

Key Points
Question
Is the presence of exfoliation syndrome, a common cause of glaucoma, associated with rare, protein-changing variants?
Findings
In this multicenter, case-control, whole-exome sequencing study that included 20 441 participants, persons with exfoliation syndrome, compared with those without exfoliation syndrome, were significantly more likely to be carriers of functionally deficient CYP39A1 alleles (odds ratio, 2.03).
Meaning
The presence of exfoliation syndrome was significantly associated with carriage of functionally deficient CYP39A1 sequence variants.
Abstract
Importance
Exfoliation syndrome is a systemic disorder characterized by progressive accumulation of abnormal fibrillar protein aggregates manifesting clinically in the anterior chamber of the eye. This disorder is the most commonly known cause of glaucoma and a major cause of irreversible blindness.
Objective
To determine if exfoliation syndrome is associated with rare, protein-changing variants predicted to impair protein function.
Design, Setting, and Participants
A 2-stage, case-control, whole-exome sequencing association study with a discovery cohort and 2 independently ascertained validation cohorts. Study participants from 14 countries were enrolled between February 1999 and December 2019. The date of last clinical follow-up was December 2019. Affected individuals had exfoliation material on anterior segment structures of at least 1 eye as visualized by slit lamp examination. Unaffected individuals had no signs of exfoliation syndrome.
Exposures
Rare, coding-sequence genetic variants predicted to be damaging by bioinformatic algorithms trained to recognize alterations that impair protein function.
Main Outcomes and Measures
The primary outcome was the presence of exfoliation syndrome. Exome-wide significance for detected variants was defined as P < 2.5 × 10−6. The secondary outcomes included biochemical enzymatic assays and gene expression analyses.
Results
The discovery cohort included 4028 participants with exfoliation syndrome (median age, 78 years [interquartile range, 73-83 years]; 2377 [59.0%] women) and 5638 participants without exfoliation syndrome (median age, 72 years [interquartile range, 65-78 years]; 3159 [56.0%] women). In the discovery cohort, persons with exfoliation syndrome, compared with those without exfoliation syndrome, were significantly more likely to carry damaging CYP39A1 variants (1.3% vs 0.30%, respectively; odds ratio, 3.55 [95% CI, 2.07-6.10]; P = 6.1 × 10−7). This outcome was validated in 2 independent cohorts. The first validation cohort included 2337 individuals with exfoliation syndrome (median age, 74 years; 1132 women; n = 1934 with demographic data) and 2813 individuals without exfoliation syndrome (median age, 72 years; 1287 women; n = 2421 with demographic data). The second validation cohort included 1663 individuals with exfoliation syndrome (median age, 75 years; 587 women; n = 1064 with demographic data) and 3962 individuals without exfoliation syndrome (median age, 74 years; 951 women; n = 1555 with demographic data). Of the individuals from both validation cohorts, 5.2% with exfoliation syndrome carried CYP39A1 damaging alleles vs 3.1% without exfoliation syndrome (odds ratio, 1.82 [95% CI, 1.47-2.26]; P < .001). Biochemical assays classified 34 of 42 damaging CYP39A1 alleles as functionally deficient (median reduction in enzymatic activity compared with wild-type CYP39A1, 94.4% [interquartile range, 78.7%-98.2%] for the 34 deficient variants). CYP39A1 transcript expression was 47% lower (95% CI, 30%-64% lower; P < .001) in ciliary body tissues from individuals with exfoliation syndrome compared with individuals without exfoliation syndrome.
Conclusions and Relevance
In this whole-exome sequencing case-control study, presence of exfoliation syndrome was significantly associated with carriage of functionally deficient CYP39A1 sequence variants. Further research is needed to understand the clinical implications of these findings.
This case-control study uses whole-exome sequencing to identify coding-sequence genetic variants associated with exfoliation syndrome, a form of glaucoma caused by abnormal fibrillar protein aggregates in the anterior eye.
Introduction
Exfoliation syndrome (also known as pseudoexfoliation syndrome) is a systemic disorder characterized by progressive accumulation of abnormal fibrillar protein aggregates manifesting clinically in the anterior chamber of the eye (Figure 1). The fibrillar protein aggregates obstruct drainage of aqueous humor and cause increased intraocular pressure, resulting in glaucoma.1 Exfoliation syndrome is distinct from true exfoliation, which is a condition linked to heat or environmental exposure–related eye injury.2 Exfoliation syndrome affected up to 70 million people in 2010 and remains a major cause of irreversible blindness worldwide.3 An association of exfoliation syndrome with increased risk of cardiovascular diseases has been reported,4 although this association could be confounded by several factors including age, sex, vascular abnormalities, and serum homocysteine levels.5
Figure 1. Eye of a Patient With Exfoliation Syndrome.

The typical white exfoliation material deposits on the surface of the lens are visible (blue arrows) with the aid of a slit lamp at 16 × magnification under white light. Exfoliation material may not be visible on unaided examination.
Although previous genome-wide association studies (GWAS) have identified 7 loci associated with exfoliation syndrome,6 it has been challenging to ascertain which genes were affected by the noncoding variants discovered through GWAS.7 The modest effect sizes of GWAS-discovered common variants limit interpretation of their clinical relevance.8 Conversely, rare, coding-sequence genetic variants often have larger effect sizes and are able to directly implicate causative genes and illuminate disease biology.9,10 Genetic studies of rare variants also have facilitated advances in the treatment of other common systemic diseases via the development of potential drug targets.11 Although more than 95% of coding-sequence variants are rare (defined as an allele frequency <1%) individually,12 their cumulative prevalence may be sufficient for detection of statistically significant differences when the aggregated rare variant counts within each gene are compared between cases and controls.13
The objective of this study was to use whole-exome sequencing to assess whether rare, coding-sequence genetic variants are related to the pathogenesis of exfoliation syndrome by directly altering protein function. Functional assays and gene expression analyses were used to characterize a single gene that surpassed exome-wide significance.
Methods
Study Design and Participants
The sequencing and analytical approaches used to identify genes associated with exfoliation syndrome are depicted in Figure 2. This case-control, whole-exome sequencing study included a discovery stage (discovery cohort) and a validation stage with 2 independently ascertained cohorts to evaluate significant findings from the discovery stage. Study participants from 14 countries were enrolled between February 1999 and December 2019. The date of last clinical follow-up was December 2019. Patients with exfoliation syndrome and those without exfoliation syndrome were enrolled after obtaining written informed consent, adhering to the tenets of the Declaration of Helsinki. All relevant local and hospital institutional review boards approved the studies.
Figure 2. Sequencing and Analytical Approaches to Identify Genes Associated With Exfoliation Syndrome.
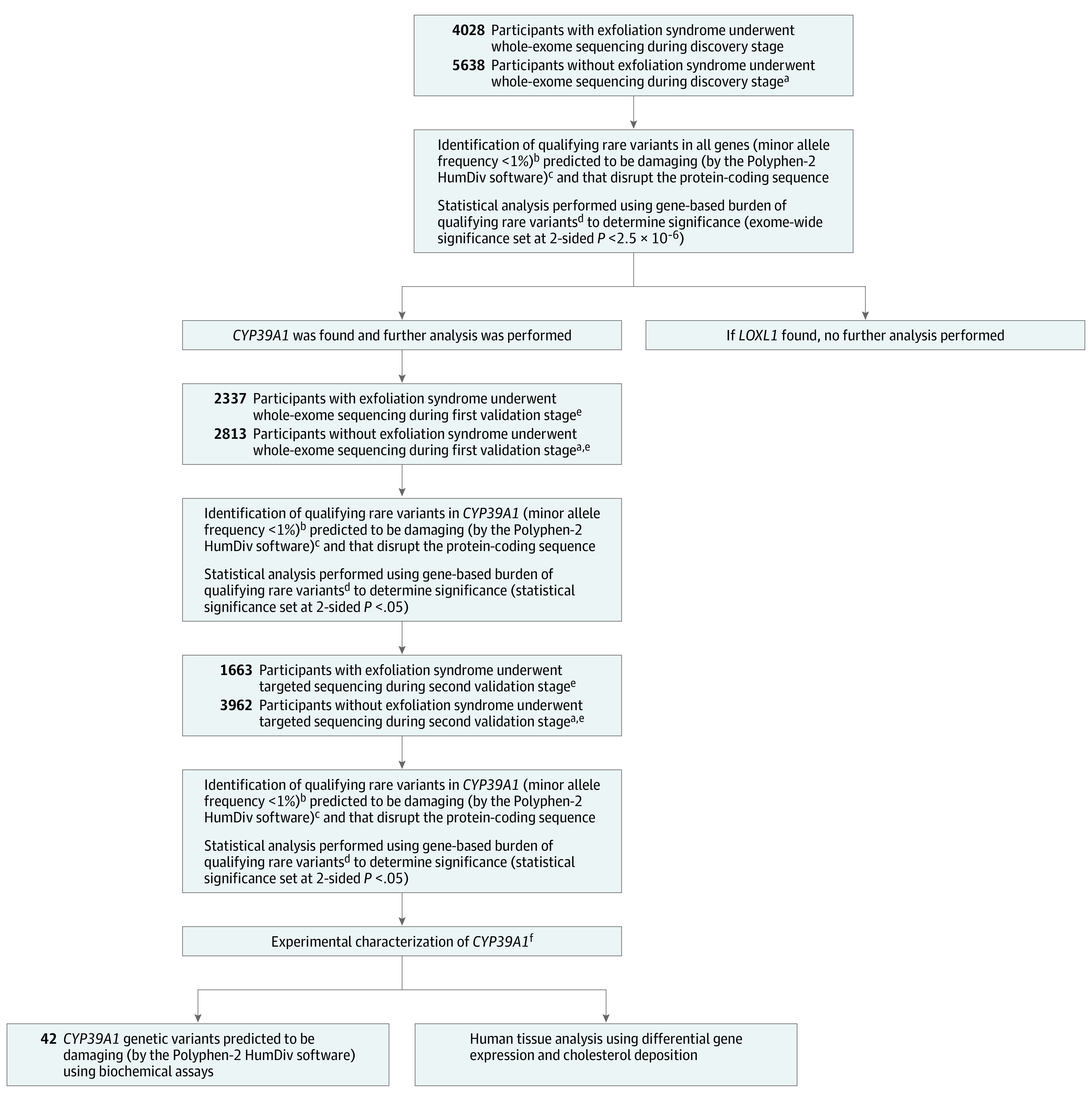
aEach participant with exfoliation syndrome was matched to at least 1 participant without exfoliation syndrome (for every individual with exfoliation syndrome, ≥1 individual without exfoliation syndrome could be recruited as a matching control). Matching was by geographical site of recruitment and self-reported ancestry.
bMost disease-causing genetic variants are maintained at low frequencies by purifying evolutionary selection.
cPolymorphism Phenotyping version 2 (Polyphen-2) is a widely used computer algorithm to identify genetic variants that damage protein function. Such variants are likely to cause disease. Alleles predicted to be benign were excluded because their inclusion could have masked disease associations caused by damaging allelic variants.
dMany medical conditions are caused by haploinsufficiency, which is a state where 1 copy of a gene has been damaged by alterations, leaving the remaining normally functioning gene copy insufficient to sustain normal function. In many of these conditions, different alterations within a given gene causes the same damaging consequences to the encoded protein product. In this study, the burden test was designed to compare the number (or burden) of damaging genetic alterations found in each gene among persons with exfoliation syndrome vs those without exfoliation syndrome.
eValidation of original discovery stage findings in 2 independently enrolled validation cohorts increases the confidence that the original observations were not false discoveries.
fGenes identified to be significantly associated with disease during the discovery and validation stages were characterized further using post hoc experimental assays to garner additional insights into disease pathogenesis.
Discovery Stage
For the discovery cohort, the enrollment criteria for individuals with exfoliation syndrome were: (1) aged older than 50 years at time of recruitment and (2) presence of exfoliation material visualized by slit lamp examination of the eye along the pupillary margin, anterior lens surface, or other anterior segment structures of at least 1 eye.14 Individuals who were younger than aged 50 years were excluded as well as those who had other secondary forms of glaucoma (such as neovascular glaucoma) or those with uveitis.
Included individuals without exfoliation syndrome were aged 60 years or older and had a complete eye examination to confirm the absence of exfoliation syndrome and other eye diseases. Because even subtle differences in geographical population structure can confound genetic analysis,15,16 individuals without exfoliation syndrome were matched to individuals with exfoliation syndrome by enrollment site and self-reported ancestry.
Validation Stage
For the 2 independently enrolled validation cohorts, the inclusion criteria for persons with exfoliation syndrome were identical to those used for the discovery cohort. Persons without exfoliation syndrome who were aged 60 years or older and who had complete eye examinations to verify absence of exfoliation syndrome were enrolled in Austria, Georgia, Greece, Japan, Poland, Romania, Russia, Spain, and Turkey. For the US study, the inclusion criteria for individuals without exfoliation syndrome were aged 50 years or older with a complete eye examination. Age-based inclusion criteria for individuals without exfoliation syndrome were not used for Canada and Germany. All individuals without exfoliation syndrome were matched to individuals with exfoliation syndrome by enrollment site (city [eg, Erlangen, Germany] or province [eg, Nova Scotia, Canada]) and self-reported ancestry (eAppendix and eFigure 1 in Supplement 1). The study details for each sample set (by location) appear in eTable 1 in Supplement 1.
Whole-Exome Sequencing and Targeted Sequencing
Genomic DNA from all participants was extracted from venous blood. Whole-exome sequencing and targeted sequencing (targeting the CYP39A1 [Refseq NM_016593] coding sequence) libraries were prepared using hybridization capture kits (Roche-Nimblegen SeqCap).17 The DNA samples from individuals with exfoliation syndrome and individuals without exfoliation syndrome were sequenced together using 2 × 151 base-pair, paired-end reads on Illumina instruments.
Postsequencing Bioinformatics Analyses
The bioinformatics procedures were applied uniformly across all samples and were blinded by case vs control status. The DNA sequence reads were mapped using Burrows-Wheeler Aligner software (version 0.7.16a-r1181). Variant calling was performed using the Genome Analysis Tool Kit (version 3.7) and the Picard (version 2.18.11) software packages (Broad Institute for both). Quality control procedures were performed using VCFtools (version 0.1.15), BCFtools (version 1.9), and PLINK (version 1.9). Variant annotation was performed using the Ensembl variant effect predictor (GRCh37).
Due diligence procedures, such as the use of information on synonymous variants (eFigure 2 and eTables 2-3 in Supplement 1), ancestry principal components (eFigures 3-4 in Supplement 1), exome-wide variant count (eFigures 5-6 in Supplement 1), singleton-variant count (eFigure 7 in Supplement 1), and excessive heterozygosity (eFigure 8 in Supplement 1), were used to avoid systematic differences in variant detection between individuals with exfoliation syndrome and those without exfoliation syndrome (eAppendix in Supplement 1).
Identification of Rare Genetic Variants Predicted to Be Damaging
Rare genetic variants predicted to have a damaging effect on protein function were identified using the following criterion: (1) minor allele frequency less than 1% across all ethnic groups studied18; (2) variants that disrupt the protein-coding sequence19 (these refer to stop-gained, start-loss, frameshift, or canonical splice-site alterations12,18,20; eAppendix in Supplement 1); and (3) missense variants predicted to be damaging by the Polymorphism Phenotyping version 2 (Polyphen-2) computer prediction algorithm.21
The filter for minor allele frequencies less than 1% was applied because genetic variants with minor allele frequencies greater than 1% were well represented by previously used GWAS genotyping arrays.22 All bioinformatics filters were prespecified and applied uniformly across the discovery and validation cohorts using PLINK (version 1.9).
Sanger Capillary Sequencing
Participants carrying CYP39A1 rare alleles were randomly selected for capillary sequencing to confirm the rare allele calls from exome sequencing, which was consistent with previous reports that used capillary sequencing to validate a subset of rare variant calls detected from whole-exome sequencing.23
Analysis of Individual CYP39A1 Variant Activity
CYP39A1 metabolizes 24(S)-hydroxycholesterol to 24(S)-7α,24-dihydroxycholesterol.24,25 The enzymatic activity of 50 CYP39A1 coding-sequence variants was tested via transfection into human embryonic kidney 293 cells. For transfection, the reference CYP39A1 complementary DNA sequence and the variants were cloned into an expression plasmid. During the transfection process, 6 μg of plasmid was transfected into 1.8 million human embryonic kidney 293 cells in a 10-cm dish using 18 μL of Lipofectamine 2000 (Thermo Fisher Scientific). Twenty-four hours later, 5 μM of 24(S)-hydroxycholesterol was added to the transfected cells, followed by a further 24-hour incubation period before harvesting. Liquid chromatography–tandem mass spectrometry was used to assay harvested cell lysates for the abundance of 24(S)-7α,24-dihydroxycholesterol as a direct readout of enzymatic activity (eAppendix and eTables 4-5 in Supplement 1). The design and validation of the experimental system appear in eFigures 9 and 10 in Supplement 1.
Expression Analysis of CYP39A1 in Eye Tissues
Quantitative real-time polymerase chain reaction was performed on a panel of eye tissues from patients with exfoliation syndrome and from matched individuals without exfoliation syndrome. Disease-specific expression was tested in an independent panel (eAppendix and eTable 6 in Supplement 1). The sample sizes were consistent with previous reports assessing differential gene expression for significant associations with disease genes.6,26
Immunohistochemical analysis was performed on age-matched donor eyes from individuals with exfoliation syndrome and those without exfoliation syndrome using antibodies against CYP39A1 on cryosections and paraffin-embedded sections via heat-induced antigen retrieval. Filipin staining for esterified and free unesterified cholesterol was performed as previously described.27 Additional staining for β1 integrin (a cell membrane marker) and apolipoprotein E and LOXL1 (both are markers found in abnormal exfoliative material28) was performed to assess co-localization. Differences in CYP39A1 protein expression were based solely on visual inspection of stained slides.
To detect unesterified free cholesterol, tissue cryosections were fixed with 4% paraformaldehyde and incubated with 250 μg/mL of filipin for 1 hour. To detect esterified cholesterol in cryosections, free cholesterol was extracted using 70% ethanol for 30 minutes, followed by conversion of esterified to unesterified cholesterol by incubation with 2 U/mL of cholesterol esterase for 3 hours at 37 °C before detection with filipin. Sections were counterstained with propidium iodide and examined under a fluorescence microscope. Filipin fluorescence was observed using a UV filter set (λex/λem = 350 nm/455 nm). As a negative control, cholesterol esterase was replaced by phosphate-buffered saline.
Statistical Analysis
An assessment of the statistical power was made (statistical power calculations appear in eTable 7 in Supplement 1). All rare genetic variants predicted to impair protein function (ie, damaging) within each gene were aggregated together for an association analysis using the burden test.10,19 The burden test evaluated whether any of the approximately 20 000 genes across the human exome bore an excess or a deficit of damaging genetic variants in individuals with exfoliation syndrome vs those without exfoliation syndrome. Because many medical conditions show a haploinsufficiency effect with allelic heterogeneity, the burden test was used to evaluate whether different damaging variants within the same gene are, in aggregate, associated with the presence of disease.
Discovery Stage
A stratified Cochran Mantel-Haenszel fixed-effects meta-analysis (without continuity correction29) was used to summarize gene-based burden test results across the exomes of the study participants from the 3 countries studied in the discovery cohort. The accuracy of this method was verified by adjusting the primary association test statistics for potential confounders (such as ancestry principal component scores and exome-wide variant count) using Firth penalized logistic regression.18,30 Exome-wide significance was specified at 2-sided P < 2.5 × 10−6 to reflect multiple testing correction for 20 000 genes.19
Validation Stage
First Cohort
The newly identified genes surpassing exome-wide significance during the discovery stage (ie, CYP39A1) were evaluated for validation. A meta-analysis using the same stratified Cochran Mantel-Haenszel fixed-effects method as described for the discovery stage was used for the association between CYP39A1 rare variant burden and the presence of exfoliation syndrome across the participants from 8 countries in the first validation cohort. The association test was adjusted for ancestry principal components and exome-wide variant count. Because only 1 gene (CYP39A1) was tested here, 2-sided P < .05 was considered statistically significant.
Second Cohort
A meta-analysis using the same stratified Cochran Mantel-Haenszel fixed-effects method across the 4 participating countries was performed in the second validation cohort. The Fisher exact test was used to evaluate the individual strata. Because only 1 gene (CYP39A1) was tested here, 2-sided P < .05 was considered statistically significant.
First and Second Cohorts
A meta-analysis using the same stratified Cochran Mantel-Haenszel fixed-effects method was used for the association between CYP39A1 rare variant burden and the presence of exfoliation syndrome across the participants from all 12 countries.
Post Hoc Analysis of CYP39A1 Biochemical Enzymatic Activity
Enzymatic activity of 50 tested CYP39A1 variants were compared with enzymatic activity of wild-type CYP39A1 using the absolute range of their experimentally measured enzymatic activity. A variant was classified as increased function if its experimentally measured enzymatic activity range was significantly higher than, and did not overlap with, that of wild-type CYP39A1. Conversely, a variant was classified as deficient if its experimentally measured enzymatic activity range was significantly lower than, and did not overlap with, that of wild-type CYP39A1. For the 34 variants that were classified as functionally deficient, their median reduction in enzymatic activity was determined from all their measured data points (34 variants measured in 3 independent biological replicates, resulting in 102 data points; eFigure 11 in Supplement 1).
Post Hoc Analysis of Gene Expression in Eye Tissues
Expression levels of assessed genes were normalized relative to the GAPDH housekeeping gene. Comparison of gene expression levels between tissues from patients with exfoliation syndrome and those without exfoliation syndrome were made using the unpaired 2-tailed t test. A 2-sided P < .05 was considered significant.
All statistical analyses were performed using R statistical software package version 3.4.3 (R Foundation for Statistical Computing).
Results
Rare Coding Variants Associated With Exfoliation Syndrome
The discovery cohort included 4028 participants with exfoliation syndrome (median age, 78 years [interquartile range, 73-83 years]; 2377 [59.0%] women) and 5638 participants without exfoliation syndrome (median age, 72 years [interquartile range, 65-78 years]; 3159 [56.0%] women) (Table). A total of 415 871 rare variants predicted to impair protein function were identified across 18 753 genes. An exome-wide significant association with exfoliation syndrome was observed at genes LOXL1 (Refseq NM_005576) and CYP39A1 (eFigure 12 in Supplement 1 and eTable 8 in Supplement 2). The association at LOXL1 was driven by rare variants that conferred protection from exfoliation syndrome (eTable 9 in Supplement 1).6 Persons with exfoliation syndrome were significantly more likely to carry damaging CYP39A1 variants compared with persons without exfoliation syndrome (52 of 4028 [1.3%] vs 17 of 5638 [0.30%], respectively; odds ratio [OR], 3.55 [95% CI, 2.07-6.10], P = 6.1 × 10−7; Figure 3).
Table. Baseline Participant Characteristics in the Discovery Cohorta.
| Characteristic | With exfoliation syndrome | Without exfoliation syndrome |
|---|---|---|
| Japanb | ||
| No. analyzed | 3806 | 4197 |
| Age, median (IQR), y | 78 (73-83) | 74 (68-79) |
| Sex, No. (%) | ||
| Female | 2280 (59.9) | 2468 (58.8) |
| Male | 1526 (40.1) | 1729 (41.2) |
| Exfoliation glaucoma, No. (%) | 2180 (57.3) | 0 |
| Self-reported ancestry | Japanese | Japanese |
| No. of participants | 3806 | 4197 |
| Singaporeb | ||
| No. analyzed | 172 | 1288 |
| Age, median (IQR), y | 79 (73-85) | 67 (63-73) |
| Sex, No. (%) | ||
| Female | 76 (44.2) | 612 (47.5) |
| Male | 96 (55.8) | 676 (52.5) |
| Exfoliation glaucoma, No. (%) | 82 (47.7) | 0 |
| Self-reported ancestry | Chinese | Chinese |
| No. of participants | 172 | 1288 |
| South Africab | ||
| No. analyzed | 50 | 153 |
| Age, median (IQR), y | 71 (67-77) | 69 (63-76) |
| Sex, No. (%) | ||
| Female | 21 (42.0) | 79 (51.6) |
| Male | 29 (58.0) | 74 (48.4) |
| Exfoliation glaucoma, No. (%) | 47 (94.0) | 0 |
| Self-reported ancestry | Black African | Black African |
| No. of participants | 50 | 153 |
Abbreviation: IQR, interquartile range.
All participants underwent whole-exome sequencing.
Indicates the country where participants were recruited.
Figure 3. CYP39A1 Association With Presence of Exfoliation Syndrome.
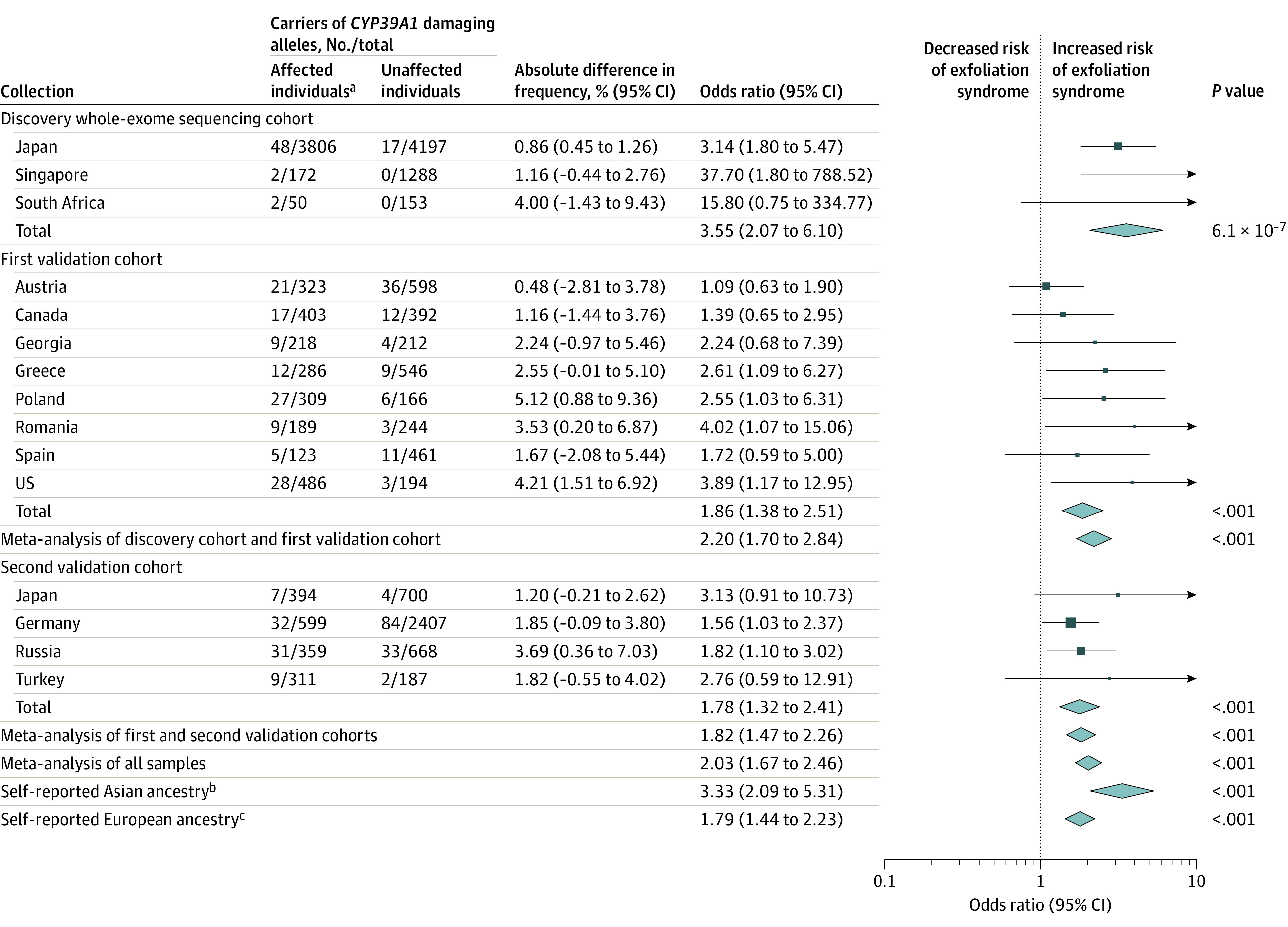
Forest plot describing the association between the burden of damaging CYP39A1 allelic variants and exfoliation syndrome among 20 441 participants from 14 countries. The height of the data marker is proportional to the size of the sample. The width of the diamond represents the 95% CI. P values were calculated using the Cochran-Mantel-Haenszel meta-analysis method. There was no meta-analysis conducted for self-reported Black African ancestry because this would simply duplicate the analysis from South Africa as reported elsewhere in the Figure. Further information on selection of the study participants based on self-reported ancestry appears in the eAppendix in Supplement 1.
aAffected individuals had exfoliation syndrome and unaffected individuals did not have it.
bIncluded participants from Japan and Singapore who self-identified as having Asian ancestry.
cIncluded participants from Austria, Canada, Georgia, Germany, Greece, Poland, Romania, Russia, Spain, Turkey, and the US.
The first validation cohort included 2337 individuals with exfoliation syndrome (median age, 74 years; 1132 women; n = 1934 with demographic data) and 2813 individuals without exfoliation syndrome (median age, 72 years; 1287 women; n = 2421 with demographic data). Persons with exfoliation syndrome were again observed to be significantly more likely to carry damaging CYP39A1 variants vs those without exfoliation syndrome (128 of 2337 [5.5%] vs 84 of 2813 [3.0%], respectively; OR, 1.86 [95% CI, 1.38-2.51]; P < .001). This association was reassessed in a second validation cohort including 1663 individuals with exfoliation syndrome (median age, 75 years; 587 women; n = 1064 with demographic data) and 3962 individuals without exfoliation syndrome (median age, 74 years; 951 women; n = 1555 with demographic data). Persons with exfoliation syndrome were observed to be significantly more likely to carry damaging CYP39A1 variants compared with persons without exfoliation syndrome (79 of 1663 [4.8%] vs 123 of 3962 [3.1%], respectively; OR, 1.78 [95% CI, 1.32-2.41], P < .001; Figure 3). Of the individuals from both validation cohorts, 5.2% of individuals (207 of 4000) with exfoliation syndrome carried CYP39A1 damaging alleles vs 3.1% of individuals (207 of 6775) without exfoliation syndrome (OR, 1.82 [95% CI, 1.47-2.26]; P < .001).
A meta-analysis of all participants confirmed that patients with exfoliation syndrome were significantly more likely to carry rare, damaging CYP39A1 variants compared with persons without exfoliation syndrome (OR, 2.03 [95% CI, 1.67-2.46], P < .001; Figure 3 and eTable 10 in Supplement 1). The heritability explained by CYP39A1 rare variant burden was estimated to be 0.2%. Complete concordance between the rare variant calls from exome sequencing and capillary sequencing was observed for 130 CYP39A1 rare variant carriers selected for capillary sequencing (eAppendix and eFigure 13 in Supplement 1).
Post Hoc Evaluation of CYP39A1 Activity of CYP39A1 Rare Coding Alleles
Sequencing of all 20 441 study participants identified 483 individuals carrying 42 unique CYP39A1 rare variants that were predicted to be damaging by Polyphen-2 (eTable 11 in Supplement 1).
Biochemical assays classified 34 of 42 damaging CYP39A1 alleles as functionally deficient (median reduction in enzymatic activity compared with wild-type CYP39A1, 94.4% [interquartile range, 78.7%-98.2%] for the 34 deficient variants). Seven variants carried by 7 individuals had enzymatic activity ranges that overlapped with that of wild-type CYP39A1. One variant (p.F175L), which appeared to have increased enzymatic activity compared with wild-type CYP39A1, was carried by 2 individuals (Figure 4). The enzymatic assays were highly reproducible when 16 deficient variants were randomly selected and retested (eFigure 14 in Supplement 1). Persons with exfoliation syndrome were significantly more likely to carry 1 of the 34 deficient variants compared with persons without exfoliation syndrome (OR, 2.02 [95% CI, 1.66-2.47]; P < .001).
Figure 4. Experimental Determination of Functional Enzymatic Activity for CYP39A1 Variants.

Box and whisker plots of functional enzymatic activity of 1 wild-type CYP39A1 variant, 3 common variants, 42 rare variants predicted to be damaging (by Polymorphism Phenotyping version 2 [Polyphen-2]; software that predicts the possible effect of amino acid substitutions on human protein function), and 5 variants predicted by Polyphen-2 to be benign. Three data points (representing 3 independent biological replicates) were plotted for all tested CYP39A1 genetic variants. The crossbars represent the median level of activity, the boxes represent the interquartile range, and the whiskers portray the range of enzymatic activity for each variant. Individual biological replicates are shown as individual data points. Amino acid substitutions are caused by coding-sequence genetic variation. The case-control distribution of all variants predicted by Polyphen-2 to be damaging appears in eTable 10 in Supplement 1. The enzymatic activity of individual variants was expressed as a proportion relative to the wild-type CYP39A1 enzymatic activity. The functional enzymatic activity of each tested CYP39A1 variant was determined by the relative 24(S)-7α,24-dihydroxycholesterol product abundance it produced with respect to wild-type CYP39A1. The product abundance was calculated by integrating the area under the 24(S)-7α,24-dihydroxycholesterol product peak (eFigure 10 in Supplement 1), averaged across 3 independent samples for each tested variant. The empirical distribution of the wild-type CYP39A1 enzymatic activity was defined using 14 independent biological replicates. The design and validation of the experimental system appear in eFigures 9 and 10 in Supplement 1.
The enzymatic activity of 3 common CYP39A1 variants and 5 CYP39A1 rare variants predicted by Polyphen-2 to be benign also were measured. All 5 benign variants had enzymatic activity comparable with wild-type CYP39A1. Two of the 3 common variants showed increased function relative to wild-type CYP39A1 (Figure 4). None of the 8 variants were significantly associated with the presence of exfoliation syndrome (eTables 12-13 in Supplement 1).
Post Hoc Differential Gene Expression of CYP39A1 in Eye Tissues
CYP39A1 expression was observed in the liver and in all normal ocular tissues analyzed (eFigure 15 in Supplement 1). CYP39A1 was evaluated with several other functionally related CYP enzymes (eFigure 16 in Supplement 1) for differences in gene expression between eye tissues affected by exfoliation syndrome (n = 23) and unaffected eye tissues (n = 23). CYP39A1 was the only CYP enzyme tested to show consistent and significant downregulation across all affected eye tissues (eg, mean reduction for ciliary body of 47% [95% CI, 30%-64%]; P < .001) compared with unaffected eye tissues (Figure 5), as well as significant downregulation in eye tissues from all disease stages compared with unaffected eye tissues (eFigure 17 in Supplement 1).
Figure 5. Expression of CYP39A1 in Eye Tissues From Participants With Self-Reported European Ancestry.
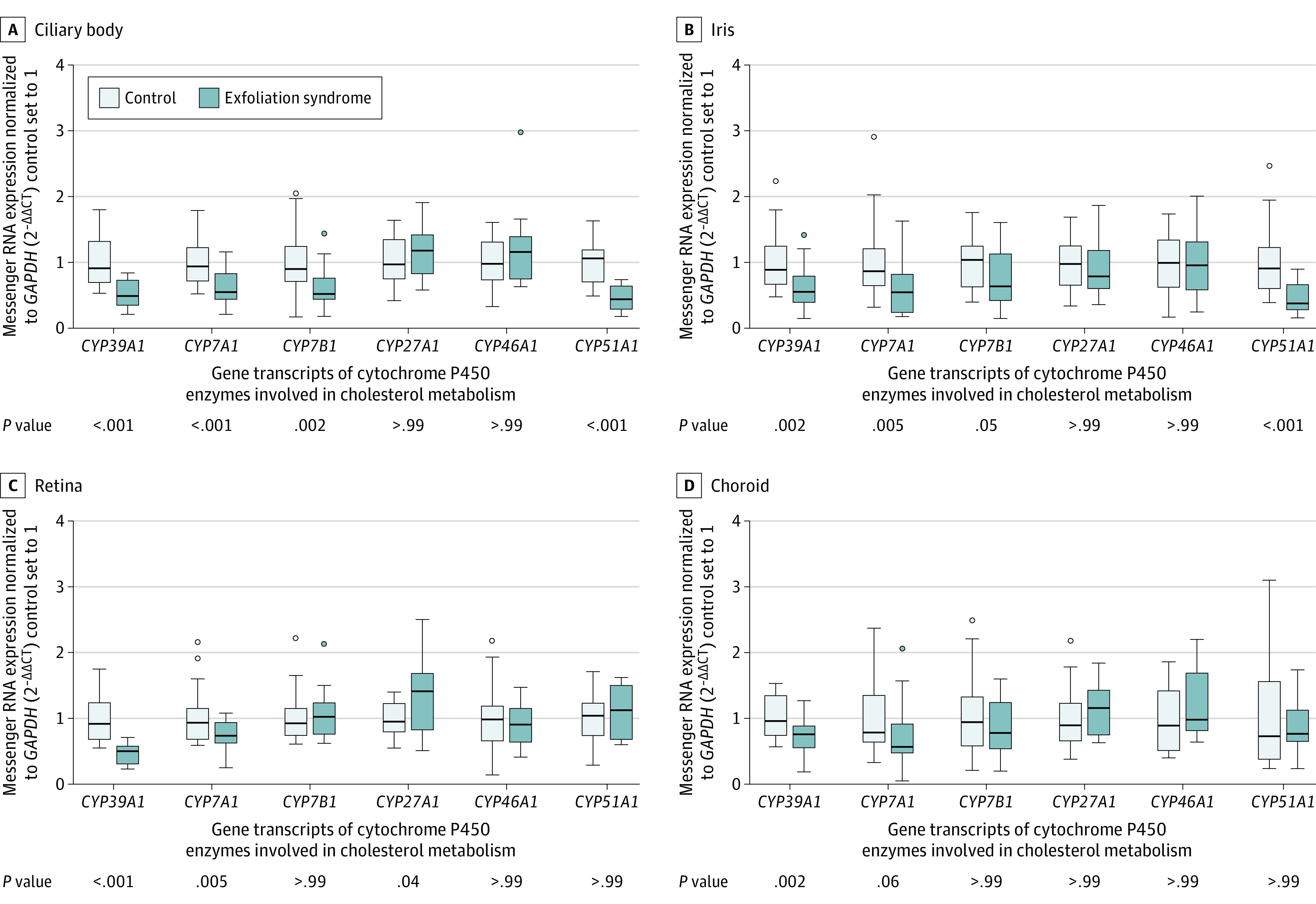
Box plots showing relative messenger RNA expression levels of cytochrome P450 enzymes involved in cholesterol metabolism. The crossbars represent median relative messenger RNA expression levels, the boxes represent the interquartile range, the whiskers extend to the most extreme observed values with 1.5 × interquartile range of the nearer quartile, and the dots represent observed values outside that range. The ocular tissues were derived from patients with late-stage exfoliation syndrome (n = 23 [10 males and 13 females]; mean age, 79.1 years [SD, 8.8 years]) and those without exfoliation syndrome (n = 23 [12 males and 11 females]; mean age, 72.7 years [SD, 8.1 years]) using quantitative real-time polymerase chain reaction. The affected tissues and the unaffected control tissues were matched by age and sex if possible. Expression levels of CYP39A1 were significantly reduced in all ocular tissues from patients with exfoliation syndrome compared with controls, with the most significant differences seen in the ciliary body and retina. In contrast, CYP7A1, CYP7B1, CYP27A1, and CYP51A1 were reduced only in specific ocular tissues derived from patients with exfoliation syndrome, whereas CYP46A1 levels were not changed. Expression levels were normalized relative to the GAPDH housekeeping gene. P values were calculated based on the unpaired 2-tailed t test. A cholesterol metabolism schematic appears in eFigure 16 in Supplement 1.
Immunohistochemical analyses using anti-CYP39A1 antibodies showed CYP39A1 localized to the ciliary epithelium, iris, choroid, and retina (eFigure 18 in Supplement 1). Visual inspection showed reduced immunostaining for CYP39A1 in the ciliary epithelium (eFigure 19 in Supplement 1) and in the retina (eFigure 20 in Supplement 1) of patients with exfoliation syndrome compared with those without exfoliation syndrome. Immunostaining for CYP39A1 was visually observed to be lower in the ciliary epithelium from an individual with exfoliation syndrome carrying a deficient CYP39A1 sequence variant (p.G410R) compared with individuals with exfoliation syndrome not carrying a deficient CYP39A1 sequence variant (eFigure 19 in Supplement 1).
Because of the role of CYP39A1 in cholesterol metabolism, potential differences in localization of cholesterol in its esterified and unesterified forms were assessed between affected and unaffected eye tissues using immunofluorescence analysis. Abnormal extracellular signals for esterified cholesterol localizing within the exfoliation material deposits on the surface membrane of the ciliary epithelium of affected eye tissues were observed. These signals showed clear co-localization with known markers of exfoliation material (such as apolipoprotein E and LOXL1).28 Conversely, these abnormal extracellular signals for esterified cholesterol were not observed in unaffected tissues (eFigure 21 in Supplement 1).
Discussion
The study of 20 441 participants found that individuals with exfoliation syndrome were significantly more likely to carry damaging CYP39A1 alleles compared with those without exfoliation syndrome. The full concordance observed between next-generation exome sequencing and capillary sequencing made it unlikely that the CYP39A1 rare alleles identified were sequencing artifacts.
Deficient CYP39A1 function impairs cellular metabolism of 24(S)-hydroxycholesterol to downstream intermediates. Because 24(S)-hydroxycholesterol regulates cellular lipid homeostasis,31 its dysregulation may lead to abnormalities in cholesterol homeostasis and transport. This may result in excess cholesterol accumulation in extracellular aggregates of exfoliation material, which represent the hallmark of the disease.
It is possible that the association between CYP39A1 deficiency and the presence of exfoliation syndrome was not detected in prior GWAS because GWAS tend to focus on common genetic variants, most of which have modest ORs for disease susceptibility. The low heritability fraction explained by CYP39A1 rare variant burden is consistent with similar studies of other complex diseases.9,18,19
The findings raise a possibility that future research efforts aimed at restoring deficient CYP39A1 function and inhibiting the formation of exfoliation material in the eye (thereby preventing complications such as blinding from its accumulation) could be an approach to assess new strategies to treat exfoliation syndrome.
Limitations
This study has several limitations. First, the deficient CYP39A1 variants were present in only a small percentage of persons with exfoliation syndrome. Thus, even if there was a causal link between these variants and exfoliation syndrome, the variants accounted for only a very small proportion of patients with the disorder.
Second, the observational design of this case-control study precluded conclusions about causal relationships.
Third, this study was insufficiently powered to assess potential interactions between CYP39A1 rare alleles and other loci.
Fourth, this study did not explore experimental perturbation of CYP39A1 in an animal model that recapitulates exfoliation syndrome. Such research is complex and could be addressed by future work.
Conclusions
In this whole-exome sequencing case-control study, presence of exfoliation syndrome was significantly associated with carriage of functionally deficient CYP39A1 sequence variants. Further research is needed to understand the clinical implications of these findings.
eAppendix
eFigure 1. Ancestry principal component analysis between HapMap reference samples of European (CEU), Chinese (CHB), Japanese (JPT), and African (YRI) ancestry and study participants recruited from Europe, Canada, and the USA
eFigure 2. Quantile-quantile plot of the discovery exome sequencing series for gene-based burden of rare synonymous alleles
eFigure 3. Principal component analysis of genetic ancestry for the discovery exome sequencing series from Japan, Singapore, and South Africa
eFigure 4. Principal component analysis of genetic ancestry for participants carrying CYP39A1 alleles predicted by Polyphen2 to be damaging
eFigure 5. Exome-wide variant count distribution for the discovery exome sequencing series
eFigure 6. Exome-wide variant count distribution for first replication series
eFigure 7. Number of singleton variants for each sample in the discovery exome sequencing analysis, analyzed by country of enrollment
eFigure 8. Ratio of heterozygous / homozygous genotypes for each sample in the discovery exome sequencing analysis, analyzed by country of enrollment
eFigure 9. Experimental design of the functional enzymatic activity assay for CYP39A1 alleles
eFigure 10. Validation of experimental design of the functional enzymatic activity assay for CYP39A1 alleles
eFigure 11. Determination of the median reduction in enzymatic activity relative to wild-type for the 34 CYP39A1 sequence variants classified as functionally deficient
eFigure 12. Quantile-quantile plot of the discovery exome sequencing results for gene-based burden of rare alleles predicted by Polyphen-2 to be damaging
eFigure 13. Capillary sequencing traces (using the Sanger method) for a selection of CYP39A1 rare alleles that were predicted by Polyphen2 to be damaging
eFigure 14. Assessment of reproducibility of the CYP39A1 functional enzymatic assay
eFigure 15. Box plots showing relative expression levels of CYP39A1 mRNA in ocular tissues derived from normal human donors using real-time PCR
eFigure 16. Schematic representation of the classic and alternative pathways of cholesterol metabolism and bile acid biosynthesis focusing on steps involving cytochrome P450 (CYP) enzymes
eFigure 17. Box plots showing relative expression levels of transcripts for CYP7A1, CYP7B1, CYP39A1, CYP46A1 and CYP51A1 in ciliary body tissues
eFigure 18. Expression of CYP39A1 protein in ocular tissues of normal human donor eyes as determined by immunofluorescence labelling of cryosections
eFigure 19. Expression of CYP39A1 protein in ciliary body tissue of normal human donor eyes and eyes affected by exfoliation syndrome as determined by immunohistochemistry on paraffin sections
eFigure 20. Immunohistochemical analysis using anti-CYP39A1 antibodies on the retina
eFigure 21. Cholesterol deposition in ciliary body tissues of the eye
eTable 1. Sample sets included in the first and second replication stages of this study
eTable 2. Association of rare synonymous variant burden in CYP39A1 with risk for exfoliation syndrome
eTable 3. List of CYP39A1 rare synonymous alleles from the discovery and first replication series
eTable 4. List of CYP39A1 variants subjected to functional enzymatic assays
eTable 5. Primer sequences for CYP39A1 allele-specific mutagenesis
eTable 6. Primers and hybridization probes used for quantitative real-time PCR
eTable 7. Statistical power calculations
eTable 8. Summary statistics for the discovery whole exome sequencing study from Japan, Singapore, and South Africa (Excel file in Supplement 2)
eTable 9. Damaging LOXL1 mutations are associated with protection from exfoliation syndrome
eTable 10. Association of damaging CYP39A1 mutations with risk for exfoliation syndrome
eTable 11. List of CYP39A1 rare coding-sequence variants (minor allele frequency <1%) that were predicted to be damaging by the Polyphen-2 bioinformatic algorithm
eTable 12. Association of three common coding-sequence polymorphisms in in CYP39A1 with risk for exfoliation syndrome
eTable 13. List of CYP39A1 rare synonymous alleles predicted to be benign by Polyphen-2 and tested for their enzymatic activity
eReferences
Excel file
References
- 1.Zenkel M, Schlötzer-Schrehardt U. The composition of exfoliation material and the cells involved in its production. J Glaucoma. 2014;23(8)(suppl 1):S12-S14. doi: 10.1097/IJG.0000000000000123 [DOI] [PubMed] [Google Scholar]
- 2.Teekhasaenee C, Suwan Y, Supakontanasan W, Tulvatana W, Ritch R. The clinical spectrum and a new theory of pathogenesis of true exfoliation syndrome. Ophthalmology. 2016;123(11):2328-2337. doi: 10.1016/j.ophtha.2016.07.030 [DOI] [PubMed] [Google Scholar]
- 3.Nazarali S, Damji F, Damji KF. What have we learned about exfoliation syndrome since its discovery by John Lindberg 100 years ago? Br J Ophthalmol. 2018;102(10):1342-1350. doi: 10.1136/bjophthalmol-2017-311321 [DOI] [PubMed] [Google Scholar]
- 4.Chung H, Arora S, Damji KF, Weis E. Association of pseudoexfoliation syndrome with cardiovascular and cerebrovascular disease: a systematic review and meta-analysis. Can J Ophthalmol. 2018;53(4):365-372. doi: 10.1016/j.jcjo.2017.10.039 [DOI] [PubMed] [Google Scholar]
- 5.Andrikopoulos GK, Alexopoulos DK, Gartaganis SP. Pseudoexfoliation syndrome and cardiovascular diseases. World J Cardiol. 2014;6(8):847-854. doi: 10.4330/wjc.v6.i8.847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aung T, Ozaki M, Lee MC, et al. Genetic association study of exfoliation syndrome identifies a protective rare variant at LOXL1 and five new susceptibility loci. Nat Genet. 2017;49(7):993-1004. doi: 10.1038/ng.3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi SH, Weng LC, Roselli C, et al. ; DiscovEHR study and the NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium . Association between titin loss-of-function variants and early-onset atrial fibrillation. JAMA. 2018;320(22):2354-2364. doi: 10.1001/jama.2018.18179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manolio TA Bringing genome-wide association findings into clinical use. Nat Rev Genet. 2013;14(8):549-558. doi: 10.1038/nrg3523 [DOI] [PubMed] [Google Scholar]
- 9.Estrada K, Aukrust I, Bjørkhaug L, et al. ; SIGMA Type 2 Diabetes Consortium . Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA. 2014;311(22):2305-2314. Published correction appears in JAMA. 2014;312(18):1932. doi: 10.1001/jama.2014.6511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Povysil G, Petrovski S, Hostyk J, Aggarwal V, Allen AS, Goldstein DB. Rare-variant collapsing analyses for complex traits: guidelines and applications. Nat Rev Genet. 2019;20(12):747-759. doi: 10.1038/s41576-019-0177-4 [DOI] [PubMed] [Google Scholar]
- 11.Kathiresan S Developing medicines that mimic the natural successes of the human genome: lessons from NPC1L1, HMGCR, PCSK9, APOC3, and CETP. J Am Coll Cardiol. 2015;65(15):1562-1566. doi: 10.1016/j.jacc.2015.02.049 [DOI] [PubMed] [Google Scholar]
- 12.Lek M, Karczewski KJ, Minikel EV, et al. ; Exome Aggregation Consortium . Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285-291. doi: 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flannick J The contribution of low-frequency and rare coding variation to susceptibility to type 2 diabetes. Curr Diab Rep. 2019;19(5):25. doi: 10.1007/s11892-019-1142-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orr AC, Robitaille JM, Price PA, et al. Exfoliation syndrome: clinical and genetic features. Ophthalmic Genet. 2001;22(3):171-185. doi: 10.1076/opge.22.3.171.2223 [DOI] [PubMed] [Google Scholar]
- 15.Mathieson I, McVean G. Differential confounding of rare and common variants in spatially structured populations. Nat Genet. 2012;44(3):243-246. doi: 10.1038/ng.1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okada Y, Momozawa Y, Sakaue S, et al. Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat Commun. 2018;9(1):1631. doi: 10.1038/s41467-018-03274-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cirulli ET, Lasseigne BN, Petrovski S, et al. ; FALS Sequencing Consortium . Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347(6229):1436-1441. doi: 10.1126/science.aaa3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flannick J, Mercader JM, Fuchsberger C, et al. ; Broad Genomics Platform; DiscovEHR Collaboration; CHARGE; LuCamp; ProDiGY; GoT2D; ESP; SIGMA-T2D; T2D-GENES; AMP-T2D-GENES . Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature. 2019;570(7759):71-76. doi: 10.1038/s41586-019-1231-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Do R, Stitziel NO, Won HH, et al. ; NHLBI Exome Sequencing Project . Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518(7537):102-106. doi: 10.1038/nature13917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat. 2016;37(3):235-241. doi: 10.1002/humu.22932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248-249. doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacArthur DG, Manolio TA, Dimmock DP, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508(7497):469-476. doi: 10.1038/nature13127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krumm N, Turner TN, Baker C, et al. Excess of rare, inherited truncating mutations in autism. Nat Genet. 2015;47(6):582-588. doi: 10.1038/ng.3303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li-Hawkins J, Lund EG, Bronson AD, Russell DW. Expression cloning of an oxysterol 7alpha-hydroxylase selective for 24-hydroxycholesterol. J Biol Chem. 2000;275(22):16543-16549. doi: 10.1074/jbc.M001810200 [DOI] [PubMed] [Google Scholar]
- 25.Stiles AR, Kozlitina J, Thompson BM, McDonald JG, King KS, Russell DW. Genetic, anatomic, and clinical determinants of human serum sterol and vitamin D levels. Proc Natl Acad Sci U S A. 2014;111(38):E4006-E4014. doi: 10.1073/pnas.1413561111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Germain M, Eyries M, Montani D, et al. Genome-wide association analysis identifies a susceptibility locus for pulmonary arterial hypertension. Nat Genet. 2013;45(5):518-521. doi: 10.1038/ng.2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curcio CA, Rudolf M, Wang L. Histochemistry and lipid profiling combine for insights into aging and age-related maculopathy. Methods Mol Biol. 2009;580:267-281. doi: 10.1007/978-1-60761-325-1_15 [DOI] [PubMed] [Google Scholar]
- 28.Sharma S, Chataway T, Burdon KP, et al. Identification of LOXL1 protein and apolipoprotein E as components of surgically isolated pseudoexfoliation material by direct mass spectrometry. Exp Eye Res. 2009;89(4):479-485. doi: 10.1016/j.exer.2009.05.001 [DOI] [PubMed] [Google Scholar]
- 29.Khera AV, Won HH, Peloso GM, et al. ; Myocardial Infarction Genetics Consortium; DiscovEHR Study Group; CARDIoGRAM Exome Consortium; and Global Lipids Genetics Consortium . Association of rare and common variation in the lipoprotein lipase gene with coronary artery disease. JAMA. 2017;317(9):937-946. doi: 10.1001/jama.2017.0972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farhan SMK, Howrigan DP, Abbott LE, et al. ; ALSGENS Consortium; FALS Consortium; Project MinE Consortium; CReATe Consortium . Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat-shock protein. Nat Neurosci. 2019;22(12):1966-1974. doi: 10.1038/s41593-019-0530-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun MY, Linsenbardt AJ, Emnett CM, et al. 24(S)-Hydroxycholesterol as a modulator of neuronal signaling and survival. Neuroscientist. 2016;22(2):132-144. doi: 10.1177/1073858414568122 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eAppendix
eFigure 1. Ancestry principal component analysis between HapMap reference samples of European (CEU), Chinese (CHB), Japanese (JPT), and African (YRI) ancestry and study participants recruited from Europe, Canada, and the USA
eFigure 2. Quantile-quantile plot of the discovery exome sequencing series for gene-based burden of rare synonymous alleles
eFigure 3. Principal component analysis of genetic ancestry for the discovery exome sequencing series from Japan, Singapore, and South Africa
eFigure 4. Principal component analysis of genetic ancestry for participants carrying CYP39A1 alleles predicted by Polyphen2 to be damaging
eFigure 5. Exome-wide variant count distribution for the discovery exome sequencing series
eFigure 6. Exome-wide variant count distribution for first replication series
eFigure 7. Number of singleton variants for each sample in the discovery exome sequencing analysis, analyzed by country of enrollment
eFigure 8. Ratio of heterozygous / homozygous genotypes for each sample in the discovery exome sequencing analysis, analyzed by country of enrollment
eFigure 9. Experimental design of the functional enzymatic activity assay for CYP39A1 alleles
eFigure 10. Validation of experimental design of the functional enzymatic activity assay for CYP39A1 alleles
eFigure 11. Determination of the median reduction in enzymatic activity relative to wild-type for the 34 CYP39A1 sequence variants classified as functionally deficient
eFigure 12. Quantile-quantile plot of the discovery exome sequencing results for gene-based burden of rare alleles predicted by Polyphen-2 to be damaging
eFigure 13. Capillary sequencing traces (using the Sanger method) for a selection of CYP39A1 rare alleles that were predicted by Polyphen2 to be damaging
eFigure 14. Assessment of reproducibility of the CYP39A1 functional enzymatic assay
eFigure 15. Box plots showing relative expression levels of CYP39A1 mRNA in ocular tissues derived from normal human donors using real-time PCR
eFigure 16. Schematic representation of the classic and alternative pathways of cholesterol metabolism and bile acid biosynthesis focusing on steps involving cytochrome P450 (CYP) enzymes
eFigure 17. Box plots showing relative expression levels of transcripts for CYP7A1, CYP7B1, CYP39A1, CYP46A1 and CYP51A1 in ciliary body tissues
eFigure 18. Expression of CYP39A1 protein in ocular tissues of normal human donor eyes as determined by immunofluorescence labelling of cryosections
eFigure 19. Expression of CYP39A1 protein in ciliary body tissue of normal human donor eyes and eyes affected by exfoliation syndrome as determined by immunohistochemistry on paraffin sections
eFigure 20. Immunohistochemical analysis using anti-CYP39A1 antibodies on the retina
eFigure 21. Cholesterol deposition in ciliary body tissues of the eye
eTable 1. Sample sets included in the first and second replication stages of this study
eTable 2. Association of rare synonymous variant burden in CYP39A1 with risk for exfoliation syndrome
eTable 3. List of CYP39A1 rare synonymous alleles from the discovery and first replication series
eTable 4. List of CYP39A1 variants subjected to functional enzymatic assays
eTable 5. Primer sequences for CYP39A1 allele-specific mutagenesis
eTable 6. Primers and hybridization probes used for quantitative real-time PCR
eTable 7. Statistical power calculations
eTable 8. Summary statistics for the discovery whole exome sequencing study from Japan, Singapore, and South Africa (Excel file in Supplement 2)
eTable 9. Damaging LOXL1 mutations are associated with protection from exfoliation syndrome
eTable 10. Association of damaging CYP39A1 mutations with risk for exfoliation syndrome
eTable 11. List of CYP39A1 rare coding-sequence variants (minor allele frequency <1%) that were predicted to be damaging by the Polyphen-2 bioinformatic algorithm
eTable 12. Association of three common coding-sequence polymorphisms in in CYP39A1 with risk for exfoliation syndrome
eTable 13. List of CYP39A1 rare synonymous alleles predicted to be benign by Polyphen-2 and tested for their enzymatic activity
eReferences
Excel file