

Abstract
Objectives:
African Americans are at greater risk for developing Alzheimer disease (AD) dementia than non-Hispanic Whites. In addition to biological considerations (e.g., genetic influences; comorbid disorders), social and environmental factors may increase the risk of AD dementia. This paper (1) Assesses neuroimaging biomarkers of amyloid (A), tau (T), and neurodegeneration (N) for potential racial differences. (2) Considers mediating effects of socioeconomic status (SES) and measures of small vessel and cardiovascular disease on observed race differences.
Methods:
Imaging measures of AT(N) (amyloid and tau positron emission tomography [PET] structural magnetic resonance imaging (MRI), and resting state functional connectivity (rs-fc) were collected from African American (n=131) and White (n=685) cognitively normal participants age 45 years and older. Measures of small vessel and cardiovascular disease (white matter hyperintensities [WMH] on MRI, blood pressure, body mass index [BMI]) and area-based socioeconomic status (SES) were included in mediation analyses.
Results:
Compared to White participants, African American participants had greater neurodegeneration, as measured by decreased cortical volumes (Cohen’s f2 = 0.05, p < 0.001). SES mediated the relationship between race and cortical volumes. There were no significant race effects for amyloid, tau, or rs-fc signature.
Interpretation:
Modifiable factors such as differences in social contexts and resources, particularly area-level SES, may contribute to observed racial differences in AD. Future studies should emphasize collection of relevant psychosocial factors in addition to the development of intentional diversity and inclusion efforts to improve the racial/ethnic and socioeconomic representativeness of AD studies.
Introduction
The prevalence of Alzheimer disease (AD) dementia is expected to continue to increase as the population of the United States (US) ages. There were an estimated 4.4 million African Americans aged 65 years and older in the US in 2016, a number projected to increase to approximately 12.1 million by 20601. Nonetheless, African Americans are significantly underrepresented in extant AD research. Recent work has called for not only greater diversity and inclusion in research studies, but also a more holistic focus on the effects of ethno-racial factors, genetics, and social determinants of health (SDOH) in studies of racial differences in AD2–4.
Race and AD Research
Many, but not all5,6 studies report two-fold7 to four-fold8 increased risk of AD dementia for African Americans compared with non-Hispanic whites, although addressing such purported differences is complex. In addition to SDOH, other factors that may contribute to increased risk include test bias and psychosocial and behavioral variables9–11, as well as comorbid disorders such as cardiovascular disease and diabetes9,12,13.
Race-related differences in the concentrations of cerebrospinal fluid (CSF) total tau and tau phosphorylated at site 181 (p-tau181) have recently been reported14,15. However, these studies did not evaluate potential racial differences using the amyloid (A), tau (T), neurodegeneration (N) framework that has been proposed for AD progression16. Moreover, these studies also did not examine the effects of cardiovascular disease and SDOH.
Interaction between Race and Social Economic Status (SES)
The interaction between SES and race in relation to AD biomarkers is poorly understood. Although it is accepted that socioeconomic factors affect health outcomes, substantial limitations exist with commonly used measurements of SES. Within AD studies, the most common approach to correcting for differences in SES is by controlling for years of education17. However, due to widespread racial residential segregation, there are fundamental differences in quality of education across race/ethnicity18–20. For example, an analysis of older Caribbean-born African American individuals compared to US-born African Americans demonstrated that differences in neuropsychological test performance between the two groups was primarily explained by higher quality of education among the Caribbean-born African American cohort22. It is also critical to account for the influence of social factors other than educational quality, such as social and contextual elements and racial residential segregation, for the risk of developing AD4,21.
Current Study
Because our earlier report on racial differences in molecular biomarkers of AD did not have sufficient data for analysis from [18F] Flortaucipir positron emission tomography (PET), we now examine the possibility of cross-sectional racial differences relative to the AT(N) framework in cognitively normal (as ascertained by a Clinical Dementia Rating® [CDR®] of 023) participants using both amyloid and tau PET and brain magnetic resonance imaging (MRI). Due to the limited number of CDR > 0 African Americans with tau PET, only CDR 0 individuals were included in the present analyses. We also focused on potentially modifiable sources related to racial differences, including the mediating effects of small vessel and cardiovascular disease as well as area-based SES. Investigating the effects of area-based SES measures represents a proxy approach to understanding an individual’s access to quality education, healthcare, and occupational and economic opportunities. The Area Deprivation Index (ADI)24,25 was employed to investigate the effects of area-based SES on the relationship between race and AD pathology.
Materials and Methods
Participants
Data from 816 community-dwelling participants (Table 1) enrolled in longitudinal studies of memory and aging at the Knight Alzheimer Disease Research Center (ADRC) at Washington University in St. Louis were used. The majority of these community-living participants were from the greater St. Louis Metropolitan area. ADRC participants were recruited for research-only purposes and did not include clinic-based patients. Recruitment for African Americans and Whites took place through presentations and outreach efforts in the community, through snowball recruitment referrals from past participants, and through physician referral.14 For this cohort, 47% were recruited via word of mouth (i.e., a relative or friend mentions our studies to them); Knight ADRC outreach efforts accounted for 30% of the recruits; referrals from not-for-profit organizations (i.e., Alzheimer’s Association) accounted for 13%; and physician referrals accounted for 10%14. The Knight ADRC also established an African American Advisory Board in 2000, which guides in the recruitment of participants of color. Although individuals are only recruited if they are eligible for all biomarker studies, lumbar puncture expectations are waived for African Americans. Hence, our cohort is ~19% African American. We continue to explore strategies for the effective recruitment and retention of people of color in our cohort to reflect the diversity of the region.
Table 1.
Participant demographics, stratified by race
| African American | White | p | |
|---|---|---|---|
| n | 131 | 685 | |
| Age (years old) (mean (SD)) | 70.64 (8.3) | 71.64 (9.0) | 0.247 |
| Sex = Male (%) | 45 (34.4) | 282 (41.2) | 0.164 |
| Education (years) (mean (SD)) | 15.36 (2.8) | 16.12 (2.5) | 0.002 |
| APOE ε4+ (%) | 54 (42.2) | 228 (33.6) | 0.082 |
| PRS (mean (SD)) | 32.23 (12.2) | 26.62 (22.7) | <0.001 |
| AD Signature Z (mean (SD)) | −0.26 (0.6) | 0.10 (0.6) | <0.001 |
| Subset of participants who had completed PET amyloid | |||
| n | 70 | 434 | |
| Age (years old) (mean (SD)) | 67.56 (38.6) | 69.62 (9.1) | 0.076 |
| Sex = Male (%) | 27 (38.6) | 166 (38.2) | 1.000 |
| Education (years) (mean (SD)) | 15.19 (2.8) | 16.06 (2.6) | 0.010 |
| APOE ε4+ (%) | 27 (39.7) | 143 (33.2) | 0.359 |
| PRS (mean (SD)) | 31.56 (11.9) | 26.72 (10.5) | <0.001 |
| Centiloid (mean (SD)) | 5.8 (14.9) | 15.8 (30.7) | 0.051 |
| Subset of participants who had completed PET tau | |||
| n | 34 | 262 | |
| Age (years old) (mean (SD)) | 68.82 (5.7) | 69.70 (8.3) | 0.552 |
| Sex = Male (%) | 15 (44.1) | 111 (42.4) | 0.992 |
| Education (years) (mean (SD)) | 15.15 (2.5) | 16.52 (2.2) | 0.001 |
| APOE ε4+ (%) | 13 (38.2) | 91 (34.7) | 0.832 |
| PRS (mean SD)) | 28.79 (11.7) | 26.73 (11.8) | 0.335 |
| Tauopathy (mean (SD)) | 1.2 (0.2) | 1.2 (0.2) | 0.761 |
| Subset of participants who had completed resting state functional MRI | |||
| n | 55 | 317 | |
| Age (years old) (mean (SD)) | 69.03 (9.03) | 71.24 (9.40) | 0.107 |
| Sex = Male (%) | 19 (34.5) | 133 (42.0) | 0.377 |
| Education (years) (mean (SD)) | 16.09 (2.61) | 15.83 (2.50) | 0.478 |
| APOE ε4+ (%) | 24 (46.3) | 97 (30.9) | 0.039 |
| PRS (mean (SD)) | 33.80 (10.9) | 26.55 (10.8) | <0.001 |
| Global rs-fc Signature | 0.91 (0.14) | 0.94 (0.15) | 0.313 |
Note. SD = standard deviation; APOE ε4+ = APOE ε44 positive; AD = Alzheimer’s disease; rs-fc = resting-state functional connectivity; PRS = polygenic risk score
Participants were included if they had at least one clinical visit that included a CDR assessment and at least one MRI. Subsets of participants had PET imaging and/or resting state functional MRI (rs-fc), cardiovascular measures (systolic blood pressure, body mass index [BMI], and white matter hyperintensities [WMH] as extracted from structural MRI), and an available home address from which to obtain their ADI26. All measures were collected within a 2-year window. Participant race, sex, and years of education were self-reported. Race was subsequently verified by genetic analysis of ancestry. There were no differences between self-reports and ancestry. This study was approved by the Washington University in St. Louis Institutional Review Board, and each participant provided signed, informed consent.
APOE Status and Polygenic Risk Scores
DNA samples were collected at enrollment and genotyped using either an Illumina 610 or Omniexpress chip. Genotyping methods were previously published27,28. To control for effects of apolipoprotein ε4 (APOE ε4) on individuals in this analysis, APOE status was converted from a genotype to a binary variable. Participants either had at least one copy of the APOE ε4 allele (“APOE ε4 positive”) or had no copies of the allele (“APOE ε4 negative”). For the polygenic risk score (PRS) for AD, weighted scores were calculated by using a logarithm of base 2 transformation on single nucleotide polymorphisms (SNPs) as reported in the International Genomics of Alzheimer’s Project (IGAP) study29. SNPs utilized for the score had either a high genotyping rate (>90%) or were reasonable proxies to the IGAP hits.
Small Vessel Disease and Cardiovascular Risk Factors
Previous work has suggested that increased cognitive risks posed by cardiovascular- and small vessel disease-related issues could explain AD disparities9,12,13. In this work, we considered three measures related to small vessel disease and cardiovascular risk factors: blood pressure (cardiovascular), BMI (cardiovascular), and WMH volumes (small vessel). Systolic blood pressure, height, and weight were collected at each annual visit. WMH volumes were estimated by applying the lesion segmentation toolbox from SPM to T2-weighted fluid attenuated inversion recovery (FLAIR) images obtained during participant MRI scanning visits30.
Socioeconomic Status (SES)
The ADI was designed to rank neighborhoods by SES and to assess neighborhood disadvantage based on 17 factors including measures of poverty, education, housing, and employment indicators from US Census Data and the 2015 American Community Survey Five Year Estimates24–26. The ADI represents a granular measure of the current socioeconomic level of the participant’s residence and can be used to rank neighborhoods at the city, state, or national level. For this analysis, we employed the 100-point national index, where 0 represents the area of least deprivation (i.e. participants have the greatest SES), and 100 represents the area of greatest deprivation. ADI was calculated at the census-tract level and was assigned based on a participant’s mailing zip code, which was collected at each annual visit. ADI was not available for all participants due to invalid address format, residence in a high-occupancy building for which ADI is not published, or the participant providing a post office box for their mailing address. Additional details regarding the ADI can be found at https://www.neighborhoodatlas.medicine.wisc.edu.
In order to compare the Knight ADRC sample to the state population, we extracted the racial composition of each census tract for the state of Missouri. We used census tract population estimates based on the American Community Survey Five Year Estimate31 and merged these populations with published ADI26.
PET Imaging of Amyloid and Tau
Imaging studies were obtained at baseline and then every 3 years thereafter. PET images were acquired within 2 years (mean = 0.6 ± 1.2 years) of MRI using methodology previously described32,33. PET data were processed using the PET Unified Pipeline (github.com/ysu001/PUP), which uses regions of interest (ROI) defined using the FreeSurfer 5.3 (Martinos Center for Biomedical Imaging, Charlestown, Massachusetts, USA) Desikan-Killiany atlas. Data were transformed into standardized uptake ratios (SUVRs) using cerebellar gray as a reference and partial volume corrected by calculating regional spread functions as part of a geometric transfer matrix framework34. PET amyloid imaging was performed with either [11C] Pittsburgh Compound B (PiB) or [18F]-Flobetapir (AV45)35,36. The time window for quantification was 30–60 minutes post-injection for PiB and 50–70 minutes for AV45. Centiloids were used to harmonize measures from these two different tracers32,37.
PET tau imaging was performed using [18F]-Flortaucipir (AV1451)38 with SUVRs calculated for the 80–100-minute post-injection window. A summary measure of tauopathy, previously defined as the arithmetic mean of the amygdala, entorhinal cortex, inferior temporal region, and lateral occipital regions based on FreeSurfer 5.3 segmentation, was calculated for each participant36.
MRI Acquisition and Pre-Processing
MRI images (including T1, T2, and blood oxygen level dependent [BOLD] rs-fc) were obtained via 3T Siemens scanners. T1-weighted scans were segmented with FreeSurfer 5.3. Previous work has identified the temporal (inferior, middle, and superior), parietal (inferior and superior), entorhinal cortex, precuneus, and hippocampus as the regions that are most affected by disease and change the earliest39. We converted volumes to Z scores separately in the left and right hemispheres relative to the entire cohort, and then averaged them to obtain an “AD Signature Region.” The AD Signature Region creates a summary metric that succinctly describes brain volume atrophy due to AD39.
BOLD rs-fc was collected as previously described, using 36 contiguous slices to obtain total brain coverage40,41. Two hundred ninety-eight functional ROIs (predefined networks included the sensorimotor, sensorimotor-lateral, cerebellar, cingulo-opercular, auditory, ventral attention, visual, auditory, salience, default mode, memory, dorsal attention, subcortical, and frontoparietal networks) were converted into a 298 × 298 rs-fc matrix for each individual. The mean time series for each ROI was calculated, and then the pairwise correlation between each ROI was calculated. The average intra- and inter-network correlation for each of the 13 previously described networks was computed yielding a 13 × 13 summary matrix for each individual42.
In order to obtain a global measure of rs-fc, values from the 13 intra-network connections42 were extracted from the 13 × 13 composite score matrix as described previously43. We performed singular value decomposition on the 13 scaled and centered rs-fc composite scores. We then multiplied the eigenvector corresponding to the first principal component by each individual’s intra-network connections (the diagonal of each individual’s composite resting state matrix) to generate a single summary value describing an individual’s global rs-fc signature40,43. Intra-network connections change with healthy aging44, as well as conversion to symptomatic AD41. Overall, a higher global rs-fc signature value indicates greater within network connections. This value decreases over time as individuals age, but decreases much more rapidly in individuals who develop symptomatic AD40,43. Similar to the “AD signature region,” the “AD global rs-fc signature” creates a summary metric that succinctly describes functional connectivity changes due to AD.
Statistical Analysis
For demographic variables, group differences between African Americans and Whites were compared using t-tests for continuous variables and Chi-square tests for categorical variables. With regard to the AT(N) criteria, we performed linear regression for four separate models. Centiloid (total cortical amyloid burden), PET-AV1451 (tauopathy burden), AD signature volume, and AD global rs-fc signature were the dependent variables for each respective model. We tested for model differences based on race after controlling for age, sex, APOE ε4 status, and PRS. There were no interactions between race and the aforementioned covariates (p’s > 0.05). This model was developed based on an a priori hypotheses. Due to the nature of many of these models, we utilized a method of heteroskedasticity consistent estimators45 to generate models that were robust to heteroskedastic covariance. We calculated the effect size for race, using Cohen’s d, in order to assess the magnitude of racial differences.
Based on previously hypothesized potential sources of racial difference, we also compared systolic blood pressure, BMI, WMH, and SES based on race. We used an analysis of covariance (ANCOVA), correcting for differences in age, sex, APOE ε4 status, and PRS, and performed post hoc Cohen’s d effect size calculations.
In order to assess whether the participants of the Knight ADRC were representative of the population of the state of Missouri with respect to SES, we extracted census demographic information for the state of Missouri utilizing the R package tidycensus46 and merged it with census tract–specific ADI values26. We calculated Cliff’s delta, rather than Cohen’s d, to compare the participant sample to the statewide population due to the non-parametric nature of ADI for African Americans living in Missouri.
Finally, we performed mediation analysis with multiple mediators using the R package {mma}47. A visualization of the hypothesized mechanisms behind the racial differences identified is shown in Figure 1. We assessed the direct effect of race, taking into account the effects of race, sex, APOE ε4 status, and PRS on the four imaging parameters. We considered four simultaneous mediators: systolic blood pressure, BMI, WMH, and SES as described by ADI. We used the multiplicative algebraic reconstruction technique (MART method), assuming a nonlinear relationship between each mediating factor and the response variables. We performed 500 iterations in order to bootstrap confidence intervals, allowing for non-parametric distributions of mediation effects. We then repeated this analysis, instead using years of education as the SES variable, as many studies employ years of education as a proxy of SES17. All analyses were performed using R.
Figure 1. Our hypothesized mechanism of mediation.
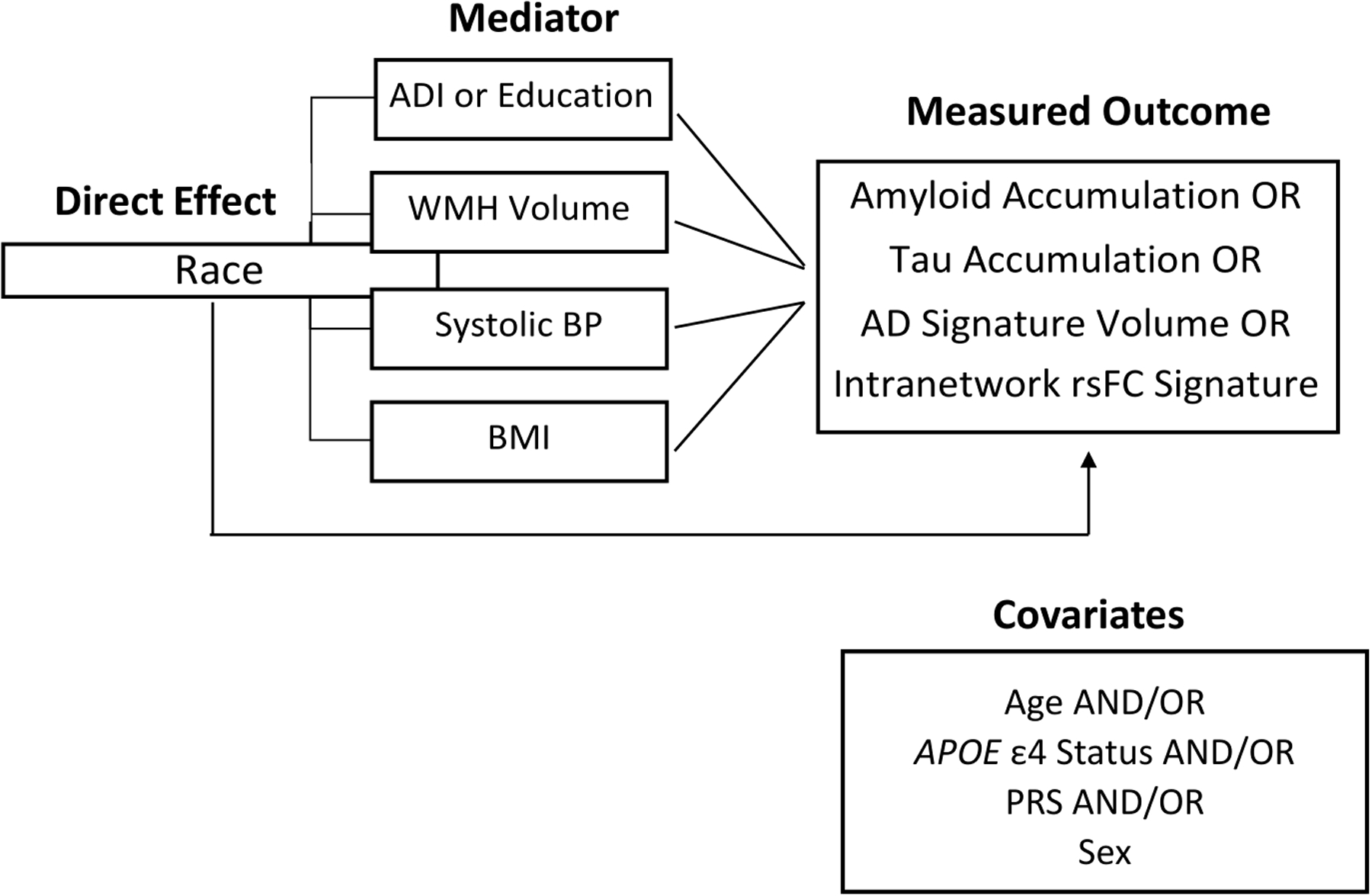
We aimed to test whether socioeconomic status (Area Deprivation Index [ADI]) or any of three small vessel and cardiovascular-related disease factors have a mediating impact of the effect of race on the AT(N) measures used throughout the analyses. In addition to forcing the four hypothesized mediators into the same model, we allowed for the consideration of four other key covariates—age, APOE ε4 status, sex, and polygenic risk score (PRS)—as possible mediators of the direct race effect.
WMH = white matter hyperintensities; BP = blood pressure; BMI = body mass index; rs-fc = resting state functional connectivity; AD = Alzheimer’s disease
RESULTS
Demographics
Overall, the sample included 685 White participants and 131 African American participants (Table 1). Whites generally reported greater levels of education compared to African American participants (p < 0.01). Additionally, African American participants had greater PRS (p < 0.001), although the two groups did not significantly differ in regard to age, sex distribution, or incidence of APOE ε4 allele presence.
Among African American participants, ADI values were not significantly different from ADI values drawn from the state of Missouri’s African American population (Cliff’s Delta: 0.0849; 95% CI: −0.0198, 0.188). However, White participants in this sample were significantly more affluent, according to ADI values, compared with values from the state of Missouri’s White population (Cliff’s Delta: 0.395; 95% CI: 0.360, 0.428; Fig 2).
Figure 2. African American participants in this study were a representative sample of the Missouri statewide African American population with respect to Area Deprivation Index (ADI).
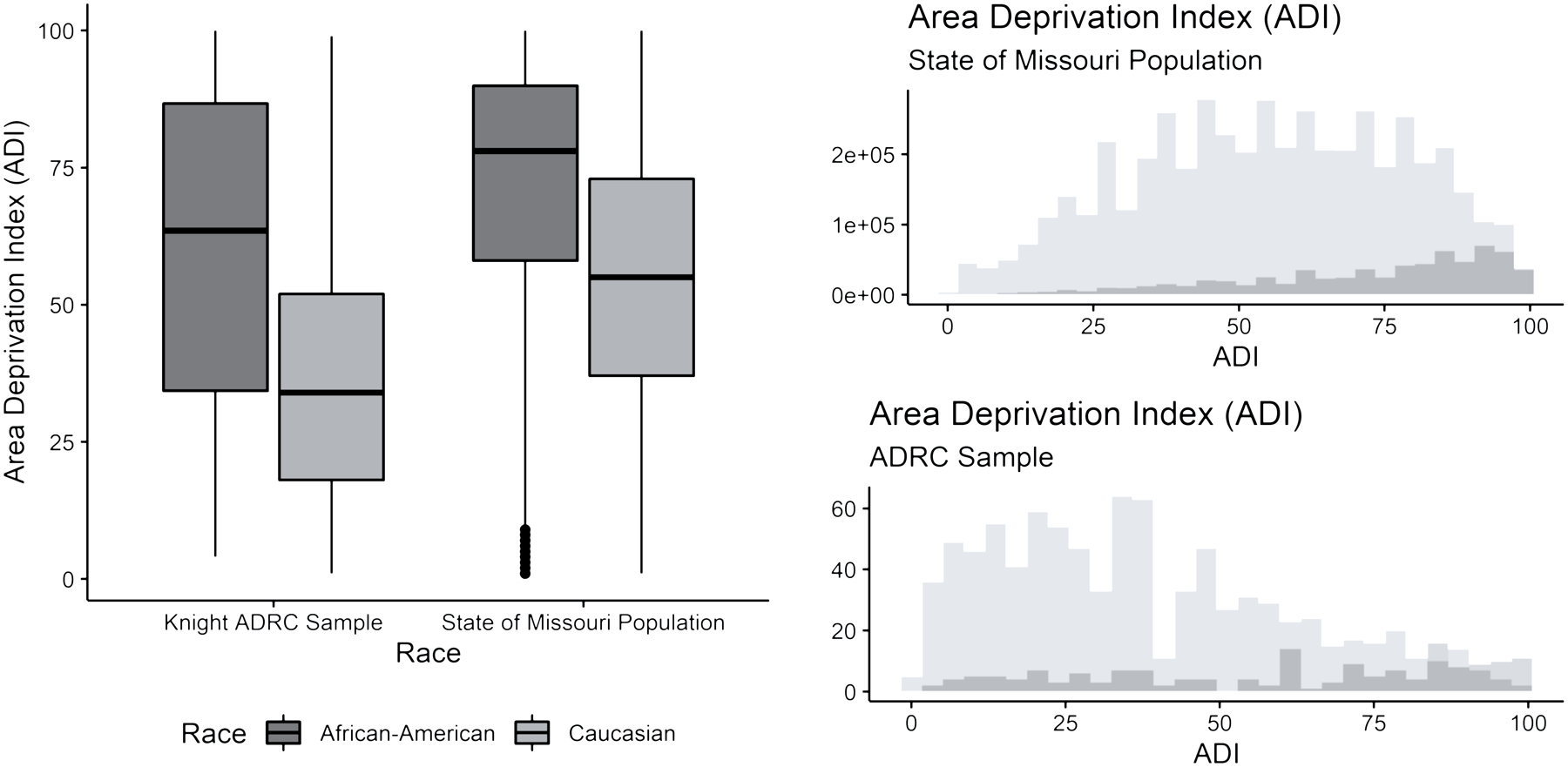
White participants in this study were significantly more affluent than the Missouri statewide White population.
ADRC = Alzheimer Disease Research Center
Amyloid
As we found previously (Morris 2019), amyloid deposition did not significantly differ by race (Cohen’s f2 = 0.008, p = 0.063). Greater amyloid burden was, however, associated with older age (Cohen’s f2 = 0.13, p < 0.001), and having at least one copy of the APOE ε4 allele (Cohen’s f2 = 0.09, p < 0.001). Regression coefficients and standard deviations are available in Supplement (Table S.1)
Tau
There was no difference between African American and White participants with respect to tau PET accumulation (Cohen’s f2 = 0.0003, p = 0.628; Fig. 3b). However, women had increased levels of tau compared to men (Cohen’s f2 = 0.05, p < 0.001), as did older individuals (Cohen’s f2 = 0.10, p < 0.001). There was also an effect of APOE ε4 status (Cohen’s f2 = 0.02, p < 0.01), and PRS (Cohen’s f2 = 0.004, p < 0.05) such that individuals with presence of at least one APOE ε4 allele and higher risk scores had greater tau burden. Full regression tables can be viewed in the supplement (Supplemental Table S.1).
Figure 3.
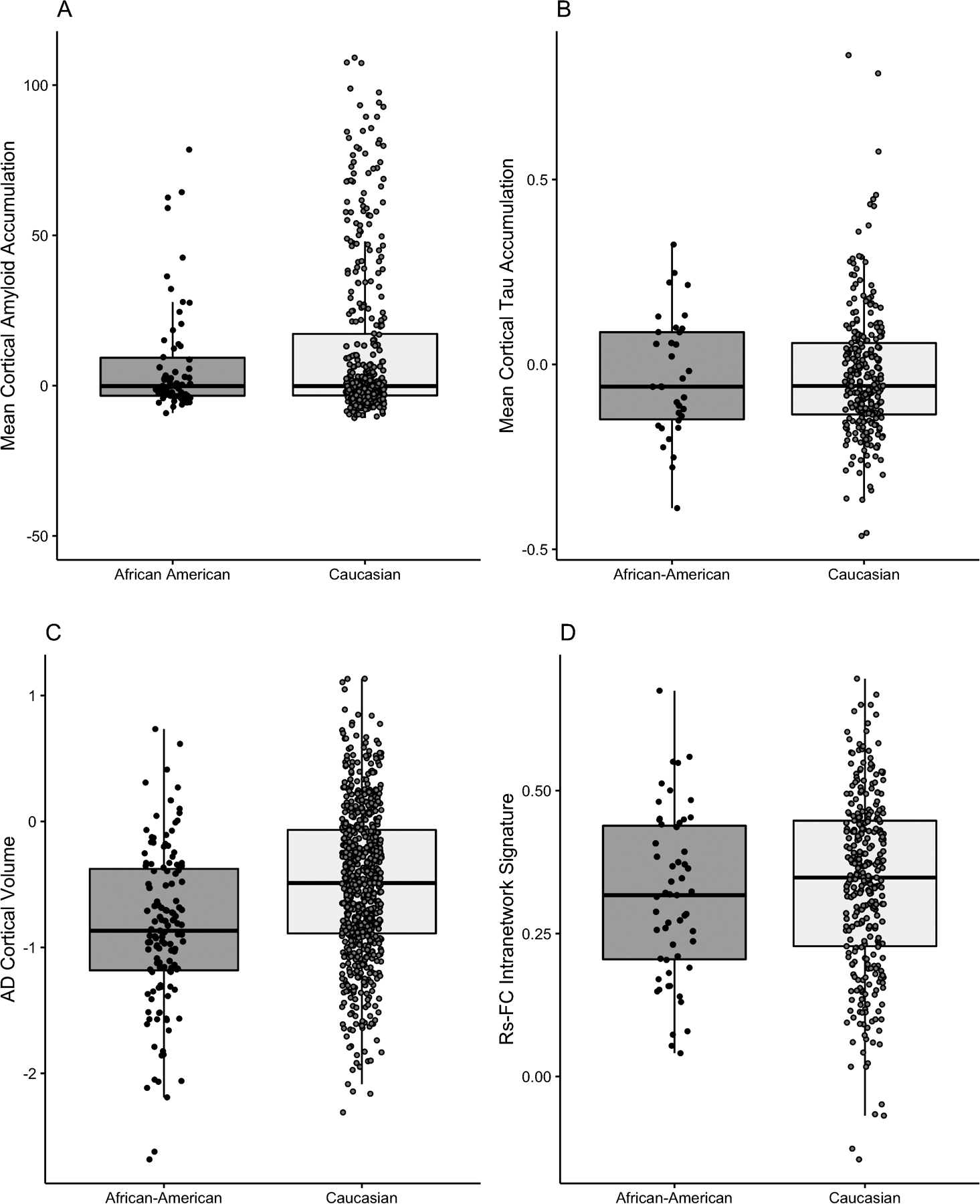
Racial differences are observable in Alzheimer disease (AD) signature volume.
Neurodegeneration
Whites had a significantly larger AD signature volume (Cohen’s f2 = 0.05, p < 0.001) compared to African American participants. There were also effects of sex (Cohen’s f2 = 0.004, p < 0.001) and age (Cohen’s f2 = 0.55, p < 0.001), as males and younger individuals had greater volumes. Conversely, there was no race effect for the rs-fc signature (Cohen’s f2 = 0.55, p = 0.003) although there was an effect of sex (Cohen’s f2 = 0.01, p < 0.01) and age (Cohen’s f2 = 0.09, p < 0.001) similar to that of the AD signature volume. Full regression tables can be viewed in the supplement (Supplemental Table S.1). The observed racial differences were robust to model selection.
Mediation Analysis
Each of the mechanisms hypothesized to explain pathological difference (ADI, blood pressure, BMI, and WMH) differed by race (Fig. 4). African Americans had significantly higher ADI (Cohen’s f2 = 0.22, p < 0.001), higher blood pressure (Cohen’s f2 = 0.02, p < 0.001), significantly higher BMI (Cohen’s f2 = 0.03, p < 0.001), and significantly greater WMH volumes (Cohen’s f2 = 0.009, p < 0.01) than Whites.
Figure 4. Racial differences exist across all four hypothesized mediating factors.
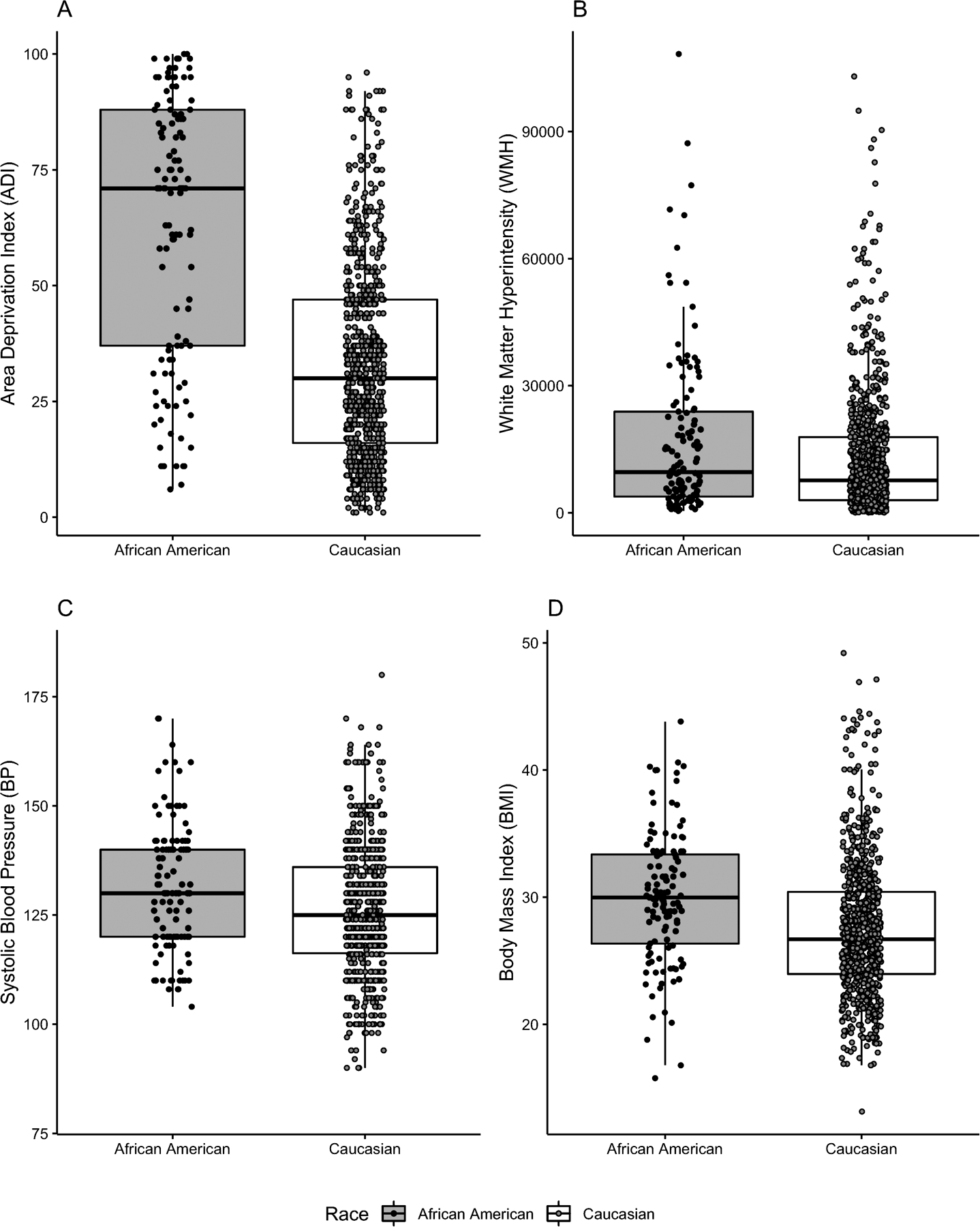
Low values of ADI signify low area deprivation, which translates to higher social economic status (SES).
In Figure 5, the effect of being African American compared to White on the AT(N) measures is shown as the total race effect. The total effect is a sum of the direct and indirect effects. The direct effect indicates the size of the effect not explained by any of the mediating factors. The indirect effect indicates the size of the effect explained by the mediating factors. For the AD cortical signature, the only biomarker to demonstrate a race effect, the estimated direct effect of race was 88% of the total effect, whereas the estimated indirect effect of the proposed mediating factors made up the remaining 12%. ADI accounted for nearly 97% of the indirect effect (95% CI: 0.010, 0.075; Fig. 5, Table 2). There were no other significant mediators. We repeated the multiple mediation analysis using education instead of ADI as the SES variable, while still including blood pressure, BMI, and WMH volumes, and found no difference in the results (Table 3).
Figure 5. Area Deprivation Index (ADI) mediates the relationship between race and Alzheimer Disease (AD) signature volume.
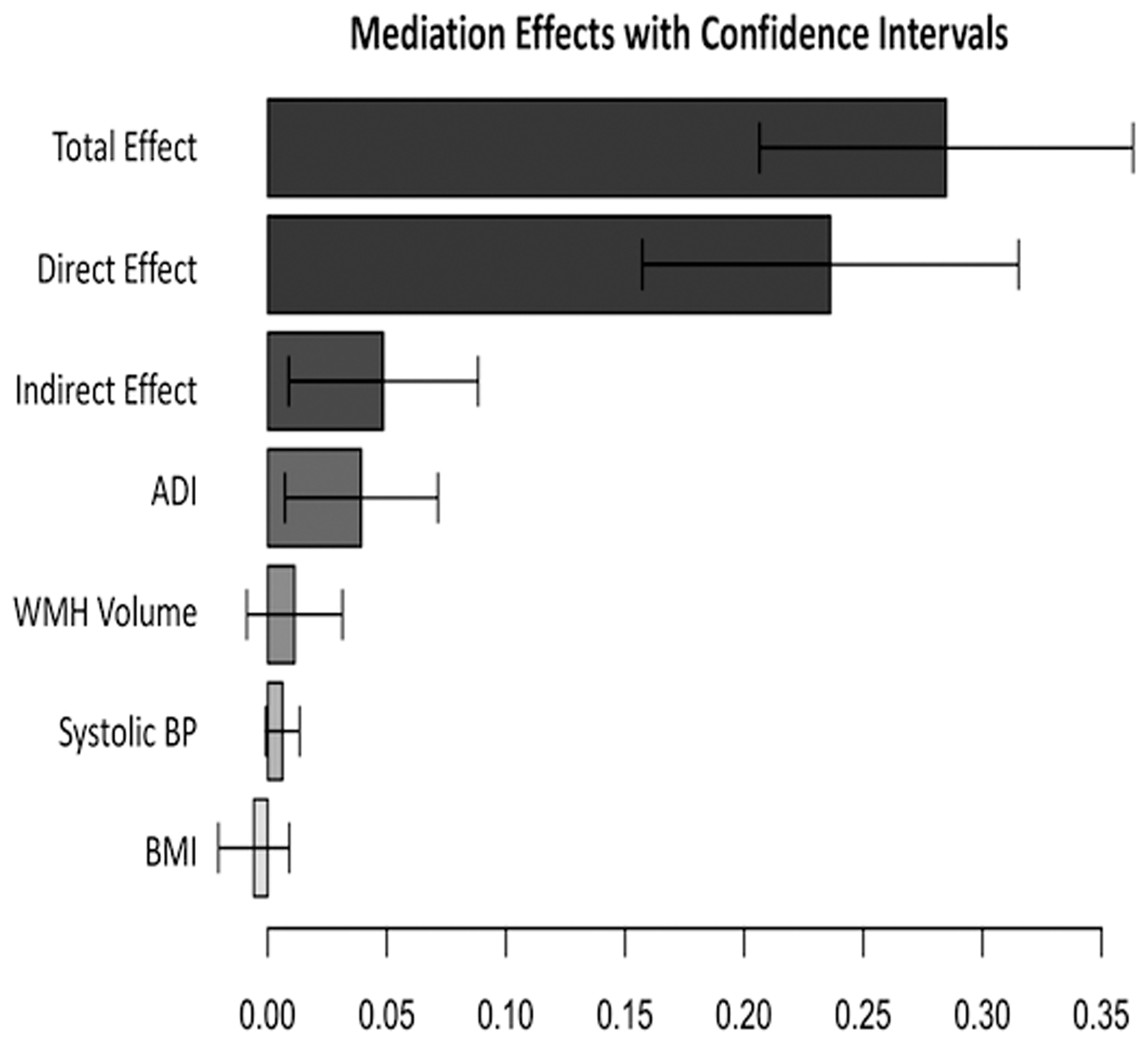
The effect of being African American compared to White on the AT(N) measures is shown as the total race effect. The total effect is a sum of the direct and indirect effects. The direct effect indicates the size of the effect not explained by any of the mediating factors. The indirect effect indicates the size of the effect explained by the mediating factors. 95% confidence intervals are also shown.
ADI = Area Deprivation Index; WMH = white matter hyperintensities; BP = blood pressure; BMI = body mass index
Table 2.
Mediation analysis results for Alzheimer Disease (AD) volume signature.
| AD Volume Signature | ||
| Total Effect | 0.265 | (0.051) |
| Direct Effect | 0.234 | (0.052) |
| Indirect Effect | 0.032 | (0.024) |
| Indirect Effect Components | ||
| ADI | 0.031 | (0.024) |
| BMI | −0.008 | (0.009) |
| Systolic BP | 0.004 | (0.004) |
| WMH | 0.004 | (0.010) |
| Age | N/A | N/A |
| Sex | −0.001 | (0.003) |
| APOE ε4 Status | N/A | N/A |
Note. ADI = Area Deprivation Index; BP = blood pressure; WMH = white matter hyperintensities; APOE ε4 = APOE ε4; PRS = polygenic risk score
For multiple mediation analysis, race served as the direct effect. ADI, BMI, BP, and WMH were the indirect effects tested in a combined model. Age, sex, APOE ε4 status, and PRS were included in the final model.
Table 3.
Mediation analysis results relying on years of education as a proxy for socioeconomic status (SES).
| AD Volume Signature | ||
| Total Effect | −0.236 | (0.063) |
| Direct Effect | −0.235 | (0.059) |
| Indirect Effect | −0.001 | (0.015) |
| Indirect Effect Components | ||
| Years Education | −0.002 | (0.004) |
| BMI | 0.011 | (0.009) |
| Systolic BP | −0.002 | (0.004) |
| WMH | −0.006 | (0.010) |
| Age | N/A | N/A |
| Sex | −0.002 | (0.004) |
| APOE ε4 Status | N/A | N/A |
Note. ADI = Area Deprivation Index; BP = blood pressure; WMH = white matter hyperintensities; APOE ε4 = APOE ε4; PRS = polygenic risk score
For multiple mediation analysis, race served as the direct effect. Education, BMI, BP, and WMH were the indirect effects tested in a combined model. Age, sex, APOE ε4 status, and PRS were included in the final model.
Discussion
We identified racial differences as, on average, African American participants were more likely to report lower levels of education and were more likely to have higher polygenic risk scores for AD. Further, cardiovascular and small vessel disease markers (WMH, systolic blood pressure, and BMI) were elevated in African Americans. There was also greater area deprivation observed among African American participants, indicating lower mean area-based SES compared to White participants. Consistent with our prior finding of lower hippocampal volumes in African Americans with a family history of AD (Morris 2019), we found racial differences in the AD signature volume, representing the neurodegeneration phase of the AT(N) framework, but not in the amyloid or tau phases as depicted by PET imaging. The absence of a difference in tau PET is in contrast to our earlier finding of lower concentrations in CSF tau and p-tau18111. Mediation analysis identified ADI as a major factor in the racial difference in AD signature volume.
The current study confirms our earlier findings (Morris 2019) that: 1) there are no racial differences in amyloid PET burden with age and APOE ε4 status influencing amyloid PET burden in both African Americans and Whites; and 2) African Americans have greater cerebral brain volume loss than do Whites. Concerning tauopathy, we previously reported lower concentrations of CSF tau and p-tau181 in African Americans as compared with Whites (Morris 2019), possibly as a function of APOE ε4 status; our findings were consistent with an earlier CSF study (Howell et al, 2017). Using tau PET rather than CSF, in the current analyses, no racial differences are apparent. We previously reported in cognitively normal older adults that there are weak associations between CSF tau and CSF p-tau181 compared to tau PET deposition, possibly reflecting the restriction of tauopathy to medial temporal lobe in these indivduals38. Lower levels of educational attainment for African Americans are unsurprising given the socio-historical context of African American participants in this sample. For example, many of these participants were school-aged or had completed their formal educations by the time Brown v. the Board of Education was decided. Biophysical cohort differences, such as increased small vessel disease and cardiovascular risk factors, have been noted to be a more prevalent comorbidity for African Americans than Whites9,12,13. We also observed that African Americans had greater ADI values, indicating lower area-based SES on average, compared to Whites.
When compared to the overall population of the state of Missouri, the African American participant median ADI value was consistent with the statewide African American population. On average, ADI values for White participants were substantially lower (suggesting less deprivation) than those of the state population. This discrepancy is likely due to the urban–rural divide in the state of Missouri. In Missouri, African American populations are largely concentrated in the two urban centers of the state (St. Louis and Kansas City); the St Louis population is the primary focus of recruitment for the Knight ADRC. The White population in Missouri is more diffuse. Therefore, the St. Louis–centric sampling of participants likely does not include many rural participants, nor less affluent Whites who comprise a large proportion of the state’s population. We note that these factors, coupled with the use of census-tract data, rather than fine-grained block data, present limitations that should be addressed in future studies. Nevertheless, our results clearly show the importance of ADI on pathological outcomes, which should be considered when recruiting participants for future studies.
Sampling bias has been highlighted as an issue in minority-focused studies3, but we observed sampling bias with respect to SES, in this instance, in the majority population cohort. Studies, particularly time-intensive neuroimaging studies, place a substantial burden on participants. Less affluent individuals may lack the resources (e.g. time off work, transportation) to participate. Efforts must be made to reduce and eliminate barriers to inclusion so that a truly representative sample can be obtained from the population.
Racial differences were observed in the AD signature volume, a marker for the neurodegeneration phase of the AT(N) framework. Multiple mediation analysis indicated that SES, rather than the cardiovascular factors considered, accounted for the largest proportion of the observable racial disparity. Perhaps in a larger cohort, additional effects of ADI and BMI could be observed. Future work should employ other possible explanatory variables when considering sources of difference in AD cortical signature volume.
It is also important to note that when we examined the effects of education, a much more commonly used SES indicator17, we found that education was not a mediating factor (Table 3). Studies that rely on years of education as a proxy for SES may be overlooking critical information available in a more robust measure of SES. Previous studies suggest that years of education mean different things for different ethno-racial groups, and our findings seem to support this assertion18,48,49,50. The mediation analysis in this study highlights the importance of SES, particularly area-level SES indicators, in understanding the development of AD pathology. These results suggest that underlying disparities in area-level socioeconomic resources may contribute substantially to observed racial differences in pathological development.
Ethno-racial differences may be manifestations of lived experiences in either relative poverty or affluence rather than underlying biological or genetic factors, especially in the context of aging51,52. Therefore, gaining an accurate understanding of SES factors is critical in future studies. SES is considered a fundamental social cause of health because greater SES, especially at the neighborhood level, determines the context in which people reside, as well as the resources and risks, such as stress, poverty, and community-level violence, embedded in their context. Health protective resources such as education and access to healthcare, in addition to adequate access to healthy diet options and places to safely recreate so that people can get physical activity, all associate with greater SES53. Future research efforts are needed not only to increase the racial diversity of study samples related to AD, but also to include a more robust account of SES indicators in order to transform our understanding of racial differences in pathological development and begin to disentangle the effects of race relative to SES on AD risk.
It is critical to develop large-scale studies to advance our understanding of what features of area-based SES are attributable to increased AD prevalence among African Americans. For example, perceptions of resource availability in neighborhoods, as well as measures of neighborhood safety and cohesion, could better characterize participants’ social contexts. In addition to collecting data to reflect area-level SES, it is also important to obtain measures of healthcare access (e.g. health insurance coverage, primary healthcare access), markers and measures of stress (e.g. allostatic load54,55, stressful life events inventories, and measures of neighborhood safety and cohesion). Future studies should also address the issue of reverse causation, through which individuals with recent symptom onset (e.g., cognitive impairment or brain atrophy) move to lower SES neighborhoods, as current ADI does not capture this effect. Similarly, it is important for future studies to collect data regarding early life ADI or other measures of childhood SES to better understand the relationship between SES and AD risk throughout the lifespan.
Conclusions
Using an amyloid and a tau PET-based approach as well as MRI to examine potential differences in AD biomarkers, we confirm findings from our earlier study (Morris 2019) that cognitively normal African Americans and White older adults do not differ in cortical amyloid burden, which is associated with age and APOE ε4 status in both groups, and that African Americans have greater cerebral volume loss than Whites. A novel finding of this study is that area-based SES, a measure of SDOH, may contribute to this volume loss, which has been proposed as a marker for neurodegeneration in the AT(N) framework. For unknown reasons, we did not find racial differences in tau, although this observation may reflect previously reported discordance in cognitively normal persons between tauopathy as measured by PET compared to CSF. We observed that SES, specifically area-based SES, may contribute to racial differences in the AT(N) framework. Future studies should continue to emphasize African American recruitment but should also expand recruitment to rural areas in order to more accurately capture the full spectrum of ADI.
Supplementary Material
Acknowledgments and Funding
This work was funded by the National Institute of Health (NIH) grants R01NR012907 (BA), R01NR012657 (BA), R01NR014449 (BA), P01AG00391 (JCM), P01AG026276 (JCM), and P01AG005681 (JCM). This work was also supported by the generous support of Barnes-Jewish Hospital, the Washington University Institute of Clinical and Translational Sciences Foundation (UL1 TR000448), the Hope Center for Neurological Disorders, the Paula and Rodger O. Riney Fund, the Daniel J. Brennan MD Fund, and the Fred Simmons and Olga Mohan Fund. The team thanks all participants involved in this study. Support for Florbetapir F18 (18F-AV-45) and Flortaucipir F18 (18F-AV-1451) was provided by Avid Radiopharmaceuticals, a wholly owned subsidiary of Eli Lilly.
Footnotes
Potential Conflicts of Interest
Dr. Benzinger is a site investigator on clinical trials with Biogen, Roche, Jaansen, and Eli Lilly. She has received research support from Eli Lilly and Avid Radiopharmaceuticals. Avid Radiopharmaceuticals provided the 18-F-AV-45 doses and assisted with scanning expenses and provided precursor and technology transfer for the 18F-AV-1451 doses which were used in this study. Dr. Benzinger does not have any financial relationships with commercial firms.
All other authors do not have any financial relationships with commercial firms.
References
- 1.Barnes LL, Bennett DA. Alzheimer’s disease in African Americans: Risk factors and challenges for the future. Health Aff. 2014;33(4):580–586. doi: 10.1377/hlthaff.2013.1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Babulal GM, Quiroz YT, Albensi BC, et al. Perspectives on ethnic and racial disparities in Alzheimer’s disease and related dementias: Update and areas of immediate need. Alzheimer’s Dement. 2019;15(2):292–312. doi: 10.1016/j.jalz.2018.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brewster P, Barnes L, Haan M, et al. Progress and future challenges in aging and diversity research in the United States. Alzheimer’s Dement. 2019;15(7):995–1003. doi: 10.1016/j.jalz.2018.07.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilkins CH, Schindler SE, Morris JC. Addressing Health Disparities Among Minority Populations. JAMA Neurol. 2020;14(9):1679–1685. doi: 10.1001/jamaneurol.2020.1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fillenbaum GG, Heyman A, Huber MS, et al. The prevalence and 3-year incidence of dementia in older Black and White community residents. J Clin Epidemiol. 1998;51(7):587–595. doi: 10.1016/S0895-4356(98)00024-9 [DOI] [PubMed] [Google Scholar]
- 6.Fitzpatrick AL, Kuller LH, Ives DG, et al. Incidence and Prevalence of Dementia in the Cardiovascular Health Study. J Am Geriatr Soc. 2004;52(2):195–204. doi: 10.1111/j.1532-5415.2004.52058.x [DOI] [PubMed] [Google Scholar]
- 7.Weuve J, Barnes LL, Mendes De Leon CF, et al. Cognitive Aging in Black and White Americans: Cognition, Cognitive Decline, and Incidence of Alzheimer Disease Dementia. Epidemiology. 2018;29(1):151–159. doi: 10.1097/EDE.0000000000000747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang MX, Stern Y, Marder K, et al. The APOE-ε4 allele and the risk of Alzheimer disease among African Americans, whites, and Hispanics. J Am Med Assoc. 1998;279(10):751–755. doi: 10.1001/jama.279.10.751 [DOI] [PubMed] [Google Scholar]
- 9.Chen HY, Panegyres PK. The role of ethnicity in Alzheimer’s disease: Findings from the C-PATH online data repository. J Alzheimer’s Dis. 2016;51(2):515–523. doi: 10.3233/JAD-151089 [DOI] [PubMed] [Google Scholar]
- 10.Fyffe DC, Mukherjee S, Barnes LL, Manly JJ, Bennett DA, Crane PK. Explaining differences in episodic memory performance among older African Americans and Whites: The roles of factors related to cognitive reserve and test bias. J Int Neuropsychol Soc. 2011;17(4):625–638. doi: 10.1017/S1355617711000476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chin AL, Negash S, Hamilton R. Diversity and disparity in dementia: the impact of ethnoracial differences in Alzheimer disease. Alzheimer Dis Assoc Disord. 2011;25(3):187–195. doi: 10.1097/WAD.0b013e318211c6c9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brickman AM, Siedlecki KL, Muraskin J, et al. White matter hyperintensities and cognition: Testing the reserve hypothesis. Neurobiol Aging. 2011;32(9):1588–1598. doi: 10.1016/j.neurobiolaging.2009.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skoog I Vascular aspects in Alzheimer’s disease. J Neural Transm Suppl. 2000;(59):37–43. doi: 10.1007/978-3-7091-6781-6_6 [DOI] [PubMed] [Google Scholar]
- 14.Morris JC, Schindler SE, McCue LM, et al. Assessment of Racial Disparities in Biomarkers for Alzheimer Disease. JAMA Neurol. 2019;76(3):264–273. doi: 10.1001/jamaneurol.2018.4249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howell JC, Watts KD, Parker MW, et al. Race modifies the relationship between cognition and Alzheimer’s disease cerebrospinal fluid biomarkers. Alzheimers Res Ther. 2017;9(1):88. doi: 10.1186/s13195-017-0315-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jack CR, Bennett DA, Blennow K, et al. A/T/N: An Unbiased Descriptive Classification Scheme for Alzheimer Disease Biomarkers.; 2016. [DOI] [PMC free article] [PubMed]
- 17.Manly JJ, Mayeux R. Ethnic differences in dementia and Alzheimer’s disease In: Critical Perspectives on Racial and Ethnic Differences in Health in Late Life.; 2004:95–144. [Google Scholar]
- 18.Walsemann KM, Gee GC, Ro A. Educational Attainment in the Context of Social Inequality: New Directions for Research on Education and Health. Am Behav Sci. 2013. doi: 10.1177/0002764213487346 [DOI] [Google Scholar]
- 19.Walsemann KM, Bell BA. Integrated Schools, Segregated Curriculum: Effects of Within-School Segregation on Adolescent Health Behaviors and Educational Aspirations. Am J Public Heal. 2010;100(9):1687–1695. doi: 10.2105/ajph.2009.179424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson RC, Schoeni RF. Early-life origins of adult disease: national longitudinal population-based study of the United States. Am J Public Health. 2011;101(12):2317–2324. doi: 10.2105/AJPH.2011.300252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glymour MM, Manly JJ. Lifecourse social conditions and racial and ethnic patterns of cognitive aging. Neuropsychol Rev. 2008;18(3):223–254. doi: 10.1007/s11065-008-9064-z [DOI] [PubMed] [Google Scholar]
- 22.Byrd DA, Sanchez D, Manly JJ. Neuropsychological test performance among Caribbean-born and U.S.-born African American elderly: the role of age, education and reading level. J Clin Exp Neuropsychol. 2005;27(8):1056–1069. doi: 10.1080/13803390490919353 [DOI] [PubMed] [Google Scholar]
- 23.Morris JC. The clinical dementia rating (cdr): Current version and scoring rules. Neurology. 1993;43(11):2412–2414. doi: 10.1212/wnl.43.11.2412-a [DOI] [PubMed] [Google Scholar]
- 24.Singh GK. Area Deprivation and Widening Inequalities in US Mortality, 1969–1998. Am J Public Health. 2003;93(7):1137–1143. doi: 10.2105/AJPH.93.7.1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kind AJH, Jencks S, Brock J, et al. Neighborhood socioeconomic disadvantage and 30-day rehospitalization: A retrospective cohort study. Ann Intern Med. 2014;161(11):765–774. doi: 10.7326/M13-2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.University of Wisconsin S of M and PH. Area Deprivation Index v2.0.
- 27.Cruchaga C, Kauwe J, Harari O, Jin S, Neuron YC-, 2013. U. GWAS of Cerebrospinal Fluid Tau Levels Identifies Risk Variants for Alzheimer’s Disease. Neuron. doi: 10.1016/j.neuron.2013.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cruchaga C, Del-Aguila JL, Saef B, et al. Polygenic risk score of sporadic late-onset Alzheimer’s disease reveals a shared architecture with the familial and early-onset forms. Alzheimer’s Dement. 2018;14(2):205–214. doi: 10.1016/j.jalz.2017.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–1458. doi: 10.1038/ng.2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ithapu V, Singh V, Lindner C, et al. Extracting and summarizing white matter hyperintensities using supervised segmentation methods in Alzheimer’s disease risk and aging studies. Hum Brain Mapp. 2014;35(8):4219–4235. doi: 10.1002/hbm.22472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Data.census.gov. 2013 – 2017 American Community Survey 5 year Estimates.
- 32.Klunk WE, Koeppe RA, Price JC, et al. The Centiloid project: Standardizing quantitative amyloid plaque estimation by PET. Alzheimer’s Dement. 2015;11(1):1–15.e4. doi: 10.1016/j.jalz.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Su Y, D’Angelo GM, Vlassenko AG, et al. Quantitative analysis of PiB-PET with FreeSurfer ROIs. PLoS One. 2013;8(11). doi: 10.1371/journal.pone.0073377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su Y, Blazey TM, Snyder AZ, et al. Partial volume correction in quantitative amyloid imaging. Neuroimage. 2015;107:55–64. doi: 10.1016/j.neuroimage.2014.11.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su Y, Blazey TM, Owen CJ, et al. Quantitative Amyloid imaging in autosomal Dominant Alzheimer’s disease: Results from the DIAN study group. PLoS One. 2016;11(3). doi: 10.1371/journal.pone.0152082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mishra S, Gordon BA, Su Y, et al. AV-1451 PET imaging of tau pathology in preclinical Alzheimer disease: Defining a summary measure. Neuroimage. 2017;161:171–178. doi: 10.1016/j.neuroimage.2017.07.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rowe CC, Jones G, Dore V, et al. Standardized Expression of 18F-NAV4694 and 11C-PiB b-amyloid PET results with the centiloid scale. J Nucl Med. 2016;57(8):1233–1237. doi: 10.2967/jnumed.115.171595 [DOI] [PubMed] [Google Scholar]
- 38.Gordon BA, Friedrichsen K, Brier M, et al. The relationship between cerebrospinal fluid markers of Alzheimer pathology and positron emission tomography tau imaging. Brain. 2016;139(8):2249–2260. doi: 10.1093/brain/aww139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L, Benzinger TL, Hassenstab J, et al. Spatially distinct atrophy is linked to β-amyloid and tau in preclinical Alzheimer disease. Neurology. 2015;84(12):1254–1260. doi: 10.1212/WNL.0000000000001401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith RX, Tanenbaum A, Strain JF, et al. Resting-State Functional Connectivity Is Associated with Pathological Biomarkers In Autosomal Dominant Alzheimer’s Disease. Alzheimer’s Dement. 2018;14(7):P1480. doi: 10.1016/j.jalz.2018.06.2512 [DOI] [Google Scholar]
- 41.Brier MR, Thomas JB, Snyder AZ, et al. Loss of intranetwork and internetwork resting state functional connections with Alzheimer’s disease progression. J Neurosci. 2012;32(26):8890–8899. doi: 10.1523/JNEUROSCI.5698-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Power JD, Cohen AL, Nelson SM, et al. Functional Network Organization of the Human Brain. Neuron. 2011;72(4):665–678. doi: 10.1016/j.neuron.2011.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wisch JK, Roe CM, Babulal GM, et al. Longitudinal Changes in Functional Connectivity in Conversion to Symptomatic AD. aanddjournal.net. 2018;4:3248. [Google Scholar]
- 44.Goh JOS. Functional Dedifferentiation and Altered Connectivity in Older Adults: Neural Accounts of Cognitive Aging. Aging Dis. 2011;2(1):30–48. [PMC free article] [PubMed] [Google Scholar]
- 45.Zeileis A Econometric computing with HC and HAC covariance matrix estimators. J Stat Softw. 2004;11:1–17. doi: 10.18637/jss.v011.i10 [DOI] [Google Scholar]
- 46.Walker K tidycensus: Load US Census boundary and attribute data as ‘tidyverse’and ‘sf’-ready data frames. R package version 0.4. 1. 2018.
- 47.Yu Q, Li B. mma: An R Package for Mediation Analysis with Multiple Mediators. J Open Res Softw. 2017;5. doi: 10.5334/jors.160 [DOI] [Google Scholar]
- 48.Walsemann K, Geronimus A, Gee G. Accumulating Disadvantage Over the Life Course. Res Aging. 2008;30(2):169. [Google Scholar]
- 49.Manly JJ, Jacobs DM, Sano M, et al. Effect of literacy on neuropsychological test performance in nondemented, education-matched elders. J Int Neuropsychol Soc. 1999;5(3):191–202. doi: 10.1017/S135561779953302X [DOI] [PubMed] [Google Scholar]
- 50.Margo RA. Race and Schooling in the South, 1880–1950.; 2013. doi: 10.7208/chicago/9780226505015.001.0001 [DOI] [Google Scholar]
- 51.Hunt JFV, Buckingham W, Kim AJ, et al. Association of Neighborhood-Level Disadvantage with Cerebral and Hippocampal Volume. JAMA Neurol. 2020;77(4):451–460. doi: 10.1001/jamaneurol.2019.4501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Powell WR, Buckingham WR, Larson JL, et al. Association of Neighborhood-Level Disadvantage With Alzheimer Disease Neuropathology. JAMA Netw open. 2020;3(6):e207559. doi: 10.1001/jamanetworkopen.2020.7559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Link BG, Phelan JC. Social conditions as fundamental causes of disease. J Health Soc Behav. 1995; Extra Issu:80–94. [PubMed] [Google Scholar]
- 54.Berger M, Sarnyai Z. “More than skin deep”: Stress neurobiology and mental health consequences of racial discrimination. Stress. 2015;18(1). doi: 10.3109/10253890.2014.989204 [DOI] [PubMed] [Google Scholar]
- 55.Geronimus AT, Thompson JP. STATE OF THE DISCIPLINE TO DENIGRATE, IGNORE, OR DISRUPT Racial Inequality in Health and the Impact of a Policy-induced Breakdown of African American Communities. 2(2004):247–279. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.