

SUMMARY
Fast-dissociating, specific antibodies are single-molecule imaging probes that transiently interact with their targets and are used in biological applications including image reconstruction by integrating exchangeable single-molecule localization (IRIS), a multiplexable super-resolution microscopy technique. Here, we introduce a semi-automated screen based on single-molecule total internal reflection fluorescence (TIRF) microscopy of antibody-antigen binding, which allows for identification of fast-dissociating monoclonal antibodies directly from thousands of hybridoma cultures. We develop monoclonal antibodies against three epitope tags (FLAG-tag, S-tag, and V5-tag) and two F-actin crosslinking proteins (plastin and espin). Specific antibodies show fast dissociation with half-lives ranging from 0.98 to 2.2 s. Unexpectedly, fast-dissociating yet specific antibodies are not so rare. A combination of fluorescently labeled Fab probes synthesized from these antibodies and light-sheet microscopy, such as dual-view inverted selective plane illumination microscopy (diSPIM), reveal rapid turnover of espin within long-lived F-actin cores of inner-ear sensory hair cell stereocilia, demonstrating that fast-dissociating specific antibodies can identify novel biological phenomena.
Graphical Abstract
In Brief
Based on single-molecule microscopy of antibody-antigen interaction, Miyoshi et al. demonstrate that fast dissociation can be a property of highly specific antibodies. Fab probes synthesized from these antibodies are useful imaging probes for multiplex super-resolution microscopy and could detect rapid turnover of actin crosslinkers in dense F-actin cores of stereocilia.
INTRODUCTION
Specific binding of antibodies is indispensable for numerous biological assays, such as western blotting, immunostaining, immunoprecipitation, and enzyme-linked immunosorbent assays (ELISA). Recently, for several new immunoassays, researchers have used antibodies as reversibly binding probes, such as Fab-based live endogenous modification labeling (FabLEM) and continuous monitoring of serum myoglobulin (Hayashi-Takanaka et al., 2011; Paek et al., 2011). In these assays, antibodies behave as single-molecule imaging probes and real-time biosensors. These assays depend on the identification of fast-dissociating yet highly specific antibodies. Antibodies for these studies were screened using a conventional affinity-based assay, usually ELISA, and then evaluated for their kinetics (Hayashi-Takanaka et al., 2011; Paek et al., 2011; Song et al., 2015). However, this strategy may overlook fast-dissociating antibodies whose affinity is too low to pass the initial screening.
We performed an initial screening assay by using single-molecule total internal reflection fluorescence (TIRF) microscopy of antibody-antigen binding, which achieved high time-resolution to detect and quantify fast-dissociating antibodies. In addition, our assay is designed to accept unpurified antibodies in hybridoma culture supernatants. Semi-automated TIRF microscopy allowed us to screen antibodies directly from thousands of hybridoma cultures. There are other valuable methods to detect antibody-antigen binding, such as label-free surface plasmon resonance (SPR) and biolayer interferometry (BLI) (Canziani et al., 2004; Fischer and Mol, 2010; Helmerhorst et al., 2012; Kamat et al., 2020; Säfsten et al., 2006; van der Merwe and Barclay, 1996). However, fast-dissociating, highly specific antibodies that have a half-life of less than a few seconds, preferable for single-molecule imaging probes, were not reported.
One goal of this study was to determine if systematic kinetic-based screens can identify fast-dissociating and yet specific monoclonal antibodies against various antigens. We established our methods by using three epitope tags, namely, FLAG-tag, S-tag, and V5-tag, and two F-actin binding proteins, namely, plastin and espin. We describe a pipeline for identifying fast-dissociating antibodies and demonstrate that fluorescently labeled Fab probes of these antibodies are highly specific imaging probes in IRIS (image reconstruction by integrating exchangeable single-molecule localization), a super-resolution microscopy technique (Kiuchi et al., 2015). IRIS reconstructs super-resolution images by using probes that bind targets with high specificity and then quickly dissociate allowing for multiplex imaging. Several Fab probes satisfied the demanding requirements of IRIS, including high specificity and quick dissociation against epitopes, and demonstrated that the two advantages of IRIS, high-fidelity imaging and multiplexity, are feasible using antibody-antigen interactions. Furthermore, we report that Fab probes work with light-sheet microscopy, such as dual-view inverted selective plane illumination microscopy (diSPIM) (Wu et al., 2013), to visualize protein dynamics in three-dimensional tissue samples. Our method has expanded the range of exchangeable imaging probes from peptide fragments, oligonucleotides, and metal ions (Kiuchi et al., 2015; Riedl et al., 2008; Schmitt et al., 2000; Schueder et al., 2017) to fast-dissociating antibodies, which are theoretically available for any antigen.
RESULTS
Development of antibody screening assays by using single-molecule TIRF microscopy
A screening assay was developed to identify fast-dissociating antibodies based on single-molecule TIRF microscopy of antibody-antigen binding (Figure 1A). We crosslinked Protein A/G to the glass surface of amine-functionalized 96-well glass-bottom plates by a bifunctional crosslinker, sulfo-SANPAH. The high affinity of Protein A/G for immunoglobulins (<1 nM dissociation constant [KD]) (Bronner et al., 2009) allowed selective capture of antibodies in hybridoma culture supernatants. EGFP-antigen molecules captured by immobilized antibodies are detected as fluorescent spots by using TIRF microscopy. Protein-A/G-dependent antibody immobilization was confirmed using DyLight488-labeled mouse immunoglobulin G1 (IgG1) (Figure 1B). The fluorescence intensity of bound fluorescent IgG1 increased with increasing concentrations of Protein A/G up to 0.1 mg/mL and then plateaued. Thus, we used Protein A/G at 0.3 mg/mL to saturate the sulfo-SANPAH on the glass surface.
Figure 1. Development of antibody screening assay.
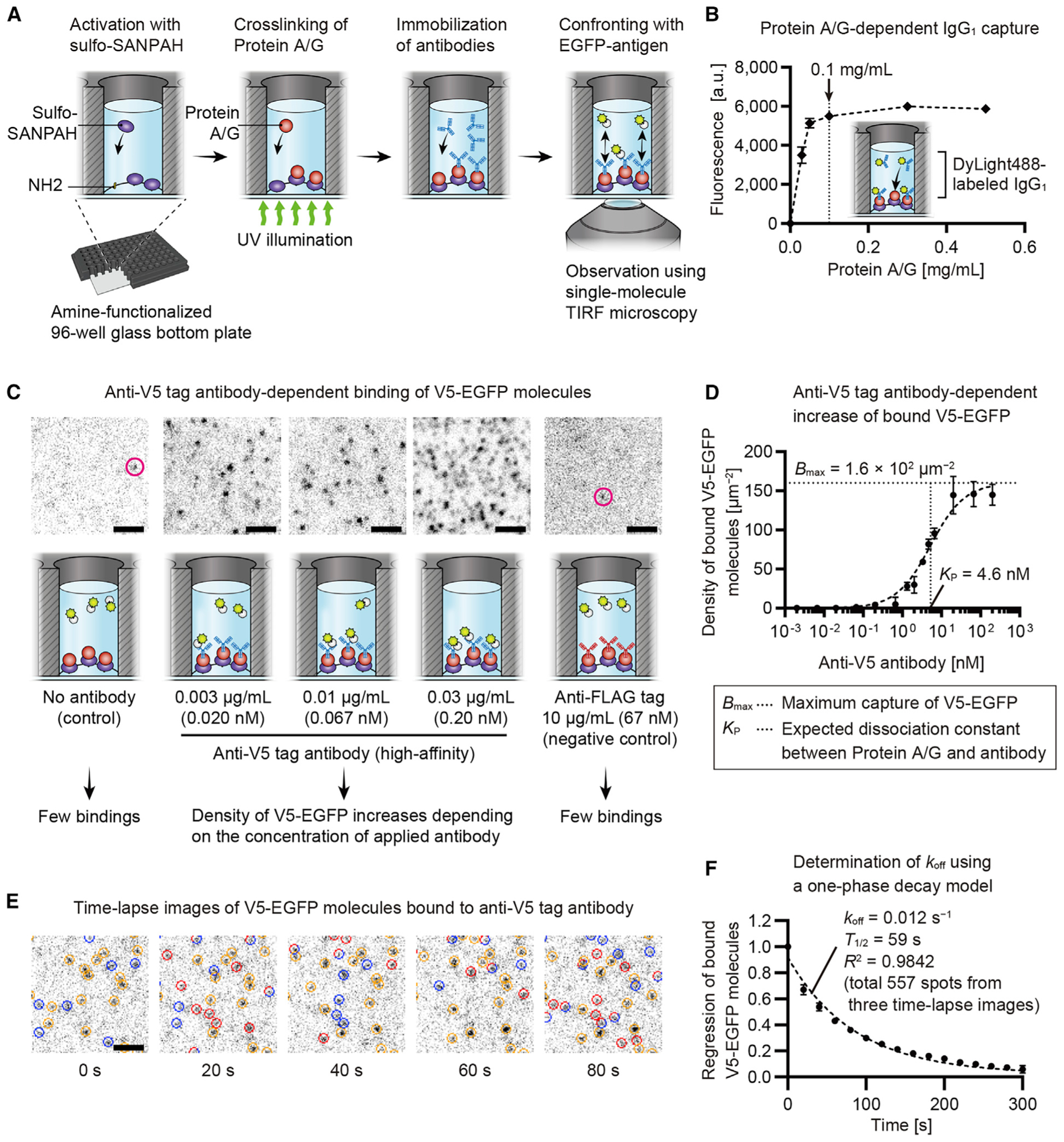
(A) Design of the screening assay. Amine-functionalized 96-well glass-bottom plates activated using sulfo-SANPAH, which links Protein A/G under UV illumination to the glass surface. Antibodies were immobilized by Protein A/G and confronted with EGFP-antigen. Binding of EGFP-antigen molecules was visualized in real-time using single-molecule TIRF microscopy.
(B) Protein A/G-dependent antibody immobilization. Glass surfaces were coated using 0–0.5 mg/mL Protein A/G and confronted with 1 μg/mL DyLight488-labeled mouse IgG1. Fluorescence intensity of bound IgG1 saturated around 0.1 mg/mL of Protein A/G. Error bars, standard deviations (SDs; n = 3).
(C) Antibody-dependent EGFP-antigen binding. High-affinity anti-V5 tag antibody immobilized and confronted with 30 nM V5-EGFP. Fluorescent spots of bound V5-EGFP appeared at 0.003 μg/mL of antibody, where a density was 0.29 ± 0.16 spots/μm2 (means ± SDs, n = 3). Few spots appeared when there was no antibody or anti-FLAG tag antibody (magenta circles, <0.02 spots/μm2; n = 3).
(D) Increased density of bound V5-EGFP with increasing concentrations of anti-V5 tag antibody. Densities were calculated using a direct count of bound molecules or using EGFP fluorescence intensities. Saturation curve illustrates binding between Protein A/G and anti-V5 tag antibody. Details are in the STAR methods. Determined Kp and Bmax were used to simulate this assay (Figure S1). The 95% confidence intervals (CIs) were Kp = 3.82 to 5.62 nM and Bmax = 1.51 × 102 to 1.70 × 102 μm−2. Error bars, SDs (n = 3).
(E) Time-lapse images of bound V5-EGFP molecules. Anti-V5 tag antibody and V5-EGFP were diluted to 0.01 μg/mL and 0.1 nM. Bound molecules are indicated by circles colored red (new binding), blue (disappears in the next frame), and orange (remains bound). Molecules that remain bound for one frame are indicated by circles composed of red and blue arcs. Time lapse, every 20 s. Exposure, 100 ms.
(F) Determination of dissociation rate (koff) and half-life (T1/2) using regression of bound V5-EGFP molecules. One-phase decay model was fit to the means of three curves (shown by means ± SDs), giving koff = 0.012 s−1 and T1/2 = 59 s. The 95% CIs were koff = 0.0110 to 0.0123 s−1 and T1/2 = 56.0 to 62.9 s. The number of tracked molecules was 557 (178, 153, and 226 spots in each curve).
Specific binding between EGFP-antigen and immobilized antibodies was confirmed using a high-affinity anti-V5 tag antibody (Southern et al., 1991) and V5-tagged EGFP (Figures 1C and 1D). Various concentrations of the anti-V5 tag antibody were applied and detected with V5-EGFP. The 30 nM concentration of V5-EGFP is the maximum concentration that our TIRF microscopy can detect against the fluorescent background of unattached V5-EGFP. Fluorescent spots of bound V5-EGFP molecules appeared when the anti-V5 tag antibody was applied at 0.003 μg/mL (Figure 1C). The density of bound V5-EGFP molecules increased with increasing concentrations of immobilized anti-V5 tag antibody (Figure 1D). Only a few V5-EGFP molecules usually bind to the control glass surface lacking antibody or to the negative control anti-FLAG tag and anti-S tag antibodies. The density of bound V5-EGFP antigen was <0.02 spots/μm2 for these negative-control experiments (n = 3 each).
Our assay can also determine the dissociation rate (koff) of antibody-antigen binding (Figures 1E and 1F). Using diluted anti-V5 antibody and V5-EGFP, we tracked the exchange of bound V5-EGFP molecules (Figure 1E; Video S1) and fit a one-phase decay model. Bound molecules were tracked in three individually prepared glass-bottom plates to confirm the reproducibility of our assay (Figure 1F). Bound molecules were slowly exchanged reflecting the slow dissociation of this high-affinity anti-V5 tag antibody, showing that koff of antibody-antigen binding and an interaction half-life were 0.012 s−1 and 59 s, respectively (95% confidence intervals [CIs] had koff = 0.0110 to 0.0123 s−1 and a half-life = 56.0 to 62.9 s). These values indicated dissociation slightly faster but consistent with koff ≈ 0.002 s−1 and a half-life ≈ 5 min of commonly used anti-epitope tag antibodies (Fujii et al., 2014). Photobleaching of EGFP under the same condition occurred at a rate constant (K) = 0.0015 s−1 and a half-life = 4.6 × 102 s (Figure S1A), which were approximately 10-fold slower than the dissociation of this anti-V5 tag antibody from V5-EGFP. Sensitivity to weak-affinity antibodies was evaluated in Figures S1B and S1C as described in the STAR methods.
Screening monoclonal antibodies against FLAG, S, and V5 tags
A total of 1,000 hybridoma culture supernatants were screened for antibodies against the FLAG tag, and 2,000 supernatants were screened against the S and V5 tags (Figure 2A). Antibodies secreted from hybridoma clones were immobilized on the glass surface and confronted with 30 nM of EGFP-antigens (FLAG-EGFP, S-EGFP, and V5-EGFP). Short time-lapse images (50–100 frames, 50-ms exposure) were acquired by scanning each candidate using a TIRF microscope with a motorized stage and auto-focusing. Affinity to EGFP-antigen was evaluated using the first few frames of time-lapse images. The criterion for a positive response was whether the density of bound EGFP-antigen molecules was greater than the density on an antibody-free glass surface. For the antibodies giving a positive response, fast-dissociating candidates were sought using the entire frame of the time-lapse images. When the dissociation speeds could not be evaluated due to excessive density of bound EGFP-antigen, follow-up scans were performed using diluted EGFP-antigen.
Figure 2. Screening of anti-epitope tag monoclonal antibodies.
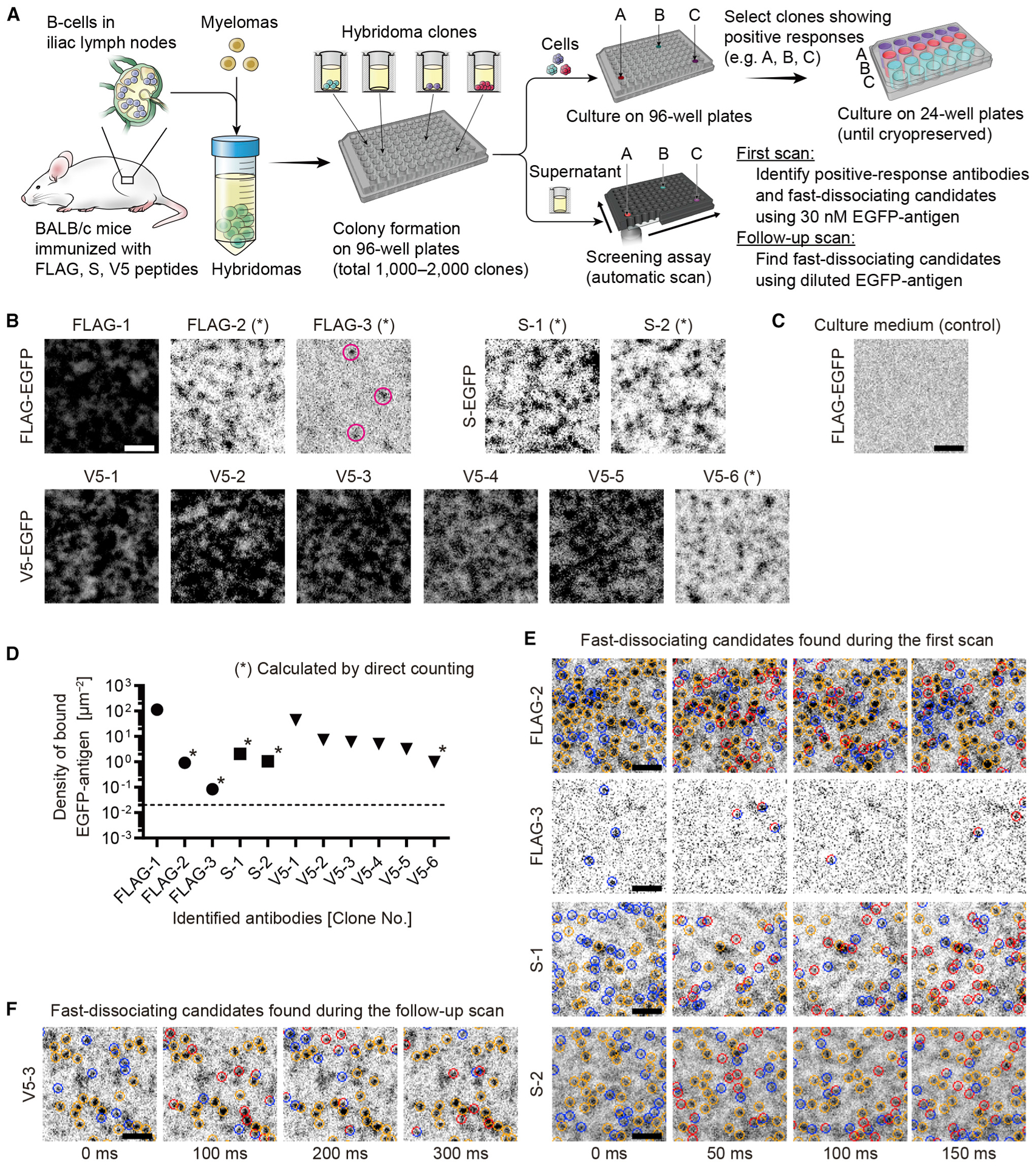
(A) Antibody screening strategy. Hybridomas developed using B cells in iliac lymph nodes and seeded in 96-well culture plates. Antibodies in culture supernatants were evaluated using the screening assay after colonies formed. During the first scan, short time-lapse images (50–100 frames at 50-ms exposure) were acquired for each candidate confronted with 30 nM of EGFP-antigen (FLAG-EGFP, S-EGFP, or V5-EGFP). Response to EGFP-antigen was evaluated using the first few frames as in (B) and to select clones for cryopreservation. When the densities of bound EGFP-antigen are sufficiently low (asterisks, B), fast-dissociating candidates were identified using the entire frames of time-lapse images (E). Follow-up scans were performed using diluted EGFP-antigen (0.1–30 nM) to identify fast-dissociating candidates not found during the first scan (F).
(B) Positive responses of antibodies to applied EGFP-antigen. Images are the first frames of time-lapse acquisition. Three anti-FLAG tag antibodies, two anti-S tag antibodies, and six anti-V5 tag antibodies responded to 30 nM of FLAG-EGFP, S-EGFP, and V5-EGFP, respectively. Magenta circles indicate a few FLAG-EGFP molecules bound to FLAG-3 antibody. For several antibodies, densities of bound EGFP-antigens were sufficiently low to evaluate dissociation speeds (asterisk).
(C) Response to antibody-free control glass surface treated with fresh culture medium. Representative binding to 30 nM FLAG-EGFP is shown. No bound FLAG-EGFP is observed in this image. Densities of bound molecules were < 0.02 spots/μm2 for FLAG-EGFP, S-EGFP, and V5-EGFP, respectively (n = 3 for each EGFP-antigen).
(D) Densities of bound EGFP-antigen molecules calculated using images in (B). Densities were calculated by direct counting the FLAG-2, FLAG-3, S-1, S-2, and V5-6 antibodies (asterisks) and estimated by fluorescence intensities for other antibodies. Means determined using three images. Dashed line is the maximum binding on antibody-free glass surface, 0.02 spots/μm2, in (C).
(E) Fast-dissociating candidates found in the first scan using 30 nM EGFP-antigen. Bound FLAG-EGFP and S-EGFP molecules were frequently exchanged. Indication of circles is similar to Figure 1E.
(F) Fast-dissociating candidate found in the follow-up scan. V5-EGFP was applied at 0.1 nM. Bound V5-EGFP molecules were frequently exchanged but seemed slightly slower than the four antibodies in (E).
Bars, 2 μm. See also Figure S1, Table S1, and Videos S2 and S3.
Eleven anti-epitope tag antibodies were identified with positive responses to antigens. Antibodies with positive responses included three anti-FLAG tag antibodies, two anti-S tag antibodies and six anti-V5 tag antibodies (Figure 2B). Negative-control glass surfaces were prepared with fresh culture medium (Figure 2C), which captured EGFP-antigen at <0.02 spots/μm2 (n = 3). Densities of bound EGFP-antigen molecules were calculated for the identified antibodies by using a direct count for five antibodies, namely, FLAG-2, FLAG-3, S-1, S-2, and V5-6 (asterisks, Figures 2B and 2D), or calculated from the fluorescence intensity of bound EGFP for other antibodies. Some of these 11 antibodies, such as FLAG-2, would have been overlooked by an ELISA because ELISA scores of these antibodies are not the highest among the candidates (asterisks, Figure S1E). Culture supernatants contained 1 to 10 μg/mL of antibodies as previously reported (Yokoyama, 2008; Figure S1F). As simulated (Figure S1D), the high densities of bound EGFP-antigen indicate high affinity (low KD) of antibodies contained in these culture supernatants.
Next, we sought fast-dissociating candidates using time-lapse images of bound EGFP-antigen molecules (Figures 2E and 2F). Four antibodies, namely, FLAG-2, FLAG-3, S-1, and S-2, showed a frequent exchange of bound EGFP-antigen (FLAG-EGFP or S-EGFP) molecules during the first scan (Figure 2E). One antibody, V5-3, showed exchange of bound EGFP-antigen (V5-EGFP) molecules in the follow-up scan (Figure 2F). These five antibodies (FLAG-2, FLAG-3, S-1, S-2, and V5-3) are the fast-dissociating candidates that we focused on in this study. In addition, most of these antibodies successfully recognized ectopically expressed epitopes in conventional immunostaining, including antibodies showing fast dissociation, such as FLAG-2, S-1, S-1, and V5-3 (Figure S1G). These results indicate that conventional immunostaining is useful for evaluating specificity of fast-dissociating antibodies except for antibodies with extremely fast dissociation kinetics, such as FLAG-3. During immunostaining using antibodies with half-lives of a few seconds, such as FLAG-2, small amounts of antibodies, perhaps due to increased affinity by bivalent binding to epitopes (Hadzhieva et al., 2017), appear to remain bound to epitopes and are stabilized and enhanced by secondary antibodies. Screening results are summarized in Table S1.
Measuring koff of anti-epitope tag antibodies
The koff values of anti-epitope tag antibodies were determined from the regression of bound EGFP-antigen molecules (Figure 3). To track bound molecules, FLAG-EGFP, S-EGFP, and V5-EGFP antigens were applied at a lower concentration than in the screening (Figure 3A). Intervals for time-lapse acquisition were adjusted to observe 10%–30% exchange of bound EGFP-antigen molecules per frame. koff and half-lives were determined by fitting one-phase decay models to the regression of bound EGFP-antigen molecules. Regression curves are shown in Figure S2. The fastest exchange was observed for FLAG-2, FLAG-3, S-1, S-2, and V5-3 antibodies with optimal intervals from 50 ms to 300 ms (Figure 3B). Fitting one-phase decay models gave koff values of 0.71, 7.5, 0.73, 0.89, and 0.31 s−1 (half-lives = 0.98, 0.093, 0.95, 0.77, and 2.2 s) for FLAG-2, FLAG-3, S-1, S-2, and V5-3, respectively. Other antibodies showed slower exchange of bound EGFP-antigen (FLAG-1 shown in Figure 3C). Kinetic parameters of the antibodies are summarized in Figure 3D and Table S2. FLAG-2, FLAG-3, S-1, S-2, and V5-3 antibodies showed the fastest dissociation rates among the anti-FLAG tag, anti-S tag, and anti-V5 tag antibodies (red markers, Figure 3D), indicating that our screening assay correctly identified fast-dissociating antibodies without initially having a precise koff determination. FLAG-1 and V5-1 antibodies showed slow dissociation with half-lives of 25 s and 24 s, respectively, similar to a conventional high-affinity anti-V5 tag antibody.
Figure 3. Determining koff for anti-epitope tag antibodies.
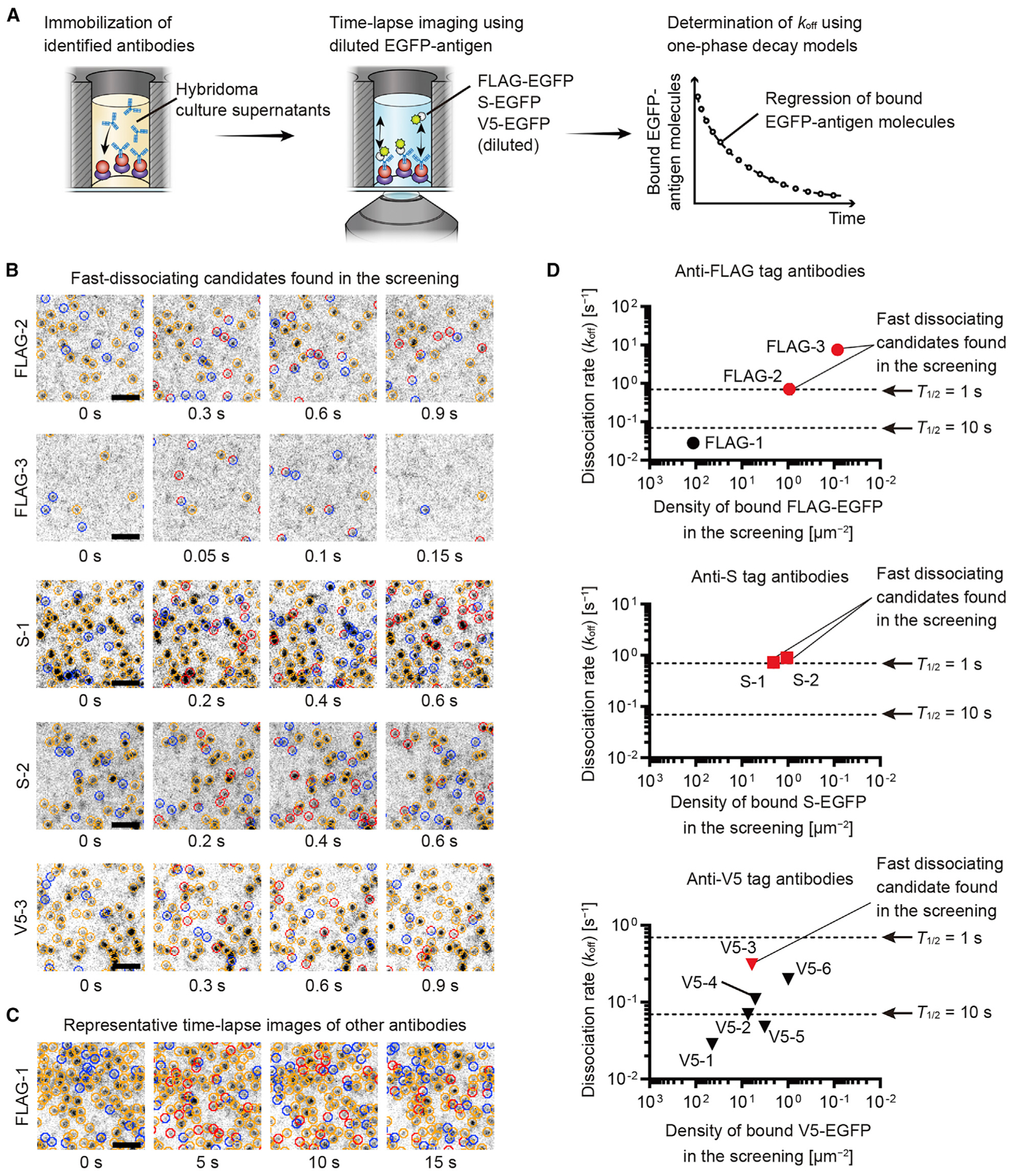
(A) koff determination for antibodies against FLAG tag, S tag, and V5 tag. Time-lapse images acquired at an interval to cause 10%–30% exchange of bound EGFP-antigen molecules per frame. One-phase decay models fit to the regression of bound EGFP-antigen to give koff and half-lives.
(B) Optimal intervals were 50 ms to 300 ms for time-lapse image acquisition of fast-dissociating candidate FLAG-2, FLAG-3, S-1, S-2, and V5-3 antibodies. Indication of circles is similar to that in Figure 1E. FLAG-EGFP, 0.3 nM for FLAG-2 and 30 nM for FLAG-3. S-EGFP, 1 nM for S-1 and 3 nM for S-2. V5-EGFP, 1 nM for V5-3.
(C) Representative time-lapse images of other antibodies (shown for FLAG-1). Circles indicate bound FLAG-EGFP molecules as described in (B). FLAG-EGFP, 0.1 nM.
(D) Kinetics of identified antibodies. koff values were plotted against the densities of bound EGFP-antigen molecules in the screening (Figure 2D). Dashed lines indicate koff = 0.693 s−1 (half-life = 1 s) and 0.0693 s−1 (half-life = 10 s). Fast-dissociating candidates from the screening showed faster koff than other antibodies (red markers). The koffs of fast-dissociating candidates were ~0.693 s−1 or faster (half-lives = 1 s or shorter) for FLAG-2, FLAG-3, S-1, and S-2 antibodies. koff was slightly slower for the V5-3 antibody at 0.31 s−1 (half-life = 2.2 s).
Bars, 2 μm. See also Figure S2, Tables S1 and S2, and Videos S4, S5, S6, S7, and S8.
Screening and characterization of monoclonal antibodies against plastin and espin
Monoclonal antibodies were developed against two proteins, namely, mouse plastin 1 (mPLS1) and espin 1 (mESPN1) (Figure 4; Figure S3A), and screened using EGFP-mPLS1 and two EGFP-fused mESPN1 fragments (Figure S3B). Anti-mPLS1 antibodies were screened using 20 nM EGFP-mPLS1 (Figure 4A). Anti-mESPN1 antibodies were twice screened using two EGFP-fused mESPN1 fragments, EGFP-N534 and EGFP-657X (Figure 4B). Full-length EGFP-mESPN1 was not used due to non-specific binding to the glass surface. Instead, we obtained EGFP-N534 and EGFP-657X after truncating EGFP-mESPN1 until the non-specific binding was suppressed (Figures S4A–S4D). EGFP-N534 and EGFP-657X were used at 5 nM and 20 nM, respectively, because non-specific binding was minimal up to these concentrations (n = 3 each, images not shown). These EGFP-antigens also had FLAG-tags for purification purposes. Thus, binding to immobilized antibodies was tested using the FLAG-2 antibody. koff values of EGFP-mPLS1, EGFP-N534, and EGFP-657X from FLAG-2 antibody were 0.98 s−1, 0.74 s−1, and 0.66 s−1 (half-lives = 0.71 s, 0.94 s, and 1.0 s), respectively, which is consistent with the fast dissociation between the FLAG-EGFP and FLAG-2 antibody (Figure S3C).
Figure 4. Screening anti-mPLS1 and anti-mESPN1 monoclonal antibodies.
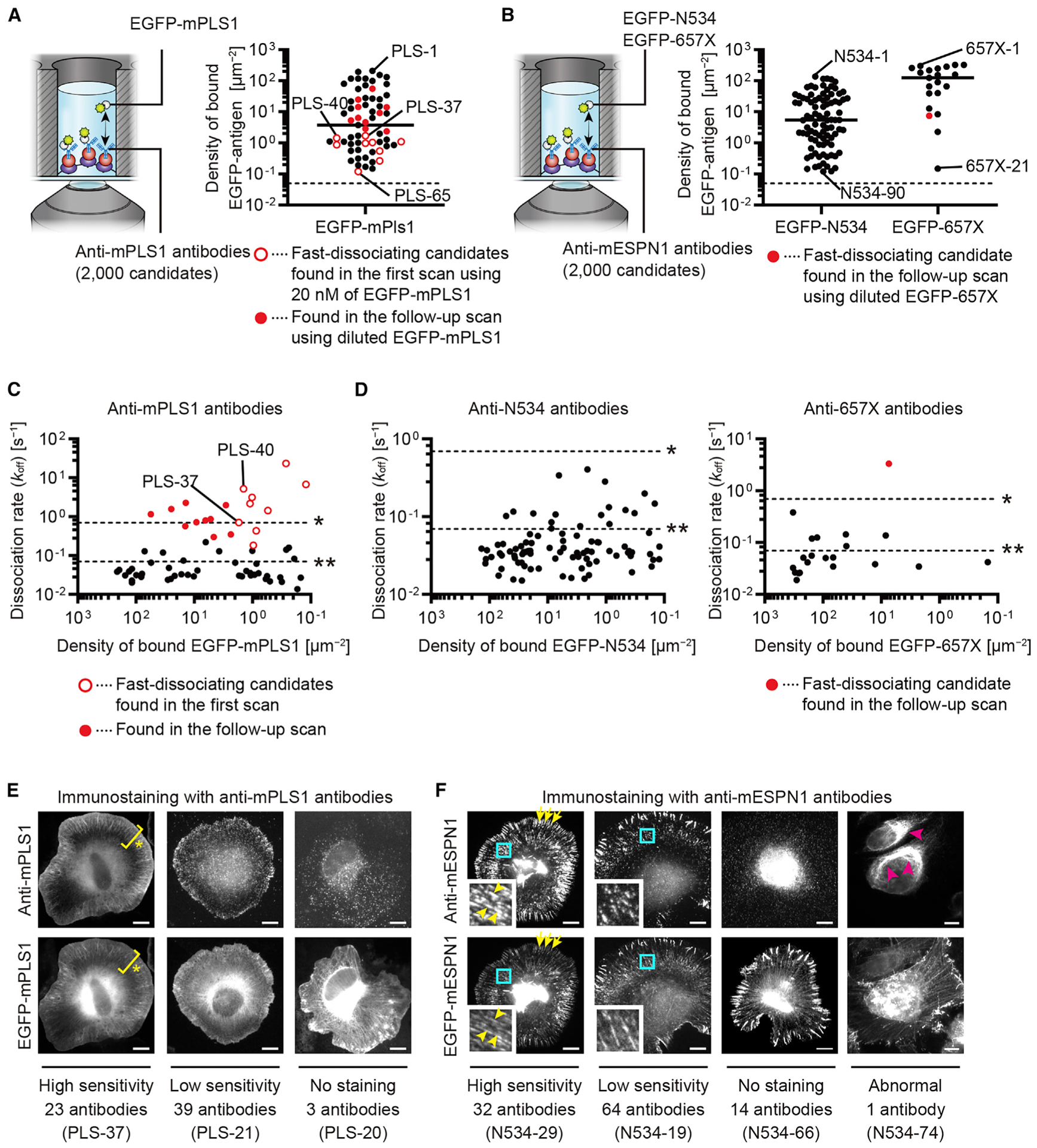
(A) Screening anti-mPLS1 antibodies. Among 2,000 candidates, 65 antibodies responded to EGFP-mPLS1 applied at 20 nM. Antibodies are plotted against densities of bound EGFP-mPLS1. Open red circles are fast-dissociating candidates found in the first scan. Closed red circles are those found in a follow-up scan with diluted EGFP-mPLS1 (0.1–20 nM). Representative images are in Figure S3D. Dashed line is maximum binding to antibody-free control glass surface, 0.05 spots/μm2 (n = 3). Markers indicate means determined using two images. Bar, median.
(B) Screening anti-mESPN1 antibodies. Among 2,000 candidates, 90 antibodies responded to 5 nM EGFP-N534 (anti-N534 antibodies), and 21 antibodies responded to 20 nM EGFP-657X (anti-657X antibodies). Follow-up scans were performed using EGFP-N534 at 0.1–5 nM and EGFP-657X at 0.1–20 nM. One anti-657X antibody showed fast-dissociation during the follow-up scan with 0.1 nM EGFP-657X (closed red circle). Dashed lines are maximum binding to antibody-free glass surface, 0.05 spots/μm2 (n = 3 for EGFP-N534 and EGFP-657X). Markers indicate means determined using two images. Bars, medians.
(C) Kinetic summary of anti-mPLS1 antibodies. koff values determined using diluted EGFP-mPLS1 (0.1–20 nM). Time-lapse images and regression curves of PLS-37 and PLS-40 are in Figures S3E and S3F. Dashed lines indicate koff = 0.693 s−1 (half-life = 1 s, asterisk) and 0.0693 s−1 (half-life = 10 s, double asterisks). Compatible with the screening, fast-dissociating candidates showed faster koff than other antibodies (open and closed red circles).
(D) Kinetic summary of anti-N534 and anti-657X antibodies. koff values determined using diluted EGFP-N534 (0.1–5 nM) or EGFP-657X (0.1–20 nM). One anti-657X fast-dissociating candidate showed faster koff than other antibodies (closed red circle). Most antibodies showed koff slower than 0.693 s−1 (half-life = 1 s), which distribute at a slower range than anti-mPLS1 antibodies.
(E) Immunostaining using anti-mPLS1 antibodies and XTC cells expressing EGFP-mPLS1 (representative images). EGFP-mPLS1 was distributed in lamellipodia (asterisks). Sensitivity of staining was different among the antibodies: 23 antibodies detected mPLS1 concordantly with EGFP tag (high sensitivity) and 39 antibodies at lower densities than EGFP tag (low sensitivity). Three antibodies did not detect mPLS1 (no staining). Bars, 10 μm.
(F) Immunostaining using anti-mESPN1 antibodies and XTC cells expressing EGFP-mESPN1 (representative images). Small panels magnify cyan rectangles. EGFP-mESPN1 was distributed on thick actin fibers induced by mESPN1 and on filopodia (yellow arrowheads and arrows). Sensitivity of staining was different: 32 antibodies detected mESPN1 concordantly with EGFP-tag (high sensitivity) and 64 antibodies at lower densities then EGFP-tag (low sensitivity). Fourteen antibodies did not detect mESPN1 (no staining). One anti-N534 antibody abnormally stained perinuclear organelles (magenta arrowheads, abnormal).
Bars, 10 μm. See also Figures S3 and S4.
We identified 65 anti-mPLS1 antibodies and 111 anti-mESPN1 antibodies from each of 2,000 monoclonal antibody candidates (Figures 4A and 4B). The anti-mPLS1 antibodies were numbered from PLS-1 to PLS-65, from high density to low density of bound EGFP-mPLS1. The anti-mESPN1 antibodies were designated N534-1 to N534-90 and 657X-1 to 657X-21 according to the densities of bound EGFP-N534 or EGFP-657X. Among the anti-mPLS1 antibodies, 9 fast-dissociating candidates were found during the first scan with 20 nM EGFP-mPLS1 (open red circles, Figure 4A), and 10 fast-dissociating candidates were found during the follow-up scan with 0.1–20 nM EGFP-mPLS1 (red circles, Figure 4A). Representative time-lapse images for the screening are shown for PLS-37 and PLS-40 (Figure S3D) because these antibodies turned out to be useful as super-resolution imaging probes (see Figure 6). Among the anti-mESPN1 antibodies, only one anti-657X antibody was a fast-dissociating candidate found during the follow-up scan with 0.1 nM EGFP-657X (red circle, Figure 4B). In the screening of anti-mPLS1 and anti-mESPN1 antibodies, the numbers of antibodies showing positive responses to EGFP-antigens were larger than those in the screening of anti-epitope tag antibodies, possibly because recombinant proteins were used to develop these antibodies, which usually show higher immunogenicity than peptides used to develop anti-epitope tag antibodies (Skwarczynski and Toth, 2016).
Figure 6. Super-resolution images acquired with anti-mPLS1 and anti-mESPN1 Fab probes.

(A) Images acquired with four anti-mPLS1 Fab probes achieving high-density labeling and XTC cells expressing FLAG-mPLS1. Counter-imaging, FLAG-2 Fab probe. Images reconstructed from 50,000 frames acquired every 50 ms. Fab probes, 1 nM. Bars, 5 μm.
(B) Magnified images of rectangles in (A). FLAG-mPLS1 was detected in lamellipodia (asterisks) and filopodia (arrowheads) by anti-mPLS1 and FLAG-2 Fab probes. Labeling density was slightly higher by PLS-37 and PLS-40 Fab probes, which were synthesized from fast-dissociating anti-mPLS1 antibodies and showed faster koff than PLS-1 and PLS-4 Fab probes (Figure S8B). Bars, 2 μm.
(C) Flow chart of anti-mPLS1 antibodies showing relationship between synthesis of successful Fab probes and immunostaining.
(D) Super-resolution images acquired with anti-mESPN1 Fab probes and XTC cells expressing mESPN1-V5. Five Fab probes achieved high-density labeling. Four Fab probes were derived from anti-N534 antibodies and one from an anti-657X antibody. Counter-imaging, V5-3 Fab probe. Images reconstructed from 60,000 frames acquired every 50 ms. Fab probes, 1 nM. Bars, 5 μm.
(E) Magnified images of rectangles in (D). mESPN1-V5 was detected on thick actin fibers by anti-mESPN1 and V5-3 Fab probe (arrowheads). Labeling density was slightly lower by N534-57 and 657X-4 Fab probes probably because of their slower koff than the other three Fab probes (asterisk, Figure S8B). Bars, 1 μm.
(E) Flow chart of anti-mESPN1 antibodies showing relationship between synthesis of successful Fab probes and immunostaining.
The koff values of these antibodies were plotted against the densities of bound EGFP-antigen molecules (Figures 4C and 4D). Time-lapse images and regression curves of PLS-37 and PLS-40 antibodies are shown in Figures S3E and S3F. koff of anti-mPLS1 antibodies ranged from 0.014 s−1 to 23 s−1 (half-lives between 30 ms and 50 s). Anti-mPLS1 antibodies identified as fast-dissociating candidates (open and closed red circles, Figure 4C) showed koff values ranging from 0.18 s−1 to 23 s−1 (half-lives between 30 ms and 3.9 s), which were higher than the koff values of other antibodies. koff values of anti-mESPN1 antibodies were slow compared with those of anti-mPLS1 antibodies. koff of anti-N534 antibodies ranged from 0.015 s−1 to 0.41 s−1 (half-lives between 1.7 s and 46 s), and anti-657X antibodies ranged from 0.019 s−1 to 3.3 s−1 (half-lives between 0.21 s and 36 s). One fast-dissociating candidate showed the fastest koff among the anti-mESPN1 antibody at 3.3 s−1 (a half-life = 0.21 s) (red circle, Figure 4D).
We tested antibody epitope recognition by using conventional immunostaining of XTC cells expressing EGFP-mPLS1 or EGFP-mESPN1 (Figures 4E and 4F). Most of the anti-mPLS1 and anti-mESPN1 antibodies successfully recognized mPLS1 and mESPN1. Some antibodies recognized these antigens at high labeling densities concordant with the fused EGFP tags, whereas other antibodies showed lower labeling densities than the fused EGFP tags (high sensitivity and low sensitivity, Figures 4E and 4F). Three anti-mPLS1 and 14 anti-mESPN1 antibodies failed in immunostaining (no staining, Figures 4E and 4F), and 1 anti-N534 antibody showed abnormal staining to perinuclear organelles (Figure 4F, magenta arrowheads). Success or failure in immunostaining was not explained by affinity or koff of these antibodies (Figures S4E and S4F).
Super-resolution imaging with anti-epitope tag Fab probes
Fluorescently labeled Fab fragments (Fab probes) of anti-epitope tag antibodies were used as imaging probes for IRIS (Figure 5A). Antibodies in hybridoma culture supernatants were collected on Protein A beads and labeled with DyLight488-maleimide with the intent to label antibodies by free cysteine residues (Huh et al., 2013). The pH of the reaction mixture was adjusted to 7.0 for higher selectivity to sulfhydryl groups of cysteine residues, although some amine groups could be labeled (Hermanson, 2013). Fab fragments for super-resolution imaging were recovered after papain cleavage. The specificity of anti-FLAG tag, anti-S tag, and anti-V5 tag Fab probes was tested using XTC cells expressing epitope-tagged actin (FLAG-actin, S-actin, and V5-actin; Figures 5B and 5C). Fab probes from FLAG-1, FLAG-2, S-1, V5-1, V5-2, and V5-3 antibodies showed specific binding to the epitope-tagged actin fibers at 1 nM (Figure 5B; hereafter, Fab probes are named after original antibodies). Other Fab probes did not show specific binding even at 10 nM and displayed non-specific binding to coverslip surface (Figure 5C, representative image for V5-6). Among the Fab probes with specific binding, three Fab probes (FLAG-2, S-1, and V5-3) showed fast exchange of bound Fab probes (Figure 5D), and their koff values were faster than those of FLAG-1, V5-1, and V5-2 Fab probes (Figures S5 and S6A; Table S3). koff values of Fab probes were similar to those of the original antibodies except for a slightly slower dissociation of the S-1 Fab probe (Figure S6B).
Figure 5. Super-resolution imaging acquired with anti-epitope tag Fab probes.
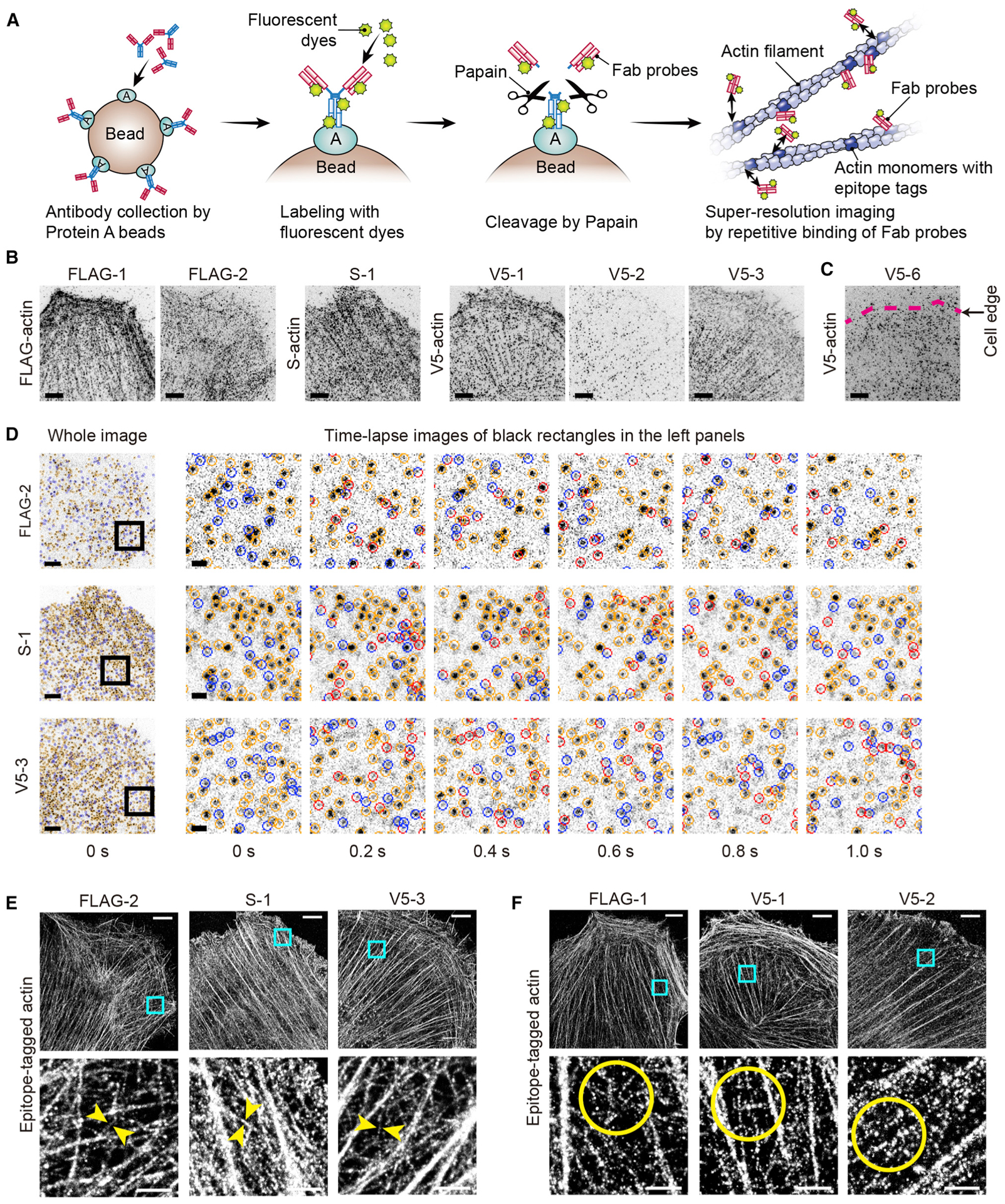
(A) Synthesis of fluorescently labeled Fab probes and imaging by their repetitive binding to epitope tags (epitope-tagged actin is shown). Details in STAR methods.
(B) Fab probes showing specific binding to epitope-tagged actin (FLAG-actin, S-actin, or V5-actin) expressed in XTC cells. FLAG-1, FLAG-2, S-1, V5-1,V5-2, and V5-3 Fab probes visualized actin fibers by specific binding to epitope tags. Images are maximum projections of 500 frames acquired every 50 ms. Fab probes, 1 nM. Bars, 5 μm.
(C) Representative image of non-specific binding of Fab probes (V5-6 Fab probe and V5-actin). Fab probe bound diffusely without visualizing actin fibers. Maximum projection of 500 frames acquired every 50 ms. Fab probe, 10 nM. Bar, 5 μm.
(D) Time-lapse images showing repetitive binding of FLAG-2, S-1, and V5-3 Fab probes. Compatible with the fast-dissociating abilities of FLAG-2, S-1, and V5-3 antibodies, bound Fab probes were frequently exchanged. Time lapse recorded every 50 ms but shown every 200 ms (i.e., every 4th frame). Fab probes, 1 nM. Bars, 5 μm and 2 μm.
(E) Super-resolution images acquired with FLAG-2, S-1, and V5-3 Fab probes. These Fab probes dissociate with half-lives of a few seconds (Figure S6). Thin actin fibers were visualized at high-density labeling (between arrowheads) reflecting the frequent binding and dissociation of these Fab probes. Images were reconstructed from 160,000 frames acquired every 50 ms. Fab probes, 1 nM. Bars, 5 μm and 1 μm.
(F) Super-resolution images acquired with FLAG-1, V5-1, and V5-2 Fab probes. These Fab probes dissociate at half-lives of nearly 10 s or longer (Figure S6). Reconstructed from 160,000 frames acquired every 50 ms as performed in (E). Thick actin fibers were visualized, and thin actin fibers were visualized only at low labeling densities (circles). Fab probes, 1 nM. Bars, 5 μm and 1 μm.
See also Figures S5–S7, Table S3, and Video S9.
The six Fab probes successfully reconstructed super-resolution images of actin fibers from 160,000 frames acquired every 50 ms (Figures 5E and 5F). Compatible with the fast dissociation and frequent exchange, FLAG-2, S-1, and V5-3 Fab probes achieved high labeling density sufficient to visualize thin actin fibers of approximately 60–80 nm thickness (arrowheads, Figure 5E). Thin fibers were visualized after acquiring 80,000–160,000 frames (Figure S7A), whereas the more slowly dissociating FLAG-1, V5-1, and V5-2 Fab probes did not visualize thin actin fibers using the same number of frames (circles, Figure 5F). Binding of FLAG-2, S-1, and V5-3 Fab probes was specific despite their rapid and short interaction with epitopes, as shown by the absence of binding to areas without actin fibers (arrowheads, Figure S7B). Specific binding was further confirmed using super-resolution imaging of XTC cells expressing actin with epitope tags at the C terminus (Figure S7C) and by the lack of a signal when using control XTC cells without epitope-tagged actin (Figure S7D). Fab probes that showed non-specific binding did not visualize actin fibers (representative images for FLAG-3, S-2, and V5-6; Figure S7E).
Super-resolution imaging with anti-mPLS1 and anti-mESPN1 Fab probes
Super-resolution images were acquired using anti-mPLS1 and anti-mESPN1 Fab probes (Figure 6). FLAG-mPLS1 and mESPN1-V5 were expressed in XTC cells to use FLAG-2 and V5-3 Fab probes as positive controls. Among the 65 anti-mPLS1 Fab probes, 4 probes, namely, PLS-1, PLS-4, PLS-37, and PLS-40, detected FLAG-mPLS1 at high labeling densities (Figure 6A). These four anti-mPLS1 Fab probes detected FLAG-mPLS1 at lamellipodia and filopodia concordantly with the FLAG-2 Fab probe (asterisks and arrowheads, Figure 6B). Slightly higher labeling densities were achieved by PLS-37 and PLS-40 Fab probes, which were synthesized from fast-dissociating antibodies (Figure S8A). Among the 111 anti-mESPN1 Fab probes, 5 probes, namely, N534-30, N534-56, N534-57, N534-60, and 657X-4, succeeded in high-density labeling of mESPN1-V5 (Figure 6D).These Fab probes detected mESPN1-V5 on thick actin fibers induced by mESPN1 expression (arrowheads, Figure 6E). All Fab probes that demonstrated high-density labeling were synthesized from antibodies with high-sensitivity immunostaining abilities and never from antibodies with low-sensitivity or those that failed immunostaining (Figures 6C and 6F).
koff values of Fab probes were determined using time-lapse images and compared with those of original antibodies (Figure S8B; Table S4). koff values from epitopes of the original antibodies were maintained for most anti-mPLS1 Fab probes. Especially, the PLS-37 Fab probe rapidly dissociated from epitopes at koff = 0.39 s−1 (a half-life = 1.8 s) as the original antibody showed. In contrast, koff of the PLS-40 Fab probe, derived from a fast-dissociating antibody, was more than 10-fold lower than that of the original antibody (magenta cross, Figure S8B). Nevertheless, koff values of PLS-37 and PLS-40 Fab probes were still higher than those of PLS-1 and PLS-4 Fab probes and may explain their high labeling density in super-resolution imaging (Figures 6A and 6B). For anti-mESPN1 Fab probes, N534-30 and N534-56 showed faster dissociation than the original antibodies at koff = 0.16 s−1 and 0.15 s−1 (half-lives = 4.4 s and 4.5 s; cyan diamonds, Figure S8B). koff values of N534-57 and 657X-4 Fab probes were the lowest among the five anti-mESPN1 Fab probes (asterisk, Figure S8B). Slow dissociation of these two Fab probes may explain their low labeling densities in imaging (Figures 6D and 6E). Altered koff values after conversion of antibodies to Fab probes may be due to the conjugation of fluorescent dyes or to conformational differences between recombinant proteins used in the screening assay and proteins in cells and tissues that Fab probes recognize or both (Orlova et al., 2003; Szabó et al., 2018; Wingfield, 2015).
Multiplex imaging of cells and tissues using Fab probes
Fast-dissociating Fab probes that can be easily washed away can be used for sequential multiplex imaging (Figures S9A and S9B). The 657X-4 Fab probe was slow to dissociate but was used because it is the only probe to recognize the C terminus of mESPN1. Peptide fragment probes, CLIP-170 and Lifeact, were used for counter-imaging of microtubules and F-actin (Kiuchi et al., 2015). Using these probes, we acquired super-resolution images of XTC cells expressing FLAG-mPLS1, mESPN1-V5, and S-tagged human histone-2B family member b (hH2Bb-S). The probes were sequentially used in the order of 657X-4, CLIP-170, V5-3, N534-56, FLAG-2, PLS-37, S-1, and Lifeact (Figure S9A). Probes could be washed away from the cells as shown for 657X and CLIP-170, CLIP-170 and V5-3, N534-56 and FLAG-2, PLS-37 and S-1, and S-1 and Lifeact (Figures S9A and S9B). In the magnified images, only anti-mESPN1 and anti-V5 tag Fab probes visualized thick actin bundles induced by mESPN1 expression, which also confirms that these probes were successfully washed away between acquisitions (Figure S9B). We also confirmed that anti-mESPN1 and anti-mPLS1 Fab probes (657X-4 and PLS-37) recognized endogenous espin and plastin in frozen tissue sections (Figure S9C). Fab probes and Lifeact conjugated with red fluorescent DyLight550 and Atto550 were used to avoid the predominantly green autofluorescence emitted from tissue samples. 657X-4 and PLS-37 Fab probes bound to stereocilia and not to the bodies of hair cells, indicating specific binding of these probes to endogenous espin and plastin.
Light-sheet microscopy to visualize protein turnover in tissue
Finally, we performed three-dimensional multiplex imaging of tissue samples by using light-sheet microscopy, diSPIM (Wu et al., 2013) (Figure 7), and demonstrated that Fab probes can be used to visualize the dynamics of molecular components in tissues. We compared protein turnover in stereocilia, organelles on the apical surface of inner ear hair cells that function as mechanical switches to convert sound vibration into electric impulses. Stereocilia have a core of unidirectionally oriented actin filaments crosslinked by many different actin-bundling proteins. The replenishment of macromolecules in the long-lived F-actin cores of stereocilia is an unresolved question (Drummond et al., 2015; Narayanan et al., 2015). Thus, we visualized the turnover of ESPIN, a crosslinker whose genetic variants are associated with the human deafness gene DFNB36 (Naz et al., 2004; Zheng et al., 2000). We used EGFP-actin to locate hair cell F-actin stereocilia cores and mESPN1-V5 to visualize the turnover of espin (Figures 7A and 7B). The distribution of mESPN1-V5 was visualized using anti-V5 tag Fab probe (V5-3) and compared with the distribution of endogenous espin and F-actin detected using the 657X-4 Fab probe and Lifeact, respectively (Figure 7C).
Figure 7. Tissue imaging using light-sheet microscopy.
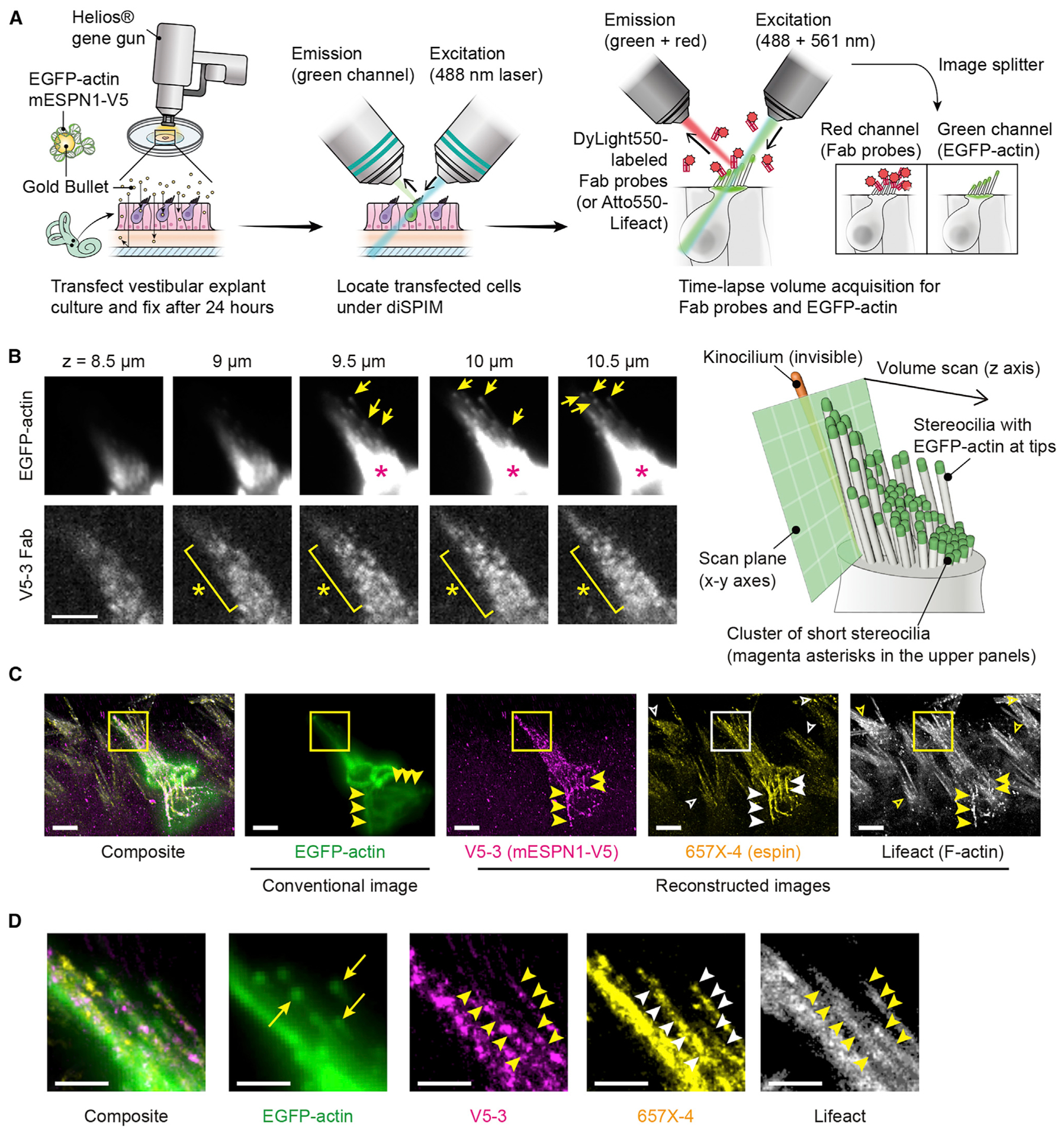
(A) Gene-gun transfection and diSPIM imaging. Vestibular explant cultures were transfected to express EGFP-actin and mESPN1-V5, fixed 24 h later. Transfected cells were located under diSPIM and imaged sequentially using DyLight550-labeled Fab probes and Atto550-labeled Lifeact, acquiring a time-lapse volume scan every 5 s (100-ms acquisitions every 0.5 μm in z, 40 total slices per volume). Images of EGFP-actin were acquired simultaneously using an image splitter for drift correction.
(B) Representative volume scans of EGFP-actin and Fab probe (V5-3 shown). EGFP-actin was detected at each stereocilium tip (arrows). The high-intensity area is a cluster of short stereocilia (magenta asterisks). Bound Fab probes detected as fluorescent spots (yellow asterisks) and used to reconstruct images in (C). Bar, 5 μm.
(C) Orthogonal projection showing conventional volume scan of EGFP-actin (162-nm bin size, 0.5-μm z-spacing) and reconstructed images of mESPN1-V5, endogenous espin, and endogenous F-actin (81-nm bin size, 0.5-μm z-spacing). Images of two Fab probes (V5-3 and 657X-4) and Lifeact were reconstructed from 720, 1,440, and 1,440 volume acquisitions, respectively. V5-3 Fab probe specifically bound to the transfected cell (V5-3, center cell), whereas 657X-4 Fab probe and Lifeact visualized stereocilia of non-transfected cells (657X-4 and Lifeact, open arrowheads). Abnormal rod-like actin fibers indicate an intact actin bundling function of overexpressed mESPN1-V5. (yellow and white arrowheads). Fab probes, 1 nM. Lifeact, 0.5 nM. Bars, 5 μm.
(D) Comparison of molecular turnover between EGFP-actin and mESPN1-V5 using individual stereocilia. Images are magnified rectangles in (C). EGFP-actin is incorporated at the tips (EGFP-actin, arrows), whereas mESPN1-V5 was detected along the entire length of stereocilia (V5-3, arrowheads). Stereocilia are also visualized using 657X-4 and Lifeact probes (arrowheads).
Bars, 2 μm. See also Figure S9.
mESPN1-V5 was detected along the entire mature stereocilia F-actin cores, whereas EGFP-actin was mainly detected at the tips (Figure 7D), indicating that the mESPN1-V5 turnover is much faster than that of actin monomers in stereocilia cores. Correspondingly, fluorescence of transiently expressed EGFP-mESPN3a recovered to half maximal levels 3.9 min after photobleaching, demonstrating with an orthologous method that espin is mobile within the stereocilia core (Figures S9D and S9E). Subsequent imaging with anti-mESPN1 Fab probe (657X-4) and Lifeact visualized stereocilia in adjacent non-transfected cells (Figure 7C), corroborating the conclusion that the V5-3 Fab probe specifically bound to the V5 tags and not to other components of stereocilia.
DISCUSSION
We directly characterized the dissociation kinetics of antibodies secreted by hybridoma clones and systematically identified fast-dissociating, specific antibodies from thousands of monoclonal candidates. These antibodies were specific enough to produce Fab probes for IRIS super-resolution microscopy (Kiuchi et al., 2015). ELISA scores of fast-dissociating antibodies were not high (Figure S1E), indicating that an initial ELISA screen would perhaps have rejected fast-dissociating antibodies due to low scores. Our kinetic screening to readily identify fast-dissociating yet specific monoclonal antibodies should facilitate their use as single-molecule imaging probes and real-time biosensors (Hayashi-Takanaka et al., 2011; Paek et al., 2011).
Our assay based on single-molecule microscopy has significant advantages for the identification of fast-dissociating antibodies. The high time resolution of current single-molecule microscopy permits the detection of short-term antibody-antigen binding of <100 ms half-lives, such as for FLAG-3 and several anti-mPLS1 antibodies. In addition, single-molecule microscopy can detect antibody-antigen binding at high sensitivity and specificity by visualizing each binding event as one fluorescent spot, enabling the survey of low-affinity antibodies. Kinetic screening of antibodies is possible with other protein-protein interaction analyses, such as SPR (Canziani et al., 2004; Kamat et al., 2020; Karlsson et al., 1991; Murray et al., 2014; Säfsten et al., 2006; Ylera et al., 2013). However, it is still challenging for SPR to achieve these advantages at the same time (Helmerhorst et al., 2012), and previous studies using SPR did not identify fast-dissociating antibodies with a 1-s half-life, such as our FLAG-2 and S-1 antibodies (Canziani et al., 2004; Kamat et al., 2020; Säfsten et al., 2006). Compared to bulk protein-protein interaction analyses, single-molecule imaging usually has two disadvantages: low throughput and the necessity to fluorescently label analytes. In our study, throughput was improved by using a programmable stage and automatic focus. The necessity to fluorescently label thousands of monoclonals was avoided by using EGFP-labeled antigens. This flipped relationship between antibodies and antigens in our assay avoided the avidity effect and allowed an accurate measure of koff (Myszka, 1999).
We show that fast dissociation is not such a rare property of a specific antibody even with a half-life on the target of a few seconds. We identified such monoclonal antibodies for three epitope tags, mPLS1, and mESPN1. Most of these fast-dissociating antibodies had sufficient specificity to detect antigens in conventional immunostaining (Figures 4, S1, and S4). FLAG-2, S-1, V5-3, and PLS-37 antibodies produced Fab probes combining fast dissociation and high specificity useful for IRIS super-resolution microscopy. Fast-dissociating antibodies can also complement other super-resolution microscopy approaches using reversible-binding probes, such as DNA-PAINT and Exchange-PAINT (Jungmann et al., 2014; Schueder et al., 2017). Compared with these multiplex imaging approaches using reversible binding oligonucleotide-labeled antibodies, Fab probes are advantageous for elevating the labeling density of imaging targets because different populations of epitopes are labeled by repeated cycles of binding and dissociation. In addition, Fab probes are potentially suitable for imaging densely packed structures, such as protein scaffolds, because Fab probes are washed away after each sequence of multiplex imaging to avoid spatial interference with the next Fab probe. Fast-dissociating antibodies were rarely available for some antigens, such as mESPN1. For such antigens, one solution is to append epitope tags, such as FLAG, S, and V5 tags. Antibody engineering might also be used to produce fast-dissociating antibodies, for example, through amino acid substitutions within the antigen recognition sites of antibodies to accelerate dissociation from epitopes (Fukunaga et al., 2018; Hugo et al., 2003).
To produce Fab probes useful for IRIS, epitopes must be accessible and uncommon endogenously in cells and tissues of interest. In this study, only a few anti-mPLS1 and anti-mESPN1 antibodies produced Fab probes useful in IRIS, whereas we obtained useful Fab probes from many of the anti-epitope tag antibodies. Compared with epitopes in protein antigens, epitope tags attached in-frame to a protein’s N or C terminus are advantageous as they are often accessible to antibodies (Angeletti, 1999). Additionally, the amino acid sequences of epitope tags are usually expressed endogenously at negligible levels, as indicated by a variety of useful specific antibodies (Brizzard, 2008; Kimple et al.,2013). In contrast, epitopes of protein antigens, such as mPLS1 and mESPN1 in this study, may be buried in its three-dimensional structure or blocked by binding partners to form a macromolecular complex and sometimes resemble amino acid sequences of endogenous proteins. These differences in the availability of epitopes may explain why immunostaining using some anti-mPLS1 and anti-mESPN1 antibodies detected mPLS1 and mESPN1 only at low labeling densities even though these antibodies showed either a high affinity, a slow koff, or both.
High labeling densities in immunostaining are hallmarks of good Fab probes for IRIS. To visualize antigens using IRIS, specific binding to epitopes must overcome the background noise from non-specific binding. High specificity and easy access to epitopes is required for Fab probes because their binding is not enhanced by other factors, such as secondary antibodies used in conventional immunostaining. Surface loop regions, β-turns, and termini are often good candidates for unique and easily accessible epitopes (Angeletti, 1999; Wisdom, 1994), and computational predictions may be helpful for selecting potential epitopes (Moreau et al., 2008). Avoiding an epitope containing lysine residues may also produce a Fab probe that successfully recognizes the epitope because during fixation, lysines can be crosslinked by paraformaldehyde and glutaraldehyde (Thavarajah et al., 2012).
Fab and peptide fragment probes were useful in three-dimensional imaging of tissue samples and can be used to visualize the dynamics of molecular components in tissues (Figure 7). We discovered that espin, an actin bundling protein encoded by the DFNB36 deafness gene ESPN, is replenished along the entire length of long-lived F-actin stereocilia cores despite its high-affinity binding to F-actin bundles (Bartles et al., 1998). This result suggests that proteins, especially actin crosslinkers, can infiltrate the dense F-actin cores of stereocilia. We hypothesize that ESPN1 is involved in routine maintenance processes, such as the repair of gaps in the F-actin cores of stereocilia (Belyantseva et al., 2009), perhaps contributing to the ability of stereocilia to tolerate a lifetime of sound vibrations. To use Fab probes in tissue imaging, background fluorescence of the tissue itself and unbound Fab probes are hurdles to detect bound Fab probes using single-molecule microscopy. Light-sheet microcopy, such as diSPIM (Wu et al., 2013), is one practical solution that decreases the thickness of illumination and thus background fluorescence. The lens design of diSPIM is fitting for tissue samples that have structures protruding from the apical surface of cells, such as stereocilia and microvilli, because excitation and emission light are not scattered by cell bodies.
Here, we describe a systematic pipeline for identifying fast-dissociating specific antibodies. Among the 1,000 to 2,000 anti-epitope tag monoclonal antibodies screened, we identified 1 anti-FLAG tag, 1 anti-S tag, and 1 anti-V5 tag antibody (FLAG-2, S-1, and V5-3) that produced fast-dissociating, highly specific Fab probes. Among the 3,000 anti-mPLS1 candidate antibodies, 1 antibody (PLS-37) retained fast dissociation after being converted to Fab fragments. Specific, yet reversible binding is a property of many reagents such as oligonucleotides, peptide fragments, and metal ions (Kiuchi et al., 2015; Riedl et al., 2008; Schmitt et al., 2000; Schueder et al., 2017). However, antibodies have an unrivaled advantage over these reagents because they can recognize almost any epitope. Our single-molecule screening assay indicates that fast-dissociating antibodies with half-lives of less than a few seconds are not so rare and can achieve high specificity useful for multiplex super-resolution microscopy to characterize the turnover of proteins, as we have demonstrated. Our approach will help researchers who require probes that have readily reversible binding properties. We speculate that fast-dissociating antibodies and their fragments will be broadly useful single-molecule real-time detectors for many biological applications and will complement the benefits of conventional, off-the-shelf antibodies.
STAR ★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Takushi Miyoshi (miyoshi.takushi.76e@kyoto-u.jp and takushi.miyoshi@nih.gov).
Materials availability
Materials (hybridomas, antibodies, and plasmids) generated in this study will be made available upon request from the Lead Contact, Takushi Miyoshi (National Institutes of Health), and the co-corresponding author, Naoki Watanabe (Kyoto University), with a completed Materials Transfer Agreement.
Data and code availability
Python scripts used in this study are available from a GitHub repository (https://github.com/takushim/tanitracer).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and transfection
Xenopus laevis XTC cells were maintained in Leibovitz’s L15 medium (Thermo Fisher Scientific) diluted to 70% with sterilized water and supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific) as previously described (Watanabe and Mitchison, 2002). HEK293 cells were cultured in Dulbecco’s modified eagle medium (DMEM; nacalai tesque, Inc) containing 10% FBS. Expi293F suspension cells (Thermo Fisher Scientific) were cultured in Expi293 expression medium (Thermo Fisher Scientific) as per the manufacturer’s instruction. Transfection of these cells was performed using polyethylenimine (PEI; Polysciences) following the manufacturer’s protocol and a previous report (Portolano et al., 2014).
Animals and tissue samples
ICR mice, from which we obtained frozen tissue sections, were purchased from Japan SLC, Inc. and euthanized at postnatal day (P) 2. Inner ears were dissected and fixed in 4% PFA in PBS for 2 h at RT. After adjusting the osmotic pressure using 30% sucrose in PBS overnight, the inner ears were embedded in Tissue-Tek O.C.T. compound (Sakura), and frozen in hexane (approximately −100°C) cooled with a cooling apparatus (UT2000F; Leica Microsystems). Frozen sections were prepared at 8-μm thickness using a Leica CM3050 S cryostat (Leica Microsystems).
Explant culture and gene-gun transfection for light-sheet microscopy (diSPIM) were performed as previously described (Belyantseva et al, 2003, 2016). Briefly, C57BL6/J mice for vestibular explant culture was purchased from The Jackson Laboratory, and dissected at P2 after euthanization. Vestibular sensory epithelia were cultured on collagen-coated glass-bottom dishes (MatTek Corporation) in DMEM/F12 medium (Thermo Fisher Scientific) supplemented with 7% FBS (Atlanta Biologicals) and 20 μg/mL Ampicillin (Sigma) overnight at 37°C with 10% CO2. Cells were transfected using Helios Gene Gun (Bio-Rad) and 1.0 μm Gold Microcarriers (Bio-Rad) coated with plasmids at 110 psi. Explant cultures were incubated up to 24 h for expression.
Explant culture and injectoporation for fluorescent recovery after photobleaching (FRAP) experiments were prepared similarly, but from P4 C57BL/6J mice following dissection and adhering the tissue to the lid of a 3.5 cm plastic tissue culture treated dish (USA Scientific) in 2 mL of DMEM/F12 with HEPES. Cultures were incubated at 37°C for 1–2 hours before being transfected by injectoporation, which was carried out essentially as previously described (Xiong et al., 2014). Briefly, plasmid DNA encoding EGFP-mESPN3a was purified with an endotoxin free maxiprep kit (QIAGEN) and the concentration was adjusted to 1 mg/ml in water before mixing 1:10 with 1% fast green dye. This mixture was loaded in a glass microinjection needle with a 2–3 μm diameter tip. The DNA was injected into the organ of Corti between the 2nd and 3rd rows of outer hair cells by applying backpressure to the needle using a PLI-100 microinjector (Harvard Apparatus) until the tissue bulged slightly. After 10–20 s of injection, the tissue was electroporated by applying three pulses at a 60-V amplitude, a 15-ms duration and a 1 s interval through platinum wire electrodes that flanked the explanted tissue. The culture was returned to the 37°C incubator for 16–24 hours before imaging.
All experiments were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and were approved by the Animal Research Committee of Kyoto University Graduate School of Medicine (No. 17555, Kyoto, Japan), the Institutional Animal Care and Use Committees of IUPUI (No. SC238R to B.J.P.), and the Animal Care and Use Committees at the NIH (No. 1263 to T.B.F.).
Development and culture of hybridoma clones
Hybridoma clones seeded in 96-well tissue culture plates were purchased from MBL for FLAG-tag and from Mediridge Co., Ltd. for S-tag, V5-tag, mPLS1, and mESPN1. Clones were developed using female BALB/c mice and a mouse iliac lymph node method (Sado et al., 2006; Sado and Okigaki, 1996). In MBL, mice were immunized three times (day 1, 8 and 22) using subcutaneous injection of 50 μg immunogen and dissected at day 24 to obtain iliac lymph nodes. In Mediridge, mice were immunized three times (day 1, 15 and 25) injecting immunogens of 100 μg, 100 μg and 40 μg, respectively, and dissected at day 28. B cells in lymph nodes were hybridized with mouse myeloma cells (P3U1; RIKEN BRC) using polyethylene glycol fusion by the manufacturers. As immunogens to inject into mice, FLAG, S, and V5 peptides were chemically synthesized and conjugated with keyhole limpet hemocyanin (KLH). Recombinant mPLS1 and mESPN1 were purified by the authors and then emulsified in Freund’s complete adjuvant. Clones seeded in 96-well plates were cultured in incubators with 5% CO2 and 37°C until stables colonies were formed. Clones for anti-FLAG antibodies were cultured in Hybridoma-SFM medium (Thermo Fisher Scientific) supplemented with 15% Hyclone Super-Low IgG FBS (GE Healthcare) containing 1 × HAT Supplement (Thermo Fisher Scientific), and 1% Stabilized Penicillin-Streptomycin Mixed Solution (nacalai tesque, Inc). Other clones were cultured in RPMI1640 medium (nacalai tesque, Inc) supplemented with 15% Hyclone Super-Low IgG FBS, 1 × HAT Supplement, 2 mM L-glutamine (Thermo Fisher Scientific), 1 mM sodium pyruvate (Thermo Fisher Scientific), and 1% penicillin-streptomycin.
ELISA assays were performed for the hybridomas by the manufacturers (FLAG-tag: MBL; S-tag and V5-tag; Mediridge). At MBL, chemically synthesized peptide antigens were diluted with PBS to 1 μg/mL and immobilized on 96-well assay plates (Thermo Fischer Scientific) by non-specific adsorption at 4°C overnight. After blocking with 1% BSA (nacalai tesque) in PBS at 4°C overnight, immobilized antigens were confronted with hybridoma culture supernatants containing candidate monoclonal antibodies for 1 h at room temperature (RT; approximately 25°C). Immobilized antibodies were confronted with horse radish peroxidase (HRP)-conjugated goat polyclonal anti-mouse IgG (H+L) Fab’ diluted 1:10000 in PBS for 1 h at RT and detected using 3,3′,5,5′-Tetramethylbenzidine dihydrochloride hydrate (Millipore Sigma) as per the manufacturer’s instruction. Light adsorption was determined at 450–620 nm using a microplate reader (TECAN). At Mediridge, chemically synthesized peptide antigens were diluted with PBS to 1 μg/mL and immobilized on 96-well assay plates (BM Equipment) by non-specific adsorption at 4°C overnight. After blocking with 0.5% skim milk (Wako) in PBS at 4°C overnight, immobilized antigens were confronted with hybridoma culture supernatants containing candidate monoclonal antibodies for 1 h at RT. Immobilized antibodies were confronted with alkaline phosphatase (AP)-conjugated goat anti-mouse IgG (Southern Biotech) diluted 1:2500 in PBS containing 0.5% skim milk for 1 h at RT. AP-conjugated secondary antibodies were detected using phenylphosphate disodium (Wako), 4-aminoantipyrine (Wako), and sodium periodate (Wako) as per the manufacturer’s instruction. Light adsorption was determined at 492 nm using a microplate reader (CORONA).
METHOD DETAILS
cDNAs and plasmid vectors
EST clones encoding mouse plastin 1 (mPLS1, NM_1033210.3) and human histone cluster 1 H2B family member b (hH2Bb, NM_021062.2) were purchased from Dharmacon. cDNA encoding transcript variant 1 of mouse espin (mESPN1, NM_207687.2) was purchased from UNITECH, Inc. cDNA encoding transcript variant 3a of mouse espin (mESPN3a, AY587570.1) was provided by James Bartles (Feinberg School of Medicine, Northwestern University). cDNA encoding rat CAP-GLY domain containing linker protein 1 (CLIP-170; NM_031745.2) was provided by Y. Mimori-Kiyosue(RIKEN, Japan). The expression vector for bacteria, pGEX-6P-1, was purchased from GE Healthcare. The expression vectors for culture cells, pEGFP-C1, pEGFP-N3, and pEGFP-actin, were purchased from Clonetech. These vectors were used to express EGFP-tagged proteins or used to epitope-tagged proteins after replacing the EGFP-sequences with oligonucleotides gactacaaagacgatgacgacaag (DYKDDDDK peptide; FLAG tag), aaagaaaccgctgctgctaaattcgaacgc cagcacatggacagc (KETAAAKFERQHMDS peptide; S tag) and ggcaaaccgattccgaacccgctgctgggcctggatagcacc (GKPIPNPLLGLDST peptide; V5 tag) with or without the first methionine. These vectors were named as pFLAG/S/V5-C1, pFLAG/S/V5-N3, and pFLAG/S/V5-actin. Among these vectors, Cytomegalovirus (CMV) promoter of pFLAG-actin vector was deleted (delCMV) (Watanabe and Mitchison, 2002) to lower cytotoxicity. For protein synthesis using HEK293 cells, the double-tagged vectors, pFLAG-EGFP-C1 and pFLAG-6P-EGFP-C1, were constructed by inserting oligonucleotide encoding FLAG tag and/or 6P-site (human rhinovirus 3C protease site) to pEGFP-C1.
Microscopy
Time-lapse images of the screening assay, cell samples, and frozen tissue sections were acquired by an Olympus IX83 inverted microscope installed with an MS-2000 Flat-Top XY Automated Stage (Applied Scientific Instrumentation), an IX3-ZDC2 Z-drift compensator (Olympus), and a UPlansApo 100 × , 1.40 NA oil objective (Olympus). The microscope for the screening assay and cell samples equipped an Evolve 512 EMCCD camera (Roper Scientific) and a Cobolt Blues 50 mW laser (473 nm; Cobolt). The microscope for frozen tissue sections equipped an ORCA-Flash4.0 V3 Digital CMOS camera (Hamamatsu), an OBIS 552 nm LS 150 mW laser (Coherent), and a custom-made cone-shaped beam splitter rotating at 12,000 rpm by a hollow shaft motor (Olympus) (Adachi et al., 2007). Devices were controlled by MetaMorph Microscopy Automation and Image Analysis Software (Molecular Devices).
Time-lapse volume scans of vestibular explant cultures were acquired by symmetrical dual-view inverted selective plane illumination microscopy (diSPIM, Applied Scientific Instrumentation) (Wu et al., 2013), equipped with 40 × Nikon CFI APO NIR objectives, 0.80 NA, 3.5 mm WD (Nikon), OBIS 488 nm LX 150 mW Laser (Coherent), and OBIS 561 nm LS 150 mW Laser (Coherent). Images were split by W-VIEW GEMINI Image splitting optics (Hamamatsu) equipped with a 561 nm laser BrightLine single-edge super-resolution/TIRF dichroic mirror (Semrock), a 525/50 nm BrightLine single-band bandpass filter (Semrock), and a 568 nm Edge-Basic best-value long-pass edge filter (Semrock) to acquire with an ORCA-Flash4.0 V2 Digital CMOS camera (Hamamatsu). The devices were controlled by Micro-Manager (Edelstein et al., 2014).
Fluorescent immunostaining of XTC cells
XTC cells were spread onto poly-L-lysine-coated coverslips and fixed with 3.7% paraformaldehyde (PFA) in cytoskeleton buffer (10 mM MES, 90 mM KCl, 3 mM MgCl2, 2 mM EGTA, pH 6.1) containing 0.5% Triton-X for 20 min and blocked with 1% bovine serum albumin (BSA; nacalai tesque, Inc) in phosphate-buffered saline (PBS) containing 0.2% Triton-X (PBS-Tx) for 1 h at RT. Cells expressing mPLS1 was fixed without Triton-X. Primary antibodies in hybridoma culture supernatants were applied without dilution, and incubated for 2 h at RT. The secondary antibody was DyLight550 Goat Anti-Mouse IgG (H+L) (Thermo Fisher Scientific) applied at 1:500 for 1 h at RT.
SDS-PAGE analyses of hybridoma culture supernatants
Recombinant proteins were denatured and reduced in the equal volume of 2 × Laemmli sample buffer (100 mM Tris-HCl, 3.4% sodium dodecyl sulfate, 10% glycerol, 0.2M dithiothreitol, 5% 2-mercaptoethanol, and 0.02% bromophenol blue, pH 6.8) at 95°C for 10 min, and then fractionated using 10% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad). Antibodies in hybridoma culture supernatants were collected using 20 μL (bed volume) of Protein A Sepharose CL-4B beads (GE Healthcare) per 1 mL of culture supernatant. Beads and supernatants were incubated at 4°C overnight, and unbound antibodies were washed away with PBS. Antibodies were eluted using equal volumes of 2 × Laemmli sample buffer and fractionated using 10% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad).
Recombinant proteins
Epitope tags fused with EGFP to screen anti-epitope tag antibodies, FLAG-EGFP, S-EGFP, and V5-EGFP, were synthesized in HEK293 cells and harvested as cell lysates. The expression vectors of these proteins were constructed by inserting the EGFP sequence into pFLAG/S/V5-C1 vectors and transfected into cells using PEI. Cells expressing FLAG-EGFP, S-EGFP, or V5-EGFP were washed with ice-cold PBS and lysed in HEPES-KCl-Tx buffer (10 mM HEPES-KOH, 90 mM KCl, 3 mM MgCl2, 0.1 mM dithiothreitol, 0.2% Triton-X, pH 7.2) supplemented with EDTA-free Protease Inhibitor Cocktail (nacalai tesque, Inc). Lysates were clarified by centrifugation at 13,000 × g for 20 min. Protein concentration was determined by measuring the fluorescence intensity of EGFP using an EnVision 2103 Multilabel Plate Reader (Parkin Elmer). Lysates of non-transfected HEK293 cells were used as control to determine fluorescence intensity of GFP.
Other EGFP-fused antigens (EGFP-antigen) and EGFP-fused CLIP-170 fragment were synthesized using Expi293F cells. The expression vector of EGFP-fused mPLS1 (EGFP-mPLS1) was constructed by subcloning mPLS1 cDNA into pFLAG-EGFP-C1 vector. The expression vectors of EGFP-fused mESPN1 fragments, such as EGFP-N534 and EGFP-657X, were constructed by subcloning the corresponding sequences of mESPN1 cDNA into pFLAG-6P-EGFP-C1 vector (Figures S4B and S4C). The expression vector of EGFP-fused CLIP-170 fragment was constructed by subcloning sequences encoding the residues 3–309 of rat CLIP-170 into pFLAG-6P-EGFP-C1 vector (Kiuchi et al., 2015). Harvested Expi293F cells were washed with ice-cold PBS and lysed in ice-cold HEPES-KCl-Tx buffer supplemented with EDTA-free Protease Inhibitor Cocktail. Lysates were briefly sonicated and clarified using centrifugation at 13,000 × g for 20 min. Synthesized proteins were collected by Anti-DYKDDDDK-tag Antibody Beads (Wako) incubated at 4°C overnight. EGFP-mPLS1 and EGFP-fused mESPN1 fragments were eluted using 1 mg/mL DYKDDDDK peptide (Wako) in HEPES-KCl-Tx buffer at 4°C overnight. EGFP-fused CLIP-170 fragment was cleaved in HEPES-KCl-Tx buffer containing 0.02 unit/μL of PreScission Protease (GE Healthcare) at 4°C overnight. Added protease was removed by incubation with small amounts of COSMOGEL GST-Accept beads (nacalai tesque, Inc) at 4°C for 2 h.
mPLS1 and mESPN1 for immunization were bacterially synthesized using BL21(DE3) cells (New England Biolabs) transformed with mPLS1 and mESPN1 cDNAs subcloned into pGEX-6P-1 vector. The cells were pre-cultured in Luria-Bertani (LB) medium containing 50 μg/mL of ampicillin at 37°C overnight and diluted into fresh medium. Protein expression was induced with 0.1 mM isopropyl-β-D-thiogalactopyranoside at OD600 = 0.6, followed by culturing at 16°C for 24 h. Harvested cells were resuspended and sonicated in buffer containing 20 mM Tris-HCl, 140 mM NaCl, 1 mM dithiothreitol, 0.5% Triton-X, pH 7.4 supplemented with EDTA-free Protease Inhibitor Cocktail. Centrifugation at 13,000 × g for 20 min was used to clarify the lysates. mPLS1 and mESPN1 in the soluble fraction were collected using GST-Accept beads incubating at 4°C for 2 h. Proteins were cleaved in PBS containing 0.02 unit/μL PreScission Protease. During the recovery of mESPN1, the PBS was supplemented with 0.05% Tween-20 to inhibit aggregation (Chen et al., 1999).
Antibody screening assay with semi-automated single-molecule TIRF microscopy
Custom-made 96-well glass bottom plates were produced with a high-density amino group coating (density of the amino group is 1.8 × 10−9 mol/μm2; available from Matsunami Glass, Inc.). The amino groups on glass surfaces were activated with 5 mM of sulfo-succinimidyl 6-(4’-azido-2′-nitrophenylamino) hexanoate (sulfo-SANPAH; ProteoChem), which were dissolved in a small amount of dimethyl sulfoxide and diluted in PBS (adjusted to pH 7.0 with HCl), for 2 h at RT. After unbound sulfo-SANPAH was washed away, Protein A/G was crosslinked on the glass surfaces by filling the wells with 0.3 mg/mL of Protein A/G (ProSpec) in PBS (pH 7.0), and by following UV illumination at 365 nm using a UV transilluminator (NIPPON Genetics) for 30 min at RT. Unreacted sulfo-SANPAH was blocked with 3% BSA in PBS (pH 7.0) at 4°C overnight. Solutions containing antibodies to evaluate, typically hybridoma culture supernatants, were applied into the wells and incubated at 4°C overnight. The control antibodies (monoclonal anti-V5 tag antibody [SV5-Pk1] [Abcam], monoclonal anti-S tag antibody [SBSTAGa] [Abcam], and monoclonal anti-FLAG M2 antibody [Sigma]) were applied diluted in PBS-Tx.
Glass-bottom plates to confirm Protein A/G-dependent antibody immobilization were prepared using 0–0.5 mg/mL of Protein A/G and confronted with the 1:30 mixture of Mouse IgG1 isotype control monoclonal antibody (MOPC-21) (DyLight 488 conjugate) (Enzo) and Mouse IgG1, kappa monoclonal [MOPC-21] isotype control (Abcam) at 1 μg/mL in PBS-Tx. After washing away unbound IgG1, the fluorescence intensity was determined using TIRF microscopy with an Evolve 512 EMCCD camera without EM Gain (5 MHz) and 473-nm laser illumination. For each Protein A/G concentration, fluorescence intensities were measured at three different wells in one assay plate.
To observe antibody-antigen binding, immobilized antibodies were confronted with EGFP-antigens after washing away unbound antibodies with HEPES-KCl-Tx buffer. EGFP-antigens were diluted at 0.1–30 nM in HEPES-KCl-Tx buffer containing 0.2 mg/mL glucose oxidase (Sigma), 0.035 mg/mL catalase (Sigma), and 0.45% glucose for oxygen scavenging (Desai et al., 1999). Bound EGFP-antigen molecules were detected using TIRF microscopy with an Evolve 512 EMCCD camera and 473-nm laser illumination. The EMCCD camera was used with EM Gain (EM = 300, Gain State 3, 10 MHz) to acquire single-molecule imaging and without EM Gain (5 MHz) to compare fluorescence intensities. The density of bound EGFP-antigen molecules was directly counted for a small amount of bound EGFP-antigen molecules and determined using the fluorescence intensity for a large amount of bound EGFP-antigen. Fluorescent spots of bound EGFP-antigen molecules were localized using our Python program combining a Gaussian-Laplacian filter (https://www.scipy.org/) and Gaussian fitting with “peak_local_max” function (scikit-image.org) followed by sub-pixel correction. Localized spots were tracked using k-nearest neighbor algorithm (scikit-image.org). koff of antibody-antigen binding was determined by fitting a one-phase decay models (non-linear regression in Prism 8.0) to the regression of bound EGFP-antigen molecules.
In the control experiments in Figure 1, anti-V5 tag antibody (SV5-Pk1) were applied at 0–10 μg/mL and confronted with 30 nM V5-EGFP. The concentration, 30 nM, is the maximum concentration at which our TIRF microscopy can detect single-molecule against the background of diffusing V5-EGFP. For each antibody concentration in Figures 1C and 1D, images to measure bound EGFP-antigen were acquired in n = 3 independently-prepared glass-bottom plates. In Figures 1E and 1F, time-lapse images were acquired using n = 3 independently-prepared glass-bottom plates. Fitting of one-phase decay models in Figure 1F was performed as previously described (Kiuchi et al., 2015) using a sufficiently large number of bound V5-EGFP molecules except that three regression curves were drawn with mean ± SD values to show the reproducibility of our assay. The photobleaching rate of EGFP (Figure S1A) was measured using XTC cells expressing EGFP-actin, which were weakly fixed in 3.7%PFA (5 min at RT) in cytoskeleton buffer and immersed in HEPES-KCl-Tx buffer containing glucose oxidase, catalase and glucose. The rate constant and half-life were determined using n = 3 cells by fitting a one-phase decay model.
Simulation of antibody screening assay
To test the sensitivity of our assay to detect weak-affinity antibodies, which ranges to KD = 10 μM among the reported antibodies (Canziani et al., 2004; Säfsten et al., 2006), we simulated the density of bound EGFP-antigen molecules (ρ) from the concentration of applied antibody ([I]0), affinity between antibody and EGFP-antigen (KD), and the concentration of EGFP-antigen ([G]0). The final equation to introduce ρ is the following two-step Hill’s equation:
| (Equation 1) |
This equation includes two constants, ρmax and KP: ρmax is the maximum density of available antigen binding sites on the glass surface (assuming that Protein A/G was fully occupied with antibodies), and KP is the dissociation constant between Protein A/G and antibody.
To introduce the Equation 1, we focus on two interactions in our screening assay (Figure 1A). One is the binding between Protein A/G crosslinked to glass surface and applied antibody. The other is the binding between antibodies immobilized by Protein A/G and applied EGFP-antigens. The binding between Protein A/G and antibodies are simulated by Hill’s equation:
| (Equation 2) |
where [P]0 is the concentration of crosslinked Protein A/G, [PI] is the concentration of Protein A/G bound to antibody, and [I] is the concentration of free antibody. The assumption that [I] ≈ [I]0 is not true when [I]0 is small compared with [P]0, but the error in this assumption was acceptable for the range [I]0 of our simulation (Figure S1B). This equation can be rewritten to use the density of immobilized antibody (ρI),
| (Equation 3) |
where ρP is the density of crosslinked Protein A/G and CP is the ratio of intact Protein A/G to total Protein A/G. CP was introduced to explain the damage of Protein A/G by the crosslinking reagent, and should be a constant since the condition of crosslinking is always consistent. The binding between immobilized antibody and applied EGFP-antigen is similarly written as:
| (Equation 4) |
where ρI is the density of immobilized antibody and λI is the ratio of intact antigen binding sites per one antibody molecule (λI = 2 at ideal condition). From the Equations 3 and 4, we obtain the following equation:
| (Equation 5) |
which is equivalent to the Equation 1 by defining ρmax = CP × λI × ρP.
The constants in the Equation 1, ρmax and KP, must be determined to calculate ρ for given [G]0 and KD. For this purpose, we utilized the saturation curve between established anti-V5 tag antibody [SV5Pk1] (Southern et al., 1991) and V5-EGFP antigen (Figure 1D). In Figure 1D, almost all intact antigen binding sites of antibodies are occupied by EGFP-antigen due to the high affinity of this antibody and a sufficient concentration of EGFP-antigen. The ratio of occupied binding sites (RV5) is calculated from the Equation 4 as:
| (Equation 6) |
since the concentration of EGFP-antigen ([G]0) is 30 nM and the affinity of this antibody is estimated to be KD ≈ 1 nM according to the KD of commonly-used monoclonal antibodies (Fujii et al., 2014). With this ratio, the Equation 1 can be rewritten as the saturation curve between Protein A/G and antibody:
| (Equation 7) |
Bmax and KP were determined to be 1.6 × 102 μm−2 and 4.6 nM (95% CIs are Bmax = 1.51 × 102 to 1.70 × 102 μm−2 and KP = 3.82 to 5.62 nM) using one-site saturation binding model in Prism 8.0 (GraphPad Software, Inc.). ρmax was close to Bmax and determined to be 1.65 × 102 μm−2 since Bmax = ρmax × 0.968.
The density of bound EGFP-antigen for weak-affinity antibodies (Figures S1B and S1C) were calculated using the Equation 1 with the constants ρmax = 1.65 × 102 μm−2 and KP = 4.6 nM determined above. The measured data between the anti-V5 tag antibody [SV5Pk1] and V5-EGFP is plotted as white circles in Figure S1B, which must be on the curve of KD = 1 nM because the constants, ρmax and KP, are determined assuming a KD of this anti-V5 tag antibody [SV5Pk1] is 1 nM. The white circles and curve are well fit for small concentration of antibody, indicating the assumption above, [I] ≈ [I]0, is available within the range of [I]0 in this simulation.
The final goal of this simulation was to estimate the lower detection limit of antibody concentration when a weak-affinity (high-KD) antibody was immobilized. The criterion to detect antibody is that the density of bound EGFP-antigen molecules exceeds their density of 0.02 spots/μm2 on control antibody-free glass surface (Figure 1C). Theoretically, our assay can detect antibodies of KD = 10 μM, the weakest affinity for a reported antibody (Canziani et al., 2004; Säfsten et al., 2006), applied at 0.20 nM (Figure S1B), which corresponds to 0.030 μg/mL assuming an IgG molecular weight of 150 kDa (Janeway et al., 2001). Since typical hybridoma culture supernatants contains 1 to 10 μg/mL of antibody (Yokoyama, 2008), our assay is expected to have an approximately 30-fold safety margin for antibodies of KD = 10 μM. In addition, we simulated the density of bound EGFP-antigen for lower concentrations of EGFP-antigen (Figure S1C). Although 3 nM of EGFP-antigen theoretically detect antibodies of KD = 10 μM applied at 3 nM (0.45 μg/mL), the maximum density of bound EGFP-antigen only reaches to 0.05 spots/μm2, which is close to the density on control antibody-free glass surface (0.02 spots/μm2). Thus, EGFP-antigen was used at approximately 30 nM in screening assays unless there were special reasons to reduce the concentration.
The difference in the densities of bound EGFP-antigen is considered to reflect the difference of dissociation constants (KD) between antibodies and EGFP-antigen. To introduce this relationship, the Equation 1 is rewritten as:
| (Equation 8) |
When we plot KD against ρ (Figure S1D), KD is similar for the same ρ within the range of [I]0 in hybridoma culture supernatant, 1–10 μg/mL (Yokoyama, 2008), which corresponds to 6.7–67 nM assuming the molecular weight of IgG is 150 kDa (Janeway et al., 2001). Thus, we used the density of bound EGFP-antigen molecules as an index reflecting the affinity (KD) of antibodies in this study.
Screening of monoclonal antibodies
Culture supernatants of the purchased hybridoma clones were applied to 96-well glass-bottom plates coated with Protein A/G. Antibodies were immobilized to the bottom of the wells and then confronted with EGFP-antigens. Anti-FLAG tag, anti-S tag, and anti-V5 tag antibodies were screened using 30 nM of FLAG-EGFP, S-EGFP, and V5-EGFP, respectively. Anti-mPLS1 antibodies were screened using 20 nM EGFP-mPLS1, and anti-mESPN1 antibodies were screened using two EGFP-fused mESPN1 fragments, EGFP-N534 and EGFP-657X, applied at 5 nM and 20 nM, respectively (see Figure S4 for the preparation of these fragments). To identify antibodies with affinity to the applied EGFP-antigens, 50–100 frames of 50-ms time-lapse images were acquired for each culture supernatant and compared with the densities of bound EGFP-antigen on control antibody-free glass surface. Time-lapse images were acquired at three different sites (anti-epitope tag antibodies) or at two different sites (anti-mPLS1 and anti-mESPN1 antibodies) in the well of each culture supernatants. The criterion, the densities of bound EGFP-antigen on control antibody-free plates, were determined using three wells in three independently prepared assay plates for each EGFP-antigen.
To maintain hybridoma clones during screening, clones seeded on the 96-well plates were subcultured by splitting into multiple non-treated 96-well plates (IWAKI) using epMotion96 (Eppendorf). Clones secreting antibodies identified in the screening assay were then subcultured into non-treated 24-well plates (IWAKI) and cryopreserved using CELLBANKER 1 (Nippon-Zenyaku). Antibody-secreting ability of identified clones were monitored during the process of subculture using the screening assay. Monoclonal populations of clones were isolated by limiting dilution using cryopreserved cells. koff and half-lives were determined from the regression of bound EGFP-antigen molecules. Briefly, immobilized antibodies were confronted with diluted EGFP-antigen, and one-phase decay models were fit to the regression of bound EGFP-antigen molecules (Figure 3). The intervals of time-lapse images were adjusted to observe a 10%–30% exchange of bound EGFP-antigen molecules per frame. We attempted to determine the koff between our antibodies and peptide analytes (FLAG, S and V5; APExBIO) using BioLayer Interferometry (BLI; Octet RED96, Sartorius), conventional streptavidin biosensors (Sartorius), and biotinylating reagents (EZ-Link NHS-PEG4-Biotin; Thermo Fisher Scientific). However, our attempts were not successful perhaps due to the low sensitivity of BLI to low molecular weight peptides as suggested by a previous study where the peptides had to be fused to T4 Lysozyme (Fujii et al., 2014). Therefore, we confirmed the reproducibility of koff values determined in our screening assay by comparing these values with koff determined using their Fab fragments (Figures S6 and S8).
Synthesis of Fluorescently labeled Fab fragments (Fab probes)
Antibodies in the hybridoma culture supernatants were collected using Protein A Sepharose CL-4B beads (GE Healthcare) at 4°C overnight. The beads were suspended in a 5-fold volume of PBS (pH 7.0) containing 10 mM ethylenediaminetetraacetic acid (PBS-EDTA; pH 7.0). DyLight 488/550 Maleimide (Thermo Fisher Scientific) was used to label antibodies via free Cysteine residues (Huh et al., 2013), which was dissolved in dimethyl sulfoxide at 10 μg/μL immediately before use and added to the bead suspension at 0.5 mM. Dye-to-protein molecular ratio was maintained at approximately 1:30 including the Protein A on the beads. The reaction was performed on a tube rotator for 2 h at RT, and unbound dye was carefully and repeatedly washed away using PBS and centrifuged at 500 × g for 2 min. The beads were incubated in digestion buffer (50 mM Tris-HCl, 10 mM cysteine-HCl, 2 mM EDTA, pH 8.0) containing 0.01 mg/mL papain (nacalai tesque, Inc) for 1 h at 37° C water bath. Cleaved fluorescently-labeled Fab fragments (Fab probes) were recovered by centrifugation at 500 × g for 2 min and supplemented by 1 μg/mL of leupeptin (Peptide Institute).
Super-resolution microscopy of cells using Fab probes
Super-resolution imaging was performed as previously reported with optimization for Fab probes (Kiuchi et al., 2015). Cell were spread on coverslips coated with poly-L-lysine and Fibronectin (Sigma), fixed with 3.7% PFA in cytoskeleton buffer containing 0.5% Triton-X for 20 min, and blocked with 3% BSA in HEPES-KCl-Tx buffer. Cells expressing mPLS1 was fixed without Triton-X. Cells expressing both mESPN1 and mPLS1 were fixed with 0.5% Triton-X for 3 min and fixed without Triton-X for 20 min. Fab probes conjugating DyLight488 were applied at 0.1 to 10 nM in HEPES-KCl-Tx buffer supplemented with oxygen scavenging mixture (0.2 mg/mL glucose oxidase [Sigma], 0.035 mg/mL catalase [Sigma], 0.45% glucose, 0.5% 2-mercaptoethanol) (Desai et al., 1999) and 1 ng/mL leupeptin. Time-lapse images were acquired every 50 ms under 473-nm TIRF or epi-fluorescence illumination. Bound Fab probes were detected using our python programs (available from the GitHub repository, https://github.com/takushim/tanitracer) and plotted on the blank image arrays of 10 nm bin size. Stage drift was corrected using phase only correlation with discrete Fourier transform (Kuglin and Hines, 1975). F-actin and microtubules were visualized using Atto488-Lifeact and EGFP-fused CLIP-170 fragments as previously described (Kiuchi et al., 2015).
Super-resolution microscopy of tissue samples using Fab probes
Frozen tissue sections were mounted on custom-ordered coverslips with a high-density amino group coating (available from Matsunami Glass, Inc.) and blocked with 3% BSA in PBS-Tx for 2 h at RT. Fab probes conjugating DyLight550 and Lifeact conjugating Atto550 (red fluorescent dyes) were used to avoid the predominantly green autofluorescence emitted from tissue samples. Time-lapse images were acquired every 50 ms (Lifeact) or every 200 ms (Fab probes) under 561-nm TIRF illumination using a custom-made cone-shaped beam splitter rotating at 12,000 rpm by a hollow shaft motor (Olympus). Bright-field images were inserted every 500 frames for drift correction. Super-resolution images were reconstructed using our python programs and plotted on the blank image arrays of 10 nm bin size. Stage drift was corrected using A-KAZE feature matching (Alcantarilla et al., 2013).
Explant cultures of vestibular sensory epithelia for light-sheet microscopy (diSPIM) were prepared from P2 mice. Gene-gun transfection was performed next day using gold particles coated with plasmids encoding EGFP-actin and mESPN1-V5. After 24-hour incubation, explant cultures were fixed with 4% PFA in PBS for 30 min at RT and blocked with 2% BSA in PBS-Tx for 1 h at RT. Transfected hair cells were located under diSPIM using distribution of EGFP-actin at stereocilia tips as a hallmark of hair cells harboring stereocilia with stable F-actin cores (Drummond et al., 2015; Zhang et al., 2012). Fab Probes conjugated with DyLight550 and Lifeact conjugated with Atto550 were applied. Time-lapse volume images were acquired every 5 s with inter-slice spacing 0.5 μm for 40 slices (total 20 μm thickness) and with 100-ms frame acquisition time. Samples were illuminated by 488-nm and 561-nm lasers simultaneously, and the emission was split into green (EGFP) and red (DyLight550) channels. Images of EGFP-actin were acquired by a conventional volume scan and also used for drift correction. Bound Fab probes were detected by the spot detection function in Imaris software (Oxford Instruments) and plotted on the blank z stack images of 81 nm bin size and 0.5 μm z-spacing. Sample drift was corrected by detecting the surface of EGFP-actin signal in Imaris (Oxford Instruments). In Figure 7, contrast of EGFP-actin image was adjusted using the gamma correction function in Imaris to show the entire structure. Contrast in other panels were linearly adjusted.
FRAP experiment with explant culture
To image the explant, a bead of silicon grease was applied around the edge of a 50 mm #1 glass coverslip, which was placed over the explant to form a chamber. The remaining media was removed from the dish lid before inverting over a Leica 40 × NA 1.1 water immersion objective on a Leica SP8 confocal microscope operating in resonant scanning mode. EGFP-mESPN3a in transfected outer hair cells was excited using the 488 line from an argon laser. Emission light collected by a photomultiplier tube. A region of the bundle was photobleached with 488 nm light, and recovery was imaged at 1 min intervals for 20 min. Imaging was done at 37°C. Integrated fluorescence intensities from equal sized regions of interest (ROIs) from bleached and unbleached areas of the bundle were measured from max intensity projections using ImageJ. The fraction recovery was calculated by dividing the bleached by the unbleached value. A single exponential curve was fit in GraphPad Prism.
QUANTIFICATION AND STATISTICAL ANALYSIS
Densities and fluorescence intensities of bound EGFP-antigen molecules are plotted as means (Figures 1B, 1D, 2D, 4A, and 4B). Standard Deviations (SDs) are indicated by error bars (Figures 1B and 1D) and listed in a supplemental table (Table S1 for Figure 2D). SDs in Figures 4A and 4B are small and hidden by markers. Numbers of replicates (n) are noted in corresponding figure legends. koff and half-lives of antibody-antigen binding were determined from regression of bound molecules (Figures 1F, S2, S3C, S3F, S6A, S8C, and S8D). Numbers of tracked molecules are noted in graphs. Best fit values were determined by fitting one-phase decay models using Prism 8.0 software. 95% CIs are listed in supplemental tables (Tables S2–S4) or noted in Figure Legends (Figures S3C, S3F, S8C, and S8D). Error bars indicating SDs are added to Figure 1F since this experiment was performed using three individually prepared glass-bottom plates. Regression of fluorescent intensity to determine the photobleaching rate was plotted as means ± SDs and fit by a one-phase decay model using Prism 8.0 software. Best fit values and 95% CIs are noted in Figure Legend with the number of replicates (Figure S1A). The fluorescence intensity in FRAP experiments were also plotted as means ± SDs and fit by a single exponential curve using Prism 8.0 software (Figure S9E). Best fit values are noted in Figure Legend with the number of replicates. ELISA was not replicated (Figure S1E). Graphs summarizing or comparing the properties of antibodies show means for densities of bound EGFP-antigens and best fit values for koff (Figures 3D, 4C, 4D, S4E, S4F, S6B, and S8B).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Horse radish peroxidase-conjugated goat polyclonal anti-mouse IgG (H+L) Fab’ | MBL | Cat#330; RRID: N/A |
| Goat Anti-Mouse IgG, Human adsorbed - Alkaline Phosphatase | Southern Biotech | Cat#1030-04; RRID: AB_2794293 |
| Mouse monoclonal anti-V5 tag antibody [SV5-Pk1] | Abcam | Cat#ab27671; RRID: AB_471093 |
| Mouse monoclonal anti-S tag antibody [SBSTAGa] | Abcam | Cat#ab24838; RRID: AB_448427 |
| Mouse monoclonal anti-FLAG M2 antibody | Sigma-Aldrich | Cat#F1804; RRID: AB_262044 |
| DyLight550 Goat Anti-Mouse IgG (H+L) | Thermo Fisher Scientific | Cat#84540; RRID: AB_10942171 |
| Mouse IgG1 isotype control monoclonal antibody (MOPC-21) (DyLight 488 conjugate) | Enzo | Cat#ADI-SAB-600-488; RRID: N/A |
| Mouse IgG1 kappa monoclonal [MOPC-21] isotype control | Abcam | Cat#ab18443; RRID: AB_1107784 |
| Custom-made anti-FLAG tag monoclonal antibodies (FLAG-1, FLAG-2 and FLAG-3) | This paper | N/A |
| Custom-made anti-S tag monoclonal antibodies (S-1 and S-2) | This paper | N/A |
| Custom-made anti-V5 tag monoclonal antibodies (V5-1 to V5-6) | This paper | N/A |
| Custom-made anti-mPLS1 monoclonal antibodies (PLS-1 to PLS-65) | This paper | N/A |
| Custom-made anti-mESPN1 monoclonal antibodies (N534-1 to N534-90; 657X-1 to 657X-21) | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Sulfo-SANPH Crosslinker | ProteoChem | Cat#c1111-100mg |
| Hyclone Super-Low IgG FBS | GE Healthcare | Cat#SH30898.03 |
| Protein A Sepharose CL-4B beads | GE Healthcare | Cat#17096303 |
| EZ-Link NHS-PEG4-Biotin | Thermo Fisher Scientific | Cat#21330 |
| DyLight 488 Maleimide | Thermo Fisher Scientific | Cat#46602 |
| DyLight 550 Maleimide | Thermo Fisher Scientific | Cat#62290 |
| Papain from papaya latex | Sigma-Aldrich | Cat#P4762 |
| 1.0 μm Gold Microcarriers | Bio-Rad | Cat#1652263 |
| DYKDDDDK tag Peptide | APExBIO | Cat#A6002 |
| S tag Peptide | APExBIO | Cat#A6007 |
| V5 Epitope tag Peptide | APExBIO | Cat#A6005 |
| Custom-made Atto488-Lifeact | Sigma-Aldrich, (Kiuchi et al., 2015) | N/A |
| Custom-made Atto550-Lifeact | Sigma-Aldrich, (Kiuchi et al., 2015) | N/A |
| Protein A/G | ProSpec | Cat#pro-646 |
| HEK293F cell lysate containing FLAG-EGFP | This paper | N/A |
| HEK293F cell lysate containing S-EGFP | This paper | N/A |
| HEK293F cell lysate containing V5-EGFP | This paper | N/A |
| Recombinant mPLS1 | This paper | N/A |
| Recombinant mESPN1 | This paper | N/A |
| Recombinant EGFP-mPLS1 | This paper | N/A |
| Recombinant EGFP-fused N534 mESPN1 fragment | This paper | N/A |
| Recombinant EGFP-fused 657X mESPN1 fragment | This paper | N/A |
| Experimental models: cell lines | ||
| Xenopus laevis XTC cells | Watanabe and Mitchison, 2002 | N/A |
| HEK293 cells | ATCC | Cat#CRL-1573 |
| Expi293F cells | Thermo Fisher Scientific | Cat#A14635 |
| P3U1 mouse myeloma cells | RIKEN BRC | Cat#JCRB0708 |
| Custom-made Hybridomas for FLAG-tag | MBL | N/A |
| Custom-made Hybridomas for S-tag | Mediridge Co., Ltd. | N/A |
| Custom-made Hybridomas for V5-tag | Mediridge Co., Ltd. | N/A |
| Custom-made Hybridomas for mPLS1 | Mediridge Co., Ltd. | N/A |
| Custom-made Hybridomas for mESPN1 | Mediridge Co., Ltd. | N/A |
| Experimental models: organisms/strains | ||
| Mouse (Slc: ICR) | Japan SLC, Inc. | Charles River Laboratories, Inc (1965) |
| Mouse (C57BL/6J) | The Jackson Laboratory | Stock No. 000664 |
| Oligonucleotides | ||
| gactacaaagacgatgacgacaag (DYKDDDDK peptide; FLAG tag) | Sigma-Aldrich | N/A |
| aaagaaaccgctgctgctaaattcgaacgccagcacatggacagc (KETAAAKFERQHMDS peptide; S tag) | Sigma-Aldrich | N/A |
| ggcaaaccgattccgaacccgctgctgggcctggatagcacc (GKPIPNPLLGLDST peptide; V5 tag) | Sigma-Aldrich | N/A |
| Recombinant DNA | ||
| Mouse plastin1 cDNA | Dharmacon | NM_1033210.3 |
| Mouse espin1 cDNA | UNITECH, Inc. | NM_207687.2 |
| Mouse espin3a cDNA | James Bartles (Feinberg School of Medicine, Northwestern University) | AY587570.1 |
| Rat CAP-GLY domain containing linker protein 1 | Y. Mimori-Kiyosue (RIKEN, Japan) | NM_031745.2 |
| Human histone cluster 1 H2B family member b cDNA | Dharmacon | NM_021062.2 |
| delCMV-EGFP-C1 plasmid vector | Watanabe and Mitchison, 2002 | N/A |
| Software and algorithms | ||
| MetaMorph Microscopy Automation and Image Analysis Software | Molecular Devices | N/A |
| Micro-Manager | Edelstein et al., 2014 | N/A |
| ImageJ | Schneider et al., 2012 | N/A |
| Prism 8.0 | GraphPad Software, Inc. | N/A |
| Custom-made python scripts | https://github.com/takushim/tanitracer | N/A |
| Imaris | Oxford Instruments | N/A |
| Other | ||
| Custom-made 96-well glass bottom plates with a high-density amino group coating | Matsunami Glass Ind., Ltd. | N/A |
| UV illumination at 365 nm using a UV transilluminator | NIPPON Genetics | MUV21-365 |
| epMotion96 | Eppendorf | Cat#5069000004 |
| Octet RED96 System (BioLayer Interferometry; BLI) | Sartorius | N/A |
| Streptavidin (SA) Biosensors | Sartorius | Cat#18-5019 |
| Custom-made coverslips with a high-density amino group coating (No. 1 thickness, ø = 25 mm) | Matsunami Glass Ind., Ltd. | N/A |
| Helios Gene Gun System | Bio-Rad | Cat#1652431 |
| Custom-made cone-shaped beam splitter rotating at 12,000 rpm by a hollow shaft motor | Olympus (Adachi et al., 2007) | N/A |
Highlights.
Antibody screening assay is developed using semi-automated single-molecule microscopy
Pipeline is established to identify fast-dissociating, highly specific antibodies
Fast-dissociating Fab probes are useful tools for multiplex super-resolution imaging
ACKNOWLEDGMENTS
We thank Dr. Akira Kakizuka, Mr. Kimiaki Tsukagami, and Mr. Seiji Suzuki (Kyoto University) for managing this collaboration and Drs. Katie Kindt, Wade Chien, and Yasuharu Takagi for valuable comments. We thank Mr. Makio Higuchi for technical support in screening hybridomas; Dr. Daisuke Taniguchi for python scripts, which provided the core algorithm of Gaussian fitting and drift correction; and Ms. Erina He for beautiful diagrams. BLI experiments were performed with support of Drs. Di Wu and Grzegorz Piszczek in the Biophysics Core, NHLBI, NIH. diSPIM images were acquired with support of the Advanced Imaging and Microscopy Resource, NIH. This work was supported by CREST grant number JPMJCR15G5 to N.W., JSPS KAKENHI grant number JP19H01020 to N.W., JSPS KAKENHI grant numbers 16J09300 and 18K16884 to T. Miyoshi, and NIDCD/NIH Extramural Research Program DC015495 to B.J.P. This research was also supported (in part) by the Intramural Research Programs of the NIH, NIDCD, DC000039 to T.B.F. and from NIBIB, NIH to H.S., J.C., and H.D.V.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2021.108708.
DECLARATION OF INTERESTS
The authors declare no conflict of interest associated with this manuscript.
REFERENCES
- Adachi K, Oiwa K, Nishizaka T, Furuike S, Noji H, Itoh H, Yoshida M, and Kinosita K Jr. (2007). Coupling of rotation and catalysis in F(1)-ATPase revealed by single-molecule imaging and manipulation. Cell 130, 309–321. [DOI] [PubMed] [Google Scholar]
- Alcantarilla PF, Nuevo J, and Bartoli A (2013). Fast Explicit Diffusion for Accelerated Features in Nonlinear Scale Spaces. In Proceedings of the British Machine Vision Conference 2013. https://doi.org/10.5244ZC.27.13. [Google Scholar]
- Angeletti RH (1999). Design of useful peptide antigens. J. Biomol. Tech 10, 2–10. [PMC free article] [PubMed] [Google Scholar]
- Bartles JR, Zheng L, Li A, Wierda A, and Chen B (1998). Small espin: a third actin-bundling protein and potential forked protein ortholog in brush border microvilli. J. Cell Biol 143, 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyantseva IA (2016). Helios® gene gun-mediated transfection of the inner ear sensory epithelium: recent updates In Auditory and Vestibular Research (Springer; ), pp. 3–26. [DOI] [PubMed] [Google Scholar]
- Belyantseva IA, Boger ET, and Friedman TB (2003). Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc. Natl. Acad. Sci. USA 100, 13958–13963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belyantseva IA, Perrin BJ, Sonnemann KJ, Zhu M, Stepanyan R, McGee J, Frolenkov GI, Walsh EJ, Friderici KH, Friedman TB, and Ervasti JM (2009). Gamma-actin is required for cytoskeletal maintenance but not development. Proc. Natl. Acad. Sci. USA 106, 9703–9708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizzard B (2008). Epitope tagging. Biotechniques 44, 693–695. [DOI] [PubMed] [Google Scholar]
- Bronner V, Tabul M, and Bravman T (2009). Rapid Screening and Selection of Optimal Antibody Capturing Agents Using the ProteOn™ XPR36 Protein Interaction Array System (Bio-Rad Laboratories, Inc.). [Google Scholar]
- Canziani GA, Klakamp S, and Myszka DG (2004). Kinetic screening of antibodies from crude hybridoma samples using Biacore. Anal. Biochem 325, 301–307. [DOI] [PubMed] [Google Scholar]
- Chen B, Li A, Wang D, Wang M, Zheng L, and Bartles JR (1999). Espin contains an additional actin-binding site in its N terminus and is a major actin-bundling protein of the Sertoli cell-spermatid ectoplasmic specialization junctional plaque. Mol. Biol. Cell 10, 4327–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai A, Verma S, Mitchison TJ, and Walczak CE (1999). Kin I kinesins are microtubule-destabilizing enzymes. Cell 96, 69–78. [DOI] [PubMed] [Google Scholar]
- Drummond MC, Barzik M, Bird JE, Zhang DS, Lechene CP, Corey DP, Cunningham LL, and Friedman TB (2015). Live-cell imaging of actin dynamics reveals mechanisms of stereocilia length regulation in the inner ear. Nat. Commun 6, 6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, and Stuurman N (2014). Advanced methods of microscope control using μManager software. J. Biol. Methods 1, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer MJE, and Mol NJ (2010). Surface plasmon resonance: methods and protocols (Springer Protocols; ). [Google Scholar]
- Fujii Y, Kaneko M, Neyazaki M, Nogi T, Kato Y, and Takagi J (2014). PA tag: a versatile protein tagging system using a super high affinity antibody against a dodecapeptide derived from human podoplanin. Protein Expr. Purif 95, 240–247. [DOI] [PubMed] [Google Scholar]
- Fukunaga A, Maeta S, Reema B, Nakakido M, and Tsumoto K (2018). Improvement of antibody affinity by introduction of basic amino acid residues into the framework region. Biochem. Biophys. Rep 15, 81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadzhieva M, Pashov AD, Kaveri S, Lacroix-Desmazes S, Mouquet H, and Dimitrov JD (2017). Impact of Antigen Density on the Binding Mechanism of IgG Antibodies. Sci. Rep 7, 3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Takanaka Y, Yamagata K, Wakayama T, Stasevich TJ, Kainuma T, Tsurimoto T, Tachibana M, Shinkai Y, Kurumizaka H, Nozaki N, and Kimura H (2011). Tracking epigenetic histone modifications in single cells using Fab-based live endogenous modification labeling. Nucleic Acids Res. 39, 6475–6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmerhorst E, Chandler DJ, Nussio M, and Mamotte CD (2012). Real-time and Label-free Bio-sensing of Molecular Interactions by Surface Plasmon Resonance: A Laboratory Medicine Perspective. Clin. Biochem. Rev 33, 161–173. [PMC free article] [PubMed] [Google Scholar]
- Hermanson GT (2013). Bioconjugate techniques (Academic Press; ). [Google Scholar]
- Hugo N, Weidenhaupt M, Beukes M, Xu B, Janson JC, Vernet T, and Altschuh D (2003). VL position 34 is a key determinant for the engineering of stable antibodies with fast dissociation rates. Protein Eng. 16, 381–386. [DOI] [PubMed] [Google Scholar]
- Huh JH, White AJ, Brych SR, Franey H, and Matsumura M (2013). The identification of free cysteine residues within antibodies and a potential role for free cysteine residues in covalent aggregation because of agitation stress. J. Pharm. Sci 102, 1701–1711. [DOI] [PubMed] [Google Scholar]
- Janeway CJ, Travers P, Walport M, and Shlomchik MJ (2001). Immunobiology, 5th edition The Immune System in Health and Disease (Garland Publishing; ). [Google Scholar]
- Jungmann R, Avendaño MS, Woehrstein JB, Dai M, Shih WM, and Yin P (2014). Multiplexed 3D cellular super-resolution imaging with DNA-PAINT and Exchange-PAINT. Nat. Methods 11, 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat V, Rafique A, Huang T, Olsen O, and Olson W (2020). The impact of different human IgG capture molecules on the kinetics analysis of antibody-antigen interaction. Anal. Biochem 593, 113580. [DOI] [PubMed] [Google Scholar]
- Karlsson R, Michaelsson A, and Mattsson L (1991). Kinetic analysis of monoclonal antibody-antigen interactions with a new biosensor based analytical system. J. Immunol. Methods 145, 229–240. [DOI] [PubMed] [Google Scholar]
- Kimple ME, Brill AL, and Pasker RL (2013). Overview of affinity tags for protein purification. Curr. Protoc. Protein Sci 73, 9.9.1–9.9.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiuchi T, Higuchi M, Takamura A, Maruoka M, and Watanabe N (2015). Multitarget super-resolution microscopy with high-density labeling by exchangeable probes. Nat. Methods 12, 743–746. [DOI] [PubMed] [Google Scholar]
- Kuglin CD, and Hines DC (1975). The phase correlation image alignment method. Proc. Int. Conf. Cybernet. Soc 1975, 163–165. [Google Scholar]
- Moreau V, Fleury C, Piquer D, Nguyen C, Novali N, Villard S, Laune D, Granier C, and Molina F (2008). PEPOP: computational design of immunogenic peptides. BMC Bioinformatics 9, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JB, Roughley SD, Matassova N, and Brough PA (2014). Off-rate screening (ORS) by surface plasmon resonance. An efficient method to kinetically sample hit to lead chemical space from unpurified reaction products. J. Med. Chem 57, 2845–2850. [DOI] [PubMed] [Google Scholar]
- Myszka DG (1999). Improving biosensor analysis. J. Mol. Recognit 12, 279–284. [DOI] [PubMed] [Google Scholar]
- Narayanan P, Chatterton P, Ikeda A, Ikeda S, Corey DP, Ervasti JM, and Perrin BJ (2015). Length regulation of mechanosensitive stereocilia depends on very slow actin dynamics and filament-severing proteins. Nat. Commun 6, 6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naz S, Griffith AJ, Riazuddin S, Hampton LL, Battey JF Jr., Khan SN, Riazuddin S, Wilcox ER, and Friedman TB (2004). Mutations of ESPN cause autosomal recessive deafness and vestibular dysfunction. J. Med. Genet 41, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlova MA, Chubar TA, Fechina VA, Ignatenko OV, Badun GA, Ksenofontov AL, Uporov IV, and Gazaryan IG (2003). Conformational differences between native and recombinant horseradish peroxidase revealed by tritium planigraphy. Biochemistry (Mosc.) 68, 1225–1230. [DOI] [PubMed] [Google Scholar]
- Paek SH, Cho IH, Seo SM, Kim DH, and Paek SH (2011). Production of rapidly reversible antibody and its performance characterization as binder for continuous glucose monitoring. Analyst (Lond.) 136, 4268–4276. [DOI] [PubMed] [Google Scholar]
- Portolano N, Watson PJ, Fairall L, Millard CJ, Milano CP, Song Y, Cowley SM, and Schwabe JW (2014). Recombinant protein expression for structural biology in HEK 293F suspension cells: a novel and accessible approach. J. Vis. Exp 16, e51897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, et al. (2008). Lifeact: a versatile marker to visualize F-actin. Nat. Methods 5, 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sado Y, and Okigaki T (1996). A novel method for production of monoclonal antibodies. Evaluation and expectation of the rat lymph node method in cell and molecular biology. Cell Biol. Int 20, 7–14. [DOI] [PubMed] [Google Scholar]
- Sado Y, Inoue S, Tomono Y, and Omori H (2006). Lymphocytes from enlarged iliac lymph nodes as fusion partners for the production of monoclonal antibodies after a single tail base immunization attempt. Acta Histochem. Cytochem 39, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Säfsten P, Klakamp SL, Drake AW, Karlsson R, and Myszka DG (2006). Screening antibody-antigen interactions in parallel using Biacore A100. Anal. Biochem 353, 181–190. [DOI] [PubMed] [Google Scholar]
- Schmitt L, Ludwig M, Gaub HE, and Tampé R (2000). A metal-chelating microscopy tip as a new toolbox for single-molecule experiments by atomic force microscopy. Biophys. J 78, 3275–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schueder F, Strauss MT, Hoerl D, Schnitzbauer J, Schlichthaerle T, Strauss S, Yin P, Harz H, Leonhardt H, and Jungmann R (2017). Universal Super-Resolution Multiplexing by DNA Exchange. Angew. Chem. Int. Ed. Engl 56, 4052–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skwarczynski M, and Toth I (2016). Peptide-based synthetic vaccines. Chem. Sci. (Camb.) 7, 842–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song HN, Kim DH, Park SG, Lee MK, Paek SH, and Woo EJ (2015). Purification and characterization of Fab fragments with rapid reaction kinetics against myoglobin. Biosci. Biotechnol. Biochem 79, 718–724. [DOI] [PubMed] [Google Scholar]
- Southern JA, Young DF, Heaney F, Baumgärtner WK, and Randall RE (1991). Identification of an epitope on the P and V proteins of simian virus 5 that distinguishes between two isolates with different biological characteristics. J. Gen. Virol 72, 1551–1557. [DOI] [PubMed] [Google Scholar]
- Szabó Á, Szendi-Szatmári T, Ujlaky-Nagy L, Rádi I, Vereb G, Szöllősi J, and Nagy P (2018). The Effect of Fluorophore Conjugation on Antibody Affinity and the Photophysical Properties of Dyes. Biophys. J 114, 688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thavarajah R, Mudimbaimannar VK, Elizabeth J, Rao UK, and Ranganathan K (2012). Chemical and physical basics of routine formaldehyde fixation. J. Oral Maxillofac. Pathol 16, 400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Merwe PA, and Barclay AN (1996). Analysis of cell-adhesion molecule interactions using surface plasmon resonance. Curr. Opin. Immunol 8, 257–261. [DOI] [PubMed] [Google Scholar]
- Watanabe N, and Mitchison TJ (2002). Single-molecule speckle analysis of actin filament turnover in lamellipodia. Science 295, 1083–1086. [DOI] [PubMed] [Google Scholar]
- Wingfield PT (2015). Overview of the purification of recombinant proteins. Curr. Protoc. Protein Sci 80, 6.1.1–6.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisdom GB (1994). Peptide antigens: a practical approach (I.R.L. Press; ). [Google Scholar]
- Wu Y, Wawrzusin P, Senseney J, Fischer RS, Christensen R, Santella A, York AG, Winter PW, Waterman CM, Bao Z, et al. (2013). Spatially isotropic four-dimensional imaging with dual-view plane illumination microscopy. Nat. Biotechnol 31, 1032–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Wagner T, Yan L, Grillet N, and Müller U (2014). Using injectoporation to deliver genes to mechanosensory hair cells. Nat. Protoc 9, 2438–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylera F, Harth S, Waldherr D, Frisch C, and Knappik A (2013). Off-rate screening for selection of high-affinity anti-drug antibodies. Anal. Biochem 441, 208–213. [DOI] [PubMed] [Google Scholar]
- Yokoyama WM (2008). Production of monoclonal antibody supernatant and ascites fluid. Curr. Protoc. Mol. Biol Chapter 11, Unit 11.10. [DOI] [PubMed] [Google Scholar]
- Zhang DS, Piazza V, Perrin BJ, Rzadzinska AK, Poczatek JC, Wang M, Prosser HM, Ervasti JM, Corey DP, and Lechene CP (2012). Multiisotope imaging mass spectrometry reveals slow protein turnover in hair-cell stereocilia. Nature 481, 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Sekerková G, Vranich K, Tilney LG, Mugnaini E, and Bartles JR (2000). The deaf jerker mouse has a mutation in the gene encoding the espin actin-bundling proteins of hair cell stereocilia and lacks espins. Cell 102, 377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Python scripts used in this study are available from a GitHub repository (https://github.com/takushim/tanitracer).