

Summary
Robust patient-derived platforms that recapitulate the cellular and molecular fingerprints of glioblastoma are crucial for developing effective therapies. Here, we describe a chemically defined protocol for 3D culture and propagation of glioblastoma in 3D gliospheres, patient-derived organoids (PDOs), mouse brain orthotopic xenografts (PDOXs), and downstream drug and immunofluorescence assays. This simple-to-follow protocol allows assessing drug sensitivity, on-target activity, and combined drug synergy. Promising therapies can then be validated in PDOXs for translation in precision medicine oncology trials.
For complete details on the use and execution of this protocol, please refer to Chadwick et al. (2020) and Patrizii et al. (2018).
Keywords: o, Organoids, glioblastoma, patient derived organoids, orthotopic patient derived xenografts, PDX, combination therapy, drug synergy, precision medicine
Graphical Abstract
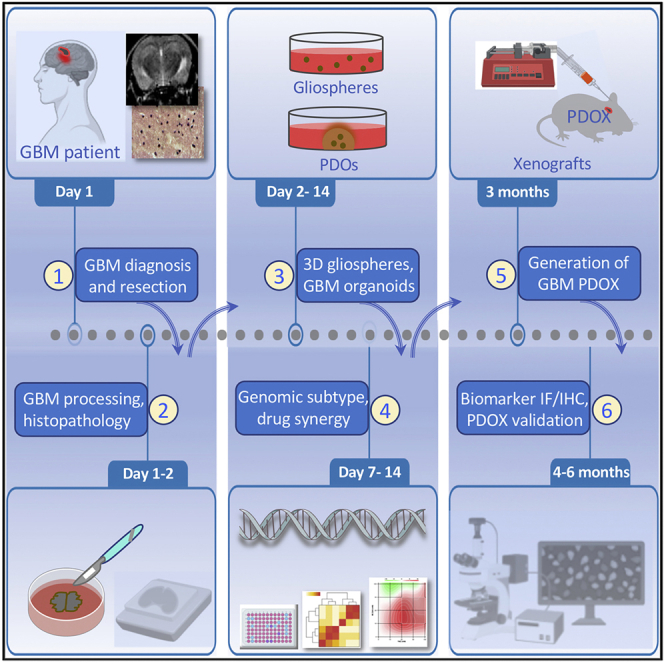
Highlights
-
•
Reproducible generation of 3D cultured glioblastoma patient-derived organoids (PDOs)
-
•
Gliospheres and PDOs can be propagated and cryopreserved for downstream applications
-
•
Engrafted cells are imaged with MRI/BLI in mouse brain orthotopic xenografts
-
•
PDOs are used to assess synergy between drug combinations for combined targeted therapy
Robust patient-derived platforms that recapitulate the cellular and molecular fingerprints of glioblastoma are crucial for developing effective therapies. Here, we describe a chemically defined protocol for 3D culture and propagation of glioblastoma in 3D gliospheres, patient-derived organoids (PDOs), mouse brain orthotopic xenografts (PDOXs), and downstream drug and immunofluorescence assays. This simple-to-follow protocol allows assessing drug sensitivity, on-target activity, and combined drug synergy. Promising therapies can then be validated in PDOXs for translation in precision medicine oncology trials.
Before you begin
-
1.
Patient-derived tissues, including glioblastoma (GBM) material and corresponding blood samples, must be obtained under an approved Institutional Review Board (IRB) protocol. Please consult with your local institutional authority board for IRB guidelines regarding the use of human specimens and biological samples.
-
2.
All personnel involved in the studies must acquire the required training and be certified to meet their local institute’s requirements for working with human subject materials. Tissue culture procedures must be completed under an appropriate Biosafety Level containment (e.g., BL2) as determined by local Biosafety Review Board for use of human tissues and viral vectors used in xenografts studies. All animal xenograft studies must be approved by the Institutional Animal Care and Use Committee (IACUC).
-
3.
GBMs are typically diagnosed clinically using MRI and histopathology according to WHO criteria (see Weller et al., 2015).
-
4.
On the same day, prior to receiving the surgical specimen, reconstitute all of the components of the tissue culture media from stock growth factors reconstituted per the manufacturer’s instructions into working media and stored accordingly (see reagents within the Key resources table below).
-
5.
Thaw Matrigel or another extracellular matrix (ECM) basement membrane material in 4°C overnight (8–12 h) and per the manufacturer’s instructions.
-
6.
When culturing organoids, put the desired number of cell culture plates in the 37°C incubator for 1–2 h prior to starting the organoid culture.
-
7.
All cell cultures must be performed using sterile/aseptic practices and under standard culture conditions (37°C and 5% CO2). Prolonged organoid cultures may be transferred to bioreactors under similar conditions after 2 weeks of initiating the 3D plate cultures.
-
8.
If xenograft studies are planned, prepare lentiviral reporters (optional to allow performing live imaging of mouse xenografts) from frozen stock and generate fresh suspensions of 3D cultures to be labeled with lentiviral reporters for engraftment in the mouse brain and generation of patient-derived orthotopic xenograft (PDOX) models.
-
9.
Age and sex matched NSG mice or alternatively other immunodeficient mice (3–4 mice that are 6–12 weeks old per patient-derived sample) are made available prior to starting the PDOX experiment. The sex of the mouse should be matched to the sex of the patient from which the tissue sample has been derived, in order to account for a male to female ratio of 1.6:1 of GBM incidence. Our results were generated using NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice obtained from the Jackson Laboratory.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BMI1 antibody (1:200) | Cell Signaling Technology | 6964 |
| GALC antibody (1:200) | Santa Cruz Biotechnology | SC-67352 |
| GFAP antibody (1:300) | Cell Signaling Technology | 3670 |
| NESTIN antibody (1:200) | Millipore | MAB5326 |
| SOX2 antibody (1:400) | Cell Signaling Technology | 23064 |
| TLX antibody (5 μg/mL) | Invitrogen | PA5-40484 |
| TUJ1 antibody (1:200) | Millipore | MAB1637 |
| P16 antibody (1:1,000) | Santa Cruz Biotechnology | SC-81157 |
| Nucleolin antibody (1–2 μg/mL) | Abcam | Ab-22758 |
| Phospho-S6 antibody (1:1,000) | Cell Signaling Technology | 2215S |
| Phalloidin (1:400) | Thermo Fisher Scientific | A12380 |
| DAPI (4′,6-diamidino-2-phenylindole, dilactate) (300 nM) | BioLegend | 422801 |
| Chemicals, peptides, and recombinant proteins | ||
| GlutaMAX-I | Gibco | 35050-061 |
| Advanced DMEM/F12 | Gibco | 12634-010 |
| Eagle’s minimum essential medium (EMEM) | ATCC | 30-2003 |
| Matrigel matrix basement membrane | Corning | 356237 |
| B27 Supplement without vitamin A | Gibco | 12587-010 |
| Recombinant human EGF | Peprotech | AF-100-15 |
| Recombinant human FGF-basic (BFGF) | Peprotech | AF-100-18B |
| Hydrocortisone stock solution (200×) | STEMCELL Technologies | 07925 |
| Penicillin and streptomycin (10,000 U/mL) | Gibco | 15140-122 |
| Sodium pyruvate | Himedia | TCL015-100mL |
| Primocin | InvivoGen | Ant-pm-1 |
| Accutase | STEMCELL | 07920 |
| 37% Formaldehyde | Fisher Bioreagents | BP-531-500 |
| Phosphate buffer saline (PBS) without calcium and magnesium | Lonza | 17-516F |
| Triton X-100 | Sigma-Aldrich | X100-100mL |
| Ketamine | Henry Schein | NDC 11695-0702-1 |
| Xylazine | Akom | NDC 59399-110-20 |
| Minimal essential medium (MEM) | Gibco | 11090-081 |
| Fetal bovine serum (FBS) | Sigma-Aldrich | F4135-500 mL |
| Bovine serum albumin (BSA), Fraction V | Sigma-Aldrich | 2930-100GM |
| Cell recovery solution (CRS) | Corning | 354253 |
| Betadine | Fisher Scientific | 19027132 |
| Hydrogen peroxide (3%) | Sigma-Aldrich | 88597-100mL |
| Drugs for testing and screening | Local pharmacy or commercial sources (e.g., Selleck) | N/A |
| Critical commercial assays | ||
| RNeasy Kit | Qiagen | 75162 |
| SuperScript IV VILO MasterMix | Invitrogen | 11756050 |
| SYBR Green Master Mix | Thermo Fisher Scientific | A25742 |
| CellTiter-Glo Assay | Promega | G7572 |
| Biological samples | ||
| Surgical specimen of tumor removed from GBM patients | Surgical facility | N/A |
| Experimental models: organisms/strains | ||
| 6- to 12-week-old NSG mice | The Jackson Laboratory | NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ |
| Other | ||
| Ultra-low attachment 96-well plates | Grenier | 655185 |
| Ultra-low attachment 384-well plates | Grenier | 781186 |
| Falcon 40 μm cell strainer | Corning | 352340 |
| IncuCyte HD system | Sartorius | N/A |
| Nikon A1R Si confocal microscope | Nikon | N/A |
| IVIS Spectrum imaging station | PerkinElmer | N/A |
| StepOne plus qPCR instrument | Applied Biosystems | 4376600 |
| Victor3 multilabel plate reader | PerkinElmer | 1420-011 |
| Stereotactic apparatus | Stoelting | 51901 |
| Stereotactic probe holder attachment | Stoelting | 51681 |
| Micromotor high-speed drill with drill bits | Stoelting | 51449 |
| Drill bit, 45 mm | Stoelting | 514551 |
| 10 μL syringe, Model 701 | Hamilton | 7635-01 |
| 30 gauge removable needle | Hamilton | 7803-07 |
| Rectal probe | Stoelting | 5060-040 |
| Vetbond tissue adhesive | 3M | 1469SB |
| Bonewax | Ethicon | W31G |
| Microdrill | Foredom | MH-170 |
| Micromotor control | Foredom | HP4-917 |
| Foot speed control | Foredom | HP4-970 |
| Homeothermic monitoring system | Harvard Apparatus | 55-7020 |
| Remote infuse/withdraw PHD ULTRA Nanomite programmable syringe pump | Harvard Apparatus | 70-3601 |
| Pura lube ophthalmic ointment | Dechra | NDC 17033-211-040 |
| Veet hair removal cream | Any commercial source (e.g., Amazon) | N/A |
| Syringe (1 mL) | BD | 309659 |
| Syringe needle (27 gauge) | BD | 305109 |
| Scalpels | BD | 371610 |
| Sodium chloride (0.9%) | Hospira | NDC 0509-4888-06 |
| Software and algorithms | ||
| ELDA software | Walter + Eliza Hall | http://bioinf.wehi.edu.au/software/elda/ |
| TCGA database | NHGRI & NCI | https://portal.gdc.cancer.gov/ |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Drug synergy software | Synergy Finder | https://synergyfinder.fimm.fi |
| Image analysis software | NIH | https://imagej.nih.gov/ij/ |
| StepOne software | Applied Biosystems | N/A |
| IVIS imaging software | PerkinElmer | N/A |
Materials and equipment
-
•
Serum-free GBM collection medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Advanced DMEM/F12 | - | ∗500 mL |
| Primocin (50 mg/mL) | 100 μg/mL | 1 mL |
| B27 (50×) | 1× | 10 mL |
| EGF (10 μg/mL) | 20 ng/mL | 1 mL |
| bFGF (10 μg/mL) | 20 ng/mL | 1 mL |
∗Note: The volume of DMEM/F12 varies depending on the number of plates used. If making 500 mL of serum-free GBM collection medium, add 1 mL of Primocin, 10 mL B27, and dilute stock solution for EGF and bFGF to a final concentration of 20 ng/mL. Media should be prepared every week and stored at 4°C.
-
•
Gliosphere basal media (GSBM)
| Reagent or resource | Final concentration | Volume |
|---|---|---|
| EMEM | - | 490 mL |
| Glutamax (100×) | 1× (2 mM of L-alanyl-L-glutamine dipeptide) | 5 mL |
| Penicillin/streptomycin (100×) | 100 U/mL of penicillin and 100 μg/mL of streptomycin | 5 mL |
Note: Preparation of gliosphere basal medium for culturing of patient-derived spheres and organoids. Media should be prepared every week and stored at 4°C.
CRITICAL: It is paramount to incorporate B27, EGF, and FGF factors freshly before starting cell culture each time. Therefore, make sure to calculate the volume of media that will be needed for each cell culture and add the required growth factor amounts accordingly.
-
•
Preparation of ketamine/xylazine anesthetics for generating mouse xenografts
| Reagent | Final concentration | Amount per mouse |
|---|---|---|
| Ketamine (100 mg/mL) | 10 mg/mL | 30 μL |
| Xylazine (20 mg/mL) | 1 mg/mL | 15 μL |
| PBS | 1× | 255 μL |
| Total | n/a | 300 μL |
Note: Mouse anesthetics solution should be prepared fresh, with the amount based on the weight of mice and is shown based on an average mouse weight of 30 g.
Step-by-step method details
Experimental timing
On day 1, collect tissue from surgery. This must be done within 60 min of tumor removal. Please refer to step 1 of tissue collection and processing for the details on the processing during day 1 and reagents used.
Patient-derived 3D culture initiation: 2–14 days
Note: Patient-derived sphere (gliospheres) formation can be achieved in 7 days.
Note: Patient-derived organoid (PDO) formation can be achieved in as early as 14 days. However, depending on the collected tumor size, seeding density, culture conditions, and the desired use, PDO culture could take up to three months.
Patient-derived orthotopic xenografts (PDOXs) after 3D culture: 2–12 weeks
Gliosphere and PDO expression profiling by qPCR or RNA seq: 8 h to 7 days
Whole-mount IF and IHC on gliosphere and PDO cultures: 2–4 days
Drug treatment and synergy assays: 5 days
Note: Refer to Figure 1 for an overview and images depicting the morphology and size of the GBM organoids and gliospheres generated using this protocol.
Figure 1.

Generation of gliospheres and PDOs from patient tissue
Morphological and histological analysis of GBM PDSs, PDOs, and PDOXs.
(A) Schematic outline for histological analysis of GBM patient-derived spheres (PDSs) also called gliospheres, patient-derived organoids (PDOs), and mouse brain patient-derived orthotopic xenografts (PDOXs).
(B) Representative GBM gliospheres in bright-field images taken using inverted microscopy on day 7 (top left panel). Representative histology analysis with H&E staining images of gliospheres (bottom left panel) and immunofluorescent (IF) staining of neuroepithelial differentiation marker GFAP with phalloidin and DAPI is shown (right panel).
(C) Representative GBM PDOs in bright-field images on day 1, day 8, and day 14 (top left panel), H&E stain of PDOs (bottom left panel) and IF staining of neuroepithelial progenitor marker Nestin with phalloidin and DAPI (right panel).
(D) Representative GBM PDOX mouse brains in H&E and IHC staining for cell proliferation marker Ki67 and neuroepithelial progenitor marker Nestin. Scale bars are 100 μm in bright-field, H&E, and IHC images in (B)–(D) and 20 μm in IF images in (B) and (C).
Tissue collection and processing
Timing: days 1–3
-
1.
Surgical specimen should be collected from the operation room within 60 min of tumor removal from GBM patients previously diagnosed using MRI and/or histology and undergoing craniotomy resection. The neuropathologist will place the GBM specimen in serum-free GBM collection medium after diagnostic confirmation of GBM using H&E staining. Pathology assistant will record the deidentified sample number and have it delivered to the tissue culture facility.
-
2.Tissue samples are divided into three fractions: first for cryofreezing, DNA and RNA extraction (for glioseq exome sequencing; Nikiforova et al., 2016), second for expression profiling to determine GBM subtypes and targets for drug testing, and third for live tissue for initiating 3D cultures to make gliospheres and PDOs.
-
a.Under the tissue culture hood, place the tissue in a Petri dish and use a sterile scalpel to cut patient GBM tissue into small cubic fragments (~1 mm3 each).
-
b.Incubate tissue fragments with Accutase at 10 mL per 75 cm2 surface area for 2–4 min at 37°C only when tissue fragments are ≥1 mm3 in size. This will dissociate tumor cells from the surrounding extracellular matrix. If fragments are smaller in size, use only 1–2 mL of Accutase in a 15 mL Falcon tube and incubate for 2 min at 37°C.
-
c.Collect fragments from the incubator and pass multiple times through a 26-gauge needle to obtain a suspension of single cells. Serial passing through the needle helps break down tissue fragments and generate tumor cell suspension.
-
d.Filter the cell suspension through a 40 μm filter placed on top of a 50 mL Falcon tube. Wash the filter with 5 mL of serum-free GBM media into the 50 mL tube.
-
e.Pellet the cells by centrifugation at 300 × g for 5 min at room temperature (20°C–21°C).
-
f.Resuspend in 500 μL to 1 mL of serum-free GBM media.
-
g.Count the number of cells using a hemocytometer.
-
h.Plate the cells (based on surface area) in multi-well ultra-low attachment plates.Note: If using a 96-well ultra-low attachment plates, seed approximately 1,000 cells per well. Alternatively, if using 6 well suspension plates, seed approximately 10,000–25,000 cells per well.
-
i.Monitor the cells during the first 2–3 days.
-
i.Check for cell clumping and formation of sphere-like structures at days 5–7 for gliospheres.
-
ii.For PDOs, monitor the Matrigel droplets for 14 days.
-
iii.At days 7–14, respectively, cells are ready for processing and downstream applications, or for cryofreezing for future use.
-
i.
-
j.At day 7 or 14, accordingly, start to propagate gliospheres or PDOs for 2–12 weeks to generate GBM cells for histological processing (see representative data in Figures 1A–1C), cryofreezing, labeling with luciferase reporters, and for engraftment into mouse brains to generate PDOXs or perform drug testing and synergy assays.
-
a.
Culture establishment, storage, and propagation
-
3.Add growth factors to GSBM (see table in Materials and equipment for reference on how to make GSBM).
-
a.B27 (50× stock concentration), 20 μL/mL final concentration.
-
b.EGF (10 μg/mL stock concentration), 20 ng/mL final concentration.
-
c.bFGF (10 μg/mL stock concentration), 20 ng/mL final concentration.
-
a.
Note: The amount of media needed is dependent upon the number of wells containing GBM cultures. For a 96-well plate, each well receives 100 μL of media. Calculate the media needed and add desired factors based on the media volume being used.
-
4.Prepare GBM Organoid Basal Media (GOBM)
-
a.GSBM supplemented with:
-
i.Hydrocortisone (200× stock concentration), 1× (50 ng/mL) final concentration.
-
ii.B27 (50× stock concentration), 20 μL/mL final concentration.
-
iii.EGF (10 μg/mL stock concentration), 20 ng/mL final concentration.
-
iv.bFGF (10 μg/mL stock concentration), 20 ng/mL final concentration.
-
i.
-
a.
Note: The amount of media needed is dependent upon the number of wells containing organoids. For a 96 well plate, each well receives 100–200 μL of media (usually 100 μL is enough to cover the gel matrix). Calculate the amount of media needed and add desired factors based on the total media volume being used.
Thawing and plating of cells
-
5.If working from previously cryo-frozen samples, thaw the vials by gentle shaking in a 37°C water bath until the cell pellets are approximately 2/3 thawed.
-
a.Add thawed cell pellets to a 15 mL canonical tube and overlay cells with 9 mL of GSBM.
-
b.Spin the sample down at room temperature (20°C–21°C) for 5 min at 300 × g.
-
c.Remove the supernatant and resuspend cells in 1 mL of GSBM.
-
d.Using a 200 μL pipette, mechanically dissociate the pellet into a single cell suspension by pipetting up and down gently multiple times.CRITICAL: Make sure to be careful and gentle when pipetting to prevent damage to the cells.
-
e.Count the cells using a hemocytometer.
-
f.Plate 1,000–5,000 cells per well in 96-well untreated/suspension plates and add 100–200 μL of fresh GSBM to each well.
-
g.Incubate at 37°C and 5% CO2.
-
h.Change half of the media every 2 days by tilting the plate to a 45°angle.CRITICAL: It is easy to aspirate the spheres/organoids when removing fluids (i.e., media, PBS, fixative) from the wells, therefore, it is critical to monitor the cells during the aspiration.Note: Tilting the plate and slowly aspirating from the surface of the media/plate interface will help minimize the number of spheres/organoids that are lost throughout the media changes and passaging processes. Alternatively, working under a dissecting microscope will help track the organoids/spheres throughout the process. Though, this might be laborious and time consuming.Note: If working from freshly dissociated cells, proceed to steps 6a–6f.
-
a.
-
6.When plating PDOs additional care and steps are required:
-
a.Refer to steps 5a–5e for details on how to thaw, prepare a single cell suspension, and count before proceeding to 6b.CRITICAL: It is important to place the desired number of plates in the incubator 1–2 h prior to starting the following steps. Additionally, it is paramount to work and keep tubes on ice from this step forward.
-
b.Place thawed Matrigel in ice.
-
c.Based on cell counts using a hemocytometer, dilute cells to 100 cell/μL.CRITICAL: Overall composition of the gel/cells and media solution should not exceed 20% cells with media and 80% Matrigel. Additionally, avoid creating bubbles to the mixture of gel/cells and media when pipetting up and down.
-
d.Prewarm a 96-well plate by placing it in the cell culture incubator.
-
e.Retrieve the 96-well plates from the incubator and add a 10 μL cell/Matrigel droplet to the middle of the well to generate the basement membrane 3D culture chamber.
-
f.Pipette slowly to generate a doom-shaped droplet chamber.Note: It is important not to touch the bottom of the plate with the pipette tip. This would help with maintaining the integrity of the gel matrix longer.
-
g.Move the plate back to the incubator and allow the Matrigel to harden.Note: The overall time for hardening of the gel varies based on the size of the Matrigel droplet. For a 10 μL droplet, it should take no more than 10 min for the gel to harden.CRITICAL: For a 96-well plate, do not leave the gel in the incubator for longer than 10 min since this could affect the cells embedded in the gel.
-
h.Once the Matrigel has solidified, overlay the droplet with enough GOBM to cover it. This is usually achieved by adding 100–200 μL per well.
-
i.Follow steps 5g and 5h.
-
j.Monitor organoid growth every other day and record images (see Methods video S1 for a typical GBM PDO growth during the first week of culture. Note the cell mobility and interaction with the ECM, which requires readjustment of focus for proper imaging) . PDOs are usually ready for analysis and use starting at 14 days (Figure 1).Methods video S1. Cellular motility of GBM PDO cells in organoid culture, related to step 6j in Step-by-step method details and Figure 1Time lapse recording of a typical GBM PDO growth during the first five days of the 3D culture after plating. Note the clusters of GBM dividing cells and the cellular motility of GBM cells, with neurite extensions, and the repeated interactions between various cells and the ECM, which forms the surrounding Matrigel droplet edge. These continuous cellular motilities might require readjustment of focus for proper time lapse imaging. This time lapse movie was taken during days 1–5 of 3D GBM organoid culture and was set to 200× playback.Download video file (2.8MB, flv)
-
a.
Passaging of gliospheres and PDO cells
-
7.Passaging cells from gliospheres: gliospheres are ready for passaging after 7 days. Pool gliospheres in 15 mL canonical tubes.
-
a.Centrifuge the tube at room temperature (20°C–21°C) for 5 min at 300 × g.
-
b.Discard the supernatant and resuspend the cells in 1 mL GSBM.
-
c.Mechanically dissociate spheres using a 200 μL pipette.CRITICAL: Make sure to be careful and gentle when pipetting to prevent damage to the sphere producing cells.
-
d.Count the cells using a hemocytometer.
-
e.Plate 1,000–5,000 cells on 96-well untreated/suspension plates.
-
f.Incubate at 37°C and 5% CO2.
-
g.Change half of the media every 2 days.
-
a.
-
8.Passaging of PDO cells: PDOs are ready for passaging after 10–14 days. When passaging organoid cells, additional steps are required:
-
a.Remove all the media from the wells.
-
b.Dissolve the Matrigel droplet chamber by adding 200 μL of ice-cold GOBM.
-
c.Scrape the bottom of the well by gently using a cell scraper/or a pipette tip (depending on culture dish used) and collect the organoid/Matrigel sample in a 15 mL canonical tube.
-
d.Centrifuge the collected tube at 300 × g for 5 min.
-
e.Remove supernatant and wash the pellet three times with ice-cold PBS.CRITICAL: You must centrifuge at room temperature (20°C–21°C) at 300 × g for 5 min between each wash in order to prevent losing any of the organoids collected from the wells.
-
f.Centrifuge one more time at 300 × g for 5 min at room temperature (20°C–21°C), remove supernatant and overlay the remaining organoid pellet/Matrigel mixture with 1 mL of cell recovery solution (CRS).
-
g.Allow the 15 mL tube to sit on ice for 30 min. Make sure that the Matrigel remnants are no longer visible.
-
h.Centrifuge one more time at 300 × g for 5 min.
-
i.Remove the supernatant and wash once with PBS.
-
j.Remove PBS by centrifugation (as described above).
-
k.Add 2 mL of GOBM.
-
l.Dissociate the organoid pellet by mechanical dissociation using a broken Pasteur pipette or with a laser pulled needle.Note: Avoid dissociating organoids into single cells, rather leave them as small fragments if possible, in order to maintain relatively similar proliferative rate without single cell outgrowth.
-
m.Count the organoids under a light microscope and split/pass at a ratio of 1:3 or 1:4 depending on organoid density.Note: It is important to determine if proliferation rate in 3D cultured PDOs reflects a relatively similar proliferation rate of the original GBM tumor (e.g., relatively similar number of Ki67+ cells) and additionally upon the formation of PDOXs (see below). The cell doubling time should be established under light and/or fluorescent microscopy (when cells are labeled with fluorescence or luciferase reporters).
-
n.Follow steps 5g and 5h for culture conditions, and media changes.CRITICAL: Since Ki67 levels are usually determined during GBM diagnosis, immunofluorescence staining for Ki67 and/or PCNA can be used to confirm the same proliferative rates of GBM cells in PDOs and PDOXs compared to original tumor. This is highly critical when using PDOs and PDOXs for drug sensitivity testing.
-
a.
Cryofreezing of cells
-
9.Freezing cells: gliospheres. Pool gliospheres in a 15 mL canonical tube.
-
a.Centrifuge for 5 min at 244 × g.
-
b.Discard the supernatant and resuspend in 1 mL of GSBM.CRITICAL: Additional care should be taken when aspirating the supernatant. Using a long 200 μL pipette might help prevent aspirating some of the spheres.
-
c.Resuspend the pellet in 1 mL of freezing mediaNote: Freezing media is made by mixing GSBM with 10% DMSO
-
d.Aliquot cells in 1.5 mL cryotubes, and store in a Mr. Frosty or similar freezing container at −80°C for up to 24–48 h.
-
e.Move the frozen vials into liquid nitrogen for long-term storage.
-
a.
-
10.Freezing cells: PDOs. When freezing organoids, additional steps are required:
-
a.Follow steps 8a–8j.
-
b.Add 2–4.5 mL of GOBM substituted with 10% DMSO to the tube containing the organoid pellet.Note: GOBM substituted with 10% DMSO is used for cryopreservation of PDOs
-
c.Add the desired volume containing the desired number of organoids to a 1.0–1.5 mL cryogenic tube.
-
d.Follow steps 9d–9e.
-
a.
Initiation of mouse brain Patient-derived orthotopic xenografts (PDOX)
Before you begin, prepare mouse anesthesia as described in Materials and equipment.
Alternatives: Alternatively, you can replace PBS with 0.9% Sodium Chloride.
-
11.
Before planning the surgical implantation of gliospheres or PDO cells in the mouse brain, GBM cells may optionally be labeled with luciferase reporters and/or fluorescent reporters (e.g., using lentiviral vectors) (see Chadwick et al., 2020 for details) to be able to monitor glioma formation in live mouse brains after formation of PDOXs.
-
12.
On the day of the mouse surgery, prepare a cell suspension from labeled gliosphere or PDO cultures and prepare the appropriate media (see Materials and equipment and Culture establishment, storage, and propagation, steps 3 and 4). Suspend the desired number of cells to be implanted in 3–5 μL of media. Use GSBM for spheres or GOBM for organoids .
Note: Detection of mouse glioma depends on the proliferative rate of originating GBM. We have detected gliomas in PDXs using luciferase and/or MRI imaging from as little as 2,000 implanted cells in as early as 2–3 weeks. Mouse brain tumors could be confirmed in all animals at 12 weeks following implantation.
-
13.
Anesthetize 6- to 12-week-old NSG mice with 100 mg/kg ketamine, 10 mg/kg xylazine solution via intraperitoneal injection with a sterile 27-gauge insulin needle.
Note: If the animal is still responsive to toe or tail pinch, an additional IP injection of no more than 25% the initial anesthetic dose can be administered. Ensure full anesthesia is achieved before making the scalp incision.
-
14.
Apply ophthalmic ointment to eyes to prevent drying throughout the procedure.
-
15.
Proceed to remove hair on scalp with the hair removal cream.
-
16.
Immobilize the anesthetized mouse on the stereotactic device (Figure 2A) by hooking incisors on the indentation on the tooth bar, securing the snout clamp, and then tightening the screws holding on either side of the head (Figure 2B).
Note: Make sure that the head is held stably and is on an even plane.
-
17.
Swab the scalp with 3 alternating cycles of betadine and 70% ethanol, making sure that the area is free of loose hair and any remaining hair removal cream.
-
18.
Using a scalpel, make a 0.5–0.75 cm longitudinal scalp incision midway in the scalp from between the eyes to between the ears.
-
19.
Swab the skull with hydrogen peroxide to identify the bregma.
Note: The bregma is the point on the surface of the skull where the sagittal and coronal sutures meet. It can be distinctly identified because when exposed to hydrogen peroxide it should adopt a pale coloration that stands out from its surroundings.
Note: Small bubbles should form on the T-shape of the bregma when hydrogen peroxide is applied. Adjust incision to make sure the bregma is in view before proceeding.
-
20.
Attach the drill to the stereotactic apparatus and center the drill tip above the bregma.
-
21.
Set up the stereotactic coordinates on the Stoelting instrument display to X = 0, Y = 0, and Z = 0 at this point.
-
22.
Set the drill 2.5 mm to the right, and 1.5 mm anterior to directions +2.5, +1.5, 0.
-
23.
Lower the drill to contact the skull, making sure to maintain 2.5 mm and 1.5 mm in the x and y directions, and gently drill through the skull.
Note: Stop as soon as you cut through the bone, it will be very quick!
-
24.
Resuspend cells in media by gently pipetting up and down before filling the Hamilton syringe with the appropriate volume of cells and media for injection.
Note: The maximum volume for infusion is 5 μL.
-
25.
Replace the drill with the Nanomite syringe pump on the stereotactic frame. Clean off the needle of the Hamilton syringe with an alcohol pad before attaching it to the infusion pump.
-
26.
Move the needle to the burr site and move the needle downward so it is just touching the brain. Reset the coordinates of the stereotactic instrument ensuring that the z coordinate is at 0 mm.
-
27.
Lower the needle slowly in the z direction to −3.5 mm.
-
28.
Allow the automated injector pump to inject the cells at a rate of 0.5 μL/min.
-
29.
Leave the needle in brain for an additional 2 min after infusion is complete before slowly withdrawing it.
-
30.
Cover the hole in the skull completely with bone wax and close the scalp incision with Vetbond adhesive.
-
31.
Remove the rectal probe from the mouse and remove the mouse from the stereotactic device. Place the mouse in a heated recovery cage. When the mouse regains consciousness and coordination, it can be returned to its original cage.
-
32.
Tumor growth can be monitored with MRI, or with bioluminescent imaging using the IVIS system if luciferase expressing cells are used (see Patrizii et al., 2018).
-
33.
Monitor mice for neurological symptoms or general signs of distress at which point mice should be euthanized and brains can be collected for histology (see the multiple steps for generating these mouse PDOXs in Figure 3 and the representative histology outcome of PDOX tumors in Figure 1D).
Figure 2.

Stereotactic device station used for establishing mouse orthotopic xenografts
(A) An image of the device and mouse surgical station.
(B) An immobilized anesthetized mouse on the stereotactic device with ophthalmic and hair cream applied, and with the incisors hooked on the tooth bar, the snout clamps secured, and the screws are holding on either side of the head.
(C) A mouse after graft implantation and during recovery while continuously placed on the blue heating pad with rectal thermal monitoring.
Figure 3.

Generation of PDOXs
Steps and key requirement for establishing mouse PDOXs from PDO 3D cultures. The steps include dissociation of PDOs, labeling with lentiviral fluorescent reporters, engrafting glioma in mouse brains, detecting PDOXs using bioluminescence imaging (BLI) and MRI imaging, and molecular assays. Ctl, control mice are compared to PDOX mice.
RNA extraction, RNA sequencing, and quantitative real-time PCR (qPCR)
-
34.
Gliospheres and PDOs could be pelleted as described above.
-
35.
RNA extraction from gliospheres, PDOs, PDOX brain, and their corresponding originating GBM is accomplished by following the RNeasy kit instructions (https://www.qiagen.com/us/products/discovery-and-translational-research/dna-rna-purification/rna-purification/total-rna/rneasy-mini-kit/#productdetails). Alternatively, if the RNeasy kit is not available, RNA extractions can be accomplished by following the Trizol RNA extraction method (http://assets.thermofisher.com/TFS-Assets/LSG/manuals/trizol_reagent.pdf).
-
36.
The resulting amount of RNA can be measured using the nanodrop/Nanovue system and RNA could be used for expression profiling using RNA sequencing and qPCR. The expression profiles can then be matched to the original GBM tissue preserved to ensure parity between the generated 3D cultures and the originating GBM.
-
37.Approximately 1 μg of RNA is used for cDNA Synthesis (explained below):
-
a.cDNA synthesis is accomplished by following Thermo Fisher Superscript IV VILO Master Mix protocol (https://assets.thermofisher.com/TFS-Assets/LSG/manuals/superscriptIV_VILO_master_mix_UG.pdf).
-
b.Resulting cDNA is used along with the desired primers for qPCR (for a list of target genes that are used to identify the expression signature of GBM subtypes, please refer to the list in Chadwick et al., 2020):
-
i.Newly synthesized cDNA is diluted using ultra-pure PCR grade water.
-
ii.A SYBR green mix including forward (F), reverse (R) primers and ultra-pure PCR grade water mixture is made fresh and following the manufacturer’s specifications.
-
iii.Use triplicate wells for each target, and cDNA is added independently with a fresh pipette tip (10 μL pipette) to prevent cross-contamination of the samples.
-
iv.The following set up (Table 1) is used on StepOne qPCR instrument using SYBR Green master mix following the manufacturer’s instructions (https://www.thermofisher.com/document-connect/document-connect.html?url=https%3A%2F%2Fassets.thermofisher.com%2FTFS-Assets%2FLSG%2Fmanuals%2FMAN0013511_PowerUp_mastermix_UG.pdf&title=VXNlciBHdWlkZTogUG93ZXJVcCBTWUJSIEdyZWVuIE1hc3RlciBNaXggLSBVbml2ZXJzYWwgMlggbWFzdGVyIG1peCBmb3IgcmVhbC10aW1lIFBDUiB3b3JrZmxvd3M=).
-
i.
-
a.
Table 1.
Cycling parameters for subtyping of GBM from PDOs, PDOXs, and original tumor
| Stage 1 | Stage 2 | Stage 3 |
Melt Curve |
|||
|---|---|---|---|---|---|---|
| Step 1 | Step 2 | Step 1 | Step 2 | Step 3 | ||
| 50°C | 95°C | 95°C | 60°C | 95°C | 60°C | 95°C |
| 2:00 min | 2:00 min | 15 s | 1:00 min | 15 s | 1:00 min | 15 s |
Stage 1 = 1×, Stage 2 = 1×, Stage 3 = 40×
Immunofluorescence (IF) and immunohistochemistry (IHC)
See Figure 1 for representative histology and IF staining of gliospheres and PDOs.
-
38.Fixation, permeabilization, and staining of gliospheres and PDOs for IF and IHC
-
a.Spheres and organoids are collected and the media or Matrigel plus media are removed respectively.Note: See steps 8a–8j for instructions on how to remove the Matrigel.CRITICAL: When aspirating/removing the media, it is better to use a glass Pasteur pipette, or a 200 μL (preferred) pipette to prevent aspiration of the spheres or organoids.
-
b.The spheres and organoids can be fixed in the wells at 37°C for 10–15 min using fresh 4% paraformaldehyde (PFA).Note: The volume of PFA used is dependent upon the plate size. For a 96-well plate, 200 μL of 4% PFA is used.
-
c.Wash the samples 3 times using 1× DPBS to remove the fixative.
-
d.Permeabilize the samples overnight (8–12 h) at 4°C by using 0.5% Triton X-100. A blocking step may be included for antibodies that generate high staining background.
-
e.Wash samples 3 times using DPBS to remove as much of the Triton X-100 as possible.
-
f.Incubate cells at room temperature (20°C–21°C) for 18–24 h with desired primary antibodies diluted per the manufacturer’s specifications. Primary antibodies against neuroepithelial progenitor markers such as NESTIN, and neuroepithelial differentiation markers such as glial fibrillary acidic protein (GFAP) (for CNS mature astrocytes and ependymal cells), galactosidase C (GALC) (for early-stage oligodendroglia), and tubulin-beta-III (TUJ1) (for neuron-specific early commitment) should be used.
-
g.For drug testing in GBM PDOs (see below), IF and/or IHC are done with primary antibodies against GBM hallmark GFAP, glioma stem cell (GSC) proteins such as the orphan nuclear receptor tailless (TLX), the induced pluripotent stem cell (iPSC) and self-renewal protein SOX2, and the self-renewal marker BMI1, and against pathway specific markers such as PI3K/mTOR marker pS6 to detect drug on-target activities and validate combination therapies (See (Mehta et al., 2015) and (Chadwick et al., 2020) for details).Note: Although this method has been successful for us, we understand that not all cultures are the same. Alternatively, optimization of the antibody staining might be necessary. In some cases, when using the appropriate controls for detecting background staining, if the desired staining cannot be detected using the times specified above, a prolonged primary antibody incubation can be performed by incubating cells for 1–2 days at 4°C or 1 day at room temperature (20°C–21°C).
-
h.After incubating for 18–24 h, wash 3 times using DPBS. Each wash should be approximately 5 min.
-
i.Treat the samples by adding desired secondary antibodies selected against the species of the primary antibody used. Make sure to dilute the latter in 5% bovine serum albumin (BSA) and per the antibody manufacturer’s specifications.
-
j.Incubate the cells at 4°C for 18–24 h and 1–2 h at room temperature (20°C–21°C) before washing 3 times with DPBSNote: Each wash should be performed for approximately 5 min, preferably with mild rocking. Antibody incubation times and temperatures can be adjusted based on the organoid/gliosphere density and the antibody optimization protocols. See problem/solution under troubleshoot for more information.
-
k.Stained specimens are now ready for whole-mount visualization and imaging.
-
a.
-
39.Step by step gliospheres and PDOs embedding for use in IHC assays
-
a.Collect spheres and organoids as described is steps 7 and 8.
-
b.Fix spheres and organoids in the well for 10–15 min at 37°C using freshly made 10% PFA.
-
c.Wash three times using DPBS. Add 250 μL DPBS per well. Make sure to wash for approximately 5 min. Remove 200 μL DBPS, leaving 50 μL in the well. Repeat this process for three times.
-
d.Add the desired quantity of Histogel to a beaker and heat up using microwave for approximately 1 min.Note: Leave the Histogel in a beaker, and in the microwave while performing the next steps.
-
e.Transfer the spheroids to a standard histology cassette mold for histological processing and let them drop to the bottom of the mold.
-
f.Get rid of any excess liquid as much as possible using a light duty tissue wipe.
-
g.Place the spheroids/organoids in the center using a p10 pipette tip.Note: Be careful not aspirate or remove the spheroids/organoids.
-
h.Add the warm Histogel to spheroids/organoids.Note: Make sure that you add enough Histogel to cover spheroids/ organoids.
-
i.Let the gel hardens for 1–2 min at room temperature (20°C–21°C).
-
j.Fill in the rest of the mold with Histogel.
-
k.Let the gel second layer hardens (about 10 min) at room temperature (20°C–21°C).
-
l.Put the samples in ice for 1 min.
-
m.The formed mold can then be utilized for histological processing.
-
a.
Drug synergy assays
Alternatives: This matrix can be used for any number of doses for each compound based on GI50. At least four serial doses should be examined to determine Bliss synergy scores.
Table 2.
Dose response assays to assess synergy between drug A (e.g., BEZ235 (BEZ)) and drug B (e.g., temozolomide (TMZ))
| Tested drug A concentration | BEZ 0.1 μM | BEZ 0.25 μM | BEZ 0.5 μM | BEZ 1 μM | |
|---|---|---|---|---|---|
| TMZ 0 BEZ 0 |
TMZ 0 BEZ 1 μM |
TMZ 0 BEZ 2.5 μM |
TMZ 0 BEZ 5 μM |
TMZ 0 BEZ 10 μM |
Tested drug B concentration |
| TMZ 10 μM BEZ 0 |
TMZ 10 μM BEZ 1 μM |
TMZ 10 μM BEZ 2.5 μM |
TMZ 10 μM BEZ 5 μM |
TMZ 10 μM BEZ 10 μM |
TMZ 1 μM |
| TMZ 100 μM BEZ 0 |
TMZ 100 μM BEZ 1 μM |
TMZ 100 μM BEZ 2.5 μM |
TMZ 100 μM BEZ 5 μM |
TMZ 100 μM BEZ 10 μM |
TMZ 10 μM |
| TMZ 250 μM BEZ 0 |
TMZ 250 μM BEZ 1 μM |
TMZ 250 μM BEZ 2.5 μM |
TMZ 250 μM BEZ 5 μM |
TMZ 250 μM BEZ 10 μM |
TMZ 25 μM |
| TMZ 500 μM BEZ 0 |
TMZ 500 μM BEZ 1 μM |
TMZ 500 μM BEZ 2.5 μM |
TMZ 500 μM BEZ 5 μM |
TMZ 500 μM BEZ 10 μM |
TMZ 50 μM |
| TMZ 1 mM BEZ 0 |
TMZ 1 mM BEZ 1 μM |
TMZ 1 mM BEZ 2.5 μM |
TMZ 1 mM BEZ 5 μM |
TMZ 1 mM BEZ 10 μM |
TMZ 100 μM |
TMZ or BEZ are indicated as 10× concentrations and used to achieve final 1× concentrations.
Alternatives: This matrix can be completed for multiple combinations of compounds. At least four serial doses of two drugs should be examined to determine Bliss synergy scores. The most promising combinations should be validated in vivo in PDOXs. For larger scale drug screening, optional 384-well ultra-low attachment tissue culture plates can be used with total media and drug of 40 μL per well. See also Figure 4 for dose distribution of drugs tested and assessed for synergy in GBM PDOs.
Table 3.
Dose response assays for assessing synergy between two targeted therapy drugs drug A (poziotinib (Pozi)) and drug B (niraparib (Nira))
| Tested drug A concentration | Pozi 0.4 nM | Pozi 1 nM | Pozi 2 nM | Pozi 4 nM | |
|---|---|---|---|---|---|
| Nira 0 Pozi 0 |
Nira 0 Pozi 4 nM |
Nira 0 Pozi 10 nM |
Nira 0 Pozi 20 nM |
Nira 0 Pozi 40 nM |
Tested drug B concentration |
| Nira 4 nM Pozi 0 |
Nira 4 nM Pozi 4 nM |
Nira 4 nM Pozi 10 nM |
Nira 4 nM Pozi 20 nM |
Nira 4 nM Pozi 40 nM |
Nira 0.4 nM |
| Nira 10 nM Pozi 0 |
Nira 10 nM Pozi 4 nM |
Nira 10 nM Pozi 10 nM |
Nira 10 nM Pozi 20 nM |
Nira 10 nM Pozi 40 nM |
Nira 1 nM |
| Nira 20 nM Pozi 0 |
Nira 20 nM Pozi 4 nM |
Nira 20 nM Pozi 10 nM |
Nira 20 nM Pozi 20 nM |
Nira 20 nM Pozi 40 nM |
Nira 2 nM |
| Nira 40 nM Pozi 0 |
Nira 40 nM Pozi 4 nM |
Nira 40 nM Pozi 10 nM |
Nira 40 nM Pozi 20 nM |
Nira 40 nM Pozi 40 nM |
Nira 4 nM |
The working concentrations of Pozi or Nira are listed based on the utilization of 10 μL from a 10× stock concentration to achieve the final 1× concentration in a total of 100 μL per well.
Figure 4.

Drug synergy assays in PDOs
Steps 1–4 show the utilization of PDOs to determine the antitumor activity of drugs A and B. Drugs are added to the media from a 10× stock to reach 1× final concentration. Treatment is continued for 3 days, based on initial determination of GI50 concentration In some cases, treatment may be continued for 7 days to see the full inhibitory effects. Effects on PDO cell viability is determined by assessing intracellular ATP levels using CellTiter-Glo (Promega) following the manufacturer’s instructions. Data are input into SynergyFinder software to determine synergy between drugs A and B.
-
40.
On the first day, plate 4,000–5,000 GBM cells per well in a 96-well plate and culture in 90 μL of media.
Note: You can culture the cells as gliospheres or organoids as PDOs. For advantages of using PDOs vs gliospheres, please refer to Chadwick et al., 2020. See steps 5b–5h for spheres and 6b–6g for PDOs.
-
41.
On day 2, prepare a stock of 10× drug, and dilute to 1× by adding 10 μL of 10× drug to each well containing 90 μL of media.
Note: A media change is not necessary to add the 10 μL of the drug of choice.
-
42.
Treat the cells with drug combinations at the desired concentrations (see Tables 2 and 3) and as suggested on the SynergyFinder site for 72 h and up to 1 week.
Note: Access to the synergy site can be acquired by following this link: https://synergyfinder.fimm.fi/synergy/20201021224844908442/
-
43.
At 72 h, add 40 μL of Promega’s Cell Titer-Glo mix to each well of the culture plate to determine cell viability by measuring intracellular ATP levels.
-
44.
Move the plate to a shaker and keep at room temperature (20°C–21°C) for 30 min.
-
45.
Transfer 60 μL from each well to an opaque plate to perform the cell viability assays based on the manufacturer’s instructions (https://www.promega.com/-/media/files/resources/protocols/technical-bulletins/0/celltiter-glo-luminescent-cell-viability-assay-protocol.pdf?la=en) using a plate reader.
-
46.
Cell viability readouts are determined by comparing treated wells to untreated, DMSO, and positive control wells. Growth inhibitory concentration (IC50) and area under the curve (AUC) values can be determined with quantitative analysis using GraphPad prism software.
Note: Generate a standard curve using 8 wells containing serial dilution of a compound that kills all cells as a positive control. Median concentration can be used to establish reproducibility among PDO wells by performing a series of dilutions starting from the highly concentrated samples (e.g., high cell numbers) to highly diluted samples. Use these standard curves to measure the AUC to determine the effect of the drug on the cell viability of GBM PDOs.
-
47.
Input the data into the SynergyFinder site to generate the respective plots.
Note: While beyond the scope of this protocol, drug sensitivity assays can be confirmed in mouse PDOXs to assess the effects of promising drugs in vivo and in particular upon crossing the blood brain barrier (BBB), which is highly desired for clinical translation for GBM patient therapy.
Note: PDOs (and PDOXs) are established using deidentified tissues and by investigators who are blinded to patient data and patient responses to therapy. Upon completion of the preclinical drug assays described in this protocol, correlation to clinical responses can be established by clinical investigators who have access to clinical data following IRB guidelines.
Expected outcomes
GBM 3D gliosphere and PDO culture can be performed in 4D printed cell culture arrays to expedite the processing (Chadwick et al., 2020) or alternatively in the traditional ultra-low attachment plates described in this protocol. Gliospheres are enriched for GSCs and therefore could be used to study GBM GSCs, while PDOs contain GSCs and differentiated cells and used for drug sensitivity testing (Chadwick et al., 2020). One to 2 days after seeding, 3D cultured cells should self-assemble to make the sphere-shaped structures in gliospheres. As the gliosphere proceeds, cells will actively proliferate and appear denser, until individual cells can no longer be resolved under low magnification of the microscope (typically day 7 for gliospheres). For PDOs, cells will move slower within the outer edge of the Matrigel droplet and start projecting extensions between multiple cell groups. PDOs become darker and denser starting at day 14, and density and interconnections continue to increase with time. Using the described protocol, we could generate long-term 3D cultures of gliospheres and PDOs for many passages during the period of 3 months of continuous 3D culture. Gliospheres and PDOs could also be regenerated upon cryofreezing and reinitiating cultures after more than a year of cryo-storage. At the early stages of gliospheres and PDO cultures, there should be very little cell death. By day 7, the majority of the gliosphere culture should appear homogeneous in distribution and density across the culture well. Between days 7–14, gliospheres typically express NESTIN (≥40%), low levels of GFAP (more detectable from GBMs with astrocytic origin), and low/undetectable TUJ1 and GALC. When differentiation of these gliospheres is induced with serum and/or culture on polyornithine-coated slides, NESTIN is downregulated and gliospheres show relatively more GFAP, TUJ1-expressing cells and rarely some GALC-expressing cells. On the other hand, GBM PDOs starting from day 14 spontaneously express NESTIN (>50%), and GFAP, TUJ1 and GALC (≥20%) without induction of differentiation. GBM PDOs appear darker under the microscope as cell density increases.
GBM PDOs contain highly heterogenous cellular subtype populations, recapitulating the key expression profiles and tumor cell phenotypes in GBM patients (Chadwick et al., 2020). When using the original GBM tissue to generate PDOs and gliospheres, it is necessary to determine which of the four main cellular neurodevelopmental states: mesenchymal (MES), astrocytic (AC), oligodendroglial precursor cells (OPC) and proneuronal progenitor cells (NPC) states (Neftel et al., 2019) are represented in the originating tissues, GBM PDOs, and gliospheres. Molecular subtype profiling should be performed with combination of exome sequencing (e.g., glioseq), RNA sequencing, and qPCR of subtype targets. CDK4, EGFR, and PDGFRA copy number variations (CNVs) and NF1 mutations influence GBM cellular state (Neftel et al., 2019) and should be determined to define GBM subtypes. Parity in expression profiles between originating GBM and PDOs remains high (≥90%) in the first 2–4 weeks of PDO cultures.
Drug testing in PDOs is done by measuring intracellular ATP levels of treated PDO cells compared with untreated cells from the same patient GBM PDOs to determine GI50 for each drug. Synergy screens of dose response matrices are used for drug combination to determine the Bliss score using the Synergy Finder application. Upon drug treatment, IF using antibodies against Ki67 and GFAP are indicative of antiproliferative (anti-Ki67) and anti-GBM specific (anti-GFAP) activity and against BMI1 and pS6 to detect on-target activities against self-renewal (anti-BMI1) and PI3K/mTOR (anti-pS6) activities respectively, and to validate combination therapy. Promising drug sensitivity responses from novel and combined drugs can then be validated in vivo in mouse PDOXs to assess the effects of crossing the BBB and tumor microenvironment (TME) for clinical translation into novel GBM therapy. Moreover, correlation of drug sensitivity profiles with clinical responses to the same drugs can be done to further establish the power of using PDOs to predict responses to therapy in precision medicine oncology clinical trials.
Limitations
We have successfully utilized this protocol to generate PDOs from multiple GBM patient-derived material. We have also generated PDOXs from the same GBMs. However, the efficiency of PDO formation and both gliosphere and PDO growth rates varies between patient samples. We have found that this variation is largely dependent on the quality of the surgically resected tissue and the proliferative nature of each tumor. As such, we recommend establishing gliospheres or PDOs for at least 1 or 2 weeks before attempting to generate PDOXs. Additionally, the use of growth factor reduced Matrigel droplets as an ECM source for PDOs during the 3D culturing process results in better efficiency. However, Matrigel is animal-derived and there is some variability among batches that may confound the phenotypic cellular responses in drug testing. We are also utilizing chemically defined synthetic ECMs as xenogeneic-free reagents (amenable to clinical translation) and examining each type for reproducibility against Matrigel. These future optimizations will be important for developing clinical-grade drug testing assays but may add unnecessary expense when such stringency may not be required. We regularly interchange xenogeneic and xenogeneic-free ECM reagents without altering the protocol, although this should be determined on a case-by-case basis by the end user. Finally, prolonged 3D culture and growth factor selection might limit the preservation of immune, endothelial, microglial, and nerve cell components of the GBM TME. While these stromal components may be reintroduced in coculture, and/or modeled in mouse PDOXs, we are currently investigating multiple conditions that reproduce the precise bidirectional crosstalk of stromal cells with tumor cells by supplementation of key growth factors to maintain the stromal components of PDOs beyond the 12-week timeline, which is the longest reported time for maintaining some components of the TME in 3D culture. By improving culture conditions, PDOs could better model tumor and TME heterogeneity and allow for improved prediction of drug sensitivity for clinical applications.
Troubleshooting
Problem 1
GBM tissue has highly necrotic areas leading to an inability to establish PDOs (step 2 in Step-by-step method details).
Potential solution
During processing of tissues, light debridement by removing dead and necrotic areas (darker in color) compared to remaining tumor (red in color) could improve the potential of establishing PDOs. The use of Primocin in collection media and neurobasal media including the use of EMEM and addition of sodium pyruvate and hydrocortisone, particularly during the first week of 3D culture establishment, improves the rates of gliosphere and PDO formation. Cultures should be closely monitored during the first three days after plating for potential bacterial and fungal infections. Regular tests should be performed to ensure that the cultures remain free of mycoplasma.
Problem 2
The genetic background of the tumor is unknown at the time of establishing PDO (step 8 in Step-by-step method details).
Potential solution
GBM PDOs are used for testing drug responses as early as 2 weeks after establishing 3D cultures. Ideally, they should be paired retrospectively upon receiving sequencing and expression profiling data of the originating tumor once completed. At a minimum, histological, genetic and IHC analysis of key markers should be established to determine the extent of PDOs representing the original tumor. This is critical as it is possible that culture heterogeneity is potentially lost when PDO lines are expanded and selection is applied, therefore, it is recommended to establish parity with the original tumor using tissue from the earliest time possible after tumor resection, and if not possible, then during the first passage of PDOs. Utilizing PDOs derived from additional patient samples with similar GBM subtypes could be considered, yet we emphasize that PDOs without the original genetic background are not true representative models for precision therapy.
Problem 3
It is easy to aspirate the spheres/organoids when removing fluids (i.e., media, PBS, fixative) from the wells (steps 7 and 8 in Step-by-step method details).
Potential solution
Tilting the plate and slowly aspirating from the surface of the media/plate interface will help minimize the number of spheres/organoids that are lost throughout the fixation and antibody staining process. Alternatively, working under a dissecting microscope will help track the organoids/spheres throughout the process. Though, this might be laborious and time consuming.
Problem 4
Prolonged incubation with primary antibody might yield unwanted background (step 38 in Step-by-step method details).
Potential solution
It is important to optimize this procedure based on sphere/organoid type, and size. It is recommended that steps 38a–38f be followed first on a trial sample before deciding to utilize the prolonged primary incubation noted above. Always make sure to have a negative control with no primary antibody added in order to check for unwanted non-specific background.
Problem 5
Immunofluorescence analysis is not clear or difficult to interpret (step 38 in Step-by-step method details).
Potential solution
Optimization of each antibody is required to achieve optimum staining. Optimization can be performed using GBM cell lines of similar genetic background to the PDOs and cultured in the same 3D sphere/organoid culture conditions. Utilizing positive staining control from known reactive cells and no primary antibody negative controls allow determining the specificity of the staining. Performing additional steps such as antigen retrieval, blocking and using longer incubations can be performed while controlling for any unwanted background staining with additional controls during staining and imaging.
Problem 6
Inconsistencies in drug responses due to intra-GBM heterogeneity (step 46 in Step-by-step method details).
Potential solution
We suggest that drug testing readouts are assessed using different experimental approaches by determining on-target activity and synergy in combination therapy using multiple PDO models with similar GBM subtypes. When considering personalized therapy, an accurate integration of exome and RNA sequencing data to identify GBM subtypes is essential. To account for spatial intratumor heterogeneity and difference between core and edge GBM cells, isolating and propagating PDOs from multiple tumor sites within the same GBM would strengthen the predictive value of PDOs in drug sensitivity assays.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hatem E. Sabaawy (sabaawhe@cinj.rutgers.edu).
Materials availability
This study did not generate new unique reagents, cell, or mouse lines.
Data and code availability
This study did not generate a new dataset.
Acknowledgments
We thank our patients for consenting to donate their tissues for use in our studies. We thank Rutgers Cancer Institute of New Jersey Biorepository Services and Histopathology Services (Shafiq Bhat, Lucyann Franciosa, Kelly Walton, and Lei Cong) for assistance with tissue acquisition and histopathology. We also thank Diane Hanahan and Dr. Parisa Javidian for access to the tissue banking protocols and clinical assessments. This project was supported in part with federal funds from the National Cancer Institute (NCI) R01 award (R01CA226746 to H.E.S.); Rutgers Cancer Institute of New Jersey Shared Resources, supported, in part, with funding from NCI-CCSG (P30CA072720); and New Jersey Health Foundation Innovation Award (ISFP 7-16 to H.E.S.).
Author contributions
Conceptualization, C.M.G., K.J., S.P., and H.E.S.; Investigation, C.M.G., K.J., L.L., X.X., R.I., and S.D.; Writing – Original Draft, C.M.G., S.P., and H.E.S.; Writing – Review & Editing, C.M.G., K.J., S.P., L.L., and H.E.S.; Funding Acquisition, H.E.S.; Supervision, H.E.S.
Declaration of interests
Rutgers University has patents pending related to single-cell-derived organoids. H.E.S. is the scientific founder of Celvive, Inc.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100345.
References
- Chadwick M., Yang C., Liu L., Moya Gamboa C., Jara K., Lee H., Sabaawy H.E. Rapid processing and drug evaluation in glioblastoma patient-derived organoid models with 4D bioprinted arrays. iScience. 2020;23:101365. doi: 10.1016/j.isci.2020.101365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta M., Khan A., Danish S., Haffty B.G., Sabaawy H.E. Radiosensitization of primary human glioblastoma stem-like cells with low-dose AKT inhibition. Mol. Cancer Ther. 2015;14:1171–1180. doi: 10.1158/1535-7163.MCT-14-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neftel C., Laffy J., Filbin M.G., Hara T., Shore M.E., Rahme G.J., Richman A.R., Silverbush D., Shaw M.L., Hebert C.M. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019;178:835–849.e1. doi: 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforova M.N., Wald A.I., Melan M.A., Roy S., Zhong S., Hamilton R.L., Lieberman F.S., Drappatz J., Amankulor N.M., Pollack I.F. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro. Oncol. 2016;18:379–387. doi: 10.1093/neuonc/nov289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrizii M., Bartucci M., Pine S.R., Sabaawy H.E. Utility of glioblastoma patient-derived orthotopic xenografts in drug discovery and personalized therapy. Front. Oncol. 2018;8:23. doi: 10.3389/fonc.2018.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller M., Wick W., Aldape K., Brada M., Berger M., Pfister S.M., Nishikawa R., Rosenthal M., Wen P.Y., Stupp R. Glioma. Nat. Rev. Dis. Primers. 2015;1:15017. doi: 10.1038/nrdp.2015.17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time lapse recording of a typical GBM PDO growth during the first five days of the 3D culture after plating. Note the clusters of GBM dividing cells and the cellular motility of GBM cells, with neurite extensions, and the repeated interactions between various cells and the ECM, which forms the surrounding Matrigel droplet edge. These continuous cellular motilities might require readjustment of focus for proper time lapse imaging. This time lapse movie was taken during days 1–5 of 3D GBM organoid culture and was set to 200× playback.
Data Availability Statement
This study did not generate a new dataset.