

Interferon response sensors are expressed in the islet of Langerhans during the course of type 1 diabetes.
Abstract
Previous results indicate the presence of an interferon (IFN) signature in type 1 diabetes (T1D), capable of inducing chronic inflammation and compromising b cell function. Here, we determined the expression of the IFN response markers MxA, PKR, and HLA-I in the islets of autoantibody-positive and T1D donors. We found that these markers can be coexpressed in the same islet, are more abundant in insulin-containing islets, are highly expressed in islets with insulitis, and their expression levels are correlated with the presence of the enteroviral protein VP1. The expression of these markers was associated with down-regulation of multiple genes in the insulin secretion pathway. The coexistence of an IFN response and a microbial stress response is likely to prime islets for immune destruction. This study highlights the importance of therapeutic interventions aimed at eliminating potentially persistent infections and diminishing inflammation in individuals with T1D.
INTRODUCTION
Type 1 diabetes (T1D) is a chronic autoimmune disease that manifests when a critical amount of β cell mass is lost. β cell death leads to insulin deficiency, meaning that patients with T1D need exogenous insulin for life. Recent studies have shown that T1D can develop throughout the first six decades of life, affecting not only children and adolescents but also adults. Forty-two percent of T1D cases occur in those aged >30 years (1). Moreover, the incidence of T1D is increasing by 2 to 3% per year (2).
Although genetic components accounts for ∼80% of T1D heritability (3), the triggering events are believed to depend on environmental factors (4). In particular, epidemiological, clinical, and pathological studies in humans support an association between viral infections and T1D (5, 6). However, there is uncertainty regarding their potential role as causative agents. Enteroviruses (EVs), and especially coxsackievirus B (CVBs), are most frequently associated with T1D (7, 8). EVs are non-enveloped, positive-sense, single-stranded RNA viruses that belong to the family Picornaviridae and preferentially replicate in the respiratory and gut mucosa (9, 10). There are six serotypes of CVB (CVB1 to CVB6), which bind to and are internalized via the coxsackie and adenovirus receptor (CAR) (11). CAR is a transmembrane protein that belongs to the tight junction protein family and is highly expressed in β cells (12, 13). CVB display pancreatic tropism (13) and infects pancreatic islets during systemic infections in infants, with weaker effects on the exocrine pancreas.
Recent studies have shown an association between EV infections and the appearance of islet autoantibodies or T1D (5, 14). More recently, The Environmental Determinants of Diabetes in the Young study analyzed the entire known virome in stool samples obtained from newborns with increased genetic risk for T1D (15). The study revealed that prolonged shedding of the same EV serotype was strongly associated with islet autoimmunity, whereas multiple, independent infections without prolonged shedding were less likely to be associated with T1D (15). EVs have also been detected in the islets of pancreatic biopsies from living adults with recent-onset T1D and in pancreatic samples from children and adults obtained at organ donation or postmortem (16, 17).
Islet cells can detect viral infections through specialized molecules that are activated at an early stage of infection. One of these is the double-stranded RNA (dsRNA) sensor protein kinase R (PKR), which belongs to the eukaryotic initiation factor 2α (eIF2α) phosphorylating family of kinases, and it is induced by interferons (IFNs) and thus play a more prominent role in immune responses than other members of this family (18). Upon viral infection, the activation of PKR leads to inhibition of general protein synthesis (19) while inducing autocrine IFN production (20). However, PKR is also activated in response to other stress signals, suggesting that it is likely to play an important role in diseases associated with chronic inflammation and cellular stress, such as T1D. Enhanced PKR gene expression was detected in human islets infected with CVBs or exposed to cytokines (21). In addition, PKR was detected in the islets of donors with and without diabetes (22) and on cells positive for the EV capsid protein VP1 (23).
It is now well accepted that an IFN-I transcriptional signature precedes the appearance of autoantibodies (24). Increased responsiveness to IFN-I has been recently identified as a feature of T1D, suggesting its potential use as a biomarker (25). In addition, a significant increase in nearly half of the IFN-stimulated genes assessed was detected in laser-captured islets obtained from living donors with recent-onset T1D (26). This includes up-regulation of the human myxovirus resistance protein (MxA) by more than twofold in insulitic islets. MxA is a key mediator of the IFN-inducible antiviral responses against a wide range of viruses, and it is often used as a surrogate marker for IFN activity. IFN-I is also a potent inducer of human leukocyte antigen I (HLA-I) molecules, a hallmark of T1D (27). HLA-I is hyperexpressed in the islets of T1D donors at the protein and RNA levels, and its expression is correlated with that of signal transducer and activator of transcription 1 (STAT1), a critical protein involved in mediating the antiviral responses to IFNs (27). In the context of T1D, it is therefore tempting to speculate that an IFN-mediated increase in HLA-I expression is correlated with abnormal antigen presentation, which would favor activation of autoreactive T cells and ultimately cause β cell destruction.
In this study, we took advantage of the availability of tissues from the Network for Pancreatic Organ Donors with Diabetes (nPOD), which provides human pancreas samples for research purposes. We present a fully quantitative, systematic study aimed at investigating the expression of IFN and microbial stress response markers, including PKR, MxA, and HLA-I, and the presence of viral footprints, such as dsRNA and VP1, in human pancreas samples obtained from nondiabetic, autoantibody-positive (AAb+), and recent-onset T1D donors. Our findings point to the existence of an antiviral signature in T1D that affects a selective group of islets and is likely to contribute to β cell destruction by increasing cellular stress, diminishing insulin production, and attracting immune cells to the islets. The results also highlight the importance of therapeutic interventions aimed at eliminating potentially persistent infections and diminishing inflammation in individuals with T1D.
RESULTS
HLA-I hyperexpression can be detected in double AAb+ subjects and is present in 6.0 to 40.5% of islets in T1D donors
HLA-I expression increases upon IFN-I stimulation, and cells with enhanced HLA-I expression might become better targets for cytotoxic T cells recognizing viral and/or autoantigens. HLA-I expression was analyzed in all of the islets from 12 nondiabetic (13,262 islets), 7 AAb+ (5035 islets), and 9 T1D (4629 islets) donors. Frozen pancreatic sections were stained by immunofluorescence. Blinded analysis was performed on each tissue section, and each islet was classified into three categories based on the staining intensity as normal, elevated, or hyperexpressed, as previously described (27). Heatmaps in which each cell was color coded according to its mean intensity value were used for visualization (Fig. 1A). Normal HLA-I expression levels were detected in all of the donors, with nearly all islets in the nondiabetic group showing normal expression (percentage of islets: mean, 95.5% and range, 63 to 100%) (Fig. 1B). The percentage of islets showing elevated HLA-I expression was higher in T1D (mean, 13.3% and range, 6.2 to 17.3%) and AAb+ (mean, 16% and range, 0.4 to 37.9%) donors than in nondiabetic donors (mean, 4% and range, 0 to 37%). HLA-I hyperexpression was exclusively present in the islets of AAb+ (mean, 10.7% and range, 0 to 52.1%) and T1D (mean, 18.1% and range, 6.0 to 40.5%) donors (Fig. 1B). The highest percentage of islets showing HLA-I hyperexpression was detected in an AAb+ donor with multiple islet autoantibodies (no. 6450; mean, 52.1%). Hyperexpression of HLA-I was detected in β cells and other islet cells, as reported previously (27). HLA-I expression did not differ in sections obtained from the head, body, or tail of the pancreas (fig. S1A).
Fig. 1. HLA-I hyperexpression is present in the islets of double AAb+ and T1D donors.
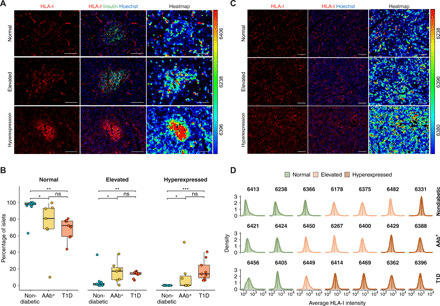
(A and C) Representative immunofluorescence images from islets (A) and exocrine tissue (C) showing normal (top), elevated (middle), and hyperexpression (bottom) of HLA-I. The right panels display heatmaps based on the relative fluorescence intensity unit of each cell. The maximum and minimum values are displayed in red and blue, respectively. Scale bars, 100 μm. (B) Box plots showing median and first and third quartile for the percentage of islets showing normal, elevated, or hyperexpression of HLA-I. The upper and lower whiskers extend from the hinges to the largest or lowest value no further than 1.5 times the interquartile range from the hinge. Data beyond the end of the whiskers are called “outlying” points. Each dot represents a donor. (D) Density plots showing the average HLA-I intensity in the exocrine tissue for a representative number of donors in each donor group. The level of expression is shown in green (normal), light orange (elevated), and dark orange (hyperexpression). *P < 0.05, **P < 0.01, and ***P < 0.001; ns, not significant.
To provide a complete overview of HLA-I expression in the exocrine and endocrine compartments, we also analyzed the exocrine tissue and classified HLA-I expression as normal, elevated, or hyperexpressed. For each section, the mean HLA-I intensity of each cell was determined. The endocrine compartments were then excluded from this analysis. Heatmaps were also used for visualization, as described above (Fig. 1C), and the pixel intensity values from all the exocrine cells were visualized as histograms (Fig. 1D). Unlike endocrine HLA-I expression, we found no differences in exocrine HLA-I expression between the three groups. Normal, elevated, or hyperexpressed exocrine HLA-I was detected in individuals from any donor group, regardless of disease status (Fig. 1D) and was not correlated with pancreatitis or immune infiltration.
MxA and PKR are expressed in a significantly higher percentage of islets in T1D donors
To investigate the islet expression of IFN response proteins, tissue sections were stained for PKR and MxA by immunofluorescence and blinded analysis was performed (Fig. 2A) in all of the islets from 12 nondiabetic (13,262 islets), 7 AAb+ (5035 islets), and 9 T1D (4629 islets) donors. PKR expression was higher in a greater percentage of islets from AAb+ (percentage of islets: mean, 5.7% and range, 0.4 to 14.7%) and T1D (mean, 10.3% and range, 4.6 to 14.6%) donors than islets from nondiabetic donors (mean, 1.5% and range, 0 to 5.4%) (Fig. 2, B and C). When present, PKR was detected mainly in α cells, although a scattered, punctuate staining pattern was also observed in β cells. PKR expression was very low in the exocrine pancreas, but it was occasionally expressed in immune cells in a few donors.
Fig. 2. MxA and PKR are expressed in a significantly higher percentage of islets in T1D donors.
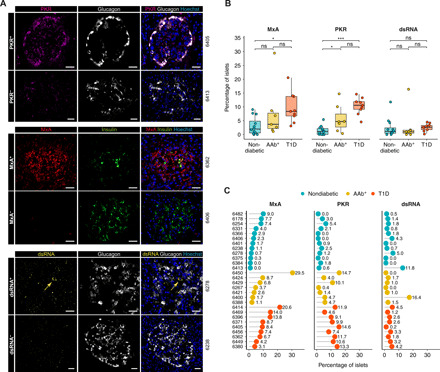
(A) Representative immunofluorescence images of islets positive (+) and negative (−) for PKR (top), MxA (middle), and dsRNA (bottom). Scale bars, 25 μm. (B) Box plots showing median and first and third quartile for the percentage of islets positive for MxA, PKR, and dsRNA. The upper and lower whiskers extend from the hinges to the largest or lowest value no further than 1.5 times the interquartile range from the hinge. Data beyond the end of the whiskers are called outlying points. Each dot represents a donor. (C) Percentage of islets positive for MxA, PKR, and dsRNA for each individual donor. *P < 0.05; ***P < 0.001; ns, not significant.
MxA, which is induced by type I and type III IFNs, was expressed in a higher percentage of islets from AAb+ (mean, 7.7% and range, 1.1 to 29.5%) and T1D (mean, 9.7% and range, 3.1 to 20.6%) donors than from nondiabetic donors (mean, 3.1% and range 0 to 9%) (Fig. 2, B and C). In positive islets, MxA was expressed in all islet cells, not just β cells. Conversely, it was not expressed in acinar cells but was only detected in fiber-like structures scattered across the pancreas. The highest percentage of PKR+ and MxA+ islets was detected in the multiple AAb+ donor no. 6450.
To investigate whether this response is a possible consequence of ongoing viral replication, we evaluated the expression of dsRNA. Only two donors presented >10% of islets that contained dsRNA-positive cells (nondiabetic no. 6413 and AAb+ no. 6400), whereas no significant differences were found between the donor groups (Fig. 2, B and C). Last, no differences in PKR, MxA, or dsRNA positivity were found between sections from the head, body, or tail of the pancreas (fig. S1B).
HLA-I, PKR, and MxA are coexpressed in a small but significant number of islets
To further evaluate whether the IFN response proteins HLA-I, MxA, and PKR and the viral replication marker dsRNA (hereafter, IFN response markers for simplification purposes) were coexpressed on the same islets, we combined data from each section and islet into a single image. Marker maps were generated for each donor, as shown in Fig. 3A. Each positive islet was assigned a color as follows: red for the expression of one marker, yellow for two markers, purple for three markers, and blue for four markers. Then, their expression levels were quantified. The mean percentage of islets that expressed one (mean nondiabetic donors versus AAb+ versus T1D, respectively; 8.7 versus 27.8 versus 28.5%, respectively), two (1.5 versus 6.0 versus 10.2%, respectively), or three (0.1 versus 1.8 versus 2.9%, respectively) markers was consistently higher in AAb+ and T1D donors than in nondiabetic donors (Fig. 3, B and C). Coexpression of four markers on the same islet was rare, being 0% in nondiabetic, 0.1% in AAb+, and 0.1% in T1D donors. The double AAb+ donor no. 6450 presented the highest percentage of islets positive for one, two, and three markers. The most common double and triple combinations were HLA-I/MxA and HLA-I/MxA/PKR, respectively (table S1).
Fig. 3. IFN response markers are coexpressed in a significant number of islets in AAb+ and T1D donors.
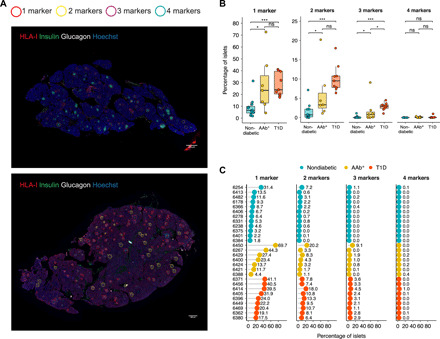
(A) Representative immunofluorescence images of entire tissue sections from a nondiabetic (top, donor no. 6278) and a T1D donor (bottom, donor no. 6362) stained for HLA-I, insulin, glucagon, and Hoechst. Each positive islet was assigned a colored circle based on the number of expressed markers as follows: red for the expression of one marker, yellow for two markers, purple for three markers, and blue for four markers. Scale bars, 290 μm. (B) Box plots showing median and first and third quartile show the percentage of islets positive for one, two, three, or four markers for each donor group. The upper and lower whiskers extend from the hinges to the largest or lowest value no further than 1.5 times the interquartile range from the hinge. Data beyond the end of the whiskers are called outlying points. Each dot represents a donor. (C) Percentage of islets positive for one, two, three, or four markers in individual donors. *P < 0.05; ***P < 0.001; ns, not significant.
Islets expressing one or more IFN response markers show decreased expression of genes that are important for islet function and insulin secretion
We examined RNA expression level in laser-captured microdissected (LCM) islets. For this, RNA was extracted from negative islets and islets positive for one or at least two IFN response markers. Pooled islets were harvested in a manner that did not differentiate between insulin-containing islets (ICIs) and insulin-deficient islets (IDIs). Because the expression profiles of islets with one or a least two markers were similar, these islets were combined for further analysis. A total of 721 unique Entrez Gene IDs with a fold change of ≥1.1 and P value of ≤0.005 were differentially expressed between islets negative for IFN response markers and islets expressing at least one marker. Pathway analysis of these genes showed the strongest enrichment for six pathways: Staphylococcus aureus infection, type 1 diabetes mellitus, allograft rejection, insulin secretion, cell adhesion molecules (CAMs), and maturity-onset diabetes of the young (MODY) (tables S2 and S3). The pathway with the lowest P value was S. aureus infection, which encodes various proteins involved in immune response and inflammation and contained genes that were up-regulated in islets that expressed at least one IFN response marker. The second pathway was type 1 diabetes mellitus, which included up-regulated HLA-I and HLA-II genes, and shared them with the allograft rejection pathway. The insulin secretion pathway included genes that were down-regulated in islets with at least one IFN response marker and encodes proteins involved in glucose transport, voltage-gated ion channels, and vesicle transport. The CAM pathway included up-regulated and down-regulated genes that encode costimulatory and cell-cell adhesion molecules. Last, the MODY pathway includes genes involved in β cell identity and maturation, which were down-regulated in islets expressing at least one marker.
Because the expression of genes involved in the insulin secretion pathway was decreased in islets that expressed at least one IFN response marker, we assessed insulin positivity for each islet and section in T1D donors and evaluated the expression of all IFN response markers, either alone or in combination. We found that HLA-I and MxA were expressed in a significantly higher percentage of ICIs than IDIs, with a similar tendency for PKR expression (Fig. 4A). In T1D donors, the majority of IDIs (67.2%) were negative for all four markers (Fig. 4B). Conversely, 45.6% of the ICIs expressed one marker. Similar results were obtained for the expression of two (23.3%) and three markers (12.3%) on the same islet, indicating that IFN response markers are more frequently coexpressed in islets that retain insulin (Fig. 4B).
Fig. 4. IFN response markers are preferentially expressed in ICIs in T1D donors.
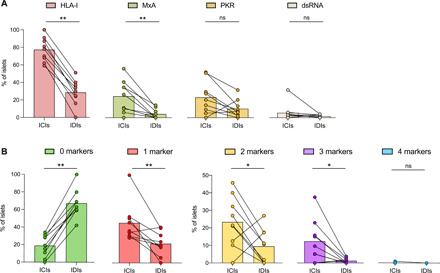
(A) Bar graphs (mean) and individual paired values are shown for the percentage of islets that express HLA-I, MxA, PKR, and dsRNA in ICIs and IDIs in T1D donors. (B) Bar graphs (mean) and individual paired values for the percentage of islets expressing none or one, two, three, or four markers in ICIs and IDIs in T1D donors. *P < 0.05; **P < 0.01; ns, not significant.
In T1D donors, the majority of islets that express HLA-I, MxA, or PKR, either alone or in combination, are highly infiltrated by T cells
Insulitis is a hallmark of T1D, and T cells are the main cell type implicated in β cell destruction. To investigate whether islets that express one or more IFN response markers were preferentially infiltrated by T cells, we stained sections for the marker CD3 (Fig. 5A). Islet T cell infiltration was quantified using machine learning and automated software analysis (4276 islets from nondiabetic donors, 1549 islets from AAb+ donors, and 1815 islets from T1D donors), and the T cell density was calculated for each islet and donor. Islets with a very high CD3+ density were mainly detected in T1D individuals and in the double AAb+ donor no. 6267 (Fig. 5B). The CD3+ cell density was significantly higher in the islets of T1D donors (mean, 62.5 CD3+ cells/mm2) than in AAb+ donors (mean, 26.6 CD3+ cells/mm2). Moreover, it was significantly greater in both of these donor groups than in nondiabetic donors (mean, 14.6 CD3+ cells/mm2) (Fig. 5C). To study the association between the expression of IFN response markers and T cell infiltration, we determined the number of islets that fulfilled the definition of insulitis [at least six CD3+ cells immediately adjacent to or within the islet (28)]. Only 0.4% of islets in nondiabetic, 2.5% in AAb+, and 6.9% in T1D donors satisfied this criterion (Fig. 5D). In nondiabetic donors, 36% of the highly infiltrated islets were positive for at least one marker, while the rest were negative (Fig. 5E). Conversely, in AAb+ donors, 53.9% were positive for one, 20.5% for two, 12.8% for three, and 2.6% for all four markers analyzed. T1D donors were similar to AAb+ donors, as 27% of the infiltrated islets were positive for one, 39.7% for two, 18.2% for three, and 0.8% for four markers (Fig. 5E). Overall, in T1D and AAb+ donors, more than 80% of the islets that fulfilled the definition of insulitis expressed at least one IFN response marker.
Fig. 5. In T1D donors, islets that express HLA-I, MxA, or PKR, either alone or in combination are highly infiltrated.
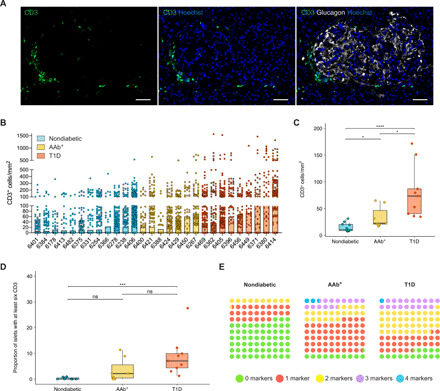
(A) Representative immunofluorescence images of a highly infiltrated islet stained for CD3, glucagon, and Hoechst. Scale bars, 50 μm. (B) Bar graph (mean) for CD3+ cells/mm2 in each donor. The dots represent individual islets. (C) Box plots showing median and first and third quartile for the distribution of CD3+ cells/mm2 for each donor group. The upper and lower whiskers extend from the hinges to the largest or lowest value no further than 1.5 times the interquartile range from the hinge. Data beyond the end of the whiskers are called outlying points. Each dot represents a donor. (D) Box plots (median, first and third quartile, and range) for the proportion of islets with at least six CD3+ cells. Each dot represents a donor. (E) Dot plots for the proportion of islets infiltrated by at least six CD3+ cells expressing none (green) or one (red), two (yellow), three (purple), or four (blue) markers for each donor group. *P < 0.05, ***P < 0.001, ****P < 0.0001, ns, not significant.
Viral footprints can be found in the islets of T1D donors and are correlated with the expression of IFN response markers
Previous reports have revealed an association between EV infections and T1D, and the EV protein VP1 has been detected in the islets of T1D subjects (16, 17, 23). To analyze the presence or absence of VP1, formalin-fixed paraffin-embedded (FFPE) sections were stained in a subgroup of donors, and VP1+ islets were quantified by automated software analysis as described in the STAR Methods. Islets (3337 from nondiabetic donors, 1136 from AAb+ donors, and 851 from T1D donors) were classified as negative, weak VP1+, or strong VP1+ on the basis of the intensity of staining (Fig. 6A). The percentage of weak VP1+ islets was significantly higher in T1D donors (47%) than in nondiabetic (12.4%) and AAb+ (5.7%) donors (Fig. 6B). Strong VP1+ cells were mainly present in the islets of T1D donors, with a higher percentage (24.2%) than in nondiabetic (1.8%) and AAb+ (1.4%) donors. Despite the high percentage of islets in which at least one VP1+ cell was detected, the mean percentage of VP1+ cells per islet was low (5.2% in T1D versus 0.13% in AAb+ versus 0.8% in nondiabetic donors).
Fig. 6. Viral footprints can be found in the islets of T1D donors and are correlated with the expression of IFN response markers.
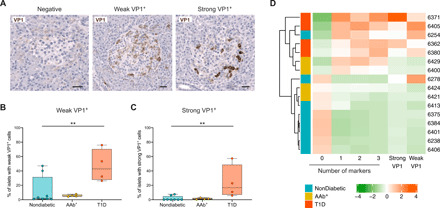
(A) Representative immunohistochemistry images of negative, weak, and strong VP1+ islets. Scale bars, 25 μm. (B and C) Box plots (median, first and third quartile, and range) for the percentage of islets with weak (B) and strong (C) VP1+ cells in each donor group. (D) Heatmap and hierarchical clustering for the percentage of islets expressing none or one, two, or three markers and weak and strong VP1. **P < 0.01.
Last, to study whether there is an association between the presence of viral protein and the expression of IFN response markers in islets, we performed a Spearman correlation analysis. The absence of markers was negatively correlated with weak (r = −0.64, P = 0.015) and strong (r = −0.75, P = 0.003) VP1 positivity. Conversely, the presence of one, two, or three IFN response markers showed moderate to strong correlations with weak (one marker: r = 0.59, P = 0.030; two markers: r = 0.63, P = 0.017; three markers: r = 0.65, P = 0.016) and strong (one marker: r = 0.69, P = 0.007; two markers: r = 0.75, P = 0.003; three markers: r = 0.76, P = 0.003) VP1 positivity. The presence of strong VP1+ cells, but not weak VP1+ cells, was correlated with islet CD3+ infiltration (strong VP1+: r = 0.71, P = 0.007; weak VP1+: r = 0.48, P = 0.08). To visualize these correlations, we constructed a heatmap using centered and scaled data for the proportion of islets expressing none or one, two, or three makers and weak and strong VP1 positivity, as shown in Fig. 6D. Hierarchical clustering separated the donors into the different clinical groups with the exception of the nondiabetic donor no. 6254, who was positive for several markers and weak VP1, and donor no. 6278, who had weak VP1 positivity but low expression of IFN response markers.
Last, we generated radar charts to summarize the expression of IFN response markers, VP1 positivity, and CD3+ infiltration in each donor (fig. S2). For each variable, data were scaled proportionally to the maximum value of the given variable across all donors.
DISCUSSION
In the present study, we systematically analyzed the expression of antiviral immune and cellular host-response factors in the islets of human organ donors with islet autoimmunity and T1D. In addition, we provided a comprehensive analysis of the association of these factors with the presence of viral footprints and immune infiltrates. We have demonstrated the presence of high levels of HLA-I expression in the islets of T1D donors (27). Here, we show that HLA-I is hyperexpressed in individuals with multiple autoantibodies. This indicates that the increase in HLA-I expression in the islets is an early phenomenon that starts before the onset of T1D and persists thereafter. In addition, we show that milder elevations in HLA-I expression can be detected in AAb+ and T1D donors, as well as in some nondiabetic donors. This elevation, without reaching hyperexpression, might be correlated with the initial level of inflammation, although this remains to be proven. Nevertheless, as previously reported, high levels of HLA-I in a subset of islets might reflect a sustained IFN response (29) in which ongoing inflammation could act as a positive feedback regulator, inducing progressively increasing responses to IFNs. Moreover, we report that HLA-I expression was also high in the exocrine pancreas in some individuals and was not correlated with pancreatitis or immune infiltration. This phenomenon does not seem to be a feature of T1D per se because it was observed in donors with and without disease. Further investigations are needed to decipher the contribution of the exocrine compartment to pancreatic inflammation in the context of T1D.
Because HLA-I hyperexpression could be a consequence of IFN-I release in islets, we evaluated the expression of MxA, which is often used as a surrogate marker of IFN production and is a key element of the antiviral response to a wide variety of viruses. Here, we show that MxA expression was significantly increased in islets from T1D donors. Moreover, HLA-I/MxA was the most common combination in islets positive for more than one marker, but some islets showed HLA-I hyperexpression without MxA expression. Almost 30% of the islets expressed MxA in the multiple AAb+ donor no. 6450, indicative of an ongoing IFN response affecting a large subset of islets. This finding, together with the hyperexpression of HLA-I molecules and the presence of IDIs, suggests that this individual might have been close to developing overt disease at the time of donation. Therefore, MxA expression might be an early phenomenon in the pathogenesis of T1D that is triggered in the initial phase of the islet immune response, while HLA-I hyperexpression might persist for longer, as previously reported (30).
In addition to MxA, we studied the expression of the dsRNA sensor PKR. This protein plays an important role in detecting viral RNA, but it is also induced in response to IFN secretion. PKR activation leads to intracellular translational arrest, which affects not only viral but also cellular mRNAs. Because of this unique feature, PKR belongs to the so-called microbial stress response in which stress-related molecules are induced during microbial sensing and activate multiple downstream signaling cascades (31). A previous study has shown the presence of PKR in the islets of donors with T1D, especially in those containing VP1+ cells (23). Here, we show that PKR expression was significantly elevated in the islets from T1D and double AAb+ donors than in nondiabetic donors. It has been proposed that infected cells may oscillate between inhibition of translation through PKR activation and the production of antiviral cytokines such as IFN to control the infection but to avoid apoptosis due to an excessively long translational arrest or due to sustained IFN release (31). Nevertheless, in these islets, PKR activation is likely to affect hormone production and perpetuate inflammatory and stress responses.
Despite the evident role of PKR as a dsRNA sensor, we only detected weak dsRNA staining in the islets of a few donors, with no significant differences between the three donor groups. However, this might not be unexpected given the capacity of EVs to establish persistence, with only minimal active replication (32, 33). We cannot exclude the possibility that the antibody that we used was unable to detect small quantities of dsRNA despite showing good performance on [polyinosinic:polycytidylic acid (poly (I:C))]–transfected cells. Another possibility is that the RNA was partially degraded. The pancreas is prone to RNA degradation due to the numerous digestive enzymes produced in the exocrine compartment, and several conditions and procedures have been reported to contribute to RNA degradation before extraction and before/during tissue processing (34). As previously reported, it is also possible that PKR is activated by IFN production (35) or cellular stress (36), independently of the presence of dsRNA.
In addition to investigating the expression of HLA-I, MxA, PKR, and dsRNA individually, we analyzed whether these markers are coexpressed on the same islets because coexpression would indicate the activation of multiple inflammatory and stress response pathways. We found that coexpression of multiple IFN response markers was more common in AAb+ and T1D donors than in nondiabetic donors. In addition, in T1D donors, these markers were more frequently expressed in ICIs than in IDIs. This suggests that, after the onset of T1D, these responses can be detected in the early phase of the destruction process, when β cells are still present, and tend to disappear following their total loss. While the overall number of islets that expressed more than one marker was small, these islets are likely to have an ongoing inflammatory response and could become perfect targets for the immune system. We previously showed that individuals with T1D have greater infiltration in the exocrine and endocrine pancreas than individuals without T1D (37). In agreement with those findings, in the current study, we detected a significantly higher percentage of infiltrated islets in AAb+ and T1D donors, but as expected, only a small proportion of these islets fulfilled the current definition of insulitis (28). In T1D and AAb+ donors, more than 80% of the islets with insulitis expressed one or more IFN response markers. This suggests that the coexpression of IFN response and microbial stress markers might contribute to the creation of an inflammatory microenvironment that could attract immune cells to the islets. These inflammatory responses are likely to be common to multiple pathogens, and therefore, they could be triggered by different viruses.
IFN-α was shown to decrease insulin synthesis and secretion in vitro, causing β cell dysfunction (38). Here, we performed transcriptomic analysis of laser-captured islets and demonstrated that islets expressing at least one IFN response marker showed decreased expression of genes important to islet function and insulin secretion, including fundamental components of vesicles and membrane fusion machinery. This suggests that the molecular machinery involved in insulin secretion might be disrupted in islets with a sustained IFN response. The gene with the lowest P value within the top six pathways was HLA-B, which was up-regulated in islets that expressed at least one marker. Moreover, genes involved in immune response and inflammation were also up-regulated. Some of the top genes in these pathways were FcγRIIA and CD40, which are expressed in antigen-presenting cells (APCs) and are involved in phagocytosis and APC activation, respectively. FcγRIIA expression increases in response to IFN-I and IFN-II (39), and its activation induces the production of proinflammatory cytokines, including IFN and tumor necrosis factor–α (40). CD40 signaling on APCs favors their maturation, induces cytokine production, and triggers T cell activation and differentiation (41). In addition, CD40 is expressed on human β cells upon cytokine treatment (42), and CD40-mediated signaling has been implicated in the early development of insulitis (43). Together, our data suggest that islets that express IFN response markers might be the perfect scenario for antigen recognition, presentation, and immune activation.
Recent studies have reported the expression of the EV capsid protein VP1 in the pancreas of more than half of the recent-onset T1D donors studied through the Exeter Archival Diabetes Biobank, in the United Kingdom, and 80% of those analyzed in the nPOD cohort (17). In the current study, weakly and strongly stained VP1+ cells were detected in all the T1D donors. A limitation of our study is that no antibodies directed against EVs worked reliably on human frozen tissue sections. Instead, we used a commercially available, validated anti-VP1 antibody that works exclusively on paraffin-embedded tissue sections (44). For this reason, we were unable to directly evaluate the coexpression of IFN response markers and VP1 on the same islet. Despite this limitation, we found that expression of the viral protein is strongly associated with the presence of IFN response markers. Another limitation is the cross-sectional nature of the study, which does not allow us to conclude whether this is a sign of a low-level, slow, and sustained infection that could induce chronic inflammation. The existence of persistent, defective infections (32) and empty viral particles containing VP1 but no viral genome have also been reported (33). Several T1D gene risk variants predispose to an exacerbated inflammatory and immune response (including IFN-I production), which might decrease the magnitude of infection and instead favor viral persistence (5). An interesting point is that we could detect IFN responses in individuals with multiple autoantibodies, but we did not find any differences in the presence of VP1+ islets between AAb+ and nondiabetic donors. A possible scenario is that in the context of autoimmunity, if antiviral immune responses are sustained for a long time and/or a few persistently infected cells remain in some islets, IFN and stress response proteins could be constantly triggered and perpetuated over time. As the disease progresses, the diabetogenic environment could make the islets more permissive to new infections, increasing the likelihood of detecting viral proteins and antiviral responses. This highlights the importance of studying these responses in longitudinal samples to further understand their temporal dynamics. As previously suggested, multiple viral infections by one or more viruses, and the potentially exacerbated and sustained immune response, could create a fertile field in which infiltrating immune cells would become activated (45).
Here, we provide a fully quantitative and systematic analysis of IFN-mediated microbial stress responses in the islets of nondiabetic, AAb+, and T1D human organ donors. We found that the IFN response markers MxA, PKR, and HLA-I are expressed in the islets of multiple AAb+ and T1D donors and that these proteins (i) can be coexpressed on the same islet, (ii) are more abundant in islets that still retain insulin, (iii) are highly expressed in islets with insulitis, and (iv) are correlated with the presence of the EV protein VP1. Targeting IFN-α, the IFN receptor, or different elements of the IFN pathway prevents or delays the onset of T1D in animal models (46, 47). In addition, a recent study has shown that Janus kinase inhibitors decrease HLA-I hyperexpression, endoplasmic reticulum stress, and β cell apoptosis in human β cells (48). Together, our data support the use of compounds targeting the IFN response pathway for potential intervention in T1D, possibly in combination with antiviral agents aimed at eliminating potential persistent infections (49). Our data additionally indicates that applying these interventions early in the disease process (before the onset of clinical T1D) might be critical to completely abrogate islet inflammation, infiltration, and, ultimately, β cell destruction.
MATERIALS AND METHODS
Study design
The objective of this study was to determine, in a fully quantitative manner, the presence or absence of an IFN response in the pancreas of individuals with and without diabetes and its possible association with the presence or absence of viral protein and islet immune infiltration. We hypothesized that these responses would be detected before the onset of T1D and that they would be associated with an increased level of infiltration in the islets. Pancreas sections from cadaveric organ donors were collected through nPOD (www.jdrfnpod.org/). Frozen sections were obtained from different regions of the pancreas (head, body, and/or tail) from nondiabetic (n = 12), recent-onset T1D (within 0 to 2 years of onset, n = 9), and single (n = 2) and double (n = 5) AAb+ donors. Overall, 280 frozen tissue sections were analyzed. Islet hormones, IFN response markers, and immune infiltration were determined in four consecutive 5-μm-thick sections. In addition, two consecutive 10-μm-thick frozen sections from the same donors were used for LCM and transcriptomics analysis. Last, an additional 4-μm-thick FFPE section was used for immunohistochemical staining of viral protein. Blinded assessment was performed in all the sections from all the donors. Only subjects with residual ICIs were included in this study. Table S4 provides a summary of the donor characteristics for each group, with extended information in table S5. All experimental procedures were approved by the Ethics Committee at the Technical University of Munich (protocol no. 215/17 S) and the Helmholtz Zentrum Munich, Institute of Diabetes Research.
Antibody validation by immunocytochemistry
The polyclonal rabbit anti-MxA (1:200; Novus Biologicals), polyclonal rabbit anti-EIF2AK2 (PKR) (1:100; Atlas Antibodies), and monoclonal mouse anti-dsRNA [J2, immunoglobulin G2A (IgG2a), 1:100; Scicons] antibodies were validated by immunocytochemistry using the human retinal pigment epithelial cell line ARPE-19 (fig. S3). Cells were incubated in Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 (DMEM/F-12) supplemented with 1% penicillin-streptomycin (10,000 U/ml) and inactivated 10% fetal calf serum at 37°C in an atmosphere containing 5% CO2. Cells were subcultured once weekly at a ratio of 1:2. To induce MxA and PKR expression, cells were stimulated with IFN-α2b (1000 U) for 24 hours before staining. The presence of dsRNA was evaluated after transfecting cells with fluorescein-labeled poly(I:C) (InvivoGen)–Lipofectamine complexes (Invitrogen) for 24 hours. Afterward, IFN-treated or untreated cells and transfected or nontransfected cells were cytospun, fixed with 1% paraformaldehyde, and permeabilized with 0.1% Triton X-100. After blocking with 2% goat serum, cells were incubated with the specified antibodies for 1 hour at room temperature. Detection was performed by incubation at room temperature for 1 hour with the following secondary antibodies: goat anti-mouse Alexa Fluor 647 (1:1000; Invitrogen) and goat anti-rabbit Alexa Fluor 555 (1:1000; Invitrogen). For nuclei visualization, Hoechst 33342 (1:5000; Invitrogen) was applied for 8 min and slides were mounted with ProLong Gold Antifade (Invitrogen).
Immunofluorescence
Pancreatic frozen sections were stained for insulin, glucagon, HLA-I, MxA, PKR, dsRNA, and CD3 by immunofluorescence. Tissue sections were fixed with 1% paraformaldehyde and blocked with 2% goat serum. The sections were then incubated with the following primary antibodies for 1 hour at room temperature: polyclonal guinea pig anti-insulin antibody (1:140; Dako), monoclonal rabbit anti-glucagon (EP3070 clone, 1:400; Abcam), monoclonal mouse anti-glucagon (K79bB10 clone, IgG1; 1:400; Abcam), monoclonal mouse anti-glucagon (181402 clone, IgG2a, 1:100; R&D Systems), monoclonal mouse anti-human HLA-ABC (W6/32 clone, IgG2a, 1:100; Dako), polyclonal rabbit anti-MxA (1:200; Novus Biologicals), polyclonal rabbit anti-EIF2AK2 (PKR) (1:100; Atlas Antibodies), monoclonal mouse anti-dsRNA (J2, IgG2a, 1:100; Scicons), and polyclonal rabbit anti-CD3 (1:300, Dako). Detection was performed by incubating the sections at room temperature for 1 hour with the following secondary antibodies: polyclonal goat anti-mouse IgG2a Alexa Fluor 488, goat anti-mouse IgG1 Alexa Fluor 555, or goat anti-mouse IgG2a Alexa Fluor 647 (1:1000; Invitrogen); F(ab′)2 fragment of goat anti-rabbit IgG (H + L) Alexa Fluor 647 (1:800; Jackson ImmunoResearch), F(ab′)2 fragment of goat anti-rabbit IgG (H + L) Alexa Fluor 555, or F(ab′)2 fragment of goat anti-rabbit Alexa Fluor 488 (1:1000; Invitrogen); and polyclonal goat anti-guinea pig IgG (H + L) Alexa Fluor 488 (1:1000; Invitrogen). Sections were counterstained with Hoechst 33342 (1:5000; Invitrogen) for 8 min and mounted with ProLong Gold Antifade (Invitrogen). Whole-tissue sections were scanned by an Axio Scan.Z1 slide scanner (Zeiss, Jena, Germany) using a 20×/0.8 numerical aperture (NA) Plan-Apochromat (a = 0.55 mm) objective lens.
LCM and RNA microarray analysis
Two consecutive frozen sections were used for LCM. From each donor, islets were picked and pooled on the basis of the number of IFN response markers (HLA-I, MxA, PKR, and/or dsRNA) expressed on the same islet (zero, one, or two or more markers). A total of 108 samples from 28 donors (50 islet pools from 12 nondiabetic donors, 26 from 7 AAb+ donors, and 32 from 9 T1D donors) were collected using LCM. After pooling the islets, RNA was extracted and a microarray was used for transcriptomics analysis. GeneSpring software (version 13.0; Silicon Genetics, Redwood, CA) was used to generate a list of genes that showed differential expression between donors and/or markers. The online Gene Set Analysis Toolkit (WebGestalt) was used to identify the pathways enriched by the differentially expressed genes.
Immunohistochemistry
FFPE sections were heated in 10 mM citrate (pH 6.0) in a pressure cooker in a microwave oven at 800 W for 20 min and then cooled at room temperature for 20 min. Sections were incubated with anti-VP1 (5D8/1 clone, 55 ng/ml; Dako) at room temperature for 1 hour, and the Dako REAL EnVision Detection System (Dako, Ely, UK) was used for antigen detection. Sections were evaluated under a Sarastro 2000 confocal laser scanning microscope (Molecular Dynamics, Sunnyvale, CA) and scanned using an Axio Scan.Z1 slide scanner (Zeiss, Jena, Germany) with a 20×/0.8 NA Plan-Apochromat (a = 0.55 mm) objective lens.
Image analysis
The numbers of insulin- and glucagon-positive islets, as well as the presence of HLA-I, MxA, PKR, and dsRNA, were assessed manually. Scanned tissue sections were visualized using ZEN Blue 2.3 software (Zeiss, Jena, Germany). The analysis was performed by researchers blinded to the study groups. Two different operators evaluated the expression of every marker and islet independently and blinded. Then, a third operator combined and revised all the analysis. Next, results from consecutive sections stained for the different markers were combined into a single image, and each islet was classified on the basis of the presence of one, two, three, or four markers, as illustrated in Fig. 3. The generated images served as a guide for LCM of islets for transcriptomic analysis.
QuPath (University of Edinburgh, Division of Pathology), an open-source software for whole-slide digital pathology (50), was used to count the number of VP1+ and CD3+ cells and determine the percentage of islets with insulitis [defined as an islet with six or more CD3+ cells immediately adjacent to or within the islet (28)]. First, the tissue area and all of the cells within that area were detected. Using machine learning, islets were automatically identified on the basis of hormone (insulin or glucagon) or hematoxylin and eosin staining, depending on the slide and staining protocol. Briefly, a manually trained random forest classifier included in QuPath, a custom script, and ImageJ (National institute of Health) were assembled in an analysis pipeline and applied to all of the corresponding tissue sections. Only islets formed by ≥30 cells were included to avoid possible detection errors derived from small artifacts. CD3+ cells and VP1+ cells were identified as areas of staining above the background level by applying optimized threshold values for intensity and cell size. Last, all images were manually checked to identify ICIs and IDIs and corrected for possible errors.
Statistical analysis
Continuous variables were compared between two independent groups using the Mann-Whitney U test, and P values were corrected for multiple comparisons using the Benjamini-Hochberg procedure. Paired samples were compared using the Wilcoxon signed-rank test. A Spearman correlation analysis was used to evaluate the correlation between the presence of viral protein and the expression of IFN response markers in islets. The significance level of two-sided P values was 0.05 for all statistical tests. Statistical analyses were performed using R version 3.6.1 and Prism version 8. Differences in gene expression between the laser-captured islets of different histological phenotypes were analyzed using Student’s t test and are presented as the fold change with the P value. Pathway enrichment within the lists of differentially expressed genes was estimated using WebGestalt software (as described by the software developer; www.webgestalt.org/) to calculate the probability that a random list with the same number of genes would contain the same or more genes from the specific pathway queried.
Acknowledgments
This research was performed with the support of the Network for Pancreatic Organ donors with Diabetes (nPOD; RRID:SCR_014641), a collaborative T1D research project sponsored by Juvenile Diabetes Research Foundation (JDRF) and The Leona M. and Harry B. Helmsley Charitable Trust. The content and views expressed in this article are the responsibility of the authors and do not necessarily reflect the official view of nPOD. The Organ Procurement Organizations (OPO), which partnered with the nPOD to provide research resources, are listed at www.jdrfnpod.org/for-partners/npod-partners/. We thank I. Kusmartseva and the nPOD team at the University of Florida, Gainesville, FL, USA for assistance with donor selection and tissue distribution and A. Ziegler (Institute of Diabetes Research, Helmholtz Zentrum Munich) for reading and providing feedback on the manuscript. Funding: nPOD is supported by JDRF (5-SRA-2018-557-Q-R) and The Leona M. and Harry B. Helmsley Charitable Trust (2018PG-T1D053). This work was supported by a JDRF grant (3-SRA-2017-492-A-N) to the nPOD-Virus working group. Author contributions: P.S.A. designed and performed experiments, analyzed and interpreted the data, and wrote the manuscript. D.B. performed experiments and analyzed the data. J.Z.-G. designed the custom scripts developed for computer-assisted software analysis, analyzed and interpreted the data using bioinformatics tools, and helped with statistical analysis. I.G. planned and supervised the LCM, which was conducted by G.N. and N.L., who share responsibility for the analysis of the gene expression data. P.A. performed experiments and analyzed the data. S.J.R. analyzed and interpreted the data and revised the manuscript. T.R.-C. designed experiments, analyzed and interpreted the data, and wrote the manuscript. T.R.-C. is the guarantor of this work and hence had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabd6527/DC1
REFERENCES AND NOTES
- 1.Thomas N. J., Jones S. E., Weedon M. N., Shields B. M., Oram R. A., Hattersley A. T., Frequency and phenotype of type 1 diabetes in the first six decades of life: A cross-sectional, genetically stratified survival analysis from UK Biobank. Lancet Diabetes Endocrinol. 6, 122–129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.CDC, National Diabetes Statistics Report 2020. Estimates of diabetes and its burden in the United States, 32 (2020).
- 3.Todd J. A., Etiology of type 1 diabetes. Immunity 32, 457–467 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Knip M., Simell O., Environmental triggers of type 1 diabetes. Cold Spring Harb. Perspect. Med. 2, a007690 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blanter M., Sork H., Tuomela S., Flodström-Tullberg M., Genetic and environmental interaction in type 1 diabetes: A relationship between genetic risk alleles and molecular traits of enterovirus infection? Curr. Diab. Rep. 19, 82 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Op de Beeck A., Eizirik D. L., Viral infections in type 1 diabetes mellitus—Why the β cells? Nat. Rev. Endocrinol. 12, 263–273 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hyöty H., Viruses in type 1 diabetes. Pediatr. Diabetes 17 (Suppl 22), 56–64 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez-Calvo T., Enteroviral infections as a trigger for type 1 diabetes. Curr. Diab. Rep. 18, 106 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Baggen J., Thibaut H. J., Strating J. R. P. M., van Kuppeveld F. J. M., The life cycle of non-polio enteroviruses and how to target it. Nat. Rev. Microbiol. 16, 368–381 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Wells A. I., Coyne C. B., Enteroviruses: A gut-wrenching game of entry, detection, and evasion. Viruses 11, 460 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marjomäki V., Turkki P., Huttunen M., Infectious entry pathway of enterovirus B species. Viruses 7, 6387–6399 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ifie E., Russell M. A., Dhayal S., Leete P., Sebastiani G., Nigi L., Dotta F., Marjomäki V., Eizirik D. L., Morgan N. G., Richardson S. J., Unexpected subcellular distribution of a specific isoform of the coxsackie and adenovirus receptor, CAR-SIV, in human pancreatic β cells. Diabetologia 61, 2344–2355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ylipaasto P., Klingel K., Lindberg A. M., Otonkoski T., Kandolf R., Hovi T., Roivainen M., Enterovirus infection in human pancreatic islet cells, islet tropism in vivo and receptor involvement in cultured islet beta cells. Diabetologia 47, 225–239 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Stene L. C., Oikarinen S., Hyöty H., Barriga K. J., Norris J. M., Klingensmith G., Hutton J. C., Erlich H. A., Eisenbarth G. S., Rewers M., Enterovirus infection and progression from islet autoimmunity to type 1 diabetes: The Diabetes and Autoimmunity Study in the Young (DAISY). Diabetes 59, 3174–3180 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vehik K., Lynch K. F., Wong M. C., Tian X., Ross M. C., Gibbs R. A., Ajami N. J., Petrosino J. F., Rewers M., Toppari J., Ziegler A. G., She J.-X., Lernmark A., Akolkar B., Hagopian W. A., Schatz D. A., Krischer J. P., Hyöty H., Lloyd R. E.; TEDDY Study Group , Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nat. Med. 25, 1865–1872 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krogvold L., Edwin B., Buanes T., Frisk G., Skog O., Anagandula M., Korsgren O., Undlien D., Eike M. C., Richardson S. J., Leete P., Morgan N. G., Oikarinen S., Oikarinen M., Laiho J. E., Hyöty H., Ludvigsson J., Hanssen K. F., Dahl-Jørgensen K., Detection of a low-grade enteroviral infection in the islets of Langerhans of living patients newly diagnosed with type 1 diabetes. Diabetes 64, 1682–1687 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Richardson S. J., Willcox A., Bone A. J., Foulis A. K., Morgan N. G., The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia 52, 1143–1151 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Pindel A., Sadler A., The role of protein kinase R in the interferon response. J. Interf. Cytokine Res. 31, 59–70 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Gal-Ben-Ari S., Barrera I., Ehrlich M., Rosenblum K., PKR: A kinase to remember. Front. Mol. Neurosci. 11, 480 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balachandran S., Roberts P. C., Brown L. E., Truong H., Pattnaik A. K., Archer D. R., Barber G. N., Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 13, 129–141 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Ylipaasto P., Kutlu B., Rasilainen S., Rasschaert J., Salmela K., Teerijoki H., Korsgren O., Lahesmaa R., Hovi T., Eizirik D. L., Otonkoski T., Roivainen M., Global profiling of coxsackievirus- and cytokine-induced gene expression in human pancreatic islets. Diabetologia 48, 1510–1522 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Jonsson A., Yngve E., Karlsson M., Ingvast S., Skog O., Korsgren O., Protein kinase R is constitutively expressed in the human pancreas. J. Histochem. Cytochem. 67, 99–105 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richardson S. J., Leete P., Bone A. J., Foulis A. K., Morgan N. G., Expression of the enteroviral capsid protein VP1 in the islet cells of patients with type 1 diabetes is associated with induction of protein kinase R and downregulation of Mcl-1. Diabetologia 56, 185–193 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Ferreira R. C., Guo H., Coulson R. M. R., Smyth D. J., Pekalski M. L., Burren O. S., Cutler A. J., Doecke J. D., Flint S., McKinney E. F., Lyons P. A., Smith K. G. C., Achenbach P., Beyerlein A., Dunger D. B., Clayton D. G., Wicker L. S., Todd J. A., Bonifacio E., Wallace C., Ziegler A.-G., A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes 63, 2538–2550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodrigues K. B., Dufort M. J., Llibre A., Speake C., Rahman M. J., Bondet V., Quiel J., Linsley P. S., Greenbaum C. J., Duffy D., Tarbell K. V., Innate immune stimulation of whole blood reveals IFN-1 hyper-responsiveness in type 1 diabetes. Diabetologia 63, 1576–1587 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lundberg M., Krogvold L., Kuric E., Dahl-Jørgensen K., Skog O., Expression of interferon-stimulated genes in insulitic pancreatic islets of patients recently diagnosed with type 1 diabetes. Diabetes 65, 3104–3110 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Richardson S. J., Rodriguez-Calvo T., Gerling I. C., Mathews C. E., Kaddis J. S., Russell M. A., Zeissler M., Leete P., Krogvold L., Dahl-Jørgensen K., von Herrath M., Pugliese A., Atkinson M. A., Morgan N. G., Islet cell hyperexpression of HLA class I antigens: A defining feature in type 1 diabetes. Diabetologia 59, 2448–2458 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campbell-Thompson M., Fu A., Kaddis J. S., Wasserfall C., Schatz D. A., Pugliese A., Atkinson M. A., Insulitis and β-cell mass in the natural history of type 1 diabetes. Diabetes 65, 719–731 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coomans de Brachène A., Dos Santos R. S., Marroqui L., Colli M. L., Marselli L., Mirmira R. G., Marchetti P., Eizirik D. L., IFN-α induces a preferential long-lasting expression of MHC class I in human pancreatic beta cells. Diabetologia 61, 636–640 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marroqui L., Dos Santos R. S., Op de Beeck A., Coomans de Brachène A., Marselli L., Marchetti P., Eizirik D. L., Interferon-α mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia 60, 656–667 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Dalet A., Gatti E., Pierre P., Integration of PKR-dependent translation inhibition with innate immunity is required for a coordinated anti-viral response. FEBS Lett. 589, 1539–1545 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Tracy S., Smithee S., Alhazmi A., Chapman N., Coxsackievirus can persist in murine pancreas by deletion of 5′ terminal genomic sequences. J. Med. Virol. 87, 240–247 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Turkki P., Laajala M., Stark M., Vandesande H., Sallinen-Dal Maso H., Shroff S., Sävneby A., Galitska G., Lindberg A. M., Marjomäki V., Slow infection due to lowering the amount of intact versus empty particles is a characteristic feature of coxsackievirus B5 dictated by the structural proteins. J. Virol. 93, e01130-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Philips T., Kusmartseva I., Gerling I. C., Campbell-Thompson M., Wasserfall C., Pugliese A., Longmate J. A., Schatz D. A., Atkinson M. A., Kaddis J. S., Factors that influence the quality of RNA from the pancreas of organ donors. Pancreas 46, 252–259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Su Q., Wang S., Baltzis D., Qu L.-K., Raven J. F., Li S., Wong A. H.-T., Koromilas A. E., Interferons induce tyrosine phosphorylation of the eIF2alpha kinase PKR through activation of Jak1 and Tyk2. EMBO Rep. 8, 265–270 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cnop M., Toivonen S., Igoillo-Esteve M., Salpea P., Endoplasmic reticulum stress and eIF2α phosphorylation: The Achilles heel of pancreatic β cells. Mol. Metab. 6, 1024–1039 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez-Calvo T., Ekwall O., Amirian N., Zapardiel-Gonzalo J., von Herrath M. G., Increased immune cell infiltration of the exocrine pancreas: A possible contribution to the pathogenesis of type 1 diabetes. Diabetes 63, 3880–3890 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimizu F., Shimizu M., Kamiyama K., Inhibitory effect of interferon on the production of insulin. Endocrinology 117, 2081–2084 (1985). [DOI] [PubMed] [Google Scholar]
- 39.Guyre P. M., Morganelli P. M., Miller R., Recombinant immune interferon increases immunoglobulin G Fc receptors on cultured human mononuclear phagocytes. J. Clin. Invest. 72, 393–397 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anania J. C., Chenoweth A. M., Wines B. D., Hogarth P. M., The human FcγRII (CD32) family of leukocyte FcR in health and disease. Front. Immunol. 10, 464 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elgueta R., Benson M. J., de Vries V. C., Wasiuk A., Guo Y., Noelle R. J., Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 229, 152–172 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klein D., Barbé-Tuana F., Pugliese A., Ichii H., Garza D., Gonzalez M., Molano R. D., Ricordi C., Pastori R. L., A functional CD40 receptor is expressed in pancreatic beta cells. Diabetologia 48, 268–276 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Vaitaitis G. M., Waid D. M., Yussman M. G., Wagner D. H. Jr., CD40-mediated signalling influences trafficking, T-cell receptor expression, and T-cell pathogenesis, in the NOD model of type 1 diabetes. Immunology 152, 243–254 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richardson S. J., Leete P., Dhayal S., Russell M. A., Oikarinen M., Laiho J. E., Svedin E., Lind K., Rosenling T., Chapman N., Bone A. J.; nPOD-V Consortium, Foulis A. K., Frisk G., Flodstrom-Tullberg M., Hober D., Hyoty H., Morgan N. G., Evaluation of the fidelity of immunolabelling obtained with clone 5D8/1, a monoclonal antibody directed against the enteroviral capsid protein, VP1, in human pancreas. Diabetologia 57, 392–401 (2014). [DOI] [PubMed] [Google Scholar]
- 45.von Herrath M. G., Fujinami R. S., Whitton J. L., Microorganisms and autoimmunity: Making the barren field fertile? Nat. Rev. Microbiol. 1, 151–157 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Marro B. S., Ware B. C., Zak J., de la Torre J. C., Rosen H., Oldstone M. B. A., Progression of type 1 diabetes from the prediabetic stage is controlled by interferon-α signaling. Proc. Natl. Acad. Sci. U.S.A. 114, 3708–3713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Q., Xu B., Michie S. A., Rubins K. H., Schreriber R. D., McDevitt H. O., Interferon-alpha initiates type 1 diabetes in nonobese diabetic mice. Proc. Natl. Acad. Sci. U.S.A. 105, 12439–12444 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Colli M. L., Ramos-Rodríguez M., Nakayasu E. S., Alvelos M. I., Lopes M., Hill J. L. E., Turatsinze J.-V., Coomans de Brachène A., Russell M. A., Raurell-Vila H., Castela A., Juan-Mateu J., Webb-Robertson B.-J. M., Krogvold L., Dahl-Jorgensen K., Marselli L., Marchetti P., Richardson S. J., Morgan N. G., Metz T. O., Pasquali L., Eizirik D. L., An integrated multi-omics approach identifies the landscape of interferon-α-mediated responses of human pancreatic beta cells. Nat. Commun. 11, 2584 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Honkimaa A., Sioofy-Khojine A.-B., Oikarinen S., Bertin A., Hober D., Hyoty H., Eradication of persistent coxsackievirus B infection from a pancreatic cell line with clinically used antiviral drugs. J. Clin. Virol. 128, 104334 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Bankhead P., Loughrey M. B., Fernández J. A., Dombrowski Y., McArt D. G., Dunne P. D., McQuaid S., Gray R. T., Murray L. J., Coleman H. G., James J. A., Salto-Tellez M., Hamilton P. W., QuPath: Open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabd6527/DC1