

Abstract
Aldosterone excess is a pathogenic factor in many hypertensive disorders. The discovery of numerous somatic and germline mutations in ion channels in primary hyperaldosteronism underscores the importance of plasma membrane conductances in determining the activation-state of zona glomerulosa (zG) cells. Electrophysiological recordings describe an electrically quiescent behavior for dispersed zG cells. Yet, emerging data indicate that in native rosette structures in situ, zG cells are electrically excitable, generating slow periodic voltage spikes and coordinated bursts of Ca2+ oscillations. We revisit data to understand how a multitude of conductances may underlie voltage/Ca2+ oscillations, recognizing that zG layer self-renewal and cell heterogeneity may complicate this task. We review recent data to understand rosette architecture and apply maxims derived from computational network modeling to understand rosette function. The challenge going forward is to uncover how the rosette orchestrates the behavior of a functional network of conditional oscillators to control zG layer performance and aldosterone secretion.
Keywords: zona glomerulosa, aldosterone, ion channels, electrical excitability, rosette
1. INTRODUCTION
Zona glomerulosa (zG) cells that assemble in rosette structures beneath the capsule of the adrenal gland produce the steroid hormone aldosterone. Aldosterone is generated from cholesterol by a series of hydroxylation and oxidation reactions catalyzed by P450 cytochromes that are located in two cellular compartments: the mitochondria and the endoplasmic reticulum. Metabolic intermediates are actively trafficked between these compartments along the cytoskeleton, whereas secretion of aldosterone into the extracellular fluid appears to be diffusion limited, without contributions from any known molecular mechanisms (1–3).
Angiotensin II (Ang II) and plasma potassium (K+) are the two major circulating secretagogues that stimulate aldosterone synthesis and secretion (1–3); they act both independently and synergistically (4). Notably, increases in extracellular K+ as small as 0.1 mM in vivo are sufficient to stimulate aldosterone production (approximately 1.3 fold), demonstrating the uncommon sensitivity of the zG cell to K+ (5). Aldosterone production also is enhanced by circulating adrenocorticotrophic hormone (ACTH) and locally produced endothelin-1 and inhibited by circulating atrial natriuretic peptide and locally produced dopamine (1–3).
Calcium (Ca2+) is the critical intracellular signal driving stimulated synthesis of aldosterone, which requires increases in Ca2+ in both the cytosol and mitochondria (6, 7). The mitochondrial uniporter facilitates transfer of cytosolic Ca2+ to the mitochondrial matrix where it increases the activity of Ca2+-dependent dehydrogenases to produce NADH (2). The subsequent action of the mitochondrial NADPH-translocase to convert NADH to NADPH generates the cofactor necessary for conversion of cholesterol to pregnenolone (mediated by CYP11A1) and deoxycorticosterone to aldosterone (mediated by CYP11B2), the early and late rate-limiting reactions in aldosterone synthesis (1–3).
Because Ca2+ is critical for aldosterone production, activities of plasma and mitochondrial membrane ion channels are key determinants of the zG activation state. Indeed, there is now abundant evidence that genetic variation in Ca2+-regulating ion channels and transporters is a driver of human hyperaldosteronism (3, 8–12), suggesting that the syndrome is a channelopathy in which autonomous zG aldosterone production is divorced from usual homeostatic control mechanisms [notably, the renin-angiotensin system (RAS)]. There are many excellent recent reviews that provide an updated inventory of the various channel and pump mutations that associate with hyperaldosteronism. Together, they provide a detailed examination of the alterations in ion channel properties (permeation, gating, and kinetics) and of the ensuing changes in functional output [membrane potential (Vm) and [Ca+]i] in electrically quiescent host cells and heterologous expressions systems (3, 8–13). In writing this review, our goal is to contribute a basic understanding of how the various ion channels that have been identified by molecular expression and/or electrophysiological currents in fully differentiated zG cells may function to produce the oscillatory electrical activity recently discovered in zG cells within native rosette structures. As such, we discuss old and new evidence challenging the long-held view that zG cells are electrically silent in situ and provide relevant information about the properties of channel classes implicated in controlling membrane voltage oscillations in native zG cells. Finally, given the heterogeneity of ion channels reported in zG cells among species, we highlight how different conductance combinations can generate similar electrical signals and how functional networks have the intrinsic capacity for intercellular communication and self-tuning. It is our hope that the synthesis of information provided here highlights both the need and the opportunity for electrophysiologists and optical imagers to elucidate how mutations in zG ion channels associated with human disease change the functional output of networks of electrically oscillating zG cells.
2. zG CELLS ARE ELECTRICALLY EXCITABLE
The Vm of zG cells is very negative at rest, approaching the K+ equilibrium potential. Recorded values consistently range from −73 to −86 mV [with one outlier at −64 mV (14)] across species (bovine, rat, mouse, human, feline), zG cell preparations (dissociated cells, intact adrenal slices), and recording techniques (sharp electrode recording, patch-clamp electrophysiology) (14–19). The Vm also follows closely, if not perfectly, the equilibrium potential for K+, depolarizing by 45–58 mV per tenfold increase in extracellular K+ concentration (versus 59 mV, predicted by the Nernst equation).
Early sharp microelectrode recordings of peripheral cells in cat adrenal slices provided the first demonstration of electrical excitability (14, 20). Spontaneous voltage fluctuations of 20–25 mV occurred in some cells; short bursts of spiking activity were evoked in others (e.g., by Ang II, K+, ACTH). However, subsequent microelectrode (15, 21) and perforated-patch recordings (22, 23) of dissociated/cultured zG cells were unable to confirm either spontaneous or evoked spiking activity. Rather, activators of aldosterone production evoked only sustained Vm depolarizations in zG cells. Nevertheless, in dissociated zG cells, hints of zG excitability remained. For example, depolarizing current injection or pharmacological blockade of outward K+ current evoked regenerative currents (21, 24) and, in perforated-patch recordings, small transient depolarizing spikes consistently overlaid the evoked analog shifts in voltage (22, 23). Together, the discrepancy between the electrical behavior of dissociated zG cells versus that in native tissue suggests the interesting possibility that the rosette organization of zG cells within the glomerulosa cell layer could promote electrical behavior.
In 2012, we reported that mouse zG cells, recorded in adrenal slices, are indeed electrically excitable (19). They exhibit recurrent (3–10 min recordings) depolarizing voltage spikes from a resting Vm of −82 mV that are large (Δ70 mV) and periodic (~2-s period). In the whole-cell, patch-clamp configuration, spiking behavior is spontaneous; Ang II and/or K+ increase activity. Recent recordings of zG intracellular Ca2+ in slices, providing a surrogate measure of cell activity, confirmed that oscillatory behavior can be evoked by Ang II or K+ in zG cells in situ (25–27) (see Section 6).
Pharmacological experiments have provided some insight into the identification of ionic conductances that control both the shape and the timing of zG spike potentials. Critical roles are assigned to low-voltage-activating, T-type Ca2+ channels and a Ca2+-activated K+ conductance that depends on T-type channel activity. Conversely, tetrodotoxin (TTX)-sensitive and TTX-insensitive Na2+ channels and high-voltage-activating Ca2+ channels have been excluded from significant participation in the mouse zG Vm spike potential; neither 60 μM TTX nor 10 μM nitrendipine alter Vm spike potentials (19).
The number of distinct channels expressed within any cell is astonishing. Yet, as previously observed for neuronal action potentials (28), a fraction of channels never participates in shaping the waveform because the kinetics and gating of other channels in the cell dominate and generate a voltage spike that precludes a significant contribution. Nonetheless, disease-causing mutations in dominant channels may allow contributions from those channels that were previously nonparticipants. Thus, identifying the conductances driving each phase of the zG Vm spike potential and determining the counterregulatory capacity of others provide a rationale for potential pharmacological targeting of specific ion channels in disease.
Like an action potential, the zG Vm spike potential can be deconstructed into its component phases (28): resting (interspike), threshold, depolarizing, peak, and repolarizing (Figure 1). Here, we highlight ion channel classes that could underlie the different component phases of the zG Vm spike potential for each, summarizing the general properties of the channel type and then the specific findings in the adrenal zG.
Figure 1.
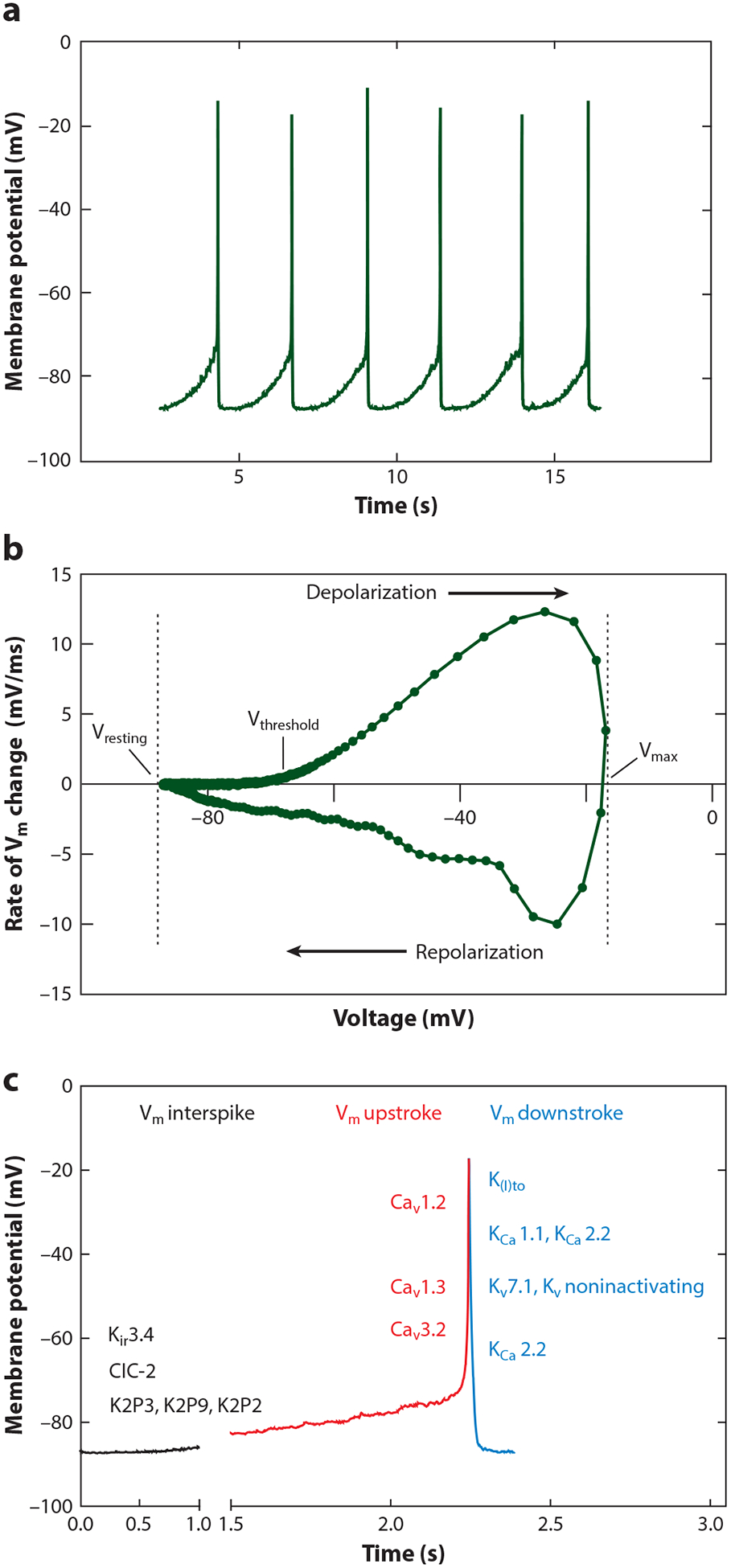
zG Vm-spike potential. (a) Current-clamp recording of spontaneous voltage oscillations from a mouse zG cell in an adrenal slice. (b) Vm phase-plane plot (dV/dt) of an averaged zG Vm-spike potential; Vresting, Vthreshold, Vmax, and depolarizing/repolarizing phases of Vm-spike potential are highlighted with arrows. (c) Ionic conductances underlying a zG Vm-spike potential waveform. Underlying molecular correlates or subfamilies are indicated on the Vm plot when known: Vm interspike (resting) = ClC (ClC-2), K2P (K2P3, K2P9: TASK; K2P2: TREK), Kir (Kir3.4); Vm upstroke (depolarizing) = LVA Ca2+ (Cav3.2), HVA Ca2+ (Cav1.3, Cav1.2); Vm downstroke (repolarizing) = Ktransient outward [K(I)to], SK (KCa 2.2), BK (KCa 1.1), Kdelayed rectifier [Kv slow (Kv7.1], Kvnoninactivating]. Abbreviations: HVA, high-voltage-activating; LVA, low-voltage-activating.
3. CONDUCTANCES UNDERLYING VREST
Multiple channels (leak K+, inward rectifying K+, and voltage-gated Cl− channels) contribute to setting the zG-interspike potential.
3.1. K2P Leak K+ Channels: TASK-1/TASK-3 and TREK-1 Channels (Encoded by Genes KCNK3, KCNK9, and KCNK2.1, respectively)
Properties:
TASK (TWIK-related acid-sensitive K+) and TREK (TWIK-related K+) channels belong to a superfamily of leak/background K+ channels that are active over a broad range of membrane potentials, including at negative resting potentials observed in zG cells. Topologically, their subunit structure is unique, with each subunit containing four transmembrane (TM) domains and two pore-forming (P) loops (4TM, 2P) that give this family the K2P moniker (29, 30). Functional channels are dimeric in structure in contrast to other types of K+ channels that are tetramers of subunits with one P-loop. Based on their sequence homology and functional characteristics, K2P channels are organized into six subfamilies (TWIK, THIK, TREK, TASK, TALK, TRESK). Homodimeric or heterodimeric K2P channels function as K+-selective channels passing currents with Goldman-Hodgkin-Katz (GHK)-like open rectification in physiological K+ gradients (29–31).
Gαq receptors uniformly inhibit TASK and TREK-1 channels, but by different mechanisms. For TASK-1/TASK-3 channels, direct actions of Gαq subunits, phosphatidylinositol 4,5-bisphosphate (PIP2) depletion, and/or diacylglycerol (DAG) on the channel protein cause channel inhibition, whereas TREK-1 channels are inhibited by PIP2 depletion and/or protein kinase C (PKC) activation (32–34).
Conversely, pH and volatile anesthetics alter these channels differentially. Extracellular acidification inhibits TASK channels (TASK-1 pK approximately 7.5; TASK-3 pK approximately 6.8), whereas intracellular acidification stimulates TREK-1 channels (pK approximately 6.0) (35). Volatile anesthetics strongly activate TASK-3 and TREK-1 but inhibit TASK-1 channels (34, 36). Protein kinases also regulate channel currents. Phosphorylation of TASK channels by protein kinase A (PKA) promotes interaction with 14–3-3b proteins, releasing channels from the endoplasmic reticulum to increase surface expression (37). By contrast, activation of Gαs-coupled receptors mediates the inhibition of TREK activity via PKA-induced phosphorylation of the channel protein (34).
Adrenal zG:
The rat, mouse, and guinea pig zG express mRNA for TASK-1 and TASK-3 (18, 38, 39) in contrast to the bovine zG that expresses mRNA for TREK-1 (40). In the whole human adrenal cortex, the expression of KCNK family genes is more diverse (TASK-1, TASK-2, TASK-3, TREK-1), with predominant mRNA expression for either TREK-1 (41) or TASK-1 (42), depending on the study. Whether this diversity or relative level of mRNA expression is attributable to mRNA captured from both the zona fasciculata (zF) and zG capsular zones remains unknown; in rodents, cortical zones differentially express mRNA for Kcnk family members (18). To date, only two studies have provided information about KCNK protein expression in the adrenal cortex. With a restricted focus on TASK subunits, they reveal predominant expression of TASK-1 in the human zG and both TASK-1 and TASK-3 in the rodent zG (43, 44). In the human adrenal cortical cell line, H295R, the reported expression of K2P transcripts (TASK-1, and/or TASK-3, and/or TREK-1) from laboratories also varies (42, 45, 46), a likely result of differences in culture media used to propagate this nonclonal cell line. In aldosterone-producing adenomas (APAs), mRNA expression of most KCNK family members is similar to that of normal adrenal tissue (i.e., TASK-1, TASK-3, TREK-1) (42), although TASK-2 mRNA is lower in APAs as a result of mutations in the TASK-2 channel promoter (47, 48). KCNK3 noncoding variants are associated with hyperaldosteronism and hypertension (49).
Functional K2P currents have been recorded in oocytes injected with rat adrenal capsular mRNA and in dispersed preparations of rat, bovine, and mouse zG cells (18, 38, 40, 50, 51). Inhibition of KCNK currents by Ang II, KCNK-directed antisense, or KCNK genetic deletion consistently depolarizes the plasma membrane of zG cells and increases aldosterone production (18, 38, 40, 42, 51). Thus, independent of the expressed subtype(s), KCNK channels play an important role in zG cells by acting at membrane voltages where most voltage-dependent K+ channels remain closed, thereby providing a major hyperpolarizing conductance that restrains aldosterone output. In addition to their expression on the plasma membrane, TASK-3 channels also localize to mitochondria where their activity depolarizes mitochondrial membrane potential and constrains aldosterone production likely by decreasing mitochondrial Ca2+ entry and NADPH generation (46). This mechanism for aldosterone regulation may also apply to other mitochondrial K+ channels located on the zG inner mitochondrial membrane.
Genetic deletion in mice of TASK channel subunits, individually or together, disrupts aldosterone regulation, yielding hyperaldosteronism that varies in magnitude and in the degree of autonomy from the RAS (18, 43, 46, 49, 51, 52). Knockout (KO) models also highlight a gender-dependent dimorphism in adrenal gland development that determines the aldosterone phenotype. In prepubertal mice, global TASK-1 channel deletion mislocalizes aldosterone synthase to the zF of the adrenal cortex, a zonation defect that corrects in adult males but not females. Global deletion of either TASK-1 or TASK-3 subunits disrupts the regulation of aldosterone output in adult mice. In some studies, this manifests as a modestly elevated aldosterone output across all salt diets (49, 52), whereas in others elevated levels are evident only on selective diets that normally suppress aldosterone (high-Na+, low-K+ content) (43, 51). The discrepancies in these reported findings could reflect differences in genetic background, and/or the reliance on plasma spot sampling versus 24-h urinary sampling. By contrast, combined global deletion of both TASK-1 and TASK-3 results in exaggerated aldosterone output and low-plasma renin (18). Across global TASK KO mouse models, blood pressure elevation is the consequence of excess aldosterone output produced by variable degrees of hypersensitivity to and/or autonomy from Ang II (43, 49, 52). When TASK-1 and TASK-3 deletion is restricted to adrenal zG cells, a mild autonomous hyperaldosteronism is observed that drives blood pressure elevation, highlighting the importance of zG-TASK channel activity to RAS dysfunction (53). However, comparison between mice with zG-specific versus global TASK channel deletion shows that the loss of extra-adrenal TASK channels also contributes to RAS dysfunction, magnifying changes in both renin (larger decreases) and aldosterone (larger increases).
3.2. Inward Rectifiers: G Protein–Gated Kir3.4 (Encoded by KCNJ5)
Properties:
Kir3.4 channels belong to the superfamily of inward rectifiers; they pass greater inward than outward current. Functionally they segregate into four subgroups: classical Kir channels (Kir2.x), Gβγ-activating Kir channels (Kir3.x), ATP-inhibited Kir channels (Kir6.x), and K+-transport channels (Kir1.x, Kir4.x–5.x, and Kir7.x) (54). Kir channels are intrinsically voltage independent, yet they conduct only a small outward current because of an asymmetric intracellular block of the open pore by magnesium (Mg2+) and polyamines (spermine, spermidine) (55). Depending on the affinity of these cationic interactions, the strength of rectification varies from weak (e.g., Kir1.1) to strong (e.g., Kir3.4). Topologically, all share a common subunit structure of two TM-spanning domains interconnected by one pore-forming loop (2TM, 1P). Functional channels are tetramers, formed by the homo- or heteromerization of four subunits, producing channels with different biophysical properties and cellular locations. Uniquely, the conductance of Kir channels (except Kir7.1) changes with extracellular K+, increasing as the square root of the extracellular K+ concentration [K+]o (54, 56). As a consequence of this extracellular K+-modifier site, [K+]o depolarization facilitated by Kir channels is greater than would be predicted from the GHK relationship (K-equilibrium potential). PIP2 is required for the maintained activity of most Kir channels (57), and activity is regulated both by PLC-mediated depletion of PIP2 and variably by protein kinases (54).
Unlike most Kir channels, Kir3.4 homomeric channels are constitutively closed. Opening requires a conformational change transduced by Gi/Go-linked G protein–coupled receptor (GPCR) activation and binding of Gβγ subunits to the channel protein (1Gβγ:1Kir-subunit) (54, 58, 59). RGS (regulators of G protein signaling) proteins deactivate Kir3.x channels by accelerating GTP hydrolysis of Gα-GTP, leading to sequestering of Gβγ-subunits. Heterotetrameric Kir3.x channels (Kir3.4/Kir3.1) display more constitutive activity than Kir3.4 homomers and remain regulated by Gβγ subunits (60). In addition, Kir3.x channels are activated by intracellular Na+ (only Kir3.2, 3.4) (61) and inhibited by intracellular acidification (54).
Adrenal zG:
Electrophysiological recordings of whole-cell and inside-out patches of zG cells (rat, bovine) demonstrate K+ channels with rectifying Kir behavior (62). Outward current is low, a consequence of intracellular Mg2+ block, and extracellular K+ augments inward current conductance. The measured conductance is small, but in zG cells with a high density of channels (rat 82% of patches, bovine 30%), the [K+]o-induced shift enhances the capacity of inward rectifiers to control basal Vm as [K+]o fluctuates physiologically. The expression of these channels in zG cells and the lack of expression of TTX-sensitive and TTX-insensitive (10 μM TTX) Na+ channels likely contribute to the exquisite sensitivity of zG cells to [K+]o both in vivo and in vitro. Ang II inhibits inward-rectifier Kir currents in both rat and bovine zG on-cell patches (63).
Because Kir3.4 mRNA or protein is virtually absent from mouse zG cells (44, 52), a member of the inward rectifier superfamily other than Gβγ-activating Kir3.x channels must carry these currents. By contrast, KCNJ5 (Kir3.4) is one of the most abundant K+ channel genes expressed in the normal human adrenal cortex (41) and is the locus of mutations in 49% of APAs worldwide (64, 65). Mutations occur mainly in regions near or within the selectivity filter to produce channels that preferentially carry depolarizing Na+ currents (41). Despite their larger size, APAs harboring KCNJ5 mutations have a lower proliferation index and a reduced expression of KCNJ5 channels than APAs carrying other mutations (65–67), suggesting that downregulation of Kir3.4 subunits and hetero-tetramerization with unaffected Kir3.x subunits may be two independent modes of protection against KCNJ5 mutational cellular toxicity (67).
3.3. ClC Voltage-Gated Cl− Channels: ClC-2 (Encoded by ClCN-2)
Properties:
ClC-2 chloride (Cl−) channels are part of a larger ClC family of channels and transporters encoded by genes CLCN1–7 and CLCNKA-B. Based on sequence homology, they segregate into three subgroups: plasma membrane voltage-gated anion channels (ClC-1–2, ClC-Ka–Kb) and two subgroups of 2Cl−/H+ antiporters that are located on endosomes and lysosomes (ClC-3–5, ClC-6–7) (68). Among voltage-gated channels, ClC channels have a unique topology. Each subunit contains 18 α-helices, most of which are membrane inserted, that wrap to form an ion permeation pore. ClC channels are dimeric; thus, they are double-barreled structures with two ion-conducting pores (protopores) (68, 69).
ClC-2:
In contrast to ClC-1 channels that open with depolarization, ClC-2s are inwardly rectifying channels that slowly activate with hyperpolarization. Channel activity occurs in bursts with a fast-gating mechanism regulating the independent opening and closing of each protopore within an activity burst, and a slow-gating mechanism that commonly terminates burst activity, regulating burst length and interburst duration. The cytoplasmic C terminus encodes two nucleotide-binding CBS (cystathionine-β-synthase) domains that impart nucleotide regulation. The cytoplasmic N terminus functions as an inactivation domain, as deletion removes hyperpolarization activation and generates constitutively open ClC-2 channels (68, 69). ClC-2 voltage gating depends on intracellular Cl− and extracellular H+. Elevation of intracellular Cl− induces a depolarizing shift in the voltage dependence of activation, permitting greater fractional opening at resting hyperpolarized voltages. Elevation of extracellular H+ induces biphasic regulation, activating (pH 6.5) and then blocking ClC-2 channels as pH becomes strongly acidic (68, 69).
Adrenal zG:
In the human adrenal cortex, ClC-2 mRNA is the most highly expressed plasma membrane Cl− channel (70); immunological detection of protein is robust and restricted to the zG layer (71). In the mouse adrenal gland, ClC-2 protein is highly expressed and functional (70). Mouse zG whole-cell Cl− currents recorded are hyperpolarization activated (negative to −60 mV) and emblematic of ClC-2 currents; they are absent from zG cells in ClC-2 KO adrenal slices (70). In human adrenocortical cell lines RNAi knock-down of native ClC-2 channels (in H295R cells) decreases CYP11B2 expression commensurate with a reduction in basal and stimulated (Ang II or K+) aldosterone production (70), whereas stable overexpression of ClC-2 channels (in HAC15 cells) increases CYP11B2 expression concomitant with cell membrane depolarization (71). Together, the estimated high intracellular Cl− concentration of zG cells (approximately 75 mM) (71), the intracellular Cl−-induced positive voltage shift in ClC-2 channel activation, and the hyperpolarized resting Vm of zG cells, combine to make ClC-2 currents a likely determinant of zG resting Vm, and hence, aldosterone production. Indeed, CLCN2 mutations that associate with early-onset familial hyperaldosteronism increase ClC-2 currents (25, 27, 70, 71), and mouse knock-in models that increase ClC-2 currents (N-terminal deletion: Clcn2op/op, Clcn2R180Q/+) display increases in plasma aldosterone and blood pressure (25, 27). Nevertheless, these mouse models of primary aldosteronism (PA) differ markedly. The Clcn2R180Q/+ mouse line carrying the human missense mutation displays a very mild phenotype (27), modeling mild PA. By contrast, the Clcn2op/op mouse line that replicates the increase in ClC-2 currents caused by each of three human N-terminal variants displays features typical of overt PA (25). Perforated patch-clamp recordings suggest that differences in ClC-2 mutant current amplitude may account for the differences in phenotypic severity (25).
4. CONDUCTANCES UNDERLYING THE DEPOLARIZING UPSTROKE
Low- and high-voltage-activating Ca2+ channels contribute to determining the peak and slope of the depolarizing phase of the zG Vm-spike potential.
4.1. Low-Voltage-Activating, T-Type Ca2+ Channels (CaV3.x Encoded by Genes CACNA1G, CACNA1H, and CACNA1I)
Properties:
Voltage-gated Ca2+ channels (CaV) separate into two main groups: high-voltage-activating (HVA: CaV1.1–1.4, L-type; CaV2.1, P/Q-type; CaV2.2, N-type; CaV2.3, R-type) and low-voltage-activating (LVA: CaV3.1–3.3, T-type) channels (72–74). T-type channels are distinguished by their negative thresholds for activation (−70 mV) and inactivation (−90 mV) that are at least 40 mV more negative than the corresponding thresholds for prototypical L-type channels. As a class, T-type channels also differ by their fast rate of inactivation and their slow rate of closing (74). Overall, these properties allow T-type Ca2+ channels to open and conduct Ca2+ under a greater driving force in response to small depolarizing stimuli. T-type current elicited by strong depolarizing stimuli is rapidly activating and transient. T-type channel inactivation is strictly voltage-dependent; Ca2+/calmodulin does not evoke channel inactivation as it does for L-type Ca2+ channels (75). Although T-type channels have a smaller single channel conductance than L-type channels (7–11 pS versus 20–30 pS) when recorded with Ba2+, their unitary conductances are similar in physiological Ca2+ (76, 77). Hence, T describes Transient, not Tiny.
T-type channels can inactivate without prior opening, displaying closed-state inactivation. Thus, the voltage of half-maximal inactivation (Vh1/2 = approximately −70 mV), which combines closed- and open-state inactivation, is approximately 20 mV more negative than the voltage of half-maximal activation (Va1/2 = approximately −50 mV). However, because the steady-state current relationships for activation and inactivation overlap, channel inactivation remains incomplete in the voltage range of −70 to −40 mV, allowing a small fraction of T-type channels to carry a steady-state window current at modest potentials (74, 78). Indeed, at −40 mV, small, sustained CaV3.2 currents elicited from recombinant channels expressed in HEK293 cells are estimated to be carried by <1% of available channels (Po = 0.006, I/Imax) (79).
Three genes encode the T-type family:
CACNA1G (CaV3.1), CACNA1H (CaV3.2), and CACNA1I (CaV3.3). Recombinant channels recorded with physiological Ca2+ (1.25 mM) show similar voltage dependence but display distinct kinetic behaviors. CaV3.3 channels are the most dissimilar; these channels open and inactivate more slowly, and they deactivate more rapidly at negative potentials than CaV3.1 and CaV3.2 isoforms, thus passing less current during the repolarization phase of the spike potential. In contrast, CaV3.1 channels recover faster from short-term inactivation, protecting them from cumulative inactivation during high-frequency stimulation (80). Mibefradil or TTA-P2 inhibits the activity of all CaV3.0 paralogs (81).
The predicted topological structure remains invariant among all CaV3.0 family members, including the CaV3.0 subgroup. The CaV3.1–3.3 genes encode an α-subunit that comprises four repeat domains (I–IV), each of which contains 6 TM segments (S1–S2) and 1 P-loop; intracellular linkers tether the domains together to produce a large α-subunit. Like KV channels, voltage is sensed by positive charges located on S4 TM segments (IS4, IIS4, IIIS4, IVS4) (74, 78).
TM domains among CaV3.0 family members are well conserved (80–90%), whereas most intracellular domain linkers are divergent (an exception is the conserved III–IV linker) (78), suggesting that structural elements in the domain linkers could dictate differential regulation of channel paralogs by well-established modulators; e.g., protein kinases and heterotrimeric G proteins. To date, the II–III domain linker has emerged as a critical determinant for both differential and shared channel regulation. For example, PKC and PKA increase whole-cell current densities of all CaV3.0 paralogs in a temperature-dependent manner (82) without changing gating. Replacement of the II–III linker with that of NaV1.4 channels (83) or CaV2.1 channels (84) generates chimeric channels that are refractory to regulation. The key phosphorylation site(s) mediating these increases remain unidentified.
The II–III linker also mediates differential regulation of channel activity. For example, (a) Ca2+/CaM-dependent protein kinase II (CaMKII) preferentially induces a hyperpolarizing shift (approximately −11 mV) in the Va1/2 of CaV3.2 channels (79, 85). Swapping the II–III linker domain between CaV3.2/CaV3.1 channels transfers CaMKII regulation from CaV3.2 to CaV3.1 chimeric channels, as does the removal of regulation caused by mutagenesis of serine 1198 on the II–III linker (86, 87). Whether additional phosphorylation sites common to both channel paralogs are also required for regulation is unknown. (b) Gα12/13 activation of Rho-kinase (ROCK) reduces peak current density of CaV3.1 and CaV3.3 (but not CaV3.2) channels without altering the current-voltage relationship, an activity change that requires ROCK-induced phosphorylation of a combination of conserved residues (serine/threonine) in the II–III linker (88). (c) Heterotrimeric Gβ2γ2-containing dimers selectively decrease whole-cell CaV3.2 current, decreasing channel open probability (functionally silencing a fraction of channels) without changing channel expression or gating (89, 90). II–III linker domain swapping between CaV3.2/CaV3.1 channels transfers regulation from CaV3.2 to CaV3.1 chimeric channels, in agreement with the preferential binding of recombinant Gβ2γ2-containing dimers to the CaV3.2 II–III intracellular loop (89).
Mass spectrometry of native CaV3.2 channels reveals many sites of phosphorylation (91). Ala-nine mutagenesis of three sites (S442, S445, T446) in the I–II linker in a region previously identified as a gating brake mimics the gating changes caused by region excision (92) and induces a large hyperpolarizing shift in the voltage dependence of activation. Whether the II–III linker regulatory mechanisms described above are distinct from, or conveyed through, the gating brake as a result of linker interactions awaits structural analysis.
Adrenal zG:
As determined by quantitative real-time polymerase chain reaction (RT-PCR) and in situ hybridization, zG cells across most species (bovine, rat, mouse) predominantly express the Cacna1h gene encoding CaV3.2 channels (93–96). By contrast, human adrenal cortex expresses mRNA for all three channel paralogs (97), although only CaV3.2 and CaV3.3 channel proteins are evident in the zG layer (96–98).
Early electrophysiological recordings of dispersed zG cells (bovine, rat, human) identified a component of Ca2+ current that was identified as T-type based on voltage dependence, kinetics, and pharmacology (23, 99–101). Although measured voltage relationships varied (likely attributable to differences in the recording charge carrier and its concentration), all studies predicted a window current to sustain the production of aldosterone (99, 101, 102). Indeed, in dissociated cells the amplitude of T-current modulated by K+ or pharmacological blockade strongly correlates with aldosterone production (23, 103, 104), enough to suggest that T-type Ca2+ channels may have a privileged role in aldosterone synthesis (102). However, the importance of T-currents in zG cells extends beyond steady-state window currents recorded in dissociated zG cells, as blocking T-type Ca2+ currents with TTA-P2 terminates electrical excitability in adrenal slices (19, 26). Levels of CaV3.2 mRNA in APAs significantly correlate with the levels of serum aldosterone in patients with PA (96).
Multiple mechanisms regulate the activity of T-type Ca2+ channels in zG cells. (a) Serotonin elicits a voltage-independent increase in T-type current via a mechanism that depends on active-Gαs and cAMP, in agreement with the molecular mechanism described above for the regulation of cloned CaV3.0 channels by PKA (105). (b) Dopamine evokes a voltage-independent inhibition of whole-cell T-current that requires the combined activity of Gβγ dimers and PKA (106), mimicking recombinant Gβ2γ2 dimer inhibition of CaV3.2 channels that requires phosphorylation of serine 1107 on the II–III linker (107). (c) By contrast, effects evoked by Ang II on T-channel gating have been variable. In calf bovine zG cells, Ang II induces a hyperpolarizing shift in the V1/2 of activation by two mechanisms that depend on the level of intracellular Ca2+. In low Ca2+, the shift in gating is mediated by active-Gαi, as pertussis toxin and anti-Gαi monoclonal antibodies dialyzed into the cell via the recording electrode block modulation (108). In high Ca2+, CaMKII catalyzes the phosphorylation of CaV3.2 channels (86, 109), molecular events that cause a shift in channel gating (85, 110) and replicate the molecular mechanism by which CaMKII modulates recombinant CaV3.2 channels reviewed above. By contrast, in adult bovine zG cells Ang II inhibits whole-cell T-current, shifting the Va1/2 (approximately +8 mV) by a mechanism that depends on active PKC, but not Ca2+. Pharmacological agents that activate PKC replicate, and those that inhibit PKC block the Ang II–induced current inhibition (111). Finally, in rat zG cells recorded in the perforated patch configuration, Ang II failed to change T-currents (23). The discrepancies among these findings remain unresolved, but they could reflect differences in paralog expression and/or specific recording conditions that permit one mechanism to prevail over the others.
4.2. High-Voltage-Activating, L-Type Ca2+ Channels (CaV1.1–1.4 Encoded by Genes CACNA1A, CACNA1B, CACNA1C, and CACNA1F)
Properties:
Unlike members of the HVA-CaV2.0 family that are primarily restricted to the central nervous system (CNS), endocrine cells express CaV1.0 family members (CaV1.2–CaV1.3). CaV1.0 family members have a predicted topology similar to that of LVA CaV3.0 channels (112). Like LVA channels, HVA genes encode a large α-subunit comprising four repeat domains, each containing 6 TM segments and 1 P-loop (4 × 6TM/1P) and a voltage sensor composed of positive charges located on each S4 segment. Unlike LVA channels, HVA channels are functional as multi-subunit structures that include one α-subunit and accessory subunits (β-, α2-, δ-subunits; CaV1.1 also contains a γ-subunit) in 1-to-1 stoichiometry. The β- and α2δ-subunits are extrinsic proteins interacting with the α-subunit pore on the intracellular (β) or extracellular (α2δ) membrane leaflets (112). β-Subunits reversibly interact with the I–II intracellular linker at the α-interaction domain. By contrast, the α2δ-subunit (formed as a single preprotein that is cleaved and maintained as a unit via disulfide bridges) is glycosylphosphatidylinositol (GPI)-anchored to the plasma membrane (113). In concert, accessory subunits increase the level of channel expression, shift the voltage dependence of activation and inactivation to hyperpolarizing voltages, and increase the rate of inactivation.
CaV1.2–1.3:
Although subunit associations (β1–4, α2δ1–4) and alternative splicing modulate the kinetics and voltage dependence of gating for both CaV1.2 and CaV1.3 channels, they have intrinsically distinct gating properties (114). With physiological recording solutions (2 mM Ca2+), prototypical Cav1.2 channels activate (−40 mV) and inactivate (−60 mV) at thresholds that are approximately 20–25 mV more depolarized than CaV1.3 channels. CaV1.3 channels open and close with faster kinetics than CaV1.2 channels, yet they inactivate with voltage (VDI) more slowly and less completely than slowly inactivating CaV1.2 channels. By opening earlier under a greater driving force and inactivating later and less completely, CaV1.3 channels support greater Ca2+ entry during an action potential-like waveform.
Ca2+ alters the biophysical properties of CaV1.2 and CaV1.3 channels. Ca2+ induces rapid Ca2+/CaM-dependent inactivation that terminates Ca2+ entry after channel opening (CaM-mediated) (75). Ca2+ also induces Ca2+-dependent facilitation that potentiates Ca2+ entry during repeated voltage-evoked openings (CaMKII-mediated) (115, 116). In contrast, cAMP and NO/cGMP signaling cascades regulate CaV1.2 and CaV1.3 channel activity voltage independently. cAMP increases current via effectors that include PKA, A-kinase-anchoring proteins, and the proteolytically cleaved distal C terminus of the α-subunit (for mechanistic details, see 112, 117, 118). cGMP decreases channel activity via effectors that vary with species and include protein kinase G and phosphodiesterases (117, 119, 120).
Adrenal zG:
The adrenal zG expresses the mRNA for CaV1.2 and CaV1.3 across species (95–98, 121); in the human adrenal cortex, the mRNA for CaV1.3 is the most abundant message (98). CaV1.2 and CaV1.3 α-subunit proteins are detected in the zG layer (67, 96, 98). Based on voltage dependence, kinetics, and pharmacology, the rat, bovine, and human zG cells express functional HVA currents that resemble prototypical CaV1.2 L-type Ca2+ channels (99–101). They activate at potentials more positive to −30 mV, with little time-dependent inactivation. However, the high concentrations of divalent cations used in the recording solutions would be expected to mask the hyperpolarized activation range of CaV1.3 α-subunits and shift them into a range where prototypical CaV1.2 α-subunits would activate (122).
In rat zG cells, ACTH increases L-type current voltage independently, an effect that is reproduced by 8-bromo cAMP (24). In bovine zG cells, Ang II inhibits L-type current, also voltage independently, by a mechanism that relies on active-Gαi/o (123). Thus, cAMP-induced regulation of cloned L-type channels reviewed above seems to describe modulation of L-type currents in zG cells.
In mouse zG cells (C57BL/6J), L-type currents are not detectable (19). However, in rat zG cells there is clear evidence that L-type currents participate in shaping evoked spike potentials. Increasing L-type current (BAYK8644, cAMP, ACTH) extends the plateau phase of the induced-spike potentials (24). Despite this, L-type current amplitude does not correlate with aldosterone production in dissociated rat or bovine zG cells. In fact, in these preparations, Ca2+ flux carried by L-type channels acts as a negative feedback modulator of aldosterone output when zG cells are strongly depolarized (23, 103, 104). In hindsight, the electrical quiescence of dissociated zG cells and the further voltage-dependent inactivation of CaV3.2 channels produced by L-type cationic flux may explain the reported inhibitory actions of L-type currents on aldosterone.
Notably, in less disrupted preparations (human adrenal slices), presumably where zG-Vm oscillates recurrently, both channel classes are important. Delivered at concentrations that retain their class selectivity, T-type and L-type blockers, alone and in combination, reduce basal and stimulated aldosterone production with the magnitude of inhibition disassociated from the age of the human donor (97). This precludes the possibility that functional L-type channels reside only in aldosterone-producing cell clusters (APCCs), which in human adrenals accumulate with age (124). e CYP11B2-expressing APCCs extend into the zF layer and harbor somatic CACNA1D gene mutations that cluster in regions of the CaV1.3 α-subunit previously associated with voltage gating (98, 125, 126). As such, wild-type CaV1.3 channels and mutant CaV1.3 channels could have an outsized role in regulating human aldosterone production.
5. CONDUCTANCES UNDERLYING THE HYPERPOLARIZING DOWNSTROKE
Ca2+-activated (SK and BK) and voltage-gated K+ channels contribute to determining the peak and the slope of the hyperpolarizing phase of the zG Vm-spike potential.
5.1. SK Channels (KCa2.1, KCa2.2, KCa2.3, Encoded by Genes KCNN1, KCNN2, and KCNN3)
Properties:
SK channels belong to a diverse superfamily of Ca2+-activated K+ channels. These K+-selective channels are distinguished from other subfamilies by their small unitary conductance (9.2 pS with symmetrical K+, 2–3 pS with normal Ringer) (127). They share a similar topology to voltage-activated K+ channels (6TM, 1P-loop per subunit, 4 subunits per channel), yet are voltage independent because of the absence of positively charged residues on TM4 (128). SK channels are inwardly rectifying: They pass greater inward than outward current (129). Submicro-molar Ca2+ (KD approximately 0.5 μM) rapidly opens the SK channel gate, binding the N-lobe of apo-calmodulin (Ca2+-free CaM) that is bound to the C terminus of each α-subunit (130). Ca2+-dependent gating is highly cooperative (Hill coefficient approximately 3–5) and regulated by the phosphorylation state of CaM, determined by the activities of casein kinase 2 and protein phosphatase 2A. In neurons, these enzymes form multimeric signaling complexes with SK2 and SK3 channels (128). Pharmacological modulators of activity (activators, inhibitors) bind directly to the channel protein. Apamin, the prototypical inhibitor of SK currents, potently targets all family members (IC50: approximately 0.04–10 nM) with SK2 channels demonstrating the highest apamin sensitivity (IC50: approximately 40 pM) (127, 129).
Adrenal zG:
H295R cells, a human adrenal cortical cell line, express mRNA for all three KCNN subtypes, and electrophysiological recordings have confirmed SK channel activity. SK inhibition by apamin or SK activation by 1-ethyl-2-benzimidazoline reciprocally changes membrane voltage (depolarizes/hyperpolarizes) and aldosterone production (stimulates/reduces), highlighting the importance of SK channel activity in zG cells (131). In the human adrenal cortex, KCNN2 mRNA is more abundant than KCNN3>KCNN4>KCNN1 (41); in the rat adrenal, Kcnn2 is one of the top 25 transcripts differentially expressed between the zG versus zF layers (132). KCNN2 protein is evident in both the zG and zF zones of the human adrenal cortex. In agreement with H295R studies and the voltage-regulation of aldosterone production, apamin (1 nM) increases basal and Ang II-stimulated, but not K+-stimulated, aldosterone output from human adrenal slices (131).
5.2. BK (Maxi-K) Channels (KCa 1.1 Encoded by KCNMA1)
Properties:
Voltage-gated BK channels belong to the Slo family of K+ channels (Slick, Slack, KSper) and are distinguished by a very large unitary conductance (200 pS in symmetrical K+ solutions). A tetramer of α-subunits forms the ion-conducting pore (129). However, unlike other voltage-gated K+ channels, these α-subunits have an additional TM segment at the N terminus (7TM + 1P-loop per subunit), and the gating charge of their voltage sensor is distributed among multiple TM segments (TM2–4), conferring only weak voltage dependence in the absence of internal Ca2+ (Va1/2 = approximately 200 mV) (133). The direct binding of Ca2+ (EC50 1–10 μM) to RCK (regulator of conductance for K+ ions) domains on the C terminus of α-subunits increases the sensitivity of BK channels to voltage, left-shifting the voltage dependence of channel activation to promote moderate opening within a physiological range of voltages (−50 to 0 mV) (133). As a result, colocalization of BK channels with voltage-gated Ca2+ channels, or at sites of intracellular Ca2+ release, originally was considered a prerequisite for intracellular activity. However, the association of BK channels with accessory β-subunits (β1–4 encoded by Kcnmb1–4 genes; 1β-subunit:1α-subunit) and leucine-rich repeat-containing proteins (γ-subunits: γ1–4) modifies gating kinetics, enhances Ca2+ sensitivity, and left-shifts the voltage dependence of activation independently of Ca2+ (134). These changes produce channels with a wide spectrum of biophysical properties that are active at modest Ca2+ concentrations. Despite such diversity, the scorpion toxins, charybdotoxin (IC50 = 2.9 nM) and iberiotoxin (IC50 = 1.7 nM), universally block conduction by occluding the BK pore (127). In most tissues, cAMP-dependent and cGMP-dependent protein kinases increase BK activity.
Adrenal zG:
The zG layer of the mouse adrenal cortex expresses BK α-subunit protein (135). Genetically deleting BK α-subunits in mice increases serum aldosterone in the absence of a rise in renin or serum K+, responses that indicate cell-autonomous hyperaldosteronism and an intrinsic role for BK channels in regulating aldosterone production (135, 136). Global deletion of accessory β-subunits in mice (Kcmb1−/− and Kcmb2−/−) also increases aldosterone production (137, 138), although the cause of hyperaldosteronism differs between the two genotypes. In the Kcmb1−/− mouse, hyperkalemia produced by insufficient K+ excretion drives aldosterone excess. By contrast, the Kcmb2−/− mouse is normokalemic, and excess aldosterone output is driven by an apparent intrinsic increase in Ang II sensitivity and Ang II autonomy, as revealed by high aldosterone on a high-Na+ diet. In agreement with an intrinsic regulatory role for BK channels in the adrenal zG, excised-patch recordings from bovine and rat zG cells exhibit BK channel activity (62). Unitary currents are large in amplitude (228 pS) and show a steep voltage-dependence (between −10 mV and +50 mV) that depends on intracellular Ca2+ >0.5 μM. BK channels are likely expressed in the human zG layer, as mRNA transcripts for both KCNMA1 and KCNMB4 are abundant (41). The coexpression of the BK α-subunit with β4-subunits would be expected to produce a channel that can gate at more hyperpolarized voltages (45% of maximal current at −50 mV in high Ca2+ >1 μM) (139).
5.3. Voltage-Gated 6-Transmembrane K+ Channels (KV Encoded by KCNA-D, KCNQ, KCNH)
Properties:
KV channels constitute the largest gene family of K+ channels; they are divided into 12 subfamilies (KV1.x–12.x) based on sequence and structural similarities and are encoded by 40 genes (140). The ion conduction pore of KV channels is formed by four α-subunits, each of which contains 6 TM segments (S1–S6) and 1 P-loop. Positively charged arginine residues on the S4 helix of each subunit confer voltage sensitivity (141). Only eight of the α-subunit subfamilies are functionally active: KV1 (KCNA1–8, Shaker), KV2 (KCNB1–2, Shab), KV3 (KCNC1–4, Shaw), KV4 (KCND1–3, Shaw), KV7 (KCNQ1–5), KV10 (KCNH1, 5), KV11 (KCNH2, 6, 7) and KV12 (KCNH8, 3, 4) (142). The other four families are nonconducting: KV5 (KCNF1), KV6 (KCNG1–4), KV8 (KCNV1–2), and KV9 (KCNS1–3) (142). Although silent, they modify channel properties by associating with KV2 subunits (140, 143). Subunit association within subfamilies (e.g., KV1, KV7) or with intracellular (KV β-subunits 1–3 or K+ channel-interacting proteins KChIP1–4) or TM ion channel regulatory proteins (KCNE1–4 or TM dipeptidylaminopeptidase-like protein: DPP4, DPP6) produces macromolecular ion channel complexes with a dizzying array of biophysical properties (140, 143).
Based on current kinetics, KV channels segregate into two functional groups: transient-outward rectifiers that activate and inactivate rapidly (Ito, A-current) and delayed-outward rectifiers that activate after a sigmoidal lag phase following a change in voltage (IKslow, M-current). Within each group there is extensive molecular diversity. For example, transient A-type current is carried by multimeric complexes that include KV4.x channels (4.1–4.3/KCHIP2–3/DPP6, 10), KV3.x channels (3.3–3.4/KCNE3), or Kv1.4 channels (KV1.4/KV1.2 heterodimers/KVβx). The molecular diversity among delayed rectifiers is even more extensive and generates currents that differ considerably in their kinetics of inactivation. Thus, while channel activation among delayed rectifiers is characteristically slow (except KV1.5, KV3.1, KV3.3), delayed rectifiers are either noninactivating (KV2.1–KV2.2; KV7.2–KV7.5; KV10.1–KV10.2; neuronal KV12.1, KV12.3 heterodimers) or inactivate on different time scales from slow to very slow, depending on their subunit associations (KV1.1–1.7; KV3.1–3.3; KV7.1/KCNE1 or KCNE3) (142).
Accessory subunit association modifies KV trafficking, gating, and kinetics. The association of KVβ1–3 subunits with KV1.x channels and KChIP1–4 with KV4.x channels increases surface expression. In addition, KVβ1–3 subunits uniformly accelerate the rate of KV1.x channel inactivation, and KChIP subunits consistently enhance the recovery of KV4.x channels from closed-state inactivation (143). By contrast, KChIP-induced changes in KV4.x voltage gating, and inactivation kinetics varies enormously among multimeric complexes that contain different individual KV4.x, KChIPx, and DPPx family members (143). Unlike KVβ and KChIP subunits that associate with members of only one subfamily and are extrinsic, KCNE proteins (except KCNE4) are membrane spanning and promiscuous, modulating the channel activity of many subfamilies (KV1, KV2, KV3, KV4, KV7, KV11) (142, 143).
Adrenal zG:
Electrophysiological recordings of dispersed zG cells reveal transient-outward and/or delayed-outward rectifier currents, the expression of which varies across species and rodent breed. In bovine zG cells an A-current is predominant (144). With intracellular Ca2+ buffered by EGTA, depolarization elicits whole-cell currents that rapidly decay (30–50 ms) and single channel openings (27 pS) in cell-attached recordings that cluster early in the records. By contrast, in rat zG cells, delayed-outward rectifier current is the dominant KV current expressed, showing demonstrable differences between strains. Sprague-Dawley rat zG cells express K+-selective delayed-outward rectifier currents that activate slowly at a Vm threshold of approximately −50 mV and show some inactivation at 2 s (62, 144); currents in Long-Evans rat zG cells activate very slowly at a Vm threshold of approximately 0 mV and remain noninactivating at 2–5 s (145). Long-Evans zG cells also express a transient-outward A-type current that is incompletely K+-selective in some cells (20%). The molecular bases for these currents remain unidentified, and the described large KV channel family diversity precludes easy assignment. Nevertheless, their electrophysiological signature varies sufficiently to conclude that the molecular components of these macromolecular complexes differ.
There are few electrophysiological recordings of KV currents in human zG cells. Of those reported, KV currents are mostly transient in normal human zG cells. However, in zG cells obtained from a patient with Cushing’s syndrome, KV currents are noninactivating (144). Whether this difference is representative remains to be determined. A comprehensive analysis of K+ channel gene expression in the human adrenal cortex provides insight into KV channels likely expressed (see 41, supplementary table 4). With a mean expression of all K+ channel mRNA transcripts determined to be 7.196 (base 2 log scale), KCNQ1 (KV7.1; 10.45) expression was the most abundant, with KCNA4 (KV1.4; 8.71) and KCNC4 (KV3.4; 7.85) also ranking within the top 13% (41). Given the caveat that these expression levels were determined from the entire adrenal cortex (zG + zF), the data suggest that both delayed rectifier currents and rapidly activating transient currents are expressed, either together or differentially in single human zG cells.
Genetic studies in mice support a role for Kcne1 in the control of aldosterone production (146). Global Kcne1 deletion increases aldosterone production without a change in renin that also is concurrent with hypokalemia. Thus, it is likely that the attendant increase in aldosterone production on a normal Na+ diet is not extrinsic to the zG, but rather is cell autonomous, arising from a primary defect in zG cells. In agreement, the mouse adrenal gland expresses Kcne1 and Kcnq genes, with the former mRNA localized to the zG by in situ hybridization (146). Thus, KCNE1-/KCNQ-mediated K+ currents may be one of the conductances limiting aldosterone output, although because of the promiscuity of KCNE1, this conclusion awaits further testing.
6. FORM AND FUNCTION
6.1. Glomerular Rosette Structure
In the adult, the zG is morphologically composed of interconnected glomeruli wrapped in a laminin β1-rich basement membrane that closely apposes the vascular endothelium. Within each glomerulus, 10–15 zG cells organize into multicellular rosette clusters. Cells are tightly packed and connected at a single contact point (147), fulfilling the structural criterion of a rosette center (148). At these centers, Ca2+-dependent cell–cell adhesion proteins (N- and K-cadherin), β-catenin and F-actin aggregate to form adherens junctions. Adherens junctions are also interspersed on lateral rosette surfaces but are absent from basal surfaces (147). Within a rosette, zG cells are nonpolarized and heterogeneous (147). They contain both Cyp11b2+ and Cyp11b2− cells, and cells likely at different stages of maturation; the three-dimensional shape of the rosette changes postnatally (147), and zG cells transit from the zG into the zF layer, transdifferentiating into bona fide fasciculata cells (149). Thus, channels expressed among zG cells within a rosette may vary significantly.
Rosettes are malleable minimum-energy structures. They use adhesive forces to increase intercellular contacts and to attain an equilibrium state of minimum intercellular surface tension (150). Thus, in a tissue layer that is devoid of gap junctions (151), and in which there is little described purinergic signaling (152), the zG-rosette structure itself may support cellular communication and provide the framework for rosette-organized activity. To date, mechanisms for information exchange within the adrenal rosette remain poorly described. However, the interaction of local electrical fields (153) (ephaptic coupling), the exchange of ions between cells within their confined shared extracellular spaces (ionic coupling/K+?) (154), or the transmission of membrane tension between coupled cells (mechanosensitive coupling) (155) are all well-established means of communication that could allow zG cells within the rosette to function as a syncytium (Figure 2).
Figure 2.
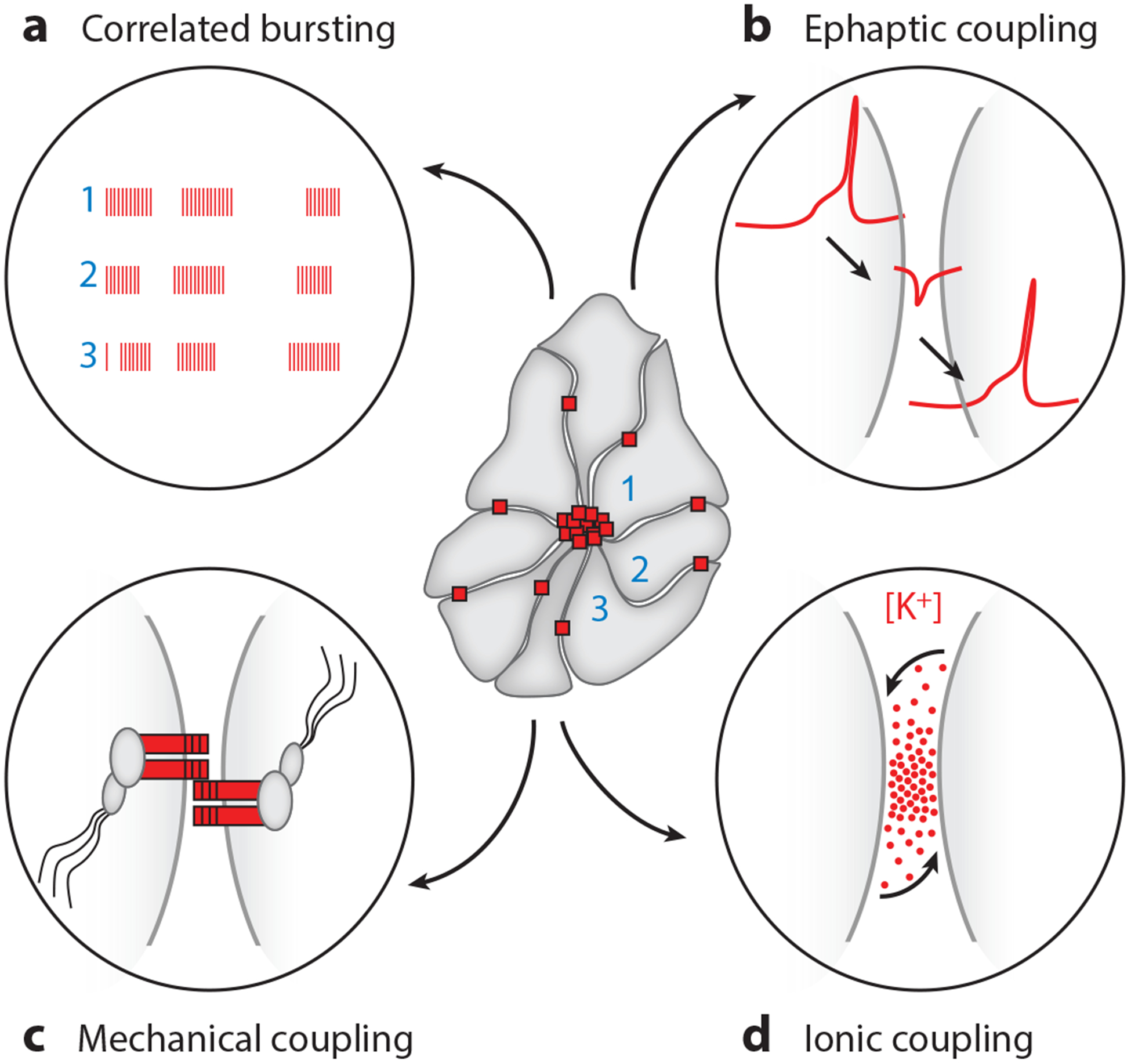
Rosette activity coordination and potential modes for intercellular communication. (Center) Two-dimensional schematic of rosette structure showing cell–cell cadherin junctions (red squares) clustered at the rosette apex and dispersed on lateral membranes. (a) Schematic of coordinated bursts of activity and Ca2+ oscillations among zG cells within a rosette. (b–d). Three potential modes of activity coupling are illustrated: (b) ephaptic, (c) mechanical, and (d) ionic.
6.2. Rosette Activity
In adrenal rosettes, active zG cells reliably generate periodic Ca2+ oscillations. These oscillations derive from plasma membrane electrical signals as endoplasmic reticulum blockers fail to halt or modify activity (26). Thus, cellular Ca2+ oscillatory activity is a good indicator of the electrical activation state of the zG layer. Within rosettes, Ang II or K+ evokes recurrent Ca2+ oscillations in zG cells that occur in stereotypic bursts of fixed event frequency and duration (26, 27). Hence, unlike many oscillators, oscillation frequency does not increase as a function of stimulus strength (28). Instead, Ang II increases only the number of evoked bursts per cell. By increasing burst number without prolonging length, Ang II controls the onset, but not the offset, of bursting activity. Notably, cellular patterns of activity are coordinated to produce an activity-based network of oscillating cells within a rosette. Indeed, based on both phase analysis, which measures fixed-activity relationships, and functional clustering analysis, which measures synchrony in relationships, functional clusters are rosette based (26). Whether activity coordination within a rosette is the consequence of a few electrical drivers that entrain followers or is the result of mutual activity modifications among members to reach a common oscillatory behavior remains unresolved. Nevertheless, the well-defined coordination of activity suggests that the rosette, and not the cell, is a unit of functional activity of the zG layer.
Surprisingly, in the unstimulated-state, wild-type zG cells display little Ca2+ oscillatory activity (25–27), in contrast to the spontaneous electrical activity of zG cells observed when recording electrical activity using the whole-cell patch-clamp technique (19). This discrepancy in behavior may relate to patch-clamp-induced depolarization (the result of applied suction and/or intracellular ion dialysis), as zG cells in slices expressing constitutively open ClC-2 channels are depolarized and spontaneously oscillate (25). Hence, in rosettes it would appear that the zG cell may not be an intrinsic oscillator (19), but rather a conditional oscillator requiring provocation to initiate electrical activity.
6.3. Diversity Within Cellular Networks and Intercellular Communication
Given multiple conductances, their isoforms and paralogs, their expressed densities, and their accessory subunit compositions, the diversity among network oscillators is daunting (28). Yet, neuronal computational modeling indicates that despite this great diversity in the intrinsic properties of network components, the output of well-balanced networks can be remarkably similar (156). This plasticity of network construction ensures stability and obviates a need to fine-tune intrinsic component properties to achieve a preferred performance output (156). These maxims when applied to the adrenal rosette provide the rationale for moving beyond the cell-centric view of aldosterone regulation to uncover fundamental rules that govern the activity of the zG rosette, a functional network of oscillating cells optimized for homeostatic control of a tissue layer that is continually self-renewing.
Lesson one:
The exact molecular identity of a channel may not matter to the output of the system. For example, adrenal zG cells express TASK-1 mRNA in humans and TASK-1 and TASK-3 mRNA in rodents. Yet, homo- and heterodimeric TASK channels share similar open-rectification properties and, with adjusted densities, can carry similar current amplitudes. Moreover, TASK-1 and TASK-3 heterodimers recapitulate the pH sensitivity of TASK-1 homodimers. Thus, under many circumstances, which TASK subunit carries current in zG cells may matter little to the level of aldosterone output.
Lesson two:
Within a cell, different sets of conductances can generate a similar Vm spike potential (156). Thus, mutational alteration of one conductance may be counterbalanced by the activity of others to suppress a mutational phenotype. In zG cells, multiple conductances mediate each phase of the zG spike potential. For example, inward rectifier K+ channels (Kir), leak K+ channels (TASK/TREK), and Cl− channels (ClC) contribute to setting the resting Vm of zG cells. Larger leak and/or smaller ClC currents could offset mutant-depolarizing inward rectifier currents, and larger SK or BK currents could compensate for larger mutant-depolarizing ClC currents resulting in little change in the zG spike potential. These compensatory changes in activity could arise from alterations in intrinsic channel properties or channel expression levels. Notably, electrically excitable cells, by permitting larger voltage excursions than electrically quiescent cells, allow the participation of more conductances and thus have a greater intrinsic capacity for self-tuning. Conversely, human mutations and KO mouse models that produce a strong aldosterone phenotype may provide the opportunity to reveal which ionic conductances cannot be offset if studied in activity-based networks of electrically excitable zG cells. Indeed, adrenal slices prepared from a mouse model of familial hyperaldosteronism type II that harbors a gain-of-function mutation (R180Q) in ClC-2 channels did show an increase in the Ca2+ oscillatory activity elicited by 20 nM Ang II. Notably, this increase was observed preferentially at 3 mM but not at 5 mM K+, suggesting that the ClC-2 mutant assumes a privileged role only at hyperpolarized resting voltages (27).
Lesson three:
The performance of a network depends less on the intrinsic properties of the individual component cells and more on their correlated values (156). Given the high turnover of the zG layer and the requisite conversion of zG to zF cells, it is likely that zG cells are heterogeneous. Indeed, electrical recordings of zG cells in slices reveal two populations of zG cells that differ in their resting Vm and frequency (19). Yet, within one rosette, cellular activity is surprisingly uniform, manifesting as bursts of spike potentials of invariant length and frequency (26, 27) that correlate among rosette members (26). Activity-correlated networks are valuable; they are better low-pass filters, screening spurious incoming noise (signal-to-noise filter), and they have an enhanced capacity for network self-tuning and a greater dynamic range. Although the mechanism(s) underlying correlated activity within the rosette remains unknown, the rosette architecture is likely the basis for coordination, obeying the architectural and industrial design axiom that form follows function.
The rosette itself is an avascular structure. Yet, the high vascular content of the zG layer and the close apposition of vascular endothelial cells to the rosette basement membrane creates a morphological geometry that allows each zG cell access to stimuli diffusing from the vasculature. Thus, although each rosette exerts local homeostatic control, putatively making each rosette an activity silo, the performance response of the zG layer to stimuli may still be achievable.
Combined, these lessons advocate for a rosette-centric view of aldosterone regulation. They position the rosette as the unit of activity of the zG layer and the locus of the primary control of aldosterone output. If correct, there remains a critical need for the development of experimental model systems (e.g., adrenal rosette organoids) to understand how to program and sustain a desired level of rosette performance.
FUTURE ISSUES.
What conductance(s) underlies the Vrest to Vthreshold transition?
What is the degree of cell heterogeneity within a rosette?
When the signal is encoded as an oscillatory burst, is the transmission of Ca2+ to Zg mitochondria more efficient?
What mechanism(s) facilitates cellular communication within a rosette?
In the zG layer, is there communication between rosettes, or are they activity silos?
How does the regulation of steroidogenesis change between a structured rosette and an aldosterone-producing cell cluster (APCC)-like unstructured cluster of zG cells?
ACKNOWLEDGMENTS
We dedicate this review to Howard Rasmussen, who was a giant in the field of cell signaling, and for whom calcium was “The King.” We acknowledge valued colleagues David T. Breaultand Mark P. Beenhakker for many thought-provoking discussions. This work was supported by the US National Institutes of Health (NIH) grants R01 HL03977 (P.Q.B. and D.A.B.) and R01 HL 138241 (P.Q.B.).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Quinn SJ, Williams GH. 1988. Regulation of aldosterone secretion. Annu. Rev. Physiol 50:409–26 [DOI] [PubMed] [Google Scholar]
- 2.Spat A, Hunyady L. 2004. Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol. Rev 84:489–539 [DOI] [PubMed] [Google Scholar]
- 3.Seccia TM, Caroccia B, Gomez-Sanchez EP, Gomez-Sanchez CE, Rossi GP. 2018. The biology of normal zona glomerulosa and aldosterone-producing adenoma: pathological implications. Endocr. Rev 39:1029–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fredlund P, Saltman S, Kondo T, Douglas J, Catt KJ. 1977. Aldosterone production by isolated glomerulosa cells: modulation of sensitivity to angiotensin II and ACTH by extracellular potassium concentration. Endocrinology 100:481–86 [DOI] [PubMed] [Google Scholar]
- 5.Himathongkam T, Dluhy RG, Williams GH. 1975. Potassium-aldosterone-renin interrelationships. J. Clin. Endocrinol. Metab 41:153–59 [DOI] [PubMed] [Google Scholar]
- 6.Aguilera G, Catt KJ. 1986. Participation of voltage-dependent calcium channels in the regulation of adrenal glomerulosa function by angiotensin II and potassium. Endocrinology 118:112–18 [DOI] [PubMed] [Google Scholar]
- 7.Capponi AM, Lew PD, Jornot L, Vallotton MB. 1984. Correlation between cytosolic free Ca2+ and aldosterone production in bovine adrenal glomerulosa cells. Evidence for a difference in the mode of action of angiotensin II and potassium. J. Biol. Chem 259:8863–69 [PubMed] [Google Scholar]
- 8.Zennaro MC, Fernandes-Rosa F, Boulkroun S. 2015. Genetic alterations in primary aldosteronism. Med. Sci 31:389–96 [DOI] [PubMed] [Google Scholar]
- 9.Scholl UI. 2017. Unanswered questions in the genetic basis of primary aldosteronism. Horm. Metab. Res 49:963–68 [DOI] [PubMed] [Google Scholar]
- 10.Monticone S, Buffolo F, Tetti M, Veglio F, Pasini B, Mulatero P. 2018. Genetics in endocrinology: the expanding genetic horizon of primary aldosteronism. Eur. J. Endocrinol 178:R101–11 [DOI] [PubMed] [Google Scholar]
- 11.Funder JW. 2019. Primary aldosteronism. Hypertension 74:458–66 [DOI] [PubMed] [Google Scholar]
- 12.Manosroi W, Williams GH. 2019. Genetics of human primary hypertension: focus on hormonal mechanisms. Endocr. Rev 40:825–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang T, He M, Hu C. 2018. Regulation of aldosterone production by ion channels: from basal secretion to primary aldosteronism. Biochim. Biophys. Acta Mol. Basis Dis 1864:871–81 [DOI] [PubMed] [Google Scholar]
- 14.Natke E Jr., Kabela E. 1979. Electrical responses in cat adrenal cortex: possible relation to aldosterone secretion. Am. J. Physiol 237:E158–62 [DOI] [PubMed] [Google Scholar]
- 15.Quinn SJ, Cornwall MC, Williams GH. 1987. Electrical properties of isolated rat adrenal glomerulosa and fasciculata cells. Endocrinology 120:903–14 [DOI] [PubMed] [Google Scholar]
- 16.Lotshaw DP. 1997. Characterization of angiotensin II-regulated K+ conductance in rat adrenal glomerulosa cells. J. Membr. Biol 156:261–77 [DOI] [PubMed] [Google Scholar]
- 17.Chen XL, Bayliss DA, Fern RJ, Barrett PQ. 1999. A role for T-type Ca2+ channels in the synergistic control of aldosterone production by ANG II and K+. Am. J. Physiol 276:F674–83 [DOI] [PubMed] [Google Scholar]
- 18.Davies LA, Hu C, Guagliardo NA, Sen N, Chen X, et al. 2008. TASK channel deletion in mice causes primary hyperaldosteronism. PNAS 105:2203–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu C, Rusin CG, Tan Z, Guagliardo NA, Barrett PQ. 2012. Zona glomerulosa cells of the mouse adrenal cortex are intrinsic electrical oscillators. J. Clin. Investig 122:2046–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matthews EK, Saffran M. 1973. Ionic dependence of adrenal steroidogenesis and ACTH-induced changes in the membrane potential of adrenocortical cells. J. Physiol 234:43–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quinn SJ, Cornwall MC, Williams GH. 1987. Electrophysiological responses to angiotensin II of isolated rat adrenal glomerulosa cells. Endocrinology 120:1581–89 [DOI] [PubMed] [Google Scholar]
- 22.Lotshaw DP. 1997. Effects of K+ channel blockers on K+ channels, membrane potential, and aldosterone secretion in rat adrenal zona glomerulosa cells. Endocrinology 138:4167–75 [DOI] [PubMed] [Google Scholar]
- 23.Lotshaw DP. 2001. Role of membrane depolarization and T-type Ca2+ channels in angiotensin II and K+ stimulated aldosterone secretion. Mol. Cell. Endocrinol 175:157–71 [DOI] [PubMed] [Google Scholar]
- 24.Durroux T, Gallo-Payet N, Payet MD. 1991. Effects of adrenocorticotropin on action potential and calcium currents in cultured rat and bovine glomerulosa cells. Endocrinology 129:2139–47 [DOI] [PubMed] [Google Scholar]
- 25.Goppner C, Orozco IJ, Hoegg-Beiler MB, Soria AH, Hubner CA, et al. 2019. Pathogenesis of hypertension in a mouse model for human CLCN2 related hyperaldosteronism. Nat. Commun 10:4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guagliardo NA, Klein PM, Gancayco CA, Lu A, Leng S, et al. 2020. Angiotensin II induces coordinated calcium bursts in aldosterone-producing adrenal rosettes. Nat. Commun 11:1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schewe J, Seidel E, Forslund S, Marko L, Peters J, et al. 2019. Elevated aldosterone and blood pressure in a mouse model of familial hyperaldosteronism with ClC-2 mutation. Nat. Commun 10:5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bean BP. 2007. The action potential in mammalian central neurons. Nat. Rev. Neurosci 8:451–65 [DOI] [PubMed] [Google Scholar]
- 29.Lesage F, Lazdunski M. 2000. Molecular and functional properties of two-pore-domain potassium channels. Am. J. Physiol. Ren. Physiol 279:F793–801 [DOI] [PubMed] [Google Scholar]
- 30.Goldstein SA, Bockenhauer D, O’Kelly I, Zilberberg N. 2001. Potassium leak channels and the KCNK family of two-P-domain subunits. Nat. Rev. Neurosci 2:175–84 [DOI] [PubMed] [Google Scholar]
- 31.Czirjak G, Enyedi P. 2002. Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J. Biol. Chem 277:5426–32 [DOI] [PubMed] [Google Scholar]
- 32.Chen X, Talley EM, Patel N, Gomis A, McIntire WE, et al. 2006. Inhibition of a background potassium channel by Gq protein α-subunits. PNAS 103:3422–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilke BU, Lindner M, Greifenberg L, Albus A, Kronimus Y, et al. 2014. Diacylglycerol mediates regulation of TASK potassium channels by Gq-coupled receptors. Nat. Commun 5:5540. [DOI] [PubMed] [Google Scholar]
- 34.Bayliss DA. 2019. Tandem pore domain potassium channels In The Oxford Handbook of Neuronal Ion Channels, ed. Bhattacharjee A, pp. 1–46. Oxford, UK: Oxford Univ. Press [Google Scholar]
- 35.Maingret F, Patel AJ, Lesage F, Lazdunski M, Honoré E. 1999. Mechano- or acid stimulation, two interactive modes of activation of the TREK-1 potassium channel. J. Biol. Chem 274:26691–96 [DOI] [PubMed] [Google Scholar]
- 36.Czirjak G, Enyedi P. 2003. Ruthenium red inhibits TASK-3 potassium channel by interconnecting glutamate 70 of the two subunits. Mol. Pharmacol 63:646–52 [DOI] [PubMed] [Google Scholar]
- 37.Plant LD, Rajan S, Goldstein SA. 2005. K2P channels and their protein partners. Curr. Opin. Neurobiol 15:326–33 [DOI] [PubMed] [Google Scholar]
- 38.Czirjak G, Fischer T, Spat A, Lesage F, Enyedi P. 2000. TASK (TWIK-related acid-sensitive K+ channel) is expressed in glomerulosa cells of rat adrenal cortex and inhibited by angiotensin II. Mol. Endocrinol 14:863–74 [DOI] [PubMed] [Google Scholar]
- 39.Bandulik S, Tauber P, Lalli E, Barhanin J, Warth R. 2015. Two-pore domain potassium channels in the adrenal cortex. Pflügers Arch 467:1027–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Enyeart JA, Danthi SJ, Enyeart JJ. 2004. TREK-1 K+ channels couple angiotensin II receptors to membrane depolarization and aldosterone secretion in bovine adrenal glomerulosa cells. Am. J. Physiol. Endocrinol. Metab 287:E1154–65 [DOI] [PubMed] [Google Scholar]
- 41.Choi M, Scholl UI, Yue P, Björklund P, Zhao B, et al. 2011. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 331:768–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nogueira EF, Gerry D, Mantero F, Mariniello B, Rainey WE. 2010. The role of TASK1 in aldosterone production and its expression in normal adrenal and aldosterone-producing adenomas. Clin. Endocrinol 73:22–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Penton D, Bandulik S, Schweda F, Haubs S, Tauber P, et al. 2012. Task3 potassium channel gene invalidation causes low renin and salt-sensitive arterial hypertension. Endocrinology 153:4740–48 [DOI] [PubMed] [Google Scholar]
- 44.Chen AX, Nishimoto K, Nanba K, Rainey WE. 2015. Potassium channels related to primary aldosteronism: Expression similarities and differences between human and rat adrenals. Mol. Cell. Endocrinol 417:141–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brenner T, O’Shaughnessy KM. 2008. Both TASK-3 and TREK-1 two-pore loop K channels are expressed in H295R cells and modulate their membrane potential and aldosterone secretion. Am. J. Physiol. Endocrinol. Metab 295:E1480–86 [DOI] [PubMed] [Google Scholar]
- 46.Yao J, McHedlishvili D, McIntire WE, Guagliardo NA, Erisir A, et al. 2017. Functional TASK-3-like channels in mitochondria of aldosterone-producing zona glomerulosa cells. Hypertension 70:347–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lenzini L, Caroccia B, Campos AG, Fassina A, Belloni AS, et al. 2014. Lower expression of the TWIK-related acid-sensitive K+ channel 2 (TASK-2) gene is a hallmark of aldosterone-producing adenoma causing human primary aldosteronism. J. Clin. Endocrinol. Metab 99:E674–82 [DOI] [PubMed] [Google Scholar]
- 48.Lenzini L, Prisco S, Gallina M, Kuppusamy M, Rossi GP. 2018. Mutations of the Twik-related acid-sensitive K+ channel 2 promoter in human primary aldosteronism. Endocrinology 159:1352–59 [DOI] [PubMed] [Google Scholar]
- 49.Manichaikul A, Rich SS, Allison MA, Guagliardo NA, Bayliss DA, et al. 2016. KCNK3 variants are associated with hyperaldosteronism and hypertension. Hypertension 68:356–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lotshaw DP. 2006. Biophysical and pharmacological characteristics of native two-pore domain TASK channels in rat adrenal glomerulosa cells. J. Membr. Biol 210:51–70 [DOI] [PubMed] [Google Scholar]
- 51.Heitzmann D, Derand R, Jungbauer S, Bandulik S, Sterner C, et al. 2008. Invalidation of TASK1 potassium channels disrupts adrenal gland zonation and mineralocorticoid homeostasis. EMBO J 27:179–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guagliardo NA, Yao J, Hu C, Schertz EM, Tyson DA, et al. 2012. TASK-3 channel deletion in mice recapitulates low-renin essential hypertension. Hypertension 59:999–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guagliardo NA, Yao J, Stipes EJ, Cechova S, Le TH, et al. 2019. Adrenal tissue-specific deletion of TASK channels causes aldosterone-driven angiotensin II-independent hypertension. Hypertension 73:407–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. 2010. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev 90:291–366 [DOI] [PubMed] [Google Scholar]
- 55.Lopatin AN, Makhina EN, Nichols CG. 1994. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature 372:366–69 [DOI] [PubMed] [Google Scholar]
- 56.Lopatin AN, Nichols CG. 1996. [K+] dependence of polyamine-induced rectification in inward rectifier potassium channels (IRK1, Kir2.1). J. Gen. Physiol 108:105–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hilgemann DW, Ball R. 1996. Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science 273:956–59 [DOI] [PubMed] [Google Scholar]
- 58.Chan KW, Sui JL, Vivaudou M, Logothetis DE. 1997. Specific regions of heteromeric subunits involved in enhancement of G protein-gated K+ channel activity. J. Biol. Chem 272:6548–55 [DOI] [PubMed] [Google Scholar]
- 59.He C, Zhang H, Mirshahi T, Logothetis DE. 1999. Identification of a potassium channel site that interacts with G protein βγ subunits to mediate agonist-induced signaling. J. Biol. Chem 274:12517–24 [DOI] [PubMed] [Google Scholar]
- 60.Krapivinsky G, Gordon EA, Wickman K, Velimirović B, Krapivinsky L, Clapham DE. 1995. The G-protein-gated atrial K+ channel IKAch is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature 374:135–41 [DOI] [PubMed] [Google Scholar]
- 61.Ho IHM, Murrell-Lagnado RD. 1999. Molecular determinants for sodium-dependent activation of G protein-gated K+ channels. J. Biol. Chem 274:8639–48 [DOI] [PubMed] [Google Scholar]
- 62.Vassilev PM, Kanazirska MV, Quinn SJ, Tillotson DL, Williams GH. 1992. K+ channels in adrenal zona glomerulosa cells. I. Characterization of distinct channel types. Am. J. Physiol 263:E752–59 [DOI] [PubMed] [Google Scholar]
- 63.Kanazirska MV, Vassilev PM, Quinn SJ, Tillotson DL, Williams GH. 1992. Single K+ channels in adrenal zona glomerulosa cells. II. Inhibition by angiotensin II. Am. J. Physiol 263:E760–65 [DOI] [PubMed] [Google Scholar]
- 64.Boulkroun S, Beuschlein F, Rossi GP, Golib-Dzib JF, Fischer E, et al. 2012. Prevalence, clinical, and molecular correlates of KCNJ5 mutations in primary aldosteronism. Hypertension 59:592–98 [DOI] [PubMed] [Google Scholar]
- 65.Lenzini L, Rossitto G, Maiolino G, Letizia C, Funder JW, Rossi GP. 2015. A meta-analysis of somatic KCNJ5 K+ channel mutations in 1636 patients with an aldosterone-producing adenoma. J. Clin. Endocrinol. Metab 100:E1089–95 [DOI] [PubMed] [Google Scholar]
- 66.Åkerström T, Crona J, Delgado Verdugo A, Starker LF, Cupisti K, et al. 2012. Comprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filter. PLOS ONE 7:e41926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang Y, Gomez-Sanchez CE, Jaquin D, Aristizabal Prada ET, Meyer LS, et al. 2019. Primary aldosteronism: KCNJ5 mutations and adrenocortical cell growth. Hypertension 74:809–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen TY. 2005. Structure and function of CLC channels. Annu. Rev. Physiol 67:809–39 [DOI] [PubMed] [Google Scholar]
- 69.Jentsch TJ, Pusch M. 2018. CLC chloride channels and transporters: structure, function, physiology, and disease. Physiol. Rev 98:1493–590 [DOI] [PubMed] [Google Scholar]
- 70.Fernandes-Rosa FL, Daniil G, Orozco IJ, Goppner C, El Zein R, et al. 2018. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat. Genet 50:355–61 [DOI] [PubMed] [Google Scholar]
- 71.Scholl UI, Stölting G, Schewe J, Thiel A, Tan H, et al. 2018. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat. Genet 50:349–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Llinas R, Yarom Y. 1981. Electrophysiology of mammalian inferior olivary neurones in vitro. Different types of voltage-dependent ionic conductances. J. Physiol 315:549–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. 1988. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci 11:431–38 [DOI] [PubMed] [Google Scholar]
- 74.Talavera K, Nilius B. 2006. Biophysics and structure-function relationship of T-type Ca2+ channels. Cell Calcium 40:97–114 [DOI] [PubMed] [Google Scholar]
- 75.Ben-Johny M, Yue DT. 2014. Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J. Gen. Physiol 143:679–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guia A, Stern MD, Lakatta EG, Josephson IR. 2001. Ion concentration-dependence of rat cardiac unitary L-type calcium channel conductance. Biophys. J 80:2742–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shorofsky SR, January CT. 1992. L- and T-type Ca2+ channels in canine cardiac Purkinje cells. Single-channel demonstration of L-type Ca2+ window current. Circ. Res 70:456–64 [DOI] [PubMed] [Google Scholar]
- 78.Perez-Reyes E 2003. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol. Rev 83:117–61 [DOI] [PubMed] [Google Scholar]
- 79.Wolfe JT, Wang H, Perez-Reyes E, Barrett PQ. 2002. Stimulation of recombinant CaV3.2, T-type, Ca2+ channel currents by CaMKIIγC. J. Physiol 538:343–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Klockner U, Lee JH, Cribbs LL, Daud A, Hescheler J, et al. 1999. Comparison of the Ca2+ currents induced by expression of three cloned α1 subunits, α1G, α1H and α1I, of low-voltage-activated T-type Ca2+ channels. Eur. J. Neurosci 11:4171–78 [DOI] [PubMed] [Google Scholar]
- 81.Martin RL, Lee JH, Cribbs LL, Perez-Reyes E, Hanck DA. 2000. Mibefradil block of cloned T-type calcium channels. J. Pharmacol. Exp. Ther 295:302–8 [PubMed] [Google Scholar]
- 82.Chemin J, Mezghrani A, Bidaud I, Dupasquier S, Marger F, et al. 2007. Temperature-dependent modulation of CaV3 T-type calcium channels by protein kinases C and A in mammalian cells. J. Biol. Chem 282:32710–18 [DOI] [PubMed] [Google Scholar]
- 83.Kim JA, Park JY, Kang HW, Huh SU, Jeong SW, Lee JH. 2006. Augmentation of CaV3.2 T-type calcium channel activity by cAMP-dependent protein kinase A. J. Pharmacol. Exp. Ther 318:230–37 [DOI] [PubMed] [Google Scholar]
- 84.Park JY, Kang HW, Moon HJ, Huh SU, Jeong SW, et al. 2006. Activation of protein kinase C augments T-type Ca2+ channel activity without changing channel surface density. J. Physiol 577:513–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lu HK, Fern RJ, Nee JJ, Barrett PQ. 1994. Ca2+-dependent activation of T-type Ca2+ channels by calmodulin-dependent protein kinase II. Am. J. Physiol 267:F183–89 [DOI] [PubMed] [Google Scholar]
- 86.Yao J, Davies LA, Howard JD, Adney SK, Welsby PJ, et al. 2006. Molecular basis for the modulation of native T-type Ca2+ channels in vivo by Ca2+/calmodulin-dependent protein kinase II. J. Clin. Investig 116:2403–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Welsby PJ, Wang H, Wolfe JT, Colbran RJ, Johnson ML, Barrett PQ. 2003. A mechanism for the direct regulation of T-type calcium channels by Ca2+/calmodulin-dependent kinase II. J. Neurosci 23:10116–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Iftinca M, Hamid J, Chen L, Varela D, Tadayonnejad R, et al. 2007. Regulation of T-type calcium channels by Rho-associated kinase. Nat. Neurosci 10:854–60 [DOI] [PubMed] [Google Scholar]
- 89.Wolfe JT, Wang H, Howard J, Garrison JC, Barrett PQ. 2003. T-type calcium channel regulation by specific G-protein βγ subunits. Nature 424:209–13 [DOI] [PubMed] [Google Scholar]
- 90.DePuy SD, Yao J, Hu C, McIntire W, Bidaud I, et al. 2006. The molecular basis for T-type Ca2+ channel inhibition by G protein β2γ2 subunits. PNAS 103:14590–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Blesneac I, Chemin J, Bidaud I, Huc-Brandt S, Vandermoere F, Lory P. 2015. Phosphorylation of the CaV3.2 T-type calcium channel directly regulates its gating properties. PNAS 112:13705–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Perez-Reyes E 2010. Characterization of the gating brake in the I-II loop of CaV3 T-type calcium channels. Channels 4:453–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schrier AD, Wang H, Talley EM, Perez-Reyes E, Barrett PQ. 2001. α1H T-type Ca2+ channel is the predominant subtype expressed in bovine and rat zona glomerulosa. Am. J. Physiol. Cell Physiol 280:C265–72 [DOI] [PubMed] [Google Scholar]
- 94.Lesouhaitier O, Chiappe A, Rossier MF. 2001. Aldosterone increases T-type calcium currents in human adrenocarcinoma (H295R) cells by inducing channel expression. Endocrinology 142:4320–30 [DOI] [PubMed] [Google Scholar]
- 95.Rossier MF, Lesouhaitier O, Perrier E, Bockhorn L, Chiappe A, Lalevee N. 2003. Aldosterone regulation of T-type calcium channels. J. Steroid Biochem. Mol. Biol 85:383–88 [DOI] [PubMed] [Google Scholar]
- 96.Felizola SJ, Maekawa T, Nakamura Y, Satoh F, Ono Y, et al. 2014. Voltage-gated calcium channels in the human adrenal and primary aldosteronism. J. Steroid Biochem. Mol. Biol 144(Part B):410–16 [DOI] [PubMed] [Google Scholar]
- 97.Yang T, He M, Zhang H, Barrett P, Hu C. 2019. L- and T-type calcium channels control aldosterone production from human adrenals. J. Endocrinol 244:237–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Scholl UI, Goh G, Stolting G, de Oliveira RC, Choi M, et al. 2013. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet 45:1050–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cohen CJ, McCarthy RT, Barrett PQ, Rasmussen H. 1988. Ca channels in adrenal glomerulosa cells: K+ and angiotensin II increase T-type Ca channel current. PNAS 85:2412–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Matsunaga H, Yamashita N, Maruyama Y, Kojima I, Kurokawa K. 1987. Evidence for two distinct voltage-gated calcium channel currents in bovine adrenal glomerulosa cells. Biochem. Biophys. Res. Commun 149:1049–54 [DOI] [PubMed] [Google Scholar]
- 101.Payet MD, Durroux T, Bilodeau L, Guillon G, Gallo-Payet N. 1994. Characterization of K+ and Ca2+ ionic currents in glomerulosa cells from human adrenal glands. Endocrinology 134:2589–98 [DOI] [PubMed] [Google Scholar]
- 102.Rossier MF. 2016. T-type calcium channel: a privileged gate for calcium entry and control of adrenal steroidogenesis. Front. Endocrinol 7:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Barrett PQ, Ertel EA, Smith MM, Nee JJ, Cohen CJ. 1995. Voltage-gated calcium currents have two opposing effects on the secretion of aldosterone. Am. J. Physiol 268:C985–92 [DOI] [PubMed] [Google Scholar]
- 104.Rossier MF, Burnay MM, Vallotton MB, Capponi AM. 1996. Distinct functions of T- and L-type calcium channels during activation of bovine adrenal glomerulosa cells. Endocrinology 137:4817–26 [DOI] [PubMed] [Google Scholar]
- 105.Lenglet S, Louiset E, Delarue C, Vaudry H, Contesse V. 2002. Activation of 5-HT7 receptor in rat glomerulosa cells is associated with an increase in adenylyl cyclase activity and calcium influx through T-type calcium channels. Endocrinology 143:1748–60 [DOI] [PubMed] [Google Scholar]
- 106.Drolet P, Bilodeau L, Chorvatova A, Laflamme L, Gallo-Payet N, Payet MD. 1997. Inhibition of the T-type Ca2+ current by the dopamine D1 receptor in rat adrenal glomerulosa cells: requirement of the combined action of the G βγ protein subunit and cyclic adenosine 3′,5′-monophosphate. Mol. Endocrinol 11:503–14 [DOI] [PubMed] [Google Scholar]
- 107.Hu C, Depuy SD, Yao J, McIntire WE, Barrett PQ. 2009. Protein kinase A activity controls the regulation of T-type CaV3.2 channels by Gβγ dimers. J. Biol. Chem 284:7465–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lu HK, Fern RJ, Luthin D, Linden J, Liu LP, et al. 1996. Angiotensin II stimulates T-type Ca2+ channel currents via activation of a G protein, Gi. Am. J. Physiol 271:C1340–49 [DOI] [PubMed] [Google Scholar]
- 109.Fern RJ, Hahm MS, Lu HK, Liu LP, Gorelick FS, Barrett PQ. 1995. Ca2+/calmodulin-dependent protein kinase II activation and regulation of adrenal glomerulosa Ca2+ signaling. Am. J. Physiol 269:F751–60 [DOI] [PubMed] [Google Scholar]
- 110.Barrett PQ, Lu HK, Colbran R, Czernik A, Pancrazio JJ. 2000. Stimulation of unitary T-type Ca2+ channel currents by calmodulin-dependent protein kinase II. Am. J. Physiol. Cell Physiol 279:C1694–703 [DOI] [PubMed] [Google Scholar]
- 111.Rossier MF, Aptel HB, Python CP, Burnay MM, Vallotton MB, Capponi AM. 1995. Inhibition of low threshold calcium channels by angiotensin II in adrenal glomerulosa cells through activation of protein kinase C. J. Biol. Chem 270:15137–42 [DOI] [PubMed] [Google Scholar]
- 112.Catterall WA. 2011. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol 3:a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dolphin AC. 2018. Voltage-gated calcium channel α2δ subunits: an assessment of proposed novel roles. F1000Research 7:1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Helton TD, Xu W, Lipscombe D. 2005. Neuronal L-type calcium channels open quickly and are inhibited slowly. J. Neurosci 25:10247–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Anderson ME, Braun AP, Schulman H, Premack BA. 1994. Multifunctional Ca2+/calmodulin-dependent protein kinase mediates Ca2+-induced enhancement of the L-type Ca2+ current in rabbit ventricular myocytes. Circ. Res 75:854–61 [DOI] [PubMed] [Google Scholar]
- 116.Jenkins MA, Christel CJ, Jiao Y, Abiria S, Kim KY, et al. 2010. Ca2+-dependent facilitation of CaV1.3 Ca2+ channels by densin and Ca2+/calmodulin-dependent protein kinase II. J. Neurosci 30:5125–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mahapatra S, Marcantoni A, Zuccotti A, Carabelli V, Carbone E. 2012. Equal sensitivity of Cav1.2 and Cav1.3 channels to the opposing modulations of PKA and PKG in mouse chromaffin cells. J. Physiol 590:5053–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Qian H, Patriarchi T, Price JL, Matt L, Lee B, et al. 2017. Phosphorylation of Ser1928 mediates the enhanced activity of the L-type Ca2+ channel CaV1.2 by the β2-adrenergic receptor in neurons. Sci. Signal 10:eaaf9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fischmeister R, Castro L, Abi-Gerges A, Rochais F, Vandecasteele G. 2005. Species- and tissue-dependent effects of NO and cyclic GMP on cardiac ion channels. Comp. Biochem. Physiol. A Mol. Integr. Physiol 142:136–43 [DOI] [PubMed] [Google Scholar]
- 120.Hofmann F, Flockerzi V, Kahl S, Wegener JW. 2014. L-type CaV1.2 calcium channels: from in vitro findings to in vivo function. Physiol. Rev 94:303–26 [DOI] [PubMed] [Google Scholar]
- 121.Szabadkai G, Horvath A, Spat A, Enyedi P. 1998. Expression of voltage-dependent calcium channel α1 subunits in rat adrenal capsular tissue and single glomerulosa cells. Endocr. Res 24:425–26 [DOI] [PubMed] [Google Scholar]
- 122.Xu W, Lipscombe D. 2001. Neuronal CaV1.3α1 L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J. Neurosci 21:5944–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Maturana AD, Casal AJ, Demaurex N, Vallotton MB, Capponi AM, Rossier MF. 1999. Angiotensin II negatively modulates L-type calcium channels through a pertussis toxin-sensitive G protein in adrenal glomerulosa cells. J. Biol. Chem 274:19943–48 [DOI] [PubMed] [Google Scholar]
- 124.Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, et al. 2015. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. PNAS 112:E4591–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Azizan EA, Poulsen H, Tuluc P, Zhou J, Clausen MV, et al. 2013. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat. Genet 45:1055–60 [DOI] [PubMed] [Google Scholar]
- 126.Fernandes-Rosa FL, Williams TA, Riester A, Steichen O, Beuschlein F, et al. 2014. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension 64:354–61 [DOI] [PubMed] [Google Scholar]
- 127.Wei AD, Gutman GA, Aldrich R, Chandy KG, Grissmer S, Wulff H. 2005. International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol. Rev 57:463–72 [DOI] [PubMed] [Google Scholar]
- 128.Adelman JP, Maylie J, Sah P. 2012. Small-conductance Ca2+-activated K+ channels: form and function. Annu. Rev. Physiol 74:245–69 [DOI] [PubMed] [Google Scholar]
- 129.Kaczmarek LK, Aldrich RW, Chandy KG, Grissmer S, Wei AD, Wulff H. 2017. International Union of Basic and Clinical Pharmacology. C. Nomenclature and properties of calcium-activated and sodium-activated potassium channels. Pharmacol. Rev 69:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, et al. 1998. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 395:503–7 [DOI] [PubMed] [Google Scholar]
- 131.Yang T, Zhang HL, Liang Q, Shi Y, Mei YA, et al. 2016. Small-conductance Ca2+-activated potassium channels negatively regulate aldosterone secretion in human adrenocortical cells. Hypertension 68:785–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nishimoto K, Rigsby CS, Wang T, Mukai K, Gomez-Sanchez CE, et al. 2012. Transcriptome analysis reveals differentially expressed transcripts in rat adrenal zona glomerulosa and zona fasciculata. Endocrinology 153:1755–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Contreras GF, Castillo K, Enrique N, Carrasquel-Ursulaez W, Castillo JP, et al. 2013. A BK (Slo1) channel journey from molecule to physiology. Channels 7:442–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gueguinou M, Chantome A, Fromont G, Bougnoux P, Vandier C, Potier-Cartereau M. 2014. KCa and Ca2+ channels: the complex thought. Biochim. Biophys. Acta 1843:2322–33 [DOI] [PubMed] [Google Scholar]
- 135.Sausbier M, Arntz C, Bucurenciu I, Zhao H, Zhou XB, et al. 2005. Elevated blood pressure linked to primary hyperaldosteronism and impaired vasodilation in BK channel-deficient mice. Circulation 112:60–68 [DOI] [PubMed] [Google Scholar]
- 136.Rieg T, Vallon V, Sausbier M, Sausbier U, Kaissling B, et al. 2007. The role of the BK channel in potassium homeostasis and flow-induced renal potassium excretion. Kidney Int 72:566–73 [DOI] [PubMed] [Google Scholar]
- 137.Larsen CK, Jensen IS, Sorensen MV, de Bruijn PI, Bleich M, et al. 2016. Hyperaldosteronism after decreased renal K+ excretion in KCNMB2 knockout mice. Am. J. Physiol. Ren. Physiol 310:F1035–46 [DOI] [PubMed] [Google Scholar]
- 138.Grimm PR, Irsik DL, Settles DC, Holtzclaw JD, Sansom SC. 2009. Hypertension of Kcnmb1−/− is linked to deficient K secretion and aldosteronism. PNAS 106:11800–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ha TS, Heo MS, Park CS. 2004. Functional effects of auxiliary β4-subunit on rat large-conductance Ca2+-activated K+ channel. Biophys. J 86:2871–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wulff H, Castle NA, Pardo LA. 2009. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov 8:982–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Long SB, Campbell EB, MacKinnon R. 2005. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science 309:903–8 [DOI] [PubMed] [Google Scholar]
- 142.Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, et al. 2005. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev 57:473–508 [DOI] [PubMed] [Google Scholar]
- 143.Pongs O, Schwarz JR. 2010. Ancillary subunits associated with voltage-dependent K+ channels. Physiol. Rev 90:755–96 [DOI] [PubMed] [Google Scholar]
- 144.Brauneis U, Vassilev PM, Quinn SJ, Williams GH, Tillotson DL. 1991. ANG II blocks potassium currents in zona glomerulosa cells from rat, bovine, and human adrenals. Am. J. Physiol 260:E772–79 [DOI] [PubMed] [Google Scholar]
- 145.Payet MD, Benabderrazik M, Gallo-Payet N. 1987. Excitation-secretion coupling: ionic currents in glomerulosa cells: effects of adrenocorticotropin and K+ channel blockers. Endocrinology 121:875–82 [DOI] [PubMed] [Google Scholar]
- 146.Arrighi I, Bloch-Faure M, Grahammer F, Bleich M, Warth R, et al. 2001. Altered potassium balance and aldosterone secretion in a mouse model of human congenital long QT syndrome. PNAS 98:8792–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Leng S, Pignatti E, Kehtani RS, Shah MS, Xu S, et al. 2020. β-Catenin and FGFR2 regulate postnatal rosette-based adrenocortical morphogenesis. Nat. Commun 11:1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Blankenship JT, Backovic ST, Sanny JS, Weitz O, Zallen JA. 2006. Multicellular rosette formation links planar cell polarity to tissue morphogenesis. Dev. Cell 11:459–70 [DOI] [PubMed] [Google Scholar]
- 149.Freedman BD, Kempna PB, Carlone DL, Shah M, Guagliardo NA, et al. 2013. Adrenocortical zonation results from lineage conversion of differentiated zona glomerulosa cells. Dev. Cell 26:666–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lecuit T, Lenne PF. 2007. Cell surface mechanics and the control of cell shape, tissue patterns and morphogenesis. Nat. Rev. Mol. Cell Biol 8:633–44 [DOI] [PubMed] [Google Scholar]
- 151.Bell CL, Murray SA. 2016. Adrenocortical gap junctions and their functions. Front. Endocrinol 7:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Burnstock G, Ralevic V. 2014. Purinergic signaling and blood vessels in health and disease. Pharmacol. Rev 66:102–92 [DOI] [PubMed] [Google Scholar]
- 153.Sperelakis N, McConnell K. 2002. Electric field interactions between closely abutting excitable cells. IEEE Eng. Med. Biol. Mag 21:77–89 [DOI] [PubMed] [Google Scholar]
- 154.Octeau JC, Gangwani MR, Allam SL, Tran D, Huang S, et al. 2019. Transient, consequential increases in extracellular potassium ions accompany channelrhodopsin2 excitation. Cell Rep 27:2249–61.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ladoux B, Nelson WJ, Yan J, Mege RM. 2015. The mechanotransduction machinery at work at adherens junctions. Integr. Biol 7:1109–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Prinz AA, Bucher D, Marder E. 2004. Similar network activity from disparate circuit parameters. Nat. Neurosci 7:1345–52 [DOI] [PubMed] [Google Scholar]