
| JWH-250 | ESI+ | 336 | 121 | 25 | 20 | 17.1 (1) |
| 188 | 16 | |||||
| MMB-FUBICA | ESI+ | 383 | 252 | 30 | 15 | 5.1 (2) |
| 109 | 32 | |||||
| 224 | 26 | |||||
| FUB-AMB | ESI+ | 384 | 253 | 30 | 20 | 6.1 (2) |
| 109 | 35 | |||||
| 324 | 15 | |||||
| PB-22 | ESI+ | 359 | 214 | 30 | 14 | 7.3 (2) |
| 144 | 35 | |||||
| 116 | 54 | |||||
| RCS-4 | ESI+ | 322 | 135 | 25 | 24 | 17.0 (1) |
| 77 | 50 | |||||
| THJ-018 | ESI+ | 343 | 215 | 30 | 18 | 8.4 (2) |
| 145 | 33 | |||||
| 90 | 53 | |||||
| THJ-2201 | ESI+ | 343 | 233 | 30 | 16 | 7.4 (2) |
| 145 | 35 | |||||
| 213 | 25 |
Note: Precursor ions are detected as [M+H]+ in ESI+ mode or [M − H]- in ESI- mode.
Table 15.
NMR data.
| Compound | Expected splitting pattern and (integral) in the aliphatic region | Expected splitting pattern and (integral) in the phenyl ring |
|---|---|---|
| FUB-AMB | d (6), m (1), m (1) | m (2), m (2) |
| Isomer 1 | d (6), m (1), m (1) | m (1), m (1), m (1), m (1) |
| Isomer 2 | d (6), m (1), m (1) | m (1), m (1), m (1), m (1) |
| Isomer 3 | t (3), m (2), m (2), m (1) | m (2), m (2) |
| Isomer 4 | t (3), m (2), m (1), s (3) | m (2), m (2) |
| Isomer 5 | t (3), q (2), s (3) | m (2), m (2) |
s = singlet, d = doublet, t = triplet, m = multiplet.
5.4.7.1.5. Results
Identification is accomplished by comparing the retention time of the analyte with that of a reference standard solution. The internal standard allows the use of the retention index as an additional identification criterion. Furthermore, the ratio of intensities of both mass transitions (precursor→product ion 1/precursor→product ion 2) of an analyte should be compared with that of a reference standard solution. Appropriate mass transitions should be selected to avoid interference between different analytes, particularly in isomers (e.g., JWH-019 and JWH-122). Hence, even co-eluting compounds can be discriminated. In some cases, recording of the product spectrum of a particular precursor (Daughter Scan; DS) may be necessary for an unambiguous identification. Caution has to be applied when identifying regioisomeric compounds.
5.4.7.1.6. Quantitation
Due to possible matrix interactions and features specific to mass spectrometers, internal standard calibration is strongly advised and matrix effects have to be explored. The use of peak area for quantitation is recommended because negative effects from peak broadening can be minimized. Generally, the most intense mass transitions (primary trace; upper product ions in the table showing mass spectro-metric data and parameters) are usually utilized for quantitation, while less intense mass transitions (secondary trace; lower product ions in the table showing mass spectrometric data and parameters) may be favoured when interferences exist. Co-eluting analytes can also be quantified simultaneously with this method. Previously characterized “herbal mixtures” or blends can be employed as precision controls. The calculation on the percentage of targeted synthetic cannabinoid in a sample is shown in section 5.4.5.

5.5. Additional analytical techniques for the analysis of synthetic cannabinoids
This section gives an overview of some additional techniques and approaches that can be applied to the analysis and identification of synthetic cannabinoids in herbal products.
5.5.1. Nuclear magnetic resonance spectroscopy
As there are a large number of structurally related synthetic cannabinoids, effective analytical tools may be required to provide the structural information necessary for their differentiation. Nuclear magnetic resonance spectroscopy (NMR), enables iden-tification as well as structural elucidation of unknown new synthetic cannabinoids. A combination of one-dimensional 1H NMR and 13C NMR and two-dimensional experiments such as 1H-1H-COSY (correlation spectroscopy), 1H-1H-NOESY (nuclear Overhauser effect spectroscopy), 1H-13C-HSQC (heteronuclear single-quantum cor-relation spectroscopy) and 1H-13C-HMBC (heteronuclear multiple-bond correlation spectroscopy) can be employed to provide unambiguous assignment of molecular structure. Furthermore, NMR can also be used for quantitative determinations. While being a powerful tool for the identification of analogues, the cost of NMR spec-trometers the need for relatively pure compounds and the technical expertise required limit its widespread application in routine analysis [[5], [6], [7],9,17].
One example is shown below to illustrate the use of NMR spectroscopy for the differentiation of FUB-AMB (MMB-FUBINACA, AMB-FUBINACA) from its other isomers.
5.5.1.1. Sample preparation and instrumentation
The sample (∼20 mg) was dissolved in deuterated Dimethyl Sulphoxide (DMSO‑d6) (0.6 mL) and 1H, 13C, COSY and HMBC NMR experiments performed using Bruker 500 MHz spectrometer.
FUB-AMB has two fluoro positional isomers in the phenyl ring and three other structural isomers (with variations at the isopropyl group) (Fig. 8). These isomers may not be differentiated from FUB-AMB based on their mass spectra. In the absence of drug reference standards for retention time or IR spectrum comparison, NMR spectroscopy can be used to confirm the identity of the compound.
Fig. 8.

Structures and expected proton NMR integrals (in red) of FUB-AMB and its isomers. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
The expected 1H NMR integrals and splitting patterns for FUB-AMB and isomers 1 to 5 are tabulated as follows:
FUB-AMB can be differentiated from its ortho and meta fluoro positional isomers 1 and 2 based on the splitting pattern of the proton signals for the phenyl ring. For FUB-AMB, the fluoro substituent is in the para position and the molecule is sym-metrical at the phenyl ring due to the free rotation of the C2′-C3′ bond. Hence, only two sets of multiplets with an integral of two will be expected since the two ortho protons and the two meta protons will be chemically equivalent, but not magnetically equivalent. A multiplet splitting pattern is observed due to long range coupling of the protons with fluorine. For isomers 1 and 2, four sets of multiplets with respective integral of one each will be expected since all the four protons in the phenyl ring are chemically and magnetically non-equivalent.
The structural isomers of FUB-AMB (isomers 3 to 5 in Fig. 4) can be distinguished from FUB-AMB through the expected NMR integral and splitting patterns in the aliphatic region of 0–5 ppm. Three sets of signals with an integral of 6, 1 and 1 were expected for FUB-AMB. Hindered internal rotation about the C2′-C3′ sigma bond causes the two methyl groups (H-4′ and H-5′) to be chemically and magneti-cally inequivalent. This results in two sets of doublets, integrated to 6H, at 0.95 and 0.97 ppm. For isomers 3 to 5, a triplet with an integral of 3 would be expected as these isomers have a terminal ethyl group (Fig. 8).
The 1H and 13C NMR assignments based on 1H, 13C, COSY and HMBC NMR experiments are tabulated in Table 16. The 1H NMR spectrum is shown in Fig. 9 (see Table 17).
Table 16.
NMR data for FUB-AMB.
| No. | Chemical shift (ppm), multiplicity, integral, and coupling constant |
|
|---|---|---|
| 1H | 13C | |
| 3 | – | 136.9 |
| 3a | – | 122.4 |
| 4 | 8.16 (d, 1H, J = 8.5 Hz, overlapping) | 121.7 |
| 5 | 7.27–7.30 (m, 1H) | 122.7 |
| 6 | 7.43–7.47 (m, 1H) | 127.0 |
| 7 | 7.79 (d, 1H, J = 8.5 Hz) | 110.5 |
| 7a | – | 140.5 |
| C=O | – | 161.8 |
| 2′ | 4.43–4.46 (m, 1H) | 57.3 |
| 3′ | 2.23–2.30 (m, 1H) | 29.9 |
| 4′ | 0.95 (d, 3H, J = 6.5 Hz) | 18.7 |
| 5′ | 0.97 (d, 3H, J = 6.5 Hz) | 19.0 |
| 1′ | – | 171.9 |
| 6′ | 3.69 (s, 3H) | 51.8 |
| NH | 8.20 (d, 1H, J = 8.0 Hz, overlapping) | – |
| NCH2 | 5.78 (s, 2H) | 51.6 |
| 1″ | – | 132.9 |
| 2’’/6″ | 7.33–7.36 (m, 2H) | 129.4 (d, JC-F = 8.3 Hz) |
| 3’’/5″ | 7.14–7.18 (m, 2H) | 115.4 (d, JC-F = 21.4 Hz) |
| 4″ | – | 161.6 (d, JC-F = 242.4 Hz) |
Fig. 9.

1H NMR spectrum of FUB-AMB.
Table 17.
K0 values for selected synthetic cannabinoids.
| Compound | K0 values (positive ion mode) |
K0 values (negative ion mode) |
|---|---|---|
| [cm2/(V∗s)] | [cm2/(V∗s)] | |
| JWH-210 | 0.9596 | – |
| JWH-081 | 0.9720 | – |
| AM-1220 | 0.9878 | – |
| JWH-019 | 0.9915 | – |
| JWH-200 | 0.9926 | – |
| JWH-122 | 0.9950 | – |
| AM-2201 | 1.0163 | – |
| JWH-250 | 1.0263 | – |
| JWH-018 | 1.0288 | – |
| AM-694 | 1.0348 | – |
| JWH-203 | 1.0455 | – |
| JWH-251 | 1.0483 | – |
| JWH-073 | 1.0658 | – |
| RCS-4 | 1.0659 | – |
| CP-55,940 | – | 0.9045 |
| CP-47,497-C8 | – | 0.9185 |
| CP-47,497 | – | 0.9354 |
1H and 13C NMR assignments of FUB-AMB.

5.5.2. Ion mobility spectrometry
Ion mobility spectrometry (IMS) is a fast and sensitive technique that is suitable for the detection of trace organics under atmospheric pressure conditions. It can be used as a rapid screening technique for many drugs of abuse including a number of synthetic cannabinoids. IMS allows for easy sampling and handling by touching the surface of the herbal mixture with a wooden rod and transferring the adherent particles distributed over the surface onto a Teflon filter for analysis. As portable IMS systems are commercially available, IMS can be used as a rapid detection technique in the field (e.g., crime scene investigations).
IMS can be operated in positive and negative ion modes. Most synthetic cannabinoids can be detected in positive ion mode while non-classical cannabinoids (e.g., CP- 47,497-C8) can be detected in negative ion mode. For switching to the negative ion mode, some of the parameters listed below have to be modified (e.g., desorber temp.: 222 °C, inlet temp.: 238 °C, drift tube temp.: 105 °C). Typical plant matrices and aromatic components of the herbal mixtures do not interfere with IMS signals of the active substances present.
Although IMS has limited selectivity, a new synthetic cannabinoid of a similar core structure will give a signal in the IMS plasmagram in the typical detection window for synthetic cannabinoids with similar core structures. However, subsequent confirmatory analysis with more sophisticated instrumentation should be carried out.
The following steps are part of a field-tested and fit-for-purpose IMS method for portable IMS systems:
5.5.2.1. Procedures
For analysis of herbal mixtures, touch the sample surface with a wooden rod. Take care that no visible particles of the plant material are on the rod after sampling. Sweep the tip of the rod several times over the Teflon filter placed in the IMS system and start analysis. To account for inhomogeneity, multiple sampling with the wooden rod is recommended.
5.5.2.2. Results
Aminoalkylindoles give sharp signals in positive ion mode within a characteristic detection window at high drift times and can be matched to reference substances by their reduced ion mobilities (K0). Non-classical cannabinoids (e.g., CP-47,497 and its homologues) can be detected with lower but sufficient sensitivity in negative ion mode within a characteristic detection window distant from the detection window for the explosives. K0 values for selected synthetic cannabinoids using the above method are as follows:
Typically, substances that exhibit differences in their K0 values < 0.025 cannot be discriminated by IMS (e.g., JWH-019/JWH-200 or JWH-073/RCS-4). As this method is only suitable as a rapid screening technique, it is essential that another method with sufficient selectivity (e.g., GC-MS, GC-IRD) be used to confirm these substances.
IMS operating conditions (positive ion mode).
Ionization source: 63Ni beta-emitting source or x-ray tube
Desorber temp.: 290 °C
Inlet temp.: 285 °C
Drift tube temp.: 235 °C
Drift flow: 300 mL/min
Sample flow: 200 mL/min
Stand-by flow: 51 mL/min
Drift gas: dried, purified air
Carrier gas: dried, purified air
Calibrant/reactant: nicotinamide
Calibrant temp.: 80 °C
Gate width: 200 μs
Desorption time: 8.0 s
Scan period: 20 ms
Number of scans: 20
Drift tube length: 6.9 cm
Threshold: 50 d.u. (for JWH-018)
FWHM: 400 μs (for JWH-018)
Note: The above conditions may be altered as long as appropriate validation is carried out.

5.5.3. Ambient ionization mass spectrometry
As synthetic cannabinoids are essentially laced onto herbal material, ambient ionization mass spectrometric techniques such as direct analysis in real time mass spectrometry (DART-MS) [44], desorption atmospheric pressure photoionization (DAPPI) [45] or desorption electrospray ionization mass spectrometry (DESI-MS) could be employed to analyse these plant materials directly without the need for extraction and sample preparation. DESI-MS could also be used in combination with TLC.
5.5.4. High-resolution mass spectrometry
Besides identification by accurate mass measurements, high-resolution mass spec-trometry (HRMS) could be used to determine the precise elemental compositions of new synthetic molecules, calculation of double bond equivalents as well as precise mass of the fragment ions. Furthermore, HRMS in conjunction with mass defect filtering enables non-targeted analysis of related compounds and analogues which could prove very useful in screening for synthetic cannabinoids [[46], [47], [48]].
5.5.5. Matrix-assisted laser desorption ionization-time of flight mass spectrometry
Matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI- TOF) provides another possibility for direct qualitative analysis of components in herbal mixtures. It offers a simple and rapid operation, allows for high throughput analysis and could be utilized as a “front screening” of seized materials [49].

6. Isolation and chemical characterization of new synthetic cannabinoids
Due to the number of synthetic cannabinoids that have emerged in recent years, it is very likely for an analyst to encounter an unknown substance in a herbal product and suspect the presence of a new synthetic cannabinoid. The identification of an unknown substance can be challenging without commercially available reference standards, reference spectra as well as relevant literature and research. Hence, in order to identify a new substance, it has to be first isolated from the herbal mixture into a pure/enriched form and then various analytical techniques could be employed to characterize the compound. The schematic diagram below illustrates a general approach towards isolation and characterization of a new synthetic cannabinoid. A case study on the isolation and characterization of two new synthetic cannabinoids is also illustrated below.
6.1. Isolation of a new compound
The first step would be to identify a suitable solvent to extract the targeted unknown cannabinoid (e.g., methanol, ethanol, ethyl acetate, acetone or isooctane) from the herbal product. Extraction should be carried out with sonication and the extract filtered. Then, the extract should be subjected to preparative/flash chromatography (e.g., silica gel column, preparative LC or TLC) to obtain a fraction containing the targeted unknown cannabinoid. This fraction should show a single spot with a TLC analysis (visualization by UV light and/or other reagents e.g., Fast Blue RR reagent, iodine, iodoplatinate). Then, the fraction containing the pure/enriched compound, should be concentrated and used for subsequent analysis aimed at characterizing the unknown cannabinoid.
6.2. Characterization of a new compound
There are a variety of techniques available for characterization of an unknown can-nabinoid. A combination of techniques such as HRMS and NMR is important for unambiguous structure elucidation. Other techniques such as IR and MS/MS may be useful to provide other structural information including differentiation between isomers or diastereomers. Techniques such as GC, LC and UV/VIS alone cannot confirm the identity of the compound as many compounds can give the same retention time or UV/VIS spectra, leading to false positive results. However, these techniques can provide useful information when the results are negative (e.g the UV spectrum of the compound is different from the reference material). Therefore, the correct combination of techniques needs to be selected in order to characterize a new compound.
With these techniques, the structure of the unknown cannabinoid could be deduced and based on this, a reference standard should be synthesized. The synthesized refer-ence standard should be analysed with the same techniques mentioned, under the same conditions. If the analysis of the synthesized reference standard yields the same results, the deduced structure of the unknown cannabinoid could be confirmed. While com-plete structural elucidation can be carried out using NMR, in cases where connectivity or stereoconfiguration cannot be ascertained by NMR experiments, proof by compari-son to a reference material synthesized in house may be relevant.
While it is not necessary to perform all the above analytical techniques for charac-terization, it is important to verify and confirm any interpretation with analysis of a synthesized standard and/or through peer review from a reputable laboratory. Collaboration with academia would also be useful as some sophisticated instru-mentation (e.g., NMR, HRMS) is not commonly available for routine use in most forensic science laboratories.

Case study: Isolation and identification of 5F-MDMB-PINACA and 5F- EDMB-PINACA in a mixture of a seized sample.
The crystalline substance from a seized exhibit was analysed using GC-MS and was suspected to contain new synthetic cannabinoids 5F-MDMB-PINACA and 5F-EDMB- PINACA based on preliminary investigation using reference MS libraries and litera-ture. Reference standards for both compounds were not available.
6.3. Isolation using semi-preparative liquid chromatography
Sample preparation: The crystalline substance (50 mg) was dissolved in acetonitrile (1 ml).
Purification:
| Semi-preparative liquid chromatography conditions | |
|---|---|
| Instrument | Shimadzu Prominence LC-20AD with an SPD-M20A PDA |
| UV-VIS detector and an SIL-10A autosampler coupled with a | |
| Shimadzu FRC-10A fraction collector | |
| Column | Agilent HPLC column Zorbax SB-C18, 250 × 9.4 mm, 5 μm |
| Flow rate | 5 mL/min |
| Mobile phase A | Water |
| Mobile phase B | Acetonitrile |
| Gradient programme | 1% B to 60% B in 5 min; |
| 60% B to 100% B in 15 min | |
| 100% B for 25 min | |
| 100% B to 1% B in 0.1 min | |
| 1% B for 4.9 min | |
| (Total run time: 50 min) | |
| Detector wavelength | 215 nm |
| Injection volume | 50 μL |
The shaded areas represent the fractions collected for the respective two peaks of interest (Fig. 10). The collected fractions were then dried and re-constituted in appropriate solvents for instrumental analysis.
Fig. 10.

HPLC separation of sample components.
6.4. Characterization of compound
GC-MS analysis (refer to method 2 in section 5.4.4 for the GC-MS conditions).
The GC-MS analyses results of the collected fractions for peak 1 and peak 2 are summarized as follows as shown in Table 18:
Table 18.
GC-MS data.
| Compound | Major ions | MS interpretation |
|---|---|---|
| Peak 1 | 131, 145, 213, 233, 246, 289, 321, 377 M+ |
MS fragmentation pattern consistent with 5F-MDMB-PINACA |
| Peak 2 | 131, 145, 233, 246, 289, 318, 335, 391 M+ |
MS fragmentation pattern consistent with 5F-EDMB-PINACA |
The MS fragmentation patterns are consistent with the proposed fragmentation path-way (refer to Scheme 5, Scheme 6). However, this is not sufficient to eliminate the possibility of isomers as they may have similar fragmentation patterns. Hence, the collected fractions for peak 1 and 2 were analysed by NMR.
Scheme 5.

Proposed mass fragmentation pathways of 5F-MDMB-PINACA.
Scheme 6.

Proposed mass fragmentation pathways of 5F-EDMB-PINACA.
NMR analysis was performed on a Bruker 500 MHz spectrometer using CDCl3 as the solvent. The NMR spectra of semi-preparative HPLC peak 1 and 2 are consist-ent with the identity of 5F-MDMB-PINACA and 5F-EDMB-PINACA respectively (Fig. 11) and unequivocally confirmed the identity of the compound.
Fig. 11.

NMR spectra of isolated peak 1 (top) and 2 (bottom).
7. Note
Operating and experimental conditions are reproduced from the original reference materials, including unpublished methods, validated and used in selected national laboratories as per the list of references. A number of alternative conditions and substitution of named commercial products may provide comparable results in many cases, but any modification has to be validated before it is integrated into laboratory routines.
Mention of names of firms and commercial products does not imply the endorsement of the United Nations.
The designations employed and the presentation of material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries.
Disclaimer
This is a republication in journal form of the United Nations Office on Drugs and Crime (UNODC), Recommended methods for the Identification and Analysis of Synthetic Cannabinoid Receptor Agonists in Seized Materials which was originally published by the United Nations and is available here: https://www.unodc.org/unodc/en/scientists/recommended-methods-for-the-identification-and-analysis-of-synthetic-cannabinoid-receptor-agonists-in-seized-materials.html. This manual is designed to provide practical guidance to national authorities and drug analysts by describing recommended methods for use in forensic laboratories for the identification and analysis of synthetic cannabinoid receptor agonists (synthetic cannabinoids) in seized materials. Peer review was organised and administered by the UNODC, further details of which can be found in the acknowledgements section.
Declaration of competing interests
The authors have no competing interests to declare.
Acknowledgements
The Laboratory and Scientific Services of the United Nations Office on Drugs and Crime (UNODC) (LSS, headed by Dr. Justice Tettey) wishes to express its appreciation and thanks to Dr. Tiong Whei Angeline Yap, Ms. Jong Lee Wendy Lim, Dr. Hui Zhi Shirley Lee and Dr. Mei Ching Ong of the Health Sciences Authority, Singapore for preparing the final draft of the present Manual.
The following experts were invited by UNODC to peer review the manual. Their valuable comments and contribution are gratefully acknowledged: Mr. Benoit Archimbault, Ms. Michelle Boileau, Mr. Adam Kerry, Ms. Lilly Ho, Ms. Josée Cloutier and Ms. Stéphanie Lessard of the Regulatory Operations and Enforcement Branch of Health Canada; Mr. Elvio Dias Botelho and Ms. Monica Paulo de Souza of the Brazilian Federal Police Forensic Chemistry Laboratory, Brazil; Prof. Niamh Nic Daéid, University of Dundee, Scotland; Prof. Franco Tagliaro, University of Verona, Italy and Dr. Dariusz Zuba, Institute of Forensic Research, Krakow, Poland. The preparation of the present Manual was coordinated by Dr. Conor Crean, staff member of LSS, and the contributions of LSS staff member Mr. Joao Rodrigues is gratefully acknowledged.
References
- 1.Auwärter V., Dresen S., Weinmann W., Müller M., Pütz M., Ferreiros N. J. Mass Spectrom. 2009;44(5):832–837. doi: 10.1002/jms.1558. [DOI] [PubMed] [Google Scholar]
- 2.Uchiyama N., Kikura-Hanajiri R., Kawahara N., Haishima Y., Goda Y. Chem. Pharm. Bull. (Tokyo) 2009;57(4):439–441. doi: 10.1248/cpb.57.439. [DOI] [PubMed] [Google Scholar]
- 3.Dresen S., Ferreiros N., Pütz M., Westphal F., Zimmermann R., Auwärter V. J. Mass Spectrom. 2010;45(10):1186–1194. doi: 10.1002/jms.1811. [DOI] [PubMed] [Google Scholar]
- 4.European Monitoring Centre for Drugs and Drug Addiction EMCDDA . 2009. Thematic Papers — Understanding the ‘Spice’ Phenomenon.www.emcdda.europa.eu/publications/thematic-papers/understanding-spice- phenomenon_en available at. [Google Scholar]
- 5.Ernst L., Schiebel H.M., Theuring C., Lindigkeit R., Beuerle T. Forensic Sci. Int. 2011;208(1–3):e31–e35. doi: 10.1016/j.forsciint.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 6.Jankovics P., Varadi A., Tolgyesi L., Lohner S., Nemeth-Palotas J., Balla J. Forensic Sci. Int. 2012;214(1–3):27–32. doi: 10.1016/j.forsciint.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 7.Kneisel S., Westphal F., Bisel P., Brecht V., Broecker S., Auwärter V. J. Mass Spectrom. 2012;47(2):195–200. doi: 10.1002/jms.2059. [DOI] [PubMed] [Google Scholar]
- 8.Lindigkeit R., Boehme A., Eiserloh I., Luebbecke M., Wiggermann M., Ernst L., Beuerle T. Forensic Sci. Int. 2009;191(1–3):58–63. doi: 10.1016/j.forsciint.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 9.Moosmann B., Kneisel S., Girreser U., Brecht V., Westphal F., Auwärter V. Forensic Sci. Int. 2012;220(1–3):e17–e22. doi: 10.1016/j.forsciint.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 10.Nakajima J., Takahashi M., Nonaka R., Seto T., Suzuki J., Yoshida M., Kanai C., Hamano T. Forensic Toxicol. 2011;29(2):132–141. [Google Scholar]
- 11.Nakajima J., Takahashi M., Seto T., Kanai C., Suzuki J., Yoshida M., Hamano T. Forensic Toxicol. 2011;29(2):95–110. [Google Scholar]
- 12.Nakajima J., Takahashi M., Seto T., Suzuki J. Forensic Toxicol. 2011;29(1):51–55. [Google Scholar]
- 13.Nakajima J., Takahashi M., Seto T., Yoshida M., Kanai C., Suzuki J., Hamano T. Forensic Toxicol. 2012;30(1):33–44. [Google Scholar]
- 14.Uchiyama N., Kawamura M., Kikura-Hanajiri R., Goda Y. Forensic Toxicol. 2011;29(1):25–37. [Google Scholar]
- 15.Uchiyama N., Kikura-Hanajiri R., Goda Y. Chemical and Pharmacetical Bulletin (Tokyo) 2011;59(9):1203–1205. doi: 10.1248/cpb.59.1203. [DOI] [PubMed] [Google Scholar]
- 16.Uchiyama N., Kikura-Hanajiri R., Ogata J., Goda Y. Forensic Sci. Int. 2010;198(1–3):31–38. doi: 10.1016/j.forsciint.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 17.Westphal F., Sonnichsen F.D., Thiemt S. Forensic Sci. Int. 2012;215(1–3):8–13. doi: 10.1016/j.forsciint.2011.03.031. [DOI] [PubMed] [Google Scholar]
- 18.Kneisel S., Bisel P., Brecht V., Broecker S., Müller M., Auwärter V. Forensic Toxicol. 2012;30(2):126–134. [Google Scholar]
- 19.Hudson S., Ramsey J. Drug Test. Anal. 2011;3(7–8):466–478. doi: 10.1002/dta.268. [DOI] [PubMed] [Google Scholar]
- 20.Tai S., Fantegrossi W.E. Current Addiction Reports. 2014;1(2):129–136. doi: 10.1007/s40429-014-0014-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tai S., Fantegrossi W.E. Current Topics in Behavioural Neurosciences. 2017;32:249–262. doi: 10.1007/7854_2016_60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tampus R., Yoon S.S., de la Peña J.B., Botanas C.J., Kim H.J., Seo J.W., Jeong E.J., Jang C.G., Cheong J.H. Biomolecules and Therapuetics (Seoul) 2015;23(6):590–596. doi: 10.4062/biomolther.2015.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howlett A.C., Barth F., Bonner T.I., Cabral G., Casellas P., Devane W.A., Felder C.C., Herkenham M., Mackie K., Martin B.R., Mechoulam R., Pertwee R.G. International union of pharmacology. XXVII. Pharmacol. Rev. 2002;54(2):161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- 24.Uchiyama N., Kawamura M., Kikura-Hanajiri R., Goda Y. Forensic Toxicol. 2012;30(2):114–125. [Google Scholar]
- 25.Ernst L., Krüger K., Lindigkeit R., Schiebel H.M., Beuerle T. Forensic Sci. Int. 2012;222(1–3):216–222. doi: 10.1016/j.forsciint.2012.05.027. [DOI] [PubMed] [Google Scholar]
- 26.UK Advisory Council on the Misuse of Drugs, Consideration of the Major Cannabinoid Agonists. 16th July 2009. www.gov.uk/government/publications/acmd-further-consideration-of-the-synthetic-cannabinoids available at. [Google Scholar]
- 27.Ginsburg B.C., McMahon L.R., Sanchez J.J., Javors M.A. J. Anal. Toxicol. 2012;36(1):66–68. doi: 10.1093/jat/bkr018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Breitbarth A.K., Morgan J., Jones A.L. Drug Alcohol Depend. 2018;192:98–111. doi: 10.1016/j.drugalcdep.2018.07.031. [DOI] [PubMed] [Google Scholar]
- 29.European Monitoring Centre for Drugs and Drug Addiction EMCDDA . EMCDDA Rapid Communication; 2018. New Psychoactive Substances in Prison.www.emcdda.europa.eu/publications/rapid-com- munications/nps-in-prison_en available at. [Google Scholar]
- 30.Norman C., Walker G., McKirdy B., McDonald C., Fletcher D., Antonides L., Sutcliffe O., Nic Daéid N., Mckenzie C. Drug Test. Anal. 2020;12(4):538–544. doi: 10.1002/dta.2767. [DOI] [PubMed] [Google Scholar]
- 31.Ford L., Berg J. Ann. Clin. Biochem. 2018;55(6):673–678. doi: 10.1177/0004563218767462. [DOI] [PubMed] [Google Scholar]
- 32.Boff B., Filho J., Nonemacher K., Schroeder S., Arbo M. Forensic Sci. Int. 2020;306:110002. doi: 10.1016/j.forsciint.2019.110002. [DOI] [PubMed] [Google Scholar]
- 33.Valoti E., Casagni E., Dell’acqua L., Pallavicini M., Roda G., Rusconi C., Straniero V., Gambaro V. Forensic Sci. Int. 2012;223(1–3):e42–e46. doi: 10.1016/j.forsciint.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 34.Banister S.D., Moir M., Stuart J., Kevin R.C., Wood K., Longworth E., Wilkinson S.M., Beinat C., Buchanan A.S., Glass M., Connor M., McGregor I.S., Kassiou M. ACS Chem. Neurosci. 2015;6(9):1546–1559. doi: 10.1021/acschemneuro.5b00112. [DOI] [PubMed] [Google Scholar]
- 35.Kavanagh P., Grigoryev A., Savchuk S., Mikhura I., Formanovsky A. Drug Test. Anal. 2013;5(8):683–692. doi: 10.1002/dta.1456. [DOI] [PubMed] [Google Scholar]
- 36.Zuba D., Byrska B., Maciow M. Anal. Bioanal. Chem. 2011;400(1):119–126. doi: 10.1007/s00216-011-4743-7. [DOI] [PubMed] [Google Scholar]
- 37.Uchiyama N., Matsuda S., Kawamura M., Kikura-Hanajiri R., Goda Y. Forensic Toxicol. 2013;31:223–240. [Google Scholar]
- 38.Logan B.K., Reinhold L.E., Xu A., Diamond F.X. J. Forensic Sci. 2012;57(5):1168–1180. doi: 10.1111/j.1556-4029.2012.02207.x. [DOI] [PubMed] [Google Scholar]
- 39.Kneisel S., Westphal F., Rösner P., Ewald A., Klein B., Pütz M., Thiemt S., Auwärter V. Toxichem Krimtech. 2011;78(1):23–35. [Google Scholar]
- 40.Lee H.Z.S., Koh H.B., Tan S., Goh B.J., Lim R., Lee J., Lim W., Tiong Whei Yap A. Forensic Sci. Int. 2019;299:21–33. doi: 10.1016/j.forsciint.2019.03.025. [DOI] [PubMed] [Google Scholar]
- 41.Etxebarria N., Zuloaga O., Olivares M., Bartolome L.J., Navarro P. Journal of Chromatography. A. 2009;1216:1624–1629. doi: 10.1016/j.chroma.2008.12.038. [DOI] [PubMed] [Google Scholar]
- 42.Wilson M.B., Barnes B.B., Boswell P.G. What experimental factors influence the accuracy of rentention projections in gas chromatography-mass spectrometry. Journal of Chromatography. A. 2014;1373:179–189. doi: 10.1016/j.chroma.2014.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D’Acampora Zellner B., Bicchi C., Dugo P., Rubiolo P., Dugo G., Mondello L. Flavour Fragrance J. 2008;23:297–314. [Google Scholar]
- 44.Musah R.A., Domin M.A., Walling M.A., Shepard J.R. Rapid Commun. Mass Spectrom. 2012;26(9):1109–1114. doi: 10.1002/rcm.6205. [DOI] [PubMed] [Google Scholar]
- 45.Kauppila T.J., Flink A., Haapala M., Laakkonen U.M., Aalberg L., Ketola R.A., Kostiainen R. Forensic Sci. Int. 2011;210(1–3):206–212. doi: 10.1016/j.forsciint.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 46.Grabenauer M., Krol W.L., Wiley J.L., Thomas B.F. Anal. Chem. 2012;84(13):5574–5581. doi: 10.1021/ac300509h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hudson S., Ramsey J., King L., Timbers S., Maynard S., Dargan P.I., Wood D.M. J. Anal. Toxicol. 2010;34(5):252–260. doi: 10.1093/jat/34.5.252. [DOI] [PubMed] [Google Scholar]
- 48.Sekula K., Zuba D., Stanaszek R. J. Mass Spectrom. 2012;47(5):632–643. doi: 10.1002/jms.3004. [DOI] [PubMed] [Google Scholar]
- 49.Gottardo R., Chiarini A., Dal Pra I., Seri C., Rimondo C., Serpelloni G., Armato U., Tagliaro F. J. Mass Spectrom. 2012;47(1):141–146. doi: 10.1002/jms.2036. [DOI] [PubMed] [Google Scholar]