

Abstract

Molecular dynamics (MD) simulations are widely used to monitor time-resolved motions of biomacromolecules, although it often remains unknown how closely the conformational dynamics correspond to those occurring in real life. Here, we used a large set of open-access MD trajectories of phosphatidylcholine (PC) lipid bilayers to benchmark the conformational dynamics in several contemporary MD models (force fields) against nuclear magnetic resonance (NMR) data available in the literature: effective correlation times and spin–lattice relaxation rates. We found none of the tested MD models to fully reproduce the conformational dynamics. That said, the dynamics in CHARMM36 and Slipids are more realistic than in the Amber Lipid14, OPLS-based MacRog, and GROMOS-based Berger force fields, whose sampling of the glycerol backbone conformations is too slow. The performance of CHARMM36 persists when cholesterol is added to the bilayer, and when the hydration level is reduced. However, for conformational dynamics of the PC headgroup, both with and without cholesterol, Slipids provides the most realistic description because CHARMM36 overestimates the relative weight of ∼1 ns processes in the headgroup dynamics. We stress that not a single new simulation was run for the present work. This demonstrates the worth of open-access MD trajectory databanks for the indispensable step of any serious MD study: benchmarking the available force fields. We believe this proof of principle will inspire other novel applications of MD trajectory databanks and thus aid in developing biomolecular MD simulations into a true computational microscope—not only for lipid membranes but for all biomacromolecular systems.
Introduction
Ever since the conception of the Protein Data Bank (PDB)1,2 and Genbank,3,4 open access to standardized and searchable pools of experimental data has revolutionized scientific research. Constantly growing and improving in fidelity due to collaborative effort,5−8 the now hundreds of databanks9 fuel the data-driven development of biomolecular structure determination,10 refinement,11 prediction,12 and design13 approaches as well as the development of drugs,14,15 materials,16,17 and more.18,19 It is clear that open data enables scientific progress that is far beyond the resources of a single research group or institute. Consequently, the call for public availability and conservation of data has extended to molecular dynamics (MD) simulation trajectories of biomolecules,20−22 and the discussion on how and by whom such databanks for dynamic structures would be set up is currently active.23−26 While there are currently no general MD databanks in operation, individual databanks are accepting contributions on nucleic acid,27 protein/DNA/RNA,28 cyclodextrin,29 G-protein-coupled receptor,30 and lipid bilayer31 simulations.
Since 2013, the NMRlipids Project (nmrlipids.blogspot.fi) has promoted a fully open collaboration approach, where the whole scientific research process—from initial ideas and discussions to analysis methods, data, and publications—is all the time publicly available.32 While its main focus has been on conformational ensembles of different lipid headgroups and on ion binding to lipid membranes,32−34 the NMRlipids Project has also built a databank31 (zenodo.org/communities/nmrlipids) containing hundreds of atomistic MD trajectories of lipid bilayers and indexed at nmrlipids.fi.
MD databanks are expected to be particularly relevant for disordered biomolecules, such as biological lipids composing cellular membranes or intrinsically disordered proteins. These, in contrast to folded proteins or DNA strands, cannot be meaningfully described by the coordinates of a single structure alone. Realistic MD simulations, however, can provide the complete conformational ensemble and dynamics of such molecules as well as enable studies of their biological functions in complex biomolecular assemblies. Unfortunately, the current MD force fields largely fail to capture the conformational ensembles of lipid headgroups and disordered proteins.32,34−37 Therefore, before they can be used to draw conclusions, the quality of MD simulations must always be carefully assessed against structurally sensitive experiments. For lipid bilayers, such evaluation is possible against NMR and scattering data.38
Here, we demonstrate the use of a pre-existing, publicly available set of MD trajectories to rapidly evaluate the fidelity of phospholipid conformational dynamics in state-of-the-art force fields. The rate at which individual molecules sample their conformational ensemble is traditionally used to assess if a given MD simulation has converged. Going beyond such practicalities, realistic dynamics are particularly desired for the intuitive interpretation of NMR experiments sensitive to molecular motions39 as well as to understand the dynamics of biological processes where molecular deformations play a rate-limiting role, such as membrane fusion.40 The here presented comprehensive comparison of dynamics between experiments and different MD models at various biologically relevant compositions and conditions is thus likely to facilitate the development of increasingly realistic phospholipid force fields.
Above all, our results demonstrate the power of publicly available MD trajectories in creating new knowledge at a lowered computational cost and high potential for automation. We believe that this paves the way for novel applications of MD trajectory databanks as well as underlines their usefulness—not only for lipid membranes but for all biomolecular systems.
Methods
Lipid Conformational Dynamics in NMR Data
We analyzed the veracity of phosphatidylcholine (PC) lipid dynamics in MD based on two quantities that are readily available from published39,41−4313C NMR experiments and directly quantifiable from atomistic MD simulations: the effective C–H bond correlation times τe and the spin–lattice relaxation rates R1.
Effective C–H Bond Correlation Times τe
In a lipid bilayer in the liquid crystalline state, each individual lipid samples its internal conformational ensemble and rotates around the membrane normal. Lipid conformational dynamics are reflected in the second-order autocorrelation functions of its C–H bonds
| 1 |
where the angular brackets
depict time average, μ⃗(t) is the unit
vector in the direction of the C–H bond at time t, and P2 is the second-order Legendre
polynomial 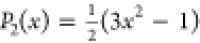 .
.
To analyze the internal dynamics of lipids, the C–H bond autocorrelation function is often written as a product
| 2 |
where gf(τ) characterizes the fast decays owing to, e.g., the internal dynamics and rotation around membrane normal, and gs(τ) the slow decays that originate from, e.g., lipid diffusion between lamellae with different orientations and periodic motions due to magic-angle spinning conditions (Figure 1). Ferreira et al.41 have experimentally demonstrated that, for all phospholipid carbons, the motional correlation times contributing to gf are well below μs and to gs well above 100 μs. This separation of timescales gives rise to the plateau g(1 μs ≲ τ ≲ 100 μs) = SCH2 illustrated in Figure 1. SCH is the C–H bond order parameter
| 3 |
where θ(t) is the angle between the C–H bond and the bilayer normal. SCH can be independently measured using dipolar coupling in 13C or quadrupolar coupling in 2H NMR experiments. Knowing the set of SCH for all the C–H bonds in a lipid is highly useful in order to evaluate its conformational ensemble.38
Figure 1.

C–H bond autocorrelation function g(τ). (A) Idealized illustration of the fast (white background) and the slow (green) mode of the correlation function in solid-state NMR experiments. The fast mode decays to a plateau on which g(τ) = SCH2, while the slow mode gives the final descent to zero. Oscillations at the slow mode region are due to magic-angle spinning. (B) Typical g(τ) obtained from an MD simulation, showing the decay toward SCH2. The gray area under the curve is equal to (1 – SCH2)τe.
As SCH describe the conformational ensemble of the lipid, the fast-decaying component gf of the C–H bond autocorrelation function intuitively reflects the time needed to sample these conformations. The complex internal dynamics containing multiple timescales can be conveniently summarized using the effective correlation time
| 4 |
which is related to the gray shaded area below the correlation function in Figure 1. The τe detects essentially an average over all the timescales relevant for the lipid conformational dynamics. Their relation to process speeds is intuitive: an increase in long-lived correlations increases τe.
Spin–Lattice Relaxation Rates R1
The C–H bond dynamics relate to R1, the spin–lattice relaxation rate, through
| 5 |
where ωH is the 1H and ωC the 13C NMR Larmor frequency and NH is the number of hydrogens covalently bonded to the carbon. The rigid dipolar coupling constant dCH ≈ – 2π × 22 kHz for the methylene bond. The spectral density j(ω) is given by the Fourier transformation
| 6 |
of the C–H bond autocorrelation function g(τ) (eq 1). Clearly, the connection between R1 and molecular dynamics is not straightforward; the magnitude of R1 does, however, reflect the relative significance of processes with timescales near the inverse of ωH and ωC. These two frequencies depend on the field strength used in the NMR experiments: Typically, R1 is most sensitive to motions with timescales of ∼0.1–10 ns. (In our experimental data,39,41−43 ωC = 125 MHz and ωH = 500 MHz, which gives (2π × 125 MHz)−1 = 1.3 ns and (2π × 625 MHz)−1 = 0.25 ns.) A change in given R1, therefore, indicates a change in the relative amount of processes occurring in a window around the sensitive timescale; inferring also the direction to which the processes changed (speedup/slowdown) requires measuring R1 at various field strengths.
Data Acquisition and Analysis
All the experimental quantities used in this work were collected from the literature sources39,41−43 cited at the respective figures.
The simulation trajectories were collected from the general-purpose open-access repository Zenodo (zenodo.org), with the majority of the data originating from the NMRlipids Project32,33 (nmrlipids.blogspot.fi). The trajectories were chosen by hand based on how well the simulation conditions matched the available experimental data (lipid type, temperature, cholesterol content, and hydration) and how precisely one could extract the quantities of interest from the trajectory (length of simulation and system size). Note that, apart from the sampling accuracy, simulation size does not affect τe and R1 (Figure 2). Table 1 lists the chosen trajectories of pure POPC (1-palmitoyl-2-oleoyl-glycero-3-phosphocholine) bilayers at/near room temperature and at full hydration; Table 2 lists the trajectories with cholesterol; and Table 3 those with varying hydration. Full computational details for each simulation are available at the cited Zenodo entry.
Figure 2.

Effective correlation times (τe, upper panel) and R1 rates (lower panel) do not markedly depend on the system size. Shown are two CHARMM36 POPC data sets that varied the size while keeping other simulation parameters fixed: ref (44) (blue, system sizes 72 and 648 lipids)45 and ref (46) (red, system sizes 200, 800, and 1800 lipids). Both data sets are shown normalized against their smallest system. The 15 datapoints shown for each system correspond, from left to right, to the carbon segments γ, β, α, g3, ..., C17/C15′, C18/C16′, cf. Figure 3.
Table 1. Analyzed Open-Access MD Trajectories of Pure POPC Lipid Bilayers at Full Hydration.
| force-field lipid/water | Nla | Nwb | Tc (K) | tanald (ns) | filese |
|---|---|---|---|---|---|
| Berger-POPC-0747/SPC48 | 256 | 10,240 | 300 | 300 | [49] |
| CHARMM3650/TIP3P51 | 256 | 8704 | 300 | 300 | [52] |
| MacRog53/TIP3P54 | 128 | 5120 | 300 | 500 | [55] |
| Lipid1456/TIP3P54 | 72 | 2234 | 303 | 50 | [57] |
| Slipids58/TIP3P54 | 200 | 9000 | 310 | 500 | [59] |
| ECC60/SPC-E61 | 128 | 6400 | 300 | 300 | [62] |
Number of POPC molecules.
Number of water molecules.
Simulation temperature. Note that the temperature varied across these openly available simulation data, but in no case was T lower than in the experiment. Thus, as dynamics slows down when the temperature drops, any overestimation of τe by MD (as typically seen in Figure 3) would get worse if the simulations were done at the experimental 298 K.
Trajectory length used for analysis.
Reference for the openly available simulation files.
Table 2. Analyzed Open-Access MD Trajectories of Cholesterol-Containing POPC Bilayers at Full Hydration.
| force-field POPC/water+cholesterol | cchola | Ncholb | Nlc | Nwd | Te (K) | tanalf (ns) | filesg |
|---|---|---|---|---|---|---|---|
| Berger-POPC-0747/SPC48 | 0% | 0 | 128 | 7290 | 298 | 50 | [63] |
| +Höltje-CHOL-1364,65 | 50% | 64 | 64 | 10,314 | 298 | 50 | [66] |
| CHARMM3650/TIP3P51 | 0% | 0 | 200 | 9000 | 310 | 500 | [67] |
| +CHARMM3668 | 50% | 200 | 200 | 18,000 | 310 | 500 | [69] |
| MacRog53/TIP3P54 | 0% | 0 | 128 | 6400 | 310 | 500 | [70] |
| +MacRog53 | 50% | 64 | 64 | 6400 | 310 | 500 | [70] |
| Slipids58/TIP3P54 | 0% | 0 | 200 | 9000 | 310 | 500 | [59] |
| +Slipids71 | 50% | 200 | 200 | 18,000 | 310 | 500 | [59] |
Bilayer cholesterol content (mol %).
Number of cholesterol molecules.
Number of POPC molecules.
Number of water molecules.
Simulation temperature.
Trajectory length used for analysis.
Reference for the openly available simulation files.
Table 3. Analyzed Open-Access MD Trajectories of PC Lipid Bilayers under Varying Hydration Level.
| force field lipid/water | lipid | nw/la | Nlb | Nwc | Td (K) | tanale (ns) | filesf |
|---|---|---|---|---|---|---|---|
| Berger-POPC-0747/SPC48 | POPC | 40 | 256 | 10,240 | 300 | 300 | [49] |
| POPC | 7 | 128 | 896 | 298 | 60 | [72] | |
| Berger-DLPC-1373/SPC-E61 | DLPCg | 24 | 72 | 1728 | 300 | 80 | [74] |
| DLPCg | 16 | 72 | 1152 | 300 | 80 | [75] | |
| DLPCg | 12 | 72 | 864 | 300 | 80 | [76] | |
| DLPCg | 4 | 72 | 288 | 300 | 80 | [77] | |
| CHARMM3650/TIP3P51 | POPC | 40 | 128 | 5120 | 303 | 140 | [78] |
| POPC | 34 | 256 | 8704 | 300 | 500 | [55] | |
| POPC | 31 | 72 | 2232 | 303 | 20 | [79] | |
| POPC | 15 | 72 | 1080 | 303 | 20 | [80] | |
| POPC | 7 | 72 | 504 | 303 | 20 | [81] | |
| MacRog53/TIP3P54 | POPC | 50 | 288 | 14,400 | 310 | 40 | [82] |
| POPC | 25 | 288 | 7200 | 310 | 50 | [82] | |
| POPC | 15 | 288 | 4320 | 310 | 50 | [82] | |
| POPC | 10 | 288 | 2880 | 310 | 50 | [82] | |
| POPC | 5 | 288 | 1440 | 310 | 50 | [82] |
Water/lipid molar ratio.
Number of lipid molecules.
Number of water molecules.
Simulation temperature.
Trajectory length used for analysis.
Reference for the openly available simulation files.
1,2-Dilauroyl-sn-glycero-3-phosphocholine.
The trajectories were analyzed using in-house scripts. These are available in ref (83), along with a Jupyter notebook outlining an example analysis run. To enable automated analysis of several force fields with differing atom naming conventions, we used the mapping scheme developed within the NMRlipids Project to automatically recognize the atoms and bonds of interest for each trajectory.
After downloading the necessary files from Zenodo, we processed the trajectory with Gromacs gmx trjconv to make the molecules whole; that is, we made sure that, for each covalent bond, the partaking atoms are from the same periodic image of the molecule. For the united atom Berger model, hydrogens were added using the Gromacs 4.0.2 tool g_protonate. We then calculated the SCH (eq 3) with the OrderParameter.py script that uses the MDanalysis84,85 Python library. The C–H bond correlation functions g(τ) (eq 1) were calculated with Gromacs 5.1.486 gmx rotacf (note that on MD timescales gs = 1 so that g = gf) after which the SCH were used to normalize the gf to obtain the reduced and normalized correlation function
| 7 |
that is, the integrand in eq 4.
The effective correlation times τe were then calculated by integrating gf′(τ) from τ = 0 until τ = t0. Here, t0 is the first time point at which gf reached zero: t0 = min{t | gf′(t) = 0}. If gf did not reach zero within tanal/2, the τe was not determined, and we report only its upper and lower estimates.
To quantify the error on τe, we first estimate the error on gf′(τ), where we account for two sources of uncertainty: gf(τ) and SCH2. Performing linear error propagation on eq 7 gives
| 8 |
Here, the ΔSCH was determined as the standard error of the mean of the SCH over the Nl individual lipids in the system.32 Similarly, we quantified the error on gf(τ) by first determining the correlation function gfm(τ) for each individual lipid m over the whole trajectory and then obtaining the error estimate Δgf(τ) as the standard error of the mean over the Nl lipids. Importantly, this gives an uncertainty estimate for gf(τ) at each time point τ.
To obtain the lower bound on τe, we integrate the function gf′(τ) – Δgf(τ) over time from τ = 0 until τ = tl. Here
| 9 |
That is, tl equals the first time point at which the lower error estimate of gf′ reached zero, or tl = tanal/2, if zero was not reached before that point.
To obtain the upper error estimate on τe, we first integrate the function gf′(τ) + Δgf(τ) over time from τ = 0 until tu = min{t0, tanal/2}. Note, however, that this is not yet sufficient, because there could be slow processes that the simulation was not able to see. Although these would contribute to τe with a low weight, their contribution over long times could still add up to a sizable effect on τe. That said, it is feasible to assume (see Figure 1A) that there are no longer-time contributions to gf than something that decays with a time constant of 10–6 s. We use this as our worst case estimate to assess the upper bound for τe, that is, we assume that all the decay of gf from the time point tu onward comes solely from this hypothetical slowest process that decays with a time constant of 10–6 s. The additional contribution to the upper bound for τe then reads
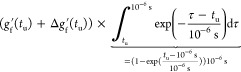 |
10 |
The R1 rates were calculated using eq 5. The spectral density j(ω) was obtained from the normalized correlation function gf′ by fitting it with a sum of 61 exponentials
| 11 |
with logarithmically spaced timescales τi ranging from 1 ps to 1 μs and then calculating the spectral density of this fit based on the Fourier transformation41
| 12 |
The R1 rate of a given C–H pair was first calculated separately for each lipid m (using eq 5 with NH = 1, and jm(ω) obtained for the normalized correlation function gf′m). The resulting Nl measurements per C–H pair were then assumed independent: their mean gave the R1 rate of the C–H pair, and the standard error of the mean its uncertainty. The total R1 rate of a given carbon was obtained as a sum of the R1 rates of its C–H pairs. When several carbons contribute to a single experimental R1 rate due to the overlapping peaks (for example, in C2 carbon in the acyl chains and the γ carbons), the R1 from simulations was obtained as an average over carbons with overlapping peaks. The segment-wise error estimates were obtained by standard error propagation, starting from the uncertainties of the R1 rates of the C–H pairs.
To gain some qualitative insight on the timescales at which the main contributions to the R1 rates arise, we also calculated “cumulative” R1 rates, R1(τ), which contained those terms of the sum in eq 12 for which τi < τ. Note that here the gf′ averaged over lipids was used; therefore, the “cumulative” R1(τ→∞) does not necessarily have exactly the same numerical value as the actual R1.
Finally, we note that the fit of eq 11 provides an alternative to estimating τe because
| 13 |
When the simulation trajectory is not long enough for the correlation function to reach the plateau, integrating gf′ gives a lower bound estimate for τe, while the sum of eq 13 includes also (some) contribution from the longer-time components via the fitting process. However, in practice, the fit is often highly unreliable in depicting the long tails of the correlation function, and thus we chose to quantify τe using the area under gf and estimate its uncertainty as detailed above.
Results and Discussion
Using open-access MD simulation trajectories, we benchmark phospholipid conformational dynamics in six MD force fields. We start with pure POPC bilayers in their liquid crystalline fully hydrated state (see Table 1 for simulation details and Figures 3 and 4 for the data) and then proceed to check the changes in dynamics when cholesterol is added to the bilayer (Table 2 and Figure 5) and when the hydration level is reduced (Table 3 and Figure 6). Our yardsticks are the effective correlations times τe (eq 4) and the R1 rates (eq 5) measured at 125 MHz 13C (500 MHz 1H) Larmor frequency; an MD model with correct rotational dynamics in a window around ∼1 ns will match the experimental R1 rates, whereas the τe reflect all the sub-μs timescales (Figure 1).
Figure 3.
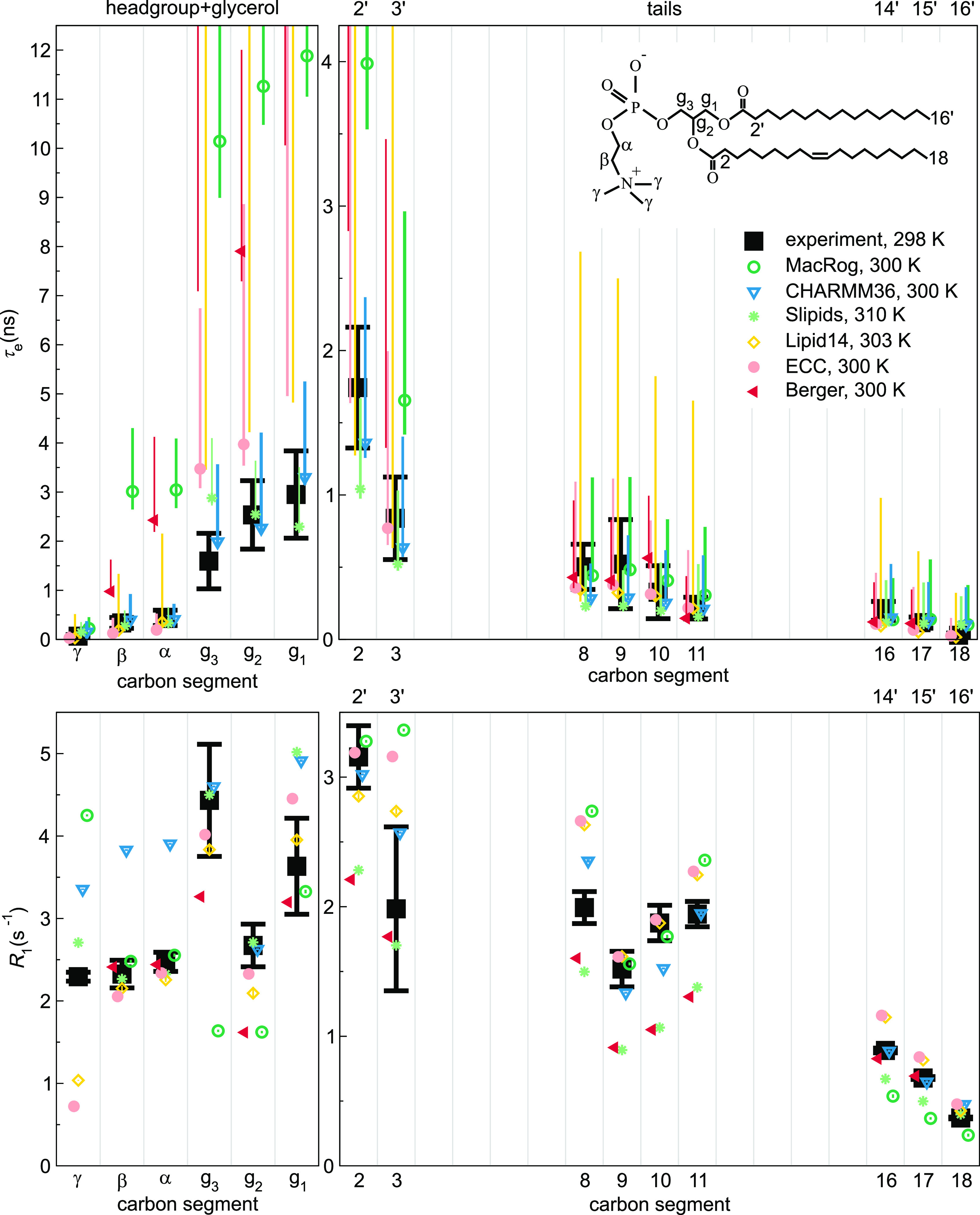
Effective correlation times (τe, top) and R1 rates (bottom) in experiments39 (black) and MD simulations (colored) of POPC bilayers in the Lα phase under full hydration. Inset shows the POPC chemical structure and carbon segment labeling. Each plotted value contains contributions from all the hydrogens within its carbon segment; the data for segments 8–11 are only from the sn-2 (oleoyl) chain, whereas the (experimentally non-resolved) contributions of both tails are included for segments 2–3 (2′–3′ in the sn-1 chain) and 16–18 (14′–16′). Simulation results are only shown for the segments for which experimental data were available. For τe, a simulation data point indicates the average over C–H bonds; however, if τe could not be determined for all bonds, only the error bar (extending from the mean of the lower to the mean of the upper error estimates) is shown. The Berger data for segments γ, C18, and C16′ are left out as the protonation algorithm used to construct the hydrogens post-simulation in united atom models does not preserve the methyl C–H bond dynamics. Table 1 provides further simulation details, while information on the experiments is available at ref (39).
Figure 4.

Contributions to the dynamics of the headgroup segments. (A) Zoom on the headgroup τe (left panel) and R1 (right). (B) “Cumulative” R1(τ) of the γ (top panel), β (middle), and α (bottom) segments. R1(τ) is obtained, as detailed in Methods, by including in the sum of eq 12 only terms with τi < τ. Consequently, at τ → ∞, the R1(τ) approaches the actual R1. (C) Prefactor weights αi from eq 11 of γ (top), β (middle), and α (bottom). Note that panels (B) and (C) show a sliding average over three neighboring data points.
Figure 5.

Effect of cholesterol on POPC conformational dynamics. (A) Experimental effective correlation times τe (top panels) and R1 rates (bottom) in 100/0 and 50/50 POPC/cholesterol bilayers at full hydration, see ref (39) for further details. (B) Change in τe (Δτe, top panels) and R1 (ΔR1, bottom), in NMR (black) and MD (color), when the bilayer composition changes from pure POPC to 50% cholesterol. Error estimates for the simulated Δτe are the maximal possible based on the errors at 0% and 50% cholesterol; for other data, regular error propagation is used. The Berger Δτe is not shown because the available open-access trajectories were too short to determine meaningful error estimates. Table 2 provides further simulation details; for segment labeling, see Figure 3.
Figure 6.
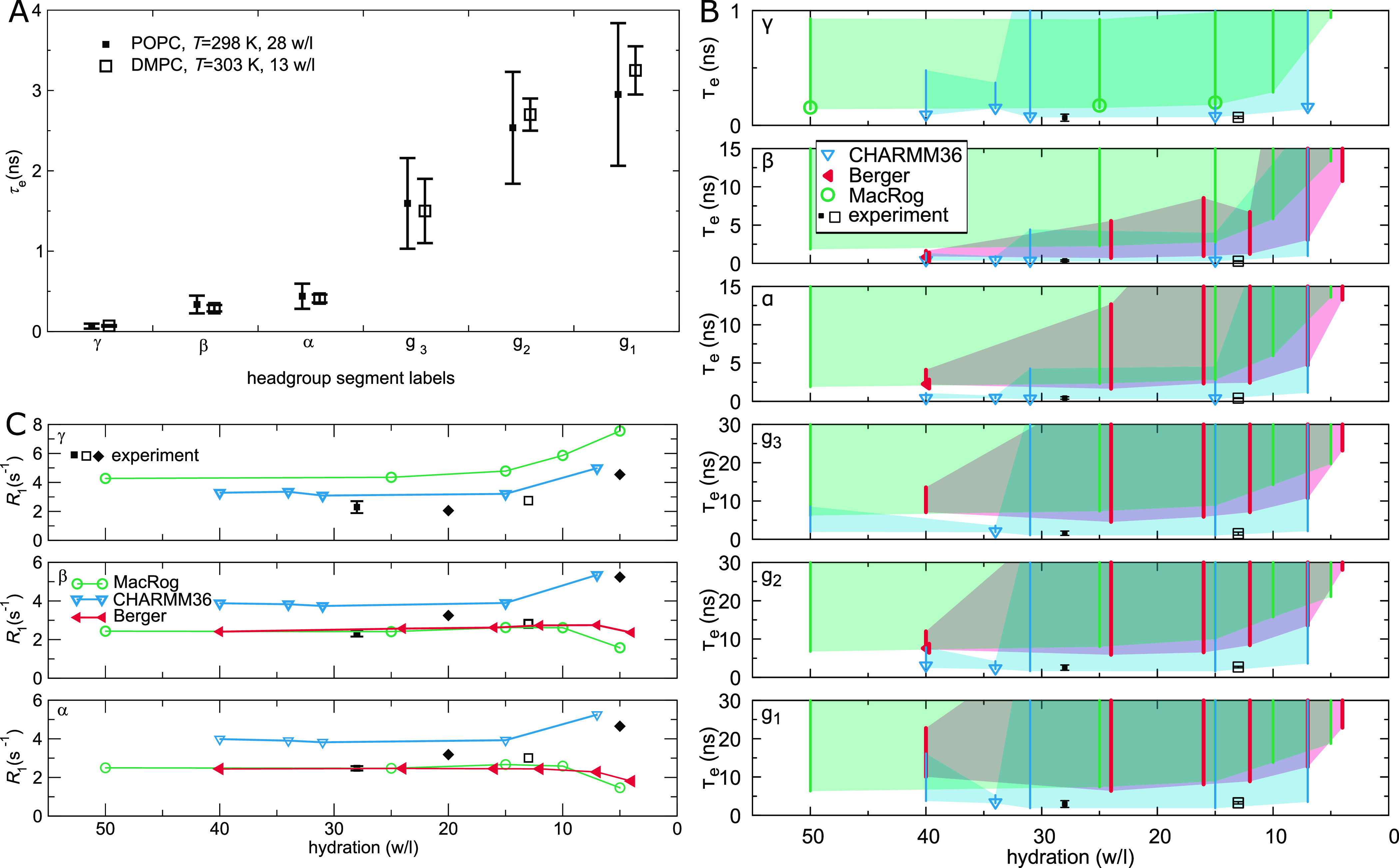
Effect of drying on PC headgroup and glycerol backbone conformational dynamics. (A) Experimental effective correlation times τe for DMPC at low hydration (from ref (42)) do not significantly differ from the τe for POPC at full hydration (from ref (39)). (B) Calculated τe for POPC at decreasing hydration in three MD models. Symbols indicate the mean of segment hydrogens if τe could be determined for all of them; otherwise, only the error bar (extending from the mean of the lower to the mean of the upper uncertainty estimates) is drawn. The area limited by the error bars shaded for visualization. Note that four Berger data points (24, 16, 12, and 4 w/l) are from DLPC. (C) 13C NMR R1 rates (at ωC = 125 MHz) of the PC headgroup segments in experiments and simulations: experiments indicate an increasing trend upon dehydration. Experimental POPC (T = 298 K) data at 28 w/l is from ref (39) (solid boxes), POPC (298 K) at 20 and 5 w/l from ref (43) (solid diamonds), and DMPC (303 K) at 13 w/l from ref (42) (open boxes). See Table 3 for simulation details.
Pure POPC at Full Hydration: Slipids and CHARMM36 Reproduce τe Excellently
The top panels of Figure 3 compare the effective correlation times τe obtained for fully hydrated POPC bilayers in experiments (black) and in six different MD force fields (color). We see that—as implied by the discussion leading to eq 10—sub-μs MD simulations typically lead to asymmetric error bars on τe; if these open-access trajectories were extended, the τe values would more likely increase than decrease. Qualitatively, every force field captures the general shape of the τe profile: dynamics slows down toward the glycerol backbone in both the headgroup and the tails.
Quantitatively, most MD simulations tend to produce too slow dynamics in the glycerol region (Figure 3). This is consistent with previous results for the Berger model41 and with the insufficient conformational sampling of glycerol backbone torsions observed in 500-ns-long CHARMMc32b287,88 simulations of a PC lipid.89
The best overall τe performance is seen in Slipids and in particular CHARMM36 (Figure 3). This is in line with CHARMM36 reproducing the most realistic conformational ensembles for the headgroup and glycerol backbone among the MD simulation force fields benchmarked here.32,34 Indeed, it is important to keep in mind that the conformational ensembles greatly differ between force fields and are not exactly correct in any of them.32,34 Consequently, the calculated τe times and R1 rates depict the dynamics of sampling a somewhat different and incorrect phase space for each model. To this end, we try to avoid overly detailed discussion on the models and rather concentrate on common and qualitative trends. That said, there are a few carbon segments in the data for which the experimental order parameters, R1, and τe are all (almost) reproduced by simulations, suggesting that both the conformational ensemble and the dynamics are correctly captured by MD in these cases. For example, Slipids performs well at the β and α and CHARMM36 at the g3, g2, and C2 segments. These are, however, exceptions.
An Excellent τe May Be Accompanied by a Poor R1, or Vice Versa
The lower panels of Figure 3 compare the experimental and simulated R1 rates under the same conditions that were used for the τe above. Notably, there are several instances where the R1 comparison distinctly differs from what was seen for τe.
There are cases where a matching R1 is accompanied by a larger-than-experimental τe. MacRog for the β, α, and g1 segments provides a prominent example of this. Such a combination suggests that MD has the correct relative weight of 1 ns-scale dynamics but has too slow long-time dynamics.
There are also cases where τe matches experiments but R1 does not, such as the β and α segments in CHARMM36. Therein, a cancellation of errors occurs in τe: the overestimation of the relative weight of 1 ns-scale dynamics is compensated by wrong dynamics at the other timescales. As CHARMM36 overall performs rather well for C–H bond order parameters, R1, and τe, we proceed to study this shortcoming on the headgroup R1 rates in some more detail.
Conformational Dynamics of PC Headgroup Segments in MD
Figure 4A zooms in on the headgroup (γ, β, and α) segments, whose τe were not clearly visible on the scale of Figure 3. We see that, for γ, no force field provides both τe and R1, but Slipids comes closest. For β and α, Slipids captures both measurables near perfectly. In other words, among the benchmarked force fields, Slipids gives the most realistic description of the conformational dynamics in the headgroup region. CHARMM36, e.g., overestimates (R1) the relative weight of timescales around ∼1 ns.
To investigate closer how the differences between force fields arise, Figure 4B shows the “cumulative” R1(τ), where the ranges of steepest increase indicate timescales that most strongly contribute to R1 rates.
For the γ segment, Figure 4B shows that, for models that overestimate the R1 rate (MacRog, CHARMM36, and Slipids, see Figure 4A), the major contribution to R1 arises at τ > 50 ps, whereas for models that underestimate R1 (Lipid14 and ECC), the major contribution comes from τ < 50 ps. This also manifests in the distribution of fitting weights (αi in eq 11) in Figure 4C: the later non-zero weights occur, the larger is the resulting R1 of γ.
For the β and α segments, Figure 4B shows that the main contribution to R1 rates arises from processes between 100 ps and 1 ns. CHARMM36 has the largest relative weights of all models in this window (Figure 4C), which explains its overestimation of R1 of β and α. All the other models have R1 rates close to experiments, but only Slipids simultaneously gives also the τe correctly. Notably, Slipids has its largest weights at τ < 100 ps. Indeed, the considerable weights at short (<10 ps) timescales in all models except MacRog and at long (>10 ns) timescales in MacRog and Berger hardly manifest in R1. However, the latter contribute heavily to τe, which is thus considerably overestimated by MacRog and Berger (Figure 3).
It would be highly interesting to identify the origins of the observed artificial timescales, particularly for the 0.1–1 ns window overpresented in CHARMM36, and propose how to correct those in the simulation models. After all, it is known that the R1 rates of mono- and disaccharides90 and proteins91 in solution agree satisfactorily with experiments when the artificially low viscosity of TIP3P water is accounted for by a simple scaling. Viscosity at the bilayer–water interface, however, remains an open question—although one which a careful comparison between spin relaxation rates of lipid headgroups in simulations and experiments might be able to answer. Nevertheless, we refrain from further analysis here as the connection between the fitted correlation times and the correlation times of distinct motional processes, such as dihedral rotations and lipid wobbling, turns out to be highly non-trivial.
Effect of Cholesterol
An essential component in cell membranes, cholesterol has various biological functions. It is well known to order the acyl chains in lipid bilayers, but its effect on the headgroup is more controversial.65,92 For example, it has been proposed that lipid headgroups reorganize to shield cholesterol from water.92 However, while acyl chains do substantially order, NMR experiments show no significant conformational changes in the headgroup upon addition of even 50% of cholesterol, which suggests that the tail and head regions behave essentially independently.32,65 In principle, the headgroups could shield cholesterol from water even without changing their conformational ensemble: by reorienting only laterally on top of the cholesterol. In this case, one would expect the rotational dynamics of headgroup segments to change when cholesterol is added.
Top panels of Figure 5A depict the experimental effective correlation times τe in pure POPC bilayers and in bilayers containing 50% cholesterol. The τe at the glycerol backbone slow down markedly when cholesterol is added. Tail segment dynamics slows down too, most notably close to the glycerol backbone. In stark contrast, the τe of the headgroup segments (γ, β, and α) remain unaffected. Furthermore, cholesterol induces no measurable change in the headgroup β and α segment dynamics at short (∼1 ns) timescales, as demonstrated by the experimental R1 rates (Figure 5A, bottom panels). That said, there is a small but measurable impact on R1 at γ. In summary, these experimental findings support the idea39 that the acyl chains and the headgroup can respond—in agreement with the relative uncoupling of the PC head and tails reported in simulations93—almost independently to changes in conditions and composition.
All four benchmarked force fields (Figure 5B) qualitatively reproduce the experimental increase in τe: Slipids and CHARMM36 give rather decent magnitude estimates, while MacRog grossly overestimates the slowdown of glycerol, C2, and C3 segments. Notably, MacRog appears to predict slowdown also for the headgroup (β and α), for which experiments detect no change. Note that, while CHARMM36 correctly shows no change in τe of the γ, β, and α segments, it does predict an erroneous ΔR1 for all three, indicating some inaccuracies in the headgroup rotational dynamics. Such inaccuracies might be reflected in the recent findings94 (obtained using CHARMM36) that the headgroups of PC-lipids neighboring (within 6.6 Å) a lone cholesterol spend more time on top of the said cholesterol than elsewhere. Interestingly, the tail ΔR1 seem to be qualitatively reproduced by all three all-atom force fields, whereas Berger fails to capture the trend at the oleoyl double bond. All these findings are in line with the general picture obtained from C–H bond order parameters:38 MD simulations capture the changes in the acyl chain region rather well but changes in and near the glycerol backbone region can be overestimated. Of the benchmarked force fields, CHARMM36 appears most realistic in reproducing the effects of cholesterol on the glycerol backbone—and Slipids on the PC headgroup—conformational dynamics.
Effect of Drying
Understanding the impact of dehydration on the structure and dynamics of lipid bilayers is of considerable biological interest. Dehydrated states are found, e.g., in skin tissue. Most prominently, the process of membrane fusion is always preceded by removal of water between the approaching surfaces, and thus the dehydration-imposed changes can considerably affect fusion characteristics, such as its rate.
Figure 6A shows how a mild dehydration affects C–H bond dynamics in the PC headgroup and glycerol backbone; the plot compares the experimental effective correlation times τe measured for POPC at full hydration and for DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) at 13 waters per lipid. The τe are the same within experimental accuracy, which suggests two conclusions. First, the headgroup (γ, β, and α) τe are rather insensitive to the chemical identities of the tails. This is analogous to what was seen experimentally when adding cholesterol (Figure 5A): structural changes in the tail and glycerol regions do not (need to) affect the headgroup dynamics. Second, a mild dehydration does not alter the τe in the headgroup and glycerol regions.
Figure 6B shows the effects of dehydration in three MD models. Combination of the unrealistically slow dynamics, especially in the glycerol backbone (Figure 3), and the relatively short lengths of the available open-access trajectories (Table 3) led to large uncertainty estimates; thus, we only point out qualitative trends here. For all headgroup and glycerol segments, the simulated τe indicate slowdown upon dehydration. This is manifested in the increase in the magnitude of the error estimate (cf. the Berger data for β and α) as well as in the increase in the lower limit of the error. For CHARMM36 the lower error estimates stay almost constant all the way until 7 w/l, whereas for Berger and MacRog, they hint that a retardation of dynamics starts already between 15 and 10 w/l.
These simulational findings suggest that experiments reducing hydration levels below 10 w/l would also show an increase in τe. This prediction is in line with the exponential slowdown of the headgroup conformational dynamics upon dehydration that was indicated by 2H NMR R1 measurements of DOPC bilayers: R1 ≈ exp( – nw/l/4).95 The slowdown was attributed to the reduced effective volume available for the headgroup95 as it tilts toward the membrane upon dehydration; such tilt is observed via changes of the lipid headgroup order parameters96 and is qualitatively reproduced by all the simulation models.32
Figure 6C shows a collection of experimental 13C NMR R1 rates for the headgroup segments at different water contents; in addition to the full hydration POPC data from Figure 3, DMPC at 13 w/l42 and POPC at 20 and 5 w/l43 are shown. Experimentally, an increasing trend with decreasing hydration is observed for all three segments, indicating changes of headgroup dynamics at short (∼1 ns) timescales. Interestingly, only CHARMM36 captures this, whereas Berger and MacRog give decreasing R1 rates for β and α.
The slowdown characteristics discussed here are of significance not only for computational studies of intermembrane interactions, such as fusion, but also when simulating a bilayer (stack) under low hydration: slower dynamics require longer simulation times for equilibration, for reliably quantifying the properties of the bilayers, and for observing rare events.
Conclusions
We have here demonstrated that open-access databanks of MD trajectories enable the creation of new scientific information without running a single new simulation. More specifically, we have benchmarked (against published NMR data39,41−43) the conformational dynamics of a wide range of phosphatidylcholine MD models using existing open-access trajectories from the Zenodo repository, in particular those belonging to the NMRlipids Databank (zenodo.org/communities/nmrlipids).
We found that every MD model captures the 13C NMR effective correlation time (τe) profile of POPC qualitatively, but that most are prone to too slow dynamics of the glycerol backbone C–H bonds (Figure 3). While no force field perfectly reproduces all the experimental data, CHARMM36 and Slipids have overall impressive τe. This is a particularly exciting finding concerning CHARMM36 as it is also known to reproduce quite well the experimental conformational ensemble.32 That said, we do find that CHARMM36 struggles with the balance of dynamics in the headgroup region: The R1 rates, sensitive for ∼1 ns processes, are too high for the γ, β, and α segments (Figure 4). In fact Slipids, which also reproduces the experimental headgroup order parameters,32 appears to outperform CHARMM36 when it comes to headgroup conformational dynamics (Figure 4).
Further, we found that when cholesterol is mixed into a POPC bilayer, MD qualitatively captures the slowdown of conformational dynamics in the tail and glycerol regions (Figure 5). However, the benchmarked force fields overestimate the changes in the ∼1 ns dynamics of the headgroup—except Slipids, which captures well the effects of cholesterol on PC headgroup conformational dynamics.
Finally, we found that, upon reducing the water content below 10 waters per lipid, MD exhibits slowdown of headgroup and backbone dynamics in qualitative agreement with experimental data. That said, only CHARMM36 (but not Berger or MacRog) qualitatively captures the experimentally detected increase in R1 rates upon dehydration (Figure 6).
While work is still needed in capturing even the correct phospholipid conformations,32 realistic dynamics will be an essential part of developing MD into a true computational microscope. Here, we gathered a set of published experimental 13C NMR data on phosphatidylcholine conformational dynamics and charted the typical features of the existing MD models against it, thus laying the foundation for further improvement of MD force fields. Importantly, our work demonstrates the potential of open-access MD trajectories in achieving such benchmarks at a reduced computational and labor cost—but it also highlights the challenges inherent in using such data: not all system permutations might readily exist (here the dehydration data for Lipid14 and Slipids were lacking, see Figure 6); the available sampling (simulation length and size) might vary, requiring extreme care with error estimation; and one has to remain aware of the subtle differences in the many simulation parameters (barostats, temperatures, characteristics of the MD engines, etc.). That said, it has not escaped our notice that a pool of well indexed and documented open-access data provides an ideal platform for automation, which in turn will facilitate faster progress in pinpointing the typical failures of the existing force fields, in identifying key differences in models describing chemical variations of the same molecule type (such as different lipid headgroups), and in developing better models through data-driven approaches.
Acknowledgments
H.S.A. gratefully acknowledges the support from Osk. Huttunen Foundation, Finnish Academy of Science and Letters (Foundations’ Post Doc Pool), Instrumentarium Science Foundation, and the Alexander von Humboldt Foundation. O.H.S.O. acknowledges the Academy of Finland (315596 and 319902) for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.0c01299.
SMILES database (ZIP)
The authors declare no competing financial interest.
Supplementary Material
References
- Crystallography: Protein Data Bank. Nat. New Biol. 1971, 233, 223.
- Burley S. K.; Berma H. M.; Bhikadiya C.; Bi C.; Chen L.; Constanzo L. D.; Christie C.; Duarte J. M.; Dutta S.; Feng Z. Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. 10.1093/nar/gky949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan E.; Carrico C. DNA Database. Science 1982, 218, 108. 10.1126/science.7123223. [DOI] [PubMed] [Google Scholar]
- Sayers E. W.; Cavanaugh M.; Clark K.; Ostell J.; Pruitt K. D.; Karsch-Mizrachi I. GenBank. Nucleic Acids Res. 2020, 48, D84–D86. 10.1093/nar/gkz956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt M. Growth of novel protein structural data. Proc. Natl. Acad. Sci. 2007, 104, 3183–3188. 10.1073/pnas.0611678104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzezinski D.; Dauter Z.; Minor W.; Jaskolski M. On the evolution of the quality of macromolecular models in the PDB. FEBS J. 2020, 287, 2685–2698. 10.1111/febs.15314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris D. J. Can you bank on GenBank?. Trends Ecol. Evol. 2003, 18, 317–319. 10.1016/S0169-5347(03)00150-2. [DOI] [Google Scholar]
- Steinegger M.; Salzberg S. L. Terminating contamination: large-scale search identifies more than 2,000,000 contaminated entries in GenBank. Genome Biol. 2020, 21, 115. 10.1186/s13059-020-02023-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigden D. J.; Fernández X. M. The 27th annual Nucleic Acids Research database issue and molecular biology database collection. Nucleic Acids Res. 2020, 48, D1–D8. 10.1093/nar/gkz1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkin A. J.; Thomas J. M. H.; Simkovic F.; Keegan R. M.; Rigden D. J. Molecular replacement using structure predictions from databases. Acta Crystallogr., Sect. D: Struct. Biol. 2019, 75, 1051–1062. 10.1107/S2059798319013962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leelananda S. P.; Lindert S. Using NMR Chemical Shifts and Cryo-EM Density Restraints in Iterative Rosetta-MD Protein Structure Refinement. J. Chem. Inf. Model. 2020, 60, 2522–2532. 10.1021/acs.jcim.9b00932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior A. W.; Evans R.; Jumper J.; Kirkpatrick J.; Sifre L.; Green T.; Qin C.; Žídek A.; Nelson A. W. R.; Bridgland A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- Huang P.-S.; Boyken S. E.; Baker D. The coming of age of de novo protein design. Nature 2016, 537, 320–327. 10.1038/nature19946. [DOI] [PubMed] [Google Scholar]
- Westbrook J. D.; Burley S. K. How Structural Biologists and the Protein Data Bank Contributed to Recent FDA New Drug Approvals. Structure 2019, 27, 211–217. 10.1016/j.str.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Mayorga K.; Madariaga-Mazon A.; Medina-Franco J. L.; Maggiora G. The impact of chemoinformatics on drug discovery in the pharmaceutical industry. Expert Opin. Drug Discovery 2020, 15, 293–306. 10.1080/17460441.2020.1696307. [DOI] [PubMed] [Google Scholar]
- Senderowitz H.; Tropsha A. Materials Informatics. J. Chem. Inf. Model. 2018, 58, 2377–2379. 10.1021/acs.jcim.8b00927. [DOI] [PubMed] [Google Scholar]
- Wan X.; Feng W.; Wang Y.; Wang H.; Zhang X.; Deng C.; Yang N. Materials Discovery and Properties Prediction in Thermal Transport via Materials Informatics: A Mini Review. Nano Lett. 2019, 19, 3387–3395. 10.1021/acs.nanolett.8b05196. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Zorin A.; Dass G.; Vu M.-T.; Xu P.; Glont M.; Vizcaíno J. A.; Jarnuczak A. F.; Petryszak R.; Ping P.; et al. Quantifying the impact of public omics data. Nat. Commun. 2019, 10, 3512. 10.1038/s41467-019-11461-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z.; Verdiguel N.; Di Costanzo L.; Goodsell D. S.; Westbrook J. D.; Burley S. K.; Zardecki C. Impact of the Protein Data Bank Across Scientific Disciplines. Data Sci. J. 2020, 19, 25. 10.5334/dsj-2020-025. [DOI] [Google Scholar]
- Feig M.; Abdullah M.; Johnsson L.; Pettitt B. M. Large scale distributed data repository: design of a molecular dynamics trajectory database. Future Gener. Comput. Syst. 1999, 16, 101–110. 10.1016/S0167-739X(99)00039-4. [DOI] [Google Scholar]
- Tai K.; Murdock S.; Wu B.; Ng M. H.; Johnston S.; Fangohr H.; Cox S. J.; Jeffreys P.; Essex J. W.; Sansom M. S. P. BioSimGrid: towards a worldwide repository for biomolecular simulations. Org. Biomol. Chem. 2004, 2, 3219–3221. 10.1039/b411352g. [DOI] [PubMed] [Google Scholar]
- Silva C. G.; Ostropytskyy V.; Loureiro-Ferreira N.; Berrar D.; Swain M.; Dubitzky W.; Brito R. M. M.. P-found: The Protein Folding and Unfolding Simulation Repository. In 2006 IEEE Symposium on Computational Intelligence and Bioinformatics and Computational Biology; IEEE: 2006; pp. 1–8.
- Hildebrand P. W.; Rose A. S.; Tiemann J. K. S. Bringing Molecular Dynamics Simulation Data into View. Trends Biochem. Sci. 2019, 44, 902–913. 10.1016/j.tibs.2019.06.004. [DOI] [PubMed] [Google Scholar]
- Abraham M.; Apostolov R.; Barnoud J.; Bauer P.; Blau C.; Bonvin A. M. J. J.; Chavent M.; Chodera J.; Čondić-Jurkić K.; Delemotte L.; et al. Sharing Data from Molecular Simulations. J. Chem. Inf. Model. 2019, 59, 4093–4099. 10.1021/acs.jcim.9b00665. [DOI] [PubMed] [Google Scholar]
- Abriata L. A.; Lepore R.; Dal Peraro M. About the need to make computational models of biological macromolecules available and discoverable. Bioinformatics 2020, 36, 2952–2954. 10.1093/bioinformatics/btaa086. [DOI] [PubMed] [Google Scholar]
- Hospital A.; Battistini F.; Soliva R.; Gelpí J. L.; Orozco M. Surviving the deluge of biosimulation data. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2020, 10, e1449 10.1002/wcms.1449. [DOI] [Google Scholar]
- Hospital A.; Andrio P.; Cugnasco C.; Codo L.; Becerra Y.; Dans P. D.; Battistini F.; Torres J.; Goñi R.; Orozco M.; et al. BIGNASim: a NoSQL database structure and analysis portal for nucleic acids simulation data. Nucleic Acids Res. 2016, 44, D272–D278. 10.1093/nar/gkv1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker G.-J.; Kawabata T.; Kurisu G. The Biological Structure Model Archive (BSM-Arc): an archive for in silico models and simulations. Biophys. Rev. 2020, 12, 371–375. 10.1007/s12551-020-00632-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mixcoha E.; Rosende R.; Garcia-Fandino R.; Piñeiro Á. Cyclo-lib: a database of computational molecular dynamics simulations of cyclodextrins. Bioinformatics 2016, 32, 3371–3373. 10.1093/bioinformatics/btw289. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Espigares I.; Torrens-Fontanals M.; Tiemann J. K. S.; Aranda-García D.; Ramrez-Anguita J. M.; Stepniewski T. M.; Worp N.; Varela-Rial A.; Morales-Pastor A.; Medel-Lacruz B.; et al. GPCRmd uncovers the dynamics of the 3D-GPCRome. Nat. Methods 2020, 17, 777–787. 10.1038/s41592-020-0884-y. [DOI] [PubMed] [Google Scholar]
- Miettinen M. S.; Ollila O. H. S. LDB: Lipid Databank from the NMRlipids Project. Biophys. J. 2019, 116, 91a. 10.1016/j.bpj.2018.11.532. [DOI] [Google Scholar]
- Botan A.; Favela-Rosales F.; Fuchs P. F. J.; Javanainen M.; Kanduč M.; Kulig W.; Lamberg A.; Loison C.; Lyubartsev A.; Miettinen M. S.; et al. Toward Atomistic Resolution Structure of Phosphatidylcholine Headgroup and Glycerol Backbone at Different Ambient Conditions. J. Phys. Chem. B 2015, 119, 15075–15088. 10.1021/acs.jpcb.5b04878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catte A.; Girych M.; Javanainen M.; Loison C.; Melcr J.; Miettinen M. S.; Monticelli L.; Määttä J.; Oganesyan V. S.; Ollila O. H. S.; et al. Molecular electrometer and binding of cations to phospholipid bilayers. Phys. Chem. Chem. Phys. 2016, 18, 32560–32569. 10.1039/C6CP04883H. [DOI] [PubMed] [Google Scholar]
- Antila H.; Buslaev P.; Favela-Rosales F.; Ferreira T. M.; Gushchin I.; Javanainen M.; Kav B.; Madsen J. J.; Melcr J.; Miettinen M. S.; et al. Headgroup Structure and Cation Binding in Phosphatidylserine Lipid Bilayers. J. Phys. Chem. B 2019, 123, 9066–9079. 10.1021/acs.jpcb.9b06091. [DOI] [PubMed] [Google Scholar]
- Robustelli P.; Piana S.; Shaw D. E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. 2018, 115, E4758–E4766. 10.1073/pnas.1800690115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques J.; Arleth L.; Lindorff-Larsen K.; Skepö M. On the Calculation of SAXS Profiles of Folded and Intrinsically Disordered Proteins from Computer Simulations. J. Mol. Biol. 2018, 430, 2521–2539. 10.1016/j.jmb.2018.03.002. [DOI] [PubMed] [Google Scholar]
- Virtanen S. I.; Kiirikki A. M.; Mikula K. M.; Iwaï H.; Ollila O. H. S. Heterogeneous dynamics in partially disordered proteins. Phys. Chem. Chem. Phys. 2020, 22, 21185–21196. 10.1039/D0CP03473H. [DOI] [PubMed] [Google Scholar]
- Ollila O. H. S.; Pabst G. Atomistic resolution structure and dynamics of lipid bilayers in simulations and experiments. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2512–2528. 10.1016/j.bbamem.2016.01.019. [DOI] [PubMed] [Google Scholar]
- Antila H. S.; Wurl A.; Ollila O. H. S.; Miettinen M. S.; Ferreira T. M.. Quasi-uncoupled rotational diffusion of phospholipid headgroups from the main molecular frame. arXiv preprint 2020, arXiv:2009.06774v1. [Google Scholar]
- Han J.; Pluhackova K.; Böckmann R. A. The Multifaceted Role of SNARE Proteins in Membrane Fusion. Front. Physiol. 2017, 8, 5. 10.3389/fphys.2017.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira T. M.; Ollila O. H. S.; Pigliapochi R.; Dabkowska A. P.; Topgaard D. Model-free estimation of the effective correlation time for C-H bond reorientation in amphiphilic bilayers: 1H–13C solid-state NMR and MD simulations. J. Chem. Phys. 2015, 142, 044905 10.1063/1.4906274. [DOI] [PubMed] [Google Scholar]
- Pham Q. D.; Topgaard D.; Sparr E. Cyclic and Linear Monoterpenes in Phospholipid Membranes: Phase Behavior, Bilayer Structure, and Molecular Dynamics. Langmuir 2015, 31, 11067–11077. 10.1021/acs.langmuir.5b00856. [DOI] [PubMed] [Google Scholar]
- Volke F.; Pampel A. Membrane Hydration and Structure on a Subnanometer Scale as Seen by High Resolution Solid State Nuclear Magnetic Resonance: POPC and POPC/C12EO4 Model Membranes. Biophys. J. 1995, 68, 1960–1965. 10.1016/S0006-3495(95)80373-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venable R. M.POPC bilayers from MD simulations run with CHARMM using C36 parameters; 2020; 10.5281/zenodo.4362554. [DOI] [Google Scholar]
- Venable R. M.; Brown F. L. H.; Pastor R. W. Mechanical properties of lipid bilayers from molecular dynamics simulation. Chem. Phys. Lipids 2015, 192, 60–74. 10.1016/j.chemphyslip.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javanainen M.Simulations of large POPC bilayers using the Charmm36 force field; 2017; 10.5281/zenodo.545941. [DOI] [Google Scholar]
- Ollila S.; Hyvönen M. T.; Vattulainen I. Polyunsaturation in Lipid Membranes: Dynamic Properties and Lateral Pressure Profiles. J. Phys. Chem. B 2007, 111, 3139–3150. 10.1021/jp065424f. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; Hermans J.. In Intermolecular forces: proceedings of the Fourteenth Jerusalem Symposium on Quantum Chemistry and Biochemistry; Pullman B., Ed.; Springer Netherlands: Dordrecht, 1981; pp. 331–342. [Google Scholar]
- Bacle A.; Fuchs P. F. J.. Berger pure POPC MD simulation (300 K - 300ns - 1 bar); 2018; 10.5281/zenodo.1402417. [DOI] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MackKerell A. D. M. Jr.; Pastor R. W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A. D. Jr.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Papadopoulos C.; Fuchs P. F. J.. CHARMM36 pure POPC MD simulation (300 K - 300ns - 1 bar); 2018; 10.5281/zenodo.1306800. [DOI] [Google Scholar]
- Kulig W.; Jurkiewicz P.; Olżyńska A.; Tynkkynen J.; Javanainen M.; Manna M.; Rog T.; Hof M.; Vattulainen I.; Jungwirth P. Experimental determination and computational interpretation of biophysical properties of lipid bilayers enriched by cholesteryl hemisuccinate. Biochim. Biophys. Acta 2015, 1848, 422–432. 10.1016/j.bbamem.2014.10.032. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Milan Rodriguesz P.; Fuchs P. F. J.. MacRog pure POPC MD simulation (300 K - 500ns - 1 bar); 2020; 10.5281/zenodo.3741793. [DOI] [Google Scholar]
- Dickson C. J.; Madej B. D.; Skjevik Å. A.; Betz R. M.; Teigen K.; Gould I. R.; Walker R. C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. 10.1021/ct4010307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollila O. H. S.; Retegan M.. MD simulation trajectory and related files for POPC bilayer (Lipid14, Gromacs 4.5); 2014; 10.5281/zenodo.12767. [DOI] [Google Scholar]
- Jämbeck J. P. M.; Lyubartsev A. P. An Extension and Further Validation of an All-Atomistic Force Field for Biological Membranes. J. Chem. Theory Comput. 2012, 8, 2938–2948. 10.1021/ct300342n. [DOI] [PubMed] [Google Scholar]
- Javanainen M.POPC with 0, 10, 20, and 30 mol-% of cholesterol at 310 K. Slipids force field; 2016; 10.5281/zenodo.3243328. [DOI] [Google Scholar]
- Melcr J.; Martinez-Seara H.; Nencini R.; Kolafa J.; Jungwirth P.; Ollila O. H. S. Accurate Binding of Sodium and Calcium to a POPC Bilayer by Effective Inclusion of Electronic Polarization. J. Phys. Chem. B 2018, 122, 4546–4557. 10.1021/acs.jpcb.7b12510. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Grigera J. R.; Straatsma T. P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. 10.1021/j100308a038. [DOI] [Google Scholar]
- Melcr J.Simulations of POPC lipid bilayer in water solution at various NaCl, KCl and CaCl2 concentrations using ECC-POPC force field; 2019; 10.5281/zenodo.3335503. [DOI] [Google Scholar]
- Ollila O. H. S.; Ferreira T.; Topgaard D.. MD simulation trajectory and related files for POPC bilayer (Berger model delivered by Tieleman, Gromacs 4.5); 2014; 10.5281/zenodo.13279. [DOI] [Google Scholar]
- Höltje M.; Förster T.; Brandt B.; Engels T.; von Rybinski W.; Höltje H.-D. Molecular dynamics simulations of stratum corneum lipid models: fatty acids and cholesterol. Biochim. Biophys. Acta 2001, 1511, 156–167. 10.1016/S0005-2736(01)00270-X. [DOI] [PubMed] [Google Scholar]
- Ferreira T. M.; Coreta-Gomes F.; Ollila O. H. S.; Moreno M. J.; Vaz W. L. C.; Topgaard D. Cholesterol and POPC segmental order parameters in lipid membranes: solid state 1H–13C NMR and MD simulation studies. Phys. Chem. Chem. Phys. 2013, 15, 1976–1989. 10.1039/C2CP42738A. [DOI] [PubMed] [Google Scholar]
- Ollila O. H. S.MD simulation trajectory and related files for POPC/cholesterol (50 mol%) bilayer (Berger model delivered by Tieleman, modified Höltje, Gromacs 4.5); 2014; 10.5281/zenodo.13285. [DOI] [Google Scholar]
- Javanainen M.POPC with 0, 10, 20, and 30 mol-% of cholesterol at 310 K. Charmm36 force field; 2016; 10.5281/zenodo.3237420. [DOI] [Google Scholar]
- Lim J. B.; Rogaski B.; Klauda J. B. Update of the Cholesterol Force Field Parameters in CHARMM. J. Phys. Chem. B 2012, 116, 203–210. 10.1021/jp207925m. [DOI] [PubMed] [Google Scholar]
- Javanainen M.POPC with 40 and 50 mol-% of cholesterol at 310 K. Charmm36 force field; 2016; 10.5281/zenodo.3238157. [DOI] [Google Scholar]
- Javanainen M.; Kulig W.. POPC/Cholesterol @ 310K. 0, 10, 40, 50 and 60 mol-% cholesterol. Model by Maciejewski and Rog; 2015; 10.5281/zenodo.3252052. [DOI] [Google Scholar]
- Jämbeck J. P. M.; Lyubartsev A. P. Another Piece of the Membrane Puzzle: Extending Slipids Further. J. Chem. Theory Comput. 2013, 9, 774–784. 10.1021/ct300777p. [DOI] [PubMed] [Google Scholar]
- Ollila O. H. S.MD simulation trajectory and related files for POPC bilayer in low hydration (Berger model delivered by Tieleman, Gromacs 4.5); 2015; 10.5281/zenodo.13814. [DOI] [Google Scholar]
- Kanduč M.; Schneck E.; Netz R. R. Hydration Interaction between Phospholipid Membranes: Insight into Different Measurement Ensembles from Atomistic Molecular Dynamics Simulations. Langmuir 2013, 29, 9126–9137. 10.1021/la401147b. [DOI] [PubMed] [Google Scholar]
- Kanduc M.MD trajectory for DLPC bilayer (Berger, Gromacs 4.5.4), nw=24 w/l; 2015; 10.5281/zenodo.16289. [DOI] [Google Scholar]
- Kanduc M.MD trajectory for DLPC bilayer (Berger, Gromacs 4.5.4), nw=16 w/l; 2015; 10.5281/zenodo.16292. [DOI] [Google Scholar]
- Kanduc M.MD trajectory for DLPC bilayer (Berger, Gromacs 4.5.4), nw=12 w/l; 2015; 10.5281/zenodo.16293. [DOI] [Google Scholar]
- Kanduc M.MD trajectory for DLPC bilayer (Berger, Gromacs 4.5.4), nw=4 w/l; 2015; 10.5281/zenodo.16295. [DOI] [Google Scholar]
- Santuz H.MD simulation trajectory and related files for POPC bilayer (CHARMM36, Gromacs 4.5); 2015; 10.5281/zenodo.14066. [DOI] [Google Scholar]
- Ollila O. H. S.; Miettinen M.. MD simulation trajectory and related files for POPC bilayer (CHARMM36, Gromacs 4.5); 2015; 10.5281/zenodo.13944. [DOI] [Google Scholar]
- Ollila O. H. S.; Miettinen M.. MD simulation trajectory and related files for POPC bilayer in medium low hydration (CHARMM36, Gromacs 4.5); 2015; 10.5281/zenodo.13946. [DOI] [Google Scholar]
- Ollila O. H. S.; Miettinen M.. MD simulation trajectory and related files for POPC bilayer in low hydration (CHARMM36, Gromacs 4.5); 2015; 10.5281/zenodo.13945. [DOI] [Google Scholar]
- Javanainen M.POPC @ 310K, varying water-to-lipid ratio. Model by Maciejewski and Rog; 2014; 10.5281/zenodo.13498. [DOI] [Google Scholar]
- Antila H. S.; Ferreira T.; Ollila O. H. S.; Miettinen M. S.. hsantila/Corrtimes: First release of the correlation time analysis codes; 2020; 10.5281/zenodo.4395592. [DOI] [Google Scholar]
- Michaud-Agrawal N.; Denning E. J.; Woolf T. B.; Beckstein O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowers R. J.; Linke M.; Barnoud J.; Reddy T. J. E.; Melo M. N.; Seal S. L.; Domanski J.; Dotson D. L.; Sebastien K.; Ian M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference. 2016; pp. 98–105.
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1-2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Schlenkrich M.; Brickmann J.; MacKerell A. D.; Karplus M.. Biological Membranes; Springer: 1996; pp. 31–81. [Google Scholar]
- Feller S. E.; MacKerell A. D. An improved empirical potential energy function for molecular simulations of phospholipids. J. Phys. Chem. B 2000, 104, 7510–7515. 10.1021/jp0007843. [DOI] [Google Scholar]
- Vogel A.; Feller S. E. Headgroup Conformations of Phospholipids from Molecular Dynamics Simulation: Sampling Challenges and Comparison to Experiment. J. Membr. Biol. 2012, 245, 23–28. 10.1007/s00232-011-9411-5. [DOI] [PubMed] [Google Scholar]
- Venable R. M.; Hatcher E.; Guvench O.; MacKerell A. D. Jr.; Pastor R. W. Comparing Simulated and Experimental Translation and Rotation Constants: Range of Validity for Viscosity Scaling. J. Phys. Chem. B 2010, 114, 12501–12507. 10.1021/jp105549s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollila O. H. S.; Heikkinen H. A.; Iwaï H. Rotational Dynamics of Proteins from Spin Relaxation Times and Molecular Dynamics Simulations. J. Phys. Chem. B 2018, 122, 6559–6569. 10.1021/acs.jpcb.8b02250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Feigenson G. W. A Microscopic Interaction Model of Maximum Solubility of Cholesterol in Lipid Bilayers. Biophys. J. 1999, 76, 2142–2157. 10.1016/S0006-3495(99)77369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauda J. B.; Roberts M. F.; Redfield A. G.; Brooks B. R.; Pastor R. W. Rotation of Lipids in Membranes: Molecular Dynamics Simulation, 31P Spin-Lattice Relaxation, and Rigid-Body Dynamics. Biophys. J. 2008, 94, 3074–3083. 10.1529/biophysj.107.121806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeb F.; Maibaum L. Spatially Resolving the Condensing Effect of Cholesterol in Lipid Bilayers. Biophys. J. 2018, 115, 2179–2188. 10.1016/j.bpj.2018.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich A. S.; Watts A. Molecular response of the lipid headgroup to bilayer hydration monitored by 2H-NMR. Biophys. J. 1994, 66, 1441–1449. 10.1016/S0006-3495(94)80934-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechinger B.; Seelig J. Conformational changes of the phosphatidylcholine headgroup due to membrane dehydration. A 2H-NMR study. Chem. Phys. Lipids 1991, 58, 1–5. 10.1016/0009-3084(91)90105-K. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.