

Abstract
Intracerebral hemorrhage (ICH) or hemorrhagic stroke is a major public health problem with no effective treatment. Given the emerging role of epigenetic mechanisms in the pathophysiology of ICH, we tested the hypothesis that a class 1 histone deacetylase inhibitor (HDACi), Entinostat, attenuates neurodegeneration and improves neurobehavioral outcomes after ICH. To address this, we employed a preclinical mouse model of ICH and Entinostat was administered intraperitoneally one-hour post induction of ICH. Entinostat treatment significantly reduced the number of degenerating neurons and TUNEL-positive cells after ICH in comparison to vehicle-treated controls. Moreover, Entinostat treatment significantly reduced hematoma volume, T2-weighted hemorrhagic lesion volume and improved acute neurological outcomes after ICH. Further, Entinostat significantly reduced the hemin-induced release of proinflammatory cytokines in vitro. Consistently, the expression of proinflammatory microglial/macrophage marker, CD16/32, was remarkably reduced in Entinostat treated group after ICH in comparison to control. Altogether, the data implicate the potential of class 1 HDACi, Entinostat, in improving acute neurological function after ICH warranting further investigation.
Keywords: ICH, epigenetics, Entinostat
1. Introduction
Intracerebral hemorrhage (ICH) is a severe subtype of stroke resulting from spontaneous rupture of blood vessels and subsequent accumulation of blood components in the brain [1]. In comparison to other stroke subtypes, ICH is associated with the highest mortality and morbidity rates. The mortality rate of ICH is approximately 40% at 1 month and approximately 74 % of the survivors remain functionally dependent 12 months after the ictus [2]. Currently, the worldwide incidence of ICH is 2 million cases per year [2], with approximately 120,000 cases per year in the United States [3-5]. However, the incidence of ICH is expected to have doubled by 2050 [6] due to aging and the spreading use of anticoagulants [7]. Despite recent advances in clinical and preclinical research, there is no effective treatment for ICH implicating the need to develop effective therapeutic strategies that could benefit the patient population.
The pathophysiological mechanisms of ICH involve both primary injury and secondary brain injury owing to the mass effect of hematoma and blood component-induced oxidative and inflammatory stress, respectively. In contrast to primary brain damage, the mechanisms underlying secondary brain damage are regarded as amenable to therapeutic intervention [8, 9]. Further, given the enormous potential of blood components in inducing neurotoxicity after ICH [10], strategies to limit the hematoma volume or enhance hematoma resolution are highly needed since it could improve functional outcomes after ICH.
Epigenetic mechanisms such as histone acetylation have been increasingly explored after several neurological disorders [11]. We previously reported that ICH results in the hypoacetylation of histones in the ipsilateral brain region in the acute phase of injury and SAHA or vorinostat, an HDACi (histone deacetylase inhibitor), improved acute neurological outcomes after ICH [12] suggesting a critical role of epigenetic mechanisms in the pathophysiology of ICH [12]. HDACi modulate the activities of histone deacetylases and regulate the acetylation status of both histone and non-histone proteins [13], resulting in the activation and/or repression of gene expression [14, 15]. SAHA is a broad spectrum and first-generation HDACi which inhibits both class 1 and class 2 HDACs (histone deacetylases) [16, 17]. However, it remains unknown which enzyme class contributes to ICH-induced brain damage. To this end, given the emerging role of class I HDACs in brain pathology [11], herein, we question whether Entinostat, a class 1 HDAC inhibitor, attenuates neurodegeneration and improves acute neurological outcomes in a pre-clinical model of ICH.
2. Results
2.1. Entinostat attenuated neurobehavioral deficits after ICH
To test whether Entinostat confers neuroprotection after ICH, a well-established and pre-clinical model of ICH was employed. Entinostat is a class 1 HDAC inhibitor (HDACi) and in a cell-free system, Entinostat inhibited class 1 HDAC enzymes, HDAC1, HDAC2 and HDAC3 with IC50 of 0.46, 1.29 and 1.57 micromoles, respectively (Figure 1). ICH was induced in male CD1 mice by striatal injection of collagenase, as detailed in the methods section. Entinostat/vehicle was administrated intraperitoneally 1 hour post-ICH. Compared to vehicle-treated control, the Entinostat-treated group exhibited significant improvement in neurological outcomes on day 1 and day 3 post-ICH. Of note, Entinostat treatment significantly reduced the sensorimotor deficits score by 34.9% and 43.8% on day 1 and day 3, respectively in comparison to controls (Figure 2A & 2B).
Figure 1.
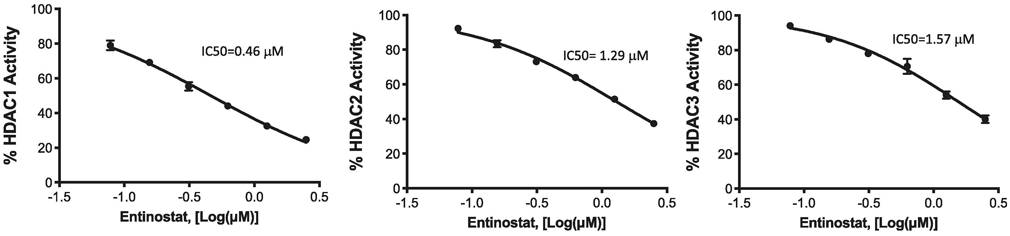
The efficacy of Entinostat in inhibiting the deacetylase activity of recombinant human class I HDACs 1, 2, and 3 was determined and the IC50 values were calculated using GraphPad.
Figure 2.

Entinostat treatment attenuated acute neurobehavioral deficits after ICH. Mice were administered with Entinostat/vehicle 1 hour post induction of ICH and subjected to composite neurobehavioral analysis as detailed in methods. Entinostat significantly reduced sensorimotor deficits after ICH on A) day 1 (n=10/group) and B) day 3 post-ICH ( ICH+ vehicle; n=9 and ICH+ Entinostat; n=10) in comparison to control.
2.2. Entinostat attenuated proinflammatory response both in vitro and in vivo
Given the emerging role of HDACi in mitigating neuroinflammatory response, we next questioned whether Entinostat attenuated hemin-induced inflammatory response in a murine macrophage cell line, RAW 264.7. Hemin is a hemoglobin metabolite, which accumulates in the brain after ICH and is partly responsible for proinflammatory activation of microglia and secondary brain damage [18]. Importantly, treatment with Entinostat significantly reduced the release of proinflammatory cytokines, TNF-α and IL-6 from RAW 264.7 cells upon hemin treatment as estimated by ELISA (Figure 3A & 3B). Consistently, treatment with Entinostat remarkably reduced the expression of CD16/32, a marker of proinflammatory microglia/macrophages in comparison to control on day 3 following ICH (Figure 4A), a time point that exhibited very prominent glial activation, a characteristic feature of neuroinflammatory response [19]. The stereotactic cell counting revealed 28.9% decrease in CD16/32 positive cells upon Entinostat treatment in comparison to control (Figure 4B).
Figure 3.

Entinostat attenuated proinflammatory response in vitro. For in vitro studies, Raw 264.7cells, a macrophage-like cell line, were pre-treated with Entinostat (0.25μM or 0.5 μM) for 1 hour and that was followed by an 18-hour co-treatment with hemin (30 μM) and the supernatant was collected. The level of proinflammatory cytokines, TNF-α (A), and IL-6 (B), were quantified using ELISA, as detailed in methods. Both TNF-α and IL-6 levels were significantly reduced with Entinostat treatment in comparison to respective control. (* p < 0.05, ** p <0.01, ***p<0.001 vs. hemin alone (control; n=4, hemin; n=8 and hemin +Entinostat; n=4).
Figure 4.

Entinostat attenuated proinflammatory activation of microglia/macrophages on day 3 post-ICH. Coronal brain sections were immunostained with M1 microglial/macrophage marker, CD 16/32 and a nuclear marker, DAPI and confocal images were captured. The number of CD 16/32 positive cells was counted and the average number of cells per 0.1mm2 is demonstrated. (B). Entinostat treatment significantly reduced the number of CD 16/32 positive cells after ICH in comparison to controls (ICH+ vehicle; n=3 and ICH+ Entinostat; n=4).
2.3. Entinostat attenuated acute neurodegeneration after ICH
ICH results in profound neurodegeneration [20] and we next questioned whether Entinostat-mediated attenuation of inflammation is concomitant with the reduction in neurodegeneration after ICH. Along these lines, the brain sections from Entinostat/vehicle-treated animals on day 3 post-ICH were subjected to Fluro-Jade B, a marker of degenerating neurons. Of note, Entinostat treatment significantly reduced Fluro-Jade B positive cells after ICH in comparison to controls (Figure 5A). The number of Fluoro-Jade B positive cells was reduced by 29.9% in comparison to controls (Figure 5B). In addition, Entinostat treatment reduced the number of apoptotic cells after ICH. Along these lines, Entinostat treatment significantly reduced TUNEL -positive cells on day 3 post-ICH (Figure 5C) and the number of TUNEL positive cells was reduced by 33.8% compared to control (Figure 5D).
Figure 5.

Entinostat treatment reduced neurodegeneration and apoptotic cell death on day 3 post-ICH . Brain sections were stained with Fluoro jade B (A) or TUNEL (C), as detailed in methods and confocal images were captured. The number of Fluoro jade B (ICH+ vehicle; n=3 and ICH+ Entinostat; n=6) or TUNEL positive cells (ICH+ vehicle; n=3 and ICH+ Entinostat; n=6) were counted and the average number of positive cells per brain section is provided (B, D). Entinostat significantly reduced the number of both Fluoro jade B and TUNEL-positive cells after ICH in comparison to control.
2.4. Entinostat attenuated hematoma and lesion volume after ICH
Hematoma volume is a key predictor of poor patient outcome after ICH [21] and brain inflammation contributes to hematoma expansion [22]. Therefore, we next questioned whether Entinostat treatment could modulate the hematoma volume after ICH. To estimate brain hematoma volume, we employed hemoglobin assay as well as SW-MRI (susceptibility-weighted magnetic response imaging), which is exquisitely sensitive in detecting brain hemorrhage [23]. Of note, Entinostat treatment significantly reduced hemoglobin content in the ipsilateral brain region (Figure 6A) and hematoma volume as estimated by SW-MRI on day 3 post-ICH in comparison to vehicle-treated controls (Figure 6B& 6C). In addition, Entinostat treatment resulted in the reduction of lesion volume as estimated by Cresyl violet staining (Figure 7A & 7B) and T2-weighted MR imaging on day 3 post-ICH in comparison to control (Figure 7C & 7D).
Figure 6.

Entinostat treatment attenuated hematoma volume on day 3 post-ICH. To assess hematoma volume, the brain hemoglobin content and SW-MRI were performed at 72 h post-ICH as described in methods. Entinostat treatment significantly reduced the (A) ipsilateral brain hemoglobin content (ICH+ vehicle; n=3 and ICH+ Entinostat; n=4) and (B&C) hematoma volume as assessed by SW-MRI (n= 4/group).
Figure 7.

Entinostat treatment attenuated brain lesion volume on day 3 post-ICH. Coronal brain sections were stained with Cresyl violet (A) and the lesion volume was estimated (B) as described in methods. Entinostat significantly reduced the lesion volume in comparison to control (ICH+ vehicle; n=3 and ICH+ Entinostat; n=4). (C) Entinostat treatment attenuated T2-weighted hemorrhagic lesion on day 3 post-ICH. Mice were subjected to T2-weighted MRI after ICH and hemorrhagic lesion volume was estimated, as detailed in methods. (D) Entinostat significantly reduced the hemorrhagic lesion volume after ICH in comparison to control (n= 4/group).
3. Discussion
Herein, we report for the first-time that administration of Entinostat improved acute neurological outcomes after ICH implicating a novel role of class I HDACs in the pathophysiology of ICH. Entinostat is a second-generation HDAC inhibitor which inhibits Class I HDACs (HDAC 1, 2 and 3) with no significant activity against class II HDACs [24]. Second generation HDACi typically contain a benzamide head group that chelates the zinc ion within the active site of HDACs, resulting in HDAC inhibition [25, 26]. In contrast to first-generation HDACi, the second generation HDACi exhibits greater HDAC selectivity and show slow on/slow off binding kinetics, thereby conferring a long-lasting effect [27]. Consistently, the improvement in neurological outcomes were evident three days post administration of Entinostat after ICH.
Neuroinflammation is a key regulator of ICH-induced brain damage and the proinflammatory response after ICH correlates with cell death, hematoma expansion, neurological deterioration, secondary brain damage and poor functional recovery [22, 28-34]. Entinostat-mediated improvement in neurological outcome after ICH was associated with a reduction in the number of proinflammatory M1 microglia/macrophages that produce largely deleterious proinflammatory cytokines [35, 36] that contribute to ICH-induced secondary brain injury and loss of neurological function [7, 37, 38]. Importantly, a recent clinical study reported an independent correlation between a reduction in M1- like macrophages and favorable outcome on day 90 post-ICH [39]. Further, Entinostat treatment significantly attenuated hemin-induced release of proinflammatory cytokines, IL-6 and TNF-α from macrophages. This is consistent with the anti-inflammatory potential of Entinostat [40-43], implicating that attenuation of neuroinflammation could partly be responsible for Entinostat-mediated neuroprotective effects after ICH. Further, Entinostat treatment attenuated ICH-induced neurodegeneration and apoptotic cell death after ICH. Apart from anti-inflammatory effects, class 1 HDACi are potent inhibitors of oxidative stress-induced neuronal death [44], which could also be contributing to Entinostat-mediated improvement in neurological outcomes after ICH warranting further investigation. Also, further studies are required to determine whether Entinostat can exert neuroprotection in a manner that is independent of epigenetic mechanisms.
In clinical practice, the neurological outcome is positively associated with the rate of hematoma absorption/resolution [2]. Therefore, strategies that could prevent hematoma expansion or attenuate hematoma volume are highly warranted and it may reduce blood-component induced oxidative as well as inflammatory neurotoxicity after ICH [10]. To this end, Entinostat-mediated attenuation of hematoma volume, an independent determinant of neurological outcomes after ICH [2] offers promising and novel strategy. Though neuroinflammation correlates with hematoma expansion after ICH [22], the precise molecular mechanism by which Entinostat attenuates hematoma volume requires further investigation. Altogether, the data suggest the efficacy of Entinostat in attenuating neurodegeneration, hematoma volume and improving acute neurological outcomes after ICH.
HDACi are in clinical trials for various pathological conditions and the expression of HDACs varies with neuropathological conditions, including stroke [45] together making HDACs a potential target for therapeutic intervention. Although, HDACi are generally well-tolerated in clinical settings, their adverse effects in humans include gastrointestinal symptoms, fatigue, thrombotic events, and cardiotoxicity [40]. Given the efficacy of broad spectrum HDACi in improving acute neurological outcomes after ICH [12], this study was designed as a proof-of-concept study to test the role of class 1 HDACs and hence, only one dose of Entinostat was used and only acute neurological outcomes were analyzed, which are study limitations HDACs comprise a large family of proteins, with 18 HDAC isoforms currently identified in humans [46] and it has become clear that individual HDAC enzyme play different biological roles in the brain [47]. Therefore, it is critical to identify the isoform of HDACs that are important for the amelioration of disease phenotypes. Therefore, future studies from our laboratory will focus on identifying the isoform of HDAC that is responsible for HDACi-mediated neuroprotection and will also determine whether HDACi can confer long-term neuroprotection and can improve neurological outcomes after ICH in a gender and age-independent manner.
4. Experimental Procedure
4.1. Induction of ICH
All animal studies were performed according to protocols approved by the Institutional Animal Care and Use Committee in accordance with the NIH and USDA guidelines. ICH was induced in adult male CD-1 mice (Charles River; 8-12 weeks), as reported previously [48-50]. Briefly, mice were anesthetized and placed on a stereotaxic head frame and a burr hole (0.5 mm) was made on the skull 2.2 mm lateral to bregma with a high-speed drill. This was followed by the injection (3 mm deep) of 0.04 U of bacterial type IV collagenase (Sigma, St. Louis, MO) into the left striatum under stereotaxic guidance using a 26-G Hamilton Syringe. Throughout the procedure, the rectal temperature was maintained at 37 ± 0.5°C.
4.2. Drug treatment
Entinostat was purchased from Sigma (St. Louis, MO, USA). Entinostat (10 mg/kg), prepared in DMSO (25mM stock), was administered intraperitoneally in a total volume of 200 μl in PBS at 1h post-induction of ICH and the control mice received an equal volume of vehicle (DMSO) in PBS. The dose and route of administration of Entinostat were based on previous studies, which demonstrated that this method increased the brain acetylation status [19, 51].
4.3. HDAC inhibition assay
The HDAC inhibition assay was performed with a Fluorogenic HDAC Assay kit (BPS Bioscience, San Diego, CA, USA). Briefly, the respective human recombinant HDAC enzymes were incubated with various concentrations of Entinostat and a pro-fluorogenic substrate. The deacetylation reaction was stopped by the addition of the HDAC Stop Solution and the fluorescence was measured using a BioTek synergy plate reader. Nonlinear regression was used to fit the data to the log (inhibitor concentration) vs. activity (%) using GraphPad.
4.4. ELISA
A mouse macrophage cell line, RAW264.7, was incubated on a 24 well plate for 48 hours in DMEM containing 5% FBS (Fetal Bovine Serum), 5% BGS (Bovine Growth Serum), and 1% Penicillin/Streptomycin. Cells were then pre-treated with Entinostat (0.25 or 0.5 μM; Sigma Aldrich, St. Louis, MO) for one hour and exposed to both Entinostat (0.25 or 0.5 μM) and hemin (30 μM) for 18 hours. Subsequently, the supernatant was collected and used for the estimation of cytokines, TNF-α and IL-6 by ELISA (Max Deluxe Kit; Bio legend, San Diego, CA), as per the manufacturer’s instructions. Following an overnight immobilization on polystyrene wells with a capture antibody and blocking for an hour, the cell culture supernatant was added to the wells and incubated at room temperature for 2 hours. Wells were then washed to remove any unbound materials, and a detection antibody solution was added and incubated at room temperature. After washing, 100 μl of Avidin- HRP solution was added for a 30-minute incubation at room temperature and the substrate solution was added to the wells and left for color development for 15 minutes. A stop solution was then added and the plate was read at 450 nm using a microtiter plate reader (Bio-Tek, Epoch).
4.5. Fluoro-Jade B staining
Brain sections were hydrated and then incubated with 0.06% potassium permanganate solution for 15 minutes. Sections were then washed with distilled water and incubated with 0.001% Fluoro-Jade B solution in acetic acid for 30 minutes. Sections were then air-dried, washed in xylene and cover-slipped using DPX mounting media. Using a Zeiss 780 upright confocal microscope, the Fluoro-Jade B staining was analyzed with an excitation wavelength of 488 nm.
4.6. TUNEL staining
To estimate the number of TUNEL positive cells, a commercially available apoptosis kit (Apoptag in situ: Millipore, S7110) was used. As per the manufacturer’s instruction, the brain sections were fixed in ethanol: acetic acid solution and then incubated in equilibration buffer. This was followed by incubation with the Terminal deoxynucleotidyl Transferase (TdT) enzyme. After an hour, the reaction was stopped using a stop buffer. Sections were washed with PBS, incubated with an anti-digoxigenin-Fluorescein conjugate solution for 30 minutes and cover-slipped using a mounting media. Using a Zeiss 780 upright confocal microscope, the fluorescence was determined and images were captured.
4.7. Estimation of Fluoro Jade B and TUNEL Positive Cells
To estimate the number of Fluoro jade B or TUNEL positive cells, we used six coronal sections (two from the collagenase injection site and two each from 0.25 mm anterior and 0.25 mm posterior to the injection site) per animal. Fluoro jade B or TUNEL positive cells in the perihematomal region in the striatum were counted manually with the aid of a fluorescence microscope and average number of cells per brain section is provided. The Fluoro jade B or TUNEL positive cells in the perihematomal region were observed starting from the edge of the hematoma to a maximum distance of approximately 500 microns.
4.8. Immunohistochemistry
Anesthetized mice were transcardially perfused with PBS and the brain was collected, placed in 4% paraformaldehyde overnight and cryoprotected in 30 % sucrose. The brain tissue was then snap frozen on liquid Nitrogen and cut into coronal sections (25 μM) using a cryostat. Brain section mounted on glass slides were incubated with 10% donkey serum for 2 hours at room temperature and exposed to respective primary antibody (anti-CD 16/32; 1:100; rat monoclonal; BD Biosciences, CA, USA; Catalogue No: 553142) overnight at 4°C. Sections were then washed and incubated with the corresponding Alexa fluor-tagged secondary antibody (Alexa Fluor 594 conjugated donkey anti-Rat IgG; 1:1000; Invitrogen/Life Technologies, USA; Catalogue No: A-21209) for one hour at room temperature and the immunostaining was visualized using a Zeiss 780 upright confocal microscope. The number of immunopositive cells in the perihematomal region was quantified as detailed earlier [49].
4.9. Neurobehavior Analysis
Mice were subjected to neurobehavioral analysis using a 24-point scale as described previously by our laboratory and others[12, 20, 52]. This comprehensive neurobehavioral testing which measures the sensorimotor deficits includes six different testings; climbing, beam walking, circling, bilateral grasp, whisker response, and compulsory circling. Each of the six different tests was then graded from 0 (no impairment) to 4 (severe impairment) and the six individual test scores were then added together to obtain the composite neurological deficit score with a maximum deficit score of 24.
4.10. Cresyl Violet staining
Brain sections were rehydrated with ethanol and stained with 0.5% Cresyl Violet solution. Sections were then dehydrated, treated with xylene and images were captured. For estimating brain lesion volume, six non-consecutive sections per mouse were subjected to cresyl violet staining and the area of the hematoma was quantified using Image J software (NIH, USA). The hematoma volume was then calculated by multiplying the sum of the areas by the interslice distance as reported earlier [53].
4.11. Magnetic Resonance Imaging (MRI)
Anesthetized mice were subjected to MRI using 7 Tesla small animal imaging system from Bruker (Bruker Biospin USR 70/20) as reported earlier [54]. Briefly, after positioning the mouse using a triplanar FLASH sequence, T2-weighted and susceptibility-weighted MR (SW-MR) images were obtained to quantify hemorrhagic lesion volume and hematoma volume, respectively as reported earlier [55]. T2 weighted images were obtained with parameters, FOV 24x24, matrix 256x256, TE=35 ms, TR=2500 ms and 20 slices with a slice thickness of 0.5 mm. The susceptibility-weighted sequences were obtained with parameters TR=624.7 ms, TE=15 ms, 35° flip angle, 24x24 FOV, 256x256 matrix, 20 slices with a slice thickness of 0.5 mm and NEX = 2. The volume of the hemorrhagic lesion and hematoma was estimated from T2 and SW-MR images, respectively using Image J software. The lesioned or hemorrhagic area was outlined with the help of Image J and volume (in mm3) was determined by integration of the lesioned area in each section over the slice thickness.
4.12. Hemoglobin assay
Hemoglobin assay was conducted as a measure of hematoma volume, as described earlier [56]. Briefly, anesthetized mice were subjected to transcardial perfusion and the ipsilateral brain tissue was collected, homogenized and centrifuged at 10,000 g for 15 min at 4°C . The supernatant was subjected to hemoglobin assay using a Hemoglobin Colorimetric Assay Kit (Cayman, Ann Arbor, USA), as per the manufacturer’s instructions.
4.13. Statistical Analysis
Mice were randomly assigned to the experimental groups, and all analyses were performed by an investigator-blinded manner. Data were analyzed using unpaired two-tailed t-test or one-way analysis of variance followed by Student-Newman-Keuls post hoc test, as appropriate and are expressed as mean ± SE. The neurobehavioral data were analyzed using the nonparametric Mann-Whitney test. A p-value of <0.05 was considered significant.
Highlights.
Class 1 HDACi, Entinostat attenuated acute neurobehavioral deficits after ICH.
Entinostat treatment reduced proinflammatory response both in vitro and in vivo.
Entinostat reduced neurodegeneration and hematoma volume after ICH.
Acknowledgments
Funding
This work was supported by grants from the National Institutes of Health (R01NS107853) and American Heart Association (14SDG18730034) to SSR.
Footnotes
Conflicts of Interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Qureshi AI, Mendelow AD, Hanley DF: Intracerebral haemorrhage. Lancet 2009, 373(9675):1632–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Asch CJ, Luitse MJ, Rinkel GJ, van der Tweel I, Algra A, Klijn CJ: Incidence, case fatality, and functional outcome of intracerebral haemorrhage over time, according to age, sex, and ethnic origin: a systematic review and meta-analysis. The Lancet Neurology 2010, 9(2):167–176. [DOI] [PubMed] [Google Scholar]
- 3.Aguilar MI, Freeman WD: Spontaneous intracerebral hemorrhage. Seminars in neurology 2010, 30(5):555–564. [DOI] [PubMed] [Google Scholar]
- 4.Broderick J, Connolly S, Feldmann E, Hanley D, Kase C, Krieger D, Mayberg M, Morgenstern L, Ogilvy CS, Vespa P et al. : Guidelines for the management of spontaneous intracerebral hemorrhage in adults: 2007 update: a guideline from the American Heart Association/American Stroke Association Stroke Council, High Blood Pressure Research Council, and the Quality of Care and Outcomes in Research Interdisciplinary Working Group. Circulation 2007, 116(16):e391–413. [DOI] [PubMed] [Google Scholar]
- 5.Ribo M, Grotta JC: Latest advances in intracerebral hemorrhage. Current neurology and neuroscience reports 2006, 6(1):17–22. [DOI] [PubMed] [Google Scholar]
- 6.Qureshi AI, Tuhrim S, Broderick JP, Batjer HH, Hondo H, Hanley DF: Spontaneous intracerebral hemorrhage. The New England journal of medicine 2001, 344(19):1450–1460. [DOI] [PubMed] [Google Scholar]
- 7.Wang J: Preclinical and clinical research on inflammation after intracerebral hemorrhage. Progress in neurobiology 2010, 92(4):463–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aronowski J, Zhao X: Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke; a journal of cerebral circulation 2011, 42(6):1781–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xi G, Keep RF, Hoff JT: Mechanisms of brain injury after intracerebral haemorrhage. The Lancet Neurology 2006, 5(1):53–63. [DOI] [PubMed] [Google Scholar]
- 10.Madangarli N, Bonsack F, Dasari R, Sukumari-Ramesh S: Intracerebral Hemorrhage: Blood Components and Neurotoxicity. Brain Sci 2019, 9(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer A, Sananbenesi F, Mungenast A, Tsai LH: Targeting the correct HDAC(s) to treat cognitive disorders. Trends in pharmacological sciences 2010, 31(12):605–617. [DOI] [PubMed] [Google Scholar]
- 12.Sukumari-Ramesh S, Alleyne CH Jr., Dhandapani KM: The Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid (SAHA) Confers Acute Neuroprotection After Intracerebral Hemorrhage in Mice. Transl Stroke Res 2016, 7(2):141–148. [DOI] [PubMed] [Google Scholar]
- 13.Glozak MA, Sengupta N, Zhang X, Seto E: Acetylation and deacetylation of non-histone proteins. Gene 2005, 363:15–23. [DOI] [PubMed] [Google Scholar]
- 14.Shahbazian MD, Grunstein M: Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem 2007, 76:75–100. [DOI] [PubMed] [Google Scholar]
- 15.Glauben R, Sonnenberg E, Zeitz M, Siegmund B: HDAC inhibitors in models of inflammation-related tumorigenesis. Cancer Lett 2009, 280(2):154–159. [DOI] [PubMed] [Google Scholar]
- 16.Dokmanovic M, Clarke C, Marks PA: Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res 2007, 5(10):981–989. [DOI] [PubMed] [Google Scholar]
- 17.Butler LM, Zhou X, Xu WS, Scher HI, Rifkind RA, Marks PA, Richon VM: The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl Acad Sci U S A 2002, 99(18):11700–11705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, Wang JZ, Su BY, Yang QW: Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. Journal of neuroinflammation 2012, 9:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joksimovic SM, Osuru HP, Oklopcic A, Beenhakker MP, Jevtovic-Todorovic V, Todorovic SM: Histone Deacetylase Inhibitor Entinostat (MS-275) Restores Anesthesia-induced Alteration of Inhibitory Synaptic Transmission in the Developing Rat Hippocampus. Mol Neurobiol 2018, 55(1):222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sukumari-Ramesh S, Alleyne CH Jr.: Post-Injury Administration of Tert-butylhydroquinone Attenuates Acute Neurological Injury After Intracerebral Hemorrhage in Mice. Journal of molecular neuroscience : MN 2016, 58(4):525–531. [DOI] [PubMed] [Google Scholar]
- 21.Broderick JP, Brott TG, Duldner JE, Tomsick T, Huster G: Volume of intracerebral hemorrhage. A powerful and easy-to-use predictor of 30-day mortality. Stroke 1993, 24(7):987–993. [DOI] [PubMed] [Google Scholar]
- 22.Leira R, Davalos A, Silva Y, Gil-Peralta A, Tejada J, Garcia M, Castillo J, Stroke Project CDGotSNS: Early neurologic deterioration in intracerebral hemorrhage: predictors and associated factors. Neurology 2004, 63(3):461–467. [DOI] [PubMed] [Google Scholar]
- 23.Halefoglu AM, Yousem DM: Susceptibility weighted imaging: Clinical applications and future directions. World J Radiol 2018, 10(4):30–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryu Y, Kee HJ, Sun S, Seok YM, Choi SY, Kim GR, Kee SJ, Pflieger M, Kurz T, Kim HS et al. : Class I histone deacetylase inhibitor MS-275 attenuates vasoconstriction and inflammation in angiotensin II-induced hypertension. PLoS One 2019, 14(3):e0213186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou N, Moradei O, Raeppel S, Leit S, Frechette S, Gaudette F, Paquin I, Bernstein N, Bouchain G, Vaisburg A et al. : Discovery of N-(2-aminophenyl)-4-[(4-pyridin-3-ylpyrimidin-2-ylamino)methyl]benzamide (MGCD0103), an orally active histone deacetylase inhibitor. J Med Chem 2008, 51(14):4072–4075. [DOI] [PubMed] [Google Scholar]
- 26.Lauffer BE, Mintzer R, Fong R, Mukund S, Tam C, Zilberleyb I, Flicke B, Ritscher A, Fedorowicz G, Vallero R et al. : Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. The Journal of biological chemistry 2013, 288(37):26926–26943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boissinot M, Inman M, Hempshall A, James SR, Gill JH, Selby P, Bowen DT, Grigg R, Cockerill PN: Induction of differentiation and apoptosis in leukaemic cell lines by the novel benzamide family histone deacetylase 2 and 3 inhibitor MI-192. Leuk Res 2012, 36(10):1304–1310. [DOI] [PubMed] [Google Scholar]
- 28.Hickenbottom SL, Grotta JC, Strong R, Denner LA, Aronowski J: Nuclear factor-kappaB and cell death after experimental intracerebral hemorrhage in rats. Stroke; a journal of cerebral circulation 1999, 30(11):2472–2477; discussion 2477-2478. [DOI] [PubMed] [Google Scholar]
- 29.Platt N, da Silva RP, Gordon S: Recognizing death: the phagocytosis of apoptotic cells. Trends in cell biology 1998, 8(9):365–372. [DOI] [PubMed] [Google Scholar]
- 30.Zhao X, Zhang Y, Strong R, Zhang J, Grotta JC, Aronowski J: Distinct patterns of intracerebral hemorrhage-induced alterations in NF-kappaB subunit, iNOS, and COX-2 expression. Journal of neurochemistry 2007, 101(3):652–663. [DOI] [PubMed] [Google Scholar]
- 31.Mayne M, Fotheringham J, Yan HJ, Power C, Del Bigio MR, Peeling J, Geiger JD: Adenosine A2A receptor activation reduces proinflammatory events and decreases cell death following intracerebral hemorrhage. Ann Neurol 2001, 49(6):727–735. [DOI] [PubMed] [Google Scholar]
- 32.Gong C, Hoff JT, Keep RF: Acute inflammatory reaction following experimental intracerebral hemorrhage in rat. Brain Res 2000, 871(1):57–65. [DOI] [PubMed] [Google Scholar]
- 33.Chu K, Jeong SW, Jung KH, Han SY, Lee ST, Kim M, Roh JK: Celecoxib induces functional recovery after intracerebral hemorrhage with reduction of brain edema and perihematomal cell death. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 2004, 24(8):926–933. [DOI] [PubMed] [Google Scholar]
- 34.Jung KH, Chu K, Jeong SW, Han SY, Lee ST, Kim JY, Kim M, Roh JK: HMG-CoA reductase inhibitor, atorvastatin, promotes sensorimotor recovery, suppressing acute inflammatory reaction after experimental intracerebral hemorrhage. Stroke 2004, 35(7):1744–1749. [DOI] [PubMed] [Google Scholar]
- 35.Liao B, Zhao W, Beers DR, Henkel JS, Appel SH: Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Experimental neurology 2012, 237(1):147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K, Hirakawa A, Takeuchi H, Suzumura A, Ishiguro N et al. : Minocycline selectively inhibits M1 polarization of microglia. Cell death & disease 2013, 4:e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J, Dore S: Inflammation after intracerebral hemorrhage. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 2007, 27(5):894–908. [DOI] [PubMed] [Google Scholar]
- 38.Carmichael ST, Vespa PM, Saver JL, Coppola G, Geschwind DH, Starkman S, Miller CM, Kidwell CS, Liebeskind DS, Martin NA: Genomic profiles of damage and protection in human intracerebral hemorrhage. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 2008, 28(11):1860–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang C, Wang Y, Hu Q, Shou J, Zhu L, Tian N, Sun L, Luo H, Zuo F, Li F et al. : Immune changes in peripheral blood and hematoma of patients with intracerebral hemorrhage. FASEB J 2020, 34(2):2774–2791. [DOI] [PubMed] [Google Scholar]
- 40.Zhang ZY, Zhang Z, Schluesener HJ: MS-275, an histone deacetylase inhibitor, reduces the inflammatory reaction in rat experimental autoimmune neuritis. Neuroscience 2010, 169(1):370–377. [DOI] [PubMed] [Google Scholar]
- 41.Lin HS, Hu CY, Chan HY, Liew YY, Huang HP, Lepescheux L, Bastianelli E, Baron R, Rawadi G, Clement-Lacroix P: Anti-rheumatic activities of histone deacetylase (HDAC) inhibitors in vivo in collagen-induced arthritis in rodents. Br J Pharmacol 2007, 150(7):862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nencioni A, Beck J, Werth D, Grunebach F, Patrone F, Ballestrero A, Brossart P: Histone deacetylase inhibitors affect dendritic cell differentiation and immunogenicity. Clin Cancer Res 2007, 13(13):3933–3941. [DOI] [PubMed] [Google Scholar]
- 43.Zhang ZY, Schluesener HJ: Oral administration of histone deacetylase inhibitor MS-275 ameliorates neuroinflammation and cerebral amyloidosis and improves behavior in a mouse model. J Neuropathol Exp Neurol 2013, 72(3):178–185. [DOI] [PubMed] [Google Scholar]
- 44.Sleiman SF, Olson DE, Bourassa MW, Karuppagounder SS, Zhang YL, Gale J, Wagner FF, Basso M, Coppola G, Pinto JT et al. : Hydroxamic acid-based histone deacetylase (HDAC) inhibitors can mediate neuroprotection independent of HDAC inhibition. J Neurosci 2014, 34(43):14328–14337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT: Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci 2009, 32(11):591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu WS, Parmigiani RB, Marks PA: Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007, 26(37):5541–5552. [DOI] [PubMed] [Google Scholar]
- 47.Baltan S, Bachleda A, Morrison RS, Murphy SP: Expression of histone deacetylases in cellular compartments of the mouse brain and the effects of ischemia. Translational stroke research 2011, 2(3):411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sukumari-Ramesh, Alleyne CH Jr., Dhandapani KM: Astrogliosis: a target for intervention in intracerebral hemorrhage? Translational stroke research 2012, 3(Suppl 1):80–87. [DOI] [PubMed] [Google Scholar]
- 49.Ft Bonsack, Alleyne CH Jr., Sukumari-Ramesh S: Augmented expression of TSPO after intracerebral hemorrhage: a role in inflammation? J Neuroinflammation 2016, 13(1):151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sukumari-Ramesh S, Alleyne CH Jr., Dhandapani KM: Astrocyte-specific expression of survivin after intracerebral hemorrhage in mice: a possible role in reactive gliosis? Journal of neurotrauma 2012, 29(18):2798–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu Y, Dou J, Wan X, Leng Y, Liu X, Chen L, Shen Q, Zhao B, Meng Q, Hou J: Histone Deacetylase Inhibitor MS-275 Alleviates Postoperative Cognitive Dysfunction in Rats by Inhibiting Hippocampal Neuroinflammation. Neuroscience 2019, 417:70–80. [DOI] [PubMed] [Google Scholar]
- 52.Rosenberg GA, Mun-Bryce S, Wesley M, Kornfeld M: Collagenase-induced intracerebral hemorrhage in rats. Stroke 1990, 21(5):801–807. [DOI] [PubMed] [Google Scholar]
- 53.Min H, Jang YH, Cho IH, Yu SW, Lee SJ: Alternatively activated brain-infiltrating macrophages facilitate recovery from collagenase-induced intracerebral hemorrhage. Mol Brain 2016, 9:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dasari R, Zhi W, Bonsack F, Sukumari-Ramesh S: A Combined Proteomics and Bioinformatics Approach Reveals Novel Signaling Pathways and Molecular Targets After Intracerebral Hemorrhage. Journal of molecular neuroscience : MN 2020, 70(8):1186–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun N, Shen Y, Han W, Shi K, Wood K, Fu Y, Hao J, Liu Q, Sheth KN, Huang D et al. : Selective Sphingosine-1-Phosphate Receptor 1 Modulation Attenuates Experimental Intracerebral Hemorrhage. Stroke 2016, 47(7):1899–1906. [DOI] [PubMed] [Google Scholar]
- 56.Wei CC, Kong YY, Li GQ, Guan YF, Wang P, Miao CY: Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO-1 signaling pathway. Sci Rep 2017, 7(1):717. [DOI] [PMC free article] [PubMed] [Google Scholar]