

Key Points
Pharmacologic targeting of ER proteostasis reduces secretion of amyloidogenic LCs from plasma cells.
Compounds that target ER proteostasis are compatible with current AL treatment strategies.
Abstract
Light chain (LC) amyloidosis (AL) involves the toxic aggregation of amyloidogenic immunoglobulin LCs secreted from a clonal expansion of diseased plasma cells. Current AL treatments use chemotherapeutics to ablate the AL plasma cell population. However, no treatments are available that directly reduce the toxic LC aggregation involved in AL pathogenesis. An attractive strategy to reduce toxic LC aggregation in AL involves enhancing endoplasmic reticulum (ER) proteostasis in plasma cells to reduce the secretion and subsequent aggregation of amyloidogenic LCs. Here, we show that the ER proteostasis regulator compound 147 reduces secretion of an amyloidogenic LC as aggregation-prone monomers and dimers in AL patient–derived plasma cells. Compound 147 was established to promote ER proteostasis remodeling by activating the ATF6 unfolded protein response signaling pathway through a mechanism involving covalent modification of ER protein disulfide isomerases (PDIs). However, we show that 147-dependent reductions in amyloidogenic LCs are independent of ATF6 activation. Instead, 147 reduces amyloidogenic LC secretion through the selective, on-target covalent modification of ER proteostasis factors, including PDIs, revealing an alternative mechanism by which this compound can influence ER proteostasis of amyloidogenic proteins. Importantly, compound 147 does not interfere with AL plasma cell toxicity induced by bortezomib, a standard chemotherapeutic used to ablate the underlying diseased plasma cells in AL. This shows that pharmacologic targeting of ER proteostasis through selective covalent modification of ER proteostasis factors is a strategy that can be used in combination with chemotherapeutics to reduce the LC toxicity associated with AL pathogenesis.
Visual Abstract

Introduction
Light chain (LC) amyloidosis (AL) is the most common systemic amyloid disease, affecting 8 to 10 people per million per year.1-5 AL pathogenesis involves the toxic extracellular aggregation of an amyloidogenic immunoglobulin LC that is secreted from a clonally expanded cancerous plasma cell. Aggregates of full-length LCs or proteolytic fragments comprising the variable domain are deposited on postmitotic tissues, including the heart and kidneys, causing organ failure and ultimately death.1-3,6-13 Current treatments for AL use chemotherapy combined with autologous stem cell replacement to eliminate AL-associated plasma cells.1,2,14-18 This reduces circulating serum populations of amyloidogenic LCs, decreasing proteolysis and toxic LC aggregation and promoting clearance of the deposited amyloid, ultimately improving organ function.1,19-21 However, ∼30% of AL patients presenting with severe cardiac or renal LC proteotoxicity are too ill at diagnosis to tolerate chemotherapy.1,22,23 Furthermore, the survival of AL patients treated with chemotherapeutics strongly correlates with reductions in amyloidogenic LCs.24-26 These results indicate that new strategies are required to alleviate LC proteotoxicity on distal tissues to improve the treatment of AL patients in the clinic.
One strategy to reduce the secretion and toxic aggregation of amyloidogenic proteins such as LCs is through the adaptive remodeling of the endoplasmic reticulum (ER) proteostasis network comprising ER chaperones (eg, BiP), folding enzymes (eg, protein disulfide isomerases [PDIs]), and degradation factors.27-29 These ER proteostasis pathways function to partition ER proteins between folding, trafficking, and degradation in a process termed ER quality control.28,30,31 Through this partitioning, ER proteostasis pathways reduce the secretion and aggregation of nonnative, aggregation-prone proteins in secretory environments, including the ER and extracellular space. In the context of AL, destabilized amyloidogenic LCs escape plasma cell ER quality control, allowing their efficient secretion into the serum.11,28,32 This increases the extracellular populations of amyloidogenic LCs available for proteolysis and/or concentration-dependent aggregation into the toxic oligomers and amyloid fibrils implicated in AL pathogenesis.
Enhancing ER proteostasis through mechanisms such as activation of the unfolded protein response (UPR)–associated transcription factor ATF6 selectively reduces the secretion and toxic aggregation of destabilized amyloidogenic proteins, including LCs.28,29,33 ATF6 induces the expression of many ER proteostasis factors, including the ATP-dependent ER HSP70 BiP and multiple PDIs.34,35 Genetic activation of ATF6 preferentially reduces secretion and subsequent aggregation of a destabilized amyloidogenic LC from HEK293T cells, without affecting the secretion of nonamyloidogenic LCs, fully assembled immunoglobulin G (IgG), or the endogenous secretory proteome.35-37 This ATF6-dependent reduction in amyloidogenic LC secretion results from increased interactions with ER chaperones and PDIs, which retain the amyloidogenic LCs within the ER and prevent their secretion to downstream secretory environments.37 This indicates that pharmacologic approaches that similarly target ER proteostasis could also reduce secretion and toxic aggregation of amyloidogenic LCs implicated in AL through an analogous mechanism.28,33
We previously used high-throughput screening to identify small-molecule ER proteostasis regulators that selectively activate the ATF6 UPR signaling pathway.38 The prioritized compound emerging from this screen, compound 147, was shown to activate ATF6 through a mechanism involving metabolic activation and covalent labeling of multiple PDIs,39 a class of proteins involved in regulating disulfide bonds within the ER.30,40,41 Importantly, 147 is nontoxic in multiple cell lines and mice,38,42-44 indicating that this compound provides unique opportunities to promote ER proteostasis remodeling for destabilized, aggregation-prone proteins in diseases such as AL.28,29 Here, we show that 147 reduces secretion of the destabilized amyloidogenic λ6a LC ALLC from AL patient–derived plasma cells without significantly affecting secretion of fully assembled IgGs from control plasma cells. However, the 147-dependent reduction in ALLC secretion is refractory to cotreatments with ATF6 inhibitors, demonstrating that this compound reduces ALLC secretion through an ATF6-independent mechanism. Instead, we show that 147 reduces ALLC plasma cell secretion through an on-target mechanism involving metabolic activation and covalent modification of ER proteostasis factors, including PDIs. Consistent with this, we show that 147 alters interactions between ALLC and ER PDIs and demonstrate that other covalent PDI inhibitors also reduce ALLC plasma cell secretion through a mechanism analogous to that observed for 147. These results indicate that pharmacologic targeting of ER proteostasis factors such as PDIs using covalent compounds like 147 represents a potential strategy to mitigate the plasma cell secretion of amyloidogenic LCs implicated in AL. Importantly, we show that 147 is compatible with chemotherapeutics used to ablate AL plasma cells (eg, bortezomib), indicating that these 2 approaches could potentially be used in combination in the treatment of AL to enhance reductions in amyloidogenic LCs in the serum and improve patient treatment.
Methods
Cell lines and culture conditions
ALMC-2, ALMC-1, and KAS-6/1 plasma cells were a gift from Diane Jelinek.45 We cultured these 3 plasma cell lines in Iscove modified Dulbecco medium GlutaMAX (Life Technologies), supplemented with penicillin/streptomycin, 5% fetal bovine serum, and 2 ng/mL of interleukin-6, following published protocols.38,45 Cells were cultured at 37°C with 5% carbon dioxide.
ELISA
The enzyme-linked immunosorbent assay (ELISA) for ALLC and fully assembled IgGs in conditioned media prepared on ALMC-2, ALMC-1, or KAS-6/1 cells was performed using an identical approach to that reported by Plate et al.38 A detailed protocol is included in the supplemental data.
Immunoblotting for ALLC
Conditioned media were collected from ALMC-1 or ALMC-2 cells as previously described.38 Media were denatured with Laemmli buffer plus 100 mM of dithiothreitol (DTT) and boiled for 5 minutes before being separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Conditioned media for nonreducing gels were prepared as above in the absence of DTT. Cell lysates were prepared as previously described38 in RIPA buffer with fresh protease inhibitor cocktail (Roche). Total protein concentration in cellular lysates was normalized using the Bio-Rad protein assay. Lysates were then denatured with Laemmli buffer plus 100 mM of DTT and boiled for 5 minutes before being separated by SDS-PAGE. Proteins were then transferred onto nitrocellulose membranes (Bio-Rad) for immunoblotting and blocked with 5% milk in tris(hydroxymethyl)aminomethane (Tris)–buffered saline and 0.5% Tween-20. Membranes were then incubated overnight at 4°C with primary antibodies. Membranes were washed in Tris-buffered saline and 0.5% Tween-20, incubated with IR-Dye–conjugated secondary antibodies, and analyzed using the Odyssey Infrared Imaging System (LI-COR Biosciences). Quantification was carried out with LI-COR Image Studio software. Of note, a second, less intense band that runs just above the oxidized ALLC dimer was observed in nonreducing gels prepared from ALMC-2 conditioned media. This band was caused by gel compression afforded by the high concentration of albumin in the media and was excluded from quantitation, as much as possible.
To measure intracellular ALLC aggregation, ALMC-2 cells were lysed in RIPA buffer as described. After centrifugation at 20 000g for 10 minutes, we collected the soluble supernatant fraction. We then resuspended the pellet in RIPA plus 2% SDS and DTT and sonicated to release aggregate protein. Supernatant and resolubilized pellets were then separated by SDS-PAGE and probed by immunoblotting as described.
ALLC immunopurification
ALLC was co-purified with ER proteostasis factors using an identical strategy to that previously reported.38 Briefly, ALMC-2 cells were treated for 30 minutes at room temperature with phosphate-buffered saline containing the reversible crosslinker dithiobis succinimidiyl propionate (500 µM; Thermo Fisher Scientific). This crosslinking reaction was quenched by addition of 100 mM of Tris (pH 7.5) for 15 minutes. Lysates were then prepared in RIPA buffer as described. Total protein concentration in cellular lysates was normalized using the Bio-Rad protein assay. Cell lysates were then subjected to preclearing with Sepharose 4B beads (Sigma-Aldrich) at 4°C for 1 hour with agitation. Lysates were then incubated overnight at 4°C with Protein A beads coupled to λ LC antibody (prepared as described in the supplemental data). After 4 washes in RIPA buffer, crosslinks were cleaved and proteins were eluted by boiling in Laemmli buffer with 100 mM of DTT. Eluates were then separated by SDS-PAGE and immunoblotted as described. Quantifications of proteins copurified with ALLC were normalized to the recovered ALLC as described previously.37
Statistical methods
Replicates for each experiment comprised cells in separate wells/dishes that were treated independently. The number of replicates and independent experiments for each figure panel are clearly stated in the figure legends. Unless otherwise noted, all P values were calculated by performing 2-tailed paired, or 2-tailed unpaired, Student t tests.
Additional methods are included in the supplemental Data.
Results
Compound 147 reduces ALLC secretion from AL patient plasma cell lines
We employed ELISA and immunoblotting to monitor the secretion of the amyloidogenic λ6a LC ALLC from ALMC-1 and ALMC-2 cells, 2 AL patient–derived plasma cell lines established from the same patient before peripheral blood stem cell transplantation and after relapse of disease, respectively.45 We found that compound 147 reduced ALLC secretion from ALMC-2 plasma cells by 50% (Figure 1A; supplemental Figure 1A). However, treatment with 147 did not significantly influence the viability of ALMC-2 cells, indicating that this reduced secretion could not be attributed to cell death (supplemental Figure 1A). Similar results were observed in ALMC-1 cells (supplemental Figure 1B). However, 147 did not significantly reduce IgG secretion or cell viability in multiple myeloma–derived KAS-6/1 cells (supplemental Figure 1C). This supports previous findings showing that 147 reduces secretion of destabilized amyloidogenic LCs.38
Figure 1.
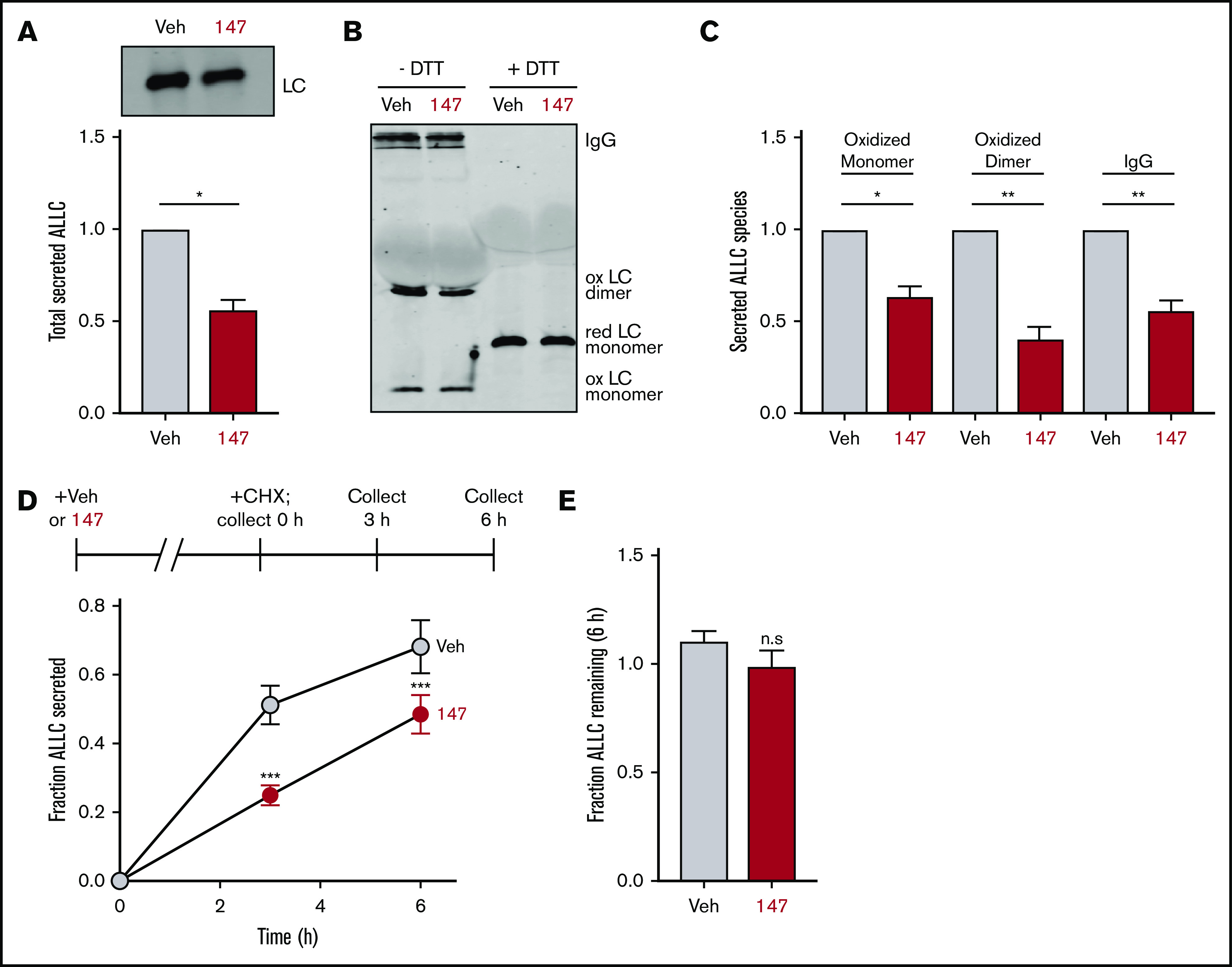
Compound 147 reduces secretion of ALLC from AL patient–derived ALMC-2 cells. (A) Representative immunoblot and quantification of ALLC in conditioned media prepared on ALMC-2 cells treated for 18 hours with vehicle (Veh) or 147 (10 µM). Error bars show standard error of the mean (SEM) for 4 independent experiments. (B) Representative nonreducing (−DTT) and reducing (+DTT) immunoblots of conditioned media prepared on ALMC-2 cells treated for 18 hours with Veh or 147 (10 µM). Fully assembled IgGs, oxidized LC dimers, oxidized LC monomers, and reduced LC monomers are indicated. (C) Quantification of nonreducing immunoblots as shown in panel B showing the relative recovery of oxidized LC monomers, oxidized LC dimers, and fully assembled IgGs. Error bars show SEM for 3 or 4 independent experiments. (D) Graph showing the fraction of ALLC secreted from ALMC-2 cells treated with Veh or 147 (10 µM) for 18 hours and then treated with cycloheximide (CHX; 50 µg/mL) at 0, 3, or 6 hours. ALLC in conditioned media and lysates were measured by ELISA. The experimental protocol is shown above. Fraction secreted was calculated as follows: fraction secreted = ALLC in media at t = 3 or 6 hours/ALLC in the lysate at t = 0 hours. Error bars show SEM for 5 replicates. (E) Graph showing the fraction of ALLC remaining from ALMC-2 cells treated for 18 hours with Veh or 147 (10 µM) and then treated with CHX (50 µg/mL), as in panel D. ALLC in conditioned media and lysates were measured by ELISA. Fraction of ALLC remaining was calculated as follows: fraction of ALLC remaining = (ALLC in media t = 6 hours + ALLC in lysate at t = 6 hours)/ALLC in lysate at t = 0 hours. Error bars show SEM for 5 replicates. *P < .05, **P < .01 (paired Student t test); ***P < .005 vs Veh (unpaired Student t test). n.s., not significant.
Amyloidogenic λ LCs are secreted in multiple conformations, including monomers, disulfide-bound dimers, and fully assembled IgGs.46 However, monomers and dimers are the predominant species associated with toxic LC aggregation and AL amyloid pathology.10,47-50 Using nonreducing SDS-PAGE, we found that ALLC dimers were generally secreted at higher levels than monomers (Figure 1B), although the relative populations of these species varied across experiments. Interestingly, we found that 147 reduced secretion of both ALLC monomers and dimers from ALMC-2 cells (Figure 1B-C). This demonstrates that this compound decreases extracellular populations of the species most associated with toxic LC aggregation. Using a cycloheximide chase assay, we found that 147 reduced the fraction of ALLC secreted by 30% (Figure 1D). However, this reduction in ALLC secretion did not correspond to a significant reduction in total ALLC over the 6-hour time course of this experiment, indicating that 147 does not significantly increase ALLC degradation (Figure 1E). This result is consistent with previous results demonstrating that both stress-independent ATF6 activation and treatment with 147 reduce ALLC secretion through a mechanism involving its increased intracellular retention in complexes bound to ER proteostasis factors, such as BiP.37,38
147 reduces intracellular ALLC in ALMC-2 cells
Although 147 did not significantly increase ALLC degradation in our cycloheximide experiment, we observed 30% to 50% reductions in intracellular ALLC in 147-treated ALMC-2 cells by ELISA and immunoblotting (Figure 2A; supplemental Figure 2A). Cotreatment of 147 with the proteasome inhibitor MG132 or the lysosome inhibitor chloroquine for 5 hours did not influence reductions in intracellular or secreted ALLC (supplemental Figure 2A-B). We also did not observe increased recovery of ALLC in cell pellets prepared from 147-treated ALMC-2 cells, indicating that 147 does not promote intracellular ALLC aggregation (supplemental Figure 2C). Furthermore, compound 147 did not alter ALLC messenger RNA levels (supplemental Figure 2D). However, [35S] metabolic labeling showed a 20% reduction in both newly synthesized ALLC and total newly synthesized protein in 147-treated ALMC-2 cells (Figure 2B-D). This indicates that the reduced intracellular levels of ALLC can in part be attributed to a decrease in translation. Previous work has shown that 147 does not reduce translation in other cells, including liver-derived HepG2 cells,38 indicating that this reduction in synthesis is not observed in all cells. Cotreatment with ISRIB, a compound that blocks translational attenuation induced by the PERK arm of the UPR,51,52 did not affect the reduction of secreted or intracellular ALLC in 147-treated ALMC-2 cells (Figure 2E-F). This suggests that the reductions in intracellular ALLC are independent of PERK signaling. Collectively, these results show that 147 reduces intracellular ALLC through a mechanism involving reduced translation and that this reduction in synthesis contributes to the reduced ALLC secreted from 147-treated ALMC-2 cells.
Figure 2.
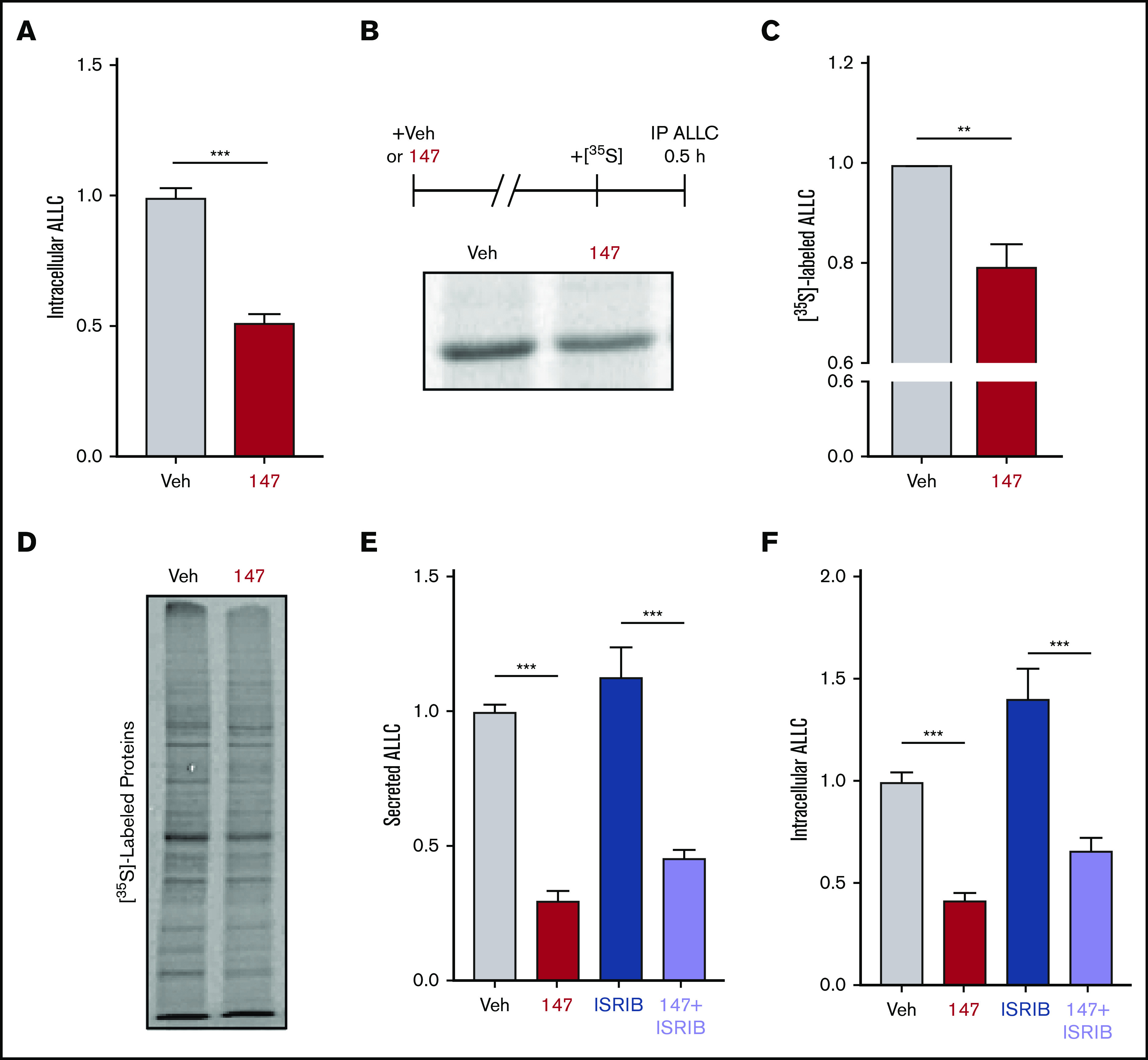
Compound 147 reduces lysate levels of ALLC in ALMC-2 cells. (A) Graph showing normalized amounts of ALLC in lysates prepared from ALMC-2 cells incubated for 18 hours with vehicle (Veh) or 147 (10 µM). ALLC was quantified by ELISA. Error bars show standard error of the mean (SEM) for 23 replicates across 5 independent experiments. (B) Representative autoradiogram of [35S]-labeled ALLC immunopurified from ALMC-2 cells treated for 18 hours with Veh or 147 (10 µM) and then labeled for 30 minutes with [35S]. The experimental protocol is shown above. (C) ALLC quantification of autoradiograms shown in panel B normalized to Veh-treated cells. Error bars show SEM for 5 independent experiments. (D) Autoradiogram of whole-cell lysates prepared from ALMC-2 cells treated for 18 hours with Veh or 147 and then metabolically labeled with [35S] for 30 minutes. (E) Quantification of ALLC in conditioned media prepared on ALMC-2 cells treated for 18 hours with Veh, 147 (10 µM), and/or ISRIB (200 nM), as indicated. ALLC was measured by ELISA and normalized to Veh-treated cells. Error bars show SEM for 14 replicates across 3 independent experiments. (F) Quantification of ALLC in lysates prepared from ALMC-2 cells treated for 18 hours with Veh, 147 (10 µM), and/or ISRIB (200 nM), as indicated. ALLC was measured by ELISA and normalized to Veh-treated cells. Error bars show SEM for 14 replicates across 3 independent experiments. **P < .01 (paired Student t test); ***P < .005 (unpaired Student t test).
147-dependent reductions in ALLC secretion are not dependent on ATF6 activation
ATF6 is activated through a mechanism involving the reduction of disulfide-linked ATF6 oligomers and increased trafficking of reduced ATF6 monomers to the Golgi (Figure 3A).53-56 In the Golgi, ATF6 is site-specifically processed by S1P and S2P, releasing the active ATF6 N-terminal bZIP transcription factor domain to localize to the nucleus and promote ATF6 transcriptional activity.57 The compound Ceapin-7 (CP7) stabilizes ATF6 oligomers within the ER, preventing ATF6 activation induced by ER stress or ATF6-activating compounds such as 147 (Figure 3A).39,58-60 We confirmed that cotreatment of ALMC-2 cells with CP7, but not the inactive analog CP5 (supplemental Figure 3A), blocked 147-dependent increases in the ATF6 target gene BiP (Figure 3B). However, cotreatment with CP7 did not block 147-dependent reductions in ALLC secretion from ALMC-2 cells (Figure 3C) or 147-dependent reductions in intracellular ALLC (Figure 3D). Similarly, cotreatment with the S1P inhibitor PF429242, a compound that blocks ATF6 activation through a mechanism distinct from CP7 (Figure 3A),61,62 also did not influence 147-dependent reductions in ALLC secretion from ALMC-2 cells (Figure 3E). Similar results were observed in ALMC-1 cells (supplemental Figure 3B). These results demonstrate that 147 reduces ALLC secretion from ALMC-2 cells through a mechanism independent of ATF6 activation.
Figure 3.
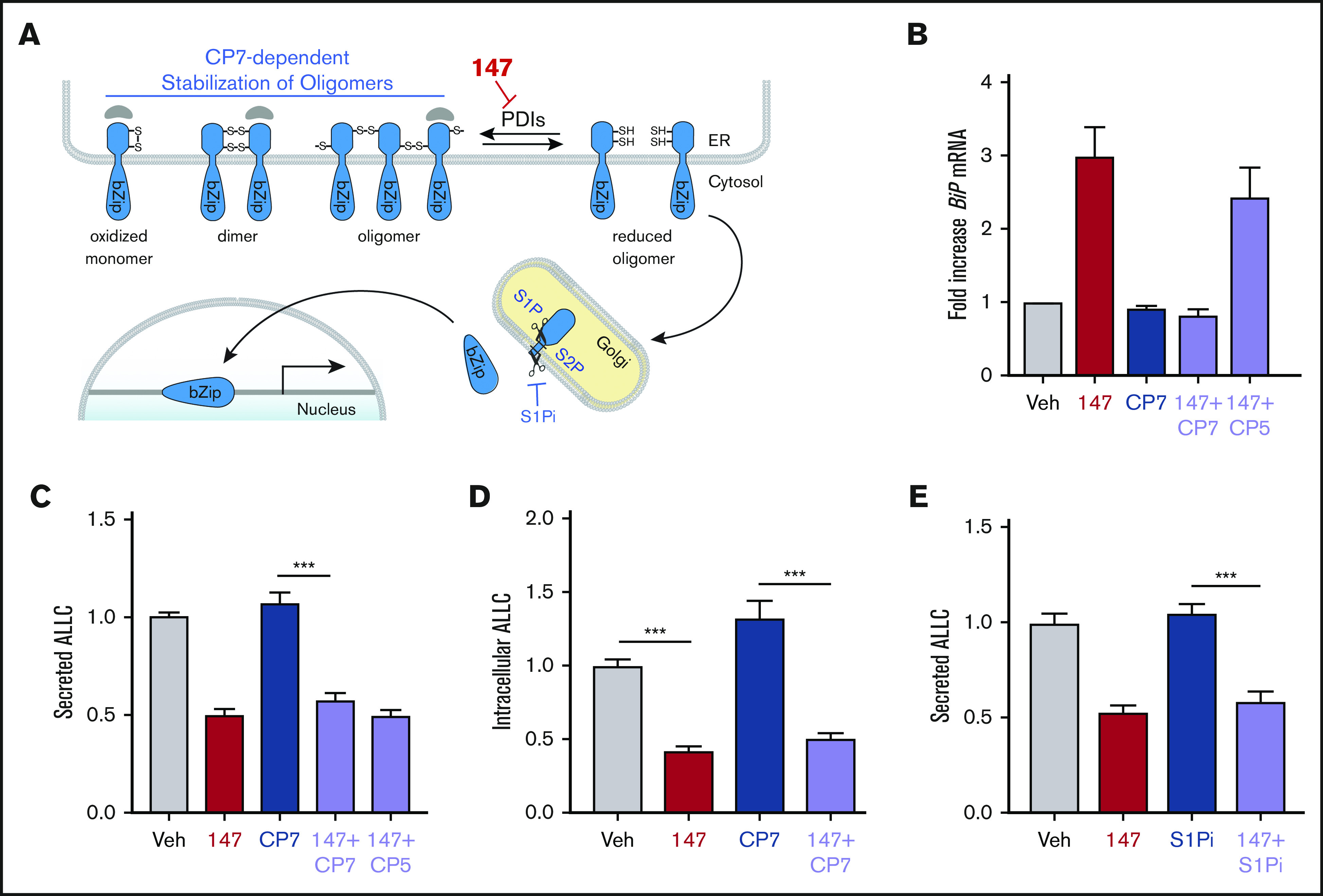
147-dependent reductions in ALLC secretion are independent of ATF6 activation. (A) Illustration showing the mechanism of 147-dependent ATF6 activation. ATF6 is retained within the ER as oxidized monomers, disulfide-bonded dimers, or disulfide-bonded oligomers that are maintained by PDIs. Compound 147 covalently modifies a subset of PDIs, increasing the population of reduced ATF6 monomers that can traffic to the Golgi and undergo proteolytic processing by S1P and S2P. This releases the N-terminal bZIP domain of ATF6 to localize to the nucleus and promote ATF6 transcriptional signaling. The specific steps of this activation mechanism inhibited by CP7 or site 1 protease (S1P) inhibitor (S1Pi) are shown. (B) Graph showing BiP expression measured by quantitative polymerase chain reaction in ALMC-2 cells treated for 5 hours with vehicle (Veh) or 147 (10 µM), CP7 (10 µM), or CP5 (10 µM), as indicated. Error bars show 95% confidence interval for 3 replicates. (C) Bar graphs showing normalized amounts of ALLC in conditioned media prepared from ALMC-2 cells treated for 18 hours with Veh or 147 (10 µM), CP7 (10 µM), or CP5 (10 µM), as indicated. ALLC was quantified by ELISA. Error bars show standard error of the mean (SEM) for 21 replicates across 4 independent experiments. (D) Bar graphs showing normalized amounts of ALLC in lysates prepared from ALMC-2 cells treated for 18 hours with Veh, 147 (10 µM), and/or CP7 (10 µM), as indicated. ALLC was quantified by ELISA. Error bars show SEM for 10 replicates across 2 independent experiments. (E) Bar graphs showing normalized amounts of ALLC in conditioned media from ALMC-2 cells treated for 18 hours with Veh or 147 (10 µM) and/or S1Pi PF429242 (10 µM), as indicated. ALLC was quantified by ELISA. Error bars show SEM for 8 replicates across 2 independent experiments. ***P < .005 (unpaired Student t test). mRNA, messenger RNA.
147 reduces ALLC secretion through an on-target mechanism involving metabolic activation and covalent protein modification
Compound 147 activates ATF6 through a mechanism involving metabolic oxidation to a quinone methide and subsequent covalent modification of ER-localized proteins, including multiple PDIs (Figure 4A).39 This activation mechanism requires the 2-amino-p-cresol moiety of the 147 A ring (Figure 4A). To define a structure-activity relationship for 147-dependent reductions in ALLC secretion, we screened 147 analogs to define their ability to reduce ALLC secretion from ALMC-2 cells.39 Compounds containing alterations to the 147 2-amino-p-cresol A ring did not reduce ALLC secretion, indicating that this ring is required for compound activity in this assay (Figure 4B). Similarly, analogs that alter the 147 linker did not substantially reduce ALLC secretion (Figure 4A; supplemental Figure 4A). However, analogs containing alterations to the 147 B ring showed varying levels of efficacy in reducing ALLC secretion (Figure 4A; supplemental Figure 4B). This structure-activity relationship is identical to that previously established for 147-dependent ATF6 activation.39 Consistent with this, we observed an inverse correlation between analog-dependent activation of an ATF6 reporter in HEK293 cells39 and reductions in ALLC secretion from ALMC-2 cells (Figure 4C). This suggests that 147 reduces ALLC secretion from ALMC-2 cells through the same mechanism required for ATF6 activation (Figure 4A).
Figure 4.
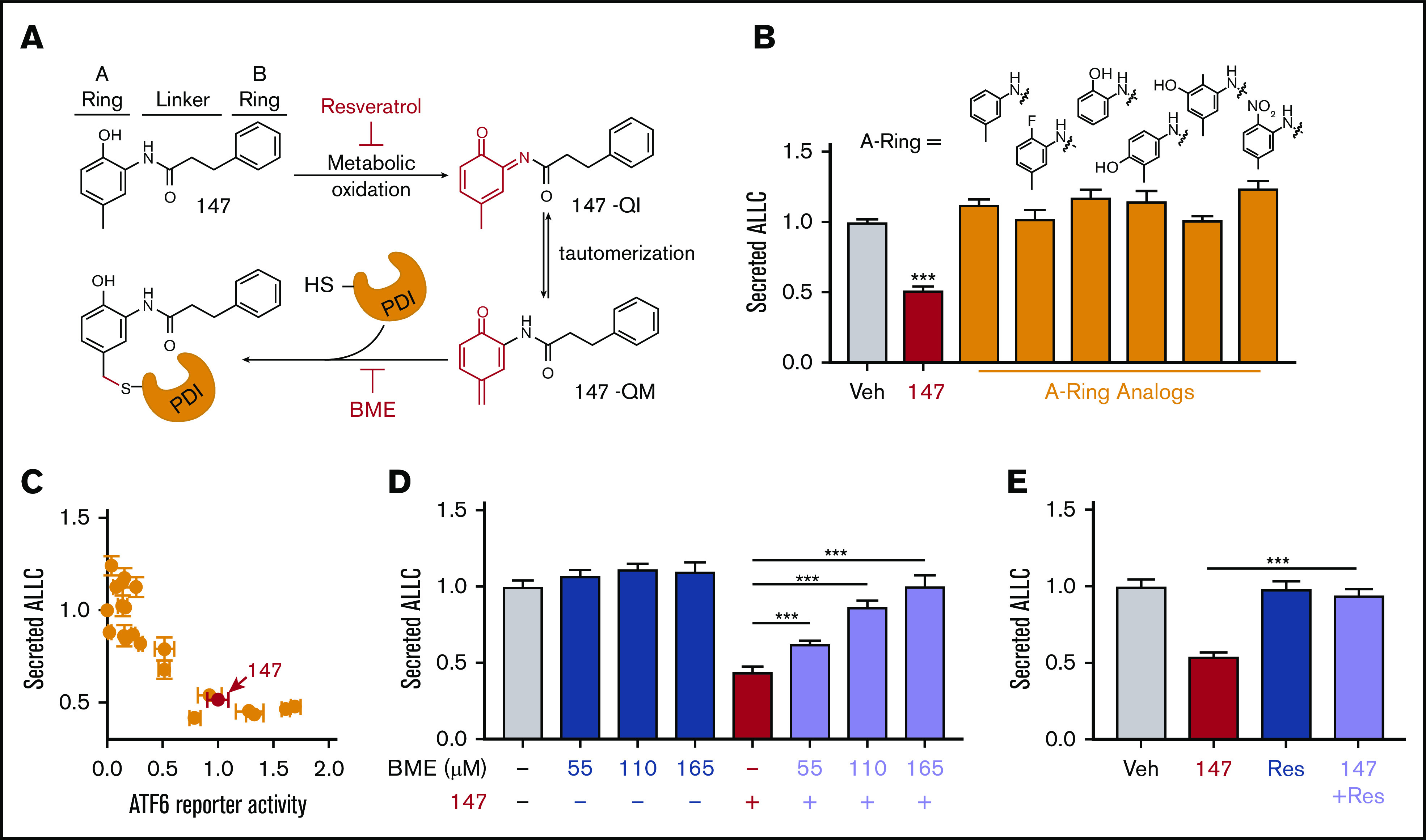
Compound 147 reduces ALLC secretion through a mechanism involving compound metabolic activation and covalent protein modification. (A) Illustration showing the metabolic activation of 147 to a quinone imide (147-QI) and quinone methide (147-QM). These reactive electrophiles covalently modify ER proteins, including multiple PDIs. The A ring, linker, and B ring of 147 are indicated. Specific steps inhibited by resveratrol (Res) and β- mercaptoethanol (BME) are also indicated. Adapted from Paxman et al.39 (B) Graph showing normalized amounts of ALLC in conditioned media from ALMC-2 cells treated for 18 hours with vehicle (Veh), 147 (10 µM), or the indicated 147 A ring analog (10 µM). ALLC was quantified by ELISA. Error bars show standard error of the mean (SEM) for 10 replicates across 2 independent experiments. (C) Plot comparing the normalized activation of an ATF6-selective luciferase reporter in HEK293 cells39 with reductions in ALLC secretion from ALMC-2 cells for 147 (10 µM) or 147 analogs containing alterations to the A ring, linker, and B ring. Error bars for the ATF6 reporter activation show SEM for 3 independent experiments. Error bars for ALLC secretion show 10 replicates across 2 independent experiments. (D) Graph showing normalized amounts of ALLC secretion from ALMC-2 cells treated for 18 hours with Veh or 147 (10 µM) and/or BME, as indicated. ALLC was quantified by ELISA. Error bars show SEM for 10 replicates across 2 independent experiments. (E) Graph showing normalized ALLC secretion from ALMC-2 cells treated for 18 hours with Veh, 147 (10 µM), and/or Res (10 µM), as indicated. ALLC was quantified by ELISA. Error bars show SEM for 9 replicates across 2 independent experiments. ***P < .005 vs Veh (unpaired Student t test; B);*** P < .005 (paired Student t test; D-E).
ATF6 activation by 147 can be inhibited by coadministration of resveratrol or BME, which block different steps of the compound activation mechanism (Figure 4A).39 Cotreatment with BME or resveratrol blocked 147-dependent reductions in ALLC secretion (Figure 4D-E). Neither BME nor resveratrol affected ALMC-2 cell viability in the absence or presence of 147 (supplemental Figure 4C-D). Similar results were observed for ALMC-2 cells treated with other active analogs of 147 (supplemental Figure 4E). Cotreatment with resveratrol or BME also inhibited 147-dependent reductions in ALLC secreted from ALMC-1 cells (supplemental Figure 4F). Interestingly, 147-dependent reductions in intracellular ALLC in ALMC-2 cells were also blocked by cotreatment with resveratrol or BME (supplemental Figure 4G). Furthermore, resveratrol inhibited translation attenuation in 147-treated ALMC-2 cells (supplemental Figure 4H). These results further demonstrate that 147 reduces ALLC secretion from ALMC-2 cells through the same mechanism required for ATF6 activation (Figure 4A).
Compound 147 alters interactions between ALLC and ER PDIs
We previously identified proteins modified by 147 using an alkyne-containing 147 analog (147-alkyne; supplemental Figure 5A-B).39 147-alkyne reduced ALLC secretion from ALMC-2 cells to levels identical to that observed for 147, indicating that these compounds have similar activities (supplemental Figure 5C). Three predominant proteins modified by 147-alkyne in ALMC-2 cells were PDIA1, PDIA4, and PDIA6 (supplemental Figure 5B).39 Importantly, addition of excess parent compound 147 competed for the labeling of these PDIs by 147-alkyne, confirming on-target activity (supplemental Figure 5D).39 However, we did not observe significant labeling of ALLC in ALMC-2 cells (supplemental Figure 5B,D).39 This suggests that 147 influences ALLC secretion from ALMC-2 cells by targeting the activity of ER proteostasis factors such as PDIs rather than by directly targeting ALLC. Consistent with this, inhibition of PDIA1 was previously shown to reduce ALLC secretion from HEK293T cells,63 and ATF6-dependent reductions in ALLC secretion from HEK293 cells correlate with increased interactions with PDIA4.37 Therefore, we defined how 147 influences interactions between ALLC and these 2 PDIs. Interestingly, 147 reduced ALLC interactions with PDIA1 by 25% while increasing interactions with PDIA4 (Figure 5A-B; supplemental Figure 5E). Both of these changes corresponded with the alterations in ALLC interactions previously implicated in reduced ALLC secretion.37,63 This suggests that 147 reduces ALLC secretion through an ATF6-independent mechanism involving altered interactions with these PDIs.
Figure 5.
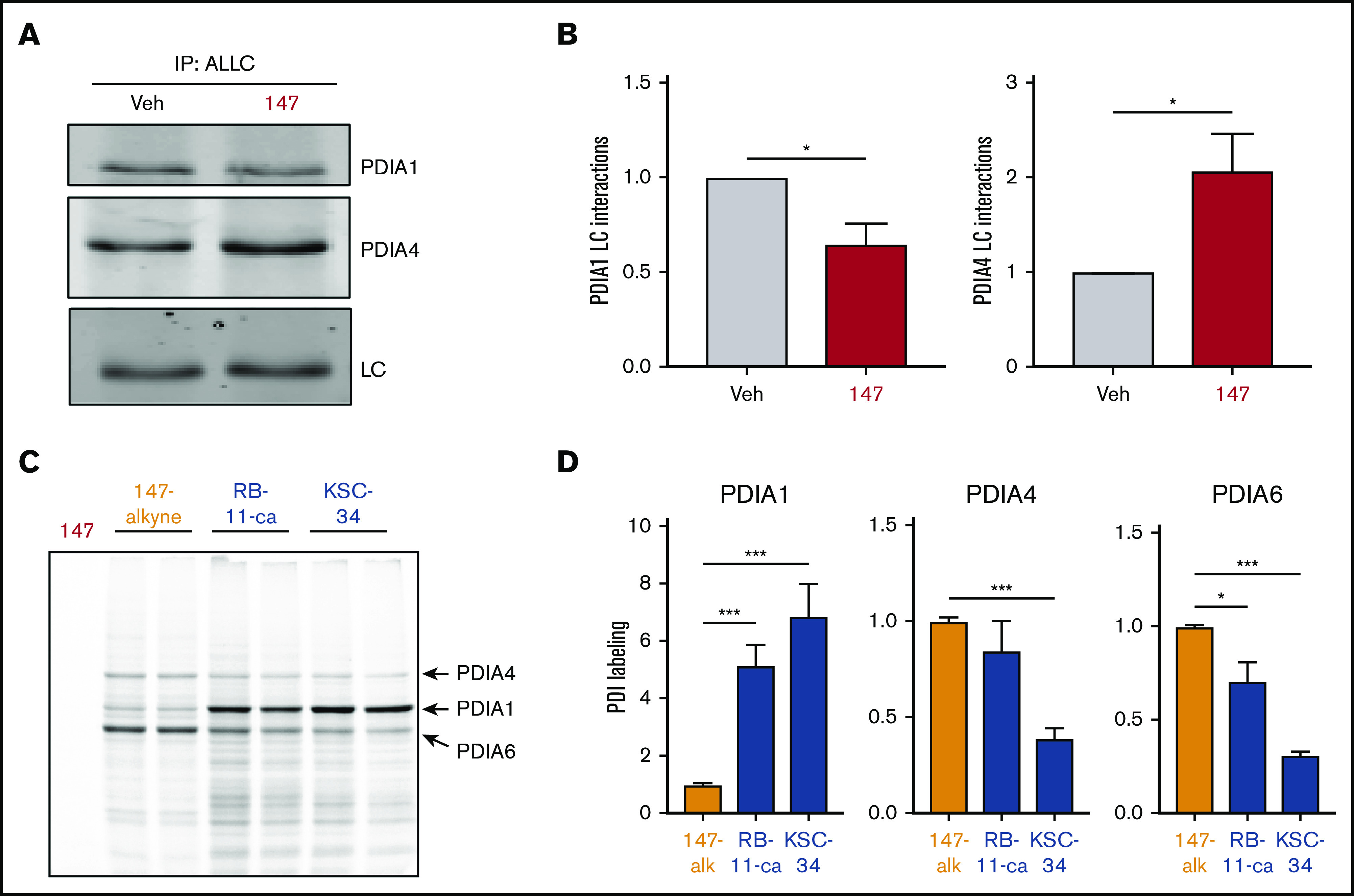
PDI interactions with ALLC are altered by 147 treatment. (A) Representative immunoblot of PDIA1 and PDIA4 in ALLC immunopurifications from ALMC-2 cells treated for 5 hours with vehicle (Veh) or 147 (10 µM). Inputs from this experiment are shown in supplemental Figure 5E. (B) Quantification of PDIA1 and PDIA4 from immunoblots as shown in panel A. The recovery of each PDI was normalized to the recovery of ALLC under each condition, allowing accurate evaluation of the interaction between these 2 proteins. Error bars show standard error of the mean (SEM) for >5 independent experiments. (C) Representative fluorescence gel showing the covalent modification of proteins in ALMC-2 cells treated for 18 hours with 147-alkyne (10 µM), RB-11-ca (30 µM), or KSC-34 (30 µM). Click chemistry was used to incorporate a TAMRA fluorophore onto the alkyne contained in each of these molecules. Cells treated with 147 are shown as control. PDI bands were assigned based on previous mass spectrometric analysis of SDS-PAGE bands excised from identical gels of 147-alkyne–treated ALMC-2 cell lysates.39 (D) Normalized quantification of PDIA1, PDIA4, and PDIA6 labeling from gels as shown in panel C. Error bars show SEM for 4 replicates across 2 independent experiments. *P < .05, **P < .01, ***P < .005 (paired Student t test).
Depletion of PDIs (including PDIA1 and PDIA4) by RNA interference induces ER stress and UPR activation,39 challenging our ability to define the specific contributions of individual PDIs in the 147-dependent reduction in ALLC secretion from ALMC-2 cells. Therefore, to define how pharmacologic targeting of PDIs influences ALLC secretion, we used the structurally distinct PDI inhibitors RB-11-ca and KSC-34 (supplemental Figure 5F).63,64 These compounds contain an alkyne moiety that allowed us to rapidly define their modification of different PDIs using an approach identical to that employed for 147-alkyne (supplemental Figure 5B). RB-11-ca and KSC-34 showed increased background labeling relative to 147-alkyne; however, both compounds labeled bands that corresponded to the 3 predominant PDIs modified by 147 (ie, PDIA1, PDIA4, and PDIA6), although to different extents (Figure 5C-D). RB-11-ca and KSC-34 modified PDIA1 to higher levels than 147-alkyne, consistent with their development as PDIA1 inhibitors.63,64 Alternatively, RB-11-ca labeled PDIA4 and PDIA6 to levels 70% to 85% of that observed for 147-alkyne, whereas KSC-34 showed lower levels of labeling for these PDIs. This is consistent with the increased selectivity of KSC-34 for PDIA1 relative to RB-11-ca.63 Collectively, these results show that KSC-34 and, more specifically, RB-11-ca covalently modify PDIs similar to those modified by 147-alkyne, providing a complementary approach to defining how covalent targeting of PDIs affects ALLC secretion from ALMC-2 cells.
Pharmacologic PDI inhibition reduces ALLC secretion from ALMC-2 cells
Both RB-11-ca and KSC-34 reduced ALLC secretion from ALMC-2 cells, as measured by ELISA (Figure 6A). RB-11-ca reduced ALLC secretion by 35%, whereas KSC-34 reduced ALLC secretion by 25%. KSC-34 similarly reduced secretion of ALLC from ALMC-1 cells (supplemental Figure 6A). Importantly, this reduction did not correspond to reductions in ALMC-1 or ALMC-2 cell viability (supplemental Figure 6A-B). Similar reductions in ALLC secretion from ALMC-2 cells were observed for RB-11-ca by immunoblotting (supplemental Figure 6C). As with 147, RB-11-ca reduced secretion of ALLC dimers and monomers, the conformations most commonly linked to AL pathogenesis (Figure 6B-C). This reduction in ALLC secretion also corresponded to reductions in intracellular ALLC (Figure 6D) and reductions in protein translation (supplemental Figure 6D), mimicking the results observed with 147. Furthermore, RB-11-ca reduced ALLC interactions with PDIA1 and increased interactions with PDIA4 (Figure 6E-F). Again, these changes were analogous to those observed with 147 (Figure 5A-B). However, the reduction in ALLC interactions with PDIA1 was greater than that observed for 147, likely reflecting the increased labeling of PDIA1 by RB-11-ca (Figure 5C-D). These results indicate that pharmacologic targeting of PDIs with other PDI inhibitors reduces ALLC secretion from ALMC-2 cells through a mechanism analogous to that observed for 147. This supports a model whereby 147 reduces ALLC secretion through an on-target ATF6-independent mechanism involving covalent modification of multiple PDIs.
Figure 6.
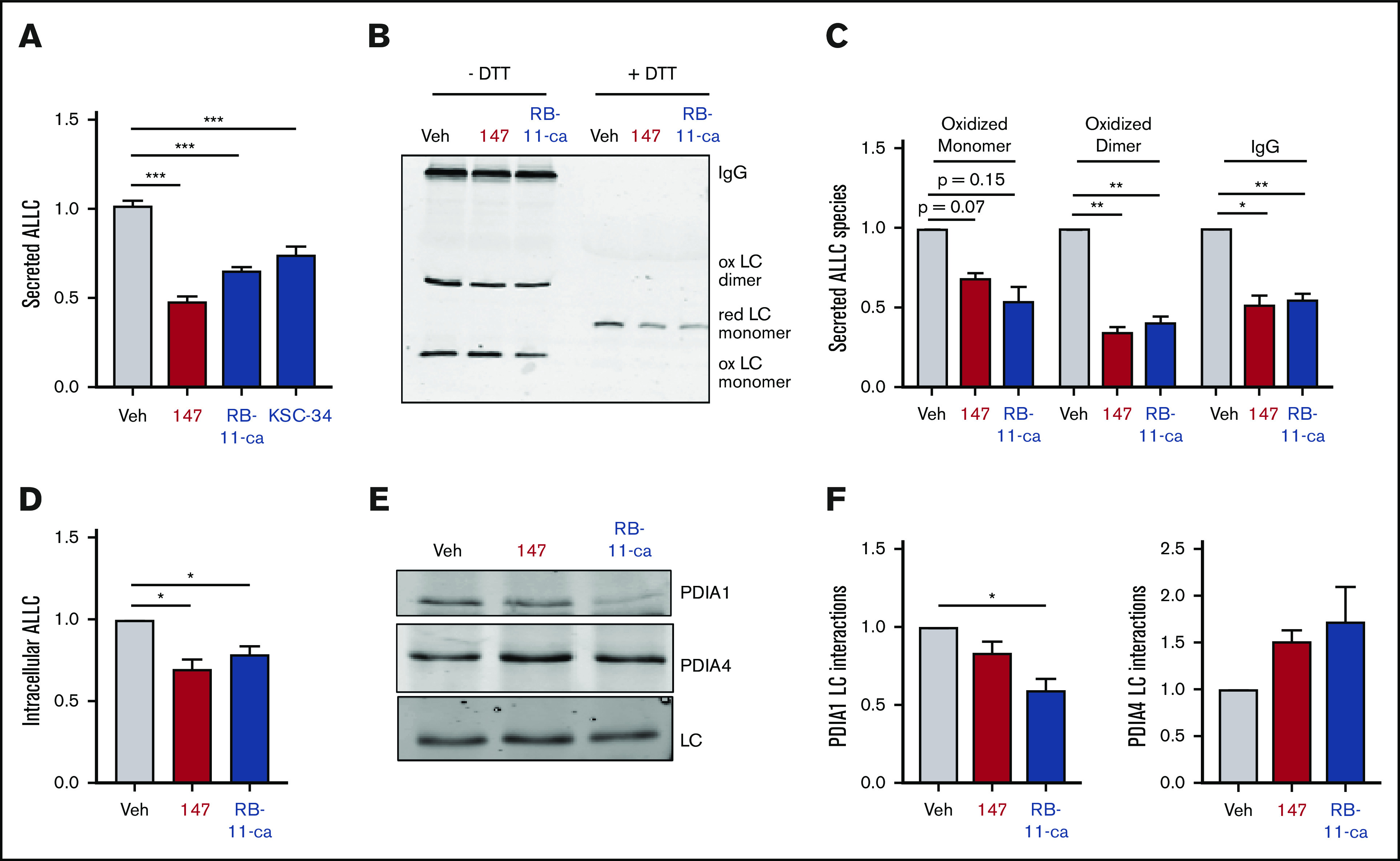
Pharmacologic targeting of PDIs reduces ALLC secretion from ALMC-2 cells. (A) Graph showing normalized ALLC secretion from ALMC-2 cells treated for 18 hours with vehicle, 147 (10 µM), RB-11-ca (30 µM), or KSC-34 (30 µM). ALLC was quantified by ELISA. Error bars show standard error of the mean (SEM) for >9 replicates across >2 independent experiments. (B) Representative nonreducing (−DTT) and reducing (+DTT) immunoblots of conditioned media prepared from ALMC-2 cells treated for 18 hours with vehicle, 147 (10 µM), or RB-11-ca (30 µM). Fully assembled IgGs, oxidized LC dimers, oxidized LC monomers, and reduced LC monomers are indicated. (C) Graph showing normalized quantification of nonreducing immunoblots as shown in panel B for oxidized LC monomers, oxidized LC dimers, and fully assembled IgGs. Error bars show SEM for >2 independent experiments. (D) Graph showing normalized quantification of ALLC in lysates prepared from ALMC-2 cells treated for 18 hours with vehicle, 147 (10 µM), or RB-11-ca (30 µM). ALLC was measured by immunoblotting. Error bars show SEM for 4 independent experiments. (E) Representative immunoblot of ALLC immunopurified from ALMC-2 cells treated for 5 hours with vehicle, 147 (10 µM), or RB-11-ca (30 µM). (F) Normalized quantification of PDIA1 and PDIA4 from immunoblots as shown in panel E. The recovery of each PDI was normalized to the recovery of ALLC under each condition, allowing accurate evaluation of the interaction between these 2 proteins. Error bars show SEM for >2 independent experiments. *P < .05, **P < .01 (paired Student t test); ***P < .005 (unpaired Student t test).
147-dependent reductions in ALLC secretion are compatible with bortezomib-induced ablation of AL plasma cells
AL is currently treated by using chemotherapeutics such as the proteasome inhibitor bortezomib to ablate the underlying plasma cell malignancy.17,18,21,65 Bortezomib induces toxicity in plasma cells through multiple mechanisms, including the inhibition of ER-associated degradation and increased ER stress.66-68 Compound 147 promotes adaptive ER proteostasis remodeling in plasma cells through both covalent modification of ER proteostasis factors (eg, PDIs) and ATF6 activation, suggesting that treatment with this compound could reduce bortezomib-induced toxicity in AL-associated plasma cells. To test this, we evaluated the viability of ALMC-2 cells cotreated with bortezomib and 147. Bortezomib induced toxicity in ALMC-2 cells with a 50% effective concentration of 45 nM (supplemental Figure 7A-B). Although 147 treatment showed a modest ∼15% reduction in ALMC-2 viability (as reported previously38), this compound did not significantly influence bortezomib-induced toxicity in these cells (supplemental Figure 7A-B). Furthermore, 147 did not increase or decrease activation of the proapoptotic caspase 3 or 7 in ALMC-2 cells cotreated with subtoxic or toxic doses of bortezomib, respectively (supplemental Figure 7C). Similarly, 147 did not influence bortezomib-induced toxicity in ALMC-1 or KAS-6/1 plasma cells (supplemental Figure 7D-E). These results indicate that 147 does not interfere with bortezomib-induced plasma cell cytotoxicity and that these 2 compounds have the potential to be used in combination to mitigate the amyloid pathology and plasma cell malignancy associated with AL pathogenesis.
Discussion
Currently, no therapeutic strategies are established that directly reduce the LC proteotoxicity limiting the treatment of AL patients in the clinic. Our results show that pharmacologic targeting of ER proteostasis using compound 147 would allow us to reduce the plasma cell secretion of amyloidogenic LCs implicated in AL. As demonstrated previously,36,38 this reduction in amyloidogenic LC secretion reduces the secreted LC populations available for proteolysis and/or concentration-dependent aggregation into toxic oligomers and amyloid fibrils.28,29 Previous results have shown that reductions in amyloidogenic LC serum concentration represent a critical determinant for dictating patient survival in AL.24-26 Therefore, the ability for compounds such as 147 to enhance reductions in serum LCs in patients treated with chemotherapeutics provides opportunities to further mitigate LC-associated proteotoxicity in AL and improve patient outcomes.
Although developed as an ATF6-activating compound, we show that 147 reduces amyloidogenic ALLC secretion through an on-target, ATF6-independent mechanism that involves metabolic activation of 147 and covalent protein modification. Although 147-dependent ATF6 activation has been shown to protect different tissues, including the liver and heart, against toxic insults,42,44 these results establish a new molecular mechanism by which this compound can also reduce secretion and toxic aggregation of amyloidogenic proteins through pharmacologic targeting of ER proteostasis regulators, such as PDIs. PDIs primarily function by regulating disulfide bonds within secretory proteins, although many PDIs also have chaperoning activity independent of their redox function.40,41 We show that 147 induces alterations in the interactions between ALLC and both PDIA1 and PDIA4. PDIA1 facilitates LC folding in the early secretory pathway,69,70 indicating that reductions in PDIA1 interactions will decrease the proper folding of ALLC into trafficking-competent conformations. Conversely, PDIA4 is implicated in the ER retention of misfolded proteins, including ALLC.37,71,72 Thus, the increased interactions between PDIA4 and ALLC induced by 147 also likely contribute to the preferential reduction of plasma cell secretion for this destabilized amyloidogenic protein. We further highlight the importance of PDIs in plasma cell LC secretion by showing that structurally distinct PDI inhibitors also reduce ALLC secretion from ALMC-2 plasma cells. Interestingly, PDI inhibitors such as KSC-34 and RB-11-ca reduced ALLC secretion from ALMC-2 cells through a mechanism analogous to that observed for 147, involving altered interactions with PDIA1 and PDIA4, reductions in intracellular ALLC, and increased translational attenuation. Although the mechanistic basis for this translational attenuation remains to be defined, our results showing 2 distinct PDI inhibitors induce similar effects suggest that this reduced translation likely results from PDI targeting. Collectively, these results further define the potential for pharmacologically targeting ER proteostasis factors such as PDIs to reduce the plasma cell secretion and subsequent toxic aggregation of amyloidogenic LCs.
Despite similarities, 147 has advantages over other available PDI inhibitors that make it particularly suitable for translational development. Compound 147 is metabolically activated to its reactive form on the ER membrane, allowing it to preferentially modify ER proteins (eg, PDIs) and minimize off-target compound activity.39 Furthermore, 147 shows reduced PDIA1 modification relative to other compounds. Because PDIA1 is important for the folding and trafficking of many other secretory proteins (eg, insulin73), this reduced PDIA1 modification will minimize potential consequences associated with strong PDIA1 inhibition. Consistent with this, the administration of 147 to mice has not been associated with any toxicity, but instead, it protects multiple tissues from diverse types of pathologic stress.42 These unique properties of 147 make it a particularly attractive compound for pharmacologically targeting ER proteostasis in diseases like AL.
Finally, we confirm that 147 does not influence bortezomib-induced toxicity in ALMC-2 cells, indicating that these 2 compounds could potentially be used in combination for the treatment of AL. This combination strategy provides the opportunity to both mitigate LC-associated proteotoxicity and ablate the underlying diseased plasma cells in patients with severe LC-associated proteotoxicity. Ultimately, our results establish the pharmacologic targeting of ER proteostasis as a potential strategy to reduce proteotoxicity in AL and reveal insights into compound-dependent reductions in amyloidogenic LC secretion that will enable the development of next-generation compounds with improved translational potential for this disease.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Evan Powers and Jessica Rosarda for critical reading of the manuscript; Kelly Chen (Scripps Research Institute), Elena Klimtchuk, and Lawreen Connors (Boston University) for experimental support; Diane Jelinek (Mayo Clinic) for providing the ALMC-2 and KAS-6/1 cell lines; Eranthie Weerapana (Boston College) for providing RB-11-ca and KSC-34; and Peter Walter (University of California, San Francisco) for providing CP7 and CP5.
This work was funded by National Institutes of Health grants DK107604 from the National Institute of Diabetes and Digestive and Kidney Diseases (R.L.W.) and AG046495 from the National Institute on Aging (R.L.W., J.W.K.), a Leukemia & Lymphoma Society Postdoctoral Fellowship (B.R.), and an American Heart Association Predoctoral Fellowship (I.C.R.).
Footnotes
For data sharing requests, e-mail the corresponding author, R. Luke Wiseman (wiseman@scripps.edu).
Authorship
Contribution: B.R., J.S.M., I.C.R., and R.L.W. conceptualized the experiments and performed formal analysis of the primary data. B.R. and J.S.M. performed the experiments; R.J.P. and J.W.K. provided resources and developed methodology used in this manuscript; B.R. and R.L.W. wrote the manuscript; R.L.W. was responsible for supervision, administration, and funding acquisition for the work described in this manuscript; and all authors were involved in the review and editing of the manuscript.
Conflict-of-interest disclosure: J.W.K. is a board member and shareholder of Proteostasis Therapeutics, Inc, Protego Biopharma, and Yumanity. R.L.W. is a board member of Protego Biopharma. R.J.P., J.W.K., and R.L.W. are included as inventors on a patent (WO20171174301A1) describing the use of ER proteostasis regulators including 147 to treat protein misfolding diseases. The remaining authors declare no competing financial interests.
Correspondence: R. Luke Wiseman, Department of Molecular Medicine, Scripps Research Institute, La Jolla, CA 92037; e-mail: wiseman@scripps.edu.
References
- 1.Merlini G, Dispenzieri A, Sanchorawala V, et al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Primers. 2018;4(1):38. [DOI] [PubMed] [Google Scholar]
- 2.Badar T, D’Souza A, Hari P. Recent advances in understanding and treating immunoglobulin light chain amyloidosis. F1000Res. 2018;7:F1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Merlini G, Comenzo RL, Seldin DC, Wechalekar A, Gertz MA. Immunoglobulin light chain amyloidosis. Expert Rev Hematol. 2014;7(1):143-156. [DOI] [PubMed] [Google Scholar]
- 4.Cohen AD, Comenzo RL. Systemic light-chain amyloidosis: advances in diagnosis, prognosis, and therapy. Hematology Am Soc Hematol Educ Program. 2010;2010:287-294. [DOI] [PubMed] [Google Scholar]
- 5.Blancas-Mejía LM, Ramirez-Alvarado M. Systemic amyloidoses. Annu Rev Biochem. 2013;82:745-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guan J, Mishra S, Shi J, et al. Stanniocalcin1 is a key mediator of amyloidogenic light chain induced cardiotoxicity. Basic Res Cardiol. 2013;108(5):378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imperlini E, Gnecchi M, Rognoni P, et al. Proteotoxicity in cardiac amyloidosis: amyloidogenic light chains affect the levels of intracellular proteins in human heart cells. Sci Rep. 2017;7(1):15661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diomede L, Romeo M, Rognoni P, et al. Cardiac light chain amyloidosis: the role of metal ions in oxidative stress and mitochondrial damage. Antioxid Redox Signal. 2017;27(9):567-582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi J, Guan J, Jiang B, et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway. Proc Natl Acad Sci USA. 2010;107(9):4188-4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brenner DA, Jain M, Pimentel DR, et al. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res. 2004;94(8):1008-1010. [DOI] [PubMed] [Google Scholar]
- 11.Blancas-Mejia LM, Misra P, Dick CJ, et al. Immunoglobulin light chain amyloid aggregation. Chem Commun (Camb). 2018;54(76):10664-10674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guan J, Mishra S, Qiu Y, et al. Lysosomal dysfunction and impaired autophagy underlie the pathogenesis of amyloidogenic light chain-mediated cardiotoxicity. EMBO Mol Med. 2015;7(5):688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kazman P, Vielberg MT, Pulido Cendales MD, et al. Fatal amyloid formation in a patient’s antibody light chain is caused by a single point mutation. elife. 2020;9:e52300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pinney JH, Smith CJ, Taube JB, et al. Systemic amyloidosis in England: an epidemiological study. Br J Haematol. 2013;161(4):525-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gertz MA. Immunoglobulin light chain amyloidosis diagnosis and treatment algorithm 2018. Blood Cancer J. 2018;8(5):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milani P, Palladini G, Merlini G. New concepts in the treatment and diagnosis of amyloidosis. Expert Rev Hematol. 2018;11(2):117-127. [DOI] [PubMed] [Google Scholar]
- 17.Manwani R, Hegenbart U, Mahmood S, et al. Deferred autologous stem cell transplantation in systemic AL amyloidosis. Blood Cancer J. 2018;8(11):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kastritis E, Dialoupi I, Gavriatopoulou M, et al. Primary treatment of light-chain amyloidosis with bortezomib, lenalidomide, and dexamethasone. Blood Adv. 2019;3(20):3002-3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dember LM, Sanchorawala V, Seldin DC, et al. Effect of dose-intensive intravenous melphalan and autologous blood stem-cell transplantation on al amyloidosis-associated renal disease. Ann Intern Med. 2001;134(9 Pt 1):746-753. [DOI] [PubMed] [Google Scholar]
- 20.Meier-Ewert HK, Sanchorawala V, Berk J, et al. Regression of cardiac wall thickness following chemotherapy and stem cell transplantation for light chain (AL) amyloidosis. Amyloid. 2011;18(suppl 1):130-131. [DOI] [PubMed] [Google Scholar]
- 21.Salinaro F, Meier-Ewert HK, Miller EJ, et al. Longitudinal systolic strain, cardiac function improvement, and survival following treatment of light-chain (AL) cardiac amyloidosis. Eur Heart J Cardiovasc Imaging. 2017;18(9):1057-1064. [DOI] [PubMed] [Google Scholar]
- 22.Wechalekar AD, Schonland SO, Kastritis E, et al. A European collaborative study of treatment outcomes in 346 patients with cardiac stage III AL amyloidosis. Blood. 2013;121(17):3420-3427. [DOI] [PubMed] [Google Scholar]
- 23.Palladini G, Hegenbart U, Milani P, et al. A staging system for renal outcome and early markers of renal response to chemotherapy in AL amyloidosis. Blood. 2014;124(15):2325-2332. [DOI] [PubMed] [Google Scholar]
- 24.Gertz MA. Immunoglobulin light chain amyloidosis: 2020 update on diagnosis, prognosis, and treatment. Am J Hematol. 2020;95(7):848-860. [DOI] [PubMed] [Google Scholar]
- 25.Kumar SK, Dispenzieri A, Lacy MQ, et al. Changes in serum-free light chain rather than intact monoclonal immunoglobulin levels predicts outcome following therapy in primary amyloidosis. Am J Hematol. 2011;86(3):251-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milani P, Basset M, Nuvolone M, et al. Indicators of profound hematologic response in AL amyloidosis: complete response remains the goal of therapy. Blood Cancer J. 2020;10(8):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959-991. [DOI] [PubMed] [Google Scholar]
- 28.Plate L, Wiseman RL. Regulating secretory proteostasis through the unfolded protein response: from function to therapy. Trends Cell Biol. 2017;27(10):722-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romine IC, Wiseman RL. Starting at the beginning: endoplasmic reticulum proteostasis and systemic amyloid disease. Biochem J. 2020;477(9):1721-1732. [DOI] [PubMed] [Google Scholar]
- 30.Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71-99. [DOI] [PubMed] [Google Scholar]
- 31.Sun Z, Brodsky JL. Protein quality control in the secretory pathway. J Cell Biol. 2019;218(10):3171-3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eisele YS, Monteiro C, Fearns C, et al. Targeting protein aggregation for the treatment of degenerative diseases. Nat Rev Drug Discov. 2015;14(11):759-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kelly JW. Pharmacologic approaches for adapting proteostasis in the secretory pathway to ameliorate protein conformational diseases. Cold Spring Harb Perspect Biol. 2020;12(5):a034108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10(11):3787-3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shoulders MD, Ryno LM, Genereux JC, et al. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013;3(4):1279-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cooley CB, Ryno LM, Plate L, et al. Unfolded protein response activation reduces secretion and extracellular aggregation of amyloidogenic immunoglobulin light chain. Proc Natl Acad Sci USA. 2014;111(36):13046-13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plate L, Rius B, Nguyen B, Genereux JC, Kelly JW, Wiseman RL. Quantitative interactome proteomics reveals a molecular basis for ATF6-dependent regulation of a destabilized amyloidogenic protein. Cell Chem Biol. 2019;26(7):913-925.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plate L, Cooley CB, Chen JJ, et al. Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. elife. 2016;5:e15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paxman R, Plate L, Blackwood EA, et al. Pharmacologic ATF6 activating compounds are metabolically activated to selectively modify endoplasmic reticulum proteins. elife. 2018;7:e37168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Appenzeller-Herzog C, Ellgaard L. The human PDI family: versatility packed into a single fold. Biochim Biophys Acta. 2008;1783(4):535-548. [DOI] [PubMed] [Google Scholar]
- 41.Wang L, Wang X, Wang CC. Protein disulfide-isomerase, a folding catalyst and a redox-regulated chaperone. Free Radic Biol Med. 2015;83:305-313. [DOI] [PubMed] [Google Scholar]
- 42.Blackwood EA, Azizi K, Thuerauf DJ, et al. Pharmacologic ATF6 activation confers global protection in widespread disease models by reprograming cellular proteostasis. Nat Commun. 2019;10(1):187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kroeger H, Grimsey N, Paxman R, et al. The unfolded protein response regulator ATF6 promotes mesodermal differentiation. Sci Signal. 2018;11(517):eaan5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stauffer WT, Blackwood EA, Azizi K, Kaufman RJ, Glembotski CC. The ER unfolded protein response effector, ATF6, reduces cardiac fibrosis and decreases activation of cardiac fibroblasts. Int J Mol Sci. 2020;21(4):1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arendt BK, Ramirez-Alvarado M, Sikkink LA, et al. Biologic and genetic characterization of the novel amyloidogenic lambda light chain-secreting human cell lines, ALMC-1 and ALMC-2. Blood. 2008;112(5):1931-1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaplan B, Ramirez-Alvarado M, Sikkink L, et al. Free light chains in plasma of patients with light chain amyloidosis and non-amyloid light chain deposition disease. High proportion and heterogeneity of disulfide-linked monoclonal free light chains as pathogenic features of amyloid disease. Br J Haematol. 2009;144(5):705-715. [DOI] [PubMed] [Google Scholar]
- 47.Andrich K, Hegenbart U, Kimmich C, et al. Aggregation of full-length immunoglobulin light chains from systemic light chain amyloidosis (AL) patients is remodeled by epigallocatechin-3-gallate. J Biol Chem. 2017;292(6):2328-2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klimtchuk ES, Gursky O, Patel RS, et al. The critical role of the constant region in thermal stability and aggregation of amyloidogenic immunoglobulin light chain. Biochemistry. 2010;49(45):9848-9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramirez-Alvarado M. Amyloid formation in light chain amyloidosis. Curr Top Med Chem. 2012;12(22):2523-2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sikkink LA, Ramirez-Alvarado M. Cytotoxicity of amyloidogenic immunoglobulin light chains in cell culture. Cell Death Dis. 2010;1(11):e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sidrauski C, Acosta-Alvear D, Khoutorsky A, et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. elife. 2013;2:e00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sidrauski C, McGeachy AM, Ingolia NT, Walter P. The small molecule ISRIB reverses the effects of eIF2α phosphorylation on translation and stress granule assembly. elife. 2015;4:e05033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nadanaka S, Okada T, Yoshida H, Mori K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol Cell Biol. 2007;27(3):1027-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nadanaka S, Yoshida H, Mori K. Reduction of disulfide bridges in the lumenal domain of ATF6 in response to glucose starvation. Cell Struct Funct. 2006;31(2):127-134. [DOI] [PubMed] [Google Scholar]
- 55.Koba H, Jin S, Imada N, et al. Reinvestigation of disulfide-bonded oligomeric forms of the unfolded protein response transducer ATF6. Cell Struct Funct. 2020;45(1):9-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oka OB, van Lith M, Rudolf J, Tungkum W, Pringle MA, Bulleid NJ. ERp18 regulates activation of ATF6α during unfolded protein response. EMBO J. 2019;38(15):e100990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ye J, Rawson RB, Komuro R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6(6):1355-1364. [DOI] [PubMed] [Google Scholar]
- 58.Torres SE, Gallagher CM, Plate L, et al. Ceapins block the unfolded protein response sensor ATF6α by inducing a neomorphic inter-organelle tether. elife. 2019;8:e46595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gallagher CM, Garri C, Cain EL, et al. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6α branch. elife. 2016;5:e11878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gallagher CM, Walter P. Ceapins inhibit ATF6α signaling by selectively preventing transport of ATF6α to the Golgi apparatus during ER stress. elife. 2016;5:e11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hay BA, Abrams B, Zumbrunn AY, et al. Aminopyrrolidineamide inhibitors of site-1 protease. Bioorg Med Chem Lett. 2007;17(16):4411-4414. [DOI] [PubMed] [Google Scholar]
- 62.Lebeau P, Byun JH, Yousof T, Austin RC. Pharmacologic inhibition of S1P attenuates ATF6 expression, causes ER stress and contributes to apoptotic cell death. Toxicol Appl Pharmacol. 2018;349:1-7. [DOI] [PubMed] [Google Scholar]
- 63.Cole KS, Grandjean JMD, Chen K, et al. Characterization of an A-site selective protein disulfide isomerase A1 inhibitor. Biochemistry. 2018;57(13):2035-2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Banerjee R, Pace NJ, Brown DR, Weerapana E. 1,3,5-triazine as a modular scaffold for covalent inhibitors with streamlined target identification. J Am Chem Soc. 2013;135(7):2497-2500. [DOI] [PubMed] [Google Scholar]
- 65.Gupta VK, Brauneis D, Shelton AC, et al. Induction therapy with bortezomib and dexamethasone and conditioning with high-dose melphalan and bortezomib followed by autologous stem cell transplantation for immunoglobulin light chain amyloidosis: long-term follow-up analysis. Biol Blood Marrow Transplant. 2019;25(5):e169-e173. [DOI] [PubMed] [Google Scholar]
- 66.Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr., Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907-4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ri M. Endoplasmic-reticulum stress pathway-associated mechanisms of action of proteasome inhibitors in multiple myeloma. Int J Hematol. 2016;104(3):273-280. [DOI] [PubMed] [Google Scholar]
- 68.Nikesitch N, Lee JM, Ling S, Roberts TL. Endoplasmic reticulum stress in the development of multiple myeloma and drug resistance. Clin Transl Immunology. 2018;7(1):e1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Borth N, Mattanovich D, Kunert R, Katinger H. Effect of increased expression of protein disulfide isomerase and heavy chain binding protein on antibody secretion in a recombinant CHO cell line. Biotechnol Prog. 2005;21(1):106-111. [DOI] [PubMed] [Google Scholar]
- 70.Lilie H, McLaughlin S, Freedman R, Buchner J. Influence of protein disulfide isomerase (PDI) on antibody folding in vitro. J Biol Chem. 1994;269(19):14290-14296. [PubMed] [Google Scholar]
- 71.Forster ML, Sivick K, Park YN, Arvan P, Lencer WI, Tsai B. Protein disulfide isomerase-like proteins play opposing roles during retrotranslocation. J Cell Biol. 2006;173(6):853-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sørensen S, Ranheim T, Bakken KS, Leren TP, Kulseth MA. Retention of mutant low density lipoprotein receptor in endoplasmic reticulum (ER) leads to ER stress. J Biol Chem. 2006;281(1):468-476. [DOI] [PubMed] [Google Scholar]
- 73.Jang I, Pottekat A, Poothong J, et al. PDIA1/P4HB is required for efficient proinsulin maturation and ß cell health in response to diet induced obesity. elife. 2019;8:e44528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.