

Abstract
O2- and O4-alkylated thymidine lesions are known to be poorly repaired and persist in mammalian tissues. To understand how mammalian cells sense the presence and regulate the repair of these lesions, we employed a quantitative proteomic method to discover regioisomeric O2- and O4-n-butylthymidine (O2- and O4-nBudT)-binding proteins. We were able to identify 21 and 74 candidate DNA damage recognition proteins for O2-nBudT- and O4-nBudT-bearing DNA probes, respectively. Among these proteins, DDB1 and DDB2 selectively bind to O2-nBudT-containing DNA, whereas three high-mobility group (HMG) proteins (i.e., HMGB1, HMGB2, and mitochondrial transcription factor A (TFAM)) exhibit preferential binding to O4-nBudT-bearing DNA. We further demonstrated that TFAM binds directly and selectively with O4-alkyldT-harboring DNA, and the binding capacity depends mainly on the HMG box-A domain of TFAM. We also found that TFAM promotes transcriptional mutagenesis of O4-nBudT and O4-pyridyloxobutylthymidine, which is a DNA adduct induced by tobacco-specific N-nitrosamines, in vitro and in human cells. Together, we explored, for the first time, the interaction proteomes of O-alkyldT lesions, and our study expanded the functions of TFAM by revealing its capability in the recognition of O4-alkyldT-bearing DNA and uncovering its modulation of transcriptional mutagenesis of these lesions in human cells.
Graphical Abstract
INTRODUCTION
Alkylated DNA lesions constitute an important class of DNA damage products arising from exposure to environmental toxicants and/or endogenous metabolites.1,2 Alkylating agents are present in the environment (e.g., in cigarette smoke and fuel combustion products), widely used in cancer chemo-therapy, and generated internally as byproducts of oxidative stress (e.g., lipid peroxidation).1,3 Alkylating agents can react with all nitrogen and oxygen atoms on nucleobases as well as backbone phosphate to generate a variety of DNA adducts.1,2 These DNA adducts, if not repaired, can block DNA replication and transcription and induce mutations in DNA and RNA, which may lead to cancer and other human diseases.1,4 For example, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and N′-nitrosonornicotine (NNN), which constitute two major tobacco-specific N-nitrosamines, are known carcinogens and their metabolites react preferentially with thymine and guanine to yield alkylated DNA adducts.5
Thymidine can be alkylated at the O2, O4, and N3 positions to yield regioisomeric O2-alkyldT, O4-alkyldT, and N3-alkyldT lesions.2 It was reported that cigarette smoking can elicit higher levels of ethylated DNA lesions in humans, where the levels of ethylated dT adducts (O2-EtdT, O4-EtdT, and N3-EtdT) in blood samples of smokers are much higher than those of nonsmokers.6 In addition, previous studies showed that the major-groove O4-alkyldT lesions are highly mutagenic during DNA replication and transcription, whereas N3-alkyldT and the minor-groove O2-alkyldT lesions strongly impede transcription in vitro and in mammalian cells.7–13 Moreover, N3-alkyldT lesions can be efficiently repaired by the transcription-coupled nucleotide excision repair pathway,10 whereas O2-alkyldT and O4-alkyldT lesions are known to be poorly repaired and persist in mammalian tissues.14,15 These studies unveiled the similarities and differences in effects of major- and minor-groove alkyldT lesions on genomic integrity. Little, however, is known about how cells respond to O2-alkyldT and O4-alkyldT lesions. We reason that a thorough investigation about how these lesions are recognized by cellular proteins will provide important insights into damage response signaling and repair of these lesions.
Previous quantitative proteomics-based affinity screening enabled the identification of novel cellular proteins that can recognize modified nucleosides in DNA and RNA or unique secondary structures of nucleic acids, and some of the identified proteins were confirmed to interact directly with these nucleic acid substrates and modulate their functions.16–19 In this study, we set out to profile comprehensively the cellular proteins that are involved in the recognition and repair of major- and minor-groove alkyldT lesions. To this end, we employed a quantitative proteomic method, based on stable isotope labeling by amino acids in cell culture (SILAC),20 to discover novel cellular proteins that can bind to O2- and O4- alkyldT lesions. We were able to identify a number of candidate DNA damage recognition proteins for O2- and/or O4-alkyldT lesions. We also demonstrated that one of these proteins, mitochondrial transcription factor A (TFAM), binds directly and preferentially with O4-alkyldT lesions and promotes transcriptional mutagenesis of these lesions in vitro and in human cells.
EXPERIMENTAL SECTION
Cell Culture.
HeLa cells (ATCC) were cultured in SILAC DMEM medium (Fisher Scientific) containing 10% dialyzed fetal bovine serum (Invitrogen) and 1% penicillin and streptomycin (Invitrogen), and the cells were maintained at 37 °C in a humidified incubator containing 5% CO2. The SILAC media were prepared by supplementing arginine- and lysine-depleted DMEM medium with unlabeled L-arginine and L-lysine (Sigma, for the light medium), or [13C6]-L-arginine and [13C6,15N2]-L-lysine (Cambridge Isotope Laboratories, for the heavy medium). The cells were cultured in heavy SILAC medium for at least 10 cell doublings to ensure complete heavy isotope incorporation.
Preparation of Nuclear Protein Lysates.
When reaching 80% confluency, HeLa cells were harvested using trypsin–EDTA (Invitrogen) and pelleted by centrifugation. The cell pellet was washed twice with phosphate-buffered saline. The nuclear proteome was prepared from heavy- and light-labeled cells using the NE-PER nuclear and cytoplasmic extraction reagents (Thermo Fisher Scientific) following the manufacturer’s guidelines. In this vein, we found that, because the centrifugation step in the nuclei isolation protocol also led to the precipitation of mitochondria, the nuclear protein lysate prepared from this method also contained mitochondrial proteins (data no shown). The protein concentrations were measured using Bradford Quick Start Protein Assay kit (Bio-Rad), and the nuclear lysate was stored at −80 °C until use.
Preparation of Damage-Bearing DNA Substrates.
The 20-mer lesion-containing oligodeoxyribonucleotides (ODNs) were prepared by ligating previously synthesized 5′-ATGGCGXGCTAT-3′ (X = O2-nBudT, O4-nBudT or O4-POBdT)8,12,21 to an 8-mer ODN (5′-GATCCTAT-3′) in the presence of a 28-mer template (5′-ATAGGATCATAG-CACGCCATTTTTTTTT-3′). The resulting ligation products were purified by denaturing polyacrylamide gel electrophoresis (PAGE), and their identities and purities confirmed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) and PAGE analyses (Figures S1–S4).
Affinity Pull-Down of O2- and O4-nBudT-Binding Proteins.
The 20-mer O2- and O4-nBudT-containing ODNs (50 pmol) were annealed individually with a 5′-biotin-conjugated complementary strand in a buffer containing 100 mM potassium acetate and 30 mM HEPES (pH 7.5) by raising the temperature to 95 °C and allowing to cool to room temperature over 3 h. The annealed double-stranded DNA was incubated with high-capacity streptavidin agarose beads (Thermo Pierce) at room temperature with shaking for 60 min. The beads were then washed three times with a buffer containing 20 mM Tris-HCl (pH 7.5), 10 mM KCl, 10 mM MgCl2, and 0.5 mM EDTA to remove any single-stranded or unbound DNA.
The DNA-bound beads were then incubated with nuclear lysates at 4 °C for 2 h in a buffer containing 20 mM Tris-HCl (pH 7.5), 10 mM KCl, 0.5 mM EDTA, and 10% glycerol. The lesion-containing probe and the control lesion-free probe were incubated with the heavy and light isotope-labeled lysates, respectively, in forward SILAC experiments, but with the light and heavy isotope-labeled lysates, respectively, in reverse SILAC experiments. After washing, two beads were combined and the bound proteins were eluted with 1% TFA in CH3CN/H2O (7:3, v/v) at 75 °C. The eluent was dried in a Speed-vac, and subjected to filter-aided sample preparation, as previously described with some modifications.22 Briefly, the dried samples were dissolved in a 400-μL solution containing 8 M urea and 50 mM ammonium bicarbonate (ABC) (pH 8.5), and transferred to an ultracentrifugal filter (10,000 MWCO, VWR International). After centrifugation at 14,000 g for 3 min, the filter devices were washed twice with 200 μL of 50 mM ABC to remove urea. After the addition of dithiothreitol and iodoacetamide for cysteine reduction and alkylation, 200 μL of 50 mM ABC containing 0.8 μg of trypsin was added to each filter device and the digestion was continued at 37 °C for 16 h. The digestion mixture was subsequently centrifuged, and the filter device was rinsed with 200 μL of 50 mM ABC and centrifuged again. The combined filtrates were dried in a Speed-vac, desalted using OMIX C18 Tips (Agilent Technologies), and then analyzed by LC-MS/MS.
Mass Spectrometry.
LC-MS/MS experiments were performed on an EASY-nLC 1200 system coupled with a Q Exactive Plus quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific), as previously described.16 The trapping (150 μm × 50 mm) and analytical (75 μm × 120 mm) columns were packed with ReproSil-Pur C18-AQ resin (5 μm and 3 μm in particle sizes, respectively, Dr. Maisch HPLC GmbH, Germany). The peptide samples were loaded onto the trapping column with 0.1% (v/v) formic acid in water at a flow rate of 3 μL/min, and resolved on the separation column with a 180-min linear gradient of 2–40% acetonitrile in 0.1% (v/v) formic acid at a flow rate of 300 nL/min. The mass spectrometer was set up in the positive-ion mode, and the spray voltage was 1.8 kV. MS/MS was recorded in a data-dependent acquisition mode in which one full MS scan was followed with 25 MS/MS scans. All proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository23 with the dataset identifier: PXD021719.
Competitive Transcription and Adduct Bypass Assay.
DNA templates for the competitive transcription and adduct bypass (CTAB) assay were prepared following published procedures.24 Briefly, the parent vector (i.e., pTGFP-T7 hha10T) was digested with Nt.BstNBI to generate a gapped plasmid by removing a 25-mer single-stranded ODN followed by filling the gap with a 13-mer lesion-free ODN (5′-AATTGAGTCGATG-3′) and a 12-mer lesion-containing ODN (5′-ATGGCGXGCTAT-3′, X = O2-nBudT, O4-nBudT or O4-POBdT) to yield the lesion-bearing plasmids. The resulting supercoiled plasmids were purified by agarose gel electrophoresis. The purified lesion-containing and lesion-free plasmids were premixed individually with the competitor vector (i.e., pTGFP-T7 Hha10-comp) at a molar ratio of 4:1 and used as DNA templates for in vitro and cellular transcription assays.
In vitro transcription was mediated by human RNA polymerase II (hRNAPII) in HeLa nuclear extract supplied in the HeLaScribe nuclear extract in vitro transcription system (Promega).24 Briefly, 50 ng of NotI-linearized DNA templates were mixed with HeLa nuclear extract, rNTP, MgCl2, and RNase inhibitor in a 25-μL transcription buffer. After incubation at 30 °C for 1 h, the transcription reaction was terminated by adding transcription stop solution, and RNA was subsequently extracted from the reaction mixture. For the in vitro transcription assay with TFAM, linearized DNA templates were preincubated with 0.5 μM recombinant TFAM protein at r.t. for 15 min before adding to the transcription reaction mixture.
For cellular transcription assay, HeLa cells in a 24-well plate at ~80% confluency were transfected with 50 ng of DNA templates and 450 ng of carrier plasmid (e.g., pGEM-T) using TransIT-2020 (Mirus), following the vendor’s recommended procedures. For the TFAM overexpression experiment, HeLa cells in a 24-well plate at ~60% confluency were transfected with 500 ng of pRK7-TFAM plasmid. After a 12-h incubation, 50 ng of DNA templates and 450 ng of carrier plasmid were cotransfected into the cells. After 24 h, the cells were harvested for RNA extraction.
The RNA products were extracted using Total RNA Kit I (Omega), and were treated twice with the DNA-free kit (Ambion) to eliminate DNA contamination. Reverse transcription was performed with M-MLV reverse transcriptase (Promega) and a mixture of oligo(dT)18 and a gene-specific primer (5′-TCGGTGTTGCTGTGAT-3′) followed by polymerase chain reaction (PCR) amplification with a pair of primers spanning the lesion site and Phusion high-fidelity DNA polymerase. The reverse transcription-PCR (RT-PCR) products were purified using E.Z.N.A. Cycle Pure Kit (Omega Bio-Tek) and stored at −20 °C until use.
For PAGE analysis, ~150 ng of RT-PCR products were treated with 5 U NcoI and 0.5 U recombinant shrimp alkaline phosphatase (rSAP) at 37 °C for 1 h in NEB buffer 3.1 followed by heating at 70 °C for 20 min to deactivate the rSAP. The resulting mixture was then treated with 5 U T4 PNK, 10 pmol cold ATP and 2.5 μCi [γ−32P]ATP at 37 °C for 30 min followed by heating at 65 °C for 20 min to deactivate the T4 PNK. To the mixture was subsequently added 2 U SfaNI, and the solution was incubated at 37 °C for 1.5 h. The reaction was then terminated by adding formamide gel-loading buffer. The resulting 32P-labeled restriction fragments were resolved on a 30% native polyacrylamide gel (acrylamide:bis-acrylamide = 19:1) and quantified by phosphorimager analysis. The relative bypass efficiency (RBE) was calculated using the following formula, RBE (%) = (lesion signal/competitor signal)/(unmodified control signal/competitor signal) × 100%.
For LC-MS/MS analysis, 3 μg of RT-PCR products were treated with SfaNI and rSAP, and then with NcoI. The resulting DNA fragments were extracted with phenol/chloro-form/isoamyl alcohol (25:24:1, v/v/v), desalted by using an HLB cartridge (Waters), and dissolved in 8 μL of H2O. After centrifugation at 15,000 g for 3 min, 7 μL of the sample was subjected to LC-MS/MS analysis following previously described conditions.10
RESULTS
Quantitative Proteomic Screening of O2-nBudT- and O4-nBudT-Binding Proteins.
Alkylation products formed at the O2 and O4 positions of thymidine (O2- and O4-alkyldT) are known to be poorly repaired,14,15 and confer adverse consequences on genomic stability.4,7–13 To understand better the mechanisms involved in the recognition and repair of these DNA lesions in human cells, we set out to employ a quantitative proteomic approach to identify the cellular proteins that are involved in the binding of these lesions. For this purpose, we synthesized 20-mer O2-nBudT- and O4-nBudT-bearing ODNs with the same sequence, and annealed them individually with the lesion-free complementary strand carrying a 5′-biotin tag. The identities and purities of the lesion-containing ODNs were confirmed by LC-MS/MS and PAGE analyses (Figures S1–S4). In this vein, we chose to use 20-mer DNA probes owing to their adequate duplex stability and their relative ease of preparation and purification. In addition, the same sequence contexts were used in our previous cellular replication experiments for these lesions.8,9,12,13 Moreover, the damage site in the probes is located 13 base pairs away from the biotin label in the complementary strand, which minimizes potential interference of streptavidin beads on protein-DNA lesion interactions.
Using these two lesion-bearing duplex DNA probes and the corresponding lesion-free DNA probe as baits, we developed a SILAC-based quantitative proteomic method to discover cellular proteins that can bind to O2- and O4-nBudT lesions (Figure 1a). To remove experimental bias, we conducted both forward and reverse SILAC experiments, as described previously.16,19 Briefly, the above-mentioned biotinylated duplex DNA probes were coupled to streptavidin beads. In the forward SILAC experiment (Figure 1a), lesion-free control and lesion-bearing DNA-bound beads were incubated with light and heavy stable isotope-labeled lysine/arginine-containing nuclear lysates, respectively. After extensive washing, the two groups of beads were combined and the proteins were eluted from the beads and subjected to tryptic digestion. The resulting peptides were then analyzed by LC-MS/MS. The reverse SILAC experiment was conducted in the same way except that the control and lesion DNA probes were incubated with heavy- and light-labeled nuclear lysates, respectively.
Figure 1.
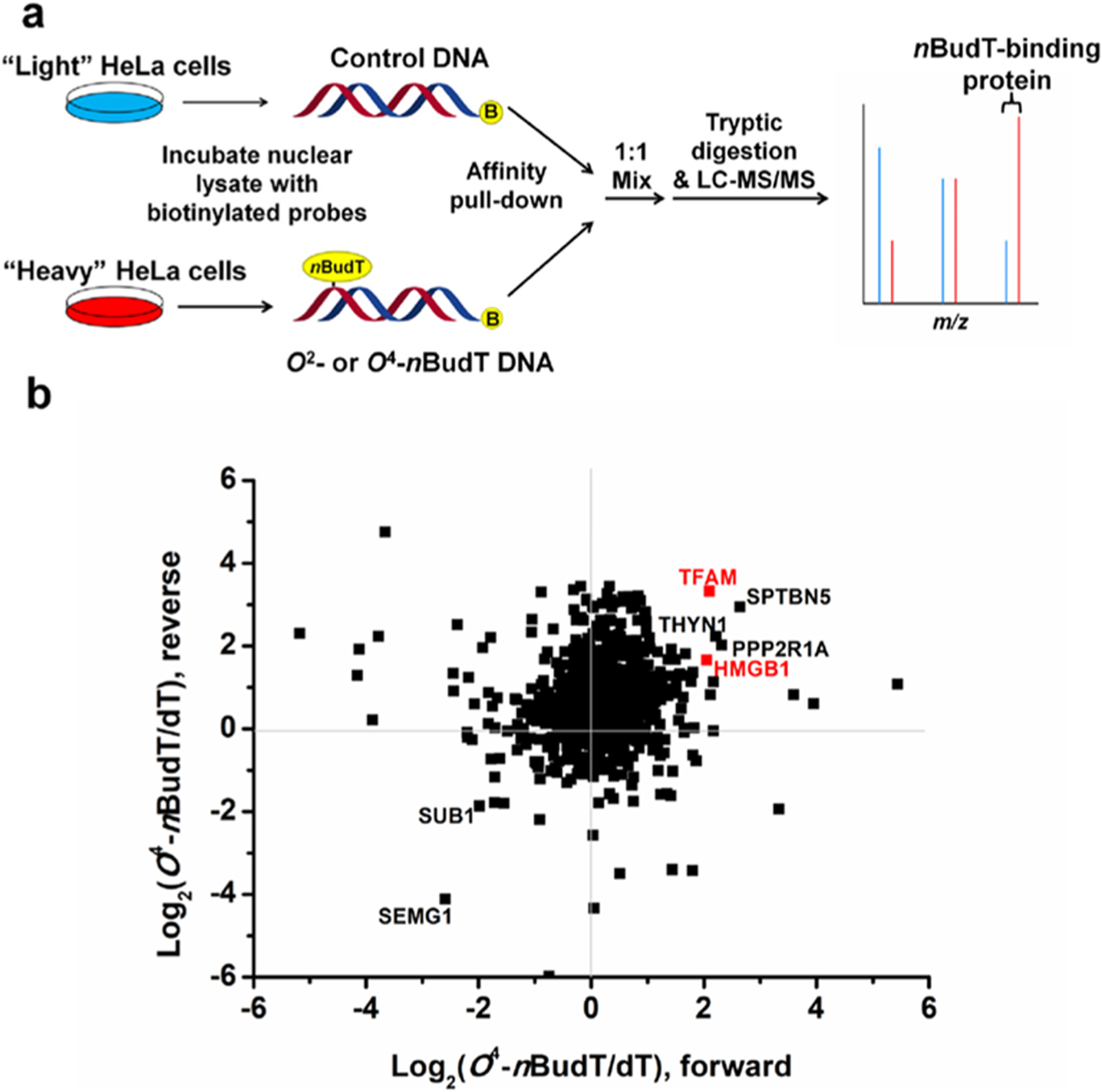
SILAC-based interaction screening for the identification of O2-nBudT- and O4-nBudT-binding proteins. (a) Schematic diagram illustrating the forward SILAC workflow, where the ‘B’ in yellow circles denotes 5′-biotin labeling. (b) Scatter plot showing the proteins identified from pull-down assays using O4-nBudT DNA relative to the lesion-free DNA with nuclear protein lysates prepared from HeLa cells.
The results from the quantitative proteomic analyses allowed for the identification of a number of proteins with preferential binding toward O2-nBudT- and/or O4-nBudT-bearing DNA over the corresponding unmodified DNA, where the protein needs to be enriched for the lesion-bearing DNA probe over the corresponding lesion-free DNA probe in both forward and reverse SILAC experiments with an average enrichment ratio of >1.5. With these criteria, 21 and 74 proteins exhibit preferential binding toward the O2-nBudT- and O4-nBudT- carrying DNA probes over the corresponding unmodified probe, respectively (Tables S1,S2). Most identified proteins are distinct for each lesion, except for PDS5A, PRPF40A, SF3B1, THOC2, and TOP1, which are enriched for both lesion-bearing DNA probes.
Among the putative O2-nBudT-binding proteins (Figure S5; Table S1), several were previously shown to be involved in DNA damage response and repair, including FUS, MSH6, PDS5A, PRKDC, and TOP1.25–28 In addition, we found that DNA damage binding protein 1 and 2 (DDB1 and DDB2) bind preferentially to minor-groove O2-nBudT-containing DNA, with the SILAC ratios (O2-nBudT/dT) being 2.0 ±0.6 and 1.8 ± 0.4 (Table S1), respectively. In this vein, DDB1 and DDB2 can form a heterodimer, which binds to UV-induced dimeric DNA photoproducts and stimulates their repair.29–31 On the other hand, several high-mobility group (HMG) box-containing proteins, including HMGB1, HMGB2, and TFAM, display stronger binding toward O4-nBudT DNA over control DNA, with the SILAC ratios (O4-nBudT/dT) being 3.7 ± 1.3, 2.3 ± 1.2, and 7.2 ± 5.4, respectively (Figure 1b; Table S2). Representative ESI-MS and MS/MS of tryptic peptides from several DNA damage-binding proteins, including DDB1, DDB2, and TFAM, are shown in Figure 2 and Figures S6–S9. We also validated the quantitative proteomic results of these proteins by Western blot analyses (Figure 3a).
Figure 2.
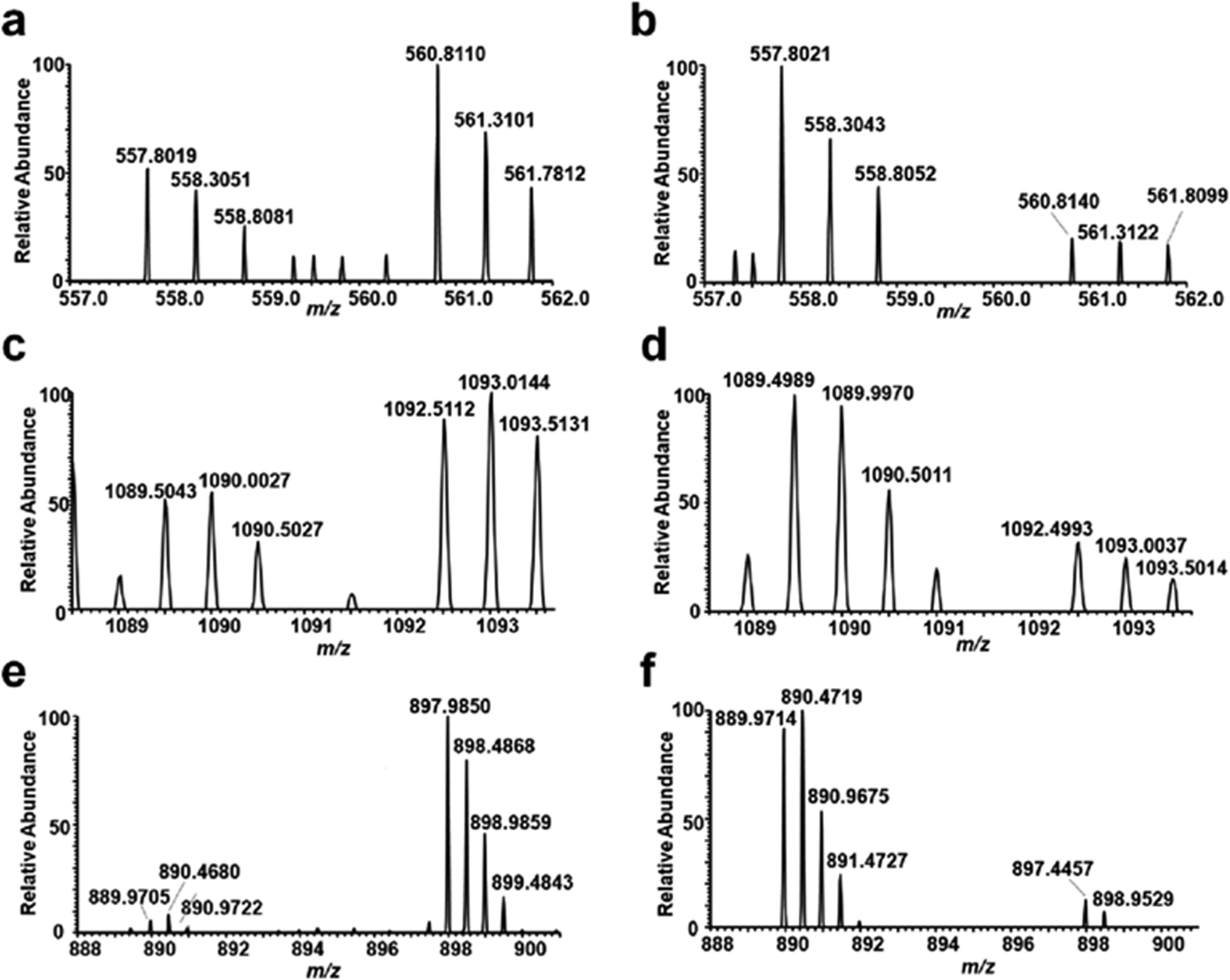
ESI-MS (a, c, and e, forward SILAC; b, d, and f, reverse SILAC) showing the [M + 2H]2+ ions of light and heavy arginine-containing peptides, EMLGGEIIPR derived from DDB1 (a, b), FNPLNTNQFYASSMEGTTR derived from DDB2 (c, d), and lysine-containing peptide FKEQLTPSQIMSLEK derived from TFAM (e, f).
Figure 3.
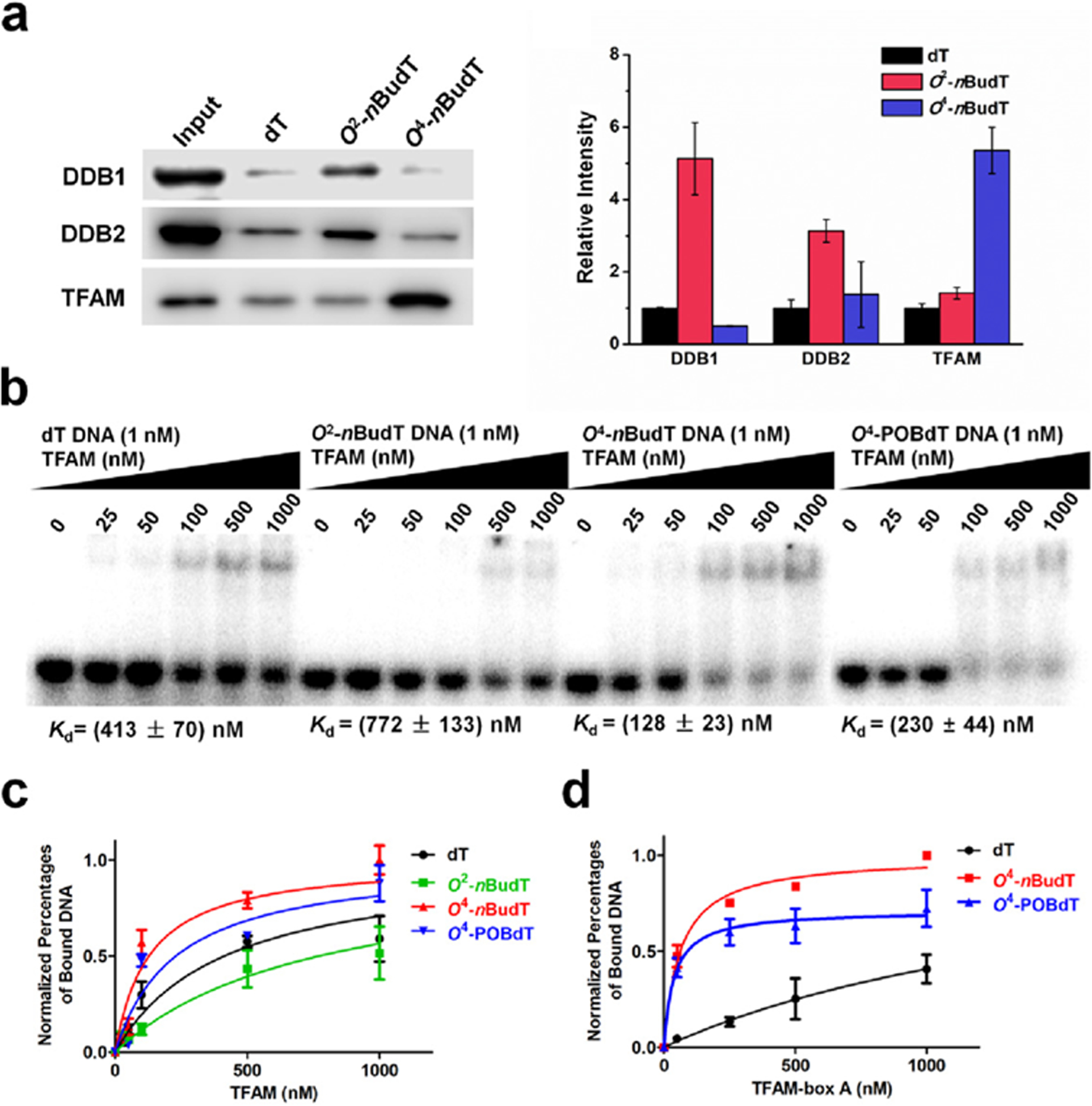
TFAM binds directly and selectively with O4-alkyldT-containing duplex DNA, and the binding capacity is derived from the HMG-box A domain of TFAM. (a) Representative Western blot images and quantitative results showing the levels of three proteins in pull-down samples with different probes (n = 3). (b) Representative gel images of EMSA for measuring the binding affinities of TFAM with lesion-free and lesion-containing DNA probes. (c) Nonlinear fitting curve of the EMSA result of TFAM (n = 3). (d) Nonlinear fitting curves for the binding of the HMG-box A domain with different DNA substrates (n = 3). Error bars represent S. D.
Among all the putative O2-alkyldT and O4-alkyldT DNA-binding proteins, TFAM exhibits the highest average SILAC ratio, and our quantitative proteomic analyses led to the identification of 24 tryptic peptides from this protein with a ~55% sequence coverage. Thus, we decided to characterize further the interactions between this protein and O-alkyldT lesions.
TFAM Binds Directly to O4-alkyldT DNA through Its HMG Box-A Domain.
One limitation of the above quantitative proteomic experiment resides in that it may also give rise to proteins that are indirectly associated with the DNA probes (i.e., through protein-protein interactions). Therefore, we next examined if TFAM can bind directly with O4-alkyldT lesions, including O4-nBudT and O4-pyridyloxobutylthymidine (O4-POBdT). O4-POBdT is one of the major DNA alkylation adducts produced in cells upon treatment with 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone, which is a model DNA alkylating agent for metabolites of NNK and NNN.21,32 Toward this end, we purified full-length recombinant TFAM protein (Figure S10) and measured its binding affinities toward lesion-free as well as the corresponding O2-nBudT-, O4-nBudT-, and O4-POBdT-containing DNA probes by using electrophoretic mobility shift assay (EMSA). The results showed that TFAM indeed exhibits higher binding affinities toward O4-nBudT and O4-POBdT DNA than lesion-free and O2-nBudT DNA, where the Kd values were 128 ± 23, 230 ± 44, 772 ± 133, and 413 ± 70 nM, respectively (Figure 3b,c).
TFAM harbors two HMG boxes (HMG-box A and HMG-box B) and a C-terminal tail (Figure S11a), where HMG-box A assumes a major role in DNA binding.33 Hence, we generated two truncated forms of TFAM proteins which harbor only HMG-box A or HMG-box B (Figure S11b), and assessed their capabilities in binding with lesion-free, O4-nBudT, and O4-POBdT DNA probes. Strikingly, we found that the binding affinities of HMG-box A of TFAM toward O4-alkyldT DNA probes were > 30-fold higher than lesion-free DNA (Figure 3d and Figure S12a), indicating that the binding selectivity of HMG-box A toward O4-alkyldT DNA is even higher than the full-length protein. In addition, the HMG-box B domain of TFAM displayed very weak binding with lesion-free and lesion-containing DNA probes (Figure S12b). Therefore, TFAM binds directly and preferentially with O4-alkyldT DNA, and the binding capacity emanates mainly from its HMG-box A domain.
TFAM Promotes the Mutagenic Transcriptional Bypass of O4-AlkyldT Lesions In Vitro and in Cells.
We next explored the functions of the interactions between TFAM and O-alkyldT lesions by investigating the effects of TFAM on the transcriptional bypass of O2-nBudT, O4-nBudT, and O4-POBdT lesions. Our previous study showed that O4-EtdT is highly mutagenic during DNA transcription, whereas O2-EtdT strongly blocks transcription in vitro and in cells.10 No study, however, has been conducted to examine the impacts of other O-alkyldT lesions on transcription and how this process is influenced by cellular proteins.
We employed our previously published method24 to incorporate O2-nBudT, O4-nBudT, and O4-POBdT lesions into the transcribed strand of a nonreplicative double-stranded plasmid, and performed the CTAB assay in vitro using HeLa cell nuclear extract and in HeLa cells. After transcription, the transcripts of interest were isolated, reversed transcribed, amplified with PCR, and the RT-PCR products were sequentially digested with NcoI and SfaNI for PAGE and LC-MS/MS analyses (Figure 4 and Figures S13,S14).
Figure 4.
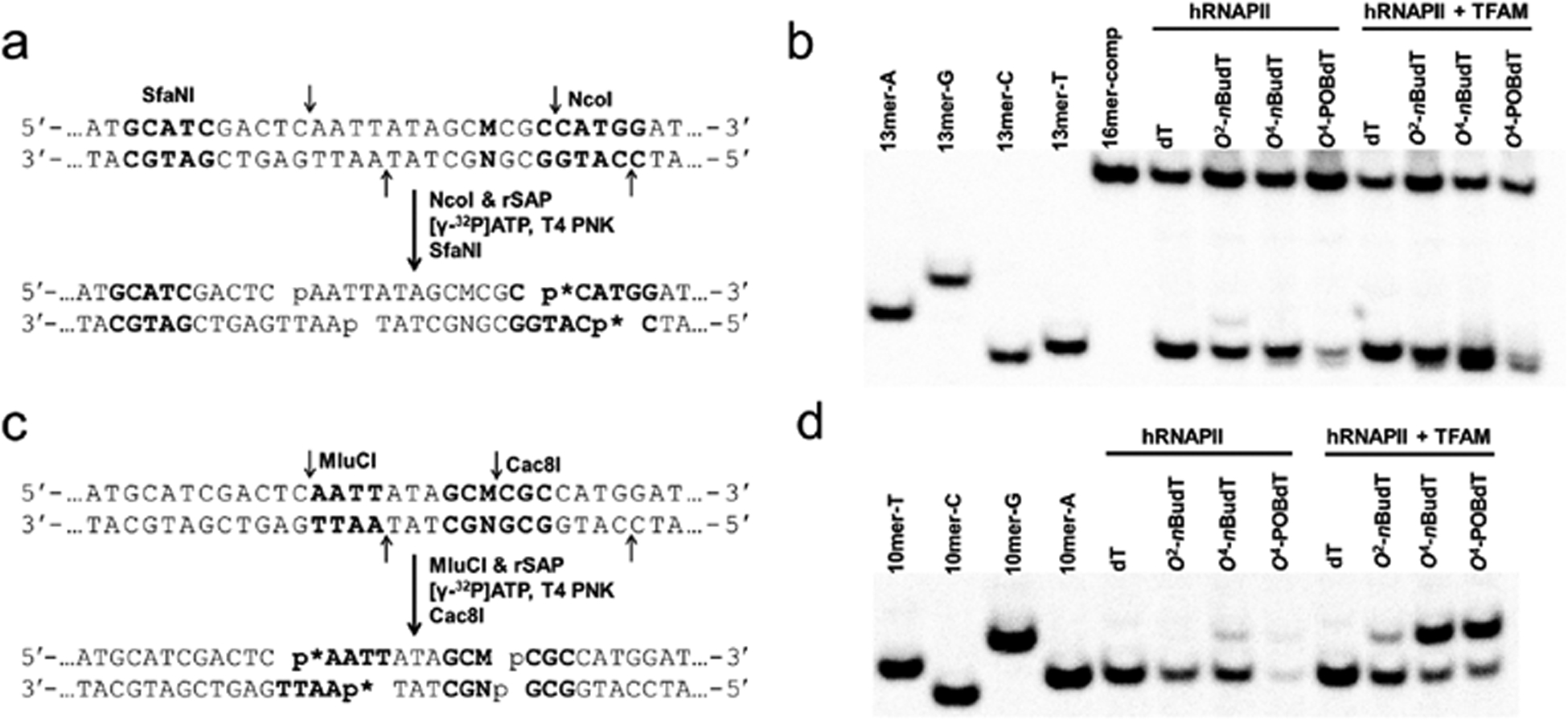
Restriction digestion and postlabeling method for determining the transcriptional bypass efficiencies and mutation frequencies of O2-nBudT, O4-nBudT, and O4-POBdT in vitro. (a) Sample preparation for restriction digestion using NcoI and SfaNI, and postlabeling assay (p* indicates 32P-labeled phosphate group). (b) Representative gel image showing the NcoI/SfaNI-treated restriction fragments of interest. ‘16mer-comp’ represents the standard ODN d(CATGGCGATATGCTAT), which corresponds to the restriction fragment arising from the competitor vector; ‘13mer-C’, ‘13mer-A’, ‘13mer-G’, and ‘13mer-T’ represent the standard ODN d(CATGGCGNGCTAT), where ‘N’ is C, A, G, and T, respectively. (c) Sample preparation for restriction digestion using MluCI and Cac8I and postlabeling assay. (d) Representative gel image showing the MluCI/Cac8I-produced restriction fragments of interest. ‘10mer-C’, ‘10mer-A’, ‘10mer-G’, and ‘10mer-T’ represent the standard ODN d(AATTATAGCM), where ‘M’ is C, A, G, and T, respectively.
The above restriction digestion with NcoI and SfaNI yields 13-mer 32P-labeled fragments, which can distinguish nonmutagenic fragment d(CATGGCGTGCTAT) (13mer-T) from the corresponding products carrying an A → U or A → C mutation, i.e., d(CATGGCGAGCTAT) (13mer-A) and d(CATGGCGGGCTAT) (13mer-G), respectively (Figure 4a,b). However, the 13-mer fragment with an A → G mutation (13mer-C) cannot be resolved from the nonmutagenic 13mer-T. Therefore, we adopted an alternative restriction digestion by using MluCI and Cac8I, which yielded 10-mer 32P labeled fragments from the opposite strand that enables the differentiation of the nonmutagenic product (AATTATAGCA, 10mer-A) from the product with an A → G mutation (AATTATAGCG, 10mer-G) (Figure 4c,d).
The quantification results from PAGE analyses showed that the RBE value of O4-nBudT in human RNA polymerase II (hRNAPII)-mediated transcription in vitro was ~53%, which was over fivefold higher than that of O4-POBdT (~10%) and also higher than that of regioisomeric O2-nBudT (~31%) (Figure 5a). PAGE analysis also allowed us to measure the mutation frequencies of these lesions. The results revealed that O2-nBudT induced ~10% A → U mutation (Figure 5b), while both O4-nBudT and O4-POBdT induced over 20% A → G mutation (Figure 5c,d).
Figure 5.
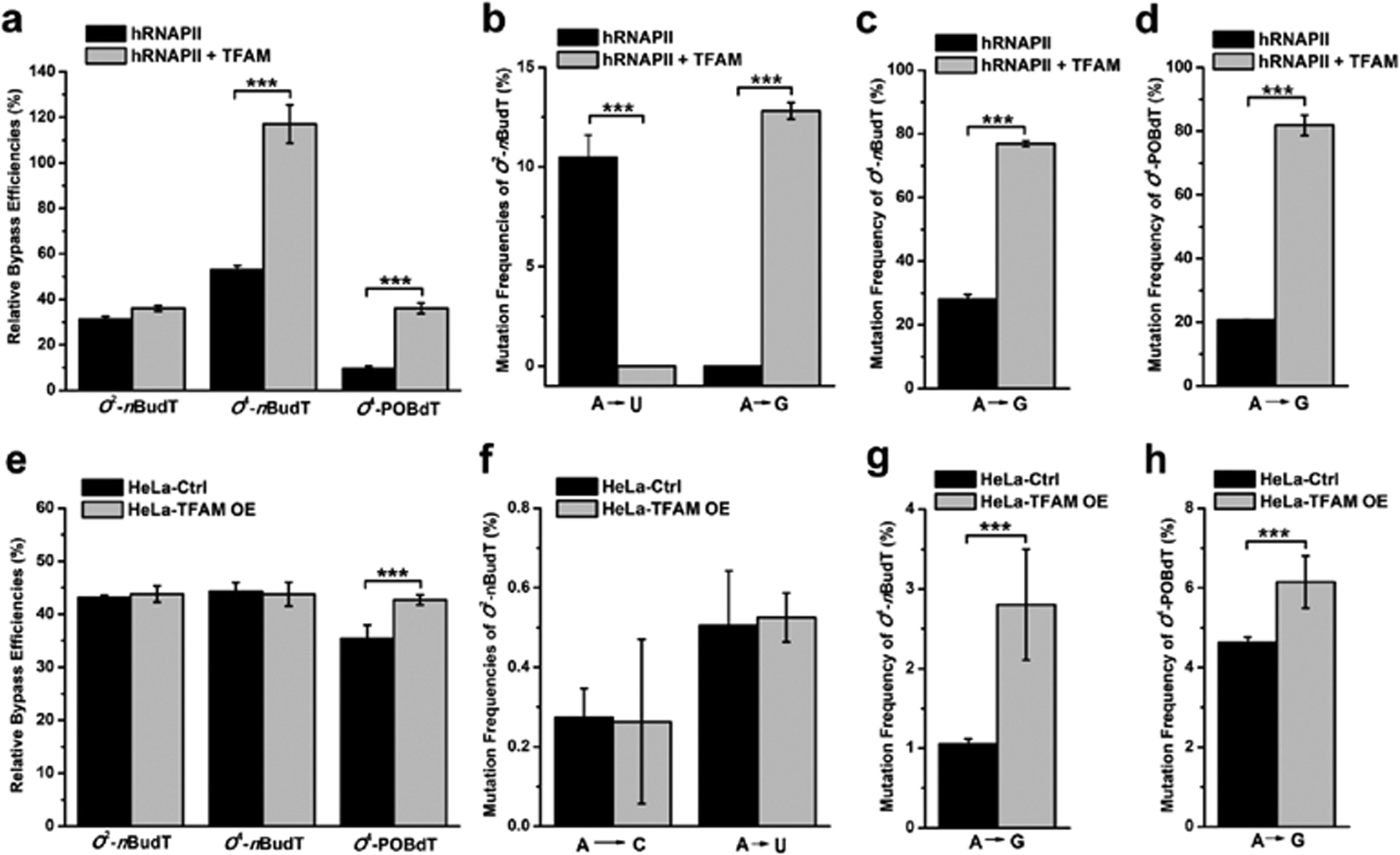
TFAM promotes the mutagenic transcriptional bypass of O4-alkyldT in vitro and in HeLa cells. (a) RBE values and (b–d) mutagenic properties of O2-nBudT, O4-nBudT, and O4-POBdT in the hRNAPII-mediated transcription system with or without the inclusion of TFAM protein. The RBE values (e) and frequencies of mutant transcripts (f–h) of O2-nBudT, O4-nBudT, and O4-POBdT in HeLa cells with or without overexpression of TFAM protein. The data represent the mean ± S.D. of results from three independent experiments. ***, p < 0.001. The p values were calculated with unpaired two-tailed Student’s t-test.
To examine how TFAM modulates the transcriptional bypass of these lesions, we preincubated DNA templates with0.5 μM recombinant TFAM for 15 min before initiating the in vitro transcription reaction. Interestingly, the RBE values of O4-nBudT and O4-POBdT increased by >2-fold in the transcription reactions containing added TFAM protein (Figure 5a), which is accompanied with a marked increase in A → G mutation (from ~20% to ~80%) (Figure 5c,d). While the inclusion of TFAM in the transcription reaction did not alter the RBE value of O2-nBudT, it changes substantially the type of mutant transcripts induced by O2-nBudT, where we observed a ~10% A → G in lieu of A → U mutation in the resulting transcripts (Figure 5b). Hence, our results reveal that the binding of TFAM on damaged DNA promotes the misincorporation of guanosine opposite the lesion site during the hRNAPII-mediated transcription. We also confirmed the identities of the above-mentioned mutagenic products by LC-MS/MS analyses (Figures S13,S14).
We next evaluated how TFAM affects the transcriptional bypass and mutagenesis of these lesions in human cells. Toward this end, we transfected DNA templates into either HeLa cells or TFAM-overexpressing HeLa cells. After a 24 h transfection, the cells were harvested for RNA extraction and the ensuing transcripts were again reverse transcribed, PCR-amplified, digested with restriction enzymes, and subjected to PAGE and LC-MS/MS analyses (Figures S15,S16). Our results showed that O2-nBudT, O4-nBudT, and O4-POBdT considerably inhibited transcription in HeLa cells, with the RBE values being ~43, ~44, and ~35%, respectively (Figure 5e and Figure S15). In addition, the frequencies of A → C and A → U mutations induced by O2-nBudT were less than 1% (Figure 5f), and the frequencies of A → G mutation were less than 5% for O4-nBudT and O4-POBdT in HeLa cells (Figure 5g,h), which were all much lower than what were observed in in vitro experiments (Figure 5b–d). In this vein, we also confirmed the mutagenic products formed from cellular transcription by LC-MS/MS analyses (Figures S14 and S16).
We also found that the overexpression of TFAM in HeLa cells elicited a slight, yet statistically significant, increase in the RBE value of O4-POBdT (From ~35% to 43%), along with elevated mutation frequencies of O4-nBudT and O4-POBdT. However, the overexpression of TFAM did not elicit any apparent alteration in the transcription bypass efficiency or mutation frequency of O2-nBudT. The results illustrated that TFAM enhances the transcriptional bypass of O4-alkyldT lesions and augments transcriptional mutagenesis of these lesions.
DISCUSSION
Alkylation represents an important and major type of DNA damage.34 In the past few years, a number of studies have been performed to investigate the cytotoxic and mutagenic properties of alkylated DNA lesions in E. coli and mammalian cells.8–10,12 However, very little is known about the cellular proteins that are involved with the recognition of these lesions. Here, we used a SILAC-based quantitative proteomic method to identify cellular proteins that can recognize alkylated DNA lesions. SILAC is a metabolic labeling approach that incoporates light or heavy stable isotope-labeled amino acids into newly synthesized proteins in live cells.20 The resulting light- and heavy-labeled proteomes are chemically identical, but can be differentiated by MS analysis. Thus, the SILAC-based quantitative proteomic method, especially with the adoption of both forward and reverse SILAC labeling experiments, facilitates unbiased interrogation of the interaction proteomes of damage-containing DNA without a priori knowledge.
Our quantitative proteomic experiments led to the identification of a number of proteins that can bind more strongly to O2-nBudT- and/or O4-nBudT-containing DNA than the corresponding unmodified DNA (Tables S1,S2). Most proteins are distinct in the two interactomes, suggesting that cells may be equipped with different damage recognition and repair systems to cope with the regioisomeric O2- and O4- alkyldT lesions.
Among the identified damage recognition proteins, many are known to be involved in DNA damage response and repair, including FUS, MSH6, PDS5A, PRKDC, TOP1, DDB1, DDB2, HMGB1, HMGB2, and TFAM.25–28 For instance, MSH6, a component of MutSα, binds to mismatched DNA to trigger DNA mismatch repair;28 PRKDC, a DNA-dependent serine/threonine kinase, is involved in double-strand break repair through the nonhomologous end-joining pathway.27 In addition, several proteins exhibit stronger binding to lesion-free DNA than alkyldT lesions (Figure 1b and Figure S5), including SUB1, SSBP1, FLII, and SEMG1. For instance, SUB1, which is known to bind to platinated DNA and assume multiple roles in transcription,18,35 showed diminished binding affinities toward both O2-nBudT- and O4-nBudT-containing DNA over the corresponding lesion-free DNA. It will be important to examine, in the future, how diminished binding to alkylated DNA modulates the functions of SUB1 and other proteins.
TFAM is a versatile protein that functions in many important cellular processes, including packaging of mitochondrial DNA into nucleoid and regulation of copy number of mitochondrial DNA.36–38 Additionally, TFAM was previously shown to bind to DNA containing cisplatin adduct and 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG), where the binding could impede the access of to 8-oxodG by the base excision repair machinery.39,40 Very recently, N6-methyl-2′-deoxyade-nosine in mitochondrial DNA was found to repress DNA binding and bending by TFAM.41 Furthermore, TFAM was shown to form cross-link with abasic sites in DNA, which stimulates DNA strand cleavage at abasic sites and is thought to modulate the turnover of mitochondrial DNA.42 Here, we expanded the functions of TFAM by revealing that it can bind directly and selectively to the major-groove O4-alkyldT lesions (Figure 3b,c).
We also showed that the HMG-box A domain of TFAM is indispensable for its binding with DNA, and the binding affinity of HMG-box A domain of TFAM toward O4-alkyldT DNA was over 30-fold higher than unmodifed DNA (Figure 3d and Figure S12a). This finding suggests that TFAM-box A perhaps can be harnessed for the enrichment and mapping of O4-alkyldT lesion in genomic DNA in the future.
As previously reported, the major-groove O4-alkyldT lesions are highly mutagenic during transcriptional bypass, whereas the minor-groove O2-alkyldT lesions induce much stronger transcriptional stalling than O4-alkyldT lesions, and both O2-alkyldT and O4-alkyldT are known to be poorly repaired in human cells.10,11 Our CTAB assay results revealed that O2-nBudT and O4-POBdT, but not O4-nBudT, strongly block hRNAPII-mediated DNA transcription (Figure 5a). O4-nBudT and O4-POBdT induce exclusively A → G mutation both in vitro and in HeLa cells (Figure 5c,d,g,h), which is in agreement with what was observed previously for O4-EtdT,10 further substantiating the distinct mispairing ability of O4-alkyldT with guanosine.43 Moreover, the ectopic overexpression of TFAM promotes the misincorporation of guanosine opposite the lesion sites during transcription, thereby augmenting mutagenic transcriptional bypass of O4-alkyldT lesions. Hence, the presence of TFAM exacerbates the transcriptional mutagenesis of O4-alkyldT lesions. It will be important to assess, in the future, how genetic depletion of TFAM alters the transcriptional mutagenesis across the O4-alkyldT lesions.
It is worth noting that, although the ectopic overexpression of TFAM in HeLa cells results in elevated A→G mutation of O4-nBudT (Figure 5g), it did not alter the transcriptional bypass efficiency of this lesion (Figure 5e). The exact reason underlying the manifestation of a consequence of TFAM overexpression on transcriptional mutagenesis, but lack of an effect on transcriptional bypass efficiency, is not clear. Nevertheless, this is not unprecedented. For instance, similar effects were observed for Ada protein on the replicative bypass of N3-MedT in Escherichia coli and for its mammalian orthologs (Alkbh2 and Alkbh3) on the transcriptional bypass of N3-EtdT in mouse embryonic fibroblast cells.11,44
The molecular mechanisms through which TFAM binding modulates the transcriptional mutagenesis of these lesions remain unclear and warrant future investigation. In this vein, previous X-ray crytal structure studies revealed that the two HMG domains of TFAM can bind to unmodified DNA and induce a U-turn to the DNA double helix.45,46 Thus, we reason that the DNA conformational changes elicited by TFAM binding may alter the recognition of the O4-alkyldT by RNA polymerase II, which may contribute to the alterations in transcriptional mutagenesis of these lesions.
Our study also identified a number of other candidate DNA damage-binding proteins, which set the stage for future assessments about their capacities in binding toward alkylated DNA lesions and their roles in the repair and transcriptional bypass of DNA lesions. A comparison of the biological functions of proteins recognizing these two types of lesions may also enable us to understand better the differential cellular responses toward the induction of major- and minor-groove alkyldT lesions.
CONCLUSIONS
In this study, we identified, by using an unbiased quantitative proteomic method, multiple candidate DNA damage-recognition proteins for O2-nBudT and/or O4-nBudT, and we demonstrated that TFAM can bind directly to O4-alkyldT lesions through its HMG box-A domain, and TFAM augments the mutagenic transcriptional bypass of O4-alkyldT lesions in vitro and in human cells. In addition, this is the first investigation about how O2-nBudT, O4-nBudT, and O4-POBdT lesions perturb transcription and how TFAM affects the transcriptional bypass and mutagenesis of these lesions. Thus, our work afforded novel insights into the roles of DNA damage-binding proteins and expanded the functions of TFAM. It will be important to explore, in the future, the mechanism of TFAM in DNA alkylation damage response and the possibility of employing TFAM-box A for the enrichment and subsequent mapping of O4-alkyldT in genomic DNA.
Supplementary Material
Funding
This study was funded by National Institutes of Health (R35 ES031707).
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.0c04224
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c04224.
Experimental conditions for LC-MS/MS database search; Western blot; generation of recombinant proteins; EMSA; LC-MS/MS results; PAGE gel images; and quantitative proteomics data (PDF)
The authors declare no competing financial interest.
Contributor Information
Xiaomei He, Department of Chemistry, University of California, Riverside, California 92521-0403, United States.
Pengcheng Wang, Department of Chemistry, University of California, Riverside, California 92521-0403, United States.
Yinsheng Wang, Department of Chemistry, University of California, Riverside, California 92521-0403, United States.
REFERENCES
- (1).Shrivastav N; Li D; Essigmann JM Carcinogenesis 2010, 31, 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Liu S; Wang Y Chem. Soc. Rev 2015, 44, 7829–7854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Fu D; Calvo JA; Samson LD Nat. Rev. Cancer 2012, 12, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).You C; Wang Y Acc. Chem. Res 2016, 49, 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hecht SS Chem. Res. Toxicol 2008, 21, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chen HJC; Wang YC; Lin WP Anal. Chem 2012, 84, 2521–2527. [DOI] [PubMed] [Google Scholar]
- (7).Jasti VP; Spratt TE; Basu AK Chem. Res. Toxicol 2011, 24, 1833–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wang P; Amato NJ; Zhai Q; Wang Y Nucleic Acids Res. 2015, 43, 10795–10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wu J; Li L; Wang P; You C; Williams NL; Wang Y Nucleic Acids Res. 2016, 44, 9256–9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).You C; Wang P; Dai X; Wang Y Nucleic Acids Res. 2014, 42, 13706–13713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).You C; Wang P; Nay SL; Wang J; Dai X; O’Connor TR; Wang Y ACS Chem. Biol 2016, 11, 1332–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhai Q; Wang P; Cai Q; Wang Y Nucleic Acids Res. 2014, 42, 10529–10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wu J; Leng J; Wang P; Li L; Wang YJ Biol. Chem 2018, 293, 8638–8644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Brent TP; Dolan ME; Fraenkel-Conrat H; Hall J; Karran P; Laval L; Margison GP; Montesano R; Pegg AE; Potter PM Proc. Natl. Acad. Sci. U. S. A 1988, 85, 1759–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bronstein SM; Skopek TR; Swenberg JA Cancer Res. 1992, 52, 2008–2011. [PubMed] [Google Scholar]
- (16).Dai X; Wang T; Gonzalez G; Wang Y Anal. Chem 2018, 90, 6380–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Dominissini D; Moshitch-Moshkovitz S; Schwartz S; Salmon-Divon M; Ungar L; Osenberg S; Cesarkas K; Jacob-Hirsch J; Amariglio N; Kupiec M; Sorek R; Rechavi G Nature 2012, 485, 201–206. [DOI] [PubMed] [Google Scholar]
- (18).Du Z; Luo Q; Yang L; Bing T; Li X; Guo W; Wu K; Zhao Y; Xiong S; Shangguan D; Wang FJ Am. Chem. Soc 2014, 136, 2948–2951. [DOI] [PubMed] [Google Scholar]
- (19).Williams P; Li L; Dong X; Wang YJ Am. Chem. Soc 2017, 139, 12426–12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ong SE; Mann M Nat. Protoc 2006, 1, 2650. [DOI] [PubMed] [Google Scholar]
- (21).Leng J; Wang Y Anal. Chem 2017, 89, 9124–9130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wiśniewski JR; Zougman A; Nagaraj N; Mann M Nat. Methods 2009, 6, 359. [DOI] [PubMed] [Google Scholar]
- (23).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; Perez E; Uszkoreit J; Pfeuffer J; Sachsenberg T; Yilmaz S; Tiwary S; Cox J; Audain E; Walzer M; Jarnuczak AF; Ternent T; Brazma A; Vizcaino JA Nucleic Acids Res. 2019, 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).You C; Wang Y Nat. Protoc 2015, 10, 1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Litwin I; Pilarczyk E; Wysocki R Genes (Basel) 2018, 9, 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lesher D-TT; Pommier Y; Stewart L; Redinbo MR Proc. Natl. Acad. Sci. U. S. A 2002, 99, 12102–12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ruis BL; Fattah KR; Hendrickson EA Mol. Cell. Biol 2008, 28, 6182–6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Bowers J; Tran PT; Joshi A; Liskay RM; Alani EJ Mol. Biol 2001, 306, 957–968. [DOI] [PubMed] [Google Scholar]
- (29).Fujiwara Y; Masutani C; Mizukoshi T; Kondo J; Hanaoka F; Iwai SJ Biol. Chem 1999, 274, 20027–20033. [DOI] [PubMed] [Google Scholar]
- (30).Wakasugi M; Shimizu M; Morioka H; Linn S; Nikaido O; Matsunaga TJ Biol. Chem 2001, 276, 15434–15440. [DOI] [PubMed] [Google Scholar]
- (31).Tang JY; Hwang BJ; Ford JM; Hanawalt PC; Chu G Mol. Cell 2000, 5, 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hecht SS Chem. Res. Toxicol 1998, 11, 559–603. [DOI] [PubMed] [Google Scholar]
- (33).Gangelhoff TA; Mungalachetty PS; Nix JC; Churchill MEA Nucleic Acids Res. 2009, 37, 3153–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Soll JM; Sobol RW; Mosammaparast N Trends Biochem. Sci 2017, 42, 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Conesa C; Acker J RNA Biol. 2014, 7, 287–290. [DOI] [PubMed] [Google Scholar]
- (36).Alam TI; Kanki T; Muta T; Ukaji K; Abe Y; Nakayama H; Takio K; Hamasaki N; Kang D Nucleic Acids Res. 2003, 31, 1640–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ekstrand MI; Falkenberg M; Rantanen A; Park CB; Gaspari M; Hultenby K; Rustin P; Gustafsson CM; Larsson NG Hum. Mol. Genet 2004, 13, 935–944. [DOI] [PubMed] [Google Scholar]
- (38).Larsson N-G; Garman JD; Oldfors A; Barsh GS; Clayton DA Nat. Genet 1996, 13, 296. [DOI] [PubMed] [Google Scholar]
- (39).Canugovi C; Maynard S; Bayne A-CV; Sykora P; Tian J; de Souza-Pinto NC; Croteau DL; Bohr VA DNA Repair 2010, 9, 1080–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Yoshida Y; Izumi H; Torigoe T; Ishiguchi H; Itoh H; Kang D; Kohno K Cancer Res. 2003, 63, 3729. [PubMed] [Google Scholar]
- (41).Hao Z; Wu T; Cui X; Zhu P; Tan C; Dou X; Hsu K-W; Lin Y-T; Peng P-H; Zhang L-S; Gao Y; Hu L; Sun H-L; Zhu A; Liu J; Wu K-J; He C Mol. Cell 2020, 78, 382–395.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Xu W; Boyd RM; Tree MO; Samkari F; Zhao L Proc. Natl. Acad. Sci. U. S. A 2019, 116, 17792–17799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Brennan RG; Pyzalska D; Blonski WJP; Hruska FE; Sundaralingam M Biochemistry 2002, 25, 1181–1185. [DOI] [PubMed] [Google Scholar]
- (44).Delaney JC; Essigmann JM Proc. Natl. Acad. Sci. U. S. A 2004, 101, 14051–14056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Rubio-Cosials A; Sidow JF; Jimenez-Menendez N; Fernandez-Millan P; Montoya J; Jacobs HT; Coll M; Bernado P; Sola M Nat. Struct. Mol. Biol 2011, 18, 1281–1289. [DOI] [PubMed] [Google Scholar]
- (46).Ngo HB; Kaiser JT; Chan DC Nat. Struct. Mol. Biol 2011, 18, 1290–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.