

Abstract
Prostate cancer (PCa) is heterogeneous harboring phenotypically diverse cancer cell types. PCa cell heterogeneity is caused by genomic instability that leads to the clonal competition and evolution of the cancer genome, and by epigenetic mechanisms that result in subclonal cellular differentiation. The process of tumor cell differentiation is initiated from a population of prostate cancer stem cells (PCSCs) that possess many phenotypic and functional properties of normal stem cells. Since the initial reports on PCSCs in 2005, there has been much effort to elucidate their biological properties including unique metabolic characteristics. In this review, we discuss the current methods for PCSC enrichment and analysis, the hallmarks of PCSC metabolism and the role of PCSCs in tumor progression.
Keywords: prostate, cancer, cancer stem cells, heterogeneity, metabolism
1. Prostate cancer hierarchy, heterogeneity and plasticity
1.1. An introduction to cancer stem cells
Cellular heterogeneity represents an omnipresent feature of most human tumors. Cancer cell heterogeneity can result, in principle, from both clonal competition and evolution (i.e., selection of the ‘fittest’ clone) driven by genetic instability inherently high in tumor cells as well as intra-clonal (subclonal) cellular maturation (differentiation) program driven by epigenetic mechanisms (1). The latter process of cell diversification is initiated from a subset of unique cancer cells that possess many phenotypic and functional properties of normal stem cells (SCs), hence cancer stem cells (CSCs). Phenotypically, CSCs, like normal SCs, may express certain unique or common markers that can be used for prospective enrichment and purification. Functionally, CSCs may share with normal SCs the two most important properties: self-renewal and differentiation. Using these two functional properties, and, in the most strict sense, a single ‘pluripotent’ CSC should be able to regenerate a tumor that histo-structurally recapitulates the parent (patient) tumor that consists of all different cancer cell types. No such a CSC has ever been reported in any tumor. In reality, CSCs are operationally defined as cell subpopulations that are enriched in tumor-regenerating and (serial) tumor-propagating activities, which measure self-renewal, and CSC-derived tumors should harbor diverse cell types present in the parent tumor (i.e., CSC differentiation) (2). CSCs should be distinguished from cancer cell-of-origin as the latter refers to the initial normal cell or the cell type that became tumorigenically transformed whereas CSCs refer to the cell population that drives clonal tumor evolution (2).
Like most human solid tumors, prostate cancer (PCa) is heterogeneous harboring many cancer cell types. PCa cell heterogeneity tends to become accentuated during disease progression and, in particular, upon treatment. This is best evidenced by PCa cells that express AR (androgen receptor), the master pro-differentiation transcription factor in normal prostatic epithelial cells, and PSA (prostate specific antigen), a downstream target of AR and the best lineage differentiation marker for differentiated human prostate luminal cells. Although most untreated primary tumors contain AR+ and PSA+ cells as the majority, AR−/lo and PSA−/lo PCa cells become gradually enriched in high-grade untreated tumors, and PSA−/lo PCa cells frequently become the predominant cell population in castration-resistant PCa (CRPC) (3). Ever since the initial reports by three independent groups (4–6), in 2005, of PCa stem cells (PCSCs), there has been now substantial evidence for the presence and functions of PCSCs, which we shall further elaborate below.
In this review, we discuss the current methods for PCSC enrichment and analysis, the hallmarks of PCSC biology and the role of PCSCs in therapy resistance, tumor progression and metastases. Relevant literature published in peer-reviewed journals up to March of 2018, was identified via systematic search of PubMed. The scientific quality of the included literature was independently assessed by all authors. Conference abstracts and other unpublished materials were not included. This review is based on the pre-clinical basic research, clinical studies and recent reviews that provide indispensable information for current understanding of the biology of PCSCs.
1.2. Current methods for PCSC enrichment and analysis
The first proof-of-principle studies for prospective identification of PCSCs exploited different phenotypes to isolate self-renewing and tumorigenic cells from patient-derived prostate tumors and cell lines (4–6). These phenotypes included expression of surface markers CD44+/α2β1hi/CD133+, expression of transporter protein ATP-binding cassette sub-family G member 2 (ABCG2) that is involved in cell detoxification and the “side population” phenotype, which is defined by Hoechst dye exclusion by tumor cells (4–6). Since then, a few other markers have been described for human PCSC populations (Table1), including CD44/CD133 (7–9), aldehyde dehydrogenase (ALDH) (10), CD166 (11), CD44+/CD24− (12). PCa cells expressing these markers have capacities to self-renew and to generate the heterogeneous tumor cell subpopulations. Using antibodies to the membrane CSC markers and fluorescence activated cell sorting (FACS) or magnetic cell sorting (MACS), marker-positive and marker-negative cells can be isolated from cancer cell lines, xenograft tumors and patient-derived specimens, and CSC properties of these cell populations can be analyzed and compared (Table 1).
Table 1.
Methods for detection and isolation of PCSC populations.
| Marker-based methods | |||||
|---|---|---|---|---|---|
| Technique | Method | Markers | Ref | Advantages | Limitations |
| Staining cell surface proteins / fluorescence detection of enzymatic activity | Fluorescence - based cell sorting, IF microscopy, IHC | CD44 | (8) | Analysis of the experimental (xenograft animal models) and clinical (patient-derived tissues, TMA) samples | These markers are not specific for PCSCs and express on other cells including normal tissues; PCSCs can be heterogeneous for the marker expression, and marker positive cells can be functionally heterogeneous (e.g. by their self-renewal properties) |
| CD133/ CD44 | (7–9) | ||||
| ALDH/CD44/α2β1 | (28) | ||||
| CD166 | (11) | ||||
| Trop2 | (213) | ||||
| Gene reporter systems | Fluorescence - based cell sorting, fluorescence microscopy, in vitro and in vivo cell tracking | PSA | (28) | In vitro and in vivo tracking of CSC populations, in vivo CSC imaging, analysis of CSC differentiation | |
| 26S proteasome activity | (14) | ||||
| NANOG | (13, 18) | ||||
| CD44 | (168) | ||||
| Marker-free methods | ||||
|---|---|---|---|---|
| Technique | Method | Ref | Advantages | Limitations |
| Sphere forming assay | Cell cultivation under non-adherent conditions accompanied by FACS analysis to confirm CSC enrichment in spheres | (214, 215) | Elucidation of molecular and physiological properties of CSCs; CSC characteristics associated with treatment resistance |
This methods cannot be used to obtain a highly enriched CSC populations; Sphere forming properties in vitro are not indicative for tumorigenicity in vivo |
| Therapy-induced CSC enrichment | Cancer cell cultivation in the presence of chemotherapeutic drugs or repetitive cell exposure to ionizing radiation | (21, 216) | ||
| Side population | Fluorescence sorting based on the Hoechst 33342 or Rhodamine 123 efflux by CSCs | (6, 23) | Various samples can be analyzed e.g. cell lines, xenograft tumors and tumor specimens | Hoechst 33342 and Rhodamine 123 are cytotoxic; absence of the standard dye concentration; low specificity |
| CSC isolation based on the cell sizes | Microfluidic techniques, FACS | (27, 217, 218) | Isolation of cell populations with different morphology, size and metabolic activity | Low specificity |
In addition to the isolation of PCSC populations based on marker expression, other strategies exploiting PCSC-specific gene reporters are being developed for real-time analysis of PCSC behavior at a single-cell level in vitro and in animal models. These systems are based on the stable expression of a reporter gene (e.g. fluorescent protein) driven by the promoters of PCSC- or lineage-specific genes such as NANOG, PSA, SOX2, and OCT4 (13–18) (Table 1). For example, Qin et al. (3) took advantage of the fact that PSA represents one of the most lineage-specific differentiation markers in human prostate epithelia and addressed the important question of whether the phenotypically differentiated (i.e., PSA+/hi) and undifferentiated (PSA−/lo) PCa cell populations might be functionally distinct. By building and employing a series of lentiviral-based lineage-tracing vectors using the PSA promoter/enhancer (i.e., PSAP), they provided concrete evidence that the PSA−/lo PCa cell population harbors authentic self-renewing cancer cells that in vitro can undergo asymmetric cell division under time-lapse videomicroscopy and in vivo can serially propagate xenograft tumors (3). Of clinical relevance, the PSA−/lo PCa cells are intrinsically resistant to antiandrogens, chemotherapeutic drugs, pro-oxidants, and radiation, and can readily regenerate and serially propagate castration-resistant xenograft tumors (3). These results evidenced that the PSA−/lo human PCa cell population harbors true PCSCs.
The methodology exploiting marker-based CSC isolation and enrichment markedly contributed to the progress in CSC research; nevertheless it is necessary to bear in mind that the majority of the above-described markers were also identified in the malignant tumors of other origins, such as breast, colorectal, ovarian and lung carcinomas and glioblastoma (9, 19, 20). It is also known that these markers are not specifically expressed in malignant tissues, but also found on the normal embryonic and adult stem cells. Therefore, further investigation of additional reliable and more CSC-specific markers is needed to improve the marker-based CSC harvesting techniques.
Since some CSC populations are described to be treatment-resistant, there is a strategy to enrich CSCs by the repetitive use of chemotherapeutics or radiation therapy (21). PCa cell cultivation in the presence of cytostatic drugs or upon radiation exposure led to the development of acquired treatment resistance and enrichment of carcinoma cells positive for CSC markers and having high tumorigenic capacities (21). Some of the treatment-resistant CSC populations are characterized by upregulation of ABC family members including ABCG2 transporter, which provides the rationale for using the side population technique for CSC isolation (22). For this, tumor cells from cell cultures or dissociated xenograft or primary human tumors are stained with Hoechst 33342 or Rhodamine 123 dyes. Further cell analysis by flow cytometry enables detection of the cells with increased dye efflux (side population) (6, 23). It is suggested that CSCs with a high expression of ABC transporters actively extrude the dye out of the cells. Unfortunately, this method has a number of limitations. First, Hoechst 33342 and Rhodamine 123 are toxic to cells. Next, an efficiency of CSC isolation based on side population might be hampered by low specificity and inconsistency of the existing staining protocols. Although carcinoma cells with acquired therapy resistance frequently demonstrate upregulation of CSC biomarkers, treatment-resistant cells do not necessarily represent a homogeneous cell population, but rather a mixture of phenotypically different cell subsets with different properties (24). A number of studies indicate that therapy might enrich CSCs by selecting for preexisting CSC populations, and/or by inducing tumor cell dedifferentiation (3, 21, 25).
CSCs can also be identified and isolated using the differences in sizes between non-CSCs and CSCs. It was described that CSCs are markedly smaller than more differentiated cells (26, 27). Recent study of Li et al. showed that a population of small PC3 PCa cells (<10 m) has a tendency of being more tumorigenic than the corresponding larger (≥20 or 30 m) cells (27). Further investigation is warranted to clarify the mechanisms controlling cell size in homeostasis and cancer and whether cell size can be effectively used for CSC identification and isolation.
Growing established cell lines or patient-derived tumor cells under serum-free and sphere-forming conditions was one of the first methods to enrich CSCs in vitro. Most of the cells cultured under these harsh conditions undergo anoikis and die. The survived cell populations are enriched in sphere-forming cells, which can sustain their proliferation independently of the cell adhesion mechanisms. This assay also allows investigation of the cancer cell populations with self-renewal properties if the sphere formation is analyzed in multiple passages, because only self-renewing cells are capable of maintaining their spherogenicity in multiple generations (3, 28). It should be noted that sphere-forming cancer cells, though frequently showing an enrichment of CSC markers, may not necessarily display enhanced tumorigenic abilities in xenograft models (29). For example, Kuch et al. (2012) showed that the capacities of carcinoma cells to form spheres cannot be used as a reliable surrogate assay to detect the tumor-initiating capacities in animals (29).
In that respect, in vivo limiting dilution assay should be performed as a standard technique to analyze the frequency of tumor-initiating cells in cancer cell populations defined by putative CSC markers. For this analysis, the same numbers of marker-positive and marker-negative cancer cells isolated either from cancer cell lines or dissociated xenograft or human tumor specimens are injected into immunodeficient mice by orthotopic or ectopic implantation. The cells are injected at different dilutions: from a few or even a single cell to thousands or millions cells, and tumor growth is monitored by caliper measurements or/and tumor bioluminescence for a few months (3, 6, 30). Analysis of tumor incidence in different doses of injected cells enables an estimate of CSC frequencies in the injected cell populations (2). To validate if putative CSC populations indeed possess the long-term self-renewal capacities in vivo, the CSCs of interest are further purified out from regenerated tumors, and then serially transplanted in mice. Cell populations that can maintain long-term self-renewal properties (CSCs) should sustain long-term tumor regeneration during serial transplantations. In contrast, cell populations with a limited self-renewal will lose their tumorigenicity upon serial xenografting (2, 31). For lineage tracing of CSC cells in vivo, a genetically modified mouse model can be established where CSC populations are labeled by using CSC specific gene reporters and monitored during tumor development in living mice (32).
New approaches that facilitate genome editing such as transcription activator-like effector nucleases (TALENs), zinc finger nucleases (ZFN), and the clustered regularly interspaced palindromic repeats/Cas9 associated (CRISPR/Cas9) technologies are expected to offer new possibilities to investigate endogenous gene functions at the genome-wide scale and to establish a direct link between the genome and cellular properties including multipotency and self-renewal.
2. Hallmarks of PCSC metabolism
2. 1. Metabolism of normal prostate and PCa epithelial cells
In contrast to other epithelial tissues, normal prostate epithelial cells have distinct glucose metabolism profiles attributed to the main function of the prostate gland: to produce and secrete fluid that protects and nourishes sperm. The luminal cell compartment of the prostate gland produces and secretes a large amount of citrate into the prostatic fluid. The high level of citrate is important for the maintenance of sperm viability by supplying energy source and by calcium chelation (33). In most normal tissues, citrate, which is produced in the mitochondria or delivered into the cells via membrane transporters, is utilized as a major substrate for energy production in the tricarboxylic acid cycle (TCA) (34). High production of citrate in the prostate gland is a consequence of the low activity of mitochondrial aconitase (m-ACNT) and subsequent inhibition of citrate oxidation (35). In normal luminal cells, suppression of m-ACNT enzymatic activity results from a high mitochondrial concentration of zinc that is maintained by a high expression level of zinc transporter proteins (35, 36). As a consequence of dampened citrate oxidation, normal prostate epithelial cells have a low level of TCA and glycolytic metabolism. Due to a relatively quiescent state, normal prostate SCs have low levels of TCA, oxidative phosphorylation (OXPHOS), and glycolysis (37) (Figure 1).
Figure 1. Metabolism of normal and tumor prostate.
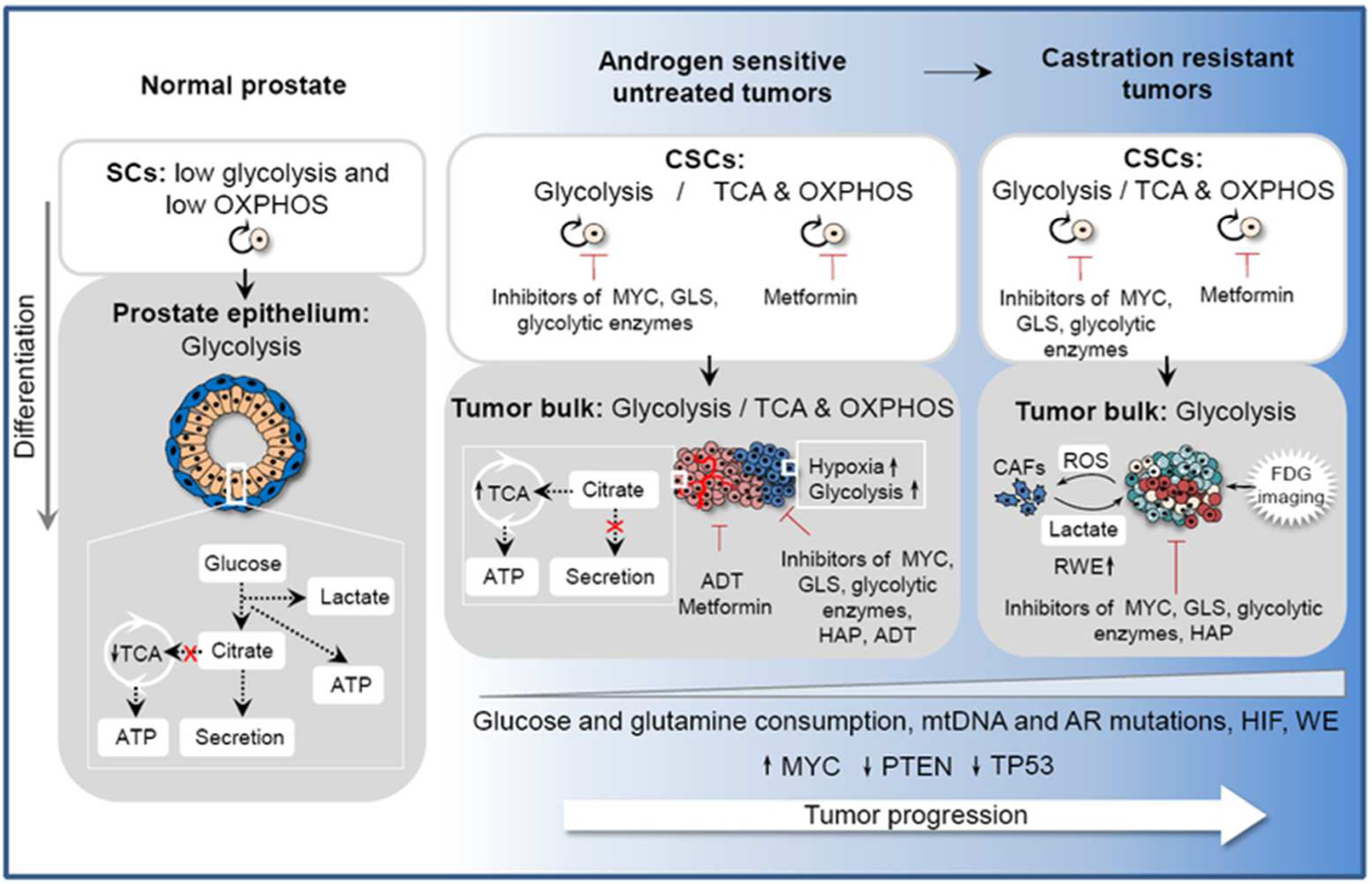
Normal prostate epithelial cells have a low level of TCA and glycolytic metabolism as a result of the impaired citrate oxidation. In contrast, prostate adenocarcinoma cells increase citrate oxidation and OXPHOS. This oxidative phenotype is controlled by AR signaling and can be affected by hypoxia. Mutations in mtDNA and tumor suppressor genes PTEN and TP53, which play a role in the metabolic shift from OXPHOS to aerobic glycolysis, can promote tumor progression. Metabolically active aggressive primary tumors and castration-resistant metastatic disease exhibit the Warburg effect and have a high level of nutrient consumption. ROS released by PCa induces oxidative stress in neighboring CAFs. In turn, CAFs secrete a high level of lactate which is utilized by PCa cells for ATP production via OXPHOS. The glycolytic features of advanced PCa can be used for clinical imaging by using positron emission tomography (PET) for glucose uptake imaging with FDG. Beside glucose, highly proliferative cancer cells require additional supplies for their biosynthesis that cannot be met by glucose consumption such as glutamine. Metabolic features of PCa can be potentially used for anti-cancer therapy to increase efficiency of conventional treatment such as chemo- or radiotherapy. TCA - tricarboxylic acid cycle; AR - androgen receptor; OXPHOS - oxidative phosphorylation; mtDNA - mitochondrial DNA; FDG - Fluoro-2-deoxyglucose; RWE - reverse Warburg effect; CAFs - cancer associated fibroblasts; ROS - reactive oxygen species; ADT - androgen deprivation therapy; HADs - hypoxia activated drugs; CAFs - cancer associated fibroblasts; WE - Warburg effect; HIF - hypoxia inducible factor signaling.
In contrast to the normal luminal cells, prostate adenocarcinoma cells contain a low level of mitochondrial zinc that abrogates the inhibition of m-ACNT activity and enables utilization of citrate in the metabolic pathways (34–36). Reprogramming of cellular energy metabolism plays an important role in tumor initiation, progression, and therapy resistance. In contrast to normal epithelial cells, PCa cells increase citrate oxidation and OXPHOS. This oxidative phenotype is controlled by AR signaling and can be affected by hypoxia (26, 38). Oncogenic mutations of mitochondrial DNA (mtDNA) and tumor suppressor genes PTEN and TP53 play a role in the metabolic shift from OXPHOS to aerobic glycolysis known as the Warburg effect (39, 40). To meet high energetic demands, fast growing tumor cells that exhibit the Warburg effect need to reprogram their metabolic pathways for a high level of nutrient consumption, and this is especially true in case of metabolically active aggressive primary tumors and castration-resistant metastatic disease. Reactive oxygen species (ROS) released by PCa into microenvironment induces oxidative stress in neighboring cancer-associated fibroblasts (CAFs). In turn, CAFs undergo aerobic glycolysis and secrete a high level of energy-rich fuels such as lactate. Consequently lactate is utilized by PCa cells for ATP production via OXPHOS (the phenomena referred to as the reverse Warburg effect) (41). The glycolytic features of advanced prostate tumors can be used in clinical setting for detection of metastatic disease and assessment of therapeutic response using positron emission tomography (PET) for glucose uptake imaging with 2-[18F]Fluoro-2-deoxyglucose (FDG) (42). In addition to increased glucose uptake, highly proliferative cancer cells require additional supplies for their biosynthesis that cannot be met by glucose consumption (Figure 1). A significant proportion of the biosynthetic needs can be covered by the metabolism of glutamine, which is the most abundant amino acid in human plasma. Similarly to the bulk of tumor cells, PCSCs can exhibit glycolytic or oxidative metabolic phenotype depending on the acquired mutations and activation of specific signaling pathways, which we shall further discuss below.
2.2. MYC - dependent metabolic reprogramming and maintenance of PCSCs
Glutamine is an important donor of nitrogen and carbon for the growth-promoting pathways. Although most normal tissues can synthesize glutamine, it becomes conditionally essential for the fast growing tissues. Tumor cells with enhanced expression of MYC oncogene, such as PCa, are particularly dependent on glutamine metabolism to sustain their viability (43–45). In these cells, deprivation of glutamine results in depression of TCA cycle, decrease in ATP level, and inhibition of cell cycle (43, 46). MYC is broadly acting transcription regulator that activates the embryonic stem cell program in human cancer and induces tumor-initiating cells (47). Recent findings indicate that targeting of MYC inhibits PCSC maintenance and tumorigenicity (48, 49). MYC contributes to metabolic adaptations of tumor cells by regulating glucose transporter GLUT1 and the enzymes involved in the glucose metabolism such as hexokinase 2 (HK2), phosphofructokinase (PFK1), enolase 1 (ENO1), and lactate dehydrogenase A (LDHA) (43).
Together with AR and well-established tumor driver the mammalian target of rapamycin, mTOR, MYC regulates expression of glutamine transporters genes SLC1A4 and SLC1A5, which are frequently overexpressed in PCa and thereby drives increased glutamine uptake (50). Through the suppression of miR-23a/b, MYC also regulates the expression levels of GLS1 glutaminase, which converts glutamine to glutamate at the first step of glutamine utilization (43). Analysis of human prostate tissues has revealed upregulation of MYC mRNA in the majority of primary PCa lesions (51). This overexpression of MYC can be attributed to MYC oncogene amplification that occurs in up to 29% of hormone-refractory recurrent prostate carcinomas (52). In addition, MYC expression can be regulated at the mRNA level as a result of APC hypermethylation and Wnt/β-catenin pathway activation, deletion of FOXP3 gene as well as TMPRSS2-ERG rearrangement that activates ERG-dependent transcription program (53–56). MYC overexpression alone is sufficient to bypass senescence and immortalize prostate cells derived from benign prostate tissue specimens (57). Transgenic expression of human MYC in the mouse prostate (i.e., Hi-Myc mice) efficiently induces the precursor lesion prostatic intraepithelial neoplasia (PIN) that rapidly progress to invasive adenocarcinoma (58). MYC-driven tumors show a MYC gene dosage-dependent phenotype, and share molecular signatures with human PCa such as loss of Nkx3.1 tumor suppressor and upregulation of Pim-1 protein kinase that increases energy metabolism and protein synthesis (58, 59). The two-step transformation of prostate basal or luminal cells from benign prostate tissues, either by MYC overexpression and AKT activation or by overexpression of MYC and knockout of the tumor suppressor phosphatase and tensin homolog (PTEN), can induce development of heterogeneous tumors from both cellular compartments (60–63). Significantly, high expression of MYC and loss of PTEN contribute to androgen-independent growth and are associated with highly invasive and metastatic prostate tumors (62–65). Interestingly, MYC is a context-dependent regulator of the expression of glutamine transporters SLC1A4 and SLC1A5, and does not regulate these transporters in PTEN wild-type tumors (50).
2.3. Androgen signaling and PCa metabolism
Dependence on androgen signaling is a hallmark of initial stages of prostate tumor development. Because of this, androgen deprivation therapy (ADT) is one of the main treatments for PCa besides prostatectomy and radiotherapy. While most of the patients initially respond to ADT, about 90% of them eventually develop androgen-refractory tumors with a fatal prognosis (66). ‘Androgen-independent’ tumor progression depends on a variety of mechanisms including AR gene amplification and gene mutation, ligand-independent activation of AR signaling pathway and alterations in the expression of AR co-regulators leading to an increase in AR-dependent transcription activity (67).
Androgen independent PCa growth and resistance to anti-androgen therapy can be conferred by COOH-terminal truncated AR variants (AR-V) (68–70). These AR variants lack ligand-binding domain but retain transcriptional activity. The most frequently occurring AR splice variants associated with poor prognosis include AR-V1, AR-V7, and AR-V567es (68, 70, 71). Studies by Antonarakis at al. demonstrated association between expression of AR splice variant-7, AR-V7 in circulating tumor cells (CTCs) and resistance of PCa patients to the new generation of antiandrogen drugs such as enzalutamide and abiraterone (72). Since then, a growing body of evidence suggested the potential application of AR-V7 detection in PCa patients’ blood as a potential biomarker for prediction of anti-androgen treatment failure (73). Recent experimental evidence suggested that AR-V7 might contribute to PCa progression through triggering epithelial-mesenchymal transition (EMT) (discussed in 3.1, below), induction of PCSC characteristics and metabolic reprogramming through increasing dependence on glutaminolysis (74, 75).
Like MYC, AR regulates expression of many genes involved in glutamine and glucose metabolism. Androgen signaling stimulates glucose consumption as well as production and export of lactate by upregulating the expression of GLUT1/GLUT3, phosphofructokinase 1 (PFK 1), monocarboxylate transporter 4 (MCT4) and calmodulin-dependent kinase kinase 2 (CAMKK2) genes (76, 77). CAMKK2 is highly upregulated in PCa, and CAMKK2-dependent activation of AMP-regulated kinase (AMPK) results in an increased PFK1 activity and promotes glucose uptake and lactate production (78). Metformin, which is the first-line drug for treating diabetes, enhances AMPK activity as a result of inhibition of the mitochondrial complex I activity (79). Metformin treatment was clinically associated with a significant decrease in early biochemical relapse and cancer-specific mortality in PCa patients (80), and also led to an increase in tumor oxygenation and response to radiotherapy in mouse xenograft models (80). Metformin combined with doxorubicin completely inhibits growth of PC3 PCa cells in xenograft models (81). Metformin treatment of docetaxel-resistant PC3 (PC3-DR) cells that manifest an increased oxidative metabolism, inhibits their proliferation and invasiveness without affecting parental PC3 cells (82). In breast and prostate cancer cells, metformin inhibits entry of glucose carbon into the TCA cycle and promotes the contribution of glutamine carbon to the TCA cycle making the cells more dependent on glutamine anaplerosis (83). However, the molecular effects of metformin on PCSCs still need to be clarified. Studies of the signaling pathways deregulated by metformin-induced metabolic stress in breast CSCs showed that metformin selectively inhibits activation of nuclear factor kappa B (NF-κB) and signal transducer and activator of transcription 3 (STAT3) - dependent transcription and expression of a number of inflammatory genes in CSCs as opposed to non-CSCs isolated from the same tumor (84). Interestingly, metformin suppresses AR-dependent expression of MYC in Hi-Myc tumors and inhibits the development of PIN and PCa lesions (85). Other studies have shown that metformin inhibits androgen dependent and androgen independent PCa by downregulation of AR mRNA levels and by inhibition of autophagy triggered by AR blockage (86, 87).
An increase in MYC copy number in tumor cells was observed as a result of ADT in PCa patients (88). Of interest, overexpression of MYC antagonizes the AR-dependent transcription program and confers the growth advantage in tumor cells in the absence of androgens. Therefore, increase in MYC expression may promote androgen-independent prostate tumor progression (65, 89).
2.4. Metabolic reprogramming of prostate tumor epigenetics
Interestingly, the MYC expression level inversely correlates with the global level of H3K27me3 repressive histone mark in human PCa and MYC-driven mouse prostate tumors (90). Decreased level of H3K27me3 correlates with a higher Gleason score and pathological stage. The basal cell compartment of normal prostate epithelium has a lower level of H3K27me3 as compared to the luminal cells (90). In support of this observation, MYC expression and MYC-dependent transcriptional program were shown to be upregulated in basal cells (91). Recent work has revealed that normal stem cells such as embryonic stem cells (ESCs) have an increased consumption of glutamine, which is utilized to maintain a high level of α-ketoglutarate (AKG). AKG is a co-factor for the Jumonji C domain-containing histone lysine demethylases as well as ten-eleven translocation (or TET) hydroxylases that regulate DNA demethylation (92, 93). In ESCs, AKG promotes histone demethylation including the H3K27me3 mark and therefore suppresses differentiation (21, 94). Targeting glutamine uptake via inhibition of ASCT2 (SLC1A5), the major glutamine transporter in cancer cells, suppresses the growth of PCa xenografts (45). Prostate tumor-initiating cell populations also exhibit a high dependence on glutaminolysis. Recent studies employed two clonal populations derived from PC3 PCa cells with different orthotopic and metastatic growth in xenograft models to characterize metabolic features associated with PCSCs (95, 96). These studies showed that PC3 CSC-like cells exhibit a high level of anaerobic glycolysis. These cells have a high level of lactate production and increased consumption of glutamine to compensate the acidification derived from the Warburg effect through the release of ammonia (96). In support of this data, other studies showed that advanced PCa have an increased glycolytic phenotype and a high activity of lactate dehydrogenase (LDH) correlates with poor clinical prognosis (26, 97). Knocking out genes that control OXPHOS, such as pyruvate dehydrogenase E1 component subunit alpha (PDHA1) or mitochondrial pyruvate carrier 1 (MPC1), results in an enrichment of PCa cells with CSC phenotype and increases therapy resistance and migration ability (98, 99).
3. The interaction between PCSCs and tumor microenvironment
3.1. Epithelial-mesenchymal transition and PCSCs
Epithelial-mesenchymal transition (EMT) is a process of switching from the epithelial cell characteristics to more migratory mesenchymal phenotype that is associated with loss of the epithelial markers (e.g. E-cadherin) and gain of mesenchymal signatures (e.g., vimentin, fibronectin, N-cadherin). The EMT process is associated with an increase in the expression levels of the transcriptional repressors of E-cadherin including SNAI1/2, SLUG, Twist, ZEB1/2 proteins and downregulation of microRNAs miR-200 and miR-34 that target ZEB1 and SNAI1 (100). Although EMT was initially identified as integral processes for embryogenesis and would healing, it was recently recognized as one of the drivers of tumor progression and metastases (100). Increasing evidences suggest that EMT plays a role in the regulation of PCSCs, metastatic ability and therapy resistance of PCa cells, reviewed in (101). A number of different factors might contribute to EMT in PCa including autocrine and paracrine signaling molecules released by tumor cells and stroma e.g. transforming growth factor beta 1 (TGF-β1), fibroblast growth factor (FGF), interleukin 6 (IL-6), hypoxia-inducible factor (HIF) and Wnt ligands as reviewed previously (102).
EMT can also be triggered by anticancer therapy including ADT, chemotherapy, and radiotherapy. Androgen withdrawal causes EMT and switch to the pro-metastatic phenotype by induction of the hypoxic stress, activation of ZEB1 mRNA expression and reduction of miR-200b (103–105). The diabetic drug metformin, which also exhibits anti-inflammatory activity, reverses TGF-β1/STAT3 pathway activated by anti-androgen treatment with enzalutamide and therefore inhibits EMT (106). Similar to androgen deprivation therapy, tissue injury induced by castration, chemotherapy and radiotherapy also inhibits or destroys tumor vasculature and promotes hypoxia and hypoxia-associated paracrine and autocrine responses in tumor cells and stroma that might trigger EMT primarily through activation of HIF - dependent gene expression (101, 107–109). HIF1 signaling is associated with the development of PCa metastases and activates transcription of genes involved in glycolytic flux and Warburg effect (110, 111). Stromal cells in the tumor microenvironment are also known to promote EMT, induce tumor cell reprogramming, and regulate the maintenance of PCSCs in their specific niches.
3.2. PCSC niche
Stemness is not a constitutive characteristic of CSCs, but rather represents a transient state that depends on the genetic, epigenetic and microenvironmental factors (1). Similar to normal SCs, the properties of CSCs such as self-renewal capacity, differentiation and therapy resistance are regulated by their cross-talk with specialized microenvironments called niches. In turn, factors produced by CSCs may also pathologically ‘transform’ normal stromal and endothelial cells hijacking them to promote tumor growth. CSC niches are formed, presumably, by multiple cellular components of the tumor microenvironment and shaped and regulated by the secreted factors such as cytokines and growth factors (112). The cellular components may include endothelial, immune and other stromal cells as well as the extracellular matrix produced by these cells. Stromal cells substantially contribute to tumor initiation and progression, become reactive even at the precancerous stage of prostatic intraepithelial neoplasia (113) and can contribute to PCSC transformation. For example, fibroblast growth factor FGF10 produced by urogenital mesenchymal cells induces cancerous transformation of CD49hi murine basal epithelial cells (114). Similar findings have been described for non-tumorigenic human basal epithelial cells that form tumors in recombination with cancer associated fibroblasts, CAFs (115). CAFs are one of the major components of tumor stroma which not only directly influence tumor cells but also influences other types of cells in form of stimulation (e.g. recruitment of endothelial cells) or inhibition (e.g. exclusion of T-cells) leading to the establishment of a tumor-permissive microenvironment (107). An activated CAF phenotype is associated with elevated microenvironmental oxidative stress that might, in turn, stimulate genetic changes in the epithelial cells and normal stromal fibroblasts and their cancer transformation (116). Based on the heterogeneity of expressed markers, CAFs have been suggested to derive from different cellular origins, e.g. activation of resident fibroblasts, recruitment of bone marrow derived progenitor cells to the tumor site, and trans-differentiation through the endothelial-mesenchymal or epithelial-mesenchymal transition as reviewed recently by Levesque and Nelson (107). In prostate tumors, CAFs induce a metabolic shift toward the mitochondrial oxidative phosphorylation called “reverse Warburg” phenotype. Intercellular contact of PCa cells and CAFs results in a mutual metabolic reprogramming in tumor cells and stroma. This includes a shift of CAFs metabolism toward glycolysis with accelerated expression of glucose transporter GLUT1 and increased lactic acid production and secretion. In turn, CAF-generated lactate stimulates mitochondrial biogenesis and aerobic metabolism in PCa cells (reverse Warburg effect) associated with a decrease in expression of GLUT1 transporter and activation of lactate upload (117–120). Notably, lactate-mediated tumor acidification has been associated with suppression of anticancer immune response mediated by the loss of cytotoxic T-cell and NK cell function, reduction of dendritic cell maturation and increased helper cell activities as reviewed by Choi and co-authors (121).
Accumulating evidences suggest that CAFs induce EMT and stemness in PCa through the production of metalloproteases (MMPs) and activation of the pro-inflammatory pathways in tumor cells involving nuclear factor-κB (NF-κB), cycloxygenase-2 (COX-2) and HIF-1 (119, 122). Inflammation gene signature was found to be highly upregulated in PCSCs, and chronic inflammation contributes to the development of aggressive prostate cancer (123). Inflammation plays a critical role in transformation of prostate epithelial cells and might determine PCa cell of origin. Cytokines and growth factors secreted by immune cells that infiltrate the injured or infected tissues can induce PCSCs. For example, recent study showed that inflammation expands the population of human CD38low luminal cells that can initiate PCa after oncogenic transformation (124). Experimental studies using murine prostate tumors demonstrated that loss of AR expression results in the activation of inflammatory STAT3 signaling pathway and induction of PCSC populations. This study may suggest the role of STAT3 signaling pathway in the development of hormone-refractory PCa upon hormone deprivation (125). Furthermore, pro-inflammatory Toll-like Receptor 9 (TLR9) and TLR9-mediated activation of NF-κB and STAT3 transcription factors is essential for the maintenance of tumorigenic prostate cells (126). Notably that TLR9+ prostate tumor cells promote expansion and activation of myeloid-derived suppressor cells (MDSC) that has immunosuppressive effects on effector CD8+ T cells (127). Accumulation of MDSC in PCa correlates with disease progression (128).
Analysis of the CSCs and CAFs in the conditional Pten deletion PCa mouse model demonstrated that signals produced by CAFs positively regulate gland forming and proliferative capacities of CSCs in vivo (129, 130). The Pten−/− mouse model recapitulates the different consequent stages of prostate tumor development, and CAFs have been shown to regulate CSCs in both androgen dependent and castration resistant (CR) tumors (129, 130). Interestingly that CAFs isolated from more aggressive CR tumors were more effective in supporting the sphere-forming properties of CSCs in vitro and formation of poorly differentiated tumors in vivo. This can be explained by the fact that interaction between tumor and microenvironment is bidirectional, and both cancer cells including CSC populations and tumor stroma mutually influence each other. Indeed, recent study showed that TGF-β1 secreted by androgen insensitive PCa cells serves for the recruitment of mesenchymal stem cells (MSC) to the tumor and their trans-differentiation into CAFs (131). Other study showed that in addition to TGF-β, other factors such as pro-inflammatory cytokine interleukin 6 (IL-6) produced by human PCa cells can activate fibroblasts. These PCa-activated fibroblasts increase PCa aggressiveness by inducing EMT, invasiveness and migration through producing matrix metalloproteinase MMPs, whereas myofibroblasts induce PCa motility by activation of serine proteases μPA/μPARs (119). CAFs synergize with other tumor stroma cells such as cancer associated macrophages (CAMs) in promoting tumor growth (132). Circulating monocytes can be attracted to tumor tissue by monocyte chemotactic protein-1 (MCP-1) produced by PCa cells or by stromal-derived growth factor-1 (SDF-1) produced by CAFs. Under exposure to IL-6 and SDF-1 released by PCa cells or CAFs, respectively, monocytes can consequently differentiate to macrophages which in turn can activate CAFs, induce de novo angiogenesis and cooperate with CAFs to increase PCa cell motility (132).
Anticancer therapy affects tumor stroma biology and its interaction with tumor cells. Recent study showed that castration- induced hypoxia activates HIF-1 and triggers autocrine TGF-β signaling in myofibroblast that promotes their activation and induction of CXCL13, an important regulator of PCa proliferation and invasion (103). Recent study showed that inhibition of CXCL13 signaling axis in prostate cancer cells impairs their tumorigenic properties (133). Tissue injury by castration, chemo- or radiotherapy also results in production of inflammatory cytokines by tumor stroma (e.g., epidermal growth factor (EGF), FGF, lymphotoxin-α (LTα), lymphotoxin-β (LTβ), IL-6, TGF-β and infiltration of immune cells (103, 107). Furthermore, DNA damage caused by cytotoxic chemotherapy in vivo induces κ light polypeptide gene enhancer in B cells 1 (NF-κB) signaling in fibroblasts which upregulates expression of wingless-type MMTV integration site family member 16B (WNT16B), activates the canonical Wnt program in PCa cells through paracrine mechanisms and promotes tumor progression (103). Taken together, these studies suggest that combination of conventional treatment with CAF-targeting therapy could decrease the effects of tumor stroma on tumor recurrence and therapy resistance and improve effectiveness of cancer treatment.
In addition to the cellular and humoral factors, physical conditions such as oxygen concentration, extracellular pH and ion strength also play a role in the CSC maintenance. Similar to CSCs, their microenvironmental niches are also dynamic in nature, and the maintenance of CSCs may be regulated by the distinct niches at the different stages of tumor development starting from tumor initiation to metastasis development (112, 134).
3.3. A -vicious cycle- of PCa bone metastases
The majority of PCa patients with advanced disease develop bone metastases (135). When the tumor spreads to the bone, it cannot be cured anymore (136, 137). This stage of disease is associated with a high risk of morbidity caused by the various complications from bone metastases (138). PCa bone metastases can produce factors that can enhance both osteoblastic and osteoclastic activities and disrupt normal bone homeostasis. Reciprocally, the supportive sites that maintain the survival and proliferation of cancer cells in the bone marrow are defined by special cytokines and growth factors produced by osteoblast, osteoclasts and bone marrow stromal cells or released from the bone matrix upon resorption. This feedback loop between the seeds (prostate tumor cells) and soil (bone microenvironment) leads to a -vicious cycle- of tumor growth as reviewed previously (139).
The bone microenvironment produces or releases various growth factors and cytokines that bind to the receptors on prostate tumor cells and regulate their growth and survival. Among them are Wnt proteins that bind to the Frizzled receptor and its LRP5/6 co-receptor and activate β-catenin - dependent gene expression, C-X-C motif chemokine ligand 12 (CXCL12) that binds to C-X-C chemokine receptor type 4 (CXCR4), as well as growth factors e.g., transforming growth factor beta (TGFβ), insulin like growth factor 1 (IGF-1), bone morphogenetic proteins (BMPs), and fibroblast growth factors (FGFs) that promote metastatic growth (140).
In turn, PCa cells in the bone can produce pro-osteolytic factors such as IL-6, IL-1, parathyroid hormone-related protein (PTHrP), and PSA that stimulate the formation of osteoclasts and promote resorption of bone matrix. Unlike bone metastases from breast cancer and myelomas, PCa metastases almost always form osteoblastic lesions (141). The factors released by PCa cells in the bone include signaling molecules affecting osteoblast proliferation or differentiation such as WNT ligands and TGFβ as well as osteoprotegerin (OPG) that regulate both bone remodeling and tumorigenesis (141, 142). OPG produced by PCa cells and by the bone marrow is an inhibitor of TNF-related apoptosis inducing ligand (TRAIL) and also an important survival factor in hormone independent PCa cells (143). Interestingly, PSA has been shown to inhibit OPG expression and increase mRNA expression of the receptor activator of nuclear factor kappa-B ligand (RANKL) that induces osteoclast differentiation (144). It is worthy mentioning that, in contrast to primary tumors that are almost always PSA positive, up to 40% of prostate metastases are negative for PSA expression (145, 146).
4. CSC evolution during tumor progression
Prostate cancer development is an evolutionary process that reflects dynamic changes of CSC phenotypes and properties over successive CSC generations (Figure 2). Luminal or basal cells in normal prostate can act as a cell of tumor origin after oncogenic transformation, however the link between the tumor initiating cells, or tumor cell of origin and PCSCs that maintain tumor growth is not yet understood (2). Prostate cancer progression is associated with the development of substantial intra-tumor heterogeneity and genomic instability that can be induced by MYC activation, loss of PTEN and mutations in DNA repair genes including BRCA2, ATM and CHEK2 (147–149). This PCSC heterogeneity fosters tumor evolution and development of metastases (Figure 2).
Figure 2. Phenotypes of CSCs, DTCs, MICs and prostate tumor progression.
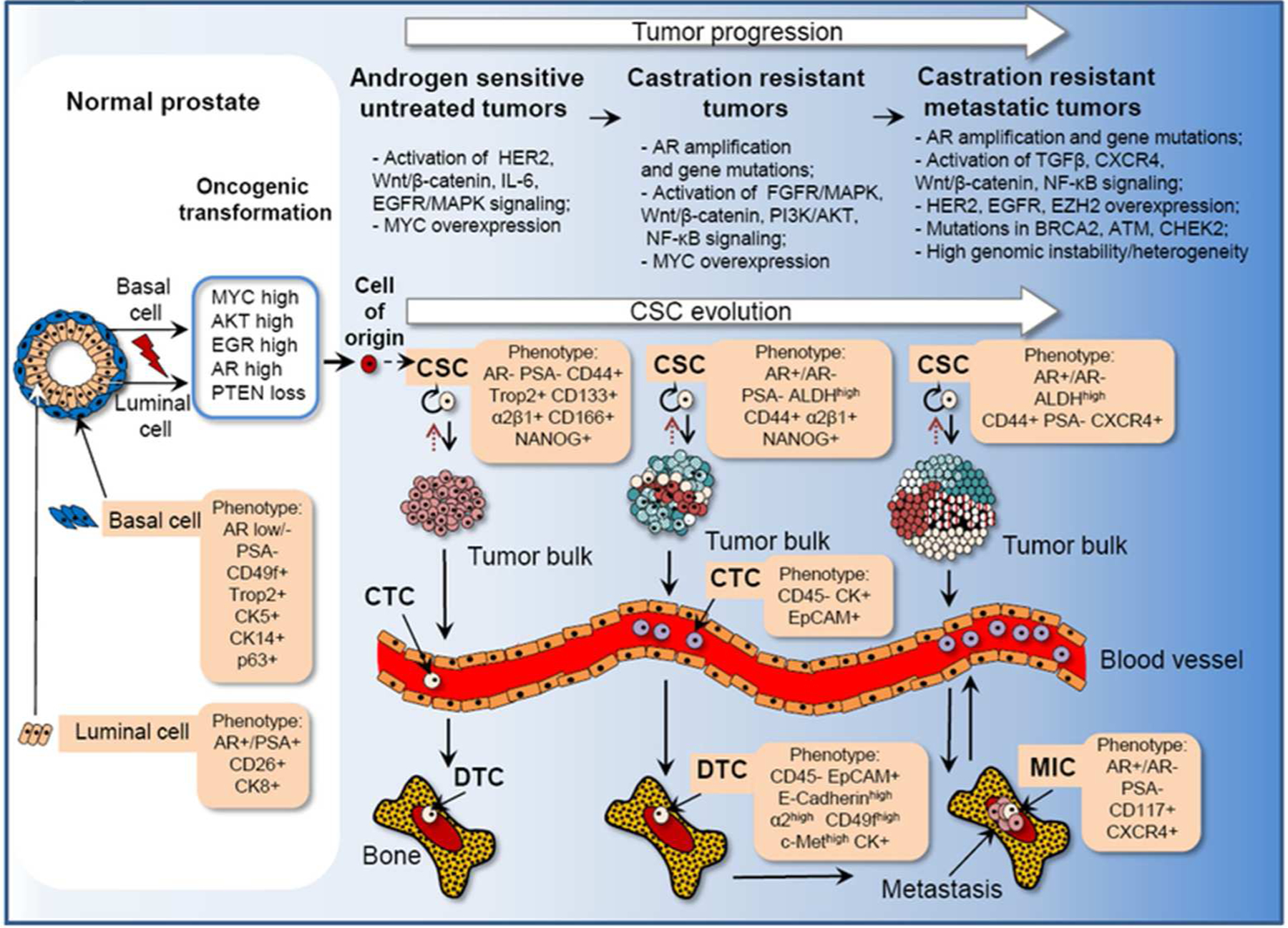
Prostate cancer development is an evolutionary process that reflects an evolving of CSCs. Luminal or basal cells in normal prostate can act as a cell of tumor origin after oncogenic transformation. The link between the tumor initiating cells, or tumor cell-of-origin and cancer stem cells (CSCs) that maintain tumor growth is not yet understood. Cancer cell-of-origin refers to the initial normal cell or the cell type that became tumorigenically transformed whereas CSCs refer to the cell population that drives clonal tumor evolution. Tumor metastases are driven by the evolved populations of CSCs at their worst. Some tumor cells with malignant potential enter the blood stream (circulating tumor cells, CTCs) and can be disseminated to the distant organs. Single prostate tumor cells can be found in bone marrow and are called disseminated tumor cells (DTCs). Single tumor cells disseminated to lymph nodes are called isolated tumor cells (ITCs, not shown). A small subset of DTCs or ITCs could give rise to metastasis - initiating cells (MICs). Metastatic spread and formation is a long-time process that might take a few years. Once developed, metastases can form secondary metastasis to distant organs. Prostate cancer progression is associated with development of substantial intra-tumor heterogeneity and genomic instability that can be induced by MYC activation, loss of PTEN and mutations in DNA repair genes including BRCA2, ATM and CHEK2. A few PCSC phenotypes have been validated so far in animal models for their tumor initiating properties e.g. PSA−/lo, ALDHhigh, NANOG+, Trop2+, CD44+CD133+, ALDHhighCD44+α2β1+. Although some of the proposed biomarkers (e.g. EpCAM, CD117, c-Met) have been correlated with prostate cancer progression and metastases, there is still a lack of solid experimental evidences that these proteins can be considered as markers of PCSCs.
Acronyms: AR - androgen receptor; CK - cytokeratin; PSA - prostate specific antigen; ALDH - aldehyde dehydrogenase; ABCG2 - ATP binding cassette subfamily G member 2; α2β1 - α2β1 Integrin; ERG1 - early growth response protein 1; PTEN - phosphatase and tensin homolog; EpCAM - epithelial cell adhesion molecule; HER2 - human epidermal growth factor receptor 2; FGF - fibroblast growth factor; MAPK - mitogen-activated protein kinase; PI3K - phosphatidylinositide 3-kinase; NF-κB - nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells; TGFβ - transforming growth factor β; EGFR - epidermal growth factor receptor; CXCR4 - C-X-C chemokine receptor type 4; EZH2 - enhancer of zeste homolog 2; BRCA2 - breast cancer type 2 susceptibility protein; ATM - ataxia telangiectasia mutated; CHEK2 - checkpoint kinase 2.
Metastatic spread is a multistep process that starts from invasion of cancer cells through the basement membrane and entering the bloodstream (intravasation). At this stage circulating tumor cells (CTCs) can be disseminated to distant organs. Some CTCs that escape anoikis, immune clearance and mechanical stresses can leave the bloodstream and enter the tissues in distant organs in a process called extravasation. At this stage, these disseminated tumor cells (DTCs) can remain dormant at distant site for a few years prior to forming macrometastasis (135, 150). Only a small subset of DTCs can become metastasis-initiating cells (MICs). Selection of DTCs toward a metastatic phenotype is driven by the microenvironmental conditions in end organs as well as by the factors from primary tumors, which define the ability of DTCs to overcome tumor latency and form a metastatic tumor (135, 151–153). Metastasis formation can be considered as an evolutionary process (Figure 2). Plasticity of genetic and epigenetic makeups and selection of the most adapted tumor cells at all stages of the metastatic cascade results in the acquisition of cell-inherent traits that favor tumor dissemination (1, 136). CSCs that possess self-renewal properties and genomic instability are considered to be an engine of tumor evolution, and tumor metastases might as be driven by the constantly evolving populations of CSCs. This can be evidenced by the fact that CTCs and DTCs express many CSC markers, and MICs also share key CSC features including ability to self-renew, establish tumors and activate CSC-specific signaling pathways (136). The phenotypes and key characteristics of CSCs, DTCs and MICs during prostate tumor progression are depicted in Figure 2 and described in Table 2.
Table 2.
The features of prostate cancer stem cells (PCSCs), circulating tumor cells (CTCs), disseminated tumor cells (DTCs) and metastasis initiating cells (MICs).
| Features╲Cells | Non-CSCs (tumor bulk) | CSCs | CTCs | DTCs | MICs |
|---|---|---|---|---|---|
| Androgen receptor expression | AR+ (luminal cells); ARlow/− (basal cells) (60, 91, 219) | AR− (primary PC) or AR−/+ (CRPC) (3, 11, 125, 220) | AR− or AR+ (221–225) | AR− or AR+ (226) | AR− or AR+ (227, 228) |
| Anrogen dependence | Primary PC: Yes CRPC: No (66, 229) | No (3, 28, 55, 220) | Yes / No (221, 230, 231) | Yes / No (228) | Yes / No (228) |
| PSA expression | Yes (luminal cells) (3, 60) | No (3) or low (232) | PSA+ or PSA− (230, 231, 233) | PSA+ or PSA− (234) | No data |
| Phenotype | Luminal or basal epithelial cells (229) | Luminal or basal epithelial cells (3, 32, 61, 62, 79, 115, 219, 235, 236) | Epithelial or Mesenchymal (EMT) (163, 222, 225, 233, 237, 238) | Epithelial (234), Mesenchymal (EMT) (163), Hematopoietic (234, 239) | No data |
| Tumorigenicity | Upon dedifferentiation (14, 232) | Yes (3, 32, 61, 62, 79, 115, 219, 235, 236) | Probably (240) | Probably (241, 242) | Yes (243, 244) |
| Metabolism | Primary PC: OXPHOS CRPC: glycolysis (Warburg effect) (26, 42) | Glycolysis (96) or/and OXPHOS (80, 245) | No data | No data | No data |
| Heterogeneity | Yes (246) | Yes (3, 32, 61, 62, 79, 115, 219, 235, 236) | Yes (222, 224, 225, 230, 233, 247) | Yes (226, 228, 234) | Yes (243, 244) |
| Cellular state | Proliferative (229) | Quiescent (3) | Proliferative or quiescent (248, 249) | Dormant or quiescent (150, 250) | No data |
| Genetic evolving | Yes (244, 251) | Yes (251) | Yes, reflection of tumor evolution (160, 161, 252) | Yes (159) (135, 151–153) | Yes (243, 244, 253) |
In addition, CSC-specific gene signatures correlate with tumor progression and development of PCa metastasis (96, 154, 155). Therefore, enumeration of CTCs in blood and of DTCs in the bone marrow has prognostic and predictive value in PCa (97, 135, 156). Metastatic tumors, CTCs, and DTCs also show varying degrees of concordance with molecular signatures of the primary tumor of origin (157–161). Recent studies, based on the analysis of a large set of RNA expression data from more than 4,000 clinical PCa specimens, demonstrated that the tumor subtype that exhibits a high expression of luminal markers EZH2, AR, MK167 and low expression of luminal markers NKX3–1, PSA, ERG, KLK2 and basal markers, e.g., TP63 and KRT5 is associated with the highest risk of progression to metastatic disease (162). Interestingly, this gene signature was highly expressed in 58% of CTCs from patients with antiandrogen therapy-resistant tumors (162). This study demonstrated that molecular characteristics of CTCs obtained by noninvasive “liquid biopsy” could be used to identify the patients at risk of metastatic disease and treatment failure. These results are consistent with previous studies showing that PSA negative (PSA−/low) prostate tumor cells are resistant to chemotherapy, androgen deprivation, and stress simulation, and exhibit long-term tumor-propagation capacities (3). Another study showed that CTCs and DTCs cells isolated from mouse xenografts of human prostate tumors exhibit increased potential to metastasize in vivo and resistance to the chemotherapeutic agents mitoxantrone and doxorubicin (163).
5. PCSCs and clinical application
Taking in account CSC properties such self-renewal, tumor-initiating and therapy resistance, CSC-related signatures can be employed in clinical practice for the different purposes e.g.: (1) to predict tumor development; (2) to identify patients with a poor prognosis who can potentially benefit from therapy intensification; (3) to visualize the most critical intratumoral areas containing CSCs that should be taken into account for the local therapy planning (surgery, radiotherapy); (4) to monitor an efficacy of therapeutic approaches administered in cancer patients; (5) to diagnose local or distant tumor recurrences after therapy; and (6) to develop novel specific anti-CSC therapeutics to effectively destroy the most aggressive and treatment-resistant CSCs.
Although a number of strategies for PCSC enrichment and analysis are considered in this review article, there are no specific PCSC markers known up to date which are already implemented in clinical practices. Nevertheless, there is growing evidence that PCSC specific characteristics are predictive of patient outcome (as illustrated in Table 3) that lends credence to PCSCs as potential clinical biomarkers and therapeutic targets.
Table 3.
Selected PCSC - specific characteristics as potential clinical biomarkers.
| Marker / parameter | Analysis | Number of analyzed patients | Clinical correlation | Ref |
|---|---|---|---|---|
| - ALDH activity; | - Fluorescence-activated cell sorting (FACS) based ALDEFLUOR analysis; | n = 39 | - ALDH activity and ALDH1A1 gene expression significantly correlates with Gleason score | (192) |
| CD166 | - Affymetrix GeneChip microarray of benign and tumor tissue for patients who underwent radical prostatectomy for prostate cancer | n = 133 | CD166/MEMD gene expression levels significantly correlate with biochemical recurrence free survival | (254) |
| CD117/c-Kit | CD117/EpCAM positive cells were analyzed in PCa patients’ blood by FACS | n = 99 | Number of CD117/EpCAM cells is higher in high-grade patients and in patients after biochemical recurrence | (255) |
| PSAlow | Illumina DASL expression microarray | n = 596 | PSA gene expression level correlates with biochemical recurrence free and overall PCa patient survival. | (3, 256) |
| CD44 | IHC staining of tissues from patients with clinically localized PCa who underwent radical prostatectomy | n = 160 | CD44v6 expression level correlates with biochemical recurrence free survival | (257) |
There is an increasing body of evidence that PCSCs possess higher chemo- and radioresistance compared to the tumor bulk (3, 21, 164). Over the last decades, different strategies have been suggested for eradicating PCSCs that are resistance to conventional therapeutic agents. One of these strategies is targeting the signaling pathways that regulate the maintenance and survival of PCSCs such as WNT/β-catenin and PI3K/AKT (164–166). Inhibition of these signaling routes results in the sensitization of the experimental tumor models to the different types of therapy (164, 167–169). Metabolic features of PCSCs can also be potentially used for anti-cancer therapy to increase efficiency of conventional treatment such as chemo- or radiotherapy (Figure 1). However, further clinical studies are warranted to understand if these drugs can increase efficiency of PCa treatment without ablating normal stem cell functions.
Another experimental approach for PCSC targeting is based on microRNAs (miRNAs) which are non-coding RNAs that regulate gene expression by translational silencing. A number of miRNAs have been implicated in regulation both normal SC and CSCs in the different tumor entities. Recent studies showed that some miRNAs could negatively regulate PCSCs. This, for example, includes miR-34a which directly regulates CD44 expression (170); miR-141 which targets multiple genes involved in tumor development and metastatic dissemination (171); miR128 which downregulates the stem cell factors NANOG, polycomb complex protein BMI-1 and Transforming Growth Factor Beta Receptor 1 (TGFBR1); miR-199a-3p which inhibits mitogenic signaling including epidermal growth factor receptor (EGFR), c-MYC and cyclin D1 (CCND1) (48); let-7 microRNA which acts as a tumor suppressor and PCSC inhibitor by targeting key oncogenes such as MYC, RAS and High Mobility Group A2 gene (HMGA2) (172) and miR-200b which inhibits PCa growth, EMT and metastases (105). Collectively, these data provide a strong rationale for development of microRNA-based therapy against PCSCs, and ongoing studies are exploring the approaches for microRNA delivery in vivo (173).
In addition to the targeting of the pro-survival and self-renewal pathways, some novel therapeutic strategies are based on the immunologic targeting of PCa- and PCSC-specific markers such as CD44, prostate stem cell antigen (PSCA) and ALDH. To target CSC marker CD44, recent studies employed phage display library to identify high affinity peptide that target CD44v6 protein isoform and can be potentially used for systemic delivery of diagnostic and therapeutic molecules in PCa patients (174). Several other studies demonstrated potential application of anti-CD44 antibodies to target CSC cells in acute myeloid leukemia (AML) and pancreatic xenograft models as reviewed in (175).
Notwithstanding its name, PSCA is not a marker of stem cells but intermediate prostate epithelial cells that co-express basal and luminal cytokeratins (176). Nevertheless, recent study showed that PSCA is also expressed in stem cells derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) mice (177). PSCA is AR-responsive gene which is highly expressed in PCa tissues and PCa bone metastases, and correlates with a poor prognosis (178, 179). This makes PSCA an attractive molecule for prostate tumor imaging and treatment. Positron emission tomography (PET) imaging with 124I-Anti-PSCA A11 minibody is currently in clinical trial for metastatic prostate cancer (ClinicalTrials.gov Identifier: NCT02092948). There are also early clinical studies in advanced castration-resistant prostate cancer patients for self-adjuvanted mRNA vaccination using prostate-cancer vaccine CV9103 which contains mRNA encoding the antigen proteins PSCA, PSA, prostate-specific membrane antigen (PSMA) and six-transmembrane epithelial antigen of prostate member 1 (STEAP1) (180). Another approach to treat localized and metastatic prostate cancers is a fully human monoclonal antibody against PSCA called AGS-PSCA which is also entered early phase clinical trial (181).
Study of Jachetti et al. on the PCSCs derived from TRAMP mouse model showed that they induce both innate and adaptive immunity. This study showed significant inhibition of tumor growth in mice immunized with PCSC-pulsed dendritic cells and therefore suggested that PCSC-derived antigens can be potentially used for the development of novel immunotherapy for prostate cancer treatment (177). As PCSCs might also express prostate antigens such as PSCA, the PCSA-targeted therapy might substantially augment tumor specific immune response and can be particularly efficient by simultaneous administration with treatment that neutralizes tumor-induced immunosuppression (177).
Studies for other tumor types such as murine squamous cell cancer, murine melanoma and human colorectal cancer demonstrated anti-tumor efficiency of other types of CSC vaccines such as dendritic cells loaded with cell lysates of ALDH positive sorted tumor cells (182, 183). These studies demonstrated that anti-tumor efficiency of the CSC-based vaccine can be significantly improved in combination with an immune checkpoint inhibitor such as anti-PD-L1 antibody which blocks the immunosuppressive effect of the tumor microenvironment (182). An ongoing Phase I/II clinical trial is testing this novel ALDH CSC-derived vaccine for patients with metastatic colorectal cancer (ClinicalTrials.gov Identifier: NCT02176746). Taking into account that ALDH is one of the clinically relevant markers of PCSCs, the results of these studies might be of high importance for the development of new anti-PCa immunotherapy. Further rigorous animal and clinical studies are warranted to demonstrate if these CSC-targeted immunotherapies alone or in combination with inhibitions of tumor immune suppression could increase the efficiency of conventional treatment approaches (175).
6. Common mechanisms underlying metastasis and therapy resistance
A growing body of evidence begins to suggest common mechanisms that underlie the development of therapy resistance and metastatic phenotypes during tumor progression (184–186). In particular, the CXCL12/CXCR4 axis is shown to regulate extravasation and dissemination of PCa cells to the bone. Furthermore, activation of the CXCL12/CXCR4 and WNT/β-catenin signaling pathways that are important regulators of PCSCs promotes tumor radioresistance and contributes to the development of skeletal metastases (21, 187–189). Aldehyde dehydrogenase (ALDH) activity can be used to enrich for PCa cells with increased tumorigenic and metastatic capability in vivo and with high radioresistance (21, 167, 190). ALDH plays an important role in the cellular response to oxidative stress and can potentially serve as one of the pro-survival mechanisms in CTCs enabling them to withstand oxidative stress in the bloodstream (191). Although several ALDH isoforms are highly expressed in PCa, only ALDH1A1 gene expression has been shown to be significantly correlated with ALDH activity and increased at high levels in advanced-stage compared to early-stage PCa and benign prostate hyperplasia (192). WNT ligands produced by tumor cells or by bone cells are acting in autocrine or paracrine fashion by enhancing tumor cell proliferation and inhibiting apoptosis (193). In addition, WNT ligands have a paracrine effect and induce osteoblastic environment promoting bone mineralization in PCa bone metastases, whereas WNT inhibitors promote an osteolytic activity (193, 194). Activation of canonical β-catenin-dependent WNT signaling is associated with malignant transformation, metastasis and radioresistance of PCa (21, 195). In different cancer models, WNT signaling pathway has been shown to regulate expression of CXCR4 and ALDH1A1 genes (21, 196, 197) whereas canonical WNT signaling pathway can be activated by CXCR4 - dependent mechanisms (198, 199). Bone-targeted therapies such as bisphosphonate drugs exhibit inhibitory effects on osteoclast-mediated bone resorption as well as on PCa cells (200, 201). Recent studies showed that bisphosphonate treatment regulates the WNT signaling pathway in PCa, e.g., by inhibition of WNT5A and FZD5 gene expression (202). TGFβ signaling activated in bone metastases regulates the pro-metastatic and pro-survival properties of PCa cells including expression of CXCR4 and increase in ALDH activity (190, 203–205). PCa cells have deregulated expression of integrins (206) that play critical roles in prostate tumor cell invasion and metastatic development (207). The PCa cell population positive for α2β1 integrin expression is enriched for tumor-initiating cells (30, 208). Recent studies showed that integrin signaling might also contribute to the resistance to androgen ablation and radiotherapy in PCa cells (209, 210).
Therefore, activation of the signaling pathways detected at the genomic and transcriptomic levels in DTCs, CTCs from the blood of the patients, and CSCs in primary tumors can be prognostic for metastasis development and may also be involved in the response of primary and metastatic tumors to therapy. Further studies are warranted to investigate the molecular mechanisms regulating metastasis-initiating and therapy-resistant prostate tumor cells and characterize their clinical relevance.
7. Summary and future directions
PCSCs that possess self-renewal properties and genomic instability might serve as a driving force of tumor evolution and metastatic dissemination. A number of recent studies demonstrated that analysis of CSCs in patients’ tumors might serve as prognostic or predictive biomarker (211, 212). In combination with other extrinsic and intrinsic parameters that might affect the properties and complexity of CSC populations, for example heterogeneity indices and hypoxia, these markers could in the future be used for patient stratification and more personalized treatment selection. However, the co-existence of multiple PCSC populations and their evolution during tumor progression and treatment could challenge the clinical application of PCSCs as prognostic biomarkers. This PCSC heterogeneity and plasticity provide a strong rationale to further development of more robust CSC-related prognostic signatures consisting of multiple markers instead of single-marker analysis, for monitoring PCSCs and CTCs during the course of treatment and for zoom in on single tumor cells analysis to identify residual PCSCs that might initiate tumor regrowth. Understanding the genetic relationship between PCSCs, CTCs, DTCs and MICs and better characterization of the MIC phenotypes and properties may be pivotal in the future for prevention of tumor cell dissemination and treatment of metastatic disease. Clinical validation of the targeting agents which are specific for PCSCs and well tolerated by normal SCs might represent a great step closer to the complete prostate cancer cure.
Acknowledgement
Work in Dubrovska lab was partially supported by grants from Deutsche Forschungsgemeinschaft (DFG) (273676790), from Wilhelm Sander-Stiftung (2017.106.1), DLR Project Management Agency (01DK17047) and BMBF (03Z1NN11). Work in Tang lab was supported, in part, by grants from the US National Institutes of Health (NIH) (R01-CA155693), Department of Defense (W81XWH-14-1-0575 and W81XWH-16-1-0575), and the Chinese Ministry of Science and Technology (MOST) grant 2016YFA0101203, and by RPCCC and NCI center grant P30CA016056. Work in Skvortsova lab was supported in part by Austrian Science Fund (FWF) (P29457), Austrian National Bank (ÖNB 17620), and Ingrid Shaker-Nessmann Cancer Research Foundation.
References
- 1.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell stem cell. 2014;14(3):275–91. [DOI] [PubMed] [Google Scholar]
- 2.Rycaj K, Tang DG. Cell-of-Origin of Cancer versus Cancer Stem Cells: Assays and Interpretations. Cancer research. 2015;75(19):4003–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qin J, Liu X, Laffin B, Chen X, Choy G, Jeter CR, et al. The PSA(−/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell stem cell. 2012;10(5):556–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer research. 2005;65(23):10946–51. [DOI] [PubMed] [Google Scholar]
- 5.Huss WJ, Gray DR, Greenberg NM, Mohler JL, Smith GJ. Breast cancer resistance protein-mediated efflux of androgen in putative benign and malignant prostate stem cells. Cancer research. 2005;65(15):6640–50. [DOI] [PubMed] [Google Scholar]
- 6.Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2-cancer cells are similarly tumorigenic. Cancer research. 2005;65(14):6207–19. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Huang Y, Jin Z, Li X, Li B, Xu P, et al. A convenient and effective strategy for the enrichment of tumor-initiating cell properties in prostate cancer cells. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2016;37(9):11973–81. [DOI] [PubMed] [Google Scholar]
- 8.Shi X, Gipp J, Dries M, Bushman W. Prostate progenitor cells proliferate in response to castration. Stem cell research. 2014;13(1):154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duan JJ, Qiu W, Xu SL, Wang B, Ye XZ, Ping YF, et al. Strategies for isolating and enriching cancer stem cells: well begun is half done. Stem cells and development. 2013;22(16):2221–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li T, Su Y, Mei Y, Leng Q, Leng B, Liu Z, et al. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Laboratory investigation; a journal of technical methods and pathology. 2010;90(2):234–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiao J, Hindoyan A, Wang S, Tran LM, Goldstein AS, Lawson D, et al. Identification of CD166 as a surface marker for enriching prostate stem/progenitor and cancer initiating cells. PloS one. 2012;7(8):e42564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hurt EM, Kawasaki BT, Klarmann GJ, Thomas SB, Farrar WL. CD44+ CD24(−) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. British journal of cancer. 2008;98(4):756–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buczek ME, Reeder SP, Regad T. Identification and Isolation of Cancer Stem Cells Using NANOG-EGFP Reporter System. Methods in molecular biology. 2018;1692:139–48. [DOI] [PubMed] [Google Scholar]
- 14.Della Donna L, Lagadec C, Pajonk F. Radioresistance of prostate cancer cells with low proteasome activity. The Prostate. 2012;72(8):868–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang B, Raviv A, Esposito D, Flanders KC, Daniel C, Nghiem BT, et al. A flexible reporter system for direct observation and isolation of cancer stem cells. Stem cell reports. 2015;4(1):155–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiagarajan PS, Hitomi M, Hale JS, Alvarado AG, Otvos B, Sinyuk M, et al. Development of a Fluorescent Reporter System to Delineate Cancer Stem Cells in Triple-Negative Breast Cancer. Stem cells. 2015;33(7):2114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torre-Healy LA, Berezovsky A, Lathia JD. Isolation, Characterization, and Expansion of Cancer Stem Cells. Methods in molecular biology. 2017;1553:133–43. [DOI] [PubMed] [Google Scholar]
- 18.Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, et al. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene. 2011;30(36):3833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hristova NR, Tagscherer KE, Fassl A, Kopitz J, Roth W. Notch1-dependent regulation of p27 determines cell fate in colorectal cancer. International journal of oncology. 2013;43(6):1967–75. [DOI] [PubMed] [Google Scholar]
- 20.Skvortsov S, Debbage P, Skvortsova I. Proteomics of cancer stem cells. International journal of radiation biology. 2014;90(8):653–8. [DOI] [PubMed] [Google Scholar]
- 21.Cojoc M, Peitzsch C, Kurth I, Trautmann F, Kunz-Schughart LA, Telegeev GD, et al. Aldehyde Dehydrogenase Is Regulated by beta-Catenin/TCF and Promotes Radioresistance in Prostate Cancer Progenitor Cells. Cancer research. 2015;75(7):1482–94. [DOI] [PubMed] [Google Scholar]
- 22.Khan MI, Czarnecka AM, Helbrecht I, Bartnik E, Lian F, Szczylik C. Current approaches in identification and isolation of human renal cell carcinoma cancer stem cells. Stem cell research & therapy. 2015;6:178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu WH, Qian NS, Li R, Dou KF. Replacing Hoechst33342 with rhodamine123 in isolation of cancer stem-like cells from the MHCC97 cell line. Toxicology in vitro : an international journal published in association with BIBRA. 2010;24(2):538–45. [DOI] [PubMed] [Google Scholar]
- 24.Freitas DP, Teixeira CA, Santos-Silva F, Vasconcelos MH, Almeida GM. Therapy-induced enrichment of putative lung cancer stem-like cells. International journal of cancer. 2014;134(6):1270–8. [DOI] [PubMed] [Google Scholar]
- 25.Vlashi E, Chen AM, Boyrie S, Yu G, Nguyen A, Brower PA, et al. Radiation-Induced Dedifferentiation of Head and Neck Cancer Cells Into Cancer Stem Cells Depends on Human Papillomavirus Status. International journal of radiation oncology, biology, physics. 2016;94(5):1198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pertega-Gomes N, Felisbino S, Massie CE, Vizcaino JR, Coelho R, Sandi C, et al. A glycolytic phenotype is associated with prostate cancer progression and aggressiveness: a role for monocarboxylate transporters as metabolic targets for therapy. The Journal of pathology. 2015;236(4):517–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Q, Rycaj K, Chen X, Tang DG. Cancer stem cells and cell size: A causal link? Semin Cancer Biol. 2015;35:191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen X, Li Q, Liu X, Liu C, Liu R, Rycaj K, et al. Defining a Population of Stem-like Human Prostate Cancer Cells That Can Generate and Propagate Castration-Resistant Prostate Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22(17):4505–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuch V, Schreiber C, Thiele W, Umansky V, Sleeman JP. Tumor-initiating properties of breast cancer and melanoma cells in vivo are not invariably reflected by spheroid formation in vitro, but can be increased by long-term culturing as adherent monolayers. International journal of cancer. 2013;132(3):E94–105. [DOI] [PubMed] [Google Scholar]
- 30.Patrawala L, Calhoun-Davis T, Schneider-Broussard R, Tang DG. Hierarchical organization of prostate cancer cells in xenograft tumors: the CD44+alpha2beta1+ cell population is enriched in tumor-initiating cells. Cancer research. 2007;67(14):6796–805. [DOI] [PubMed] [Google Scholar]
- 31.Beck B, Blanpain C. Unravelling cancer stem cell potential. Nature reviews Cancer. 2013;13(10):727–38. [DOI] [PubMed] [Google Scholar]
- 32.Chua CW, Shibata M, Lei M, Toivanen R, Barlow LJ, Bergren SK, et al. Single luminal epithelial progenitors can generate prostate organoids in culture. Nature cell biology. 2014;16(10):951–61, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ford WC, Harrison A. The role of citrate in determining the activity of calcium ions in human semen. International journal of andrology. 1984;7(3):198–202. [DOI] [PubMed] [Google Scholar]
- 34.Mycielska ME, Patel A, Rizaner N, Mazurek MP, Keun H, Patel A, et al. Citrate transport and metabolism in mammalian cells: prostate epithelial cells and prostate cancer. BioEssays : news and reviews in molecular, cellular and developmental biology. 2009;31(1):10–20. [DOI] [PubMed] [Google Scholar]
- 35.Costello LC, Liu Y, Franklin RB, Kennedy MC. Zinc inhibition of mitochondrial aconitase and its importance in citrate metabolism of prostate epithelial cells. The Journal of biological chemistry. 1997;272(46):28875–81. [DOI] [PubMed] [Google Scholar]
- 36.Franklin RB, Feng P, Milon B, Desouki MM, Singh KK, Kajdacsy-Balla A, et al. hZIP1 zinc uptake transporter down regulation and zinc depletion in prostate cancer. Molecular cancer. 2005;4:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu WY, Hu DP, Xie L, Li Y, Majumdar S, Nonn L, et al. Isolation and functional interrogation of adult human prostate epithelial stem cells at single cell resolution. Stem cell research. 2017;23:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR, et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch. Oncogene. 2014;33(45):5251–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell reports. 2014;8(5):1461–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hopkins JF, Sabelnykova VY, Weischenfeldt J, Simon R, Aguiar JA, Alkallas R, et al. Mitochondrial mutations drive prostate cancer aggression. Nature communications. 2017;8(1):656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pertega-Gomes N, Vizcaino JR, Attig J, Jurmeister S, Lopes C, Baltazar F. A lactate shuttle system between tumour and stromal cells is associated with poor prognosis in prostate cancer. BMC cancer. 2014;14:352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jadvar H Is There Use for FDG-PET in Prostate Cancer? Seminars in nuclear medicine. 2016;46(6):502–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(21):6479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW, et al. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(23):8983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Q, Hardie RA, Hoy AJ, van Geldermalsen M, Gao D, Fazli L, et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. The Journal of pathology. 2015;236(3):278–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(48):18782–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell stem cell. 2008;2(4):333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu R, Liu C, Zhang D, Liu B, Chen X, Rycaj K, et al. miR-199a-3p targets stemness-related and mitogenic signaling pathways to suppress the expansion and tumorigenic capabilities of prostate cancer stem cells. Oncotarget. 2016;7(35):56628–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Civenni G, Malek A, Albino D, Garcia-Escudero R, Napoli S, Di Marco S, et al. RNAi-mediated silencing of Myc transcription inhibits stem-like cell maintenance and tumorigenicity in prostate cancer. Cancer research. 2013;73(22):6816–27. [DOI] [PubMed] [Google Scholar]
- 50.White MA, Lin C, Rajapakshe K, Dong J, Shi Y, Tsouko E, et al. Glutamine Transporters Are Targets of Multiple Oncogenic Signaling Pathways in Prostate Cancer. Molecular cancer research : MCR. 2017;15(8):1017–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koh CM, Bieberich CJ, Dang CV, Nelson WG, Yegnasubramanian S, De Marzo AM. MYC and Prostate Cancer. Genes & cancer. 2010;1(6):617–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nupponen NN, Kakkola L, Koivisto P, Visakorpi T. Genetic alterations in hormone-refractory recurrent prostate carcinomas. The American journal of pathology. 1998;153(1):141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun C, Dobi A, Mohamed A, Li H, Thangapazham RL, Furusato B, et al. TMPRSS2-ERG fusion, a common genomic alteration in prostate cancer activates C-MYC and abrogates prostate epithelial differentiation. Oncogene. 2008;27(40):5348–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quinn DI, Henshall SM, Sutherland RL. Molecular markers of prostate cancer outcome. European journal of cancer. 2005;41(6):858–87. [DOI] [PubMed] [Google Scholar]
- 55.Wang L, Liu R, Li W, Chen C, Katoh H, Chen GY, et al. Somatic single hits inactivate the X-linked tumor suppressor FOXP3 in the prostate. Cancer cell. 2009;16(4):336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yegnasubramanian S, Haffner MC, Zhang Y, Gurel B, Cornish TC, Wu Z, et al. DNA hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer research. 2008;68(21):8954–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gil J, Kerai P, Lleonart M, Bernard D, Cigudosa JC, Peters G, et al. Immortalization of primary human prostate epithelial cells by c-Myc. Cancer research. 2005;65(6):2179–85. [DOI] [PubMed] [Google Scholar]
- 58.Ellwood-Yen K, Graeber TG, Wongvipat J, Iruela-Arispe ML, Zhang J, Matusik R, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer cell. 2003;4(3):223–38. [DOI] [PubMed] [Google Scholar]
- 59.Beharry Z, Mahajan S, Zemskova M, Lin YW, Tholanikunnel BG, Xia Z, et al. The Pim protein kinases regulate energy metabolism and cell growth. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(2):528–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park JW, Lee JK, Phillips JW, Huang P, Cheng D, Huang J, et al. Prostate epithelial cell of origin determines cancer differentiation state in an organoid transformation assay. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(16):4482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang D, Lin K, Lu Y, Rycaj K, Zhong Y, Chao HP, et al. Developing a Novel Two-Dimensional Culture System to Enrich Human Prostate Luminal Progenitors that Can Function as a Cell of Origin for Prostate Cancer. Stem cells translational medicine. 2017;6(3):748–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stoyanova T, Cooper AR, Drake JM, Liu X, Armstrong AJ, Pienta KJ, et al. Prostate cancer originating in basal cells progresses to adenocarcinoma propagated by luminal-like cells. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(50):20111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer cell. 2016;29(4):536–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer cell. 2011;19(5):575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bernard D, Pourtier-Manzanedo A, Gil J, Beach DH. Myc confers androgen-independent prostate cancer cell growth. The Journal of clinical investigation. 2003;112(11):1724–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013;32(49):5501–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saraon P, Drabovich AP, Jarvi KA, Diamandis EP. Mechanisms of Androgen-Independent Prostate Cancer. Ejifcc. 2014;25(1):42–54. [PMC free article] [PubMed] [Google Scholar]
- 68.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer research. 2009;69(6):2305–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.He Y, Lu J, Ye Z, Hao S, Wang L, Kohli M, et al. Androgen receptor splice variants bind to constitutively open chromatin and promote abiraterone-resistant growth of prostate cancer. Nucleic acids research. 2018;46(4):1895–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. The Journal of clinical investigation. 2010;120(8):2715–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PloS one. 2011;6(4):e19059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. The New England journal of medicine. 2014;371(11):1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Qu F, Xie W, Nakabayashi M, Zhang H, Jeong SH, Wang X, et al. Association of AR-V7 and Prostate-Specific Antigen RNA Levels in Blood with Efficacy of Abiraterone Acetate and Enzalutamide Treatment in Men with Prostate Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(3):726–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kong D, Sethi S, Li Y, Chen W, Sakr WA, Heath E, et al. Androgen receptor splice variants contribute to prostate cancer aggressiveness through induction of EMT and expression of stem cell marker genes. The Prostate. 2015;75(2):161–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shafi AA, Putluri V, Arnold JM, Tsouko E, Maity S, Roberts JM, et al. Differential regulation of metabolic pathways by androgen receptor (AR) and its constitutively active splice variant, AR-V7, in prostate cancer cells. Oncotarget. 2015;6(31):31997–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Karacosta LG, Foster BA, Azabdaftari G, Feliciano DM, Edelman AM. A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and the androgen receptor in prostate cancer progression. The Journal of biological chemistry. 2012;287(29):24832–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vaz CV, Marques R, Alves MG, Oliveira PF, Cavaco JE, Maia CJ, et al. Androgens enhance the glycolytic metabolism and lactate export in prostate cancer cells by modulating the expression of GLUT1, GLUT3, PFK, LDH and MCT4 genes. Journal of cancer research and clinical oncology. 2016;142(1):5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. The EMBO journal. 2011;30(13):2719–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. The Journal of clinical investigation. 2001;108(8):1167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zannella VE, Dal Pra A, Muaddi H, McKee TD, Stapleton S, Sykes J, et al. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(24):6741–50. [DOI] [PubMed] [Google Scholar]
- 81.Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer research. 2011;71(9):3196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ippolito L, Marini A, Cavallini L, Morandi A, Pietrovito L, Pintus G, et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget. 2016;7(38):61890–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fendt SM, Bell EL, Keibler MA, Davidson SM, Wirth GJ, Fiske B, et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer research. 2013;73(14):4429–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hirsch HA, Iliopoulos D, Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(3):972–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Akinyeke T, Matsumura S, Wang X, Wu Y, Schalfer ED, Saxena A, et al. Metformin targets c-MYC oncogene to prevent prostate cancer. Carcinogenesis. 2013;34(12):2823–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nguyen HG, Yang JC, Kung HJ, Shi XB, Tilki D, Lara PN Jr., et al. Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene. 2014;33(36):4521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang Y, Liu G, Tong D, Parmar H, Hasenmayer D, Yuan W, et al. Metformin represses androgen-dependent and androgen-independent prostate cancers by targeting androgen receptor. The Prostate. 2015;75(11):1187–96. [DOI] [PubMed] [Google Scholar]
- 88.Kaltz-Wittmer C, Klenk U, Glaessgen A, Aust DE, Diebold J, Lohrs U, et al. FISH analysis of gene aberrations (MYC, CCND1, ERBB2, RB, and AR) in advanced prostatic carcinomas before and after androgen deprivation therapy. Laboratory investigation; a journal of technical methods and pathology. 2000;80(9):1455–64. [DOI] [PubMed] [Google Scholar]
- 89.Barfeld SJ, Urbanucci A, Itkonen HM, Fazli L, Hicks JL, Thiede B, et al. c-Myc Antagonises the Transcriptional Activity of the Androgen Receptor in Prostate Cancer Affecting Key Gene Networks. EBioMedicine. 2017;18:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pellakuru LG, Iwata T, Gurel B, Schultz D, Hicks J, Bethel C, et al. Global levels of H3K27me3 track with differentiation in vivo and are deregulated by MYC in prostate cancer. The American journal of pathology. 2012;181(2):560–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang D, Park D, Zhong Y, Lu Y, Rycaj K, Gong S, et al. Stem cell and neurogenic gene-expression profiles link prostate basal cells to aggressive prostate cancer. Nature communications. 2016;7:10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439(7078):811–6. [DOI] [PubMed] [Google Scholar]
- 93.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466(7310):1129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Peitzsch C, Cojoc M, Hein L, Kurth I, Mbert K, Trautmann F, et al. An epigenetic reprogramming strategy to re-sensitize radioresistant prostate cancer cells. Cancer research. 2016. [DOI] [PubMed] [Google Scholar]
- 95.Celia-Terrassa T, Meca-Cortes O, Mateo F, Martinez de Paz A, Rubio N, Arnal-Estape A, et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. The Journal of clinical investigation. 2012;122(5):1849–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Aguilar E, Marin de Mas I, Zodda E, Marin S, Morrish F, Selivanov V, et al. Metabolic Reprogramming and Dependencies Associated with Epithelial Cancer Stem Cells Independent of the Epithelial-Mesenchymal Transition Program. Stem cells. 2016;34(5):1163–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Heller G, Fizazi K, McCormack R, Molina A, MacLean D, Webb IJ, et al. The Added Value of Circulating Tumor Cell Enumeration to Standard Markers in Assessing Prognosis in a Metastatic Castration-Resistant Prostate Cancer Population. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(8):1967–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li X, Han G, Li X, Kan Q, Fan Z, Li Y, et al. Mitochondrial pyruvate carrier function determines cell stemness and metabolic reprogramming in cancer cells. Oncotarget. 2017;8(28):46363–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhong Y, Li X, Ji Y, Li X, Li Y, Yu D, et al. Pyruvate dehydrogenase expression is negatively associated with cell stemness and worse clinical outcome in prostate cancers. Oncotarget. 2017;8(8):13344–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166(1):21–45. [DOI] [PubMed] [Google Scholar]
- 101.Montanari M, Rossetti S, Cavaliere C, D’Aniello C, Malzone MG, Vanacore D, et al. Epithelial-mesenchymal transition in prostate cancer: an overview. Oncotarget. 2017;8(21):35376–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lo UG, Lee CF, Lee MS, Hsieh JT. The Role and Mechanism of Epithelial-to-Mesenchymal Transition in Prostate Cancer Progression. International journal of molecular sciences. 2017;18(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ammirante M, Shalapour S, Kang Y, Jamieson CA, Karin M. Tissue injury and hypoxia promote malignant progression of prostate cancer by inducing CXCL13 expression in tumor myofibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(41):14776–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sun Y, Wang BE, Leong KG, Yue P, Li L, Jhunjhunwala S, et al. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer research. 2012;72(2):527–36. [DOI] [PubMed] [Google Scholar]
- 105.Williams LV, Veliceasa D, Vinokour E, Volpert OV. miR-200b inhibits prostate cancer EMT, growth and metastasis. PloS one. 2013;8(12):e83991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu Q, Tong D, Liu G, Xu J, Do K, Geary K, et al. Metformin reverses prostate cancer resistance to enzalutamide by targeting TGF-beta1/STAT3 axis-regulated EMT. Cell death & disease. 2017;8(8):e3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Levesque C, Nelson PS. Cellular Constituents of the Prostate Stroma: Key Contributors to Prostate Cancer Progression and Therapy Resistance. Cold Spring Harbor perspectives in medicine. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Riemann A, Schneider B, Gundel D, Stock C, Gekle M, Thews O. Acidosis Promotes Metastasis Formation by Enhancing Tumor Cell Motility. Advances in experimental medicine and biology. 2016;876:215–20. [DOI] [PubMed] [Google Scholar]
- 109.Byrne NM, Nesbitt H, Ming L, McKeown SR, Worthington J, McKenna DJ. Androgen deprivation in LNCaP prostate tumour xenografts induces vascular changes and hypoxic stress, resulting in promotion of epithelial-to-mesenchymal transition. British journal of cancer. 2016;114(6):659–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vila IK, Yao Y, Kim G, Xia W, Kim H, Kim SJ, et al. A UBE2O-AMPKalpha2 Axis that Promotes Tumor Initiation and Progression Offers Opportunities for Therapy. Cancer cell. 2017;31(2):208–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Diedrich JD, Rajagurubandara E, Herroon MK, Mahapatra G, Huttemann M, Podgorski I. Bone marrow adipocytes promote the Warburg phenotype in metastatic prostate tumors via HIF-1alpha activation. Oncotarget. 2016;7(40):64854–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell stem cell. 2015;16(3):225–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tuxhorn JA, Ayala GE, Smith MJ, Smith VC, Dang TD, Rowley DR. Reactive stroma in human prostate cancer: induction of myofibroblast phenotype and extracellular matrix remodeling. Clinical cancer research : an official journal of the American Association for Cancer Research. 2002;8(9):2912–23. [PubMed] [Google Scholar]
- 114.Lawson DA, Zong Y, Memarzadeh S, Xin L, Huang J, Witte ON. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(6):2610–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Taylor RA, Toivanen R, Frydenberg M, Pedersen J, Harewood L, Australian Prostate Cancer B, et al. Human epithelial basal cells are cells of origin of prostate cancer, independent of CD133 status. Stem cells. 2012;30(6):1087–96. [DOI] [PubMed] [Google Scholar]
- 116.Chan JS, Tan MJ, Sng MK, Teo Z, Phua T, Choo CC, et al. Cancer-associated fibroblasts enact field cancerization by promoting extratumoral oxidative stress. Cell death & disease. 2017;8(1):e2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell cycle. 2010;9(16):3256–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pistore C, Giannoni E, Colangelo T, Rizzo F, Magnani E, Muccillo L, et al. DNA methylation variations are required for epithelial-to-mesenchymal transition induced by cancer-associated fibroblasts in prostate cancer cells. Oncogene. 2017;36(40):5551–66. [DOI] [PubMed] [Google Scholar]
- 119.Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer research. 2010;70(17):6945–56. [DOI] [PubMed] [Google Scholar]
- 120.Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer research. 2012;72(19):5130–40. [DOI] [PubMed] [Google Scholar]
- 121.Choi SY, Collins CC, Gout PW, Wang Y. Cancer-generated lactic acid: a regulatory, immunosuppressive metabolite? The Journal of pathology. 2013;230(4):350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Giannoni E, Bianchini F, Calorini L, Chiarugi P. Cancer associated fibroblasts exploit reactive oxygen species through a proinflammatory signature leading to epithelial mesenchymal transition and stemness. Antioxidants & redox signaling. 2011;14(12):2361–71. [DOI] [PubMed] [Google Scholar]
- 123.Birnie R, Bryce SD, Roome C, Dussupt V, Droop A, Lang SH, et al. Gene expression profiling of human prostate cancer stem cells reveals a pro-inflammatory phenotype and the importance of extracellular matrix interactions. Genome biology. 2008;9(5):R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu X, Grogan TR, Hieronymus H, Hashimoto T, Mottahedeh J, Cheng D, et al. Low CD38 Identifies Progenitor-like Inflammation-Associated Luminal Cells that Can Initiate Human Prostate Cancer and Predict Poor Outcome. Cell reports. 2016;17(10):2596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schroeder A, Herrmann A, Cherryholmes G, Kowolik C, Buettner R, Pal S, et al. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer research. 2014;74(4):1227–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Moreira D, Zhang Q, Hossain DM, Nechaev S, Li H, Kowolik CM, et al. TLR9 signaling through NF-kappaB/RELA and STAT3 promotes tumor-propagating potential of prostate cancer cells. Oncotarget. 2015;6(19):17302–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Won H, Moreira D, Gao C, Duttagupta P, Zhao X, Manuel E, et al. TLR9 expression and secretion of LIF by prostate cancer cells stimulates accumulation and activity of polymorphonuclear MDSCs. Journal of leukocyte biology. 2017;102(2):423–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hossain DM, Pal SK, Moreira D, Duttagupta P, Zhang Q, Won H, et al. TLR9-Targeted STAT3 Silencing Abrogates Immunosuppressive Activity of Myeloid-Derived Suppressor Cells from Prostate Cancer Patients. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21(16):3771–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Adisetiyo H, Liang M, Liao CP, Jeong JH, Cohen MB, Roy-Burman P, et al. Dependence of castration-resistant prostate cancer (CRPC) stem cells on CRPC-associated fibroblasts. Journal of cellular physiology. 2014;229(9):1170–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liao CP, Adisetiyo H, Liang M, Roy-Burman P. Cancer-associated fibroblasts enhance the gland-forming capability of prostate cancer stem cells. Cancer research. 2010;70(18):7294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Barcellos-de-Souza P, Comito G, Pons-Segura C, Taddei ML, Gori V, Becherucci V, et al. Mesenchymal Stem Cells are Recruited and Activated into Carcinoma-Associated Fibroblasts by Prostate Cancer Microenvironment-Derived TGF-beta1. Stem cells. 2016;34(10):2536–47. [DOI] [PubMed] [Google Scholar]
- 132.Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene. 2014;33(19):2423–31. [DOI] [PubMed] [Google Scholar]
- 133.Garg R, Blando JM, Perez CJ, Abba MC, Benavides F, Kazanietz MG. Protein Kinase C Epsilon Cooperates with PTEN Loss for Prostate Tumorigenesis through the CXCL13-CXCR5 Pathway. Cell reports. 2017;19(2):375–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Voog J, Jones DL. Stem cells and the niche: a dynamic duo. Cell stem cell. 2010;6(2):103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Weckermann D, Polzer B, Ragg T, Blana A, Schlimok G, Arnholdt H, et al. Perioperative activation of disseminated tumor cells in bone marrow of patients with prostate cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27(10):1549–56. [DOI] [PubMed] [Google Scholar]
- 136.Peitzsch C, Tyutyunnykova A, Pantel K, Dubrovska A. Cancer stem cells: The root of tumor recurrence and metastases. Semin Cancer Biol. 2017;44:10–24. [DOI] [PubMed] [Google Scholar]
- 137.Mehlen P, Puisieux A. Metastasis: a question of life or death. Nature reviews Cancer. 2006;6(6):449–58. [DOI] [PubMed] [Google Scholar]
- 138.Msaouel P, Pissimissis N, Halapas A, Koutsilieris M. Mechanisms of bone metastasis in prostate cancer: clinical implications. Best practice & research Clinical endocrinology & metabolism. 2008;22(2):341–55. [DOI] [PubMed] [Google Scholar]
- 139.Sturge J, Caley MP, Waxman J. Bone metastasis in prostate cancer: emerging therapeutic strategies. Nature reviews Clinical oncology. 2011;8(6):357–68. [DOI] [PubMed] [Google Scholar]
- 140.Vela I, Gregory L, Gardiner EM, Clements JA, Nicol DL. Bone and prostate cancer cell interactions in metastatic prostate cancer. BJU international. 2007;99(4):735–42. [DOI] [PubMed] [Google Scholar]
- 141.Logothetis CJ, Lin SH. Osteoblasts in prostate cancer metastasis to bone. Nature reviews Cancer. 2005;5(1):21–8. [DOI] [PubMed] [Google Scholar]
- 142.Coxon JP, Oades GM, Colston KW, Kirby RS. Advances in the use of bisphosphonates in the prostate cancer setting. Prostate cancer and prostatic diseases. 2004;7(2):99–104. [DOI] [PubMed] [Google Scholar]
- 143.Holen I, Croucher PI, Hamdy FC, Eaton CL. Osteoprotegerin (OPG) is a survival factor for human prostate cancer cells. Cancer research. 2002;62(6):1619–23. [PubMed] [Google Scholar]
- 144.Nadiminty N, Lou W, Lee SO, Mehraein-Ghomi F, Kirk JS, Conroy JM, et al. Prostate-specific antigen modulates genes involved in bone remodeling and induces osteoblast differentiation of human osteosarcoma cell line SaOS-2. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12(5):1420–30. [DOI] [PubMed] [Google Scholar]
- 145.Bernacki KD, Fields KL, Roh MH. The utility of PSMA and PSA immunohistochemistry in the cytologic diagnosis of metastatic prostate carcinoma. Diagnostic cytopathology. 2014;42(7):570–5. [DOI] [PubMed] [Google Scholar]
- 146.Sheridan T, Herawi M, Epstein JI, Illei PB. The role of P501S and PSA in the diagnosis of metastatic adenocarcinoma of the prostate. The American journal of surgical pathology. 2007;31(9):1351–5. [DOI] [PubMed] [Google Scholar]
- 147.Shoag J, Barbieri CE. Clinical variability and molecular heterogeneity in prostate cancer. Asian journal of andrology. 2016;18(4):543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mateo J, Boysen G, Barbieri CE, Bryant HE, Castro E, Nelson PS, et al. DNA Repair in Prostate Cancer: Biology and Clinical Implications. European urology. 2017;71(3):417–25. [DOI] [PubMed] [Google Scholar]
- 149.Taylor RA, Fraser M, Livingstone J, Espiritu SM, Thorne H, Huang V, et al. Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nature communications. 2017;8:13671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Morgan TM, Lange PH, Porter MP, Lin DW, Ellis WJ, Gallaher IS, et al. Disseminated tumor cells in prostate cancer patients after radical prostatectomy and without evidence of disease predicts biochemical recurrence. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(2):677–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lam HM, Vessella RL, Morrissey C. The role of the microenvironment-dormant prostate disseminated tumor cells in the bone marrow. Drug discovery today Technologies. 2014;11:41–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Day KC, Lorenzatti Hiles G, Kozminsky M, Dawsey SJ, Paul A, Broses LJ, et al. HER2 and EGFR Overexpression Support Metastatic Progression of Prostate Cancer to Bone. Cancer research. 2017;77(1):74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Karlsson T, Lundholm M, Widmark A, Persson E. Tumor Cell-Derived Exosomes from the Prostate Cancer Cell Line TRAMP-C1 Impair Osteoclast Formation and Differentiation. PloS one. 2016;11(11):e0166284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu W, Yin B, Wang X, Yu P, Duan X, Liu C, et al. Circulating tumor cells in prostate cancer: Precision diagnosis and therapy. Oncology letters. 2017;14(2):1223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mazzoleni S, Jachetti E, Morosini S, Grioni M, Piras IS, Pala M, et al. Gene signatures distinguish stage-specific prostate cancer stem cells isolated from transgenic adenocarcinoma of the mouse prostate lesions and predict the malignancy of human tumors. Stem cells translational medicine. 2013;2(9):678–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lorente D, Olmos D, Mateo J, Bianchini D, Seed G, Fleisher M, et al. Decline in Circulating Tumor Cell Count and Treatment Outcome in Advanced Prostate Cancer. European urology. 2016;70(6):985–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Erho N, Crisan A, Vergara IA, Mitra AP, Ghadessi M, Buerki C, et al. Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PloS one. 2013;8(6):e66855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. The Journal of clinical investigation. 2005;115(6):1503–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Holcomb IN, Grove DI, Kinnunen M, Friedman CL, Gallaher IS, Morgan TM, et al. Genomic alterations indicate tumor origin and varied metastatic potential of disseminated cells from prostate cancer patients. Cancer research. 2008;68(14):5599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jiang R, Lu YT, Ho H, Li B, Chen JF, Lin M, et al. A comparison of isolated circulating tumor cells and tissue biopsies using whole-genome sequencing in prostate cancer. Oncotarget. 2015;6(42):44781–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Punnoose EA, Ferraldeschi R, Szafer-Glusman E, Tucker EK, Mohan S, Flohr P, et al. PTEN loss in circulating tumour cells correlates with PTEN loss in fresh tumour tissue from castration-resistant prostate cancer patients. British journal of cancer. 2015;113(8):1225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.You S, Knudsen BS, Erho N, Alshalalfa M, Takhar M, Al-Deen Ashab H, et al. Integrated Classification of Prostate Cancer Reveals a Novel Luminal Subtype with Poor Outcome. Cancer research. 2016;76(17):4948–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pavese JM, Bergan RC. Circulating tumor cells exhibit a biologically aggressive cancer phenotype accompanied by selective resistance to chemotherapy. Cancer letters. 2014;352(2):179–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira SM, Garcia-Echeverria C, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(1):268–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shahi P, Seethammagari MR, Valdez JM, Xin L, Spencer DM. Wnt and Notch pathways have interrelated opposing roles on prostate progenitor cell proliferation and differentiation. Stem cells. 2011;29(4):678–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhang K, Guo Y, Wang X, Zhao H, Ji Z, Cheng C, et al. WNT/beta-Catenin Directs Self-Renewal Symmetric Cell Division of hTERT(high) Prostate Cancer Stem Cells. Cancer research. 2017;77(9):2534–47. [DOI] [PubMed] [Google Scholar]
- 167.Peitzsch C, Cojoc M, Hein L, Kurth I, Mabert K, Trautmann F, et al. An Epigenetic Reprogramming Strategy to Resensitize Radioresistant Prostate Cancer Cells. Cancer research. 2016;76(9):2637–51. [DOI] [PubMed] [Google Scholar]
- 168.Yun EJ, Zhou J, Lin CJ, Hernandez E, Fazli L, Gleave M, et al. Targeting Cancer Stem Cells in Castration-Resistant Prostate Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22(3):670–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Dubrovska A, Elliott J, Salamone RJ, Kim S, Aimone LJ, Walker JR, et al. Combination therapy targeting both tumor-initiating and differentiated cell populations in prostate carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(23):5692–702. [DOI] [PubMed] [Google Scholar]
- 170.Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nature medicine. 2011;17(2):211–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Liu C, Liu R, Zhang D, Deng Q, Liu B, Chao HP, et al. MicroRNA-141 suppresses prostate cancer stem cells and metastasis by targeting a cohort of pro-metastasis genes. Nature communications. 2017;8:14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Albino D, Civenni G, Dallavalle C, Roos M, Jahns H, Curti L, et al. Activation of the Lin28/let-7 Axis by Loss of ESE3/EHF Promotes a Tumorigenic and Stem-like Phenotype in Prostate Cancer. Cancer research. 2016;76(12):3629–43. [DOI] [PubMed] [Google Scholar]
- 173.Wen D, Peng Y, Lin F, Singh RK, Mahato RI. Micellar Delivery of miR-34a Modulator Rubone and Paclitaxel in Resistant Prostate Cancer. Cancer research. 2017;77(12):3244–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Peng Y, Prater AR, Deutscher SL. Targeting aggressive prostate cancer-associated CD44v6 using phage display selected peptides. Oncotarget. 2017;8(49):86747–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Pan Q, Li Q, Liu S, Ning N, Zhang X, Xu Y, et al. Concise Review: Targeting Cancer Stem Cells Using Immunologic Approaches. Stem cells. 2015;33(7):2085–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tran CP, Lin C, Yamashiro J, Reiter RE. Prostate stem cell antigen is a marker of late intermediate prostate epithelial cells. Molecular cancer research : MCR. 2002;1(2):113–21. [PubMed] [Google Scholar]
- 177.Jachetti E, Mazzoleni S, Grioni M, Ricupito A, Brambillasca C, Generoso L, et al. Prostate cancer stem cells are targets of both innate and adaptive immunity and elicit tumor-specific immune responses. Oncoimmunology. 2013;2(5):e24520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhao Z, Zeng G, Ma W, Ou L, Liang Y. Peripheral blood reverse transcription PCR assay for prostate stem cell antigen correlates with androgen-independent progression in advanced prostate cancer. International journal of cancer. 2012;131(4):902–10. [DOI] [PubMed] [Google Scholar]
- 179.Liu L, Li E, Luo L, Zhao S, Li F, Wang J, et al. PSCA regulates IL-6 expression through p38/NF-kappaB signaling in prostate cancer. The Prostate. 2017;77(14):1389–400. [DOI] [PubMed] [Google Scholar]
- 180.Kubler H, Scheel B, Gnad-Vogt U, Miller K, Schultze-Seemann W, Vom Dorp F, et al. Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: a first-in-man phase I/IIa study. Journal for immunotherapy of cancer. 2015;3:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Morris MJ, Eisenberger MA, Pili R, Denmeade SR, Rathkopf D, Slovin SF, et al. A phase I/IIA study of AGS-PSCA for castration-resistant prostate cancer. Annals of oncology : official journal of the European Society for Medical Oncology. 2012;23(10):2714–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hu Y, Lu L, Xia Y, Chen X, Chang AE, Hollingsworth RE, et al. Therapeutic Efficacy of Cancer Stem Cell Vaccines in the Adjuvant Setting. Cancer research. 2016;76(16):4661–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ning N, Pan Q, Zheng F, Teitz-Tennenbaum S, Egenti M, Yet J, et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer research. 2012;72(7):1853–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hara T, Iwadate M, Tachibana K, Waguri S, Takenoshita S, Hamada N. Metastasis of breast cancer cells to the bone, lung, and lymph nodes promotes resistance to ionizing radiation. Strahlentherapie und Onkologie : Organ der Deutschen Rontgengesellschaft [et al. ]. 2017. [DOI] [PubMed] [Google Scholar]
- 185.Zakaria R, Platt-Higgins A, Rathi N, Crooks D, Brodbelt A, Chavredakis E, et al. Metastasis-inducing proteins are widely expressed in human brain metastases and associated with intracranial progression and radiation response. British journal of cancer. 2016;114(10):1101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Rofstad EK. Radiation sensitivity in vitro of primary tumors and metastatic lesions of malignant melanoma. Cancer research. 1992;52(16):4453–7. [PubMed] [Google Scholar]
- 187.Trautmann F, Cojoc M, Kurth I, Melin N, Bouchez LC, Dubrovska A, et al. CXCR4 as biomarker for radioresistant cancer stem cells. International journal of radiation biology. 2014;90(8):687–99. [DOI] [PubMed] [Google Scholar]
- 188.Sun YX, Schneider A, Jung Y, Wang J, Dai J, Wang J, et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2005;20(2):318–29. [DOI] [PubMed] [Google Scholar]
- 189.Nandana S, Tripathi M, Duan P, Chu CY, Mishra R, Liu C, et al. Bone Metastasis of Prostate Cancer Can Be Therapeutically Targeted at the TBX2-WNT Signaling Axis. Cancer research. 2017;77(6):1331–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Pelger RC, van der Pluijm G. The aldehyde dehydrogenase enzyme 7A1 is functionally involved in prostate cancer bone metastasis. Clinical & experimental metastasis. 2011;28(7):615–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Singh S, Brocker C, Koppaka V, Chen Y, Jackson BC, Matsumoto A, et al. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free radical biology & medicine. 2013;56:89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Le Magnen C, Bubendorf L, Rentsch CA, Mengus C, Gsponer J, Zellweger T, et al. Characterization and clinical relevance of ALDHbright populations in prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(19):5361–71. [DOI] [PubMed] [Google Scholar]
- 193.Sottnik JL, Hall CL, Zhang J, Keller ET. Wnt and Wnt inhibitors in bone metastasis. BoneKEy reports. 2012;1:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hall CL, Kang S, MacDougald OA, Keller ET. Role of Wnts in prostate cancer bone metastases. Journal of cellular biochemistry. 2006;97(4):661–72. [DOI] [PubMed] [Google Scholar]
- 195.Murillo-Garzon V, Kypta R. WNT signalling in prostate cancer. Nature reviews Urology. 2017;14(11):683–96. [DOI] [PubMed] [Google Scholar]
- 196.Holland JD, Gyorffy B, Vogel R, Eckert K, Valenti G, Fang L, et al. Combined Wnt/beta-catenin, Met, and CXCL12/CXCR4 signals characterize basal breast cancer and predict disease outcome. Cell reports. 2013;5(5):1214–27. [DOI] [PubMed] [Google Scholar]
- 197.Aman A, Piotrowski T. Wnt/beta-catenin and Fgf signaling control collective cell migration by restricting chemokine receptor expression. Developmental cell. 2008;15(5):749–61. [DOI] [PubMed] [Google Scholar]
- 198.Hu TH, Yao Y, Yu S, Han LL, Wang WJ, Guo H, et al. SDF-1/CXCR4 promotes epithelial-mesenchymal transition and progression of colorectal cancer by activation of the Wnt/beta-catenin signaling pathway. Cancer letters. 2014;354(2):417–26. [DOI] [PubMed] [Google Scholar]
- 199.Zhao S, Wang J, Qin C. Blockade of CXCL12/CXCR4 signaling inhibits intrahepatic cholangiocarcinoma progression and metastasis via inactivation of canonical Wnt pathway. Journal of experimental & clinical cancer research : CR. 2014;33:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Corey E, Brown LG, Quinn JE, Poot M, Roudier MP, Higano CS, et al. Zoledronic acid exhibits inhibitory effects on osteoblastic and osteolytic metastases of prostate cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2003;9(1):295–306. [PubMed] [Google Scholar]
- 201.Tuomela JM, Valta MP, Vaananen K, Harkonen PL. Alendronate decreases orthotopic PC-3 prostate tumor growth and metastasis to prostate-draining lymph nodes in nude mice. BMC cancer. 2008;8:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Thiele S, Rauner M, Goettsch C, Rachner TD, Benad P, Fuessel S, et al. Expression profile of WNT molecules in prostate cancer and its regulation by aminobisphosphonates. Journal of cellular biochemistry. 2011;112(6):1593–600. [DOI] [PubMed] [Google Scholar]
- 203.Ao M, Franco OE, Park D, Raman D, Williams K, Hayward SW. Cross-talk between paracrine-acting cytokine and chemokine pathways promotes malignancy in benign human prostatic epithelium. Cancer research. 2007;67(9):4244–53. [DOI] [PubMed] [Google Scholar]
- 204.Fournier PG, Juarez P, Jiang G, Clines GA, Niewolna M, Kim HS, et al. The TGF-beta Signaling Regulator PMEPA1 Suppresses Prostate Cancer Metastases to Bone. Cancer cell. 2015;27(6):809–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Caley MP, Kogianni G, Adamarek A, Gronau JH, Rodriguez-Teja M, Fonseca AV, et al. TGFbeta1-Endo180-dependent collagen deposition is dysregulated at the tumour-stromal interface in bone metastasis. The Journal of pathology. 2012;226(5):775–83. [DOI] [PubMed] [Google Scholar]
- 206.Goel HL, Li J, Kogan S, Languino LR. Integrins in prostate cancer progression. Endocrine-related cancer. 2008;15(3):657–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Harryman WL, Hinton JP, Rubenstein CP, Singh P, Nagle RB, Parker SJ, et al. The Cohesive Metastasis Phenotype in Human Prostate Cancer. Biochimica et biophysica acta. 2016;1866(2):221–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Liu C, Kelnar K, Vlassov AV, Brown D, Wang J, Tang DG. Distinct microRNA expression profiles in prostate cancer stem/progenitor cells and tumor-suppressive functions of let-7. Cancer research. 2012;72(13):3393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lu H, Wang T, Li J, Fedele C, Liu Q, Zhang J, et al. alphavbeta6 Integrin Promotes Castrate-Resistant Prostate Cancer through JNK1-Mediated Activation of Androgen Receptor. Cancer research. 2016;76(17):5163–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Goel HL, Sayeed A, Breen M, Zarif MJ, Garlick DS, Leav I, et al. beta1 integrins mediate resistance to ionizing radiation in vivo by inhibiting c-Jun amino terminal kinase 1. Journal of cellular physiology. 2013;228(7):1601–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Krause M, Dubrovska A, Linge A, Baumann M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Advanced drug delivery reviews. 2017;109:63–73. [DOI] [PubMed] [Google Scholar]
- 212.Butof R, Dubrovska A, Baumann M. Clinical perspectives of cancer stem cell research in radiation oncology. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology. 2013;108(3):388–96. [DOI] [PubMed] [Google Scholar]
- 213.Goldstein AS, Lawson DA, Cheng D, Sun W, Garraway IP, Witte ON. Trop2 identifies a subpopulation of murine and human prostate basal cells with stem cell characteristics. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(52):20882–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Antonarakis ES, Carducci MA, Eisenberger MA. Novel targeted therapeutics for metastatic castration-resistant prostate cancer. Cancer letters. 2010;291(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells-perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer research. 2006;66(19):9339–44. [DOI] [PubMed] [Google Scholar]
- 216.Hanrahan K, O’Neill A, Prencipe M, Bugler J, Murphy L, Fabre A, et al. The role of epithelial-mesenchymal transition drivers ZEB1 and ZEB2 in mediating docetaxel-resistant prostate cancer. Molecular oncology. 2017;11(3):251–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Bortolomai I, Canevari S, Facetti I, De Cecco L, Castellano G, Zacchetti A, et al. Tumor initiating cells: development and critical characterization of a model derived from the A431 carcinoma cell line forming spheres in suspension. Cell cycle. 2010;9(6):1194–206. [DOI] [PubMed] [Google Scholar]
- 218.Li D, Fu Z, Chen R, Zhao X, Zhou Y, Zeng B, et al. Inhibition of glutamine metabolism counteracts pancreatic cancer stem cell features and sensitizes cells to radiotherapy. Oncotarget. 2015;6(31):31151–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell of origin for human prostate cancer. Science. 2010;329(5991):568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Germann M, Wetterwald A, Guzman-Ramirez N, van der Pluijm G, Culig Z, Cecchini MG, et al. Stem-like cells with luminal progenitor phenotype survive castration in human prostate cancer. Stem cells. 2012;30(6):1076–86. [DOI] [PubMed] [Google Scholar]
- 221.Podolak J, Eilers K, Newby T, Slottke R, Tucker E, Olson SB, et al. Androgen receptor amplification is concordant between circulating tumor cells and biopsies from men undergoing treatment for metastatic castration resistant prostate cancer. Oncotarget. 2017;8(42):71447–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Crespo M, van Dalum G, Ferraldeschi R, Zafeiriou Z, Sideris S, Lorente D, et al. Androgen receptor expression in circulating tumour cells from castration-resistant prostate cancer patients treated with novel endocrine agents. British journal of cancer. 2015;112(7):1166–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Nakazawa M, et al. Androgen Receptor Splice Variant 7 and Efficacy of Taxane Chemotherapy in Patients With Metastatic Castration-Resistant Prostate Cancer. JAMA Oncol. 2015;1(5):582–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Scher HI, Graf RP, Schreiber NA, McLaughlin B, Jendrisak A, Wang Y, et al. Phenotypic Heterogeneity of Circulating Tumor Cells Informs Clinical Decisions between AR Signaling Inhibitors and Taxanes in Metastatic Prostate Cancer. Cancer research. 2017;77(20):5687–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Scher HI, Lu D, Schreiber NA, Louw J, Graf RP, Vargas HA, et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016;2(11):1441–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Carlsson A, Kuhn P, Luttgen MS, Dizon KK, Troncoso P, Corn PG, et al. Paired High-Content Analysis of Prostate Cancer Cells in Bone Marrow and Blood Characterizes Increased Androgen Receptor Expression in Tumor Cell Clusters. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(7):1722–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Shahriari K, Shen F, Worrede-Mahdi A, Liu Q, Gong Y, Garcia FU, et al. Cooperation among heterogeneous prostate cancer cells in the bone metastatic niche. Oncogene. 2017;36(20):2846–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer cell. 2017;32(4):474–89 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.English HF, Santen RJ, Isaacs JT. Response of glandular versus basal rat ventral prostatic epithelial cells to androgen withdrawal and replacement. The Prostate. 1987;11(3):229–42. [DOI] [PubMed] [Google Scholar]
- 230.Miyamoto DT, Lee RJ, Stott SL, Ting DT, Wittner BS, Ulman M, et al. Androgen receptor signaling in circulating tumor cells as a marker of hormonally responsive prostate cancer. Cancer discovery. 2012;2(11):995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Leon-Mateos L, Casas H, Abalo A, Vieito M, Abreu M, Anido U, et al. Improving circulating tumor cells enumeration and characterization to predict outcome in first line chemotherapy mCRPC patients. Oncotarget. 2017;8(33):54708–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Nouri M, Caradec J, Lubik AA, Li N, Hollier BG, Takhar M, et al. Therapy-induced developmental reprogramming of prostate cancer cells and acquired therapy resistance. Oncotarget. 2017;8(12):18949–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Markou A, Lazaridou M, Paraskevopoulos P, Chen S, Swierczewska M, Budna J, et al. Multiplex Gene Expression Profiling of In Vivo Isolated Circulating Tumor Cells in High-Risk Prostate Cancer Patients. Clinical chemistry. 2017. [DOI] [PubMed] [Google Scholar]
- 234.Guzvic M, Braun B, Ganzer R, Burger M, Nerlich M, Winkler S, et al. Combined genome and transcriptome analysis of single disseminated cancer cells from bone marrow of prostate cancer patients reveals unexpected transcriptomes. Cancer research. 2014;74(24):7383–94. [DOI] [PubMed] [Google Scholar]
- 235.Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell. 2014;159(1):163–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wang X, Kruithof-de Julio M, Economides KD, Walker D, Yu H, Halili MV, et al. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009;461(7263):495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Satelli A, Batth I, Brownlee Z, Mitra A, Zhou S, Noh H, et al. EMT circulating tumor cells detected by cell-surface vimentin are associated with prostate cancer progression. Oncotarget. 2017;8(30):49329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Xu L, Mao X, Guo T, Chan PY, Shaw G, Hines J, et al. The Novel Association of Circulating Tumor Cells and Circulating Megakaryocytes with Prostate Cancer Prognosis. Clinical cancer research : an official journal of the American Association for Cancer Research. 2017;23(17):5112–22. [DOI] [PubMed] [Google Scholar]
- 239.Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. The Journal of clinical investigation. 2011;121(4):1298–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159(1):176–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Shiozawa Y, Berry JE, Eber MR, Jung Y, Yumoto K, Cackowski FC, et al. The marrow niche controls the cancer stem cell phenotype of disseminated prostate cancer. Oncotarget. 2016;7(27):41217–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Ricci E, Mattei E, Dumontet C, Eaton CL, Hamdy F, van der Pluije G, et al. Increased expression of putative cancer stem cell markers in the bone marrow of prostate cancer patients is associated with bone metastasis progression. The Prostate. 2013;73(16):1738–46. [DOI] [PubMed] [Google Scholar]
- 243.Morin F, Beauregard JM, Bergeron M, Nguile Makao M, Lacombe L, Fradet V, et al. Metabolic Imaging of Prostate Cancer Reveals Intrapatient Intermetastasis Response Heterogeneity to Systemic Therapy. European urology focus. 2017. [DOI] [PubMed] [Google Scholar]
- 244.Gundem G, Van Loo P, Kremeyer B, Alexandrov LB, Tubio JMC, Papaemmanuil E, et al. The evolutionary history of lethal metastatic prostate cancer. Nature. 2015;520(7547):353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Margel D, Urbach DR, Lipscombe LL, Bell CM, Kulkarni G, Austin PC, et al. Metformin use and all-cause and prostate cancer-specific mortality among men with diabetes. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(25):3069–75. [DOI] [PubMed] [Google Scholar]
- 246.Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature. 2017;541(7637):359–64. [DOI] [PubMed] [Google Scholar]
- 247.Pixberg CF, Raba K, Muller F, Behrens B, Honisch E, Niederacher D, et al. Analysis of DNA methylation in single circulating tumor cells. Oncogene. 2017;36(23):3223–31. [DOI] [PubMed] [Google Scholar]
- 248.Stott SL, Lee RJ, Nagrath S, Yu M, Miyamoto DT, Ulkus L, et al. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Science translational medicine. 2010;2(25):25ra3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Lindsay CR, Le Moulec S, Billiot F, Loriot Y, Ngo-Camus M, Vielh P, et al. Vimentin and Ki67 expression in circulating tumour cells derived from castrate-resistant prostate cancer. BMC cancer. 2016;16:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Decker AM, Jung Y, Cackowski FC, Yumoto K, Wang J, Taichman RS. Sympathetic Signaling Reactivates Quiescent Disseminated Prostate Cancer Cells in the Bone Marrow. Molecular cancer research : MCR. 2017;15(12):1644–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Cooper CS, Eeles R, Wedge DC, Van Loo P, Gundem G, Alexandrov LB, et al. Analysis of the genetic phylogeny of multifocal prostate cancer identifies multiple independent clonal expansions in neoplastic and morphologically normal prostate tissue. Nature genetics. 2015;47(4):367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Lack J, Gillard M, Cam M, Paner GP, VanderWeele DJ. Circulating tumor cells capture disease evolution in advanced prostate cancer. Journal of translational medicine. 2017;15(1):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Cho WJ, Oliveira DS, Najy AJ, Mainetti LE, Aoun HD, Cher ML, et al. Gene expression analysis of bone metastasis and circulating tumor cells from metastatic castrate-resistant prostate cancer patients. Journal of translational medicine. 2016;14:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Kristiansen G, Pilarsky C, Wissmann C, Kaiser S, Bruemmendorf T, Roepcke S, et al. Expression profiling of microdissected matched prostate cancer samples reveals CD166/MEMD and CD24 as new prognostic markers for patient survival. The Journal of pathology. 2005;205(3):359–76. [DOI] [PubMed] [Google Scholar]
- 255.Kerr BA, Miocinovic R, Smith AK, West XZ, Watts KE, Alzayed AW, et al. CD117(+) cells in the circulation are predictive of advanced prostate cancer. Oncotarget. 2015;6(3):1889–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Nakagawa T, Kollmeyer TM, Morlan BW, Anderson SK, Bergstralh EJ, Davis BJ, et al. A tissue biomarker panel predicting systemic progression after PSA recurrence post-definitive prostate cancer therapy. PloS one. 2008;3(5):e2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Tei H, Miyake H, Harada K, Fujisawa M. Expression profile of CD44s, CD44v6, and CD44v10 in localized prostate cancer: effect on prognostic outcomes following radical prostatectomy. Urologic oncology. 2014;32(5):694–700. [DOI] [PubMed] [Google Scholar]