

Ability of biomass-burning smoke particles to glaciate clouds increases as the plume undergoes atmospheric dilution and aging.
Abstract
Ice-nucleating particles (INPs) in biomass-burning aerosol (BBA) that affect cloud glaciation, microphysics, precipitation, and radiative forcing were recently found to be driven by the production of mineral phases. BBA experiences extensive chemical aging as the smoke plume dilutes, and we explored how this alters the ice activity of the smoke using simulated atmospheric aging of authentic BBA in a chamber reactor. Unexpectedly, atmospheric aging enhanced the ice activity for most types of fuels and aging schemes. The removal of organic carbon particle coatings that conceal the mineral-based ice-active sites by evaporation or oxidation then dissolution can increase the ice activity by greater than an order of magnitude. This represents a different framework for the evolution of INPs from biomass burning where BBA becomes more ice active as it dilutes and ages, making a larger contribution to the INP budget, resulting cloud microphysics, and climate forcing than is currently considered.
INTRODUCTION
Biomass burning occurs globally year-round, releasing complex mixtures of organic and inorganic gaseous and particulate components, minerals, ash, and elemental carbon (soot) to the atmosphere, often in major episodic wildfire events that greatly perturb the Earth-cloud-climate system (1). This burning of plant material occurs naturally in forest fires or can be initiated by prescribed burns or by accidental human activity. With the growth of drought-stricken regions, wildfires are expected to increase in extent and severity and occur in regions not historically prone such as the southeastern United States (2–5). Biomass-burning aerosol (BBA) represents a large fraction of global particulate matter, contributing three-quarters of the total carbonaceous aerosol burden and more than one-third of total black carbon (BC) emissions (6, 7). BBA composition is complex, including organic carbon, elemental carbon, tarballs, minerals, ash, and inorganic salt phases, and this composition determines the aerosol properties and their effects on the atmosphere and climate systems (8–13).
BBA can directly influence the Earth’s radiative balance through the light-absorbing properties of the black and brown carbon present and the light-scattering effects of the aerosol. Indirectly, BBA can affect cloud formation and precipitation through its ability to act as cloud condensation nuclei (CCN) (14–16) or ice-nucleating particles (INPs). Cloud glaciation, which requires INPs to catalyze heterogeneous ice nucleation at temperatures warmer than −35°C, affects the structure, lifetime, precipitation, and radiative properties of clouds. Accurate modeling of the Earth-climate system therefore requires achieving a much more complete understanding of heterogeneous ice nucleation and the sources and properties of different types of INPs (17–19). Considering the sheer mass of BBA emitted globally and the potential impacts of INPs on many atmospheric processes, the intersection of these two topics requires greater attention.
The fraction of BBA particles that are INPs and their ice-nucleating abilities and freezing temperatures greatly vary depending on the type of fuel and combustion conditions (20–25). The first direct evidence of INPs released during biomass-burning events was only reported about 10 years ago, wherein INPs were detected in the smoke of 9 of 21 biomass fuels tested in laboratory studies (22). The authors estimated that BBA is an important source of INPs, which leads to atmospheric INP concentrations that can significantly alter cloud properties on a regional scale. Further experiments detected INPs in the smoke of 13 of 22 fuels tested, with the highest concentrations found during intense flaming combustion (23). Prenni et al. (25) sampled ambient air downwind of prescribed burns and wildfires and detected high number concentrations of INPs during flaming-phase combustion. In other field measurements, INP concentrations were elevated during biomass-burning events, but the authors noted that soil lofted due to the intense fires may have contributed to the measured INPs (20).
Most recently in the study of Jahn et al. (26), we demonstrated that new crystalline mineral phases produced during biomass combustion (i.e., not from lofted soil) are present in both the aerosol and remaining bottom ash. We concluded that these minerals are the major source of ice nucleants in BBA, inducing immersion freezing at temperatures up to −13°C, well above the temperatures at which graphitic soot particles can nucleate ice (26). This suggests that the common assumption that the lofting of already existing dust and soil particles or the presence of ice-active carbonaceous soot particles are the major sources of INPs often found in BBA is inaccurate or incomplete.
The vast majority of studies of INPs from biomass burning have only examined freshly emitted aerosol particles or smoke intercepted of unknown atmospheric age. Yet, studies on the atmospheric processing of mineral-based INPs have revealed notable changes in ice-nucleation activity (INA) in some cases (27–30), suggesting that atmospheric processing may change the mineral-based INPs emitted in BBA. Although we recently established that carbonaceous soot cannot explain the ice nucleation we observe in a variety of nascent BBA from different fuels, it has been established that oxidative aging of graphitic soot surfaces can promote INA (31). These previous aging studies on single-component systems likely do not apply to BBA because of its complex heterogeneous composition, and only a few studies have examined authentic aged BBA. The photochemical aging of wood BBA in a chamber reactor was shown to have no effect on its INA, although the INA of this particular unaged BBA was quite low to begin with, only inducing immersion freezing at −35°C (24). Conversely, wildfire smoke aerosol that traveled 1600 km had the highest ice-active particle number fraction out of all fires in one field study, suggesting that atmospherically aged BBA does retain its ice-nucleating ability or perhaps even experiences an enhancement in INA (25). Together, these findings suggest that there are strong yet unconstrained possibilities for alteration of the ice-nucleating ability of the mostly mineral-based INPs in BBA through atmospheric aging processes.
We have essentially no understanding of the susceptibility of these largely mineral-based biomass-burning INPs to atmospheric chemical aging processes or to what extent aging might alter these critical properties required for modeling the interactions of BBA with cloud systems and the hydrological cycle. Aerosol particles have atmospheric lifetimes of a week or more, and the composition and properties of BBA change rapidly as the smoke plume dilutes and mixes with external reactants and oxidants. We therefore sought to investigate how the immersion freezing ice-nucleating ability of BBA is altered during atmospheric transport such as through evaporation and (photo)oxidation mechanisms by performing controlled simulated aging of BBA produced from the combustion of authentic fuels. We found that evaporation through dilution and exposure to oxidants often results in an increase in the INA of BBA, suggesting that biomass-burning plumes likely have more extensive effects on cloud microphysical properties and climate over larger spatial extents as the smoke is transported through the atmosphere than previously recognized.
RESULTS
Authentic biomass fuels collected within the United States were burned, and the emissions from this open combustion were injected into a large Teflon smog chamber reactor (see fig. S1 for an experimental schematic). The fuels selected are representative of those commonly burned in the western and southeastern United States during wildfires and prescribed burns. The size distribution and chemical composition of the resulting aerosol particles were analyzed online, and particles were also collected on various substrates for subsequent offline analysis. After the completion of each burn and chamber filling, a 10-min waiting period allowed initial mixing of the aerosol in the chamber. Then, a “fresh” aerosol filter sample was collected from the chamber for 2 hours. Four types of aging were conducted: (i) no external perturbation or oxidants added (“time aging”); (ii) hydroxyl radical (·OH) oxidation with added nitrogen oxides (NOx); (iii) removal of organic aerosol (OA) and other semivolatile components using a thermodenuder before injection into the chamber, followed by ·OH oxidation; and (iv) injection of ozone with no ultraviolet (UV) photolysis (dark ozonolysis). After the simulated aging was completed, another mixing period of 10 to 20 min took place, and a separate “aged” filter sample was collected for the last 2 hours of the experiment. The BBA was extracted by vortexing each filter in filtered water to assess the immersion freezing INA using a novel microfluidic device (see Methods for additional details) (32). The INA of fresh BBA between different experiments varies due to natural burn-to-burn variability that results from different combustion conditions at the microscale; previous experiments on the ice-nucleating ability of BBA have also observed this natural variability (22, 23). The discussion of changes in INA with simulated atmospheric aging below always compares the fresh and aged aerosol within the same chamber experiment to account for this variability.
Enhancement of ice activity following time aging and evaporation of OA
The immersion-mode INA of the “time aged” BBA collected between 4 and 6 hours after it remained in the chamber following injection and dilution of the emissions was notably increased for sawgrass and cutgrass BBA compared to BBA collected during the first 2 hours of each experiment (Fig. 1A). These time aging experiments were conducted with no external oxidants added or other perturbations to compare to the stronger forced perturbations used in subsequent experiments; the BBA experiences appreciable evaporation just by dilution of the nascent smoke into the chamber. This increase in INA induced by several hours of holding the aerosol in the Teflon chamber is unexpected given that any oxidants present in the nascent BBA would be quickly depleted—the elemental O:C (a measure of the OA oxidation state) for time aging experiments increased on the order of 30%, while other more aggressive aging regimes increased the OA oxidation state by >90%. The increase in INA (ns) here is larger than observed when external perturbation and/or oxidants were applied, as presented below.
Fig. 1. Ice-active surface site density (ns) plotted versus freezing temperature.
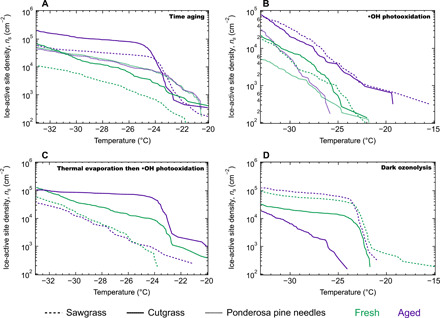
Each panel shows one type of aging of BBA produced from combustion of cutgrass, sawgrass, or ponderosa pine. Fresh samples (before external perturbation or time aging) are shown in green, and aged samples following several hours of chamber aging are shown in purple. (A) Time aging experiments revealed substantial increases in INA along with evaporation of organic carbon aerosol. (B) Hydroxyl radical photooxidative aging caused slight increases in INA (additional experiments shown in fig. S2). (C) Thermal evaporation of the BBA followed by hydroxyl radical photooxidation revealed mixed effects on INA. (D) Ozonolysis without photochemistry resulted in no observed changes or a prominent decrease in INA in one case, along with substantial increases in the OA mass loading. A subset of these experiments shown with 95% confidence intervals is provided in fig. S3, and the ice-active site density normalized to the mass concentration of BC aerosol (a conserved nonvolatile tracer) rather than aerosol surface area is provided in fig. S9.
Aerosol composition measurements indicate substantial evaporative loss of OA, seen through the decrease in the OA-to-BC (a conserved tracer) mass ratio (OA:BC) by up to 20% (Fig. 2A). BBA is well known to contain OA of intermediate to low volatility (their saturation vapor pressure) that can experience considerable evaporation through smoke plume dilution (33, 34), and vapor wall losses in Teflon chambers promote evaporation in laboratory experiments. The OA is heterogeneously distributed throughout the complex BBA and is often nonuniformly mixed in individual submicrometer particles that also contain soot, inorganic salts, and/or mineral phases (26). The evaporation of semivolatile OA observed here would reveal more of the ice-active surface sites already present in the BBA, allowing them to interact directly with water and nucleate ice that cannot occur when the sites are concealed. This explains the large increase in ice activity observed in the aged BBA and the sharp increase in the ice-active site density (ns) over a narrow temperature range, which indicates that the ice nucleants in both fresh and aged BBA have similar properties. BBA produced by the combustion of ponderosa pine needles contains many fewer Si-containing mineral particles compared to the grass fuels, which could explain why an increase in INA was not observed for this fuel type (26).
Fig. 2. Exemplary evolution of submicrometer aerosol chemical composition in the four types of simulated atmospheric aging explored.
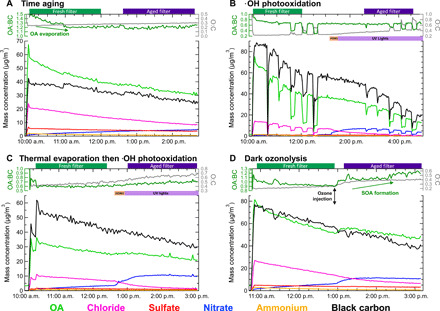
Each panel shows SP-AMS chemical composition measured for one type of aging. Mass concentration (trace color corresponds to chemical component) in SP-mode is plotted in the lower portion; the OA:BC ratio—a measure of the gain or loss of OA versus the conserved BC tracer that only undergoes chamber wall loss—is plotted on the upper left axis. A measure of OA oxidation state, the O:C atomic ratio from EI-mode measurements, is plotted on the upper right axis. OA:BC and O:C are unreliable during the first few minutes of the experiment while the chamber is filling. Collection times of fresh and aged filters for INP analysis are shown by green and purple bars, respectively, on the top of each panel. (A) Cutgrass time aging experiment with considerable evaporation of OA observed in the OA:BC ratio. (B) Sawgrass photooxidation experiment; HONO injection is shown by the orange bar and UV illumination by the purple bar. Sudden decreasing mass concentration and increasing O:C occurred when the aerosol particles were passed through a heated thermodenuder before entering the SP-AMS. (C) Sawgrass experiment where the BBA was subjected to thermal desorption at 250°C before injection into chamber. HONO injection is shown by the orange bar, and UV illumination is shown by the purple bar. (D) Cutgrass dark ozonolysis experiment, with 350 ppb of ozone injected at the labeled time. The formation of SOA is indicated through the increase in OA:BC following ozone injection.
Enhancement of ice activity through photooxidative aging
Consistent increases in INA following hydroxyl radical (·OH) aging under high NOx conditions were demonstrated across most temperatures for all fuel types (Fig. 1B), although the changes were smaller than in our time aging experiments with no added oxidants. Similar to the time aging experiments, the ponderosa pine needle BBA had the smallest increase in INA, only increasing at temperatures below −29.5°C. The hydroxyl radical is an important and powerful atmospheric oxidant and was generated by UV photolysis of nitrous acid (HONO) injected into the chamber. The increase in INA could be due to oxidation of graphitic soot particle surfaces, oxidation of mineral surfaces, or changes in the OA components that are prone to ·OH oxidation. BBA that is dominated by BC and does not contain many mineral species as identified by transmission electron microscopy (TEM)/energy-dispersive x-ray (EDX) particle analysis tends to have low INA that does not increase with photooxidation. This is presumably because there are limited mineral-based ice-active sites to uncover; see fig. S4 for further explanation.
To understand how changes to the OA components could explain the observed increases in INA, we compared the volatility of fresh and aged biomass-burning OA by measuring the mass fraction remaining following thermal desorption at different temperatures up to 200°C (fig. S5) (35). The aged aerosol consistently lost less OA mass than the fresh aerosol at every desorption temperature, indicating that aged BBA is less volatile than the fresh aerosol, similar to previous findings (36, 37). This supports what was also observed in time aging experiments where the more volatile OA present at the beginning of the experiments evaporated, uncovering ice-active surface sites and increasing the INA of aged BBA samples. Thermodenuder analysis also showed that the carbon oxidation state of the remaining OA components increased as indicated by an increased oxygen-to-carbon mass ratio following aging, corresponding to their decreased volatility (Fig. 2B). Because oxidation can also increase volatility through the organic carbon backbone fragmentation channel, the less-volatile OA observed here is the result of components that gained more oxygenated functional groups while avoiding fragmentation (38).
Oxidation of organic carbon molecules tends to increase hydrophilicity and water solubility such that the removal of OA from ice-active sites may be promoted by dissolution following oxidative aging. This mechanism would be highly relevant for immersion-mode freezing in mixed-phased clouds—the dominant heterogeneous ice nucleation mechanism that we studied here where the INP is immersed in a cloud droplet before nucleating ice. Liquid droplets containing these INPs may exist long enough before experiencing freezing conditions such that the more viscous, low-volatility organic compounds also dissolve and reveal ice-active sites in addition to the prompt removal of the more water-soluble oxidized organic carbon.
Alteration of ice activity from photooxidation following thermal evaporation of semivolatile aerosol components
No consistent trend in INA was observed following the photooxidation of BBA that had first been subjected to thermal desorption (Fig. 1C). In these experiments, most OA was removed by passing the biomass emissions through a thermodenuder heated to 250°C before injection into the smog chamber. A smaller amount of organic mass entered the chamber compared to unheated BBA; refer to Fig. 2C for an example of aerosol composition. This method allowed us to examine the INA of the initial aerosol population stripped of most of the OA. The subsequent addition of the hydroxyl radical directly targets the oxidative aging of the aerosol composed of the remaining very low volatility organic carbon components, inorganics, minerals, and graphitic soot.
In the cutgrass experiment, there was a sharp increase in the ice-active site density specifically at −23°C following thermal evaporation then oxidation. This suggests an enhancement in a specific type of ice-active site because of the large increase in INA in a narrow temperature range. One possible type is the mineral phases present in BBA, which could more effectively nucleate ice upon exposure to the hydroxyl radical, especially because the removal of the more volatile OA in the thermodenuder would expose more of the mineral surfaces. Another candidate ice-active site in BBA is graphitic soot, whose surfaces could also become exposed after the removal of the more volatile OA. ·OH oxidation could also produce surface hydroxyl groups on the graphitic surfaces that will interact strongly with water through hydrogen bonding. The molecular dynamics simulations of Lupi and Molinero (39) revealed that OH-modified graphitic surfaces were more effective at inducing freezing than unmodified graphitic surfaces. Increased hydrophilicity of soot particles through atmospheric aging has been demonstrated experimentally to increase INA at cirrus cloud temperatures of <−40°C (31, 40–42). While authentic combustion soot particles are not typically ice active at mixed-phase cloud temperatures of >−35°C (31, 43–45), it is possible that the soot particles emitted from some types of biomass fuel combustion are ice active at warmer temperatures than soot from fossil fuel combustion, and/or that these BBA soot particle surfaces are more susceptible to enhancements in INA through oxidation.
In the sawgrass experiment, the denuded fresh and aged aerosol had nearly the same INA. The BBA from the cutgrass and sawgrass experiments shown in Fig. 1C had similar BC content (48 and 53%, respectively) following evaporation in the thermodenuder before entering the chamber, and so the observed differences in INA are not simply due to BC. While the trends in INA for the thermally denuded aerosol vary by experiment, it is noteworthy that these experiments with most OA removed still result in ice-active site densities above background levels and within the same orders of magnitude as unaltered fresh BBA. This further supports the idea that ice-active sites in BBA active under immersion freezing conditions are not organic carbon based. Our previous analysis of the BBA and ash produced from these same fuels concluded that the production of new mineral phases from the biomass combustion itself is the major source of the ice nucleants, and that fuels that produced the most soot had the weakest or even unmeasurable INA at temperatures warmer than −25°C (26). These observations are supported by a recent study of prescribed burns and wildfires that estimated that BC contributed at most 10% to observed INP concentrations (44).
Impairment of ice activity from production of secondary OA by dark ozonolysis
The INA of BBA subjected to dark ozonolysis was lower than that of fresh aerosol in some cases, likely due to the production of secondary OA (SOA) that covered ice-active surface sites (Fig. 1D). In this aging mechanism, ~300 ppb (parts per billion) of ozone was injected into the chamber with no UV light illumination. Previous studies have shown that dark ozonolysis reliably produces SOA mass and the largest increase in total OA from biomass-burning emissions, making this aging mechanism a good test for the effects of SOA production and OA particle coatings on ice activity (46, 47). Note that some ·OH is still generated without UV photolysis, produced by the ozonolysis of unsaturated organic molecules such as monoterpenes or alkenes (46, 48). Dark ozonolysis did result in the greatest SOA production compared to other aging mechanisms, with the OA:BC mass ratio increasing by up to 25%, as shown in Fig. 2D.
The fresh and aged BBA had essentially the same ice-nucleating abilities following dark ozonolysis or even a decrease in INA for the cutgrass experiment (Fig. 1D). We attribute this to increasing SOA coating amount and thickness on the BBA, counteracting the effects of the evaporation and oxidation plus dissolution of OA observed in the time aging and ·OH aging experiments. These results also indicate that the SOA coatings are not sufficiently removed when the aerosol is extracted into water for immersion freezing analysis, perhaps because the SOA generated during dark ozonolysis is less fragmented and of lower water solubility and therefore remains concealing the ice-active sites. The already existing primary hydrocarbon-like OA likely only partially coats the highly heterogeneous BBA particles, and SOA production may also act to more completely coat the complex aerosol through condensation of oxidized OA. SOA of an oxidation state similar to the O:C of the aged BBA in our experiments is often found to phase separate from the aqueous phase and adopt a core-shell morphology with the organic phase on the outside (49).
Analysis of individual BBA particles
The collected BBA was analyzed at a single-particle level using TEM to investigate the potential particle types that are responsible for the observed ice activity and how these respond to the simulated aging. Many mineral-based particles coated in organic carbon phases were observed. Figure 3A shows a fractal soot particle that is agglomerated with an iron-based mineral as determined by EDX spectroscopy. The particle in Fig. 3B contains potassium chloride salts and a mineral core made of magnesium, aluminum, silicon, and oxygen, also surrounded by an organic coating that appears to have partially evaporated under the vacuum of the TEM. The particle in Fig. 3C has several inorganic salt phases surrounded by a dense organic carbon coating. These particle types and mixtures are all representative of fresh BBA filter samples, where heterogeneous ice nucleation was observed above −25°C. Note that the small <500-nm size of the mineral components suggests that these were formed during combustion, as the majority of minerals lofted by mechanical action from soil dust or biomass ash would be supermicrometer. In our previous work, the most crystalline mineral phases were present in all samples of both aerosol and ash from the tall grass fuels, which also contain more INPs and have higher INA than BBA from wood fuels (26). As most mineral particles in the BBA were found to be submicrometer, these potential INPs will have longer lifetimes versus gravitational settling, undergo atmospheric transport over longer distances, and exert more extensive effects on cloud microphysics over larger spatial scales than the much larger supermicrometer lofted ash particles would (26).
Fig. 3. TEM images of fresh BBA.
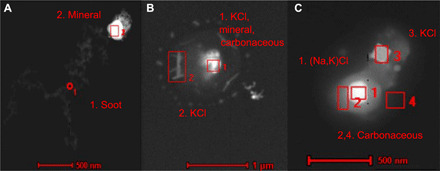
(A) and (B) are from ponderosa pine needles and (C) is from sawgrass BBA collected on substrates indicating the presence of OA coatings around mineral-containing particles. Particle (A) is a fractal soot particle agglomerated with an iron-based mineral in the region indicated by the box. Particle (B) has a mixed core in region 1 composed of KCl, minerals, and carbonaceous material, with additional KCl in region 2 and a carbonaceous OA coating that appears gray surrounding the entire particle. Particle (C) contains several inorganic salt phases in regions 1 and 3 and is also surrounded by an OA coating that appears gray in regions 2 and 4. EDX spectra of the boxed regions are provided in fig. S6.
DISCUSSION
Simulated atmospheric aging indicates that the INA and INP number concentration of authentic BBA would generally increase as the aerosol undergoes atmospheric transport, although plume dilution must also be accounted for. The evaporation of organic coatings off the BBA reveals mineral-based ice-active sites, increasing the ice-nucleating ability of the BBA during most experiments. Simulations based off of FLAME III biomass-burning experiments estimated that 35% of the loss of OA after combustion was due to organic evaporation driven by loss of semivolatile OA following smoke dilution into the chamber and vapor partitioning to the chamber walls (50). Fresh BBA is composed of 20 to 90% organic carbon compounds, a substantial fraction of which are known to evaporate upon dilution from near-fire to dispersed plume aerosol concentrations, but overall evaporation rates and partitioning depend on many factors such as aerosol mass loading, fuel type and combustion conditions, and dilution rate (33). These factors also contribute to the variability in INA among other studies of INPs from biomass burning.
Loss of OA through natural evaporation was observed at the beginning of nearly all chamber experiments, and in the time aging and ·OH photooxidation experiments, the OA:BC ratio remained fairly constant throughout the rest of these experiments. Therefore, these two aging mechanisms both resulted in similar trends in INA. The additional chemical oxidation during ·OH photooxidation experiments likely increased the water solubility of OA particle coatings, allowing for easier dissolution during immersion-mode experiments and uncovering of ice-active surface sites. However, more SOA mass was generated during the latter part of the ·OH aging experiments compared to the time aging experiments where total OA only decreased in time. This may account for the smaller increase in ice-active site density following ·OH photooxidation by not resulting in as extensive an uncovering of ice-active sites as in the time aging experiments with more substantial evaporation of OA. Direct oxidation of mineral surfaces within the BBA may have also led to the increased INA observed in aged BBA produced from mineral-rich grass fuels, but this mechanism could not be directly tested.
Even greater SOA production such as from dark ozonolysis (that avoids OA fragmentation by photolysis) further conceals ice-active sites on the particles, resulting in no increase or even a decrease in INA with aging. Previous literature has shown that SOA coatings can decrease the INA of other INPs in some instances. For example, Arizona test dust and the mineral dust illite were coated with SOA created from the ozonolysis of α-pinene, and aerosol mass fraction percentages of SOA as low as 17% were found to decrease the deposition-mode INA of these atmospheric mineral dust proxies (27). However, the immersion-mode INA of desert mineral dusts did not significantly change when coated in SOA produced from α-pinene ozonolysis (51). Here, SOA produced by the dark ozonolysis of authentic biomass-burning smoke appears to be concealing the ice-active sites in BBA, presumably because the biomass-burning SOA is more effective at covering the ice-active sites compared to previous terpene SOA coating experiments. The SOA produced here may also be less volatile and more viscous than the proxy SOA created through α-pinene ozonolysis and therefore remain concealing ice-active sites during immersion-mode freezing. These explanations are just reasonable predictions based off of previous knowledge of how the deposition and immersion-mode freezing of mineral particle systems respond to surface coatings, but they are supported by our immersion freezing experiments and aerosol composition data (28, 29). While different studies demonstrate variable degrees of SOA production or OA loss in biomass-burning plumes, our results are relevant for plumes that undergo net SOA production, such as those from larger fires in more polluted areas (52, 53).
Figure 4 illustrates a new framework to understand how INPs from biomass burning and their chemical composition and INA coevolve during plume dilution and atmospheric processing based on these findings. Evaporation of OA occurs to different extents following the emission and dilution of the smoke and leads to an increased availability of ice-active surface sites. Photooxidation of organic compounds changes the chemical composition to produce more oxidized OA with increased water solubility such that, when the particle is immersed in a cloud droplet before undergoing immersion freezing, the OA can dissolve more readily to reveal ice-active sites. Condensation of SOA, driven by more oxidized and less volatile OA produced from evaporated organic carbon (that avoids the fragmentation channel), conceals ice-active sites on the particles and decreases the apparent INA. Authentic biomass-burning plumes undergo varying degrees of evaporation, oxidation, and recondensation of OA (47, 54, 55). These processes compete simultaneously to different degrees, depending on fuel composition, combustion conditions, dilution ratios, and atmospheric conditions surrounding biomass plumes that entrain background air and reactants into the plume as it dilutes and spreads (53). Therefore, all these processes must be taken into account when considering the INA of BBA during or following atmospheric aging.
Fig. 4. Schematic representation of the atmospheric coevolution of BBA composition and INA.
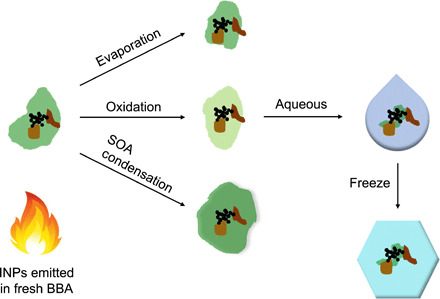
The particle contains minerals depicted in shades of brown, soot depicted in black, and OA depicted in green. Evaporation of OA leads to increased availability of ice-active sites. Oxidation of OA changes its chemical composition to be more oxidized and water-soluble organic carbon such that, when the particle is immersed in a droplet, the OA can dissolve more readily to reveal ice-active sites. SOA condensation from oxidation and condensation of organic carbon onto the particle conceals ice-active sites. This schematic represents the different BBA aging processes that may occur in the atmosphere, which are not ordinarily decoupled as depicted and can occur simultaneously or in competition to varying degrees.
This work is the first to investigate how INPs released by the combustion of several different biomass fuels evolve under different simulated atmospheric aging schemes. A strong enhancement of the INA through some types of atmospheric aging was found. INA increased more after aging in BBA produced by the combustion of grassy fuels—which have a much higher mineral content—than other fuels like pine needles and woods that produce BBA with higher OA and BC content instead. These differences can be attributed to the observed changes in particle properties and composition that lead to an uncovering of preexisting ice-active surface sites that we recently demonstrated are mostly mineral phases produced by the biomass combustion itself (13, 26). TEM images of the BBA collected in these experiments also show mineral phases and their mixtures with other particle components such as BC soot and salt phases that are often coated by organic carbon.
Here, we show how the INA of BBA can be enhanced through some types of relevant atmospheric aging mechanisms. Unlike many previous studies that have shown that the INA of mineral dust particles can sometimes be impaired through atmospheric aging, here, we find that some types of aging actually enhance the INA of BBA (27–29, 56). The different effects of aging for these two mineral-containing aerosol systems are due to the presence of primary components emitted in the nascent BBA that can conceal the ice-active particle surfaces such as the mineral phases. Aging can lead to the partial removal of these coatings, thus increasing the INA. Mineral dust particles do not typically contain primary particle coatings that resist dissolution such as the hydrocarbon-like tar material common in BBA (8, 11, 57). For atmospheric mineral dust, coatings are acquired through atmospheric aging, thus concealing or chemically altering ice-active surface sites with soluble inorganic components or oxidized SOA that sometimes lead to a decrease in INA.
Dilution, evaporation, and exposure of the BBA to oxidants often resulted in an increase of ice-active site density greater than an order of magnitude even at temperatures above −25°C and an increase in the onset freezing temperature of up to 8°C. This process would likely be enhanced under many atmospherically relevant scenarios, as OA evaporation is driven more rapidly and extensively by the continual dilution of the smoke plume during transport that proceeds for several days or even weeks for submicrometer aerosol; most of the mineral particles observed in the BBA that are the likely source of ice nucleants were submicrometer in size. Removal of these OA coatings can also make reactive mineral and halide salt phases available for direct reaction with atmospheric reactants, such as the activation of chlorine as ClNO2(g) and HCl(g) from Cl−(aq) through reactive uptake of N2O5(g) that we have recently demonstrated in BBA (58, 59).
The implications of this new framework for the coevolution of BBA composition and INA are that the emitted INPs will make important contributions to the distribution of atmospheric INPs over larger spatial and temporal extents and at warmer cloud temperatures than previously understood. The effective concentration of INPs emitted in BBA will increase during plume dilution as particle coatings experience net evaporation, until dilution of the aerosol particle numbers overcomes this effect. Previous estimates of the INP concentration in fresh BBA emitted during wildfires include 106 to 1015 INPs/m2 of burned land (active at −30°C) based on a variety of tested fuels, and 5 × 1010 to 1 × 1012 INPs/m2 of burned grassland (active at −25°C) based on our recent report on INPs emitted by combustion of tall grasses. This estimate of INP emissions results in an area of 104 km2, with a 5-km plume height having INP concentrations elevated above typical background levels due to biomass burning and likely sufficient to modify cloud microphysics, from just 1 m2 of burned land (22, 26). Our results presented here show that atmospheric aging enhances the ice-active site density by up to a factor of 27 at −25°C. Therefore, atmospheric aging of BBA could increase the area affected by INPs by at least 1 decade, causing an estimated area of 105 km2 by 5 km in altitude to be affected for each 1 m2 of burned grassland. These observed increases in ice activity in terms of both INP concentration and freezing temperature are substantial considering the vast areas of biomass consumed in wildfires, especially in recent years. Fully understanding the impacts of this newly understood dynamic evolution of the INPs in BBA requires investigation using chemical transport models that properly account for the evolution of BBA composition during atmospheric transport (33, 60).
METHODS
A small portion of each biomass fuel was placed into a partially enclosed galvanized steel pan and lit from the side with a butane lighter. The remaining biomass fuel was gradually added to maintain flaming phase combustion, until a total of 0.5 kg was burned. The fuels used were giant cutgrass (Zizaniopsis miliacea), obtained at the UF/IFAS Center for Aquatic and Invasive Plants in Florida, USA; ponderosa pine needles (Pinus ponderosa) obtained at the Klamath Basin National Wildlife Refuge Complex in Tulelake, California, USA; sawgrass (Cladium jamaicense), obtained at the Loxahatchee National Wildlife Refuge in Florida, USA; and birch and fatwood logs, purchased locally. The smoke emissions were injected and diluted using Dekati eductor diluters (Dekati DI-1000) into a 12 m3 Teflon smog chamber (26, 59). Before each experiment, the chambers were purged overnight using filtered clean air and UV lights until particle number concentrations were <50 cm−3. No artificial circulation mechanism was used in the chamber apart from some possible natural convection due to temperature gradients. The only times when the air within the chamber is turbulent is during aerosol injection and injection of HONO or ozone vapor; during these times, we did not collect filters for INA analysis. For the BBA present in these chamber experiments, given the small particle size (geometric mean diameter ~200 nm) and low concentration (<105 #/cm3), gravitational settling is not significant and there is also no significant associated coagulation under the experimental time scales (61). Particle wall loss was the dominant loss mechanism and is mainly driven by electrostatic forces close to the Teflon chamber walls, and there are no differences in the prevailing particle loss mechanisms or rates between the fresh and aged time periods that would affect our results.
Online particle analysis included a scanning mobility particle sizer (SMPS; TSI Inc., DMA model 3082 and CPC model 3775) for aerosol size distribution measurements between 8 and 749 nm in mobility diameter, and a soot-particle aerosol mass spectrometer (SP-AMS; Aerodyne Inc.) for submicrometer particle chemical characterization. The SP-AMS uses an infrared (IR) laser to measure refractory BC soot, and the instrument was operated by switching the IR laser on/off every 60 s (62). During both modes, a 600°C tungsten thermal vaporizer was used to vaporize nonrefractory aerosol components: organic carbon aerosol (OA), nitrate, sulfate, chloride, and ammonium. Laser-on (BC) mode was used to obtain the mass concentrations shown in Fig. 2, and the O:C mass ratio was determined from laser-off (EI) mode. For volatility analysis, the aerosol was alternated between passing through a heated thermodenuder (centerline residence time of 23 s) or through an unheated bypass line before being sampled by the SP-AMS. Gas monitors included a chemiluminescent NOx analyzer (Advanced Pollution Instrumentation Inc., model 200A) and an ozone analyzer (Teledyne, model T400). Particles were collected onto copper formvar TEM grids (carbon type B, 400 mesh, Ted Pella #01754-F) for offline electron microscopy analysis. TEM/EDX measurements were acquired at the Environmental Molecular Sciences Laboratory (EMSL) at Pacific Northwest National Laboratory (PNNL) using a Titan 80-300 scanning/TEM equipped with an Si(Li) detector at an accelerating voltage of 300 keV.
Particles were collected for ice nucleation analysis on polycarbonate filters (GE Healthcare 111103, Nuclepore 50-nm pore size) using an inline 44-mm filter holder and refrigerated until use. Immediately before analysis, particles were extracted off of the filter by vortexing the filter in a polypropylene Falcon tube with 3 ml of high-performance liquid chromatography (HPLC)–grade water (Sigma-Aldrich HPLC Plus #34877) that was prefiltered (Anotop 25 Plus 0.02-μm pore size, Whatman #6809-4102), as this results in the lowest level of background freezing (63). The suspension was then filled into a custom microfluidic chip, where 600 uniformly sized isolated 6-nl droplets are produced and can be tested simultaneously with a background freezing temperature for filtered water of <−33°C (32). The chip was placed atop a thermoelectric cooling element and cooled at 1°C min−1, with droplet freezing detected using the observed change in grayscale value. For some earlier experiments presented in the Supplementary Materials, the droplet freezing assay was performed using a conventional droplet-on-substrate method and 0.1-μl droplets (63). Filtered water background freezing occurred well below the droplet freezing temperature observed of the BBA samples, and the frozen fraction spectra of handling filter blanks were slightly higher than filtered water background freezing but still much lower than most BBA samples; see fig. S7 for additional information. The ice-active surface site density (ns) was calculated using the total surface area of the collected aerosol samples based off of the total air volume passed through the filters and an averaged aerosol size distribution measured by SMPS throughout the fresh or aged BBA collection period (64). To ensure that the observed trends in INA with aging were not simply due to artifacts in how the aerosol surface area distribution (used to calculate ns) changes with time across each experiment, we normalized the INA to the aerosol mass concentration of BC, as shown in fig. S9. This method of normalizing the INA to a conserved nonvolatile aerosol tracer reveals the same trends in INA for all four aging mechanisms as are shown in Fig. 1. Changes in the aerosol size distribution and surface area therefore do not explain the observed changes in the INA following various types of simulated atmospheric aging.
Acknowledgments
For assisting with electron microscopy analysis, we thank L. Kovarik, S. China, and the staff at the Environmental and Molecular Sciences Laboratory at Pacific Northwest National Laboratory. For providing us with authentic biomass fuels, we thank S. Freitas at the Klamath Basin National Wildlife Refuge Complex, S. Enloe at the University of Florida UF/IFAS Center for Aqueous and Invasive Plants, J. Glueckert at the Loxahatchee National Wildlife Refuge, and B. Wyatt at the Savannah National Wildlife Refuge. Funding: This work was supported by the NSF (CHE-1554941, AGS-1552608, and CBET-1804737) and an NSF Graduate Research Fellowship to M.J.P. L.G. Jahl and B.B.B. were partially supported by Steinbrenner Fellowships from CMU. T.A.B. was partially supported by collaborative student grant from the Department of Mechanical Engineering at CMU. Author contributions: Conceptualization: R.C.S.; investigation: L.G. Jahl, T.A.B., M.J.P., L.G. Jahn, K.P.C., B.B.B., S.G., and R.C.S.; formal analysis: L.G. Jahl, T.A.B., M.J.P., L.G. Jahn, K.P.C., W.D.F., and R.C.S.; writing: L.G. Jahl and R.C.S. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabd3440/DC1
REFERENCES AND NOTES
- 1.Fromm M., Peterson D., Di Girolamo L., The primary convective pathway for observed wildfire emissions in the upper troposphere and lower stratosphere: A targeted reinterpretation. J. Geophys. Res. Atmos. 124, 13254–13272 (2019). [Google Scholar]
- 2.Stevens-Rumann C. S., Kemp K. B., Higuera P. E., Harvey B. J., Rother M. T., Donato D. C., Morgan P., Veblen T. T., Evidence for declining forest resilience to wildfires under climate change. Ecol. Lett. 21, 243–252 (2018). [DOI] [PubMed] [Google Scholar]
- 3.O’Dell K., Ford B., Fischer E. V., Pierce J. R., Contribution of wildland-fire smoke to US PM2.5 and its influence on recent trends. Environ. Sci. Technol. 53, 1797–1804 (2019). [DOI] [PubMed] [Google Scholar]
- 4.McClure C. D., Jaffe D. A., US particulate matter air quality improves except in wildfire-prone areas. Proc. Natl. Acad. Sci. U.S.A. 115, 7901–7906 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams A. P., Abatzoglou J. T., Gershunov A., Guzman-Morales J., Bishop D. A., Balch J. K., Lettenmaier D. P., Observed impacts of anthropogenic climate change on wildfire in California. Earth’s Future 7, 892–910 (2019). [Google Scholar]
- 6.Bond T. C., Doherty S. J., Fahey D. W., Forster P. M., Berntsen T., DeAngelo B. J., Flanner M. G., Ghan S., Kärcher B., Koch D., Kinne S., Kondo Y., Quinn P. K., Sarofim M. C., Schultz M. G., Schulz M., Venkataraman C., Zhang H., Zhang S., Bellouin N., Guttikunda S. K., Hopke P. K., Jacobson M. Z., Kaiser J. W., Klimont Z., Lohmann U., Schwarz J. P., Shindell D., Storelvmo T., Warren S. G., Zender C. S., Bounding the role of black carbon in the climate system: A scientific assessment. J. Geophys. Res. Atmos. 118, 5380–5552 (2013). [Google Scholar]
- 7.Reddy M. S., Boucher O., A study of the global cycle of carbonaceous aerosols in the LMDZT general circulation model. J. Geophys. Res. 109, D14202 (2004). [Google Scholar]
- 8.Adachi K., Sedlacek A. J. III, Kleinman L., Springston S. R., Wang J., Chand D., Hubbe J. M., Shilling J. E., Onasch T. B., Kinase T., Sakata K., Takahashi Y., Buseck P. R., Spherical tarball particles form through rapid chemical and physical changes of organic matter in biomass-burning smoke. Proc. Natl. Acad. Sci. U.S.A. 116, 19336–19341 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silva P. J., Liu D.-Y., Noble C. A., Prather K. A., Size and chemical characterization of individual particles resulting from biomass burning of local Southern California species. Environ. Sci. Technol. 33, 3068–3076 (1999). [Google Scholar]
- 10.Li J., Pósfai M., Hobbs P. V., Buseck P. R., Individual aerosol particles from biomass burning in southern Africa: 2, Compositions and aging of inorganic particles. J. Geophys. Res. Atmos. 108, 8484 (2003). [Google Scholar]
- 11.Sedlacek A. J. III, Buseck P. R., Adachi K., Onasch T. B., Springston S. R., Kleinman L., Formation and evolution of tar balls from northwestern US wildfires. Atmos. Chem. Phys. 18, 11289–11301 (2018). [Google Scholar]
- 12.Reid J. S., Koppmann R., Eck T. F., Eleuterio D. P., A review of biomass burning emissions part II: Intensive physical properties of biomass burning particles. Atmos. Chem. Phys. 5, 799–825 (2005). [Google Scholar]
- 13.Vassilev S. V., Baxter D., Vassileva C. G., An overview of the behaviour of biomass during combustion: Part I. Phase-mineral transformations of organic and inorganic matter. Fuel 112, 391–449 (2013). [Google Scholar]
- 14.Petters M. D., Carrico C. M., Kreidenweis S. M., Prenni A. J., DeMott P. J., Collett J. L. Jr., Moosmüller H., Cloud condensation nucleation activity of biomass burning aerosol. J. Geophys. Res. 114, D22205 (2009). [Google Scholar]
- 15.Lathem T. L., Beyersdorf A. J., Thornhill K. L., Winstead E. L., Cubison M. J., Hecobian A., Jimenez J. L., Weber R. J., Anderson B. E., Nenes A., Analysis of CCN activity of Arctic aerosol and Canadian biomass burning during summer 2008. Atmos. Chem. Phys. 13, 2735–2756 (2013). [Google Scholar]
- 16.Engelhart G. J., Hennigan C. J., Miracolo M. A., Robinson A. L., Pandis S. N., Cloud condensation nuclei activity of fresh primary and aged biomass burning aerosol. Atmos. Chem. Phys. 12, 7285–7293 (2012). [Google Scholar]
- 17.DeMott P. J., Prenni A. J., Liu X., Kreidenweis S. M., Petters M. D., Twohy C. H., Richardson M. S., Eidhammer T., Rogers D. C., Predicting global atmospheric ice nuclei distributions and their impacts on climate. Proc. Natl. Acad. Sci. U.S.A. 107, 11217–11222 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seinfeld J. H., Bretherton C., Carslaw K. S., Coe H., DeMott P. J., Dunlea E. J., Feingold G., Ghan S., Guenther A. B., Kahn R., Kraucunas I., Kreidenweis S. M., Molina M. J., Nenes A., Penner J. E., Prather K. A., Ramanathan V., Ramaswamy V., Rasch P. J., Ravishankara A. R., Rosenfeld D., Stephens G., Wood R., Improving our fundamental understanding of the role of aerosol-cloud interactions in the climate system. Proc. Natl. Acad. Sci. U.S.A. 113, 5781–5790 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan J., Wang Y., Rosenfeld D., Liu X., Review of aerosol-cloud interactions: Mechanisms, significance, and challenges. J. Atmos. Sci. 73, 4221–4252 (2016). [Google Scholar]
- 20.McCluskey C. S., DeMott P. J., Prenni A. J., Levin E. J. T., McMeeking G. R., Sullivan A. P., Hill T. C. J., Nakao S., Carrico C. M., Kreidenweis S. M., Characteristics of atmospheric ice nucleating particles associated with biomass burning in the US: Prescribed burns and wildfires. J. Geophys. Res. Atmos. 119, 10458–10470 (2014). [Google Scholar]
- 21.DeMott P. J., Petters M. D., Prenni A. J., Carrico C. M., Kreidenweis S. M., Collett J. L. Jr., Moosmüller H., Ice nucleation behavior of biomass combustion particles at cirrus temperatures. J. Geophys. Res. Atmos. 114, D16205 (2009). [Google Scholar]
- 22.Petters M. D., Parsons M. T., Prenni A. J., DeMott P. J., Kreidenweis S. M., Carrico C. M., Sullivan A. P., McMeeking G. R., Levin E. J. T., Wold C. E., Collett J. L., Moosmüller H., Ice nuclei emissions from biomass burning. J. Geophys. Res. 114, D07209 (2009). [Google Scholar]
- 23.Levin E. J. T., McMeeking G. R., DeMott P. J., McCluskey C. S., Carrico C. M., Nakao S., Jayarathne T., Stone E. A., Stockwell C. E., Yokelson R. J., Kreidenweis S. M., Ice-nucleating particle emissions from biomass combustion and the potential importance of soot aerosol. J. Geophys. Res. Atmos. 121, 5888–5903 (2016). [Google Scholar]
- 24.Chou C., Kanji Z. A., Stetzer O., Tritscher T., Chirico R., Heringa M. F., Weingartner E., Prévôt A. S. H., Baltensperger U., Lohmann U., Effect of photochemical ageing on the ice nucleation properties of diesel and wood burning particles. Atmos. Chem. Phys. 13, 761–772 (2013). [Google Scholar]
- 25.Prenni A. J., Demott P. J., Sullivan A. P., Sullivan R. C., Kreidenweis S. M., Rogers D. C., Biomass burning as a potential source for atmospheric ice nuclei: Western wildfires and prescribed burns. Geophys. Res. Lett. 39, L11805 (2012). [Google Scholar]
- 26.Jahn L. G., Polen M. J., Jahl L. G., Brubaker T. A., Somers J., Sullivan R. C., Biomass combustion produces ice-active minerals in biomass-burning aerosol and bottom ash. Proc. Natl. Acad. Sci. U.S.A. 117, 21928–21937 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Möhler O., Benz S., Saathoff H., Schnaiter M., Wagner R., Schneider J., Walter S., Ebert V., Wagner S., The effect of organic coating on the heterogeneous ice nucleation efficiency of mineral dust aerosols. Environ. Res. Lett. 3, 025007 (2008). [Google Scholar]
- 28.Sullivan R. C., Petters M. D., Demott P. J., Kreidenweis S. M., Wex H., Niedermeier D., Hartmann S., Clauss T., Stratmann F., Reitz P., Schneider J., Sierau B., Irreversible loss of ice nucleation active sites in mineral dust particles caused by sulphuric acid condensation. Atmos. Chem. Phys. 10, 11471–11487 (2010). [Google Scholar]
- 29.Sullivan R. C., Miñambres L., Demott P. J., Prenni A. J., Carrico C. M., Levin E. J. T., Kreidenweis S. M., Chemical processing does not always impair heterogeneous ice nucleation of mineral dust particles. Geophys. Res. Lett. 37, L24805 (2010). [Google Scholar]
- 30.Kulkarni G. R., Sanders C., Zhang K., Liu X., Zhao C., Ice nucleation of bare and sulfuric acid-coated mineral dust particles and implication for cloud properties. J. Geophys. Res. Atmos. 119, 9993–10011 (2014). [Google Scholar]
- 31.Mahrt F., Marcolli C., David R. O., Grönquist P., Meier E. J. B., Lohmann U., Kanji Z. A., Ice nucleation abilities of soot particles determined with the Horizontal Ice Nucleation Chamber. Atmos. Chem. Phys. 18, 13363–13392 (2018). [Google Scholar]
- 32.Brubaker T., Polen M., Cheng P., Ekambaram V., Somers J., Anna S. L., Sullivan R. C., Development and characterization of a “store and create” microfluidic device to determine the heterogeneous freezing properties of ice nucleating particles. Aerosol Sci. Tech. 54, 79–93 (2020). [Google Scholar]
- 33.Hodshire A. L., Akherati A., Alvarado M. J., Brown-Steiner B., Jathar S. H., Jimenez J. L., Kreidenweis S. M., Lonsdale C. R., Onasch T. B., Ortega A. M., Pierce J. R., Aging effects on biomass burning aerosol mass and composition: A critical review of field and laboratory studies. Environ. Sci. Technol. 53, 10007–10022 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Robinson A. L., Donahue N. M., Shrivastava M. K., Weitkamp E. A., Sage A. M., Grieshop A. P., Lane T. E., Pierce J. R., Pandis S. N., Rethinking organic aerosols: Semivolatile emissions and photochemical aging. Science 315, 1259–1262 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Cain K. P., Pandis S. N., A technique for the measurement of organic aerosol hygroscopicity, oxidation level, and volatility distributions. Atmos. Meas. Tech. 10, 4865–4876 (2017). [Google Scholar]
- 36.Lim C. Y., Hagan D. H., Coggon M. M., Koss A. R., Sekimoto K., De Gouw J., Warneke C., Cappa C. D., Kroll J. H., Secondary organic aerosol formation from the laboratory oxidation of biomass burning emissions. Atmos. Chem. Phys. 19, 12797–12809 (2019). [Google Scholar]
- 37.Hennigan C. J., Miracolo M. A., Engelhart G. J., May A. A., Presto A. A., Lee T., Sullivan A. P., McMeeking G. R., Coe H., Wold C. E., Hao W.-M., Gilman J. B., Kuster W. C., de Gouw J., Schichtel B. A., Collett J. L., Kreidenweis S. M., Robinson A. L., Chemical and physical transformations of organic aerosol from the photo-oxidation of open biomass burning emissions in an environmental chamber. Atmos. Chem. Phys. 11, 7669–7686 (2011). [Google Scholar]
- 38.Kroll J. H., Donahue N. M., Jimenez J. L., Kessler S. H., Canagaratna M. R., Wilson K. R., Altieri K. E., Mazzoleni L. R., Wozniak A. S., Bluhm H., Mysak E. R., Smith J. D., Kolb C. E., Worsnop D. R., Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol. Nat. Chem. 3, 133–139 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Lupi L., Molinero V., Does hydrophilicity of carbon particles improve their ice nucleation ability? J. Phys. Chem. A 118, 7330–7337 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Koehler K. A., DeMott P. J., Kreidenweis S. M., Popovicheva O. B., Petters M. D., Carrico C. M., Kireeva E. D., Khokhlova T. D., Shonija N. K., Cloud condensation nuclei and ice nucleation activity of hydrophobic and hydrophilic soot particles. Phys. Chem. Chem. Phys. 11, 7906–7920 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Mahrt F., Kilchhofer K., Marcolli C., Grönquist P., David R. O., Rösch M., Lohmann U., Kanji Z. A., The impact of cloud processing on the ice nucleation abilities of soot particles at cirrus temperatures. J. Geophys. Res. Atmos. 125, e2019JD030922 (2020). [Google Scholar]
- 42.Mahrt F., Alpert P. A., Dou J., Grönquist P., Arroyo P. C., Ammann M., Lohmann U., Kanji Z. A., Aging induced changes in ice nucleation activity of combustion aerosol as determined by near edge X-ray absorption fine structure (NEXAFS) spectroscopy. Environ. Sci. Process. Impacts 22, 895–907 (2020). [DOI] [PubMed] [Google Scholar]
- 43.Kanji Z. A., Ladino L. A., Wex H., Boose Y., Burkert-Kohn M., Cziczo D. J., Krämer M., Overview of ice nucleating particles. Meteorol. Monogr. 58, 1.1–1.33 (2017). [Google Scholar]
- 44.Schill G. P., DeMott P. J., Emerson E. W., Rauker A. M. C., Kodros J. K., Suski K. J., Hill T. C. J., Levin E. J. T., Pierce J. R., Farmer D. K., Kreidenweis S. M., The contribution of black carbon to global ice nucleating particle concentrations relevant to mixed-phase clouds. Proc. Natl. Acad. Sci. U.S.A. 117, 22705–22711 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanji Z. A., Welti A., Corbin J. C., Mensah A. A., Black carbon particles do not matter for immersion mode ice nucleation. Geophys. Res. Lett. 47, e2019GL086764 (2020). [Google Scholar]
- 46.Tkacik D. S., Robinson E. S., Ahern A. T., Saleh R., Stockwell C., Veres P., Simpson I. J., Meinardi S., Blake D. R., Yokelson R. J., Presto A. A., Sullivan R. C., Donahue N. M., Robinson A. L., A dual-chamber method for quantifying the effects of atmospheric perturbations on secondary organic aerosol formation from biomass burning emissions. J. Geophys. Res. Atmos. 122, 6043–6058 (2017). [Google Scholar]
- 47.Ahern A. T., Robinson E. S., Tkacik D. S., Saleh R., Hatch L. E., Barsanti K. C., Stockwell C. E., Yokelson R. J., Presto A. A., Robinson A. L., Sullivan R. C., Donahue N. M., Production of secondary organic aerosol during aging of biomass burning smoke from fresh fuels and its relationship to VOC precursors. J. Geophys. Res. Atmos. 124, 3583–3606 (2019). [Google Scholar]
- 48.Paulson S. E., Orlando J. J., The reactions of ozone with alkenes: An important source of HOx in the boundary layer. Geophys. Res. Lett. 23, 3727–3730 (1996). [Google Scholar]
- 49.Gorkowski K., Donahue N. M., Sullivan R. C., Aerosol optical tweezers constrain the morphology evolution of liquid-liquid phase-separated atmospheric particles. Chem 6, 204–220 (2020). [Google Scholar]
- 50.Bian Q., May A. A., Kreidenweis S. M., Pierce J. R., Investigation of particle and vapor wall-loss effects on controlled wood-smoke smog-chamber experiments. Atmos. Chem. Phys. 15, 11027–11045 (2015). [Google Scholar]
- 51.Kanji Z. A., Sullivan R. C., Niemand M., Demott P. J., Prenni A. J., Chou C., Saathoff H., Möhler O., Heterogeneous ice nucleation properties of natural desert dust particles coated with a surrogate of secondary organic aerosol. Atmos. Chem. Phys. 19, 5091–5110 (2019). [Google Scholar]
- 52.Shrivastava M., Cappa C. D., Fan J., Goldstein A. H., Guenther A. B., Jimenez J. L., Kuang C., Laskin A., Martin S. T., Ng N. L., Petaja T., Pierce J. R., Rasch P. J., Roldin P., Seinfeld J. H., Shilling J., Smith J. N., Thornton J. A., Volkamer R., Wang J., Worsnop D. R., Zaveri R. A., Zelenyuk A., Zhang Q., Recent advances in understanding secondary organic aerosol: Implications for global climate forcing. Rev. Geophys. 55, 509–559 (2017). [Google Scholar]
- 53.Hodshire A. L., Bian Q., Ramnarine E., Lonsdale C. R., Alvarado M. J., Kreidenweis S. M., Jathar S. H., Pierce J. R., More than emissions and chemistry: Fire size, dilution, and background aerosol also greatly influence near-field biomass burning aerosol aging. J. Geophys. Res. Atmos. 124, 5589–5611 (2019). [Google Scholar]
- 54.Garofalo L. A., Pothier M. A., Levin E. J. T., Campos T., Kreidenweis S. M., Farmer D. K., Emission and evolution of submicron organic aerosol in smoke from wildfires in the Western United States. ACS Earth Space Chem. 3, 1237–1247 (2019). [Google Scholar]
- 55.Donahue N. M., Epstein S. A., Pandis S. N., Robinson A. L., A two-dimensional volatility basis set: 1. Organic-aerosol mixing thermodynamics. Atmos. Chem. Phys. 11, 3303–3318 (2011). [Google Scholar]
- 56.Cziczo D. J., Froyd K. D., Gallavardin S. J., Moehler O., Benz S., Saathoff H., Murphy D. M., Deactivation of ice nuclei due to atmospherically relevant surface coatings. Environ. Res. Lett. 4, 044013 (2009). [Google Scholar]
- 57.Pósfai M., Gelencsér A., Simonics R., Arató K., Li J., Hobbs P. V., Buseck P. R., Atmospheric tar balls: Particles from biomass and biofuel burning. J. Geophys. Res. Atmos. 109, D06213 (2004). [Google Scholar]
- 58.Goldberger L. A., Jahl L. G., Thornton J. A., Sullivan R. C., N2O5 reactive uptake kinetics and chlorine activation on authentic biomass-burning aerosol. Environ. Sci. Process. Impacts 21, 1684–1698 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Ahern A. T., Goldberger L., Jahl L., Thornton J., Sullivan R. C., Production of N2O5 and ClNO2 through nocturnal processing of biomass-burning aerosol. Environ. Sci. Technol. 52, 550–559 (2018). [DOI] [PubMed] [Google Scholar]
- 60.Bian Q., Jathar S. H., Kodros J. K., Barsanti K. C., Hatch L. E., May A. A., Kreidenweis S. M., Pierce J. R., Secondary organic aerosol formation in biomass-burning plumes: Theoretical analysis of lab studies and ambient plumes. Atmos. Chem. Phys. 17, 5459–5475 (2017). [Google Scholar]
- 61.Mahfouz N. G. A., Donahue N. M., Primary ion diffusion charging and particle wall loss in smog chamber experiments. Aerosol Sci. Tech. 54, 1058–1069 (2020). [Google Scholar]
- 62.Onasch T. B., Trimborn A., Fortner E. C., Jayne J. T., Kok G. L., Williams L. R., Davidovits P., Worsnop D. R., Soot particle aerosol mass spectrometer: Development, validation, and initial application. Aerosol Sci. Tech. 46, 804–817 (2012). [Google Scholar]
- 63.Polen M., Brubaker T., Somers J., Sullivan R. C., Cleaning up our water: Reducing interferences from nonhomogeneous freezing of “pure” water in droplet freezing assays of ice-nucleating particles. Atmos. Meas. Tech. 11, 5315–5334 (2018). [Google Scholar]
- 64.Vali G., Quantitative evaluation of experimental results an the heterogeneous freezing nucleation of supercooled liquids. J. Atmos. Sci. 28, 402–409 (1971). [Google Scholar]
- 65.Vali G., Revisiting the differential freezing nucleus spectra derived from drop-freezing experiments: Methods of calculation, applications, and confidence limits. Atmos. Meas. Tech. 12, 1219–1231 (2019). [Google Scholar]
- 66.Polen M., Lawlis E., Sullivan R. C., The unstable ice nucleation properties of Snomax bacterial particles. J. Geophys. Res. Atmos. 121, 11666–11678 (2016). [Google Scholar]
- 67.Maynard A. D., Estimating aerosol surface area from number and mass concentration measurements. Ann. Occup. Hyg. 47, 123–144 (2003). [DOI] [PubMed] [Google Scholar]
- 68.Buonanno G., Dell’Isola M., Stabile L., Viola A., Uncertainty budget of the SMPS-APS system in the measurement of PM1, PM2.5, and PM10. Aerosol Sci. Tech. 43, 1130–1141 (2009). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabd3440/DC1