

Abstract
KIF1A is a molecular motor for membrane-bound cargo important to the development and survival of sensory neurons. KIF1A dysfunction has been associated with several Mendelian disorders with a spectrum of overlapping phenotypes, ranging from spastic paraplegia to intellectual disability. We present a novel pathogenic in-frame deletion in the KIF1A molecular motor domain inherited by two affected siblings from an unaffected mother with apparent germline mosaicism. We identified 8 additional cases with heterozygous, pathogenic KIF1A variants–ascertained from a local data lake. Our data provide evidence for expansion of KIF1A-associated phenotypes to include hip subluxation and dystonia as well as phenotypes observed in only a single case: gelastic cataplexy, coxa valga, and double collecting system. We review the literature and suggest that KIF1A dysfunction is better understood as a single neuromuscular disorder with variable involvement of other organ systems than a set of discrete disorders converging at a single locus.
Keywords: KIF1A, Germline Mosaicism, in-frame deletion, literature review, genocentric, data lake
Kinesin Family Member 1A (KIF1A) is a kinesin-3 molecular motor that is highly expressed in the brain and spinal cord (Okada, Yamazaki, Sekine-Aizawa, & Hirokawa, 1995), and plays an important role in the development and survival of sensory neurons. Kinesin-3 motors transport membrane-bound cargo and are highly processive, adapted to long-distance intracellular and axonal transport along microtubule tracks (Huo et al., 2012; Soppina et al., 2014). KIF1A cargo includes postsynaptic proteins important for synaptic plasticity and transmission, learning, and memory. These include GRIP, GIT1, and AMPA receptors, as well as tropomyosin receptor kinase A (TRKA, encoded by NTRK1)–a transmembrane receptor for neurotrophins, a family of signaling proteins essential for neuronal development, differentiation, synaptic function and plasticity, and survival (Huganir & Nicoll, 2013; Lee et al., 2003, 2015; Reichardt, 2006; Shin et al., 2003). Furthermore, kinesin-3 motors are known to dimerize (Okada et al., 1995) upon contact with cargo (Soppina et al., 2014), greatly increasing motor processivity. However, cooperative aggregation of monomeric KIF1A has been shown in vivo to retain the capacity for processive transport (Oriola, Roth, Dogterom, & Casademunt, 2015; Schimert, Budaitis, Reinemann, Lang, & Verhey, 2019).
Amino acid residues 1 to 365 of the KIF1A protein constitute the kinesin motor domain. If the kinesin motor domain is unable to bind properly to microtubules, its movement along neurites is impaired, leading to reduced peripheral localization of KIF1A cargo vesicles on neurite distal projections (Hamdan et al., 2011; Lee et al., 2003, 2015). Certain heterozygous variants that affect the motor domain have been previously shown to disrupt transport along neurites and cause a variable spectrum of clinical phenotypes (Lee et al., 2015).
Monoallelic dysfunction of KIF1A–caused by both loss-of-function and missense variants–is associated with multiple Mendelian phenotypes. De novo missense variants in KIF1A have been reported to cause autosomal dominant mental retardation 9 (MRD9; MIM# 614255), characterized by a variable expression of lower limb spasticity, hyperreflexia, intellectual disability, hypotonia, ataxia, microcephaly, cerebellar atrophy, nystagmus, and optic atrophy. De novo missense variants have also been reported as a cause of PEHO syndrome (MIM# 260565) (Langlois et al., 2016) with overlapping clinical features similar to those previously reported in MRD9. Furthermore, heterozygous variants in KIF1A have been reported to cause hereditary spastic paraplegia (HSP), defined by bilateral lower-extremity spasticity and weakness associated with hyperreflexia and extensor plantar responses (Pennings et al., 2020; Ylikallio et al., 2015). HSP is a genetically heterogeneous disease that can be “pure” or “complicated”. In the complicated cases additional neurological symptoms are found, such as seizures, intellectual disability, and peripheral neuropathy among others (Hedera, 2000). Regardless of phenotype, most of the reported monoallelic cases were molecularly diagnosed with de novo missense variants impacting the KIF1A kinesin motor domain. Among monoallelic missense cases–even those within the kinesin motor domain–there is considerable phenotypic variability. However, pathogenic, heterozygous, loss-of-function variants seem to typically appear only in conjunction with spastic paraplegia without cognitive defects (Pennings et al., 2020), with the notable exception of one case with reportedly all of the typical features of Rett Syndrome (Wang et al., 2019). Overall, monoallelic cases are typically missense variants in the kinesin motor domain and generally present with a more severe and syndromic phenotype than biallelic cases (Klebe et al., 2012).
Here, we use familial exome sequencing (ES) to describe a family with two affected siblings apparently inheriting a heterozygous in-frame deletion in the kinesin motor domain of KIF1A from an unaffected parent with germline mosaicism. Furthermore, we report the association of novel phenotypes with monoallelic KIF1A dysfunction, ascertained through analysis of additional cases diagnosed by a single clinical testing laboratory (Yang et al., 2013) and through the Baylor Hopkins Center for Mendelian Genomics (Posey et al., 2019).
Two siblings–a 12-year-old boy (PERU1) and a 2-year-old girl (PERU2) of a non-consanguineous family–initially presented at the Clinical Genetics Department of Instituto Nacional del Niño, Lima, Peru (Figure 1). The patients have unaffected parents and two other unaffected siblings. There is a family history of a 1st-trimester spontaneous abortion of unknown cause. Both patients had an unremarkable prenatal history. The boy had delayed psychomotor development. At 18 months old he developed episodes compatible with gelastic cataplexy. He has been in physical therapy since age 3 and started walking at 8 years old. Physical examination revealed short stature (>2SD), microcephaly (>2.5SD), mild camptodactyly, scoliosis, bilateral hip subluxation and pes planus. Initial neurological examination revealed axial hypotonia, appendicular hyperreflexia and ataxia. Intellectual disability was noted with an absence of verbal communication and inability to follow verbal commands. Auditory evoked potentials were normal. Ophthalmological examination revealed strabismus, nystagmus and congenital polar posterior cataracts with left pale optic nerve. Brain magnetic resonance imaging (MRI) showed cerebellar and cerebral atrophy (Figure 1). Renal imaging showed a double collecting system in the left kidney. Upon the last follow-up at 15 years of age, he remained non-verbal, incontinent, and started to show mild spasticity of the lower limbs.
Figure 1 – Peruvian Index Family and KIF1A Kinesin Motor Domain Non-truncating Variants.
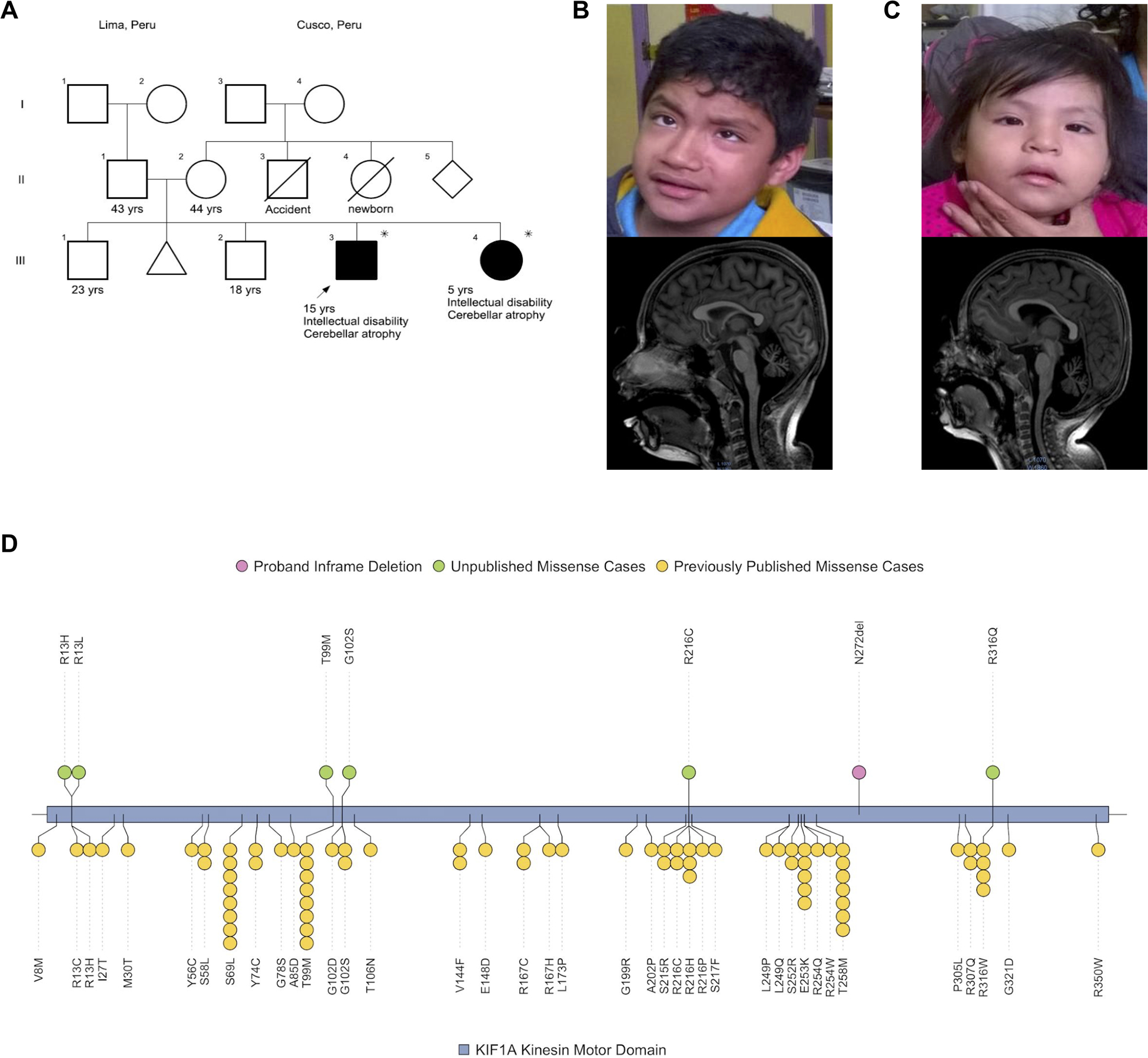
A) Siblings affected with dominant KIF1A-related disorder inherited from a germline mosaic, unaffected parent. Asterisk indicates possession of pathogenic p.Asn272del variant. Both the proband (PERU1) and unaffected sister (PERU2) present with strabismus and nystagmus. B-C) Brain MRIs obtained at 11 years of age for subject PERU1 (B) and 1 year of age for subject PERU2 (C) revealed severe cerebellar atrophy. D) The p.Asn272del (N272del) variant observed in subjects PERU1 and PERU2 is shown in pink. Other putatively pathogenic missense variants in previously unpublished cases (Table 1) within the kinesin motor domain from local cases are shown in green. Previously published pathogenic non-truncating variants in the motor domain are shown at the bottom in yellow.
The affected sister (PERU2) presented with global developmental delay, microcephaly (>2.5SD), short stature (>2SD), ataxia, axial hypotonia, and lower-limb hyperreflexia with a positive bilateral Babinski sign. She was unresponsive to verbal commands. Ophthalmological examination was positive for strabismus, nystagmus and bilateral pale optic nerves. Brain MRI reported diffuse severe cerebellar atrophy associated with compensatory basal cistern and 4th ventricle widening, with suggestive signs of cerebral atrophy (Figure 1). On follow-up at 3 years and 8 months of age the patient was non-ambulatory and non-verbal, and right hip subluxation was noted. At 5 years and 5 months, she was unable to walk without assistance, remained non-verbal and incontinent, and a diagnosis of developmental delay/intellectual disability (DD/ID) was made.
No sequence variants known to be associated with neurodevelopmental phenotypes segregated in the index family. However, ES detected a heterozygous in-frame deletion variant in KIF1A–NC_000002.11:g.241722510_241722512del, NM_004321.6:c.816_818delCAA(p.Asn272del)–which was shared by both affected siblings (supported by 26/59 reads in the proband and 26/54 reads in the sister). This variant leads to a loss of a single amino acid (p.Asn272del) in the kinesin motor domain. This variant was detected via ES from blood samples obtained from subjects PERU1 and PERU2 and confirmed with Sanger sequencing, but was not detected via ES or Sanger sequencing in the unaffected brother (Individual III.2). The variant was not detected in Sanger sequencing of parental blood samples, but present in only one read in the ES results of the mother and father (out of 81 and 56 reads, respectively)–indicative of a likely technical error due to barcode hopping. Non-paternity was ruled out through identity-by-descent analysis. This de novo variant was not seen in internal controls–as previously described (Hansen et al., 2019)–affects a highly conserved residue (GERP++ = 3.54, as obtained from the UCSC Genome Browser) and has a very low MTR score (21bp window)–0.34–indicative of a region constrained for nonsynonymous variation in control populations (Davydov et al., 2010; Kent et al., 2002; Traynelis et al., 2017).
Due to the extremely low probability of recurrence of identical de novo variation in two siblings, and the absence of the variant at any meaningful allele fraction in either parental sample, we hypothesized the variant to be mosaic in one of the two parental germlines. A paternal sperm sample was negative for the variant as assayed via Sanger sequencing. Droplet Digital PCR (ddPCR)–a sensitive assay for detecting low-level mosaicism–was then performed on the paternal sperm sample and blood samples from the proband, affected sister, unaffected brother, and both parents (Supp. Figure S1). ddPCR again confirmed the presence of the variant in the proband and affected sister but failed to detect the variant at any level in any of the remaining samples: paternal sperm, as well as blood samples for the father, mother, and unaffected brother. Thus, we suspect the variant to be inherited maternally and mosaic in the maternal germline at an unknown allele fraction. However, we cannot rule out the possibility of paternal germline mosaicism and inheritance, as the variant could be present at a level below the threshold of sensitivity for ddPCR in the paternal sperm sample tested; furthermore, one sperm sample will not necessarily capture the full extent of putative mosaicism in a male germline.
Thus, in the index family, we found a novel pathogenic in-frame deletion, causing a unique p.Asn272del deletion in the kinesin motor domain. There are many reports of pathogenic variants in this domain associated with variable Mendelian disease traits including MRD9 and SPG30 (MIM# 610357) (Pennings et al., 2020). This observation of an in-frame deletion is striking as all previously reported pathogenic variants causing MRD9 have been missense variants in the motor domain. Nonetheless, further functional studies are warranted to fully characterize the role of this variant on kinesin motor activity or other aspects of KIF1A function.
Next, we investigated the phenotypes of these two affected siblings compared to previously published and unpublished cases. We utilized HARLEE (Hansen et al., 2019) to search across >20,000 ES samples from the Baylor Hopkins Center for Mendelian Genomics and a clinical diagnostic lab (see supporting information) for cases solved by KIF1A and identified 8 such cases (Table 1, Figure 1). Phenotypes not previously known to associate with monoallelic KIF1A dysfunction were identified in multiple patients, including dystonia (4/10, 40%) and hip subluxation (3/10, 30%). Other, potentially novel phenotypes only seen in a single individual (1/10, 10%) include gelastic cataplexy and double collecting system (both present only in PERU1), as well as coxa valga (bilateral), and skin abnormalities (hypertrichosis and cutis marmorata).
Table 1 – Phenotypic expansion of KIF1A clinical manifestation.
Molecular diagnosis and phenotypic information is presented for reach patient. Phenotypes are further categorized into seven high-level categories: head and neck, central nervous system, peripheral nervous system, musculoskeletal, urinary system, gastrointestinal system, and skin/integument. Phenotypes are indicated as either positive, negative, or not-phenotyped (or reported) for each patient. For research-consented cases, individual-level phenotypic information is presented. For the four other clinical cases, phenotypic information is presented in aggregate. The summary column indicates how many patients were positive for a given phenotype. All genomic positions are based on NC_000002.11, and amino acid changes are predicted from transcript NM_004321.6.
| RESEARCH OR CLINICAL | RESEARCH | RESEARCH | RESEARCH | RESEARCH | RESEARCH | RESEARCH | CLINICAL | CLINICAL | CLINICAL | CLINICAL | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample ID | PERU1 | PERU2 | BG1 | BAB11796 | BG2 (Lee et al. patient 6) | BG3 (Lee et al. patient 8) | BG4 | BG5 | BG6 | BG7 | |
| MOLECULAR | |||||||||||
| Chromosome | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | |
| Genomic Position | 241722506 | 241722506 | 241727535 | 241727527 | 241724483 | 241723197 | 241737132 | 241737132 | 241724480 | 241715279 | |
| Genomic Variant | CTTG>C | CTTG>C | G>A | C>T | T>G | C>T | C>T | C>A | G>A | C>T | |
| Amino acid change | p.Asn272del | p.Asn272del | p.Thr99Met | p.Gly102Ser | p.Ser215Arg | p.Glu253Lys | p.Arg13His | p.Arg13Leu | p.Arg216Cys | p.Arg316Gln | |
| Zygosity | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | Heterozygous | |
| Inheritance | Parental germline mosaic | Parental germline mosaic | de novo | de novo | de novo | de novo | de novo | de novo | de novo | de novo | |
| Sex | Male | Female | Male | Male | Male | Female | Female | Female | Female | Female | |
| Age of symptoms onset | 6 months | 6 months | 6 months | 6 months | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | |
| Age at most recent phenotyping | 15 yrs | 5 yrs | 9 yrs | 14 yrs | 2 yrs | 4 yrs | 19 yrs | 9 yrs | 8 yrs | 7 yrs | |
| Additional Diagnoses | NA | NA | NA | (Family history of consanguinity) | Phenylketonuria | NA | NA | NA | de novo, likely pathogenic variant in FZD2 | NA | |
| PHENOTYPES | Summary | ||||||||||
| Head and neck | ✚ | ✚ | ✚ | ✚ | ✚ | ✚ | NP | 6 / 10 | |||
| Dysmorphic features | Hypertelorism, short philtrum, prognathia | Hypertelorism, epicanthus, down turned mouth corners | Heavy eyebrows, synophrys, large palpebral fissures | Bushy eyebrow, short philtrum | NP | ✚ | NP | 5 / 10 | |||
| Microcephaly | ✚ | ✚ | – | – | ✚ | ✚ | NP | 4 / 10 | |||
| Optic atrophy | Pale optic disc | Pale optic disc | ✚ | – | ✚ | ✚ | NP | 5 / 10 | |||
| Cataract | ✚ | – | ✚ | – | NP | NP | NP | 2 / 10 | |||
| Visual impairment, others such as cortical visual impairment | – | – | Cortical | – | ✚ | Cortical | NP | 3 / 10 | |||
| Strabismus | ✚ | ✚ | ✚ | – | NP | NP | NP | 3 / 10 | |||
| Nystagmus | ✚ | ✚ | – | – | NP | NP | NP | 2 / 10 | |||
| Central nervous system | ✚ | ✚ | ✚ | ✚ | ✚ | ✚ | 4 / 4 | 10 / 10 | |||
| Hypotonia | Infantile | Infantile | Infantile | NP | Infantile | Infantile | 3 / 4 | 8 / 10 | |||
| Delayed psychomotor development | ✚ | ✚ | ✚ | ✚ | ✚ | ✚ | 3 / 4 | 9 / 10 | |||
| Intellectual disability | ✚ | ✚ | ✚ | Mild | ✚ | NP | 3 / 4 | 8 / 10 | |||
| Poor or absent speech | ✚ | ✚ | ✚ | ✚ | ✚ | NP | NP | 5 / 10 | |||
| Seizures | – | – | ✚ | – | ✚ | ✚ | NP | 3 / 10 | |||
| Structural brain malformation on MRI | Cerebellar and cerebral atrophy | Cerebellar and cerebral atrophy | Thin corpus callosum and periventricular white matter | Arachnoid cyst | ✚ | ✚ | NP | 6 / 10 | |||
| Cerebellar ataxia/dysmetria | ✚ | ✚ | – | – | NP | NP | NP | 2 / 10 | |||
| Cerebral atrophy | ✚ | ✚ | ✚ | – | ✚ | ✚ | NP | 5 / 10 | |||
| Cerebellar atrophy | ✚ | ✚ | ✚ | – | ✚ | ✚ | NP | 5 / 10 | |||
| Spasticity (Upper/lower) | Lower limb, started at 15 yrs | – | ✚ | ✚ | ✚ | NP | 2 / 4 | 6 / 10 | |||
| Dystonia | – | – | ✚ | – | NP | ✚ | 2 / 4 | 4 / 10 | |||
| Hypertonia | – | – | ✚ | – | NP | NP | 3 / 4 | 4 / 10 | |||
| Hyperreflexia | ✚ | ✚ | – | – | NP | NP | NP | 2 / 10 | |||
| Upper motor neuron findings (extensor plantar response, ankle clonus) | – | ✚ | – | – | NP | NP | NP | 1 / 10 | |||
| Developmental regression | – | – | – | – | NP | ✚ | 1 / 4 | 2 / 10 | |||
| Progressive disorder | ✚ | – | – | – | NP | NP | NP | 1 / 10 | |||
| Other neuropsychiatric disorder | – | – | – | Autism spectrum | NP | NP | 1 / 4 ADHD | 2 / 10 | |||
| Peripheral nervous system | ✚ | ✚ | ✚ | ✚ | NP | NP | NP | 4 / 10 | |||
| Weakness | ✚ | ✚ | ✚ | ✚ | NP | NP | NP | 4 / 10 | |||
| Hyporeflexia/areflexia | – | – | – | ✚ | NP | NP | NP | 1 / 10 | |||
| Distal sensory loss | NP | NP | NP | ✚ | NP | NP | NP | 1 / 10 | |||
| Allodynia | – | – | – | – | NP | NP | NP | 0 / 10 | |||
| Axonal neuropathy on EMG/NCS | NP | NP | NP | ✚ | NP | NP | NP | 1 / 10 | |||
| Muscle paralysis | – | – | – | – | NP | NP | NP | 0 / 10 | |||
| Musculoskeletal | ✚ | ✚ | ✚ | ✚ | ✚ | NP | 2 / 4 | 7 / 10 | |||
| Short stature | ✚ | ✚ | – | – | NP | NP | 1 / 4 | 3 / 10 | |||
| Joint contractures including camptodactyly | Camptodactyly | – | ✚ | – | NP | NP | NP | 2 / 10 | |||
| Scoliosis | ✚ | – | ✚ | Mild | NP | NP | 1 / 4 | 4 / 10 | |||
| Hip subluxation | Bilateral | Right side | Positive, unspecified laterality | – | NP | NP | NP | 3 / 10 | |||
| Coxa valga/vara | – | – | Bilateral coxa valga | NP | NP | NP | NP | 1 / 10 | |||
| Foot anomaly (pes planus/clubfoot) | Pes planus | Talus valgus | – | Clubfoot | Clubfoot | NP | NP | 4 / 10 | |||
| Muscle atrophy | – | – | – | ✚ | NP | NP | NP | 1 / 10 | |||
| Cataplexy | Gelastic | – | – | NP | NP | NP | NP | 1 / 10 | |||
| Urinary system | ✚ | ✚ | NP | – | NP | ✚ | 1 / 4 | 4 / 10 | |||
| Sphincter disturbances | Incontinent | Incontinent | NP | – | NP | NP | NP | 2 / 10 | |||
| Neurogenic bladder | – | – | – | – | NP | NP | 1 / 4 Neurogenic bowel and bladder | 1 / 10 | |||
| Hydronephrosis | – | – | – | – | NP | ✚ | NP | 1 / 10 | |||
| Structural anomalies | Left duplicated collecting system | – | – | – | NP | NP | NP | 1 / 10 | |||
| Gastrointestinal system | – | – | GERD, constipation | – | NP | NP | NP | 1 / 10 | |||
| Skin/Integument | – | – | Hypertrichosis, cutis marmorata | – | NP | NP | NP | 1 / 10 | |||
Abbreviations are as follows: + = positive; – = negative; NP = not phenotyped or reported
We surveyed the literature for all suspected pathogenic heterozygous KIF1A variants reported at the time of preparing this manuscript (Supp. Table S1). A total of 55 unique KIF1A heterozygous variants have been reported in 99 individuals. Local subjects BG2 and BG3 were discovered to have been previously published in a different study (Lee et al., 2015). Variants affecting 17 amino acid residues have been reported in two or more unrelated individuals–including 16 identical variants–suggesting the presence of mutational hotspots. The most frequently reported variant in the literature is p.Thr99Met, accounting for 9 cases (9.1%). Most distinct variants occurring in our local cases (6 out of 9) have been previously reported: p.Arg13His, p.Thr99Met, p.Gly102Ser, p.Ser215Arg, p.Arg216Cys and p.Glu253Lys. Moreover, all 8 of the distinct amino acid residues altered in these 10 patients have been mutated in previously published cases. Variants p.Asn272del, p.Arg13Leu and p.Arg316Gln are novel. The most consistent phenotype related to the previously reported cases is spasticity, observed in 83/99 cases (83.8%). Forty-six out of 55 unique variants (83.6%) are missense variants, with the remaining 9/55 (16.4%) consisting of frameshift variants, nonsense variants and gene deletions. Subjects PERU1 and PERU2 thus constitute the only pathogenic in-frame deletion observed so far.
Out of these 99 individuals harboring pathogenic heterozygous variants, 29 (29%) developed a pure HSP phenotype, and 70 (71%) have additional CNS involvement (ID, developmental delay, brain abnormalities, ADHD, or ASD). Of this group of 70 patients with CNS involvement, 67 (95.7%) of them harbor variants in the motor domain, 64 of which are missense. Of the 29 individuals with pure HSP, 19 (65.5%) harbored a variant in the motor domain.
Mechanisms that explain why some heterozygous variants cause pure HSP and others cause HSP with CNS involvement are not well understood. After reviewing previous cases, we discovered that 23 unique variants were responsible for the 29 reported pure HSP cases. Only 3/23 (13%) of these variants (p.Ser69Leu, p.Arg167Cys, p.Thr258Met) have also been reported to cause HSP with CNS involvement, and these CNS phenotypes are relatively less severe: mild ID, ADHD, and/or minor brain anomalies such as thin corpus callosum (Citterio et al., 2015; Lee et al., 2015; Pennings et al., 2020; Ylikallio et al., 2015). One exception is variant p.Thr258Met, which has been linked not only to mild ID but also to moderate ID with optic nerve atrophy (Cheon et al., 2017). The remaining set of variants have only been reported with CNS involvement; out of these variants causing CNS involvement, the variant associated with the most severe phenotype is p.Glu253Lys–also present in subject BG3–associating with severe developmental delay (non-ambulatory, non-verbal), optic nerve atrophy, seizures, and cerebellar atrophy (Esmaeeli Nieh et al., 2015; Lee et al., 2015; Muir et al., 2019; Samanta & Gokden, 2019). Thus, there is a striking distinction between variants that tend to cause pure HSP vs those causing the vast majority of HSP cases with CNS involvement, indicative of variant-specific phenotypes in heterozygous KIF1A dysfunction. Future functional studies should investigate molecular mechanisms underlying variant-specific phenotypes. One hypothesis is that the spectrum of reported KIF1A pathogenic variants may differentially impact kinesin motor domain processivity, and that distinct neuronal cell types have differential sensitivity to impaired processivity.
Furthermore, KIF1A variants have been reported to cause a variety of ‘distinct’ disorders exhibiting significant phenotypic overlap, suggesting a variable spectrum of clinical presentation associating with a common etiology rather than meaningfully distinct clinical diagnoses. For example, KIF1A heterozygous variants have been reported as causal for both “pure” and “complicated” AD HSP, MRD9, PEHO syndrome, and optic atrophy (Cheon et al., 2017; Esmaeeli Nieh et al., 2015; Lee et al., 2015; Pennings et al., 2020; Raffa et al., 2017; Samanta & Gokden, 2019). Some clinical features are shared between these entities: lower-extremity spasticity, hypotonia, intellectual disability, cerebellar and optic atrophy, hyperreflexia, and convulsion/seizures. Indeed, there are some individuals with KIF1A-related disorders with a “complicated” HSP where their clinical features are similar to those seen in both MRD9 and PEHO syndrome (Langlois et al., 2016). Moreover, even among patients with the same variant there is variable expressivity of phenotype (Langlois et al., 2016; Lee et al., 2015), including in some cases intrafamilial variability of disease severity and progression (Citterio et al., 2015).
Given the inherent similarity between KIF1A-associated clinical entities, as well as the degree of phenotypic variability of KIF1A dysfunction evident at a gene, domain, and even variant level, we suggest classifying cases based on genotype–contextualized by phenotype–rather than the traditional model of classifying cases based on phenotype–contextualized by genotype. We suggest that KIF1A dysfunction is better understood as a phenotypic spectrum rather than a set of distinct–albeit overlapping–clinical entities. Thus, emphasis should be placed on patient variant alleles and specific phenotypes rather than syndromic diagnoses such as MRD9, “pure” vs. “complicated” HSP, etc.
Recently reported novel KIF1A-associated phenotypes include behavioral traits such as autism spectrum disorder (ASD) and attention-deficit/hyperactivity disorder (ADHD) (Kurihara et al., 2020; Tomaselli et al., 2017). One local case (BG4) harboring the same variant as reported by Tomaselli et al. (p.Arg13His) also presented with ADHD. Another patient reported by Kurihara et al. with variant p.Arg13Cys presented with ADHD and ASD, suggesting that substitutions of amino acid residue 13 are more likely to cause ASD and ADHD. Our phenotypic analysis of local cases (Table 1) revealed recurrent, previously unreported phenotypes: hip subluxation, dystonia, and hypertonia. Furthermore, urological abnormalities other than incontinence are observed across three patients: neurogenic bladder/bowel, double collecting system, and hydronephrosis of unspecified etiology–which can be caused by neurogenic bladder or double collecting system. While neurogenic bladder has been previously reported (Okamoto et al., 2014; Urtiaga Valle, Fournier Gil, Ramiro León, & Martínez Menéndez, 2019), double collecting system is a novel phenotype. However, without resolving whether the hydronephrosis of unspecified etiology is caused by a double collecting system, we cannot rule out double collecting system as an incidental finding. We also identified previously unreported phenotypes occurring in single patients: cataplexy, coxa valga (bilateral) and skin abnormalities including hypertrichosis and cutis marmorata. Taken together, our cases and review of the literature reveal a phenotypic expansion of KIF1A-related dysfunction.
In summary, we report a novel pathogenic in-frame deletion (p.Asn272del) in the kinesin motor domain of KIF1A causing a global developmental delay syndrome with skeletal, ocular and significant neuromuscular involvement, including variable expressivity of scoliosis, gelastic cataplexy and double collecting system. Furthermore, this variant occurs in two affected siblings and is likely mosaic in the maternal germline, as blood samples from both parents, in addition to a paternal sperm sample, did not detect the variant at any significant allele fraction. We discuss the range of overlapping Mendelian syndromes reported to associate with KIF1A dysfunction and argue that they are better understood by a genocentric diagnostic model–where genotype is contextualized by phenotype (Hansen et al., 2019). Finally, we present evidence supporting the association of KIF1A dysfunction with hip subluxation and dystonia–observed in multiple patients–as well as additional phenotypic findings observed in one patient which may or may not be incidental, including cataplexy, coxa valga, and double collecting system.
Supplementary Material
Acknowledgements
This work was supported in part by grants UM1 HG008898 from the National Human Genome Research Institute (NHGRI) to the Baylor College of Medicine Center for Common Disease Genetics and UM1 HG006542 from the NHGRI/National Heart, Lung, and Blood Institute (NHLBI) to the Baylor Hopkins Center for Mendelian Genomics. AWH was supported in part by NIH T32 GM08307-26 and the Cullen Foundation. TM is supported by the Uehara Memorial Foundation. JEP was supported by NHGRI K08 HG008986. DP was supported by Clinical Research Training Scholarship in Neuromuscular Disease partnered by the American Academy of Neurology (AAN), American Brain Foundation (ABF) and Muscle Study Group (MSG), and the International Rett Syndrome Foundation (IRSF grant #3701-1). We thank Fritz Sedlazeck, Medhat Mahmoud, and Moez Dawood for their insight with regards to data analysis. We thank the patients and their families for participating in this research.
Footnotes
Editorial Policies and Ethical Considerations
Written informed consent for all individuals in the index family were obtained under a protocol approved by the Instituto Nacional del Niño, Lima Peru committee on Human Research. Patients were counseled by a team of treating physicians, clinical geneticists, neurologists and ophthalmologists where clinical records and images were obtained. Sample BAB11796 was collected after written informed consent in conjunction with the Baylor Hopkins Center for Mendelian Genomics (CMG) (H-29697) study with approval by the institutional review board at Baylor College of Medicine. Other clinical samples (BG1-BG7) were from the Baylor College of Medicine clinical testing laboratories, now incorporated as the Baylor Genetics Laboratories (BG); these data were studied in aggregate in accordance with protocol H-41191 with approval by the institutional review board at Baylor College of Medicine. Subject BG1 was re-contacted and consented to publication of individual-level phenotypic information, also under protocol H-29697. Individual-level phenotypes for subjects BG2 and BG3 were previously published (Lee et al., 2015). Subjects BG4, BG5, BG6 and BG7 were lost to contact, thus no consent was obtained to publish individual-level phenotypes.
In silico prediction data was utilized for the variant identified in Individuals PERU1 and PERU2: MTR and GERP++. MTR scores for amino acid residues were obtained online (http://mtr-viewer.mdhs.unimelb.edu.au/) (Traynelis et al., 2017). GERP++ scores were obtained via the UCSC genome browser GERP track, with specific methodology detailed online (http://genome.ucsc.edu/cgi-bin/hgTracks?hgt_tSearch=track+search) (Davydov et al., 2010; Kent et al., 2002). The final GERP++ score was derived by calculating the mean GERP score across the length of the variant.
Declaration of Interests
JRL has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals and is a co-inventor on multiple US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from the chromosomal microarray analysis and clinical exome sequencing offered in the Baylor Genetics Laboratory (http://baylorgenetics.com).
Data Availability
All novel variants are available on ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/): accession number SCV001192885 for PERU1 and PERU2; SUB8007095 for BAB11796; SCV000807298.2 for BG3; SCV000245497.1 for BG4; SCV000807297.1 for BG7; and submissions SUB7940508, SUB7940532, and SUB7940554 for BG1, BG5, and BG6.
References
- Cheon CK, Lim SH, Kim YM, Kim D, Lee NY, Yoon TS, … Lee JR (2017). Autosomal dominant transmission of complicated hereditary spastic paraplegia due to a dominant negative mutation of KIF1A, SPG30 gene. Scientific Reports, 7(1), 1–11. 10.1038/s41598-017-12999-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citterio A, Arnoldi A, Panzeri E, Merlini L, D’Angelo MG, Musumeci O, … Bassi MT (2015). Variants in KIF1A gene in dominant and sporadic forms of hereditary spastic paraparesis. Journal of Neurology, 262(12), 2684–2690. 10.1007/s00415-015-7899-9 [DOI] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, & Batzoglou S (2010). Identifying a High Fraction of the Human Genome to be under Selective Constraint Using GERP++. PLoS Computational Biology, 6(12), e1001025 10.1371/journal.pcbi.1001025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esmaeeli Nieh S, Madou MRZ, Sirajuddin M, Fregeau B, Mcknight D, Lexa K, … Sherr EH (2015). De novo mutations in KIF1A cause progressive encephalopathy and brain atrophy. Annals of Clinical and Translational Neurology, 2(6), 623–635. 10.1002/acn3.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Araki Y, Lin DT, Yoshizawa Y, Higashi K, … Michaud JL (2011). Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. American Journal of Human Genetics, 88(3), 306–316. 10.1016/j.ajhg.2011.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AW, Murugan M, Li H, Khayat MM, Wang L, Rosenfeld J, … Gibbs RA (2019). A Genocentric Approach to Discovery of Mendelian Disorders. American Journal of Human Genetics, 105(5), 974–986. 10.1016/j.ajhg.2019.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedera P (2000). Hereditary Spastic Paraplegia Overview. GeneReviews. [Google Scholar]
- Huganir RL, & Nicoll RA (2013). AMPARs and synaptic plasticity: The last 25 years. Neuron, 80(3), 704–717. 10.1016/j.neuron.2013.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo L, Yue Y, Ren J, Yu J, Liu J, Yu Y, … Feng W (2012). The CC1-FHA tandem as a central hub for controlling the dimerization and activation of kinesin-3 KIF1A. Structure, 20(9), 1550–1561. 10.1016/j.str.2012.07.002 [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, & Haussler a. D. (2002). The Human Genome Browser at UCSC. Genome Research, 12(6), 996–1006. 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebe S, Lossos A, Azzedine H, Mundwiller E, Sheffer R, Gaussen M, … Stevanin G (2012). KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: Distinct phenotypes according to the nature of the mutations. European Journal of Human Genetics, 20(6), 645–649. 10.1038/ejhg.2011.261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara M, Ishiura H, Bannai T, Mitsui J, Yoshimura J, Morishita S, … Tsuji S (2020). A Novel de novo KIF1A Mutation in a Patient with Autism, Hyperactivity, Epilepsy, Sensory Disturbance, And Spastic Paraplegia. Intern Med, 59(6), 839–842. 10.2169/internalmedicine.3661-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois S, Tarailo-Graovac M, Sayson B, Drögemöller B, Swenerton A, Ross CJD, … Van Karnebeek CDM (2016). De novo dominant variants affecting the motor domain of KIF1A are a cause of PEHO syndrome. European Journal of Human Genetics, 24(6), 949–953. 10.1038/ejhg.2015.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JR, Shin H, Ko J, Choi J, Lee H, & Kim E (2003). Characterization of the movement of the kinesin motor KIF1A in living cultured neurons. Journal of Biological Chemistry, 278(4), 2624–2629. 10.1074/jbc.M211152200 [DOI] [PubMed] [Google Scholar]
- Lee JR, Srour M, Kim D, Hamdan FF, Lim SH, Brunel-Guitton C, … Michaud JL (2015). De novo mutations in the motor domain of KIF1A cause cognitive impairment, spastic paraparesis, axonal neuropathy, and cerebellar atrophy. Human Mutation, 36(1), 69–78. 10.1002/humu.22709 [DOI] [PubMed] [Google Scholar]
- Muir AM, Myers CT, Nguyen NT, Saykally J, Craiu D, De Jonghe P, … Mefford HC (2019). Genetic heterogeneity in infantile spasms. Epilepsy Research, 156(June), 106181 10.1016/j.eplepsyres.2019.106181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Yamazaki H, Sekine-Aizawa Y, & Hirokawa N (1995). The neuron-specific kinesin superfamily protein KIF1A is a uniqye monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell, 81(5), 769–780. 10.1016/0092-8674(95)90538-3 [DOI] [PubMed] [Google Scholar]
- Okamoto N, Miya F, Tsunoda T, Yanagihara K, Kato M, Saitoh S, … Kosaki K (2014). KIF1A mutation in a patient with progressive neurodegeneration. Journal of Human Genetics, 59(11), 639–641. 10.1038/jhg.2014.80 [DOI] [PubMed] [Google Scholar]
- Oriola D, Roth S, Dogterom M, & Casademunt J (2015). Formation of helical membrane tubes around microtubules by single-headed kinesin KIF1A. Nature Communications, 6, 1–8. 10.1038/ncomms9025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennings M, Schouten MI, van Gaalen J, Meijer RPP, de Bot ST, Kriek M, … Kamsteeg E-J (2020). KIF1A variants are a frequent cause of autosomal dominant hereditary spastic paraplegia. European Journal of Human Genetics, 28(1), 40–49. 10.1038/s41431-019-0497-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey JE, O’Donnell-Luria AH, Chong JX, Harel T, Jhangiani SN, Coban Akdemir ZH, … Centers for Mendelian Genomics. (2019). Insights into genetics, human biology and disease gleaned from family based genomic studies. Genetics in Medicine, 21(4), 798–812. 10.1038/s41436-018-0408-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffa L, Matton MP, Michaud J, Rossignol E, Decarie JC, & Ospina LH (2017). Optic nerve hypoplasia in a patient with a de novo KIF1A heterozygous mutation. Canadian Journal of Ophthalmology, 52(5), e169–e171. 10.1016/j.jcjo.2017.02.021 [DOI] [PubMed] [Google Scholar]
- Reichardt LF (2006). Neurotrophin-regulated signalling pathways. Philosophical Transactions of the Royal Society B: Biological Sciences, 361(1473), 1545–1564. 10.1098/rstb.2006.1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta D, & Gokden M (2019). PEHO syndrome: KIF1A mutation and decreased activity of mitochondrial respiratory chain complex. Journal of Clinical Neuroscience, 61, 298–301. 10.1016/j.jocn.2018.10.091 [DOI] [PubMed] [Google Scholar]
- Schimert KI, Budaitis BG, Reinemann DN, Lang MJ, & Verhey KJ (2019). Intracellular cargo transport by single-headed kinesin motors. Proceedings of the National Academy of Sciences of the United States of America, 116(13), 6152–6161. 10.1073/pnas.1817924116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Wyszynski M, Huh KH, Valtschanoff JG, Lee JR, Ko J, … Kim E (2003). Association of the kinesin motor KIF1A with the multimodular protein liprin-α. Journal of Biological Chemistry, 278(13), 11393–11401. 10.1074/jbc.M211874200 [DOI] [PubMed] [Google Scholar]
- Soppina V, Norris SR, Dizaji AS, Kortus M, Veatch S, Peckham M, & Verhey KJ (2014). Dimerization of mammalian kinesin-3 motors results in superprocessive motion. Proceedings of the National Academy of Sciences of the United States of America, 111(15), 5562–5567. 10.1073/pnas.1400759111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaselli PJ, Rossor AM, Horga A, Laura M, Blake JC, Houlden H, & Reilly MM (2017). A de novo dominant mutation in KIF1A associated with axonal neuropathy, spasticity and autism spectrum disorder. Journal of the Peripheral Nervous System, 22(4), 460–463. 10.1111/jns.12235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis J, Silk M, Wang Q, Berkovic SF, Liu L, Ascher DB, … Petrovski S (2017). Optimizing genomic medicine in epilepsy through a gene-customized approach to missense variant interpretation. Genome Research, 27(10), 1715–1729. 10.1101/gr.226589.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urtiaga Valle S, Fournier Gil B, Ramiro León MS, & Martínez Menéndez B (2019). Usefulness of exome sequencing in the study of spastic paraparesis and cerebellar atrophy: De novo mutation of the KIF1A gene, a new hope in prognosis. NEUROLOGÍA. 10.1016/j.nrl.2018.07.001 [DOI] [PubMed] [Google Scholar]
- Wang J, Zhang Q, Chen Y, Yu S, Wu X, & Bao X (2019). Rett and Rett-like syndrome: Expanding the genetic spectrum to KIF1A and GRIN1 gene. Molecular Genetics and Genomic Medicine, 7(11), 1–8. 10.1002/mgg3.968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, … Eng CM (2013). Clinical whole-exome sequencing for the diagnosis of mendelian disorders. The New England Journal of Medicine, 369(16), 1502–1511. 10.1056/NEJMoa1306555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylikallio E, Kim D, Isohanni P, Auranen M, Kim E, Lönnqvist T, & Tyynismaa H (2015). Dominant transmission of de novo KIF1A motor domain variant underlying pure spastic paraplegia. European Journal of Human Genetics, 23(10), 1427–1430. 10.1038/ejhg.2014.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.