

Sequencing-based technology reveals popping characteristics in maize, establishing a benchmark for popcorn genetics, and supporting a gradual selection process for popping traits.
Keywords: EigenGWAS, GWAS, maize adaptation, popping traits, quality traits, tropical maize landrace
Abstract
Popcorn (Zea mays L. var. Everta) is the most ancient type of cultivated maize. However, there is little known about the genetics of popping-related traits based on genotyping-by-sequencing (GBS) technology. Here, we characterized the phenotypic variation for seven popping-related traits in maize kernels among 526 CIMMYT inbred lines (CMLs). In total, 155 083 high-quality single nucleotide polymorphism (SNP) markers were identified by a GBS approach. Several trait-associated loci were detected by genome-wide association study for color, popping expansion volume, shape, pericarp, flotation index, floury/vitreous, and protein content, explaining a majority of the observed phenotypic variance, and these were validated by a diverse panel comprising 764 tropical landrace accessions. Sixty two of the identified loci were recognized to have undergone selection. On average, there was a 55.27% frequency for alleles that promote popping in CMLs. Our work not only pinpoints previously unknown loci for popping-related traits, but also reveals that many of these loci have undergone selection. Beyond establishing a new benchmark for the genetics of popcorn, our study provides a foundation for gene discovery and breeding. It also presents evidence to investigate the role of a gradual loss of popping ability as a by-product of diversification of culinary uses throughout the evolution of teosinte–to–modern maize.
Introduction
Maize originated in Mexico about 9000 years ago (Matsuoka et al., 2002; Piperno et al., 2009). Maize landraces named “ancient indigenous” include Chapalote, Palomero Toluqueño, Arrocillo and Nal-Tel, all originating from Mexico, directly from Teosinte (Zea species), and are known for their ability to pop (Wellhausen et al., 1952). Since the earliest wild and cultivated forms of maize are all popcorn types, popcorn (Zea mays L. var. Everta) from Mexico is considered to be the most ancient maize type, and its genome sequence should contain the most comprehensive sample of genomic variation of maize (Vielle-Calzada et al., 2009). In 2009, Mexican scientists sequenced the popcorn landrace called Palomero Toluqueño (popcorn from the Toluca area, west of Mexico City, in the state of Mexico) (Vielle-Calzada et al., 2009), but these data could not be used to study popcorn genome characteristics and structural variation, due to the short contigs generated by the sequencing technology available at that time. Therefore, to our knowledge, there has been no genomic characterization of popcorn until now.
The Maize HapMap (version 2) shows that genomic structural variation is enriched near the regions associated with traits (Suo et al., 2012; Liu et al., 2016a). Thus, the discovery of quantitative trait loci (QTL) by genome-wide association analysis is not only beneficial for the analysis of the genetic structure of complex traits, but also for the in-depth study of genomic structural variation (Huang et al., 2010). Still, many questions about the process of maize domestication remain unclear because modern maize specimens do not represent the full range of past diversity. This is due to the abandonment of unproductive landraces, genetic drift, on-going natural selection, and recent breeding activities. The complex evolutionary history of maize (Zea mays L. ssp. mays;Matsuoka et al., 2002; Chia et al., 2012; Kistler et al., 2018) still needs to be clarified by studies of genetic diversity in materials that bridge the gap between teosinte and modern maize. This could include materials such as archaeological cob remains from different stages in the domestication process (Ramos-Madrigal et al., 2016), as well as the most ancient types of maize (Vielle-Calzada et al., 2009).
Popcorn plays a key role in the history and spread of maize, so it is critical to understand the genetic basis of an ancestral trait such as popping, which is shared with the crop’s closest common ancestor. However this trait was subsequently lost, as other kernel types were selected in Mexico during domestication, primarily for diverse culinary uses.
Currently, there are several methods for phenotyping popping-related traits in popcorn, as well as other races of maize with similar properties. Popping volume, defined as the popped volume per 100 kernels, is the most important popping-related trait, which has been measured in all popping-related publications (Robbins and Ashman, 1984; Dofing et al., 1990, 1991; Daros et al., 2002; Lu et al., 2003; Babu et al., 2006; Li et al., 2007a, 2008, 2009). In addition, flake size, popping rate, percentage of unpopped expansion (defined as the flake volume per popped kernel), the ratio of popped versus unpopped kernels, and the number of unpopped kernels per 200 kernels after popping, were evaluated in a few studies (Babu et al., 2006; Li et al., 2007a, 2008, 2009). Even though there has been considerable work on popping characteristics, the different measurement conditions and standards have led to conflicting results and only served to increase the difficulty of further research. Some of the methods employed a specific amount of vegetable or animal oil to facilitate the cooking of the popcorn kernels in containers. Other methods utilized a microwave oven, simulating the way popcorn is cooked in small grocery stores, fast food restaurants, homes, etc. For example, different sample quantities of kernels (weights and numbers of kernels) were chosen to measure popping volume, including 40 g (Daros et al., 2002), 75 g (Robbins and Ashman, 1984; Lu et al., 2003), 100 kernels (Li et al., 2007a, 2008, 2009), 150 g (Dofing et al., 1991), and 200 kernels (Babu et al., 2006). Different popping machines were also used, combined with different treatments: (i) Cretors 1100-W popper (Cretors Co., Chicago, IL) using 50 g of partially hydrogenated soybean oil (Dofing et al., 1990, 1991) or 25 ml peanut oil (Robbins and Ashman, 1984; Lu et al., 2003) to pop each sample; (ii) BZ-99 popping machine (Shanghai Duoli Food machine building company, Shanghai, China; Li et al., 2007a, 2008, 2009); and (iii) microwave ovens with a variety of different powers, a 700 W Litton (Dofing et al., 1990) and 900 W power (Daros et al., 2002; Babu et al., 2006). A comprehensive dissection of popping characteristics would require that a standard and widely accepted popping measurement protocol be developed.
The genetic variation of popcorn has been studied using bi-parental populations and simple sequence repeat (SSR) markers (Babu et al., 2006; Li et al., 2007a, 2007b, 2008, 2009; see Supplementary Table S1 at JXB online). In these studies, both the restricted diversity of the parents and low marker density limited a comprehensive dissection of popcorn genetic variation. Current sequencing technologies are both high throughput and highly accurate, thus providing an opportunity to more thoroughly dissect the popcorn genome, and reveal the genomic variation that underlies the evolution and domestication of maize. Here we apply conventional genotyping-by-sequencing (cGBS), which is a technically simple, highly multiplexed approach (Elshire et al., 2011) that has been applied to many crops (Poland et al., 2012; Lu et al., 2013; Morris et al., 2013; Schmutz et al., 2014), and used to study the genetic architectures of many phenotypic traits in maize (Riedelsheimer et al., 2012; Romay et al., 2013).
A number of studies have focused on popping volume and the genetic relationship between this popping characteristic and yield (Dofing et al., 1991; Daros et al., 2002; Babu et al., 2006; Li et al., 2008). However, efforts to unravel information on the ability of quality traits to contribute towards superior popping expansion volume have been very limited. Quality traits are critical to determine the basis for popping ability in popcorn. For example, starch and protein contents have been reported to greatly influence popping ability (Brunson, 1937; Haugh et al., 1976; Robbins and Ashman, 1984; Da Silva et al., 1993; Park and Maga, 2002). Zhou et al. (2016) analyzed the relationship between quality traits and popping ability by transferring the O2 gene into two popcorn inbred lines: they found that the o2 gene reduced the crude protein content and the expansion volume. Li et al. (2020) recently reported the genome sequence and annotation of a South African QPM line K0326Y, identifying a mutation of o2 that suggests a potential role in vitreous endosperm formation. Vázquez-Carrillo et al. (2019) found a positive correlation between protein and expansion volume, a negative correlation between starch or floury/vitreous ratio and expansion volume, and a negative correlation between protein content and percentage of unpopped kernels and popped kernel size. These findings on popping quality traits are purely phenotypic correlations, and the specific genetic mechanism underlying these traits, for example, the number and map locations of the genes that affect popping quality characteristics throughout the genome, as well as the interactions among them, are still not well understood, and require further study.
Therefore, in this study, to characterize the genetic basis of popping-specific traits, we performed the following: (i) conducted GWAS on seven popping-related traits in 526 CIMMYT (The International Maize and Wheat Improvement Center) inbred lines (CMLs) using 155 083 high-quality cGBS SNPs; (ii) validated the expansion-volume-related loci in a diverse set of 764 tropical landrace populations; and (iii) tested if the loci associated with popping-related traits had undergone selection. The results of this study create a new resource for popcorn genetics to dissect the origin and evolution of maize.
Materials and methods
A panel of CIMMYT inbred lines (CMLs) and the genotyping platform
A set of 529 CMLs released by CIMMYT’s Maize Breeding Program from 1960 to 2017 (http://hdl.handle.net/11529/10246) was used as an association mapping panel. Field experiments were conducted at Mexico, including those at Agua Fria, El Batan, HA, Palmira, RL, and Tlaltizapan, based on the adaptation groups. Each accession was planted in a single 1 m row with 30 cm spacing between rows (Supplementary Table S2). DNA was extracted from leaf tissues for each sample using the Cetyl Trimethyl Ammonium Bromide (CTAB) method (Murray and Thompson, 1980). This panel was genotyped for single nucleotide polymorphisms (SNPs) using conventional genotyping-by-sequencing (cGBS) at the Institute for Genomic Diversity, Cornell University, Ithaca, NY, USA (http://hdl.handle.net/11529/10423). In total, there were 955 690 SNPs retained, and their physical coordinates were derived from the maize reference genome version B73 AGPv2 (https://www.maizegdb.org/genome/genome_assembly/B73%20RefGen_v2). Imputation was performed with Beagle (V4.1; Browning and Browning, 2007, 2016), using the following parameters: window=50 000, overlap=3000, iterations=5, and clusters=0.005. From this, a smaller dataset of 155 083 SNPs that met the filtering criteria of call rate (CR)≥0.4, heterozygosity≤0.1, minor allele frequency (MAF)≥0.05, and Mendelian-transmitted markers was used for GWAS; three samples (CML342, CML529, and CML539) with a missing data rate ≥90% were removed (Fig. 1). Linkage disequilibrium decay was calculated using PopLDdecay (Zhang et al., 2019) with the filtered dataset.
Fig. 1.

Basic information summarized from cGBS genotyping. Two data sets are used: unfiltered (blue) and filtered (pink). (A) SNP minor allele frequency; (B) SNP heterozygosity; (C) overall missing rate for 955 690 SNPs; (D) missing rate for each of the 529 CMLs (in numerical order), where asterisks indicate the three samples deleted from analysis due to high missing rate (i.e. CML342, CML529, and CML539).
Measurements of popping-related traits
The CMLs were planted at different locations in Mexico according to their adaptation, and the resulting seeds were harvested and deposited in the CIMMYT Germplasm Bank. For popping evaluations, the methods from two previous studies were used (Erazo-Barradas, 2009; Rangel et al., 2011). A detailed description of the procedure for popping and phenotyping popcorn seed accessions stored in CIMMYT’s maize collection is available at http://hdl.handle.net/11529/10548274. The sample quantity was 30 g of kernels. Kernels with 13% moisture content were used, to promote gelatinization of the endosperm cells during the popping process, and to ensure that the maximum efficiency was obtained in expansion (Erazo-Barradas, 2009; Rangel et al., 2011). The kernels were placed in a controlled environment of a germination chamber with the following conditions: 70% relative humidity and 21.1 °C for 10 d in order to reach the 13% moisture content (Hallauer, 2000). The samples were then popped individually using a microwave oven (110 V; output power 1000 W; consumed power 1400 W) set at 70% power for a cooking time of 2 min 45 s.
The predominant color of the popped kernels, denoted as color, was identified empirically and recorded as (1) cream or (2) white (Costich et al., 2020). The popping expansion volume (PEV) refers to the absolute volume of 30 g of popped kernels, and was measured using a graduated cylinder (Erazo-Barradas, 2009; Costich et al., 2020). The shape of the popped kernels, denoted as shape, was described with a qualitative 1–5 scale as follows: (1) mushroom or ball shape; (2) flake shape a little more extended (less rounded); (3) unilateral expansion; (4) bilateral expansion; and (5) multilateral or “butterfly”-shape (Costich et al., 2020). The degree of pericarp retention, denoted as pericarp, was measured with a quantitative 1–5 scale, as follows: (1) popped kernels retaining between 81–100% of the pericarp, (2) 61–80% pericarp retention, (3) 41–60%, (4) 21–40%, and (5) 0–20% (Costich et al., 2020) pericarp retention. Flotation index (IF) is the number of kernels that floated in a NaNO3 solution [ρ=1.250±0.001 (1.251 to 1.249) mg ml-1]. Floury/Vitreous was defined as the ratio of floury to vitreous endosperm types. Six kernels per replicate were randomly selected for scanning. The resulting images were analyzed and the areas of floury and vitreous endosperm were measured by WinSEEDLE (Regent Instruments Canada Inc.). As a result, the ratio of the area of floury versus vitreous endosperm was calculated for each kernel, and the average value was used for further analyses. The percentage of protein was evaluated using a near-infrared spectrometer (NIRs) FOSS 6500 (FOSS NIRSystems, Inc., Silver Spring, MD, USA). Spectra were collected between 400 nm and 2500 nm, registering the absorbance values log(1/R) at 4 nm intervals for each sample (Rosales et al., 2011). These four popping specific traits (color, PEV, shape, and pericarp) and three quality traits (IF, floury/vitreous, and protein content) were measured at CIMMYT Headquarters (El Batán, Mexico) in the Maize Nutrition Laboratory “Evangelina Villegas”, with two replications (Table 1).
Table 1.
Description of the seven kernel traits measured for this study.
| Trait Name | Full Name | Description | Ranges, Units of Measurement |
|---|---|---|---|
| Color | Kernel Color | The predominant color of the popped kernels. | White, Cream |
| PEV | Popping Expansion Volume | The absolute volume of 30 grams of popped kernels. | ml |
| Shape | Popcorn Shape | The shape of popped kernels. | Scale of 1–5 |
| Pericarp | Pericarp Retention | The amount of pericarp remaining after popping. | Scale of 1–5 |
| IF | Flotation Index | The number of kernels that floated in a NaNO3 solution. | Scale of 1–5, 1: (0–12 kernels), 2: (13–37 kernels), 3: (38–62 kernels), 4: (63–87 kernels), 5: (88–100 kernels) |
| Floury/Vitreous | Floury/Vitreous | The ratio of floury to vitreous area in the endosperm. | Range is 0.28–4.01 |
| Protein | Protein | The percentage of protein in the kernels. | Range is 8.61–16.48% |
Estimation of genotypic values and trait heritability
For all seven traits, best linear unbiased predictions (BLUPs) were calculated by fitting the following random model using the “lme4” package in R (De Boeck et al., 2011):
where Yij is the trait observation for entry i in replication j; Entryi is the random effect of entry i; Repj is the random effect of replication j; and εij is the residual effect. The graphical display of trait correlation was done with the corrplot() function by Pearson correlation coefficient from the R package “corrplot” (Wei and Simko, 2013). Variance components, i.e. and for genotype and residual effects were estimated from analysis of variance (ANOVA) with QTL IciMapping V4.2.23 (Meng et al., 2015). Broad-sense heritability (H2) of each trait was estimated as:
where r is the number of replications (=2 in this study).
Identification of genomic loci using Genome Wide Association Study (GWAS)
To minimize false positives and increase statistical power, population structure and cryptic relationships were considered. An iterative usage of Fixed and random model Circulating Probability Unification (FarmCPU; Liu et al., 2016b) performed by Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool (MVP; https://github.com/XiaoleiLiuBio/MVP/), was used for the association analysis, where the first three principal component analysis (PCA) values (eigenvectors) were included as fixed effects in the mixed model to correct for stratification (Price et al., 2006). To determine the cutoff level for declaring the significance of loci, the P-value threshold was determined using permutation tests with reshuffling of the PEV trait 1000 times. After log10 transformation, a –log10(P-value) threshold of 6.46 was used for an experimental type I error rate of 0.05. To balance the false positives and false negatives, the whole-genome –log10(P-value) significance cutoff was finally set at 5. The total phenotypic variation explained by all of the significant SNPs was estimated by the coefficient of determination R2 from multiple linear models using the “lm” function in R (Chambers and Hastie, 1992).
Functional annotations of the target SNPs were performed using SnpEff (Cingolani et al., 2012). The maize B73 reference V2 gene annotation was downloaded (as a gff3 file) from the Maize Genetics and Genomics Database (MaizeGDB; https://www.maizegdb.org/assembly). Based on the genome annotation, SNPs were categorized as being located in coding regions (i.e. overlapping with a coding exon), splice sites (within 2 bp of a splicing junction), 5′ UTRs and 3′ UTRs, intron, upstream and downstream regions, and intergenic regions. SNPs in coding regions were further grouped into synonymous SNPs (not causing amino acid changes) or non-synonymous SNPs (causing amino acid changes, including stop gain and stop loss). Functional enrichment analysis of the annotated genes was performed using the ClueGO plug-in for Cytoscape 3.6.1 (Bindea et al., 2009).
Identification of genomic loci that have undergone selection
Identification of potential selective signals during maize domestication and improvement through genome-wide association studies of eigenvectors was implemented using the bottom-up searching strategy EigenGWAS (Chen et al., 2016), which is a single-marker regression approach based on principal component analysis. EigenGWAS identifies regions of the genome underlying population genetic differentiation in any genetic data where the underlying population structure is unknown, or where the interest is in assessing divergence along a gradient. Using 155 083 high-quality SNPs to generate a genetic relationship matrix, the top ten eigenvalues and their corresponding eigenvectors (i.e. Ev1 to Ev10) were calculated. SNP effects, nearly equivalent to fixation index (Fst) (Wright, 1951), could be estimated by regressing each SNP for a selected eigenvector. To exclude the effect of genetic drift in selection loci mapping, the p-value was adjusted by a genomic control factor (Devlin & Roeder, 1999), and consequently the corrected p-value, PGC, was used for detecting the loci under selection. The SNPs with the top 5% of PGC values (i.e. PGC<0.0266) were considered to have undergone selection.
Validation of genomic loci in a panel of tropical landrace populations
A total of 764 heterogeneous and heterozygous tropical landraces originating from 20 countries was used in validation (Supplementary Table S3; Li et al., 2019), all of which can be found in the CIMMYT Gemplasm Bank collection (http://mgb.cimmyt.org/gringlobal/search.aspx). The origins of these landraces represent diverse ecological regions including lowland tropical, sub-tropical/mid-altitude, and highland tropical subgroups, which were planted in Celaya and Tlaltizapan in 2016 (Table S3). Tunable genotyping-by-sequencing (tGBS®, Ott et al., 2017) provided higher SNP calling accuracy, especially at heterozygous sites, with less missing data than conventional genotyping-by-sequencing (cGBS) with the same number of reads per sample (Ott et al., 2017). This was achieved by two strategies: (i) two restriction enzymes were used in tGBS to digest the genomic DNA with overhangs in an opposite orientation, which ensures only double-digested fragments are amplified and sequenced; and (ii) additional adjustable selective nucleotides of primers were used in PCR amplification: this offers an additional genome reduction. The 12 selfed seeds of F1 from a single ear for each accession that was selected to be representative of the accession, were grown in the greenhouse, and the DNA from the seedlings bulked for tGBS sequencing in Data2Bio.
In total, 0.31 terabases (Tb) of sequence data from 2.5 billion quality-trimmed reads were generated via tGBS® (Ott et al., 2017). We identified 3 713 115 SNPs after alignment to the reference genome B73 AGPv3 (https://www.maizegdb.org/). Of the 3 713 115 tGBS SNPs, 65 540 were retained after filtering for MAF and heterozygosity. The 65 540 tGBS SNPs were imputed without a reference panel using Beagle (V4.1). To further capture the genetic variation across the maize genome, the 65 540 SNPs from tGBS were also imputed to 359 618 high-quality SNPs using maize HapMap V3 as a reference panel (https://www.maizegdb.org/genome/genome_assembly/B73%20RefGen_v3). Finally, the union of the two SNP sets (N=414 124 SNPs) was filtered by MAF and heterozygosity under the same criteria as aforementioned, resulting in 355 442 high-quality SNPs which were retained for validation.
To conduct tGBS, the restriction enzymes NspI and BfuCI were used to digest the genomic DNA; while the single restriction enzyme ApeKI had been used to generate the cGBS data (Elshire et al., 2011). Consequently, tGBS and cGBS identified very different sets of polymorphisms in the two datasets (CMLs versus tropical landraces). Because the CMLs were genotyped using the cGBS platform, and SNPs were called by B73 AGPv2, to validate the significant loci associated with the seven traits identified by CMLs, the physical positions of B73 AGPv2 were converted to those of B73 AGPv3 by http://ensembl.gramene.org/Oryza_sativa/Tools/AssemblyConverter?db=core. An extended haplotype homozygosity (EHH) test was conducted for the target SNPs within a 2 Mb region, identifying long and frequent haplotypes as implemented in the R package “rehh” (Gautier and Vitalis, 2012). Color, PEV, shape, and pericarp for each accession were measured at CIMMYT Headquarters (El Batán, Mexico), and were used to determine the extreme phenotypes, and to validate the QTLs identified in the CML population (Supplementary Table S3).
Results
Genotypic data analysis and population structure
The average MAF for the final selected dataset (n=355 442 SNPs) was 0.22 and the heterozygosity was 0.02. The heterozygosity of more than 98% of the SNPs was lower than 0.05 (Fig. 1A, B). The 155 083 SNPs are relatively evenly distributed across the 10 chromosomes (Supplementary Fig. S1), and average missing rate was 0.58. (Fig. 1C). In addition, the decay of linkage disequilibrium (LD) with physical distance between SNPs occurs at only 1.5 kb (decaying to r2 of 0.1) (Fig. S2). Principal component analysis revealed that there was a moderate population structure (Fig. S3): the panel included 30 tropical highland, 215 sub-tropical/mid-altitude, and 284 tropical lowland inbred lines, three samples with high missing rate were removed (Fig. 1D). Clear clustering based on adaptation was observed, while the tropical lowland group overlapped partially with the sub-tropical/mid-altitude group (Fig. S3). The tropical highland and tropical lowland populations were relatively scattered, indicating that there exists broad genetic variation within this set of 526 CMLs (Fig. S3). The passport information of 526 CMLs is shown in Table S2. Further information about these lines can be found at this site: https://data.cimmyt.org/dataset.xhtml?persistentId=hdl:11529/10246.
Phenotypic correlation in the panel of CIMMYT inbred lines
The correlations of BLUP values are presented in Fig. 2. Most traits were continuously and normally distributed, and hence presumed to exhibit quantitative inheritance (Supplementary Fig. S4). A wide range of phenotypes was observed, ranging from 50 ml to 680 ml for PEV, 0.28 to 4.01 for floury/vitreous, and 8.61–16.48% for protein (Table S4). A number of significant pairwise correlations were observed. For example, PEV was consistently, significantly, and positively correlated with pericarp (r=0.50, P<0.01) and popcorn shape (r=0.48, P<0.01), and significantly and negatively correlated with IF (r=–0.35, P<0.01), Floury/Vitreous (r=–0.39, P<0.01), and protein content (r=–0.25, P<0.01), confirming previously reported correlations between PEV and kernel quality traits (Vázquez-Carrillo et al., 2019). Therefore, harder kernels, with a higher proportion of vitreous endosperm, and less protein content, will produce a greater expansion volume (PEV), more pericarp retention and a more “butterfly”-like shape. In addition, the broad-sense heritability (H2) of the seven popping-related traits ranged from 0.63 for floury/vitreous to 1 for color (Fig. 2), since the two replicates for color measurements were exactly the same.
Fig. 2.

The pairwise correlations (Pearson’s r) among the seven traits based on the BLUP values. The value marked under each trait name in the diagonals was the heritability in broad sense for the corresponding trait. Correlations are represented as colored circles, blue indicating negative correlation, and red indicating positive correlation.
Genomic loci affecting popping-related traits in CIMMYT inbred lines
Each of the inbred lines was genotyped via cGBS, and SNPs were called using standard methodologies (see Methods). Subsequently, a GWAS was performed independently for each of the seven examined traits (i.e. color, PEV, shape, pericarp, IF, floury/vitreous, and protein content) using FarmCPU based on 155 083 SNPs (Supplementary Fig. S5). There was a significant peak on chromosome 6 for color with P value of 2.25E-38 (Table 2, Table S5, Fig. S5). The top SNP, S6_82019628, is a synonymous variant in PSY1 (GRMZM2G300348), encoding a phytoene synthase. PSY1 has been previously reported to reduce the carotenoid pigment content in maize endosperm, owing to a 378 bp InDel upstream of its transcription start site, and an SNP in its fifth exon that results in a Thr-to-Asn substitution (Fu et al., 2013). This example highlights that our genotypic and phenotypic data can be analyzed via GWAS to yield functionally informative results about maize physiology.
Table 2.
The identified SNPs associated with popping-related traits within annotated gene regions based on GWAS
| Trait | SNP | Effect | P-value | Alleles | Annotation | GeneID | Gene Name | Description | Previous study |
|---|---|---|---|---|---|---|---|---|---|
| Color | S6_44382110 | 0.061 | 1.096E-09 | T/A | synonymous_variant | GRMZM2G086294 | zp15 | Zein-beta Precursor (Zein-2)(16 kDa zein) (Zein clone 15A3) | |
| Color | S6_82019628 | 0.14 | 2.25E-38 | A/G | synonymous_variant | GRMZM2G300348 | y1 | Phytoene synthase, chloroplastic Precursor (EC 2.5.1.32) | |
| Color | S6_86338050 | 0.038 | 1.33E-06 | C/T | 5_prime_UTR_variant | GRMZM2G082874 | uwm3035 | plant-specific domain TIGR01589 family protein | |
| Floury/Vitreous | S4_210389724 | 0.069 | 2.19E-06 | G/A | intron_variant | GRMZM2G040559 | uwm10774 | tubulin alpha-6 chain | |
| PEV | S1_55451772 | -15.95 | 2.34E-06 | T/C | upstream_gene_variant | GRMZM2G056424 | uwm46912 | photosystem II 11 kD protein | qPEV1-1 |
| PEV | S1_253277924 | 23.48 | 4.40E-06 | A/T | upstream_gene_variant | GRMZM5G837732 | CBL-interacting serine/threonine-protein kinase 15 (LOC100285495), mRNA | qPV1-3 | |
| PEV | S1_253277948 | 23.48 | 4.40E-06 | C/T | upstream_gene_variant | GRMZM5G837732 | CBL-interacting serine/threonine-protein kinase 15 (LOC100285495), mRNA | qPV1-3 | |
| PEV | S2_36136426 | 25.052 | 2.88E-06 | C/T | intron_variant | GRMZM2G030598 | kelch motif family protein | ||
| PEV | S3_162805619 | 23.61 | 9.64E-06 | C/T | downstream_gene_variant | GRMZM2G133398 | vp1 | Regulatory protein viviparous-1 | |
| PEV | S3_213803780 | -20.67 | 7.27E-06 | T/C | splice_region_variant&intron_ variant | GRMZM2G047129 | alpha-L-fucosidase 2 | qBPV3-1 | |
| PEV | S5_175731057 | 29.74 | 1.14E-06 | A/G | synonymous_variant | GRMZM2G024739 | cry1 | cryptochrome 1 | |
| IF | S5_160833678 | 0.15 | 9.26E-06 | A/C | synonymous_variant | GRMZM2G077989 | RS21-C6 protein | ||
| IF | S7_106078238 | -0.26 | 9.63E-07 | A/C | missense_variant | GRMZM2G046600 | uwm17845 | catalytic/ hydrolase | |
| IF | S9_153853384 | -0.28 | 3.39E-06 | G/A | upstream_gene_variant | GRMZM2G171466 | nascent polypeptide-associated complex alpha subunit-like protein | ||
| Protein | S7_165744712 | -0.11 | 4.21E-06 | C/A | upstream_gene_variant | GRMZM2G054032 | uwm10642 | F-box protein | |
| Shape | S5_190238066 | -0.14 | 2.18E-06 | T/C | upstream_gene_variant | GRMZM2G169558 | protein kinase APK1A | ||
| Shape | S8_136138158 | 0.14 | 2.97E-06 | A/G | 3_prime_UTR_variant | GRMZM2G100229 | calmodulin binding protein |
GWAS with these polymorphisms identified 162 SNPs distributed across the genome that were significantly (-log10P>5) associated with popping-related traits with P-values ranging from 9.88E-06 to 2.25E-38 (Supplementary Table S5). Fifteen SNPs related to PEV identified from the GWAS were found to be located within previously reported QTLs for popping expansion volume in maize (Table S5). For example, S1_55451772 was located at qPEV1-1 (Li et al., 2008), S1_253277924 and S1_253277948 were in the region of qPV1-3 (Li et al., 2007a, 2009), and S3_213803780 was within qBPV3-1 (Li et al., 2009). Of the 162 significant SNPs, 15% were annotated in intergenic regions, 43% SNPs were in the upstream and downstream regions, 12% SNPs were in the intron regions, and 30% were in coding regions, splice sites, 3’UTR, and 5’ UTR regions (Table S5, Fig. 3A). In total, 118 known genes were mapped by the significant SNPs. Following Gene Ontology analysis we found that most genes are involved in metabolic and biosynthetic processes (Table S5, Fig. 3B). The functions of most of these genes (n=101) have not been defined (Table S5). Therefore, the functional annotations of the remaining 17 are displayed in Table 2. For example, ZP15, related to color, is a zein-beta precursor; UWM46912 encodes a photosystem II 11 kDa protein; regulatory protein viviparous-1 (VP1) and cryptochrome 1 (CRY1) affect PEV; and UWM10642, which encodes an F-box protein, is associated with protein content (Table 2).
Fig. 3.

Gene annotation information and gene ontology for significant SNPs identified by GWAS. (A) Gene annotation; (B) gene ontology terms.
Pleiotropic genes play important roles in understanding correlations among phenotypes (Chen and Lübberstedt, 2010), but loci simultaneously controlling multiple popping-related traits have seldom been reported (Li et al., 2008). To dissect the genetic architecture of the correlations across different traits, we analyzed the association networks among the seven examined traits. We found that floury/vitreous and pericarp shared a common significant association, SNP S7_82404867 (Supplementary Table S5). This finding was consistent with the correlation pattern of the traits, as shown in Fig. 2: a significant correlation was observed between PEV and pericarp (r=0.5, P<0.01), PEV and protein (r=–0.25, P<0.01), and floury/vitreous and pericarp (r=–0.15, P<0.01). PEV was connected to pericarp and protein content by SNPs S2_96341835 and S2_217692527. These results suggest that the popping-related traits might be genetically co-regulated.
To better understand the popping-related traits, we estimated the proportion of the phenotypic variance explained by significant SNPs, which averaged 43% across the seven traits, ranging from 28–83% (Fig. 4 and Supplementary Fig. S6). For the most important trait, PEV, 58 associated signals explained 48.84% of the phenotypic variance. For color, 26 significant loci explained 83% of the phenotypic variance. For the other five traits, associated SNPs explained less than half of the total observed phenotypic variance (Fig. S6).
Fig. 4.
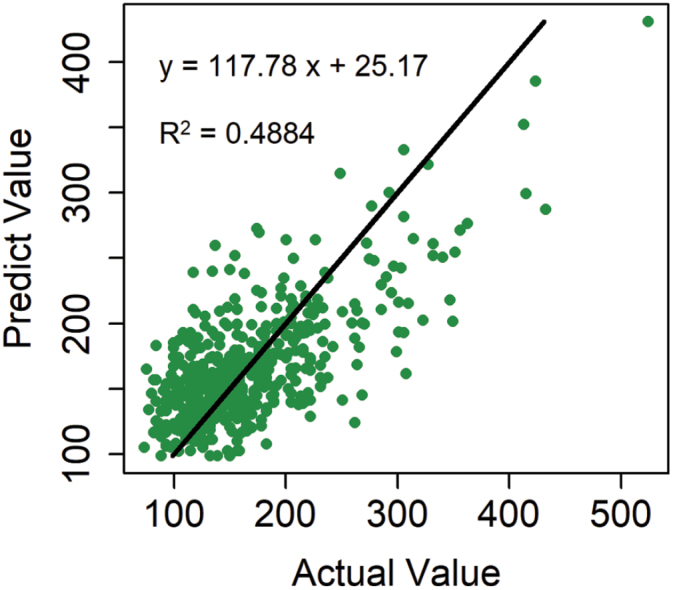
Prediction of the observed phenotype of PEV (popping expansion volume) by the significant SNPs associated with PEV. The predicted value indicates the performance of the PEV predicted by identifed QTL, and observed value indicates the phenotype measured.
Identification of popping-related loci
EigenGWAS was conducted to examine the selection loci for 155 083 SNPs based on 526 CMLs for the top ten largest eigenvectors (Fig. 5). The mean of genetic relatedness across CMLs was –0.0018, indicating that the effective sample number was 547.76 for the collection, and the effective number of genome segments was 651.34. As a result, we identified 60 677 loci (top 5%) under selection, distributed on 10 chromosomes, with the highest number of significant loci (i.e. 11 211 SNPs) on chromosome 1. Of 162 popping-related loci, 62 were identified to have undergone selection (Figs 6–8). PEV had the most significant loci with 28 under selection, with six for shape and two for pericarp (Figs 6, 7). These results indicated that most of the popping-related loci had undergone selection. Distribution of the SNPs varied considerably across different traits, with PEV having the highest numbers of SNPs (n=58), and protein content, the lowest (n=12 SNPs; Figs 6, 7). Of the 48.84% of the phenotypic variation explained by QTLs controlling PEV (Fig. 4), 20.90% could be explained by SNPs associated with PEV, exhibiting evidence of selection (Supplementary Fig. S7). For color, this proportion is two-thirds (61.44% of 83.19%), around half for shape (16.09% of 27.64%), 37% for protein (12.59% of 34.13%), a quarter for IF (10.97% of 44.37%), and less than 10% for pericarp and floury/vitreous (Fig. 4, Fig. S7). To evaluate the variation for alleles that promote popping in non-popcorn CML inbreds, we calculated the accumulated frequencies of favorable alleles that promote popping for each CML. The results showed that on average, CMLs contain 55.27% of the alleles that promote popping, with 507 lines harboring over 50% of the alleles (Fig. S8).
Fig. 5.

Miami plots from EigenGWAS. The upper panels show data for –log(PGC) and lower panels, for Fst for Ev1-Ev10, based on 526 CIMMYT inbred lines with 155 083 SNPs. Dots in blue are associated with seven popping-related traits, with their detailed information listed in Table S5.
Fig. 6.

The SNPs and gene number identified by GWAS and EigenGWAS for seven traits.
Fig. 7.

Distribution of 162 significant SNPs for seven popping-related traits. For each SNP, its physical position (Mb) and related trait name are shown on the two sides of its located chromosome. Previously reported regions that associated with PEV are indicated in blue. Loci with trait names in red and italics have undergone selection.
Fig. 8.

Genome-wide Manhattan plot and linkage disequilibrium (LD) blocks. Data are from S2_93806542 in the region from 70 to 90 Mb on chromosome 2, and S6_63434708 in the region from 80 to 100 Mb on chromosome 6. The horizontal grey dashed line indicates genome-wide significant thresholds -log10 (p) value of 5 (A), and EHH (extended haplotype homozygosity) for intervals in the length of 20 Mb with S2_93806542 and S6_63434708 in the middle when selecting 100 lowest PEV (B and D) and 100 highest PEV (C and E) accessions in 764 landrace populations.
Validation of popping-related loci in tropical landraces
To validate the popping-related loci, EHH analysis of two PEV-related loci (S2_93806542 and S6_63434708) was conducted in the set of 764 landrace populations. We narrowed down the candidate region on chromosome 2 to a 7 Mb region with 16 genes from 91 435 963 bp to 98 827 228 bp by linkage disequilibrium analysis (Fig. 8A). We evaluated the EHH values for 100 accessions with the largest PEV and 100 accessions with the lowest PEV (Supplementary Table S2) EHH tests showed that the S2_93806542-A haplotypes extended further than the reference S2_93806542-T haplotypes, depicting the long-range haplotype homozygosity across the region of S2_93806542-A haplotypes. Large differences in PEV were observed between the two tails (100 accessions each) of the 764 landraces for the S2_93806542-A haplotype and the S2_93806542-T haplotype groups (Fig. 8B, C). Similarly, there were large differences in PEV between the S6_63434708-C and S6_63434708-A haplotypes in the same 100 top and bottom ranking accessions (Fig. 8D, E). These data from a separate set of populations validate the associations observed between the SNPs and PEV in the CMLs.
Discussion
A large scale genetic study on popping-related traits by whole genome sequencing
At present, genetic studies of popping-related traits are limited to conventional SSR markers. For example, Lu et al. (2003) used 259 SSR markers and 160 selfed plants of the backcrossing families (BC1S1) derived from popcorn and dent maize parents, and detected four QTLs related to PEV on chromosomes 1, 3, and 5. Using a total of 101 SSR markers to detect QTLs in 194 F3 plants derived from the popcorn inbred A-1–6 and flint maize inbred V273, Babu et al. (2006) detected four, four, and five QTLs related to popping rate, PEV, and unpopped kernel rate, respectively. From 2007 to 2009, 10 QTLs associated with PEV, eight QTLs related to flake size, and seven QTLs associated with popping rate, were identified by 193 SSR markers using the 259 F2:3 and 220 BC2F2 derived from popcorn inbred N04 and dent corn inbred Dan232 (Li et al., 2007a, b, 2008, 2009; Supplementary Table S1). As we know, SSRs are normally non-genic markers, while many SNPs are intragenic (Shi et al., 2013). Therefore, the genomic loci identified by cGBS are more likely to represent variations in the causal genes of popping traits. As a result of our study, 162 popping-related loci were identified for seven popping-related traits by cGBS, which comprehensively incorporated most of the previously known quantitative loci for popping expansion volume (Fig. 7, Table S5). A large proportion of the phenotypic variation was explained by the identified SNPs associated with PEV (Figs 4, S6), which indicates that the additive model can accurately predict PEV. In view of this result, it is helpful to improve the accuracy of prediction and accelerate the breeding cycle by incorporating numerous small-effect QTLs in genomic selection. A synergy among popping-related traits was also uncovered, which indicates that the popping characteristic is a complex trait, determined by many factors. Due to the different sequencing platforms and versions of the reference genome employed in this study, of the 162 popping SNPs identified by the cGBS platform using B73 AGPv2 as a reference genome, only five loci could be projected to the tGBS platform using B73 AGPv3 as reference genome, and only two out of five loci could be validated in the set of 764 tropical landrace populations. The deciphered and validated genetic architecture of popping-related traits will provide a valuable resource for future molecular physiological studies and for popcorn breeding programs.
Kernel traits underwent selection during maize improvement
To further understand the history and spread of maize, Ramos-Madrigal et al. (2016) characterized the draft genome of a 5310-year-old archaeological cob found during excavations in the Tehuacan Valley of Mexico. Many genes associated with key domestication traits, such as teosinte glume architecture 1 (TGA1), zea agamous-like1 (ZAGL1), sugary1 (SU1), and waxy 1 (WX1), existed in the ancestral state, sharply contrasting with the ubiquity of derived alleles in extant landraces. Their findings suggest that much of the evolution during domestication has been gradual, and they encourage further paleogenomic research to address provocative questions about the world’s most productive cereal. The most ancient form of maize was a popcorn (Zea mays L. var. Everta), and as such it is an ideal material for researchers to access a vast archive of paleogenomic data, permitting detailed investigations of prehistoric, as well as recent, selective pressures, and ultimately achieving a new understanding of the formation of maize landraces and improved maize lines.
Recently, Li et al. (2019) showed that the domestication process of maize was at least partly driven by environmental parameters. These included soil pH at 5 cm depth, and rainfall in 1143 maize accessions from diverse ecological adaptation zones, including tropical lowland, highland, sub-tropical/mid-altitude, and temperate, covering major ecotypes of maize resources developed during domestication and breeding. Due to the lack of replicated quantitative measurements of popping characteristics for these 1143 accessions, two groups (popcorn versus non-popcorn) were identified, and these qualitative phenotypes were used to map the popping-related loci and test if they had undergone selection (Li et al., 2019). With this very limited description of phenotypic variation of popping performance, Li et al. (2019) failed to find popping-related loci under selection.
To improve the analysis, in this study a comprehensive, quantitative measurement of kernel traits was performed. As a result, we were able to identify 162 SNPs associated with the kernel traits that we measured, of which 62 (38%) exhibit evidence of having been under selection. For seven traits, more than 30% of the significant SNPs exhibit evidence of selection, except for the percentage of pericarps. Those traits (and the percentage of SNPs) are color (46%), PEV (48%), shape (40%), IF (38%), protein (33%), and floury/vitreous (31%; Fig. 6). Given the effects of environmental and climate factors on maize adaptation (Li et al., 2019), a different number of selection loci has been detected on seven traits. The reason is perhaps due to the varying selection pressures presented by the environment and climate change, and different preferences of different human groups. In total, 42–66% of the alleles for kernel traits that promote popping were found across CMLs (Supplementary Fig. S8). It suggests that selection of popping characteristics was gradual, which is consistent with the findings of Ramos-Madrigal et al. (2016) for other traits. The results shown in this study, therefore, provide a foundation for further paleogenomic and molecular biology research and pinpoint the importance of environment and climate for popcorn adaptation.
Perspectives
In this study, the 162 identified loci that we identified incorporated all of the previously known loci associated with popping-related traits. Beyond that, 147 loci were reported here for the first time. We discovered that non-popcorn inbreds (CMLs) contain variation for alleles that promote popping. Thus, our study represents a new benchmark for genetics in popcorn, and moving forward, should be helpful in dissecting both evolutionary and functional hypotheses. Specifically, it will guide efforts to determine the genetic basis of popcorn physiology, evolution, and history, assist gene discovery and breeding, and provide a more detailed understanding of the role of gradual popcorn domestication during the evolution of teosinte–to–popcorn-to-diversified modern maize. Genome sequences contain all kinds of information that control and influence biological functions. In this study, genetic variation was aligned to the B73 reference genome which has its own unique selection history. The development of the other gold-standard temperate maize genomes, Mo17 (Sun et al., 2018) and W22 (Springer et al., 2018), and especially the tropical maize genome SK (Yang et al., 2019) and quality protein maize (QPM) maize genome K0326Y (Li et al., 2020), provide valuable resources for us to increase the proportion of potentially useful genomic variation, and to characterize the phenotypic variation of popping-related traits in a more comprehensive framework in the near future.
Supplementary Data
The following supplementary data are available at JXB online.
Fig. S1. The SNP density of 155 083 SNPs on 10 chromosomes.
Fig. S2. Genome-wide average linkage disequilibrium (LD) decay over physical distance.
Fig. S3. Principal component analysis (PCA) of 155 083 SNPs based on 526 CMLs for GWAS.
Fig. S4. The phenotypic distribution of the seven traits.
Fig. S5. Manhattan plots and QQ (Quantile-Quantile) plots for six traits.
Fig. S6. Prediction of the observed performance for six traits using the significant SNPs associated with the corresponding traits.
Fig. S7. Prediction of the observed performance for seven traits using the common significant SNPs identified by GWAS and EigenGWAS.
Fig. S8. The frequency distribution of popping-related SNPs that promote popping in CMLs.
Table S1. Summary of the popping-related loci identified in previous studies.
Table S2. The information on the seven traits measured in two replications for 526 CMLs.
Table S3. The information and phenotype for the 764 maize tropical landrace populations.
Table S4. Estimates of variance components for six traits in this study.
Table S5. The significant SNPs associated with popping-related traits in 526 CMLs.
Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2016YFD0100303). Research activities carried out by CIMMYT Germplasm Bank Maize Collection staff (CZE and DEC) were supported by the CGIAR Genebank Platform funding. The CGIAR Research Program MAIZE (CRP-MAIZE) also supported this research. CRP-MAIZE receives support from the Governments of Australia, Belgium, Canada, China, France, India, Japan, Korea, Mexico, Netherlands, New Zealand, Norway, Sweden, Switzerland, United Kingdom, United States, and the World Bank. The views expressed do not necessarily reflect those of these funders.
We thank the CIMMYT Germplasm Bank’s “Popcorn Team” who, in 2014, began to reveal the global diversity in popcorn that is present in the maize collection, by carrying out a phenotypic survey of popping traits in hundreds of accessions identified in the bank database as “popcorn”. This includes Braulio Torres, J. Dennis Baldwin, and J. Alex Velazquez, who worked alongside two of the co-authors of this paper, NA and CZ.
Glossary
Abbreviations
- BLUP
Best linear unbiased prediction
- cGBS
conventional genotyping-by-sequencing
- CIMMYT
The International Maize and Wheat Improvement Center
- CML
CIMMYT Maize Line
- EHH
Extended haplotype homozygosity
- GWAS
Genome-wide association study
- IF
Flotation index
- LD
Linkage disequilibrium
- MAF
Minor allele frequency
- o2
opaque-2
- PEV
Popping expansion volume
- tGBS
tunable genotyping-by-sequencing
Author Contribution
HHL designed the experiments and managed the project; JL, HHL, and DL analyzed the data; JL, CZE, VTP, AR, NPR, JW, ASV, and NCAS performed the phenotyping; DEC directed the phenotyping of the accessions in the Maize Collection of the CIMMYT Germplasm Bank, and mentored JL during her internship in CIMMYT; JL and HHL wrote the manuscript; DEC and PSS revised the manuscript; and all authors read and approved the final version of the manuscript.
Conflict of interest
There are no conflicts of interest.
Data Availability
The data supporting the findings of this study are available from The Global Popcorn Project http://hdl.handle.net/11529/10548274.
References
- Babu R, Nair SK, Kumar A, Rao HS, Verma P, Gahalain A, Singh IS, Gupta HS. 2006. Mapping QTLs for popping ability in a popcorn x flint corn cross. Theoretical and Applied Genetics 112, 1392–1399. [DOI] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z, Galon J. 2009. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning SR, Browning BL. 2007. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. American Journal of Human Genetics 81, 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning BL, Browning SR. 2016. Genotype imputation with millions of reference samples. American Journal of Human Genetics 98, 116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunson AM 1937. Popcorn breeding. Yearbook Agricultural 1, 395–404. [Google Scholar]
- Chambers JM, Hastie TJ. 1992. Statistical models in S: Wadsworth & Brooks/Cole Advanced Books & Software Pacific Grove, CA. [Google Scholar]
- Chen GB, Lee SH, Zhu ZX, Benyamin B, Robinson MR. 2016. EigenGWAS: finding loci under selection through genome-wide association studies of eigenvectors in structured populations. Heredity 117, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Lübberstedt T. 2010. Molecular basis of trait correlations. Trends in Plant Science 15, 454–461. [DOI] [PubMed] [Google Scholar]
- Chia JM, Song C, Bradbury PJ, et al. 2012. Maize HapMap2 identifies extant variation from a genome in flux. Nature Genetics 44, 803–807. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms. SnpEff Fly. SnpEff Fly 6, 80– 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costich DE, Zavala C, Torres Morales B. 2020. “The Global Popcorn Project”, https://hdl.handle.net/11529/10548274, CIMMYT Research Data & Software Repository Network, V1; Methods_for_Phenotyping_Popping_Traits_in_Maize.pdf
- Devlin B, Roeder K. 1999. Genomic control for association studies. Biometrics 55, 9971004. [DOI] [PubMed] [Google Scholar]
- Da Silva W, Vidal B, Martins M. 1993. What makes popcorn pop? Nature 362, 417.8464472 [Google Scholar]
- Daros M, Junior ATdo A, Pereira MG. 2002. Genetic gain for grain yield and poppoing expansion in full-sib recurrent selection in popcorn. Crop Breeding and Applied Biotechnology 2, 339-344. [Google Scholar]
- De Boeck P, Bakker M, Zwitser R, Nivard M, Hofman A, Tuerlinckx F, Partchev I. 2011. The estimation of item response models with the lmer function from the lme4 package in R. Journal of Statistical Software 39, 1–28. [Google Scholar]
- Dofing SM, Thomas-Compton MA, Buck JS. 1990. Genome x popping method interaction for expansion volume in popcorn. Crop Science 30, 62-65. [Google Scholar]
- Dofing SM, D’Croz-Mason N, Thomas-Compton MA. 1991. Inheritance of expansion volume and yield in two popcorn x dent corn varieties. Crop Science 31, 715-718. [Google Scholar]
- Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE. 2011. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6, e19379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erazo-Barradas M 2009. Evaluation of popping expansion traits in a maize (Zea mays L.) population. [Mauricio Erazo-barradas Graduate Theses and Dissertations]. Iowa State University, USA. [Google Scholar]
- Fu Z, Chai Y, Zhou Y, Yang X, Warburton ML, Xu S, Cai Y, Zhang D, Li J, Yan J. 2013. Natural variation in the sequence of PSY1 and frequency of favorable polymorphisms among tropical and temperate maize germplasm. Theoretical and Applied Genetics 126, 923–935. [DOI] [PubMed] [Google Scholar]
- Gautier M, Vitalis R. 2012. rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 28, 1176–1177. [DOI] [PubMed] [Google Scholar]
- Hallauer AR 2000. Specialty Corns. CRC Press. USA. [Google Scholar]
- Haugh C, Lien R, Hanes R, Ashman R. 1976. Physical properties of popcorn. Transactions of the ASAE 19, 168–0171. [Google Scholar]
- Huang X, Wei X, Sang T, et al. 2010. Genome-wide association studies of 14 agronomic traits in rice landraces. Nature Genetics 42, 961–967. [DOI] [PubMed] [Google Scholar]
- Kistler L, Maezumi SY, Gregorio de Souza J, et al. 2018. Multiproxy evidence highlights a complex evolutionary legacy of maize in South America. Science 362, 1309–1313. [DOI] [PubMed] [Google Scholar]
- Li CS, Xiang XL, Huang YC, et al. 2020. Long-read sequencing reveals genomic structural variations that underlie creation of quality protein maize. Nature Communications, 11: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Chen GB, Rasheed A, et al. 2019. Identifying loci with breeding potential across temperate and tropical adaptation via EigenGWAS and EnvGWAS. Molecular Ecology 28, 3544–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YL, Dong YB, Cui DQ, Wang YZ, Liu YY, Wei MG, Li XH. 2008. The genetic relationship between popping expansion volume and two yield components in popcorn using unconditional and conditional QTL analysis. Euphytica 162, 345–351. [Google Scholar]
- Li YL, Dong YB, Niu SZ, Cui DQ. 2007a. QTL for popping characteristics in popcorn. Plant Breeding 126, 509–514. [Google Scholar]
- Li YL, Dong YB, Niu SZ, Cui DQ. 2009. Identification of QTL for popping characteristics using a BC2F2 population and comparison with its F2:3 population in popcorn. Agricultural Sciences in China 8, 137–143. [Google Scholar]
- Li YL, Niu SZ, Dong YB, Cui DQ, Wang YZ, Liu YY, Wei MG. 2007b. Identification of trait-improving quantitative trait loci for grain yield components from a dent corn inbred line in an advanced backcross BC2F2 population and comparison with its F2:3 population in popcorn. Theoretical and Applied Genetics 115, 129–140. [DOI] [PubMed] [Google Scholar]
- Liu H, Wang X, Wei B, et al. 2016a. Characterization of genome-wide variation in four-row wax, a waxy maize landrace with a reduced kernel row phenotype. Frontiers in Plant Science 7, 667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Huang M, Fan B, Buckler ES, Zhang Z. 2016b. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genetics 12, e1005767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HJ, Bernardo R, Ohm HW. 2003. Mapping QTL for popping expansion volume in popcorn with simple sequence repeat markers. Theoretical and Applied Genetics 106, 423–427. [DOI] [PubMed] [Google Scholar]
- Lu F, Lipka AE, Glaubitz J, Elshire R, Cherney JH, Casler MD, Buckler ES, Costich DE. 2013. Switchgrass genomic diversity, ploidy, and evolution: novel insights from a network-based SNP discovery protocol. PLoS Genetics 9, e1003215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y, Vigouroux Y, Goodman MM, Sanchez G J, Buckler E, Doebley J. 2002. A single domestication for maize shown by multilocus microsatellite genotyping. Proceedings of the National Academy of Sciences, USA 99, 6080–6084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L, Li H, Zhang L, Wang J. 2015. QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. The Crop Journal 3, 269–283. [Google Scholar]
- Morris GP, Ramu P, Deshpande SP, et al. 2013. Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proceedings of the National Academy of Sciences, USA 110, 453–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray MG, Thompson WF. 1980. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Research 8, 4321–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott A, Liu SZ, Schnable JC, Yeh CT, Wang KS, Schnable PS. 2017. tGBS® genotyping-by-sequencing enables reliable genotyping of heterozygous loci. Nucleic Acids Research 45(21): e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Maga JA. 2002. Effects of storage temperature and kernel physical condition on popping qualities of popcorn hybrids. Cereal Chemistry 79, 572. [Google Scholar]
- Piperno DR, Ranere AJ, Holst I, Iriarte J, Dickau R. 2009. Starch grain and phytolith evidence for early ninth millennium B.P. maize from the Central Balsas River Valley, Mexico. Proceedings of the National Academy of Sciences, USA 106, 5019–5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland JA, Brown PJ, Sorrells ME, Jannink JL. 2012. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One 7, e32253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics 38, 904–909. [DOI] [PubMed] [Google Scholar]
- Ramos-Madrigal J, Smith BD, Moreno-Mayar JV, Gopalakrishnan S, Ross-Ibarra J, Gilbert MTP, Wales N. 2016. Genome sequence of a 5310-year-old maize cob provides insights into the early stages of maize domestication. Current Biology 26, 3195–3201. [DOI] [PubMed] [Google Scholar]
- Rangel RM, do Amaral Júnior AT, Júnior SdPF. 2011. Associação entre características agronômicas e capacidade de expansão em população de milho pipoca sob seleção recorrente (Association between agronomical traits and popping expansion in a popcorn population under recurrent selection). Ciência e Agrotecnologia 35, 225–233. [Google Scholar]
- Riedelsheimer C, Czedik-Eysenberg A, Grieder C, Lisec J, Technow F, Sulpice R, Altmann T, Stitt M, Willmitzer L, Melchinger AE. 2012. Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nature Genetics 44, 217–220. [DOI] [PubMed] [Google Scholar]
- Robbins W, Ashman R. 1984. Parent-offspring popping expansion correlations in progeny of dent corn× popcorn and flint corn× popcorn crosses. Crop Science 24, 119–121. [Google Scholar]
- Romay MC, Millard MJ, Glaubitz JC, et al. 2013. Comprehensive genotyping of the USA national maize inbred seed bank. Genome Biology 14, R55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales A, Galicia L, Oviedo E, Islas C, Palacios-Rojas N. 2011. Near-infrared reflectance spectroscopy (NIRS) for protein, tryptophan, and lysine evaluation in quality protein maize (QPM) breeding programs. Journal of Agricultural and Food Chemistry 59, 10781–10786. [DOI] [PubMed] [Google Scholar]
- Schmutz J, McClean PE, Mamidi S, et al. 2014. A reference genome for common bean and genome-wide analysis of dual domestications. Nature Genetics 46, 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Huang S, Fu D, Yu J, Wang X, Hua W, Liu SY, Liu GH, Wang, HZ. 2013. Evolutionary dynamics of microsatellite distribution in plants: insight from the comparison of sequenced brassica, Arabidopsis and other angiosperm species. PLoS One, 8, e59988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer NM, Anderson SN, Andorf CM, Ahern KR, Bai F, Barad O, Barbazuk WB, Bass HW, Baruch K, Ben-Zvi G. 2018. The maize W22 genome provides a foundation for functional genomics and transposon biology. Nature Genetics 50, 1282. [DOI] [PubMed] [Google Scholar]
- Sun S, Zhou Y, Chen J, et al. 2018. Extensive intraspecific gene order and gene structural variations between Mo17 and other maize genomes. Nature Genetics 50, 1289–1295. [DOI] [PubMed] [Google Scholar]
- Suo C, Xu H, Khor CC, et al. 2012. Natural positive selection and north-south genetic diversity in East Asia. European Journal of Human Genetics 20, 102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez-Carrillo MG, Santiago-Ramos D, Figueroa-Cárdenas JD. 2019. Kernel properties and popping potential of Chapalote, a Mexican ancient native maize. Journal of Cereal Science, 86, 69–79. [Google Scholar]
- Vielle-Calzada JP, de la Vega OM, Hernández-Guzmán G, Ibarra-Laclette E, Alvarez-Mejía C, Vega-Arreguín JC, Jiménez-Moraila B, Fernández-Cortés A, Corona-Armenta G, Herrera-Estrella L. 2009. The Palomero genome suggests metal effects on domestication. Science 326, 1078-1078. [DOI] [PubMed] [Google Scholar]
- Wei T, Simko V. 2013. Corrplot: visualization of a correlation matrix. R package version 0.73230, 11. [Google Scholar]
- Wellhausen EJ, Roberts LM, Hernandez X, Mangelsdorf PC. 1952. Races of maize in Mexico. Their origin, characteristics and distribution 19, Bussey Institution of Harvard University, USA. [Google Scholar]
- Wright S 1951. The genetical structure of populations. Annals of Eugenics 15, 323354. [DOI] [PubMed] [Google Scholar]
- Yang N, Liu J, Gao Q, et al. 2019. Genome assembly of a tropical maize inbred line provides insights into structural variation and crop improvement. Nature Genetics 51, 1052–1059. [DOI] [PubMed] [Google Scholar]
- Zhang C, Dong SS, Xu JY, He WM, Yang TL. 2019. PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Shi QL, Dong YB, Li YW, Deng F, Ma ZY, Qiao DH, Li YL. 2016. Effect of opaque-2 gene on grain, quality and popping characteristics of popcorn. Journal of Henan Agricultural Sciences, 45:24–28 (in Chinese). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available from The Global Popcorn Project http://hdl.handle.net/11529/10548274.