

Summary
Parkinson’s disease is characterized by the loss of dopaminergic neurons in the substantia nigra leading to disabling deficits. Dopamine neuron grafts may provide a significant therapeutic advance over current therapies. We have generated midbrain dopamine neurons from human embryonic stem cells and manufactured large-scale cryopreserved dopamine progenitors for clinical use. After optimizing cell survival and phenotypes in short-term studies, the cell product, MSK-DA01, was subjected to an extensive set of biodistribution, toxicity, and tumorigenicity assessments in mice under GLP conditions. A large-scale efficacy study was also performed in rats with the same lot of cells intended for potential human use and demonstrated survival of the grafted cells and behavioral amelioration in a 6-hydroxydopamine lesioned rats. There were no adverse effects attributable to the grafted cells, no obvious distribution outside the brain and no cell overgrowth or tumor formation, thus paving the way for a future clinical trial.
Keywords: Dopamine neurons, human embryonic stem cells, transplantation, Parkinson’s disease, cell therapy, preclinical study, GMP, human pluripotent stem cells, biodistribution studies, toxicity studies, tumorigenicity studies
Graphical Abstract
Introduction
Parkinson’s disease is one of the most common neurodegenerative diseases worldwide. Its cardinal features include motor symptoms such as bradykinesia, rigidity, resting tremor and postural instability, though nonmotor symptoms are often also present. The current mainstay of pharmacologic therapy consists of augmentation of dopamine levels in the brain via dopamine supplements or agonists or by inhibiting dopamine degradation. Treatment is symptomatic but not long-lasting, and it has no neuroprotective effect. Cell therapy with grafts of human fetal tissue from the ventral mesencephalon have been carried out successfully, with multiple reports of long-term benefits. But placebo controlled double blind studies in the US have failed to reach their primary endpoints (Freed et al., 2001; Olanow et al., 2003). Nonetheless graft survival was shown by Fluorodopa PET scans (Freed et al., 2011) and autopsy studies (Kordower et al., 1995; Li et al., 2016), and subgroup analysis suggested benefit in some patients (Freed et al., 2011). While the clinical results of fetal tissue transplantation were variable, there were enough successes (up to complete cessation of anti-PD therapy in successfully grafted patients) to warrant improved trial design and development of a new European Union-funded study. This new trial, TRANSEURO (NCT01898390), has just finished accrual of 11 patients, randomly selected from a large observational cohort to receive human fetal grafts (Barker et al., 2019). While much will be learnt from this study, fetal tissue-based studies in humans are fraught with enormous logistical hurdles and ethical concerns.
Human embryonic stem cell- (hESC) or induced pluripotent stem cell- (iPSC) derived dopamine neurons provide several advantages over the use of human fetal tissue. They are readily expandable at the undifferentiated state to generate large scale cell banks. Robust differentiation protocols can then be applied to yield, large cryopreserved lots of differentiated cells that are subjected to stringent quality control measures after development of the critical release criteria. Cell composition and phenotype of ESC or iPSC-derived dopamine cells can be further validated pre-grafting. Ethical and logistical hurdles are also significantly reduced in comparison to the use of fetal tissue. There are at least two patients known to have received iPSC-derived dopamine cells, a patient in a recent trial in Japan (Cyranoski, 2018), and a patient in the US on a compassionate basis (Cyranoski, 2018; Schweitzer et al., 2020). Preclinical data have also been reported (Doi et al., 2020) .
We have previously demonstrated the in vitro derivation of midbrain dopamine neurons (Perrier et al., 2004), then developed an enhanced specification approach based on floor-plate derivation of authentic midbrain dopamine neurons that express key lineage-defining transcription factors such as FOXA2 and LMX1A and PITX3 as well as tyrosine hydroxylase (TH) upon maturation. Grafted cells survived, integrated into the host tissue and rescued the behavioral abnormalities of Parkinsonian rodents (Kriks et al., 2011).
Encouraged by these results, we elected to translate our findings towards a clinical trial. In a first step, we further enhanced the robustness of the differentiation and adapted the dopamine neuron differentiation conditions towards clinical compatibility under near current good manufacturing practice (cGMP). The resulting dopamine neuron differentiation protocol and its developmental rationale are presented in the companion article (Kim et al., 2020). Building on those results, we report here the large-scale manufacture of cryopreserved clinical-grade cells, named MSK-DA01. We also present the design and results of toxicity, bio-distribution, tumorigenicity and efficacy studies in rodents.
Results and discussion
Protocol Overview
The initial step in the transition of the protocol of dopamine neuron differentiation to clinical compatibility entailed the elimination of the mouse fibroblast feeders and the Knock-Out Serum replacement product (KSR) in the original protocol to feeder-free conditions using Essential-8 Medium for expansion, and Neurobasal medium for differentiation. However, those changes resulted in a near complete loss of TH+ midbrain dopamine (mDA) neurons co-expressing FOXA2 and in the low expression of additional dopamine and midbrain markers such as EN1. As detailed in the accompanying article (Kim et al., 2020), we addressed this problem by modifying timing and concentration of the WNT agonist, CHIR99021 with treatment at 0.7 µM from day 0 to 3, 7.5 µM from day 4 to day 9, at 3µM from day 10 to 11 to dynamically control WNT signaling levels during differentiation (“Chir-Boost” protocol). This strategy improved rostro-caudal patterning of the SHH-induced ventral FOXA2+ population with minimal contamination of anterior diencephalic or posterior hindbrain lineages or non-neural contaminant and led to the successful derivation of authentic midbrain dopamine neurons with very low run-to-run variability. We next developed standard operating procedures (SOP) for the new differentiation conditions and transiently embedded members of our team in the institutional GMP facility (Cell Therapy and Cell Engineering Facility or CTCEF) to achieve a seamless transfer of the SOP to the facility staff and to initiate the next steps.
Manufacturing and quality assurance of MSK-DA01 cells
Master banks of undifferentiated WA09 (H9) cells (passage 28) were obtained from the WiCell Research Institute. The cells were manufactured under cGMP conditions to establish working cell banks (WCB) in E8 medium that were further certified by Waisman Biomanufacturing. The cells passed a battery of tests including cell authentication by STR, karyotype, human ES marker expression, adventitious virus, and mouse antibody testing (Table S1). After transfer to our CTCEF, the cells were thawed and expanded for two or three passages prior to differentiation. Cells passed quality control analysis for morphology, cell identity (NANOG expression ranged from 95.8–98.1% by flow cytometry, criteria set at ≥ 90%), viability (ranging 83.8–90.8%, criteria≥70%, by AO/PI staining), sterility and mycoplasma tests.
For manufacturing of dopamine neuron progenitors (MSK-DA01), we first identified and performance-tested lots of all the reagents required. After completing the engineering run of our process in the CTCEF to evaluate product yield and quality, we manufactured four lots of MSK-DA01. Three of the four lots yielded at least 2 billion cells (2.0 – 2.8 × 109 cells), stored in > 200 vials each. Only lot 3 was more limited in size (1.1 × 109 cells) as some of the cells were used for extended culture to allow for adventitious agent testing. An overview of experimental design and quality assurance is shown in Figure 1A.
Figure 1. Testing of MSK-DA01 prior to preclinical efficacy and safety studies.
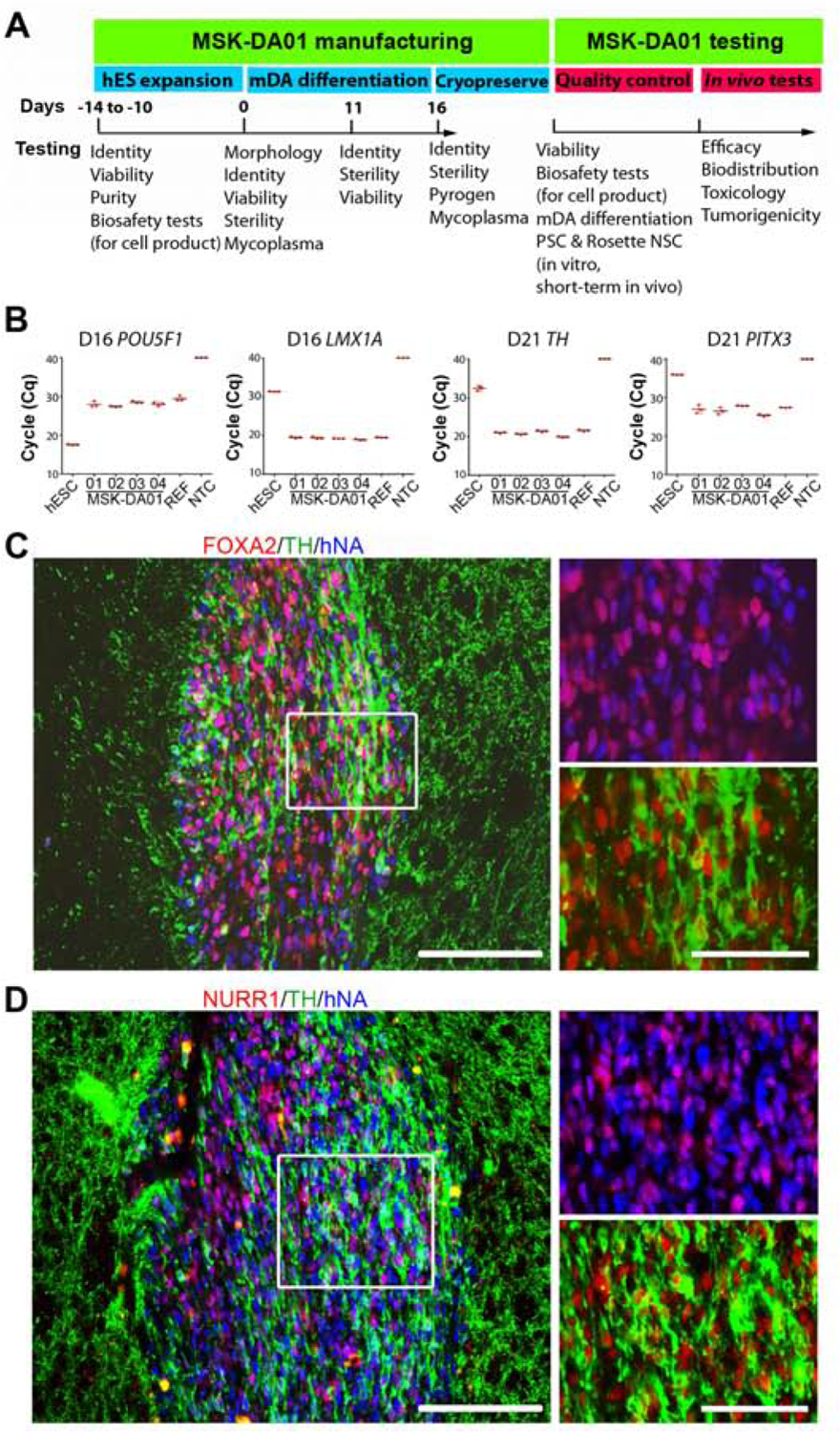
(A) Schematic diagram showing testing undertaken at different steps of manufacture and application of MSK-DA01. (B) Cycle quantification (Cq) values of POU5F1, LMX1A, PITX3 and TH in four lots (01–04) of cryopreserved MSK-DA01 assessed by qRT-PCR. PITX3 and TH were assessed at five days post thaw. WA09 (ES), reference (REF, a previous successful batch) and negative control (NTC, no template control) were included in the analysis. Data are represented as mean±SD (n=3/group). (C) Representative section through a graft 3 weeks post transplantation, immunostained for FOXA2, TH and human nuclear antigen (hNA). (D) Graft immunohistochemistry for NURR1, TH and human nuclear antigen (hNA) showing co-expression in most human cells. Rectangles indicate areas of magnification shown in the right panels. Scale bars=100 µm in left panel and 50 µm in right panels.
The differentiation process at the CTCEF was monitored at several timepoints using in process analyses of cell morphology, sterility and viability at day 11 and day 16. For cell identity, gene expression of a list of key markers (41 genes + one internal control gene (ACTB) (Kim et al., 2020) was measured at days 0, 11, 16, and extended to day 21(Figure S1A). The list consisted of genes involved in mDA development (such as FOXA2, LMX1A, EN1, TH) (Ferri et al., 2007; Simon et al., 2001; Deng et al., 2011) and genes marking possible cell impurities during rostral-caudal patterning (such as FOXG1 for forebrain, HOXB2 for hindbrain) or during differentiation (such as POU5F1 for pluripotent stem cells (PSC) or PAX6 for early neuroepithelial, rosette-stage neural stem cell (NSC)). A previous small-scale lot (NB072715SZ), shown to function in vivo, served as a reference. The data demonstrated high expression of genes involved in mDA development, and minimal expression of non-mDA fates. POU5F1 expression was undetectable as of day 11 by immunohistochemistry (IHC) (data not shown). At the end of differentiation at day 16, the cells passed sterility, pyrogen and mycoplasma testing and were cryopreserved using STEM-CELLBANKER and a controlled rate freezer into 8 million-cell vials. Viability after thaw ranged from 79.6 to 87.2% (criteria>70%; n=6 vials/lot). Additional testing on cryopreserved lots involved markers nominated as release criteria at day 16 including gene expression for LMX1A and POU5F1 (Figure 1B) and gene expression for TH and PITX3 by qRT-PCR at day 21, 5 days after further differentiation of cryopreserved day 16 product (Figure 1B). IHC for co-expression of FOXA2 and TH co-expression was also carried out at day 21(Figure S1B). We established validated intracellular flow cytometry assays for expression of key proteins of thawed cells, including FOXA2 (criteria>85%), PAX6 (criteria <5%) and NANOG (criteria<0.2%). Data on thawed cells from MSK-DA01 showed a high percentage (90.1–94.4%) of FOXA2, a floor plate marker that is maintained throughout the development of mDA neurons, very low percentage of PAX6+ (0.1–0.2%) and undetectable NANOG+ cells (Figure S1C). We did not observe significant differences among the lots or loss of marker expression in long-term cryopreserved cells.
Since contamination with undifferentiated PSCs is a major concern for any PSC-based products, in addition to our standard, flow cytometry-based release assay for NANOG and qRT-PCR based assay for POU5F1, we tested carefully for POU5F1+ cells in our product at day 16 using several orthogonal assays. MSK-DA01 was tested alone or in samples spiked with WA09-hPSCs in a dilution series ranging from 0.001% to 1% undifferentiated PSC, to determine the limit of detection of the various assays. First, we used an automated high content microscope (Operetta) to detect and quantitate immunostained POU5F1+ cells (Figure S1D). The quantified percentage of POU5F1+ is highly similar to the percentage of spiked PSCs (Figure S1E). The automatedly detected POU5F1+ cells were subjected to validation by manual observation, and false positive signals were excluded. At the lowest spiked percentage tested (0.001% PSC spike), five POU5F1+ cells were identified out of 191,570 cells (0.0026%), confirming the sensitivity of the analysis. Using high content imaging, we could not find evidence for the presence of any POU5F1 cells in MSK-DA01. For qRT-PCR-based detection of POU5F1 mRNA, we observed a 2,000-fold decrease in expression from undifferentiated cells to MSK-DA01. However, there was evidence for persistent low POU5F1 mRNA expression compared with negative control, which is not consistent with the absence of POU5F1+ cells in MSK-DA01 described above. We then suspected that our release POU5F1 primer set (Fig. 1B) may have detected known POU5F1 pseudogenes which may limit sensitivity (Liedtke et al., 2007). Therefore, we implemented an additional primer sets optimized to avoid pseudogene detection (Liedtke et al., 2007) using both ddPCR (data not shown) and qRT-PCR assays. We analyzed POU5F1 expression in a set of samples including MSK-DA01, fibroblasts (negative control), and 100%, 1%, 0.1%, 0.01%, 0.001%, 0.0001%, and 0.00001% hPSCs spiked into MSK-DA01 (Figure S1F). The data suggest that the limit of detection for this assay reaches at least low as 0.001%. MSK-DA01 showed levels not significantly different from the fibroblast negative control, confirming that POU5F1 mRNA was undetectable in MSK-DA01.
We next tested each of the four lots of MSK-DA01 for in vivo survival and mDA differentiation. We injected 400,000 cells (n=4–6 mice per lot) unilaterally into the striatum of severe combined immunodeficient/nonobese diabetic NSG (NOD.Cg-PrkdcscidIl2rgtmWjl/SzJ) mice and assessed for survival and phenotypes of grafted cells three weeks later. We observed robust human cell survival and formation of well-defined grafts in the striatum, at the site of injection. The human dopamine neurons in the graft were identified by co-expression of human cytoplasmic marker (STEM121) and TH (Figure S1G). FOXA2 was expressed in the majority of human cells (84.3±10.3%). TH+ cells in the graft were co-labeled with human nuclear antigen (hNA) and FOXA2 (Figure 1C). Most human cells also expressed NURR1 and PITX3 (Figure 1D and Figure S1H), which are critical for the development and maintenance of midbrain dopamine neurons (Kadkhodaei et al., 2009; Munckhof et al., 2003; Smidt et al., 2004). No grafted human cells expressed NKX2.1, indicating the cells did not differentiate into hypothalamic fates (Ohyama et al., 2005). POU5F1, a marker of PSC was absent while PAX6 was present very rarely in short-term grafts as well as in vitro assays, indicating the cells will unlikely form teratoma or neural rosettes in the brain. There were no significant differences in graft volume and percentage of TH-expressing cells among the four lots of MSK-DA01 three weeks after grafting. We selected lot 2 to perform all studies, as this lot resulted in the highest average graft volume (0.187 mm3) and percentage of TH+ cells (15.91%) at 3 weeks post grafting. Since cells are mDA progenitors when grafted, they are expected to complete their maturation in vivo over time.
Rescue of motor deficits in 6-hydroxydopamine induced Parkinsonian rats by MSK-DA01
We next proceeded to perform a long-term efficacy study, with a goal of analyzing the impact of MSK-DA01 on animal behavior in 6-hydroxydopamine (OHDA)-lesioned nude rats (NIH-Foxn1rnu) as well as graft survival and differentiation. We performed the study in our lab under near good lab manufacturing practice (GLP) conditions, as adequate expertise was not available at any external GLP-certified location. We induced hemiparkinsonism in nude rats by stereotactic injection of 6-OHDA in the median forebrain bundle (Kriks et al., 2011). Amphetamine-induced rotational testing was performed at 4 and 6 weeks (± 1 week). Rats exhibiting >6 rotations/minute were selected for inclusion in the efficacy study. 44 rats were separated randomly into a cell group (n=28, male:female M:F=1:1) and a vehicle group (n=16, M:F=1:1). Freshly thawed MSK-DA01 cells (day 16) passed an empirical pre-established viability threshold (>70%) and were suspended at a concentration of 100,000±10,000 cells/µl. Animals received 400,000 cells or vehicle in 4 µl via stereotactic injection at four deposits along a single track in the striatum. Cell viability showed some decrease at the end of grafting (less than 5 hours from thaw time), though remained above 70% (Figure S2A). There was no correlation between graft volumes and the time of injection after initial cell thawing (r=−0.2797, P=0.1577). We replaced one female in the cell group due to a premature withdrawal of the injection needle during the procedure, resulting in a total of 29 rats receiving graft injections (14 M and 15 F rats).
Animals underwent behavioral testing at 2, 4, 5, 6.5 and 8 months (± 1 week) post grafting (Figure 2A). There was a significant decrease in rotation scores at 5 months post grafting. Rotations per minute were down to −1.4 (median for all rats) at 8 months after transplantation as compared to a median of +14.6 in the vehicle group.
Figure 2. MSK-DA01 rescued the motor asymmetry in the preclinical efficacy study.
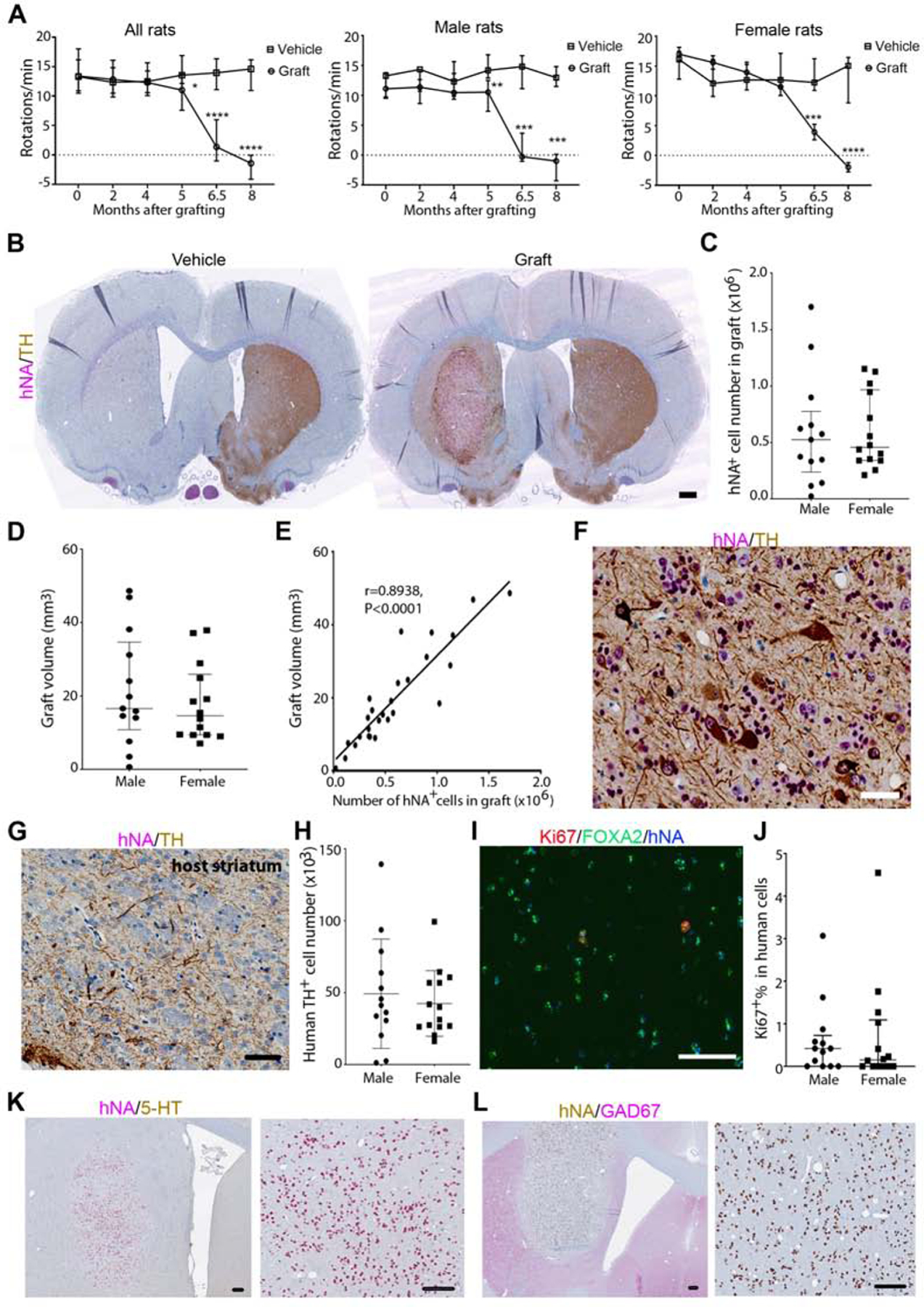
(A) Number of amphetamine-induced rotations per minute at different timepoints post-grafting in the entire animal group. Male and female groups are also plotted separately. (B) Immunohistochemistry (IHC) for human nuclear antigen (hNA) and TH in representative brain sections from vehicle or grafted animals. Note the almost complete absence of endogenous TH+ processes in the unilateral 6-OHDA lesioned striatum that received vehicle. The grafted brain shows evidence of TH reinnervation in the putamen beyond the graft itself. (C) Stereological estimation of hNA+ cell number in the MSK-DA01 graft. (D) Stereological estimation of graft volume. (E) Correlation between human cell number and graft volume in all animals. (F) mature human dopamine neurons (hNA+TH+) in the graft. (G) IHC for TH shows graft-derived processes in the host striatum. (H) Stereological estimation of TH+ cell number in MSK-DA01 graft. (I) IHC for Ki67, FOXA2 and hNA in the graft. (J) Percentage of Ki67+ expressing human cells in male or female rats. (K) IHC for 5-hydroxy-tryptamine (5-HT) shows absence of serotonergic neurons in the graft. (L) IHC for GAD67 indicates the absence of GABA-ergic neurons in the graft. Scale bars=500 µm in B; 50 µm in F,G; 85 µm in I, 100 µm in K,L. Data are represented as median with interquartile range in A, C–D and mean±SD in H, J. Graft group, n=27 (M, n=13; F, n=14); Vehicle control group, n=14 (n=7 for each gender). *P<0.05, ** P<0.01, *** P<0.001, ****, P<0.0001 compared with the vehicle group.
Animals were euthanized and their brains were analyzed by independent pathologists at a contractor site (Charles River Laboratories). In the vehicle group, there were no human cells and no dopaminergic innervation in the lesioned striatum. Grafted animals showed human TH+ cells at the injection site (Figure 2B). Stereological estimates (physical dissector) of the total number of human cells showed a median of 0.53 million cells in male rats and 0.46 million cells in female rats (Figure 2C). Graft volume analysis showed a median of 16.60 mm3 in males and 14.62 mm3 in females, a difference that was not statistically significant (Figure 2D). There was a strong correlation and linear regression between human cell number and graft volume in all animals (Figure 2E). Large well-differentiated human dopamine neurons were observed in the graft (Figure 2F), extending their processes into the surrounding tissues (Figure 2G; Figure S2F). Human dopamine neurons (hNA+TH+) cells amounted to an average of 49,250 cells in males and 42,480 cells in females (Figure 2H). Two male rats had small grafts (23,484 and 115,092 surviving human cells respectively) and a correspondingly low number of TH cells (1,250 and 2,500 cells); these animals were also the only two rats that did not show significant behavioral recovery, suggesting that less than 2500 grafted human TH+ cells cannot support recovery of motor asymmetry in rats. As expected, the number of human TH+ cells correlated with the total number of human cells in all animals (Figure S2B). The percentage of TH+ cells in hNA+ cells was not different between genders (Figure S2C).
To evaluate the proliferation of grafted cells, we performed IHC for hNA and Ki67 (Figure 2I). The percentage of Ki67 in hNA+ cells was very low in both male (average at 0.61%) and female (average at 0.68%) animals (Figure 2J). There was no obvious correlation between the percentage of Ki67+ cells and the number of hNA+ cells (Figure S2D) or graft volume (data not shown). Around 67.78% hNA+Ki67+ cells expressed FOXA2 (Figure 2I). IHC for 5-hydroxytryptamine (5-HT) and Glutamic acid decarboxylase (GAD) was negative (Figure 2K, L), suggesting the absence of human serotonergic or GABAergic neurons, respectively.
Two female grafted rats developed a flank mass at 22 and 32 weeks after transplantation but remained well. The rats were euthanized at the scheduled time and underwent a necropsy. The masses were identified histologically as mammary fibroadenomas (Figure S2E) and had no human cells (hNA) (data not shown); they were ruled as a spontaneous benign tumor, known to occur in laboratory rats (Percy et al., 2001). In addition, four rats had to be terminated earlier than scheduled (Table S2). Those rats were not included in the data analysis described above due to the discrepancy in the endpoints. However, full necropsy was performed independently by the Veterinary Pathology Services at our institution, including histological evaluation of an extensive list of organs (>30). The cause of death of three animals was infection in multiple organs while one male rat had trauma due to an attack by its cage mate; all deaths were ruled unrelated to the grafts. Two of these animals, which were from the graft group, exhibited surviving human TH neurons in the striatum (Figure S2F), with absent POU5F1 (Figure S2G), low number of Ki67 cells (Figure S2H), and no evidence of abnormal graft histology (Figure S2I).
Biodistribution testing shows grafted MSK-DA01 cells remain confined to the brain
We conducted large scale biodistribution, toxicology and tumorigenicity studies under GLP conditions. The studies were designed by our team and conducted at an outside contracted facility (WuXI-Apptec, St Paul, MN). MSK-DA01 cells were grafted into 6–8week-old normal NSG mice.
We used two doses for the biodistribution (n=10 per dose group, M:F=1:1) and toxicology studies (n=20 per dose group, M:F=1:1): a lower dose of 200,000 cells in 2 µl and a higher dose of 400,000 cells in 4 µl; a third group received vehicle only. An additional mouse group (n=10, M:F=1:1) was added to the study as a potential source of replacement should any mouse die or require termination. Our target dose in rats was 400,000 cells and we recognize that this dose and volume are too large for a mouse brain. However, to maximize our chances to reveal a potential safety signal, we used the maximally feasible dose of 400,000 cells. The animals were separated randomly and there was no significant difference in weight among all the groups in both studies. On each day of grafting, MSK-DA01 cells were thawed and reconstituted to 100,000±10,000 cells/µl. Cell viability at initial thaw, pre, and post-transplantation exceeded our pre-set threshold of > 70% (Figure S3A). A few animals did not have any graft, probably due to technical reasons, and were not included in the analysis (see methods and Table S4).
Biodistribution was assessed in an array of tissues collected on days 30 and 180, by qPCR for human-specific Alu repeat sequences. The collected tissues included liver, blood, lungs with mainstem bronchi, bone marrow, axillary lymph nodes, brain near the injection site, spinal cord, spleen, heart, kidneys and any visible abnormality. Sensitivity and specificity of the qPCR assay were determined prior to the start of the study. The limit of quantification (LOQ) was found to be 0.303 genome copies for all samples with the exceptions that LOQ was 1.515 genome copies for brain and spinal cord tissues at day 30 due to sample dilution. On both days 30 and 180, high levels of human DNA were detected in the grafted brain tissue (Figure 3A). At 30 days, all non-brain tissues including the spinal cord were negative for human DNA with the exception of sporadic very low positive signals in kidney, liver, lymph nodes or whole blood. Those signals were at marginally higher levels than LOQ, ranging from 0.432– 1.472 genome copies (Figure 3A). On day 180, most non-brain tissues were found to be negative for human DNA (Figure 3A). One male animal exhibited marginally higher than LOQ genome copies (0.571) in a lymph node sample. Spinal cord testing was negative in all animals except for one female in the low dose group which tested marginally higher than the LOQ. At both time points, in non-brain tissues, the median value of human DNA genome copies in the graft group was at LOQ which is the same as in the vehicle group without statistical difference (Figure 3A). These data suggest that the grafted MSK-DA01 cells were confined to the brain and rarely trafficked to areas outside the brain, including the spinal cord. The presence of very low levels of human genome copies in non-brain tissues in sporadic animals may be related to suboptimal cell injection. Cells can leak from the injection needle into the spinal fluid if an injection reaches the ventricle or if there is backflow upon withdrawal of the needle. Traumatic injections can also potentially expose cells to the systemic circulation or the subarachnoid space. However, since the detected levels of human DNA were very low, and not elevated over time up to 180 days, these results were considered accidental and not clinically relevant.
Figure 3. Biodistribution and graft analysis.
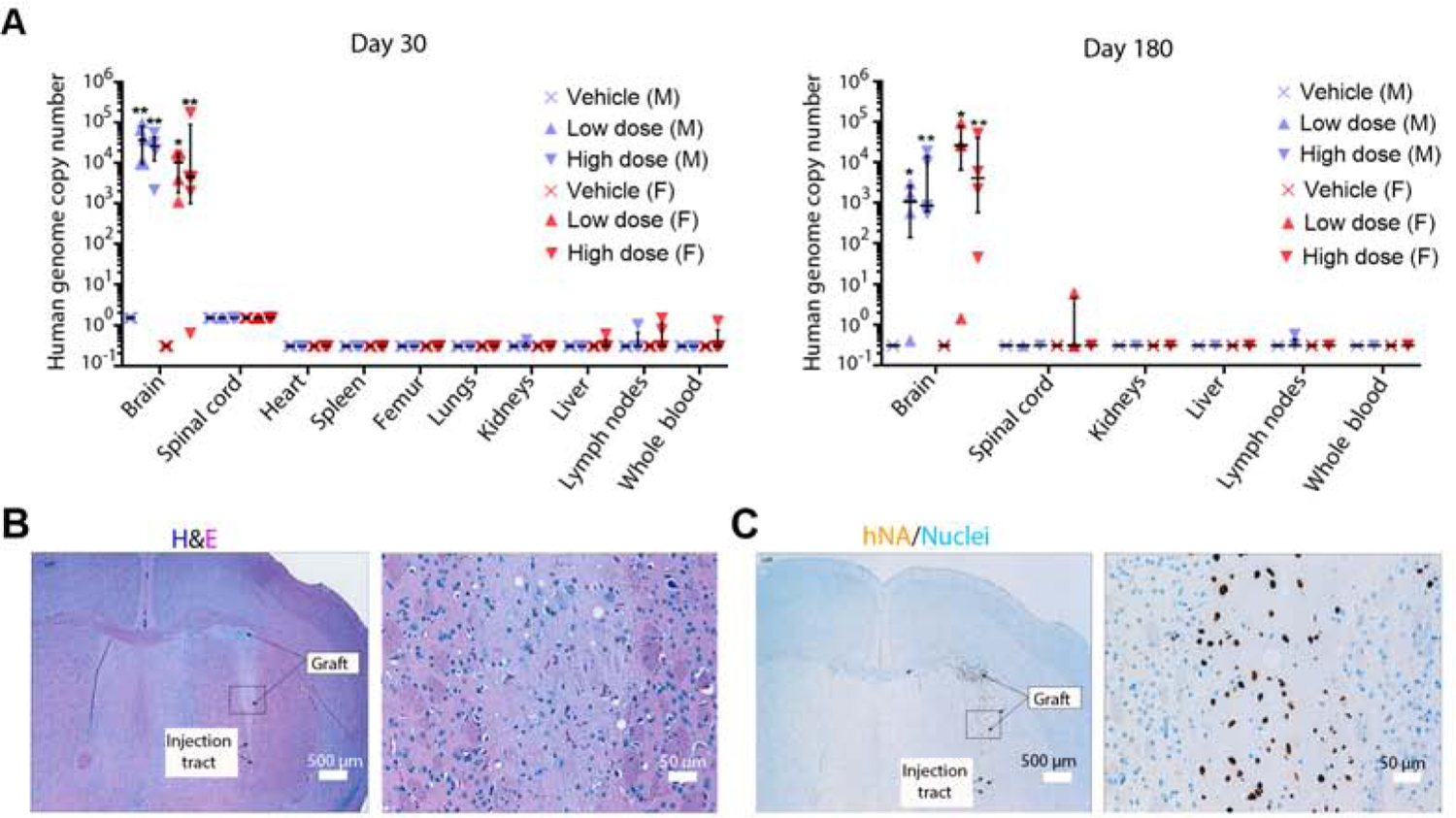
(A) Human genome copy number in the brain and other organs was estimated by DNA qPCR on days 30 and 180 post grafting, in the biodistribution study. All vehicle tissues are at the limit of quantification for the human DNA genome copy number for each tissue. Data represented as median with interquartile range. M= male; F= female. n=4–5 per group. *P<0.05, ** P<0.01 compared with vehicle group. (B) H&E staining of a representative section of a graft on day 180. Neuronal cell bodies and neuropil are visible in the magnified image on the right. (C) Immunohistochemistry for hNA in an adjacent section highlights the human cells, with a magnified image on the right. Hematoxylin nuclearstain. Scale bar values are indicated on the images.
No obvious adverse effects of MSK-DA01
We performed standard hematology and clinical chemistry tests on the animals in the toxicology study and found no clinically relevant alterations. We also documented body weights throughout the study and found no differences among groups or genders (data not shown).
Pathological macroscopic and microscopic assessments.
Macroscopic and histopathological assessments of all major organs were conducted on all animals in the toxicology study on days 30 and 180. Assessments for each animal were performed by a board-certified veterinary pathologist who was blinded to the group status.
The injection tract was observable histopathologically in most cases across all the groups. It showed a small degree of gliosis and post-traumatic changes (Figure S3D). Grafted cells, recognized by H&E and IHC with anti-hNA, were mainly located in the striatum (Figure 3B, C). Grafted cell clusters, with none to rare mitotic figures, were supported by moderate amounts of eosinophilic vacuolated neuropil with fine vasculature. No evidence of vascular invasion or pathologically significant lesions was seen. A few animals exhibited suboptimal injection with human cells outside the striatum, but with differentiation into neural tissues without abnormal structures, similar to the grafts in the striatum. We also analyzed H&E stained sections from the brain tissue outside the graft area, including medulla/pons, cerebellum and cerebral cortex. Those tissues were normal in all groups.
For non-brain tissues, most organs examined appeared normal. There were a few abnormalities and lesions but those were devoid of human cells, as listed in Table S3.
Unexpected deaths or termination were not related to the grafted cells in biodistribution and toxicology studies.
Animals were monitored regularly by independent veterinarians. Four mice were dead or terminated before the scheduled time point (Table S2). The causes of death included surgical or anesthesia complications and severe multi-organ lympho-histiocytic inflammation associated with the presence of bacterial emboli (coccoid bacteria) within small vessels and tissues throughout the body. All other animals remained in normal health. No graft related symptoms were observed.
The tumorigenicity study did not reveal any tumor formation in MSK-DA01 grafts.
An independent tumorigenicity study was designed to detect the presence of any pluripotent stem cell contaminants in MSK-DA01, capable of abnormal proliferation or formation of a teratoma.
We first performed a pilot spiking study to determine the capacity of hESC to form teratomas, following their injection in NSG mice brains. We prepared cell suspensions consisting of 100% mDA cells, mDA spiked with 0.1%, 1% or 10% hESC and 100% hESC. Before grafting, the expression of pluripotent cell markers in the undifferentiated hESC was confirmed with >94% SSEA4+, >94.7% POU5F1+SOX2+ and <2% SSEA1+ by flow cytometry. Pluripotent POU5F1+ cells were negative in the 100% mDA cells and increased with the percentage of spiked undifferentiated hESC (Figure S4A, B). Animals were injected with the different cell groups into the striatum (450,000 cells per animal), and the brains analyzed 4 months later. All animals in the 10% and 100% hESC group as well as two (out of 6) mice in the 1% spiking group formed teratomas (Figure S4C).
Having determined that a 0.1% hESC concentration does not form teratomas, we designed the tumorigenicity study for MSK-DA01 as follows. The study involved five groups of NSG mice (M:F=1:1) that received intra-striatal injections of 400,000 cells of 1) 100% MSK-DA01(n=44), 2) MSK-DA01 spiked with 0.01% undifferentiated hESC (n=44), 3) MSK-DA01 spiked with 0.1% undifferentiated hESC (n=44), 4) 100% hESC (n=24), or 5) 4 µl vehicle solution (n=24). The proportion of POU5F1+ cells and POU5F1 expression in each preparation was confirmed by flow cytometry and qRT-PCR (data not shown). The end time point was set as 266 days post grafting. Animals with no surviving grafts were excluded (see Table S4).
Teratomas were observed in most mice in the 100% ES group (8/11 M, 10/11 F) (Figure 4A), confirming adequacy of our positive control. No tumors were observed in the grafts of the 100% MSK-DA01, 0.01% hESC or 0.1% hESC groups (Figure 4B–D). Those grafts, visualized by H&E and IHC with anti-hNA, were characterized by the pathologist as a large cluster of cells with neural cell bodies scattered uniformly within abundant neuropil and merged smoothly with surrounding host neuropil. No abnormal structures or vascular invasion were observed.
Figure 4. No tumor formation was observed in the MSK-DA01 tumorigenicity study.
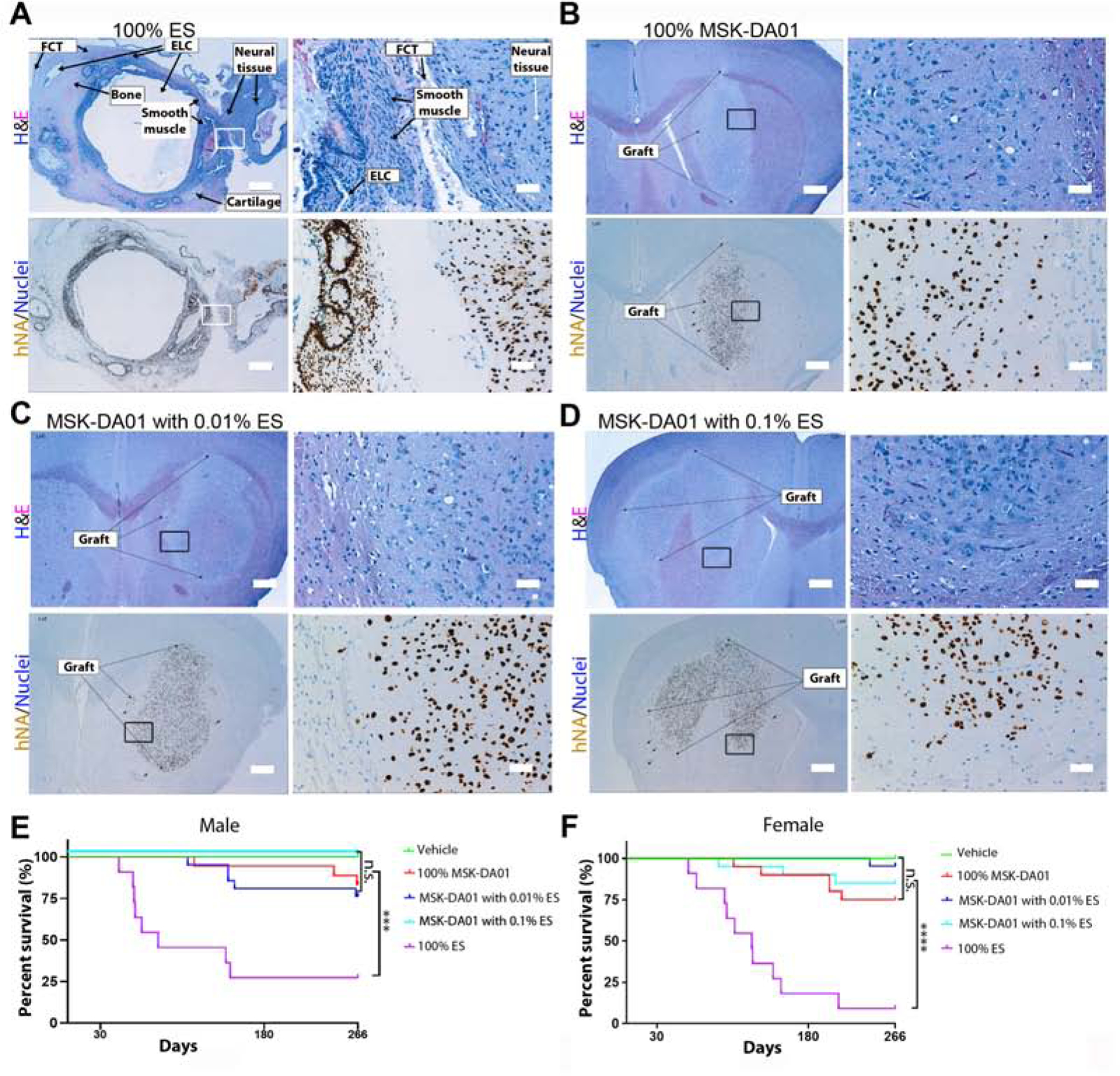
(A) Teratoma formed in the brain by intra-striatal injection of 100% cells: H&E stain in the upper panels highlights the various tissue components, and IHC for hNA in the lower panels. Teratoma is composed of multiple epithelial-lined cysts (ELC), smooth muscle, fibrous connective tissue (FCT), cartilage, bone and neural tissues. The rectangles outline the regions shown on the right. (B) Representative sections of brain grafts in the 100% MSK-DA01 by H&E and IHC staining. (C) Representative sections of brain grafts in the MSK-DA01 spiked with 0.01% ES, and (D) 0.1% ES. The rectangle outlines the regions shown on the right. (E, F) Survival curves of male (M) and female mice (F) in the vehicle, 100% MSK-DA01, and spiking groups. n=12 M or F in the vehicle group; n= 18 M and 20 F in MSK-DA01; n=21 M or F in the 0.01% ES; n=17 M and 20 F in the 0.1% ES; n=11 M or F in the 100% ES groups. ***P<0.001; ****P<0.0001; n.s.: not significant. Scale bars =500 µm in the left panels and 50 µm the in the right panels in each set.
Early death/termination occurred in most animals in the hESC group, likely related to teratoma formation. Sporadic early death/termination also occurred in the 100% MSK-DA01 (3 M and 5 F), MSK-DA01 spiked with 0.01% ES (5 M and 1 F), MSK-DA01 with 0.1% ES (3 F) groups (Figure 4A). Upon necropsy, no obvious cause of death was found, with an exception of a primary neoplasm and chronic peritonitis in 2 separate animals. Those lesions did not contain any human cells and were not considered related to the grafts. In the MSK-DA01 and spiking groups, the histology of the brain grafts was as expected in grafts of a similar age and considered not related to the cause of death. We performed a survival analysis of all animals in the tumorigenicity study. Median survival was 83 days (M) and 124 days (F) in the 100% hESC group (Figure 4E, F). There was no statistically significant difference upon comparison of survival curves between the vehicle group, MSK-DA01 and the two spiking groups, while all those groups differed significantly from the 100% hESC group (Figure 4E, F). This suggests that the deaths were likely spurious and unrelated to the MSK-DA01 grafts.
Analysis of MSK DA-01 grafts over time.
We conducted an analysis of grafts over time, including graft volumes, cell density, proliferation rates, and neural differentiation. We included animals from the safety studies that received 100% mDA cells or 100% undifferentiated hESC group that were sacrificed on day 30, 180 and 266. Graft volumes were obtained by contouring all hNA+ cells in the striatum and immediate vicinity (see methods). In the MSK-DA01 group, median graft volume increased about 8-fold from 0.122 mm3 on day 30 to 0.987 mm3 on day 266 (Figure 5A). In comparison, median volume was 10.650 mm3 in the 100% hESC group on day 266, almost 11-fold larger. In addition, there was no statistically significant difference in MSK-DA01 graft volumes between days 180 and 266, suggesting that the grafts may have reached their steady state in terms of expansion, by 6 months. In the 100% hESC group, the grafts in the four animals that survived to day 266 had a much smaller volume than the grafts in the animals that died early, suggesting that graft volume may have contributed to the early deaths in the teratoma group (Figure S5A).
Figure 5. Graft evolution over time.
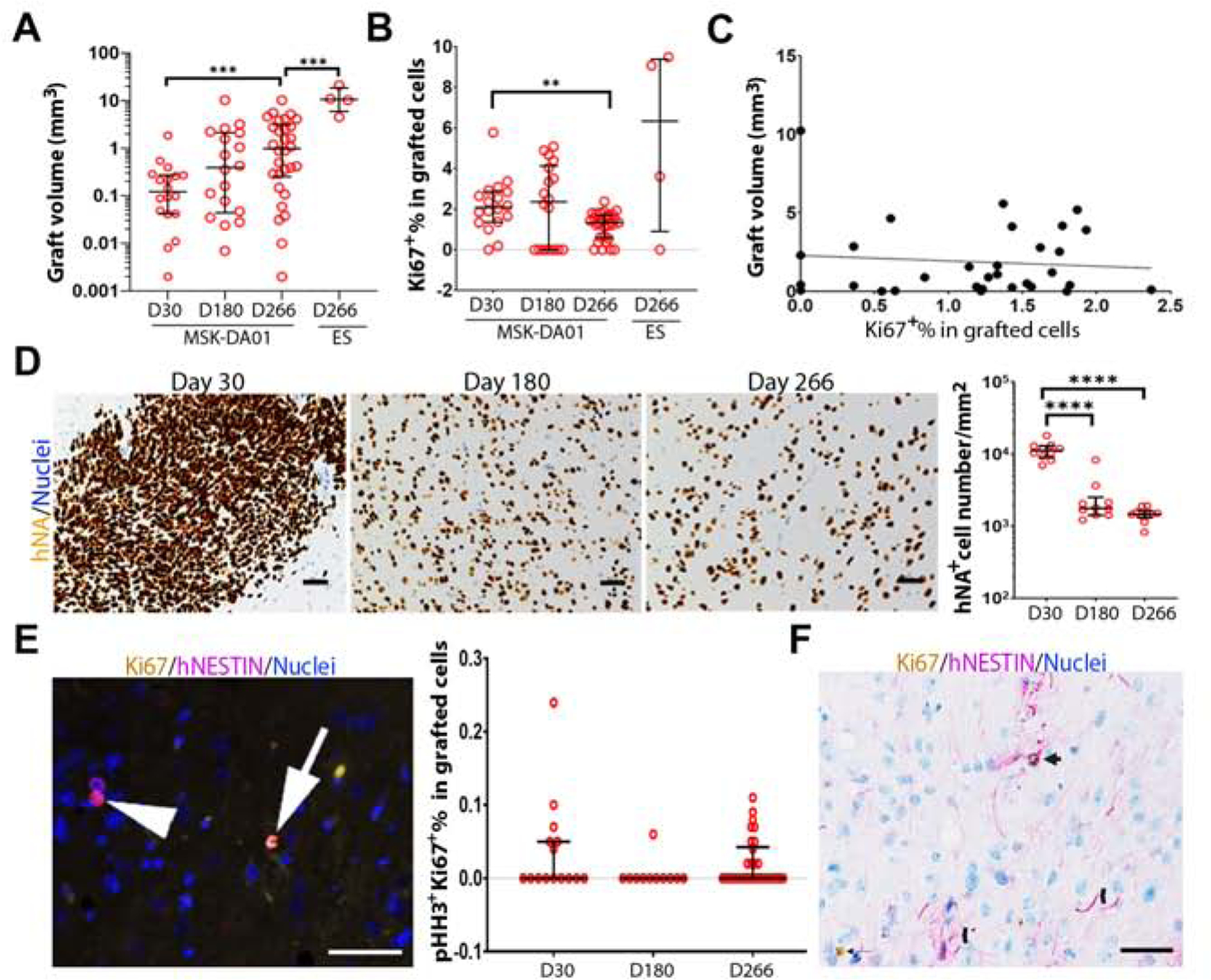
(A) Graft volumes for MSK-DA01 on day 30 (n=18), 180 (n=18), 266 (n=30) and 100% ES on day 266 (n=4). (B) Percentage Ki67 in the MSK-DA01 grafts on different days and in the 100% ES group on day 266. (C) There is no clear correlation between the graft volume and the percentage of Ki67 in MSK-DA01 grafts at day 266 (r =−0.09608, P=0.6071, n=30). (D) Representative immunohistochemical images of MSK-DA01 grafts at different timepoints, immunostained for hNA. Hematoxylin nuclear stain. Graph showing the decreasing density of human cells per mm2 over time. n=10 mice/group. (E) The presence of pHH3+Ki67+ cell (arrow), pHH3−Ki67+ cell (arrowhead) in the MSK-DA01 graft (left panel). The percentage of pHH3+Ki67+ in MSK-DA01 grafted cells on day 30 (n=15), 180 (n=11), 266 (n=30) post grafting (right panel). Data represented as median with interquartile range. Each circle represents one animal. (F) Some hNESTIN+ cells expressed Ki67 (arrow). NESTIN+ processes were observed in the graft (chevron). The data are represented as median with interquartile range. *P<0.05, ***P<0.001, ****P<0.0001. Scale bars=50 µm.
The expansion of graft volume could potentially be caused by cell proliferation and/or cell dispersion into host tissue. To evaluate the evolution of cell proliferation rates over time, we analyzed the percentage of Ki67 positive cells in MSK-DA01 and 100% hESC on days 30, 180, and 266. In the hESC group, the percentage of Ki67 was 25.84% (median) on timepoints “less than day 90” and declined to 6.34% (median) by day 266, likely related to teratoma differentiation (Figure S5B). In comparison, the percentage of Ki67 in MSK-DA01 grafted cells was already very low (2.09%, median) on day 30 and significantly decreased to 1.33% (median) on day 266 (Figure 5B). In addition, there was no clear correlation between the proliferation rate and graft volume (Figure 5C), similar to what was observed in the efficacy study. Those data suggest that the proliferation rate was not the main contributor to the expansion of graft volume. Instead, cell dispersion was observed as the grafts matured, especially in the day 180 and 266 grafts. Therefore, we evaluated the cell density in the grafts at day 30, 180, 266 (Figure 5D) and found it to have significantly decreased over time, from 11,211 cells/mm2 graft area (median) at day 30 to 1,773 cells/mm2 graft area (median) at day 180 (Figure 5D). There is no further decrease in cell density from day 180 to 266, suggesting that cell dispersion may have reached steady state. Histological analysis of the contoured grafts suggests the inclusion of host cells as individual human neurons spread out locally into the host tissue. These data suggest that traditional contouring of cells overestimates the space occupied by a human graft, which tends to expand by dispersion of its individual cells into the surrounding host striatum. Of note, none of the grafts showed any mass effect on surrounding structures.
To further evaluate the proliferation rate and the phenotype of the dividing cells, we performed immunohistochemistry for phosphohistone3 (pHH3) co-labeled with Ki67 at day 30, 180 and 266 post grafting. pHH3 expression is limited to cells undergoing mitosis in the G2/M phase (Williams and Stoeber, 2012). We found rare pHH3+Ki67+ cells with a median value of 0% and an interquartile range (IQR) up to 0.05% at all time points (Figure 5E), suggesting that only rare Ki67+ cells are actively dividing in G2/M phase. In the efficacy study, we found that the majority of Ki67+ cells expressed FOXA2 (Figure 2I, median of 67.78%). To further explore the phenotypes of Ki67+ cells, we also performed co-labeling studies with Ki67 that revealed 0–12.33% Ki67+ cells expressing hGFAP while 0–10% are hNESTIN+ (Figure 5F). There were no TuJ1+ cells in Ki67+ cells except in one animal, and no OLIG2/Ki67 co-labeled cells. Ki67-labeled cells were scattered within the graft with no clusters seen. Quantifying multiple cell subtypes using co-labels is suboptimal especially when target cells (Ki67+) are rare and the number of sections limited.
Immunohistochemistry demonstrated that majority of cells in the grafts were of the neuronal lineage with TUJ1+ cells representing 80% to 100% of hNA+ cell population in graft with no significant difference from day 30 to 266 (Figure S5C). We analyzed the composition of the graft in experiments performed with research grade cells and identified GFAP+ cells (average of 10.08%) and NESTIN+ positive cells (average of 4.06%). In separate analyses of graft composition, we performed immunohistochemistry labeling for COL1A1 (Tiklová et al., 2020), transthyretin (TTR)(Doi et al., 2020) which have been reported in the literature as contaminants of in vivo grafts and found both to be negative (Figure S5D). Similarly, immunostains for smooth muscle actin, cytokeratin 8/18, GATA4, AFP, and β-catenin were all completely negative.
Clinical relevance
Here we present an arduous journey extending over nearly a decade to develop a cell product based on a successful previously published protocol for the derivation of dopamine neurons from human ES cells (Kriks et al., 2011). The initial steps involved a new approach to pattern dopamine neuron fate from human pluripotent stem cells, based on “boosting” of WNT levels (biphasic treatment), leading to an improved protocol that increases levels of midbrain marker expression in the differentiated cell types while avoiding both anterior (diencephalic) and posterior (hindbrain) contaminants as detailed in the companion manuscript (Kim et al., 2020). Kim et al. also demonstrate that the new protocol enables mDA neuron derivation with minimal batch to batch variability and under culture conditions suitable for GMP manufacturing. Robust cryopreservation and cell stability data are also obtained, as well as proof of concept evidence for efficacy. Here we describe the path taken to develop a portfolio of preclinical data that enable submission to the regulatory agency for clinical translation. Our data demonstrate that the biodistribution, toxicology, and tumorigenicity profiles as well as the efficacy results, are supportive of clinical trial initiation. In parallel to this work, we established a team including neurologists, neurosurgeons, transplant experts, ethicists and patient advocates to develop a clinical trial draft. Clinical application brings a set of additional challenges, ranging from the method of delivery into the brain, to patient eligibility, selection of primary and exploratory endpoints and future cell product enhancements (Parmar et al., 2020). Accurate stereotactic delivery into the striatum is not technically challenging per se, but each needle or cannula insertion in the brain carries a very small risk of hemorrhage. In addition, the overarching goal is to innervate the majority of the putamen, and since the dopamine progenitors are not highly migratory, our operative design involves three trajectories into each putamen from a pre-coronal path, and three cell deposits along the axis of the needle, starting with the deepest deposit. We anticipate bilateral injections in the same setting to reduce the need for a second procedure under anesthesia. To enhance safety, we propose imaging the patient after completing the injections in the first hemisphere. Our center, like some others, affords access to an intraoperative magnetic resonance imaging (MRI) system. By this method, the quality of cell delivery will be ensured due to the accuracy of the injections, the prevention of reflux by the design of the cannula, and the possibility of monitoring for potential hemorrhage in real-time. Such factors are very difficult to account for in rodent surgery. We did however test viability of the cells after thaw and dwelling on ice for 5–8 hours then passage through the intended surgical cannula. We found no negative impact on viability or cell phenotype. Another important factor concerns the difficulty in handling very small fluid volumes in the microliter range. There are currently no approved devices for cell injections in the brain, so most teams adapt existing cannulas that are compatible with their stereotactic navigation system. Our clinical design will call for a limited dose escalation (one low dose of 0.9 million cells per hemisphere, and a higher dose of 2.7 million cells divided in 3 tracts per hemisphere), both of which will require the delivery of very small volumes of 3 or 9 microliters per track for the low and high dose groups respectively. Great care must be afforded to avoid cell settling and to minimize mechanical injury to cells during the cannula loading phase. Based on our preclinical efficacy study, we expect the low dose to result in the survival of about 100 × 103 mDA neurons, which is the lowest dose correlated with clinical improvement in fetal tissue studies (Mendez et al., 2005). The higher dose aims at a more complete restoration of mDA neuron cells numbers and re-innervation.
The question of immunosuppression in the setting of the proposed allografts was also debated among our group and external consultants. Historically, immunosuppression for fetal allografts in the CNS was considered unnecessary by some or required for a short term, as the brain was deemed to be immunologically privileged, in comparison to other organs. However, experimental and some clinical data demonstrate the development of allo-immunization to graft-derived antigens, that can be associated with symptoms and graft rejection (Krystkowiak et al., 2007; Morizane et al., 2013). It was also recently demonstrated that grafting major histocompatibility complex (MHC)-matched allogeneic neural cells in the brain of non-human primates reduces the immune response by suppressing the accumulation of microglia and lymphocytes into the grafts. This rejection response was reduced by immunosuppressants (Morizane et al., 2017), leading to enhanced graft survival. Furthermore, tacrolimus-based immunosuppression is sufficient to allow for survival of human iPSC-derived mDA neurons and functional recovery in non-human primate model of PD (Kikuchi et al., 2017). Consequently, we elected to design an immunosuppressive regimen consisting of a short course of prednisone and a maximum of one year of tacrolimus, a calcineurin inhibitor that is a commonly used and well-tolerated immunosuppressive agent. Future cell therapy products may rely on autologously derived iPSCs when manufacturing and cost parameters allow, banks of human leukocyte antigen (HLA)-typed iPSCs or engineered “universal” ES lines that are gene-edited to delete major HLA class I or II loci or to express immunosuppressive molecules such as PD-L1, thus becoming less visible to the immune system (Lanza et al., 2019).
In terms of patient eligibility, an important consideration is the fact that patients with very advanced Parkinson’s symptoms are not likely to benefit from this therapy. This is particularly the case for patients who no longer show a robust response to L-Dopa or who suffer from late-onset cognitive symptoms unlikely to benefit from an mDA graft. While it is customary and ethically appropriate to limit novel untested therapies in early phase trials to patients with no remaining options for their disease, one can argue that the corollary of this approach presents other ethical challenges. Namely, patients who are not likely to benefit from cell therapy should not be exposed to its potential risks. This concern is not limited to Parkinson’s disease but extends to other neurodegenerative diseases and beyond. Thus, we believe that early stage cell therapy trials should include patients with significant symptoms of Parkinson’s disease who have retained a capacity to respond to dopamine but no longer show satisfactory results. While a typical Phase 1 trial has to focus on feasibility, safety and tolerability as primary endpoints, the inclusion of secondary endpoints and exploratory objectives that permit a window into the potential efficacy and impact of the graft on the disease is critical. Therefore, in addition to safety, we will assess evidence for graft survival and efficacy by measuring 18 Fluoro-Dopa uptake in the striatum by PET imaging and changes in clinical rating scales (UPDRS part III). In addition, we will monitor impact on non-motor symptoms as well as an array of instruments that measure quality of life, drug intake, dyskinesia, activities of daily living, and neuropsychological parameters. Lastly, the work described here was supported by New York State’s stem cell initiative in 2013. The grant awarded to our team mandated targeted progress towards readiness for submission of an IND (investigational new drug) application over 4 years and concluded in 2017, with the production of nearly 1,000 vials of clinically qualified frozen dopamine progenitor cells, as described here. The next major and rate-limiting hurdle required the identification of a sponsor to further support the development of the product into a clinical therapy. This is a time-consuming step that presents its own sets of challenges but potentially the reward of bringing scientific discovery from the bench to the bedside.
Limitations of study
The study was designed to be responsive to the regulatory environment in the US. Key considerations discussed included approval of the human ES cell source, characterization of cell banks, details of manufacturing, animal numbers in each study, cell numbers injected in the animals, duration of observations in the animals and choice of animal species. The types of analyses performed on the grafts had to be limited to robust readouts, that are familiar to available pathologists and that are available as GLP-validated assays by an independent contract research organization (CRO). As an example, multiple co-labeling of cells in order to more fully characterize the graft phenotypes proved to be challenging. Similarly, functional in vivo readout was limited to amphetamine-induced rotations given the large number of animals to be tested and given the need for automated quantification which is difficult to achieve using alternative behavioral readouts such as the cylinder or stepping tests. Financial considerations were also important as outsourcing this type of data collection and analysis is very costly. Unlike usual scientific investigation, data analysis and interpretation are delivered to the investigating team all at once, without an opportunity to respond to early results with modifications or fine tuning. Alterations to existing experiments, additional new tests or antibody immunostaining require a formal process of amendments, approvals and pilot studies, and therefore are undertaken with serious consideration and caution. Therefore, investigators have to dedicate significant time and effort for pre-planning and must prioritize analyses and readouts that are most relevant to achieve the goal of clinical application, even if emerging scientific questions remain unaddressed.
STAR Methods
Resource availability
Lead contact
Further information and requests for resources and datasets should be directed to Viviane Tabar (Tabarv@mskcc.org).
Materials availability
The study did not generate new unique reagents.
Data and code availability
All datasets are available upon request.
Experimental model and subject details
Cell lines
WA09 (H9) hESC are supplied by the WiCell Research Institute and from a cell bank manufactured at Waisman Biomanufacturing Center, University of Wisconsin.
Animals
All animal procedures and maintenance at Memorial Sloan Kettering Cancer Center (MSKCC) and WuXi Apptec were approved by our Institutional Animal Care and Use Committee (IACUC) and following NIH guidelines. The animals were acclimated for at least five days to laboratory conditions before the procedures.
NOD.Cg-Prkdcscid IL2rg tm1Wjl mice (NSG mice, The Jackson laboratory) were 7–8 weeks old at the beginning of the studies. The short-term in vivo study was performed at MSKCC. The biodistribution, toxicity and tumorigenicity studies were performed at WuXi Apptec following GLP regulations. An equal number of males and females were used at the beginning of study. Athymic nude rats (NIH-Foxn1rnu, 8–10 weeks old, Charles Rivers Laboratory) were subjected to 6-OHDA lesioning and MSK-DA01 cell grafting in the efficacy study. In all studies, animals were randomized into the different groups.
To reduce the observation bias, the staff who performed the experiments were blinded to group assignments in all procedures. The exclusion criteria for animals in the studies are listed in Table S4. Animals without any brain grafts in the cell group due to technical issues during transplantation were not included in the graft analyses.
Method details
Cell manufacturing.
cGMP manufacturing of MSK-DA01 cells, which were differentiated from hES cells (WA09 FRS at passage 48, XX), was performed by members of the SKI Stem Cell Research Facility (SCRF) at the GMP facility named Cell Therapy and Cell Engineering Facility (CTCEF) at MSKCC, directed by Dr. Isabelle Rivière. WA09 cells were thawed and expanded on Geltrex™ coated flask/dish in Essential 8 basal medium with supplement for 10–14 days. Cells were split by Dispase (Stem cell technologies) every 3–5 days in between at ratio usually between 1:4 and 1:6. For MSK-DA01 differentiation, single cells WA09 were washed and plated on Geltrex™ at 400,000 cells/cm2 in Neurobasal (NB) with N2 and B27 (without vitamin A) containing 2mM L-glutamine, 10.8µM SB431542, 250nM LDN193189 (LDN), 0.7µM CHIR99021, and 500ng/mL SHH with 10µM Y-27632. Media was replaced daily, except without Y-27632. On day 4, the same media was used except with 7.5µM CHIR99021. Same media was replaced on day 6. On day 7 SB, LDN and SHH were withdrawn from the medium. Same media was changed on day 9. On day 10, NB (with B27) with 2mM L-glutamine, 20ng/ml BDNF, 20ng/ml GDNF, 200µM Ascorbic acid (AA), 500µM Dibutyryl-cAMP, 1ng/ml TGFβ3 and 3µM CHIR99021 was added. On day 11, cells were dissociated to single cells with Accutase for 30–40 minutes, and replated on poly-ornithine (15µg/ml, Sigma Aldrich), fibronectin (1µg/ml, Akron Biotech) and laminin (2µg/ml, Trevigen)-coated plates at 800,000 cells/cm2 using the same medium as day 10. On day 12, the medium was switched to include 10µM DAPT but CHIR99021 was withdrawn. Complete media changes were performed daily until harvest at day 16. At day 16 of differentiation, MSK-DA01 cells were dissociated with Accutase for 30–40 minutes and filter through 40 µm cell strainer. Cell pellets were resuspended at a cell density of 8 million cells/ml of STEM-CELLBANKER™ in cryotubes before being placed in a controlled rate freezer (ThermoFisher) to cryopreserve cell product. Cryopreserved vials were stored in the secured GMP Facility freezer and monitored 24/7 by Datatron system.
Biosafety tests on cell products
Cell viability was determined with Acridine Orange/Propidium Iodide (AO/PI) Staining Solution on Nexcelom cellometer (Nexcelom Biosciences, CS-01060). Mycoplasma (by Lonza MycoAlert™ PLUS Mycoplasma Detection System at cGMP standard) and pyrogen (by limulus amebocyte lysate assay at USP standard) were tested in CTCEF while sterility was tested by BD gram stain kit (BD biosciences) and 14-day growth assessment in the SCRF at MSKCC. Adventitious virus tests consisted of in vitro tests on MRC-5, Vero, A549, and NIH-3T3, and in vivo tests for the presence of inapparent viruses in suckling and adult mice and embryonated hen’s eggs. MAP was performed by testing the sera for the presence of antibodies to murine virus four weeks after injecting MSK-DA01 into viral free mice under GLP regulation. Adventitious virus and MAP were tested by Bio Reliance (Rockville, Maryland, USA) and Indiana University vector production facility (Indianapolis, IN, USA).
qRT-PCR analysis for cells.
RNA was extracted from 3 × 106 cell pellets by RNeasy Mini Kit (Qiagen). The residues of DNA were wiped out by RNase-Free DNase Set (Qiagen). After quantification of RNA using Nanodrop 2000 (Fisher), RNA samples were processed to cDNA by RT2 First Strand Kit (Qiagen). qRT-PCR was performed with 2x RT2 SYBR Green Mastermix (Qiagen) and RT2 Primer Assays (Qiagen) on Eppendorf Mastercycler Realplex. The primers to detect POU5F1 and avoid pseudogenes (Forward sequence: GATGGCGTACTGTGGGCCC; Reverse sequence: TGGGACTCCTCCGGGTTTTG; Probe Sequence: CCAAGGCGGCTTGGAGACCT Fluorophore: FAM) is applied (Liedtke et al., 2007). The procedures followed the manufacturer’s instructions. No template control is set up as negative control. Cq values of the interested genes were determined and normalized by actin mRNA.
Flow cytometry.
For each antibody set analysis, 3×106 cells were used. For analysis with anti-OCT3/4/NANOG/SOX2, or anti-PAX6, cells were fixed with Cytofix buffer (BD Biosciences) for 20 minutes at room temperature. For analysis with anti-FOXA2, cells were fixed with 1.6% PFA for 30 minutes at 37° C. After washing with 1x Perm/Wash buffer (BD Biosciences) twice, the cell pellet was suspended in 300 µl 1x Perm/Wash buffer (BD Biosciences). The cells were evenly separated into three tubes which were unstained, isotype control, and antibody. The cells were incubated with buffer only (unstained), isotype control (same volume as antibody) or antibody (20 µl PE anti-hNANOG/PerCP-Cy5.5 anti-Oct3/4/Alexa 647 anti-SOX2, 5µl Alexa 647 anti-PAX6, or 5µl Alexa 488 anti-human FOXA2) for 30 minutes in dark at room temperature, respectively. After washing with 1x Perm/Wash buffer (BD Biosciences) twice, cells were resuspended in 300 µl DPBS (Life technologies) and passed through 35 µm cell strainer, respectively. Samples were analyzed through flow cytometry (FACS Aria III, BD Biosciences).
Cell preparation for transplantation.
On the same day of transplantation cells were thawed in a 37°C water bath for two and half minutes with gentle swirling. The cells were suspended in transplantation medium (2 mM L-glutamine, 0.2mM ascorbic acid, 0.1% Kedbumin (Kedrion Biopharma) in Neurobasal medium). After centrifugation at 200xg for 5 min, cell pellets were resuspended in transplantation medium to adjust the alive cell concentration to 100,000 ± 10,000 cells/µl.
Stereotaxic brain injections.
Isoflurane anesthesia was performed for all the surgeries. For lesioning, 6-OHDA solution (3.6 mg/ml in 0.2% ascorbic acid and 0.9% saline, Sigma) was stereotactically injected into two sites to establish unilateral medial forebrain bundle lesions of the nigro-striatal pathway (2.5µl, Tooth bar set at −2.4, AP −4.4 mm, ML −1.2, VL −7.8; 3 µl Tooth bar set at +3.4, AP −4.0 mm, ML −0.8, VL −8.0). Cells were suspended in transplantation medium to 100,000 ± 10,000 cells/µl and animals received 400,000 cells each in all studies, except for the low dose groups of the biodistribution and toxicology studies which received 200,00 cells. Stereotactic coordinates for the striatal injections in all mice were: AP: +0.5mm, ML: −1.9mm; VL: −3.2 and −3.0 mm in mice, and AP: +1mm, ML: −2.8, VL: −4.7, −4.6, −4.5 and −4.4 mm in rats. The cells or vehicle solution were injected at the rate of 1 µl/min by motorized stereotaxic injector (Model 53311, Stoelting company, IL, USA). The syringe was kept in place for 5 minutes then slowly withdrawn at 0.5–1 mm/min. Surgeons were blinded to groups and graft content. Cell grafting was performed within 8 hours after cell preparation. Postoperative cares including pain management, were performed to ensure the full recovery after surgery.
Animal monitoring.
For the efficacy study, cage-side general observations were performed once daily. Body weight was noted weekly until termination, and daily for 3 days post-surgery. Veterinary care was administered as needed.
For safety studies, mortality/moribundity was checked twice a day. Cage side general health observation were performed daily. Signs of illness, toxicity, unusual or abnormal appearance were noted. Clinical observations were performed prior and post dosing, daily for the first five days, and weekly after until termination. Body weight was collected on randomization assignment day, on day 0 prior to surgery, twice weekly thereafter, one day prior to termination and at termination (fasted).
Amphetamine-induced rotation testing.
Amphetamine-induced rotation tests were performed before transplantation, and 2, 4, 5, 6.5, 8 months after transplantation. The rats were injected intraperitoneally with a D-Amphetamine in saline (Sigma, 5mg/kg). After 10 minutes, the rotation behavior was recorded for 40 minutes and the total rotations were automatically counted by Ethovision XT 11.5. The data were presented as (Ipsilateral-contralateral) rotations per minute.
Animal termination and necropsy and tissue collection.
For short-term in vivo and scheduled termination of efficacy study, animals were anesthetized by intraperitoneal injection of pentobarbital (300 mg/kg). Then the mice were transcardially perfused with 0.1M PBS and following with 4% PFA (Electron microscopy biosciences). In efficacy study, prior to perfusion, bone marrow was extracted, and bone marrow smear was performed and fixed with methanol. The other tissues were post-fixed with 10% formalin (Leica). A gross necropsy for the external body surface, all orifices, cranial cavity and contents, thoracic cavity and contents, and abdominal cavity and contents was conducted. Animal tissues were archived, including any gross lesion or tumor, heart, axillary lymph nodes-axillary, bone marrow smear, kidneys, spleen, brain, liver, spinal cord, femur (with marrow), lungs with mainstem bronchi). A full necropsy was performed by the Veterinary Pathology Services at MSKCC in two animals with a mass on the flank, and in four animals that had to be terminated early. For the GLP studies at WuXi Apptec, necropsies were performed by their pathologists. Most tissues were preserved in 10% formalin. Bone marrow smears from one femur were fixed in methanol which was only examined if deemed appropriate based on examination of marrow sections from the sternum. The brain sections included sections of grafts, medulla/pons, cerebellum cortex, and cerebral cortex. Thyroid/parathyroid was examined only if present on routine sections. All the extracted organs from all animals with scheduled termination in toxicology study were examined histologically. For the tumorigenicity study, brain tissues or other tissues which were suspicious in gross examination from all animals that were terminated on schedule were examined histologically. In the animals with early death/termination, the extracted organs were examined histologically. For any tumor or lesion, four slides from the suspected region were submitted for IHC staining with anti-hNA to determine if it is of human origin. The rest of tissues were retained in formalin for possible future analysis.
Tissue lists: Any gross lesion or tumor; Kidneys; Skin; Adrenal glands; Lacrimal glands; Spleen; Aorta (thoracic); Liver; spinal cord; Bone marrow smear; Lungs with mainstem bronchi; Sternum (with marrow); Brain; Lymph nodes, axillary; Stomach; Cecum; Lymph nodes, mesenteric; Testes; Colon; Mammary gland – female; Thymus; Duodenum; Ovaries; Thyroid/parathyroid; Epididymis; Pancreas; Tongue; Esophagus; Pituitary gland; Trachea; Eyes; Prostate; Urinary bladder; Femur (with marrow); Rectum; Uterus, cervix, oviducts; Gallbladder; salivary glands (mandibular); Vagina; Heart; Sciatic nerve; Ileum; Seminal vesicles; Jejunum; Skeletal muscle
Cell and tissue processing, immunohistochemistry (IHC) and image processing.
All procedures and analyses were processed blindly. For IHC on cells, cells were fixed in 4% PFA for 20 minutes. The primary antibodies included anti-tyrosine hydroxylase (TH) (1:400), FOXA2(1:100), LMX1A (1:100) and POU5F1 (1:100). The images were captured at 200 X magnification by Perkin-Elmer Operetta™ High Content Microscope. For calculation of the percentage of POU5F1+ cells out of all DAPI+ cells, at least 10,000 cells were analyzed per group. For the short-term in vivo study, brain tissues were dissected and post-fixed with 4% PFA for 12 hours, then changed to 30% sucrose in 0.01M PBS for 24 hours. The tissues were embedded in O.C.T (Sakura Finetek USA, Inc.) and cryosectioned at 30 µm. The primary antibodies included STEM 121 (1:500), anti-TH (1:400), anti-hNA (1:100), anti-FOXA2 (1:100), anti-NURR1 (1:50), LMX1A (1:100), PITX3 (1:100). The cells or sections were washed with 0.1M PBS (Invitrogen) for 3 times of 5 min, then blocked with 1%BSA-0.3% Triton-PBS for 1hr. The primary antibodies were incubated overnight at 4°C. After washing with 0.01M PBS for 3 times of 5 min, the cells or tissue sections were incubated with appropriate fluorescent (Alexa 488, 568, 647) conjugated second antibodies (1:400, ThermoFisher Scientific) for 1 hour at room temperature. After rinsing with PBS, the nuclei were counterstain by DAPI. The slides were examined under a fluorescent microscope (Olympus BX51). Representative pictures were taken with a Hamamatsu camera. The image for the whole graft is a collage of individual images.
In the efficacy study, all the brain tissues for scheduled termination were submitted to Charles Rivers Laboratories for processing for Immunohistochemistry and stereological analysis. The brain tissues were embedded in paraffin and the paraffin sections were immunostained on a Ventana Discovery Ultra. For stereology analysis, sections were stained for TH (Abcam, 1:750)/hNA (Abcam, 1:250) (see stereology analysis section). For further phenotyping of grafted cells, three single sections evenly spaced throughout the region of interest were stained with antibodies to hNA(1:250) /hydroxytryptamine (5-HT, 1:4000), hNA(1:100)/glutamic acid decarboxylase (GAD67,1:16000) and hNA(1:25)/Ki67 (abcam, 1:100). Positive and negative controls were included during the process. hNA/5-HT and hNA/GAD double immunostained slides were scanned at 200X magnification using the Hamamatsu Nanozoomer whole slide scanner and imported into the Visiopharm software. The sections stained with hNA/Ki67 were imaged using an Olympus fluorescent microscope BX53 equipped with an Olympus DP80 camera and associated Cell Sens software (Version 1.15). Representative images (at least 15 random images per animal) were captured at 400X magnification and uploaded into the Visiopharm software. The graft regions were outlined manually, using the hNA positive cells as a guide for the region of interest (ROI). Image analysis algorithms were created to identify single and double stained cells and the percentage of human antigen positive cells that were dual positive for each of the other markers was determined.
The brains of early death/termination in efficacy study were sliced coronally, embedded in paraffin and sectioned at 8 µm throughout 6 mm brain slices which covered 3 mm rostral and caudal to the injection site. Immunohistochemistry was performed on at five evenly spaced brain sections throughout graft region per set of staining with a Discovery XT processor (Ventana Medical Systems) in Molecular Cytology Core at MSKCC. The primary antibodies are anti-TH (1:500, Millipore), anti-hNA (1:100, Clonetech), anti-Oct-4 (1:200), anti-Ki67 (1:100). The signals were visualized by DAB (counterstained with hematoxylin). Positive and negative isotype controls were included in the procedures for quality control. Sections were scanned by Mirax Slide Scanner. The pictures were taken by Panoramic Viewer (#DHistech, Budapest, Hungary) software 1.15.2.
For two animals with a mass on the flank, the masses were collected and embedded in paraffin or O.C.T (Sakura Finetek USA. Inc.). Paraffin sections at 8 µm and cryo-sections at 30 µm were collected. The paraffin sections were stained with hematoxylin and eosin (H&E) for histological characterization of the masses. Cryo-sections were stained with anti-hNA with the same method of IHC as described above. Human Glioblastoma tissue sections were used as positive control for human cells. Negative controls were also included as quality control. The slides were examined under a fluorescent microscope (Olympus BX51). Representative pictures were taken with a Hamamatsu camera.
In the toxicology and tumorigenicity studies, the brain tissues were processed in WuXi Apptec. Brain slices at 2mm were collected which covered 1mm rostral and caudal to the injection site. Both brain slices were embedded into same paraffin block and processed to 8 µm coronal sections. All sections were collected. From each graft, the serial of six sections which are equally spaced throughout the whole tissue which is approximately 800 µm were selected for staining. One set of sections were stained with H&E while other set of sections were stained with anti-hKi67, anti-hNA (Clonetech), and anti-Tuj1 antibodies. In addition, a set of three sections, which were 1–2 mm rostral from the rostral end of the graft, 2–5 mm caudal from caudal end of the graft and 1–2 mm through the cerebellum, were stained with H&E and anti-hNA. Positive and negative control were included. For calculating the percentage of hKi67+ or Tuj1+ cells out of grafted cells, six evenly-spaced sections throughout the graft region were selected. The set of slides for hKi67+ and Tuj1+ staining which were closed to hNA staining were selected. The cells were counted in random fields of view in graft, up to a total of 1000 grafted cells on one slide. The percentage of Ki67 or Tuj1= 100 % x (sum of Ki67 or Tuj1-positive graft cells counted in 6 brain sections)/(sum of total graft cells counted in 6 brain sections).
To analyze the mitosis cells, the immunofluorescent anti-pHH3 labeling was performed was performed on six sections per animal (Day 30, 7 males, 8 females; Day 180, 4 males, 7 females; Day 266, 15 males, 15 females). Since anti-pHH3 is not human specific, co-labeling with human specific Ki67 was applied to identify human pHH3 positive cells. To further characterize Ki67+ cells, hKi67/Tuj1, hNESTIN/Ki67, hGFAP/Ki67, OLIG2/hKi67 immunostainings were performed on six sections per animal at day 266 (n=30, M:F=1:1). All the staining was performed at Premier Laboratory. The positive cells were evaluated by a board-certified pathologist. At least 1000 grafted cells were analyzed per animal.
To detect the presence of other cell types or possible contaminated cells in graft, additional immunofluorescent staining of hNA(1:100)/NESTIN (1:200, Abcam), hNA/GFAP (1:500, DAKO), hNA/hCOL1A1 (1:100, R&D systems), hNA/transthyretin (TTR, 1:50, Abcam) on 30 µm cryosections of the grafts 6 months post transplantation of research grade cells (n=3).
Stereology analysis.
In efficacy study, stereological analysis was performed by Charles Rivers Laboratory after initial discussions with the investigators of this study. The coronal brain slices were made over 6 mm which covered 3 mm rostral and caudal to the injection site. The slices were weighed before and after processing. The thickness of brain slices was measured to determine the sampling interval (T) as 500 µm for systematic uniform sampling (SURS) to have 8–10 physical dissectors throughout the grafted area. The tissues were embedded in paraffin and sections were sliced at a 5 µm thickness. For each tissue block, the physical dissectors pair sections were collected from a random start number. All the physical dissector pair sections in cell group and only three dissector pairs (sampling level 3, 5, 7) in vehicle group were stained.
Slides for physical dissector analysis were stained with TH (Abcam)/hNA(Abcam), scanned at 20X magnification using the Hamamatsu Nanozoomer whole slide scanner (Version. 3.1.9) and imported into the Visiopharm software. An algorithm was created to identify hNA positive neurons to create a ROI and guide proportionator sampling for counting of hNA-positive cells, in which three independent samplings of 10 fields were performed. For counting of TH/hNA double-positive cells, SURS sampling (Fractionator) of 8% of the ROI was performed. The initial sampling had high coefficient of error (CE) estimates and were re-sampled at 16% to improve CE. High magnification (20X) matching fields were captured for each sampling and a unique counting feature (hNA-positive nucleus) was used to count the hNA-positive cells and hNA/TH double-positive cells (perinuclear TH+ with hNA+ nucleus) within an unbiased counting frame. Additionally, point probes were placed over the ROI at low magnification, with an area per point of 270,158.5 µm2, to determine the volume of the graft according to Cavalieri’s principle (Gundersen, 1986; Gundersen et al., 2013). All stereology data were imported into the calculator tool of the Visiopharm software and estimates of total number of hNA positive cells, total number of hNA/TH dual positive cells, and total hNA graft volume were calculated.
For each animal, 3-dimensional tissue shrinkage during processing was estimated based on the weight of the brain slices before (Wpre) and after processing (Wpost) before paraffin embedding. The formula is: 3D global shrinkage = 1 – (Wpost/Wpre) (Gundersen et al., 2013). The estimated total graft volume was calculated by the Visiopharm calculator tool according to the Cavalieri method using the following equation: V = ƩP x A(p) x T; where ƩP was the number of points intersecting the tissue, A(p) was the area per point (recorded by the software), and T was the sectioning interval (Howard et al., 2005). The CE was also calculated for each animal (Løkkegaard et al., 2004). CE for graft volume is 0.052±0.069 for male and 0.034±0.012 for female. The volume was then corrected for each animal using the estimated 3D global shrinkage (Gundersen et al., 2013).
The number of hNA single positive cells was estimated by the Visiopharm calculator tool using the following equation: N = ƩQ/2 × 1/ssf. The number of hNA/TH dual positive cells was estimated by the using the following equation: N = ƩQ/2 × 1/ssf x 1/asf, where ΣQ was the sum of the Proportionator weighted counts (divided by 2 due to counting in both directions of the dissector), ssf was the sectioning sampling fraction (section thickness divided by sectioning interval) (Gardi et al., 2008), and asf was the area sampling fraction (percentage of tissue sampled) (Gundersen et al., 2013). The CE was also calculated for each animal (Gardi et al., 2008, Løkkegaard et al., 2004). CE for hNA+ cell number estimation is 0.132±0.056 for male and 0.1±0.067 for female. CE for hNA+TH+ cell number estimation is 0.188±0.191 for male and 0.129±0.025 for female. In addition, the precision and efficiency were determined for each estimate by the PROBE (precision range of an optimally balanced estimator) equation: CV2 (coefficient of variation) / CE2 to prove the design optimal (Gundersen, 1986; Gundersen et al.,2013).
For short-term in vivo analysis (n=4–6/lot), we performed stereological estimation by Stereo Investigator (MBF Bioscience, Vermont). The number of cells was determined using the optical fractionator probe. The volume of graft was analyzed using the Cavalieri estimation function.
Real time quantitative PCR for biodistribution study.
The tests were performed at WuXi Apptec under GLP conditions. A selected panel of tissues (any gross lesion or tumor, liver, blood (in EDTA), lungs with mainstem bronchi, bone marrow (femur or sternum), lymph nodes, axillary, brain, spleen, heart, spinal cord, kidneys) was collected. Whole tissue for small tissues and a randomly selected portion of each homogenized whole tissue were subjected to DNA extraction. For brain tissue, a 2mm x 2mm x 2mm tissue cube which covers the cell injection site was collected. qPCR with a human Alu-specific primer were used to detect human DNA. The PCR assay’s sensitivity was based on human DNA isolated from MSK-DA01 of ≤50 genome copies (approximately 165 pg DNA/µg gDNA). For the qPCR assay validation, DNA yield and the lowest non-interference dilution (evaluated by spiking with 50 genome copies of positive control cells) were determined. At the lowest non-interference dilution, the DNA from each naïve tissue was confirmed to be absent of any quantifiable amount of the target DNA (aka less than the Limit of Quantification, LOQ) in qPCR assay. The LOQ was determined to be about 0.30303 genome copies. The positive threshold was 0.30303 genome copies/µg x dilution factor. Therefore, the positive threshold is 1.51515 (the dilution factor is 5) for brain and spinal cord tissues in male mice and spinal cord tissues in female mice at the end time point of day 30 while 0.30303 for other tissues and time points. Each sample was run in triplicate with positive control (spiked naïve tissue) and negative control (buffer).
Graft volume estimation for graft over time in safety studies
12 evenly-spaced sections throughout the region of interest were selected for hNA staining and graft volume calculation later. The area of hNA+ cell clusters were measured by NIS-Elements AR software (Version 4.20.02). The estimated graft volume was estimated as V = A1T1 + A2T1 + A3T1 + A4T1 + A5T1 + A6T2 + A7T1 + A8T1 + A9T1 + A10T1 + A11T1 + A12T1. V, estimated volume; T1, the sampling interval; A(n), the area in slide (n) (Brown, 2017).
Cell density analysis for graft over time in safety studies.
The tests were performed at WuXi Apptec under GLP conditions. The hNA stained sections (graft greater than 0.09 mm2) for graft volume estimation was used in cell density analysis. Five random animals of each sex at the scheduled study end-point were selected for cell density analysis. For each animal, the brain section with the greatest cross-sectional area in each animal was selected for assessment. When the graft area on that section was less than 0.45 mm2, the section which is next to it will also be included. Under 200 x magnification, the total number of human cells (hNA+) were counted in five randomly selected 300 µm x 300 µm regions were counted. The data were represented as the number of human cells in graft per mm2.
Quantification and statistical analysis
Normality test was applied by Shapiro-Wilk normality test (the method of Royston). Data in normal distribution were represented as mean±SD while otherwise as median with interquartile range. The number of cases in groups were specified in figure legends and methods. For the unpaired data that passed the normality test, one-way ANOVA was applied, and unpaired t-test was used between two groups with equal SDs. Welch’s correction was applied for the data with unequal SDs. For the non-paired data that do not fit a normal distribution, Mann-Whitney test was used. For the parametric paired data, paired t test was performed between two groups when the differences between paired values are consistent. Otherwise, Wilcoxon matched-pairs signed rank test was applied. Correlation analysis was performed by Pearson correlation or nonparametric Spearman correlation accordingly to the data. The survival curve comparison was performed by logrank (Mantel-Cox) test. Data exclusion criteria were specified in section of results and methods accordingly. Probability (p) values of less than 0.05 were considered statistically significant. All statistical analyses were analyzed by GraphPad Prism 8.3.0. A statistical consultant, Certara (Princeton, NJ), oversaw the analysis of Ki67, graft volume and neuronal cell counts in the safety, toxicity and tumorigenicity studies.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa 647 anti-human Pax6 | BD Pharmingen | Cat# 562249, RRID:AB_11152956 |
| Alexa Fluor 488 goat anti-human FoxA2/HNF3β | R&D systems | Cat# IC2400G, RRID:AB_2801552 |
| PE anti-hNanog, PerCP-Cy5.5 anti-Oct3/4, Alexa 647 anti-Sox2 | BD biosciences | Cat# 560589, RRID:AB_2722505 |
| STEM101 (anti-human nuclei, anti-hNA) | Clontech | Cat# Y40400 |
| Anti-hNA | Abcam | Cat# ab190710 |
| anti-tyrosine hydroxylase (Anti-TH) | Pel freeze | Cat# P40101–150, RRID:AB_2313713 |
| anti-TH | Pel freeze | Cat# P60101–150, RRID:AB_461070 |
| anti-TH | Millipore | Cat# AB152, RRID:AB_390204 |
| anti-TH | Abcam | Cat# AB112, RRID:AB_297840 |
| Anti-FoxA2 | R&D system | Cat# AF2400, RRID:AB_2294104 |
| Anti-Lmx1a | Millipore | Cat# ab10533, RRID:AB_10805970 |
| anti-Oct-4 | Cell signaling | Cat#2840 RRID:AB_2167691 |
| Anti-STEM 121 (human specific) | Clontech | Cat#Y40410, RRID:AB_2801314 |
| Anti-Nurr1 (NGFI-beta) | Perseus (PPMX) | Cat#PP-N1404–00, RRID:AB_2251476 |
| Anti-Pitx3 | Sigma | Cat#HPA044639, RRID:AB_10964194 |
| Anti-Ki67 | Abcam | Cat#AB16667, RRID:AB_302459 |
| Anti-serotonin (anti-5HT) | Sigma | Cat#S5545, RRID:AB_477522 |
| Anti-GAD67 | Abcam | Cat#Ab26116, RRID:AB_448990 |
| Anti-human specific Ki67 (anti-hKi67) | Agilent | Cat#GA62661–2, RRID:AB_2687921 |
| Anti-Ki67 | Abcam | Cat#ab15580, RRID: AB_443209 |
| anti-Tuj1 | Sigma | Cat#T2200, RRID:AB_262133 |
| Anti-Phospho-Histone H3 (Ser10) (pHH3) | Cell signaling | Cat#9701, RRID: AB_331535 |
| Anti-STEM123 (human specific) | Clontech | Cat#Y40420, RRID: AB_2833249 |
| Sheep Anti-human Col1A1 | R&D | Cat# AF6220, RRID:AB_10891543 |
| Mouse Anti-Nestin human specific | EMD Millipore | Cat# MAB5326, RRID: AB_2251134 |
| Anti-Prealbumin antibody | Abcam | Cat# ab92469, RRID:AB_10563814 |
| Chemicals, Peptides, and Recombinant proteins | ||
| STEM-CELLBANKER™ | Amsbio | Cat#11890 |
| Gletrex™ | Life technologies | Cat#A1413202 |
| Essential 8 basal medium and supplement | Life technologies | Cat#A15169, Cat#A15171 |
| Neurobasal medium | Life technologies | Cat#21103049 |
| N2 | Stem Cell Technologies | Cat#21103049 |
| B27 (without vitamin A) | Life technologies | Cat#15750078 |
| L-glutamine | Life technologies | Cat#25030 |
| SB431542 | Tocris Bioscience | Cat#1614 |
| LDN193189 | Reprocell | Cat#04–0074 |
| CHIR99021 | Reprocell | Cat#04–0004-10 |
| SHH | R&D systems | Cat#464-SH |
| Y-27632 | Tocris Bioscience | Cat#1254 |
| BDNF | R&D systems | Cat#248-GMP |
| GDNF | R&D systems | Cat#212-GMP |
| L- Ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Ascorbic acid) | Sigma-Aldrich | Cat#A8960 |
| N6, 2’-O-Dibutyryladenosine 3’,5’-cyclic monophosphate sodium salt (Dibutyryl-cAMP) | Sigma-Aldrich | Cat#D0627 |
| TGFβ3 | R&D systems | Cat#243-GMP-025 |
| DAPT | Tocris Bioscience | Cat#2634 |
| Acridine Orange/Propidium Iodide (AO/PI) Staining Solution | Nexcelom Biosciences | Cat#CS2–0106 |
| 6-OHDA | Sigma-Aldrich | Cat#162957 |
| Experimental Models: Cell Lines | ||
| Human: WA09 FRS at passage 48, XX | Wicell | RRID:CVCL_9773 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.Cg-Prkdcscid IL2rg tm1Wjl | The Jackson Laboratory | Cat#005557, RRID:IMSR_JAX:005557 |
| Rat: NIH-Foxn1rnu | Charles river | Cat#316 |
| Software and Algorithms | ||
| Ethovision XT | Noldus Information Technology Inc., USA | 11.5 |
| Visiopharm software | Visiopharm A/S, Danmark | 6.2.0.2089 |
| GraphPad Prism | Graphpad software | 8.3.0 |
Acknowledgments
This work was supported by the New York Stem Cell Science initiative NYSTEM C028503 (Co-PI: LS and VT). Protocol development and transition to the cGMP facility was performed at the Sloan Kettering Institute’s Stem Cell Research Facility which is supported by NYSTEM C029153 (MT). We would like to extend our gratitude to the staff for the technical assistance of the staff of the following facilities at Memorial Sloan Kettering: The Cell Therapy and Cell Engineering Facility, the SKI Stem Cell Research Facility, the Anti-tumor Assessment Core Facility, the Research Animal Resource Center, the Center of Comparative Medicine & Pathology, the Molecular and Cytology Core facility and the Investigational Products Facility. We also extend our thanks to Hilarie C. Tomasiewicz, Justin Riceberg, Eveline Marie Gutzwiller, Carl Campos and Phil Clark for their assistance with animal surgery. We also thank Matthew Kohn and Kathy Chou at NYSTEM for their support and assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
VT and LS are scientific co-founders and consultants of BlueRock Therapeutics. MT, SI, and SM are currently employed at BlueRock Therapeutics Inc. Other authors declare no competing interests. MSK-DA01 is the subject of a patent, US2018 / 0094242 A1, filed by Memorial Sloan Kettering Cancer Center.
References
- Barker RA, Farrell K, Guzman NV, He X, Lazic SE, Moore S, Morris R, Tyers P, Wijeyekoon R, Daft D, et al. (2019). Designing stem-cell-based dopamine cell replacement trials for Parkinson’s disease. Nat. Med 25, 1045–1053. [DOI] [PubMed] [Google Scholar]
- Cyranoski D (2018). ‘Reprogrammed’ stem cells implanted into patient with Parkinson’s disease. Nat. News https://www.nature.com/articles/d41586-018-07407-9.
- Doi D, Magotani H, Kikuchi T, Ikeda M, Hiramatsu S, Yoshida K, Amano N, Nomura M, Umekage M, Morizane A, et al. (2020). Pre-clinical study of induced pluripotent stem cell-derived dopaminergic progenitor cells for Parkinson’s disease. Nat. Commun 11, 3369 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed CR, Greene PE, Breeze RE, Tsai W-Y, DuMouchel W, Kao R, Dillion S, Winifield H, Culver S, Trojanowsk JQ, et al. (2001). Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N. Engl. J. Med 344, 710–719. [DOI] [PubMed] [Google Scholar]
- Freed CR, Zhou W, and Breeze RE (2011). Dopamine Cell Transplantation for Parkinson’s Disease: The Importance of Controlled Clinical Trials. Neurotherapeutics 8, 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadkhodaei B, Ito T, Joodmardi E, Mattsson B, Rouillard C, Carta M, Muramatsu S, Sumi-Ichinose C, T., Nomura DM, Chambon P, et al. (2009). Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J. Neurosci 29, 15923–15932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TW, Piao J, Kriks S, Chung S, Betel D, Choi SJ, Mosharov EV, Irion S, Tomishima M, Tabar V, et al. (2020). Biphasic activation of WNT signaling enhances the derivation of midbrain dopamine neurons for translational use. Submitted [DOI] [PMC free article] [PubMed]
- Kordower JH, Freeman TB, Snow BJ, Vingerhoets FJG, Mufson EJ, Sanberg PR, Hauser RA, Smith DA, Nauert GM, Perl DP, et al. (1995). Neuropathological evidence of graft survival and striatal reinnervation after the transplantation of fetal mesencephalic tissue in a patient with parkinson’s disease. N. Engl. J. Med 332, 1118–1124. [DOI] [PubMed] [Google Scholar]
- Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, et al. (2011). Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystkowiak P, Gaura V, Labalette M, Rialland A, Remy P, Peschanski M, and Bachoud-Lévi AC (2007). Alloimmunisation to donor antigens and immune rejection following foetal neural grafts to the brain in patients with Huntington’s disease. PLoS One 2, 4–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza R, Russell DW, and Nagy A (2019). Engineering universal cells that evade immune detection. Nat. Rev. Immunol 19, 723–733. [DOI] [PubMed] [Google Scholar]
- Li W, Englund E, Widner H, Mattsson B, Van Westen D, Lätt J, Rehncrona S, Brundin P, Björklund A, Lindvall O, et al. (2016). Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc. Natl. Acad. Sci. U. S. A 113, 6544–6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke S, Enczmann J, Waclawczyk S, Wernet P, and Kögler G (2007). Oct4 and Its Pseudogenes Confuse Stem Cell Research. Cell Stem Cell 1, 364–366. [DOI] [PubMed] [Google Scholar]
- Mendez I, Rosario S-P, Cooper O, Vinuela A, Ferrari D, Bjorklund L, Dagher A, and Isacson O (2005). Cell type analysis of functional fetal dopamine cell suspension transplants in the striatum and substantia nigra of patients with Parkinson’s disease. Brain 128, 1498–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizane A, Doi D, Kikuchi T, Okita K, Hotta A, Kawasaki T, Hayashi T, Onoe H, Shiina T, Yamanaka S, et al. (2013). Direct comparison of autologous and allogeneic transplantation of IPSC-derived neural cells in the brain of a nonhuman primate. Stem Cell Reports 1, 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizane A, Kikuchi T, Hayashi T, Mizuma H, Takara S, Doi H, Mawatari A, Glasser MF, Shiina T, Ishigaki H, et al. (2017). MHC matching improves engraftment of iPSC-derived neurons in non-human primates. Nat. Commun 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munckhof P. Van Den, Luk KC, Ste-Marie L, Montgomery J, Blanchet PJ, Sadikot AF, and Drouin J (2003). Pitx3 is required for motor activity and for survival of a subset of midbrain dopaminergic neurons. Development 130, 2535–2542. [DOI] [PubMed] [Google Scholar]
- Ohyama K, Ellis P, Kimura S, and Placzek M (2005). Directed differentiation of neural cells to hypothalamic dopaminergic neurons. Development 132, 5185–5197. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Goetz CG, Kordower JH, Stoessl AJ, Sossi V, Brin MF, Shannon KM, Nauert GM, Perl DP, Godbold J, et al. (2003). A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann. Neurol 54, 403–414. [DOI] [PubMed] [Google Scholar]
- Parmar M, Grealish S, and Henchcliffe C (2020). The future of stem cell therapies for Parkinson disease. Nat. Rev. Neurosci 21, 103–115. [DOI] [PubMed] [Google Scholar]
- Perrier AL, Tabar V, Barberi T, Rubio ME, Bruses J, Topf N, Harrison NL, and Studer L (2004). Derivation of midbrain dopamine neurons from human embryonic stem cells. Proc. Natl. Acad. Sci. U. S. A 101, 12543–12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer JS, Song B, Herrington TM, Park TY, Lee N, Ko S, Jeon J, Cha Y, Kim K, Li Q, et al. (2020). Personalized iPSC-derived dopamine progenitor cells for Parkinson’s disease. N. Engl. J. Med 382, 1926–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smidt MP, Smits SM, Bouwmeester H, Hamers FP, Linden A.J. Van Der, Hellemons AJ, Graw J, and Burbach JP (2004). Early developmental failure of substantia nigra dopamine neurons in mice lacking the homeodomain gene Pitx3. Development 131, 1145–1155. [DOI] [PubMed] [Google Scholar]
- Tiklová K, Nolbrant S, Fiorenzano A, Björklund ÅK, Sharma Y, Heuer A, Gillberg L, Hoban DB, Cardoso T, Adler AF, et al. (2020). Single cell transcriptomics identifies stem cell-derived graft composition in a model of Parkinson’s disease. Nat. Commun 11, 2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GH, and Stoeber K (2012). The Cell Cycle and Cancer. J. Pathol 226, 352–364. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets are available upon request.