

Chromatin modulators with antagonistic functions on epithelial-mesenchymal interconversion regulate common pan-cancer genes.
Abstract
Epithelial-mesenchymal transition (EMT) is a developmental process hijacked by cancer cells to modulate proliferation, migration, and stress response. Whereas kinase signaling is believed to be an EMT driver, the molecular mechanisms underlying epithelial-mesenchymal interconversion are incompletely understood. Here, we show that the impact of chromatin regulators on EMT interconversion is broader than that of kinases. By combining pharmacological modulation of EMT, synthetic genetic tracing, and CRISPR interference screens, we uncovered a minority of kinases and several chromatin remodelers, writers, and readers governing homeostatic EMT in lung cancer cells. Loss of ARID1A, DOT1L, BRD2, and ZMYND8 had nondeterministic and sometimes opposite consequences on epithelial-mesenchymal interconversion. Together with RNAPII and AP-1, these antagonistic gatekeepers control chromatin of active enhancers, including pan-cancer-EMT signature genes enabling supraclassification of anatomically diverse tumors. Thus, our data uncover general principles underlying transcriptional control of cancer cell plasticity and offer a platform to systematically explore chromatin regulators in tumor-state–specific therapy.
INTRODUCTION
Epithelial-mesenchymal transition (EMT) is a developmental program activated during gastrulation and neural crest formation (1). Epithelial-mesenchymal plasticity allows cancer cells to undergo functional adaptations during the invasion-metastasis cascade (2). Evidence correlates tumor EMT with the acquisition of migration properties (2), chemotherapy resistance, and selective chemotherapeutic vulnerabilities (3, 4). Aberrations in the transforming growth factor–β (TGF-β) signaling is causal to tumor EMT (5, 6). In breast cancer cells, both TGF-β and platelet-derived growth factor receptor A receptor tyrosine kinase (RTK) signaling govern EMT and mesenchymal-epithelial interconversion (7, 8). Moreover, other EMT-regulating RTK axes were described (9, 10), suggesting a wider role for kinases in governing EMT. As many RTK pathways centrally rely on RAS signaling cascade, it is not surprising that RAS cooperates with multiple pathways to control EMT (11–13).
Tumor EMT appears to be a cellular state rather than a unidirectional cell fate decision (2), which fits the definition of a homeostatic process. Notably, TGF-β drives dichotomous programs such a tumor suppressive “lethal EMT” and an oncogenic EMT program in cooperation with transcriptional regulators (5). Chromatin regulation is a pivotal mechanism for establishing and maintaining cell identity in metazoans through transcriptional regulation, but its role in tumor EMT remains restricted to individual factors (14). In multipotent cells, a bivalent chromatin configuration of developmental and differentiation genes occurs at poised chromatin, which is simultaneously decorated by activating and repressive histone marks. Upon cell fate changes, bivalent chromatin is resolved as either active or repressed (e.g., with monovalent potential) (15). The extent of bivalent chromatin marks in multipotent cells may be numerically reduced by inhibiting antagonistic cellular signaling such as pro–self-renewal and differentiation cues, indicating that polymerase II elongation is pivotal to regulate poised genes (16, 17). In epithelial cancer cells, the mesenchymal transcription factor ZEB1 bears a bivalent chromatin configuration, suggesting that epithelial-mesenchymal plasticity involves chromatin regulation (18). While it is becoming increasingly evident that EMT is a continuum between states (14), how the EMT metastability is controlled under convergent activating and repressing cues remains unresolved.
Chromatin regulators are pervasively mutated in human cancers, but their mechanism of action is often context dependent (19, 20). For instance, the chromatin writer Polycomb repressive complex 2 (PRC2), which regulates one of the bivalent chromatin marks, dichotomously regulates EMT. PRC2 methyltransferase enhancer of zeste homolog 2 (EZH2) cooperates with SOX4 to support breast cancer cell EMT, whereas PRC2 inhibition by EED deletion or EZH2 pharmacological inhibition promotes EMT in Kras-driven lung cancer cells. In both instances, alterations in PRC2 function support tumorigenesis through different mechanisms (12, 21). EZH2 positively regulates EMT through transcriptional repression of epithelial genes (21), whereas PRC2 inhibition leads to cell fate changes by amplifying RAS-driven transcription (12, 22). Thus, modulating PRC2 function may be used as a tool to uncover cancer-relevant mechanisms of cellular interconversion, including EMT.
In our previous studies, we developed a synthetic reporter for genetic tracing of EMT homeostasis in multiple cellular models (23). Here, we combine pharmacological boosting of homeostatic EMT in lung cancer cells by EZH2 inhibition, fate mapping by genetic tracing, and a large-scale CRISPR interference (CRISPRi) screen for genes involved in RTK signaling and epigenome regulation to identify factors required for the proper regulation of EMT. These screens uncovered a large number of chromatin regulators and a more limited number of kinome genes as regulators of EMT interconversion. We found several members of the SWI/SNF chromatin remodeling complex (ARID1A, SMARCB1, and SMARCE1); of writer complexes such as KMT2A, DOT1L, and EPC1; and of reader complexes such as BRD2 and ZMYND8, whose loss affects epithelial-mesenchymal homeostasis. Also, cellular fitness and migration properties were regulated by these factors, but the outcome of their deletion on cellular properties was pleiotropic (i.e., nondeterministic). Nevertheless, despite the diverse phenotypic outcome of their loss on EMT, ARID1A, DOT1L, BRD2, and ZMYND8 occupy a common set of enhancers and promoters of genes that are critically associated with EMT in multiple cancer types, beyond the context of specific tissues. Hence, antagonistic regulation of active chromatin governs the fidelity of cell fate decisions and the plasticity of cancer cells during homeostatic regulation or in response to stress.
RESULTS
Phenotypic CRISPRi screens in non–small cell lung cancer cells to identify key kinome and epigenome regulators of EMT homeostasis
The mesenchymal genetic tracing vector #1 (MGT#1) synthetic reporter for mesenchymal program genetic tracing was designed by stitching potential cis-regulatory elements. MGT#1 is activated during proneural-to-mesenchymal transition in glioblastoma and in mesenchymal cells of multiple tissue cancers, at least in part, because of its response to broadly expressed transcription factors, including nuclear factor κB (23). To screen for regulators of MGT#1 expression as a proxy for homeostatic EMT, we scored for cell states in a large panel of human cancers cell lines and chose A549 and H1944 cells, which are both KRAS-mutant and display a quasi-mesenchymal and a quasi-epithelial gene expression program, respectively (Fig. 1, A and C). We reasoned that GSK126 would amplify the homeostatic EMT and verified this by correlating EMT markers, EZH2-targeted posttranslational modification H3K27me3 and MGT#1 expression (Fig. 1D).
Fig. 1. A dominant role for chromatin factors as regulators of EMT homeostasis revealed by phenotypic CRISPRi screens.
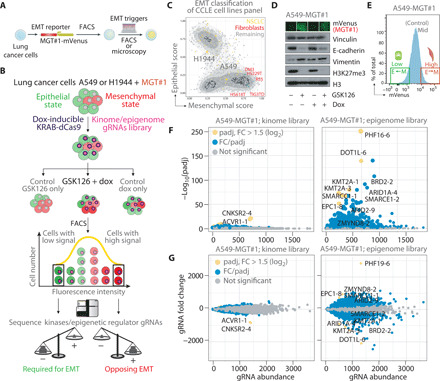
(A) Schematic of EMT reporter lung cancer cell line generation. (B) Diagram of phenotypic CRISPRi screen. (C) Selection of quasi-epithelial H1944 and quasi-mesenchymal A549 cell lines by epithelial and mesenchymal gene expression scoring of human NSCLC cell lines (see Materials and Methods). Lung fibroblasts (mesenchymal) are shown for comparison. CCLE, Cancer Cell Line Encyclopedia. (D) Immunoblotting of samples from intermediate time point of the CRISPRi screen. MGT#1 fluorescence micrograph (above) was taken before lysis. (E) Gating strategy for FACS purification of MGT#1-high and MGT#1-low populations. (F) Scatter plot of differential enrichment analysis for sgRNAs from the indicated libraries in the “GSK126 + dox” arm of the A549-MGT#1 screen. Fold change (FC) and significance were calculated by comparing MGT#1-mVenus–high and MGT#1-mVenus–low populations, upon performing all the relevant quality controls (see Materials and Methods and figs. S2 and S3). Yellow denotes gRNAs significant by FC and P value. Blue dots are significant by P value (i.e., potential hits) above the nonsignificant gray dots. Dot size represents the absolute log2FC value. (G) MA plot of gRNA abundance (x axis) and difference in gRNA abundance (y axis) in the GSK126 + dox arm of the A549-MGT#1 screen. Dot color and labels are consistent with (F), whereas size is fixed.
In agreement with previous data showing that PRC2 inhibition through either genetic means or EZH2 inhibitor induces EMT (12), GSK126 led to a moderate activation of the reporter in Kras-driven A549 and H1944 lung cancer cells, whereas TGF-β induces a more robust reporter induction (fig. S1A). More broadly, we established a correlation between the mesenchymal status of established cell lines and MGT#1 expression by quantitative polymerase chain reaction (qPCR) (fig. S1A). Using mVenus as a proxy of EMT conversion, RNA sequencing (RNA-seq) on fluorescence-activated cell sorting (FACS)–sorted MGT#1-high A549 and H1944 cells confirmed the activation of a mesenchymal gene program (fig. S1, B to E). Assay for transposase-accessible chromatin using sequencing (ATAC-seq) profiling of FACS-sorted MGT#1-high A549 cells revealed that GSK126 induces mild chromatin accessibility at the same genomic loci extensively remodeled by TGF-β signaling (fig. S1, F to I). Both treatments induce the chromatin remodeling at the integration locus of the MGT#1 reporter (fig.S1G), indicating that EZH2 inhibition by GSK126 sets the ground for EMT conversion.
Next, we exploited this system to identify regulators of EMT homeostasis among kinome and epigenome genes. We transduced both A549 and H1944 with the MGT#1 reporter. Subsequently, we introduced a Tet-inducible KRAB-dCas9 and a library of single-guide RNAs (sgRNAs) targeting the full complement of the human kinome (543 genes and 5901 sgRNAs in total; up to 10 sgRNAs per gene) or a selected set of genes involved in epigenome regulation (457 genes and 4980 sgRNAs) in both cell lines. To maintain the representation of each library, we infected at least 1000 cells per sgRNA, at a low multiplicity of infection rate (<0.3). Moreover, each library included the same set of sgRNAs targeting essential and nonessential genes that serve as control for the screening procedure. This system allows the systematic knockdown of individual genes in single cells upon doxycycline (dox) activation of the KRAB-dCas9 repressor (Fig. 1B). After prolonged EZH2 inhibition by GSK126 treatment (11 days), we FACS-purified non–small cell lung cancer (NSCLC) cells that were either enhanced or impaired in their ability to support the expression of the fluorescent reporter, thereby reflecting a more epithelial or mesenchymal phenotype, respectively (Fig. 1E). By comparing A549-MGT#1-low and H1944-MGT#1-low to their MGT#1-high counterpart, we found that multiple sgRNAs in the epigenome library but only a minor fraction of the kinome sgRNAs were enriched or depleted in either one of the two states in both cell lines (Fig. 1, F and G, and figs. S2 and S3). In the absence of GSK126, dox-induced depletion of chromatin regulators affected a lower number of gRNAs, suggesting that transcriptional repression of these genes generally affected lung cancer cell EMT and confirming that EZH2 inhibition amplifies physiological EMT. Overall, our screen indicates that multiple chromatin regulators are potentially involved in EMT, whereas most human kinases are dispensable for homeostatic or GSK126-driven EMT. Essential and nonessential gene set enrichment analysis (fig. S2G) and the fact that no gRNA scored as significant in the absence of dox treatment (fig. S2, E and F) supported the overall high quality of the screen. In support of the specificity of this screen, and in line with our previous observation that Polycomb can antagonize EMT (12), gRNAs targeting Polycomb-associated proteins PHF19 (24, 25) and L3MBT2 (26) scored among the top negative regulators of EMT (Fig. 1F and table S1). The results also align with the well-established antagonism between the Polycomb and the mixed-lineage leukemia (MLL) or SWI/SNF complex in development and cancer: gRNAs targeting MLL and SWI/SNF members scored as required for EMT in both A549 and H1944. These include KMT2A, SMARCB1, and SMARCE1 (table S1). ARID1A is a key SWI/SNF member that is among the most frequently mutated genes in cancer, indicating that synthetic genetic tracing coupled with CRISPRi uncovers critical pathways converging onto EMT homeostasis. Together, this suggests that chromatin regulation is a dominant control of cellular identity metastability.
Genetic loss of potential EMT regulators phenocopies CRISPRi screen
To identify robust chromatin regulators of EMT, we decided to independently validate the loss of function CRISPRi screen through a panel of knockout (KO) cell lines for a selected number of hits identified in the CRISPRi screen. As multiple hits may be selected on the basis of significance or fold change (FC), we shortlisted candidates on the basis of their function as chromatin regulators, including remodelers of the BRG1/BRM-associated factor (BAF) complex (ARID1A), writers (KMT2A and DOT1L), readers (BRD2 and ZMYND8), and scaffolds of writer complexes (EPC1). The selection included both potential positive regulators of EMT (e.g., ARID1A, BRD2, DOT1L, and KMT2A) and potential barriers (EPC1 and ZMYND8). As control for the epigenome, we selected ARID2, which is a SWI/SNF member whose loss strongly affects the polybromo-associated BAF (PBAF) complex but falls below both significance and FC thresholds in our screen. As hits from the kinome screen potentially required for EMT in A549 cells, we selected ACVR1, previously proposed to promote EMT in A549 cells (27), and CNKSR2, a scaffold component of the RAS complex, which promotes EMT upon GSK126 (12). For all of these candidates, expression changes during EMT in A549 and H1944 were insufficient to explain their functional performance in the screen, indicating that their function and not simply their expression contributes to EMT. Thus, we created KO lines by nucleofecting sgRNAs and Cas9 in A549-MGT#1 cells, which is expected to result in the ablation of each protein in a large number of cells and defined a gating strategy to perform EMT fate mapping at a single-cell level (Fig. 2A). The efficient deletion of each candidate was validated by targeted sequencing, and only highly effective KO polyclonal lines were retained for assessing the role of candidate genes on MGT#1 expression (fig. S4, A to F). For all tested candidates, we observed either an increase or decrease in MGT#1 reporter expression, concordant with the predictions on the basis of the CRISPRi screen (Fig. 2B). Notably, deletion of BRD2, DOT1L, KMT2A, and ARID1A significantly impaired MGT#1 expression, and ZMYND8 and EPC1 display the inverse effect, whereas only minor changes were detected upon ARID2 deletion compared to either control or other mutant cells (Fig. 2B). The same effect was observed also with a stronger EMT driver such as TGF-β1 (fig. S4G), thereby validating the CRISPRi screen in a broad range of candidates by genetic deletion.
Fig. 2. Genetic loss of potential EMT regulators phenocopies CRISPRi screen.
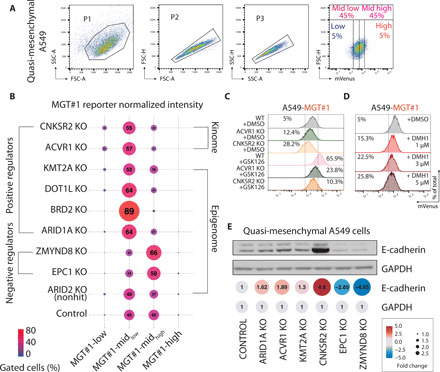
(A) Gating strategy for assessing fluctuations in EMT by FACS analysis of MGT#1 expression. SSC-A, side scatter area; FSC-A, forward scatter area. (B) Bubble plot of the FACS analysis of MGT#1 expression in A549-MGT#1 carrying individual KO mutations. (C) FACS analysis of MGT#1 fluorescence intensity in A549-MGT#1 cells with the indicated genotypes and treatments. WT, wild type; DMSO, dimethyl sulfoxide. (D) FACS analysis of MGT#1 fluorescence intensity in A549-MGT#1 cells with the indicated treatments. (E) Immunoblot of E-cadherin expression in A549-MGT#1 carrying individual KO mutations. Quantification of normalized protein band intensity is shown.
To correlate genetic deletion, MGT#1 expression, and cell identity, we first focused follow-up validation on the kinome hits. CNKSR2 and ACVR1 are required for EMT in quasi-mesenchymal A549 cells. CNKSR2 is a scaffold protein involved in RAS-dependent signaling, and RAS-driven EMT was previously shown to occur indirectly through the Hippo pathway (13). To genetically validate that loss of CNKSR2 and ACVR1 affects EMT homeostasis, we established both clonal and polyclonal A549 lines using CRISPR-Cas9. In all cases, this resulted in lower basal levels of MGT#1 expression and enhanced epithelial features compared to the parental (Fig. 2C). GSK126 partly rescued the MGT#1 expression indicating that CNKSR2 and ACVR1 KO cells are acutely impaired but still capable of executing EMT. Notably, ACVR1 loss of function effect could be phenocopied by the ACVR1/ALK2 inhibitor DMH1 in a dose-dependent manner (Fig. 2D), and MGT#1 activation by GSK126 was blunted by DMH1 cotreatment (fig. S4, H and I), indicating that SMAD1/5/9 phosphorylation by bone morphogenetic protein (BMP) type I receptor activation inversely correlates with epithelial phenotype in these cells. Last, we extended the validation to the remaining chromatin regulators by using immunoblotting of E-cadherin expression as a proxy for epithelial cell identity. Both EPC1 and ZMYND8 KO led to a marked reduction in E-cadherin expression as opposed to deletion of CNKSR2, ACVR1, DOT1L, and ARID1A (Fig. 2E).
Thus, our phenotypic CRISPRi strategy uncovered known and novel EMT regulators. These include the kinase adaptor CNKSR2, which is a novel player in the RAS/mitogen-activated protein kinase (MAPK)–dependent signaling directly contributing to EMT and a broad range of chromatin modifiers.
Phenotypic consequences of genetic loss of individual EMT regulators
Epithelial-mesenchymal conversion can be activated directly by microenvironmental signals or indirectly, as a result of response to stress (2, 14). To investigate whether the hits in our screen directly or indirectly regulate EMT, we investigated the phenotypic consequences of individual hit depletion on cellular fitness. First, we conducted a lung cancer–wide gene essentiality analysis on 60 NSCLC adenocarcinoma cell lines functionally dissected by the cancer dependency map project (28). Using two independent ranking scores for essentiality by CRISPR-Cas9 (CERES) or RNA interference (RNAi) (DEMETER2), we compared the essentiality of either the kinome or the epigenome genes for all NSCLC adenocarcinoma cell line fitness in genome-wide screens. Both the kinome and the epigenome libraries contain a limited number of genes whose depletion affects NSCLC cell fitness, but—in agreement with our essentiality analysis—the hits selected in our screen are statistically less required than the median of well-established essential genes (Fig. 3A). To validate this analysis in our set of wild-type and EMT regulator mutants’ NSCLC cells, we next performed parallel longitudinal profiling of cell proliferation. Consistent with the predictions of pooled screens, we observed moderate changes in individual polyclonal cell line proliferation in all genotypes (Fig. 3B). The proliferation assay was corroborated by a colony formation assay, which is generally used as a proxy of the ability of normal and tumor cells to recover from cell stress. Also in this case, we observed analogous responses between wild-type and EMT regulator mutant cells (Fig. 3C). Selectively, BRD2 KO showed a moderate but significant defect as compared to wild-type cells, and an opposite trend was observed for ARID1A and EPC1, in addition to ACVR1, in the proliferation assay (Fig. 3B). ARID1A and EPC1 also showed a moderate but significant increase in fitness in the colony assay (Fig. 3C). However, overall, none of the EMT regulators are critically required for cell fitness, suggesting that they directly regulate EMT.
Fig. 3. Context-dependent cellular phenotypes by genetic loss of individual EMT regulators.
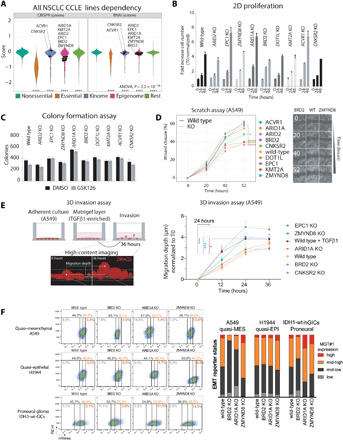
(A) Violin plot showing the distribution of the dependency for proliferation of all CCLE NSCLC cell lines for individual genes within the indicated families. The plot shows the phenotypic dependency calculated by the DepMap project. CRISPR (Avana) CERES score and RNAi DEMETER2 score are displayed in the left and right panels, respectively. Selected genes are labeled within their families. ANOVA, analysis of variance. (B) Bar plot of control and mutant A549-MGT#1 cell proliferation in parallel longitudinal analysis. Statistics: Significant by t test and Holm-Sidak post hoc test (P < 0.05; n = 4), BRD2, EPC1, ARID1A, and ACVR1 KOs versus control. (C) Bar plot of control and mutant A549-MGT#1 ± GSK126 cell colony formation assay. Statistics: Significant by t test and Holm-Sidak post hoc test (P < 0.05; n = 3) in DMSO group: ARID2, ARID1A, DOT1L, and ACVR1 KOs; GSK126: EPC1, ARID1A, BRD2, DOT1L, KMT2A, and ACVR1 KOs. (D) Left: Line plot of parallel longitudinal high-content wound healing analysis of A549-MGT#1 cells with the indicated genotypes under homeostatic conditions. Each dot represents the mean in each time point. Statistics: Two-way ANOVA and Dunnet post hoc test (n = 4). Asterisks denote significance for the indicated comparison. Antagonistic regulators of EMT and motility in A549 cells are shown to the right. (E) Left: Schematic representation of three-dimensional (3D) invasion assay. Right: Migration depth of DRAQ5-stained nuclei for each time point and clone normalized to time point T = 0 hours from high-content imaging. Statistical analysis for time point 24 hours shows corrected multiple t test (*P < 0.05; ***P < 0.001; n = 4). (F) FACS analysis (left) and quantification (right) of MGT#1 expression in lung and brain tumor cells with the indicated genotypes.
It is well established that phenotypic changes in epithelial cancer cell identity are linked to changes in cell fitness and migration properties (2, 14). Next, we tested whether individual genotypes display private migration properties in a wound healing assay, in which cell polarization toward the cell-free zone is assessed. ZMYND8- and EPC1-depleted cells healed the wound significantly better than BRD2, CNKSR2, KMT2A, and ACVR1 KO cells (Fig. 3D). DOT1L loss, which blunts EMT in our screen, appeared to increase cell motility, thereby behaving like ZMYND8 and EPC1 in the scratch assay. Hence, DOT1L may contribute to epithelial/mesenchymal identity and migratory properties through different mechanisms. Last, in a live-cell invasion assay, ZMYND8- and EPC1-depleted cells also invaded extracellular matrix as efficiently as TGF-β1–treated cells (Fig. 3E), consistent with the view that loss of these chromatin modulators led to acquisition of mesenchymal features and enhanced migration and invasion.
Last, we next tested whether the genetic deletion of selected chromatin regulators confers predictable outcomes in other cellular contexts. To this end, we deleted the pro-EMT BRD2 and ARID1A factors and the anti-EMT ZMYND8, in quasi-epithelial lung cancer cells (H1944) and in glioma-initiating cells (IDH1-wt-GICs) that undergo proneural-to-mesenchymal transition upon external signaling stimulation or therapy-induced stress (23). Consistent with phenotypic consequences in quasi-mesenchymal A549 cells, loss of BRD2 and ZMYND8 in both H1944 and IDH1-wt-GICs decreased and increased MGT#1 expression, respectively (Fig. 3F). ARID1A loss mildly affected H1944 phenotype and induced EMT in IDH1-wt-GICs, indicating that prediction based on the phenotypic screen in NSCLC cells may have significant but opposite outcomes in different contexts (Fig. 3F). Thus, we conclude that the individual regulators identified in our screen have a direct impact on EMT as their loss affects cell plasticity but not general fitness. They also have context-dependent consequences on epithelial-mesenchymal interconversion, as their effects are tissue-specific.
Antagonistic chromatin regulators mark active promoters and enhancers in lung cancer cells
To examine whether the phenotypic behavior of individual EMT-regulator mutants could be attributed to regulation of downstream genes, we performed a genome-wide occupancy analysis. We generated chromatin immunoprecipitation sequencing (ChIP-seq) profiles for ZMYND8, BRD2, ARID1A, and DOT1L in A549 cells under homeostatic and pro-EMT conditions (i.e., GSK126 and TGF-β1). At first, we focused on genomic loci bound by ZMYND8 as the most prominent EMT barrier factor per our experiments. Notably, however, after k-means partitioning of the ZMYND8 ChIP-seq profiling into three clusters, we observed limited differences in cluster intensities and, most notably, that ZMYND8 largely shares binding with the EMT promoters BRD2, ARID1A, and DOT1L (Fig. 4A and table S2). The most obvious difference between the three clusters was a variable enrichment of the active chromatin mark H3K27ac. No specific enrichment was observed at these genomic loci for the poised chromatin mark H3K27me3 or the nonspecific control for immunoprecipitation (Fig. 4A). ChIP-seq quality was assessed by analyzing technical replica and biological features of individual profiles (fig. S5A). For each locus where an annotated gene could be assigned, Gene Ontology (GO) analysis of cluster I to III genes revealed distinct enrichment for terms attributable to EMT homeostasis (Fig. 4B). Clusters I and III were characterized by GO terms associated with epithelial and mesenchymal cell functions, respectively, such as cadherin binding, actin binding, and ion channel activity (Fig. 4B). Consistently, cluster I and III genes are basally high and low in both A549 and H1944, and cluster III genes are up-regulated upon TGF-β–driven EMT (fig. S5B). All clusters were markedly enriched for genes encoding for DNA binding, chromatin, and transcriptional regulatory proteins, which are broadly considered as key regulators of cell identity and fate changes (Fig. 4B). This is in line with the GO of genes associated with bivalent histone marks (15), with the difference that clusters I to III are largely devoid of H3K27me3 and are preferentially decorated by H3K27ac, which corresponds to active chromatin.
Fig. 4. Antagonistic chromatin regulators bind to core set of accessible targets in lung cancer cells.
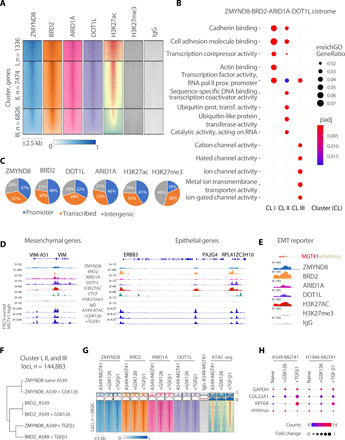
(A) Heatmap of signal intensity for ZMYND8, BRD2, ARID1A, DOT1L, acetyl- and trimethyl-H3K27, and immunoglobulin G (IgG) occupancy at the indicated genomic loci. k-means clustering was used to partition chromatin occupancy into three clusters according to ZMYND8. The number of genes included in each of the clusters and of chromatin regions is indicated to the left and below the heatmap, respectively. (B) Top five GO terms per core set of Cluster I, II, and III genes (color, adjusted P < 0.05; size, gene ratio). (C) Pie charts showing the genomic distribution of the indicated ChIP-seq peaks. Note that the ZMYND8, BRD2, DOT1L, and ARID1A binding mode mirrors the enhancer-decorating mark H3K27ac. (D) IGV view of the indicated ChIP-seq tracks for known epithelial and mesenchymal markers. For each track, scale values are indicated to the left. (E) IGV view of ZMYND8, BRD2, ARID1A, DOT1L, acetyl- and trimethyl-H3K27, and IgG occupancy at the MGT#1 reporter loci. (F) Dendrogram showing hierarchical clustering of the indicated ChIP-seq tracks for loci from (A). Note the dominant effect of TGF-β1 on the clustering. (G) Density plot (above) and heatmap (below) of the indicated ChIP-seq tracks for TGF-β–regulated loci significant by DESeq2 (padj < 0.05). (H) Bubble plot showing the expression data for the selected genes in the indicated conditions. Bubble size and color indicate FC compared to control and normalized expression per sample, respectively.
The binding profile of antagonistic chromatin regulators of EMT suggests that their function may involve the fine-tuning of a common set of cis-regulatory elements. To identify the type of cis-regulatory elements and chromatin bound by the chromatin gatekeepers, we annotated genome-wide each profile to all human coding and noncoding mRNAs. This analysis showed that ZMYND8 (anti-EMT), BRD2 (pro-EMT), ARID1A (pro-EMT), and DOT1L (pro-EMT) are equally associated with promoter regions and intragenic and intergenic loci (Fig. 4C). Because we included well-established markers for active cis-regulatory elements (i.e., H3K27ac and ATAC) and poised chromatin (H3K27me3), we conclude that nonpromoter elements are bona fide enhancers. In support of this conclusion, despite that BRD2 also colocalizes with and is required for CCCTC-binding factor (CTCF) boundary maintenance (29), we observed low CTCF binding to cluster I, II, or III, as compared to robust CTCF binding sites identified by CTCF ChIP-seq in A549 cells (Fig. 4D and fig. S5, C and D). Consistent with their antagonistic potential, BRD2 (pro-EMT) and ZMYND8 (anti-EMT) occupy the same regulatory elements surrounding genes with opposite fate during EMT (Fig. 4D). Moreover, in addition to their binding to endogenous regulatory elements, ZMYND8, BRD2, ARID1A, and DOT1L occupy the MGT#1 reporter with an enrichment profile akin to H3K27ac and ATAC-seq (Fig. 4E). Globally, EMT induction through GSK126 or TGF-β did not notably modify the binding profile of ZMYND8, BRD2, ARID1A, and DOT1L, as assessed by ChIP-seq (fig. S5D). Yet, TGF-β drove remodeling of selected loci, which was followed by increased loading of all factors (Fig. 4, F to H and fig. S5E). Moreover, deletion of BRD2 or ZMYND8 induced local changes in chromatin binding of the other regulator at endogenous and reporter loci, as gauged by ChIP-qPCR (fig. S5F). This suggests that the qualitative distribution of each chromatin modulator is associated with the cell identity, and their levels may be locally regulated by state-specific cues. Overall, our data indicate that chromatin regulators whose loss has opposite functional consequences on cell fate decisions occupy a common cistrome in human lung cancer cells, suggesting that they contribute to regulation of dichotomous signaling.
A meta-EMT program emerges from pan-cancer analysis of the antagonistic chromatin regulators’ cistrome
Given that antagonistic chromatin modulators’ binding profile correlates with cell identity features, we next examined the relevance of their functional antagonism and EMT regulation in human cancers by studying the expression pattern of their target genes across all human cancers included in the The Cancer Genome Atlas (TCGA) dataset.
First, we developed a dual-scoring method (see Materials and Methods) to select 74 patients with bona fide epithelial and mesenchymal lung adenocarcinoma (LUAD; fig. S6A). Using the full tumor transcriptome, non-negative matrix factorization (NMF) clustering independently and robustly separated these patients in two groups, consistent with our dual-scoring strategy. Metagene expression of well-established epithelial or mesenchymal cell identity markers and markedly different survival profiles are consistent with the notion that these patients bear two biologically distinct tumor states (fig. S6, B and C). Notably, clustering of the patients with epithelial or mesenchymal LUAD into biologically distinct groups was maintained when hierarchical clustering was limited to the antagonistic EMT core gene network, regardless of the cluster I, II, or III (fig. S6D). Notably, the metagene recurrent across these patients included the well-established epithelial or mesenchymal markers. Together with the minimal binding differences observed in ChIP-seq, this supports that clusters I, II, and III are part of a unique cistrome, which includes cancer relevant meta-EMT regulators.
To test whether the meta-EMT signature identifies molecular similarities that connect tumors originated in different tissues, we extended our analysis to other tumors of the pan-cancer TCGA dataset (table S4). Using our dual-scoring stratification, we extended our analysis to 1081 patients with cancer from anatomically and histologically distinct diseases. Unsupervised hierarchical clustering of the ZMYND8-BRD2-ARID1A-DOT1L cistrome (i.e., cluster I + II + III) gene expression in these tumors yielded three to five groups (Fig. 5A; fig. S7, A to C; and table S4).
Fig. 5. Antagonistic chromatin regulators targets include a pancancer meta-EMT signature influencing molecular tumor classification.
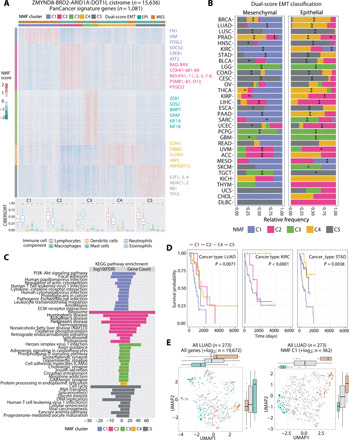
(A) Top: Heatmap showing 1081 epithelial and mesenchymal LUAD biopsies defined by the dual scoring analysis is clustered into five clusters by NMF consensus on the basis of cluster I + II + III genes (see Materials and Methods). Selected EPI and MES metagenes are highlighted to the right. Bottom: Composition of the tumor immune infiltrate. For each NMF cluster, boxplots show the proportion of six major classes of immune cells (from CIBERSORT). Lymphocytes represent the reference, and the other five classes are compared to lymphocytes. Significance is indicated by asterisks (Wilcoxon rank sum test, adjusted P < 0.05). (B) Relative frequencies of each NMF cluster in the MES and EPI patients of 32 cancer types, respectively; significance of enrichment was calculated by Fisher’s exact test and highlighted with asterisks (see Materials and Methods): *P < 0.05; **FDR (false discovery rate) < 0.05. (C) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways displayed were significantly associated with NMF clusters C1 to C5. Two panels referred to the log10(FDR) and gene count ratio. (D) Kaplan-Meier survival analysis for five NMF clusters patients of LUAD, KIRC, and STAD. Time is indicated in days. Different patient groups are compared using the log-rank test, for which the P value is shown. (E) Uniform manifold approximation and projection (UMAP) clustering of patients with LUAD based on either all genes (left) or signature genes from NMF cluster C1 (right). The latter represents the meta-mesenchymal pan-cancer signature genes.
The proportion of anatomically and histologically diverse cancer types within TCGA cohort was consistent with expectations: the largest fraction of patients with either an epithelial or mesenchymal profile being assigned to carcinomas of the breast (BRCA), lung [adeno (LUAD) and squamous (LUSC)], prostate (PRAD), head-and-neck (HNSC), kidney (KIRC), and stomach (STAD). The lymphomas, instead, were almost entirely excluded (fig. S8A). By NMF, the distribution of these cancers within the five clusters followed, in many cases, a supraclassification pattern (see Materials and Methods), with NMF C1 being predominantly composed of a homogeneous mesenchymal mix of different cancer types (P < 0.01, Fisher’s exact test) and epithelial cancers significantly enriched in NMF C2-C4-C5. Cluster C4 appeared as the most epithelial NMF cluster (P < 0.01, Fisher’s exact test; fig. S8, B and C). As an exception, NMF C3 is the only group largely enriched in one cancer type, the brain tumors (Fig. 5B and fig. S8, B and C).
Metagenes associated to each cluster also supported this supraclassification (Fig. 5A and table S4). In the mesenchymal group of patients, the metagene signature returned well-established EMT drivers, including TGF-β, tumor necrosis factor (TNF), and RAS signaling pathways, as well as GO terms of migration (Fig. 5C and figs. S8D and S9). We termed this signature the meta-mesenchymal module, as it identifies mesenchymal tumors independently of the tissue of origin. Although the epithelioid tumors were allotted into multiple groups, the NMF C4 and either NMF C2 or C5, the broad program downstream of the HNF4A transcription factor emerged as a common theme. The main drivers of subepithelioid classifications were, on the one hand, metabolic pathway activation [C2; mTOR (mammalian target of rapamycin) and oxidative phosphorylation] and, on the other hand, cell cycle and DNA repair genes (Fig. 5C, figs. S8D and S9, and table S5).
Whereas lymphocytes and macrophages are by far the largest fraction of immune cell infiltrates, CIBERSORT estimates the ratio between adaptive and innate immunity to be specific for each cluster, further underscoring their peculiar biology (Fig. 5A). The NMF C1 and C4 appear to be opposite with respect to their cell identity features, adaptive/innate immune cell ratio, and survival. NMF C4 patients are epithelial and have a significantly higher infiltration of lymphocytes [padj (adjusted P) < 0.01, Wilcoxon rank sum test; see Materials and Methods], and—at least in the case of LUAD, kidney (KIRC), and stomach (STAD) cancers—these patients are characterized by a favorable prognosis when compared to NMF C1 patients (Fig. 5D), which are instead mesenchymal and bear a larger number of innate immune cells. Consistently, in LUAD, KIRC, and STAD, differential expression and upstream regulator analyses of druggable nodes indicate that acute inflammatory pathways are dominant in the mesenchymal state, whereas potential druggable nodes emerged in the epithelial state (fig. S8E). From an immunotherapy perspective, the NMF C5 appears the most interesting one as it contains the largest fraction of DNA mismatch repair genes and a positive lymphocytes-to-macrophages ratio but a nonobvious survival trend (Fig. 5, A to D), which raises the interesting possibility that these tumors may bear potentially higher neoantigen load and qualify for immune checkpoint blockade therapy.
To assess subtype reproducibility, we compiled an independent set of cancer expression profiles from the International Cancer Genome Consortium (ICGC) cohort (see Materials and Methods). The ICGC validation cohort was highly reproducible for TCGA C1, C2, and C4 (fig. S7, D to G) and considering that this dataset is much narrower in cancer types and devoid of brain tumors (e.g., the main C3 component)—this supported a five-group classification of patients with pan-cancer based on the meta-EMT signature.
Using tumor classification by dimensionality reduction, the meta-MES signature improved partitioning of patients with epithelial and mesenchymal LUAD into coherent molecular subtypes of LUAD (Fig. 5E). This approach holds increased power of stratification compared to single/few biomarkers and is simpler than integrative analyses of multiple epigenome layers (30).
A subset of meta-EMT signature genes is regulated by RNA polymerase II elongation and converge on the activator protein 1 (AP-1) transcriptional network
Because the metagene signatures herewith identified bring together tumors of different origins, we hypothesized that these genes may be regulated by conserved transcriptional mechanisms. The functional antagonism between regulators occurs at enhancers and promoters, which led us to test whether regulation of the meta-EMT program occurs through RNA polymerase II (RNAPII). Hence, we immunoprecipitated RNAPII and assessed its genome-wide distribution in the homeostatic state and in EMT driven by GSK126, which precedes chromatin remodeling (fig. S1). Consistent with the ATAC-seq and H3K27ac biding profiles, RNAPII qualitative occupancy was also similar in naїve and GSK126 conditions (Fig. 6A). Notably, the fraction of transcriptional start site (TSS) associated with genes of the meta-EMT signature appears to have a similar weight as the other active promoters in the overall distribution, suggesting that RNAPII and the antagonistic gatekeepers both converge on broadly regulating active chromatin.
Fig. 6. Antagonistic chromatin regulators control meta-EMT activity via enhancers and promoters regulated by RNAPII elongation and converging on the AP-1 transcriptional network.
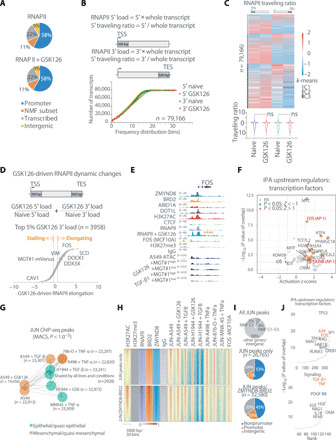
(A) Pie charts showing the genomic distribution of the RNAPII in naïve and GSK126-treated A549. The NMF signatures genes from Fig. 5A are highlighted as a subset of promoters (orange). (B) Cumulative distribution plot of the genome-wide RNAPII loading at 5′ and 3′ of each transcript, as defined above. (C) Heatmap (above) and violin plot (below) of RNAPII traveling ratio at 5′ and 3′ of each transcript for naïve and GSK126-treated cells. Clustering was performed using k-means, with k = 3, and the “ns” denotes no significance by one-way ANOVA and Sidak post hoc test. (D) Relative RNAPII traveling ratio between naïve and GSK126-treated cells on selected genes, as defined above. A ±0.5 threshold was decided on the MGT#1-mVenus reporter (more stringent), and selected examples are shown. (E) IGV view of FOS. The asterisks denote changes in elongation as determined in (D). (F) Upstream regulator analysis by IPA on the genes passing the FC in (D). The AP-1 transcription factor components are highlighted in red. (G) Giraph plot showing the distance between JUN ChIP-seq peak lists. Colors denote the known state of the cell line in which ChIP-seq was performed. (H) Heatmap of signal intensity for the indicated ChIP-seq profiles for all JUN peaks in (G). Direct overlap with genomic loci in Fig. 4A was used to partition chromatin occupancy into the indicated clusters. (I) Pie charts showing the genomic distribution of all JUN peaks in (G), above. NMF C1 genes were annotated when a JUN peak was close to the gene (−2.5 and +0.5 kb). The below charts are referred to the two clusters in (H). (J) Upstream regulator analysis by IPA on the genes annotated in (I) as direct JUN/ZMYND8-BRD2 targets.
The RNAPII elongation inhibitor flavopiridol can rescue both the transcriptional amplification of inflammatory genes driven by GSK126 in lung cancer cells (22) and the global RNAPII pausing release driven by ARID1A loss in ovarian cancer cells (31). Hence, we next investigated whether RNAPII dynamics change during GSK126-driven EMT, either globally or locally. To this end, for each human gene, we calculated the extent of RNAPII loading at TSSs (5′ TSS), coding regions, and termination sites [3′ transcription end site (TES)]. Globally, we observed that RNAPII accumulation occurred with higher frequency at 5′ TSS than at 3′ TES of human genes, which is consistent with the well-established role for proximal promoter RNAPII pausing, which contributes to maintain transcriptional homeostasis. GSK126-driven EMT did not affect this mechanism at a global level (Fig. 6, B and C). To investigate whether local changes occur at selected genes, we computed the differential RNAPII loading at both ends of each gene and, compared to the naїve condition, we observed that GSK126-driven EMT was accompanied by selective and quantitative changes in RNAPII accumulation (Fig. 6C). To gain insights into the biological significance of these changes, we focused on the selected fraction of transcripts with high load of RNAPII at either side of each gene (Fig. 6D). This analysis uncovered changes in RNAPII levels upon GSK126-induced EMT at 3958 genes (of 79,166; ~5%), which are characterized by high RNAPII load at their 3′ end. Multiple genes showed a positive score (>0.5 log2FC; table S6), a threshold that was defined on the basis of the MGT#1-mVenus genetic reporter (i.e., positive genomic locus for GSK126-driven activation). Changes in score appeared to correlate with increased and decreased RNAPII elongation. Upstream regulator analysis of the GSK126-elongating (>0.5 traveling) and stalling promoters (<−0.5 traveling) pointed to c-Myc and activator protein 1 (AP-1) transcriptional networks as critical nodes connecting genes that increase RNAPII during EMT. We focused on AP-1 as FOS and JUNB targets were independently enriched in the dataset [ingenuity pathway analysis (IPA), P = 7.62 × 10−9 and 0.01, respectively] and FOS itself was in the top tier of RNAPII elongating genes upon GSK126-driven EMT in A549 (Fig. 6, D to F, and fig. S10A).
The canonical AP-1 complex is a pioneer transcriptional regulator formed by a diverse set of FOS/JUN-like homo- and heterodimers (32). The expression pattern of canonical AP-1 factors in patients’ biopsies of LUAD, KIRC, and STAD suggests that c-JUN/JUNB and FOSL1/2 are bona fide canonical AP-1 transcription factor configurations in mesenchymal states (fig. S10, B and C). This holds true in A549-MGT#1 or H1944-MGT#1 exposed to EMT triggers (e.g., GSK126 and TGF-β1; fig. S10D). JUN is a pioneer factor for mesenchymal and stress response gene activation in glioblastoma (GBM) (33) and is activated at protein level in response to various EMT triggers such as GSK126, TGF-β1, and TNF-α in cancer cell lines of LUAD (A549 and H1944), KIRC (786-O and A498), STAD (MNK45), and GBM (IDH-wt-GICs; fig. S10E). Under these conditions, ChIP-seq for JUN as a surrogate mark for AP-1 binding revealed an average of ~25,000 binding sites, largely shared among multiple cell lines and states (Fig. 6G). k-means clustering of JUN peaks uncovered genomic regions containing 4303 genes whose chromatin is decorated with H3K27ac and simultaneously bound by the antagonistic chromatin modulators and JUN in multiple cell lines and states (Fig. 6H). While JUN binding distribution is predominantly distal to promoters, those genes that could be attributed to JUN as direct targets owing to promoter proximity (i.e., ±2.5 kb from TSS) included 37% of the metagenes enriched in mesenchymal tumors (135/362 of NMF C1; Fig. 6I and fig. S10F). Upstream regulators analysis confirmed that genes bound by all factors are significantly associated with AP-1 and generally linked these genes to pathways repeatedly associated with EMT (Fig. 6J) and specifically with the patients’ NMF cluster C1 (Fig. 5C and figs. S8D and S9). This underscores RNAPII and AP-1 as pivotal regulators of the pan-cancer EMT program.
Overall, our data reveal that the transcriptional control of EMT homeostasis across multiple human cancers is characterized by general principles, where antagonistic chromatin modulators co-occupy active chromatin and regulate epithelial cancer cell metastability. This additional layer of regulation integrates the well-established bivalent regulation of poised chromatin.
DISCUSSION
The epigenome plays a key role in the initiation and progression of human cancers and offers therapeutic opportunities (20). However, targeting chromatin factors that have pleiotropic effects on cell proliferation and cell identity poses specific challenges, as exemplified by how targeting PRC2 in NSCLC enhances cancer cell plasticity and sets the ground for tumor progression (12, 22). Here, we exploited the cellular plasticity facilitated by PRC2 inhibition in lung cancer cells to uncover factors involved in the phenotypic regulation of epithelial cell identity in solid tumors. By combining functional genetic screens with a phenotypic reporter (23), we uncovered known and novel players governing EMT in lung cancer cells.
The use of a transcriptional reporter might have facilitated the discovery of chromatin factors over the kinases, more often operating in the cytoplasm. Moreover, CRISPRi using KRAB-dCas9 has limited discovery power at both per guide and per gene levels (34). Thus, future improvement in the screening procedures is needed to tackle the detection limits of true positive hits.
The hits in the kinome screen were indeed limited to the discovery of the RAS scaffold CNKSR2 and the tyrosine kinase receptor of the TGF-β/BMP superfamily, ACVR1. CNKSR2 is understudied and determining whether it plays a critical role in one or more tumor types will require further investigation. Instead, ACVR1 is a potential driver of pediatric gliomas (35). Our data showing that ACVR1 loss or inhibition by DMH1 traps cells in their epithelial state and MAPK, MAPK kinase 1/2 (MEK1/2), c-Jun N-terminal kinase (JNK), or phosphatidylinositol 3-kinase (PI3K) inhibitors as potentially valid therapeutic options in various epithelial cancers (fig. S8E) may open to rationale combination of these compounds.
The epigenome screen revealed a functional antagonism between chromatin modulators that occurs at active chromatin including metagenes juxtaposing multiple cancers according to their epithelial-mesenchymal identity. This indicates that our findings potentially have broad implications in solid tumors. In particular, the pan-cancer EMT signature genes bring together the patients with lung, kidney, and stomach cancers with mesenchymal features and poor overall survival. Whereas their epithelial counterparts appear potentially sensitive to MAPK, MEK1/2, JNK, or PI3K inhibitors, these tumors appear to have undergone a “malignant EMT,” enriched in proinflammatory pathways and infiltrated by myeloid cells that pose hurdles to immunotherapy. In these settings, targeting the epigenome may involve neoadjuvant therapeutic strategies aiming to restore a noninflammatory status or amplifying tumor inflammation through a targeted agent followed by hitting acquired vulnerabilities (12).
The layer of chromatin regulation identified in our screen likely contributes to maintaining metastability between two states, thereby promoting cell plasticity before more discrete cell fate decisions take place. Whereas TGF-β signaling potently drives EMT, the effect of EZH2 inhibition rather promotes a “partial EMT.” In this setting, the binding of antagonistic modulators and their higher load at chromatin governing the EMT process in metastable cells is reminiscent of how Polycomb and Thritorax complexes control embryonic stem cell lineage priming (17). In both cases, two antagonistic complexes bind to the same targets, whose transcriptional activation relies on intracellular signaling and RNAPII elongation [(17) and this study]. Yet, the antagonistic regulation herein reported takes effect at active chromatin rather than inside poised one, and the cooperative transcription factors involved are broad regulators such as Myc and AP-1, rather than lineage-specific ones. Noteworthy, both Myc and AP-1 complexes involve selective homo- and heterodimerization and selective activation or repression of their targets. Unlike Myc, AP-1 is believed to act as pioneer transcription factor, which argues in favor of its ability to reconcile the broadness of the EMT with heterogeneous tissues and states. The future combination of our synthetic reporter and various EMT models may help to elucidate the biochemical events regulating cell homeostasis at active chromatin.
The functional antagonism by bromodomain-containing ZMYND8 and BRD2 in regulating EMT is interesting given that both chromatin readers recognize histone acetylation but their mechanism of action on gene regulation is markedly different. BRD2 is a positive regulator of developmental gene transcription and is required for the maintenance of genome architecture (29, 36). In contrast, ZMYND8 negatively regulates gene expression by preventing enhancer activation (37, 38). Our data show that both factors occupy largely overlapping genomic regions, which are characterized by the activating histone mark H3K27ac and transposase-accessible chromatin, thereby featuring enhancers. Our finding that ZMYND8 is a negative regulator of EMT is in keeping with the previous data supporting its role as a negative regulator of metastatic gene expression (37). However, given that ZMYND8 specificity for its targets depends on preexisting chromatin configuration and antagonistic function with other bromodomain-containing proteins, it is likely that it controls cell identity in a cell type–, developmental stage–, and disease-specific manner. The comparison between TGF-β and GSK126-driven EMT suggests that antagonistic fluctuations between cell epithelial-mesenchymal fates occur in the absence of (or preceding) substantial chromatin remodeling. The identification of multiple negative regulators of EMT in the NuA4 histone acetyltransferase complex such as EPC1, ING3, DMAP1, and BRD8 aligns well with the SGF29 from the SAGA complex to negatively regulate proneural-to-mesenchymal glioblastoma transition (23). These findings spark interest in the role of histone acetylation as a bivalent chromatin regulatory mark. Moreover, similar to a previous link between PRC1 and BRD2 (39), it may be interesting to investigate whether KMT2A, which is part of the MLL1/COMPASS complex, links the regulation of acetylated chromatin to H3K4, H3K27, and DNA methylation, which are fine-tuned by the MLL2/COMPASS complex (40).
Last, our epigenome screen uncovered a context-dependent role for the SWI/SNF member ARID1A (BAF complex) in EMT. ARID1A is intensively studied because of its frequent loss-of-function mutations in human cancers (19). Unlike SMAD4 loss that drives irreversible and deterministic alterations in BMP signaling, ultimately promoting invasion and metastasis in colon cancer (41), ARID1A loss impairs mesenchymal conversion in quasi-mesenchymal lung cancer cells, is neutral in quasi-epithelial lung cancer cells, and promotes the opposite outcome in proneural GICs (Fig. 3F). In a recent independent report, ARID1A loss in neuroblastoma cells promotes the adrenergic-to-mesenchymal transition (42). We interpreted this evidence as the loss of chromatin modulators to be nondeterministic in that these may oversee antagonistic and potentially stochastic fluctuations between cell states so that their loss will result in context-dependent outcomes. Cell state changes will be influenced by the intersection between alterations in the chromatin modulator and the preexisting genetics, tumor differentiation stage, and microenvironment, notably inflammation. This level of complexity underscores the challenges in targeting chromatin factors in cancer. SWI/SNF-mutant cancers selectively depend on a functional PRC2 (43) and EZH2 inhibition as targeted therapy in these tumors is in a clinical trial (NCT03213665). Our functional screen identified members of the BAF complex (e.g., ARID1A, SMARCB1, and SMARCE1) but not of the PBAF complex (e.g., ARID2) as required to execute EMT under homeostasis and in response to EZH2 inhibition. However, ARID1A deletion in EZH2-inhibited lung cancer cells was not detrimental to cell fitness, which suggests that genetic deletion alone may be insufficiently predictive of therapeutic outcome. Alternative to targeting the functional antagonism between SWI/SNF and PRC2, selective targeting of intra-SWI/SNF dependencies based on synthetic lethality is supported by several systematic studies using in vitro cell proliferation as a proxy for tumorigenic potential (28, 43–45). Our system may help incorporating phenotypic cell fate changes in the prioritization of druggable dependencies that are developed on the basis of their effect on tumor cell proliferation but are likely to also affect their plasticity.
Integrated molecular profiling in human cancers uncovered pan-cancer patterns attributed to the tumor cell of origin (30), but therapeutic targeting of tumor states is still unattainable. Since the original discovery of bivalent domains in pluripotent stem cells (15) and their correlation with transcriptional alterations in human cancers (46), there has been growing interest in targeting chromatin regulators as an anticancer therapy. Our data support the antagonistic chromatin regulation in human cancers as a mechanism to fine-tune epithelial-mesenchymal cancer cell interconversion. Whereas targeting pleiotropic regulators poses therapeutic challenges, synthetic genetic tracing of cell fate transitions provides a simple, robust, and scalable approach to link phenotypic responses and targeted perturbations, thereby offering additional control over prioritization of combinatorial treatments that may overcome cancer cell plasticity.
MATERIALS AND METHODS
MGT#1 reporter generation
The MGT#1 synthetic reporter is described elsewhere (23). Briefly, mesenchymal GBM tumors were compared to all other GBM subtypes, and differentially enriched genes and general transcription factors were used as the framework for discovery of mesenchymal-specific cis-regulatory DNA (23). In turn, selected candidate cis-regulatory elements were stitched together and cloned upstream of an mVenus fluorescent reporter in a lentiviral backbone. The vector showed concordant expression with epithelial-mesenchymal status in multiple cell types. An algorithm for automated generation of synthetic reporters from gene signatures will also be described elsewhere.
Cell lines and cell culture
A549, H1944, and H2122 human lung cancer cell lines (R. Bernards laboratory, Netherlands Cancer Institute (NKI), Amsterdam, The Netherlands) were cultured in RPMI medium (Gibco) supplemented with 10% fetal bovine serum (FBS, from Gibco) and penicillin and streptomycin (100 U/ml; Gibco) at 37°C in a 5% CO2 and 95% air incubator.
Human glioma cells (-IDH1-wt-GICs) were propagated as described in (33). RHB-A medium (Takara) supplemented with epidermal growth factor (20 ng/ml; R&D Systems), basic fibroblast growth factor (20 ng/ml; R&D Systems), heparin (1 μg/ml; Sigma-Aldrich), 1% penicillin and streptomycin (Gibco), and PDGF-AA (20 ng/ml; R&D Systems). Human GICs were cultured at 37°C in a 5% CO2, 3% O2, and 95% humidity incubator.
A498 kidney carcinoma cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) supplemented with 10% FBS and penicillin and streptomycin (100 U/ml; Gibco). Renal cell carcinoma (786-0; K. Schmidt-Ott laboratory, Max Delbrück Center for Molecular Medicine, Berlin, Germany) and MKN45 (S. Minucci laboratory, European Institute of Oncology, Milan, Italy) were kept in the presence of RPMI (Gibco) with 10% FBS (Gibco) and penicillin and streptomycin (100 U/ml; Gibco).
The MCF7 and MDA-231 breast cancer cell lines (provided by the R. Bernards laboratory, NKI) were cultured in DMEM (Gibco) supplemented with 10% FBS and penicillin and streptomycin (100 U/ml; Gibco). For experiments in CNKSR2 and ACVR1 KO (Fig. 2C), cells were treated with 5 μM GSK126 (GlaxoSmithKline), kept at low confluency, and cultured for 5 days. In fig. S4G, cells were cultured for 10 days ± hTGF-β1 (5 ng/ml; R&D Systems). In Fig. 2D, A549 cells were treated with 1, 3, and 5 μM DMH1 (Cayman Chemical) for 2 days. In fig. S4H, A549 cells were treated with hBMP4 (10 ng/ml, R&D Systems) and GSK126 (5 μM) for a total of 5 days. DMH1 1 μM was added for the last 3 days. To measure MGT#1 reporter, fluorescence in viable cells was identified by FACS using Calcein UltraBlue AM (Biozol) and Zombie NIR (BioLegend).
Lentivirus production and infection
Lentivirus was produced by transfecting human embryonic kidney–293 T cells using FuGENE HD (Promega) as described (33). Infected cells with MGT#1 reporter were sorted by FACS for low levels of MGT#1 expression.
CRISPRi screening procedure
The CRISPRi library was designed as previously described (34). Briefly, gRNAs targeted −50 to +300 relative to the TSS of annotated genes (APPRIS). For the CRISPRi screens, A549-MGT#1 ± GSK126 ± dox cells were sorted on a MoFlo Astrios. We aimed at a library representation of 1000× (>6 million cells) in the 10% of the lowest (dim) and 10% of the highest (bright) cells within each population. The mid population was also sorted and included in the screen analysis, as control. Cells were lysed for 10 min at 56°C in AL lysis buffer + proteinase K buffer (QIAGEN) followed by DNA extraction using AMPure beads (Agencourt) and ribonuclease A treatment. PCR amplification and barcode tagging of the CRISPRi libraries were done essentially as described, including PCR buffer composition (47). For each sample, in PCR1, we used 20 μg of DNA divided over 10 parallel reactions, including from input controls, whereas the plasmid library needed 0.1 ng of DNA in PCR1. Parallel PCR1 reactions were mixed together, and 5 μl was used as a template for PCR2. We used Phusion Polymerase (New England Biolabs), GC buffer, and 3% dimethyl sulfoxide in both PCR1 and PCR2. Primers are available upon request.
Library concentrations were measured, and barcoded libraries were pooled and sequenced on an Illumina HiSeq 2500 sequencing. Reads were mapped to the in silico library with a custom script (available upon request) to generate read counts, which were subsequently used as input for SeqMonk. We used a custom genome for SeqMonk analysis (available upon request). Aligned reads to the custom genome were used as input for Fig. 1 and figs. S2 and S3 (table S1).
Differential enrichment analysis was performed for sgRNAs using the R/Bioconductor package DESeq2 v1.16.1 (PMID: 25516281) with standard parameters. Genes exhibiting a false discovery rate (FDR) ≤ 0.05 and a log2FC ≥1.5 or ≤−1.5 were considered differentially enriched. Normalized rlog-transformed gene counts were calculated using the DESeq2’s rlog() function. We ran two independent CRISPRi screens in A549 and one additional screen in H1944.
Flow cytometry analysis
Validation by FACS analysis of MGT#1 was performed by seeding 2 × 105 cells on a well of a six-well plate for each condition and harvested 24 hours later for analysis. Flow cytometric analysis was conducted on a Fortessa five-laser analytical cytometer (BD Biosciences). Gating was set on live and single cells using forward scatter and side scatter. As gating strategy, we divided wild-type cells into four subpopulations of 5% low, 45% mid-low, 45% mid-high, and 5% high MGT#1 expression. The same gates were applied to all further conditions to determine shift of reporter expression. Comparisons between KO and control in Fig. 3F were done with respective controls but different gating due to different instrument setups. Fluorescence intensity of the reporter was detected with the following settings for mVenus: excitation, 488 nm; emission, 530 nm (30-nm bandpass filter).
Live-cell imaging
For live-cell imaging, human lung and breast adenocarcinoma transduced with the MGT#1 reporter and treated with GSK126 (5 μM for 5 days), TGF-β1 (5 ng/ml for 5 days), or vehicle were seeded out on 384-well plates in FluoroBrite DMEM (Gibco) supplemented with 10% FBS. Imaging was performed on high-content imaging platform (Operetta CLS, PerkinElmer) using 488-nm excitation and a 470- to 540-nm emission filter (confocal mode, 40× water objective). Maximum projections from 5-μm z stacks of MGT#1 reporter intensities and DRAQ5-stained nuclei are shown (fig. S1A).
Electroporation
Genome editing of all cell lines described was performed by electroporation using the Amaxa 4D-Nucleofector Kits and the specific program. Briefly, 2 × 105 cells were counted and resuspended in 20 μl of the respective buffers and supplement in a 16-well Nucleocuvette strip. For A549, H1944 electroporation was performed with SF nucleofection buffer (Lonza) and using CM130 and FF-120 programs, respectively. Glioma cell lines were electroporated in buffer P3 and using CA-138 program. Gene KOs were generated using the Synthego Gene Knockout Kits (single guides and V2 kit) following the manufacturer’s instructions.
Genomic DNA extraction and T7E1 calculation
Editing efficiency was estimated by T7 endonuclease I assay. Genomic DNA was extracted from control and edited cells using the spin tissue isolation kit (Invisorb). Target regions were PCR-amplified with the Phusion Flash High-Fidelity PCR Master Mix (Thermo Fisher Scientific) according to the manufacturer protocol. PCR products were denatured at 95°C for 5 min and reannealed at −2°C/s temperature ramp to 85°C, followed by a −0.1°C/s ramp to 25°C. The heterocomplexed PCR product (200 ng) was incubated with 5 U of T7E1 enzyme (New England Biolabs) and buffer 2 at 37°C for 30 min. Products from mismatch assays were electrophoresed on 2% agarose gel. The percentage of editing was analyzed by Sanger sequencing and calculated using the ICE (inference of CRISPR edits) webtool provided by Synthego.
Western blot
Western blotting analysis was performed using standard methods. Whole-cell extracts were prepared in radioimmunoprecipitation assay buffer [50 mM tris (pH 8.0), 50 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS] containing protease inhibitor cocktail (cOmplete, Roche) and phosphatase inhibitor cocktail (Thermo Fisher Scientific). Equal amounts of protein, as determined by the Micro BCA Protein Assay Kit (Pierce), were resolved on NuPage Novex 4 to 12% bis-tris gels (Invitrogen) or NuPAGE Novex 7% tris-acetate protein depending on the protein size and transferred onto nitrocellulose membranes (0.2 μm, Whatman).
Membranes were blocked in phosphate-buffered saline with 0.1% Tween 20 (PBST) 5% bovine serum albumin (BSA) for 1 hour, incubated with primary antibodies in PBST 1% BSA overnight at 4°C and with secondary antibodies coupled to horseradish peroxidase for 45 min in PBST 1% BSA. Bands were visualized using an enhanced chemiluminescence detection reagent (GE Healthcare). Primary antibodies used against the following antigens were as follows: anti-vimentin [D21H3 rabbit monoclonal antibody (mAb); Cell Signaling Technology, #5741], anti-vinculin (mouse clone h-VIN1; Sigma-Aldrich, #V9131), anti–E-cadherin (24E10 rabbit mAb; Cell Signaling Technology, #3195), anti-claudin (rabbit mAb; Cell Signaling Technology, #4933), anti-tubulin (DM1A mouse mAb; Sigma-Aldrich, #T9026), anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (rabbit mAb; Santa Cruz Biotechnology, #sc-25778), anti-H3K273me [rabbit polyclonal antibody (pAb); Cell Signaling Technology, #9733], anti-total H3 (rabbit mAb; Cell Signaling Technology, #AB1791), anti-Arid1a (D2A8U rabbit mAb; Cell Signaling Technology, #12354), anti-Brd2 (D89B4 rabbit mAb; Cell Signaling Technology, #5848), anti-DOT1L (D1W4Z rabbit mAb; Cell Signaling Technology, #77087), anti-ZMYND8 (rabbit pAb; Atlas Antibodies, #HPA020949), anti-ARID2 (mouse mAb; Santa Cruz Biotechnology, #sc-166117), anti–phospho-SMAD1/5/9 (rabbit pAb; Cell Signaling Technology, #9511), and anti–c-JUN (60A8 rabbit mAb; Cell Signaling Technology, #9165).
Copy number–normalized MGT#1 reporter expression
Human lung and breast adenocarcinoma transduced with the MGT#1 reporter were treated with GSK126 (5 μM for 5 days), TGF-β1 (5 ng/ml for 5 days), or vehicle. To perform lentiviral copy number normalization, gDNA was extracted using a spin tissue isolation kit (Invisorb). Relative amounts of reporter integration sites into the genome of target cells were assessed by qPCR, using mVenus (MGT#1) specific primers and N2 primers targeting a genomic region in chromosome 13 for input normalization between samples. gDNA (5 ng) was amplified using respective primers and the Power SYBR Green PCR Master Mix (Life Technologies) in a total reaction volume of 10 μl in quadruplicate. Relative DNA amounts of MGT#1 were normalized over N2 levels to calculate copy number abundance for each sample.
Expression levels of MGT#1 reporter in corresponding samples were assessed in quadruplicate by qPCR using the One-Step TB Green PrimeScript RT-PCR Kit II (Takara) with an input of 2-ng total RNA using mVenus (MGT#1) specific primers and GAPDH primers for normalization. Relative reporter expression of MGT#1 was calculated by normalizing over GAPDH expression. Final normalization as presented in fig. S1A was done by first correcting GAPDH-normalized MGT#1 expression for each condition (± treatment) divided over N2-normalized copy number abundance from the respective untreated control and then setting H2122 as a reference, by calculating the FC increase of copy number–normalized reporter expression.
Proliferation, colony formation assay, wound healing assay, and three-dimensional Matrigel invasion assay
Proliferation assay was performed by seeding, using a TECAN SPARK20M plate reader, 5 × 104 cells in a 24-well plate in triplicate and counted after 24 and 48 hours post seeding. For colony formation assay, 500 cells were plated for each KO and control cell line in triplicate. Colony counting was performed using ImageJ Colony Counter plug-in. For the wound healing assay, each cell line analyzed was seeded in 96-well plates as six replicas until confluence and then wounded using a narrow pipette tip. Longitudinal whole-well imaging with 2-hour intervals starting from wound delivery was performed using the Operetta high-content screening platform (PerkinElmer). Area of wound size for each time point was calculated in a defined imaging window by using the Fiji plugin MRI_Woundhealing_Tool. Wound closure rates for selected time points 20, 40, and 52 hours were normalized on the respective wound area at time point 0 hours and are represented as mean values ± SD of minimum three independent replicas using GraphPad Prism.
For three-dimensional Matrigel invasion assay, control cells (with and without TGF-β1 pretreatment; 5 ng/ml for 5 days) and CRISPR-Cas9–KO A549 LUAD cell lines were grown on 96-well plates as a monolayer to 30 to 50% confluence in three replicate wells. Cells were starved in serum-free medium for 12 hours before a monolayer was overlaid with TGF-β1–enriched (5 ng/ml) Matrigel and supplemented with complete medium plus 5 μM DRAQ5 to label nuclei. Using a high-content imaging platform (Operetta CLS, PerkinElmer), z stacks of DRAQ5 channel with 40-μm thickness were imaged every 12 hours to monitor invasion into the Matrigel layer. In-built image analysis building blocks facilitated DRAQ5-labeled nuclei detection and derivation of positional and morphologic properties for each single nucleus at each time point. Mean values for geometric centers of all nuclei from three replicate wells (total nuclei number 1500 to 3000) were normalized to time point T0 for each respective following time points to calculate invasion depth.
ATAC sequencing
ATAC-seq was performed on naїve, GSK126 (5 μM)–, or TGF-β1 (5 ng/ml)–treated cells FACS-sorted for MGT#1-mVenus high expression. A total of 60,000 cells were sorted in biological replica and centrifuged; the pellet was gently resuspended in 50 μl of ATAC mix [25-μl 2× tagmentation DNA (TD) buffer, 2.5-μl 891 transposase and 22.5-μl nuclease-free water from Nextera DNA Library Prep, Illumina]. Cells were incubated for 60 min at 37°C with moderate shaking (500 to 800 rpm), lysed in proteinase K and AL buffer (QIAGEN); DNA was purified using 1.8× AMPure XP beads (Beckman Coulter). Library prep was made using primers compatible with Nextera Illumina. Each library was individually quantified using Qubit 3.0 Fluorometer (Life Technologies) and profiled on a TapeStation High Sensitivity D1000 ScreenTape (Agilent). The multiplexed libraries were sequenced on a HiSeq 4000 in a 2× 75–base pair (bp) mode.
ChIP and qPCR
ChIP-seq was performed in biological duplicate on 8 × 106 A549 cells treated for 11 days ± GSK126 (5 μM) or ± TGF-β1 (5 ng/ml) adapting the ChIPmentation protocol (46). For c-JUN ChIP-seq experiments, A498, 786-0, and MKN45 cell lines were pretreated for 6 hours with TNF-α (10 ng/ml; Enzo Life Sciences), while H1944 and A549 were treated with GSK126 and TGF-β1 for 11 days.
Briefly, cells were fixed with 1% formaldehyde (Alfa Aesar) for 10 min at room temperature, and the cross-linking was subsequently quenched with Glycine.
The chromatin preparation was conducted following the SimpleChIP Enzymatic Chromatin IP Kit (Magnetic Beads, Cell Signaling Technology, #9003S) protocol. The chromatin was sheared using the Bioruptor (15 cycles, 15-s on and 30-s off), and the ChIP was performed with the SimpleChIP Enzymatic Chromatin IP Kit and using the following antibodies: anti-H3K27me3 (Millipore, #07-449), anti-H3K27ac (Abcam, #ab4729), anti-Arid1a (Cell Signaling Technology, #12354), anti-Brd2 (Cell Signaling Technology, #5848), anti-DOT1L (Cell Signaling Technology, #77087), anti-ZMYND8 (Polyclonal Atlas, #HPA020949), anti–c-JUN (Cell Signaling Technology, #9165), anti-PolII (Rockland, #600-401-HB3), or with rabbit immunoglobulin G (IgG) as control (Cell Signaling Technology). The ChIP reactions were further bound with ChIP-Grade Protein G Magnetic Beads (Cell Signaling Technology, #9006) and tagmented in 50 μl of transposition mix containing 25 μl of TAPS–N,N′-dimethylformamide (DMF) buffer [80 mM TAPS, 40 mM MgCl2, and 50% (v/v) DMF], 0.625 μl in-house–produced hyperactive Tn5 enzyme, and 24.4 μl of nuclease-free water. Tagmentation reactions were incubated for 10 min at 37°C and subsequently stopped by adding 5 μl of 10% SDS. The tagmented bead-bound chromatin-antibody complexes were washed twice with TET buffer (0.2% Tween 20, 1 mM EDTA, and 10 mM tris-HCl), eluted in 1× ChIP elution buffer by incubating 30 min at 65°C with gentle vortexing (1200 rpm). The reverse cross-linking was performed by adding 6 μl of 5 M NaCl and 2 μl of proteinase K (Cell Signaling Technology, #10012) and incubation for 2 hours at 65°C. The DNA was purified by means of spin columns and eluted in 50 μl of DNA elution buffer. To assess the optimal number of PCR cycles for efficient library amplification, 2 μl of each sample was taken as template for Ct-determining qPCR using the KAPA HiFi HotStart ReadyMix (Roche) and 1xEvaGreen Dye (Biotium). The whole probe amplification was performed in 50-μl qPCR reaction with 20 μl of ChIP DNA. Primers were previously described. Each library was individually quantified by means of Qubit 3.0 Fluorometer (Life Technologies) and profiled on a TapeStation High Sensitivity D1000 ScreenTape (Agilent). The multiplexed libraries were sequenced either on the Illumina NextSeq 500 using a Mid Output V2 75 cycles chemistry kit in a 1× 75-bp mode or on the NovaSeq S4 in a 2× 150-bp mode.
To assess the occupancy of BRD2 and ZMYND8 on the specific target loci (MGT#1, NCL, and background SALL2 3′ end), ChIP was performed as described above in a panel of A549 wild-type, BRD2-, ARID1A-, and ZMYND8-KO cells. Quadruplicates of the ChIP-ed and input DNA were analyzed by real-time qPCR using the SYBR Green PCR Master Mix (Life Technologies) in the ViiA 7 Real-Time PCR System. Primer sequences are available upon request.
RNA-seq of MGT#1-expressing cells
Briefly, naїve, GSK126 (5 μM)–, or TGF-β1 (5 ng/ml)–treated cells (11 days) were sorted by FACS for MGT#1-mVenus high and low expression.
The RNA was extracted from the cells using the TRIzol Reagent (Invitrogen, #15596026) with subsequent isopropanol precipitation and AMPure XP bead purification. The quantity and quality profiling of the extracted RNA was conducted using the Qubit RNA HS Assay Kit (Invitrogen) and the High Sensitivity RNA ScreenTape System (Agilent, #5067-5581). The SMARTer Stranded Total RNA-Seq Kit v2-Pico Input Mammalian (Takara Bio, #634413) was used for to generate the RNA-seq libraries from 10 ng of total RNA according to the manufacturer’s protocol. The final libraries were quantified using the Qubit dsDNA HS kit (Invitrogen) and the KAPA Library Quantification Kit (Roche, #7960204001). The appropriate library size distribution was determined by the TapeStation High Sensitivity D1000 ScreenTape kit (Agilent). Pooled libraries were sequenced on the Illumina NextSeq 500 platform in a 1× 75-bp dual-indexed mode. The demultiplexing was performed using the bcl2fastq conversion software (v2.20.0). The reads were aligned to a custom genome (sLCRs containing GRCh38) using STAR_2.6.0c. HTSeq was used to generate the counts of the uniquely assigned reads for each gene (parameters: htseq-count –m intersection-nonempty -a 10 -i gene_id -s reverse -f bam). The RNA-seq data analysis was conducted in R v3.6.0 applying the DESeq2 pipeline (http://bioconductor.org/packages/release/bioc/html/DESeq2.html) on raw prefiltered counts (>50 in at least two samples) for assessing the differentially expressed genes (baseMean > 5, padj < 0.05, log2FC ± 1.5) between the control and treatment conditions. DESeq2 rlog transformation was applied on the prefiltered count data for normalization. enrichGO and the dotplot function from the “clusterProfiler” R package (https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html) were used to conduct and visualize the GO enrichment analysis of the GSK126 and TGF-β1–induced genes (baseMean > 5, padj < 0.05, log2FC > 1.5) in A549 and H1944.
ChIP-seq and ATAC-seq analyses
To perform a quantitative analysis, ChIP-seq and ATAC-seq annotation files (.bam) were loaded in SeqMonk (www.bioinformatics.babraham.ac.uk/projects/seqmonk/). ATAC-seq–positive regions were identified by unbiased analysis of read counts on the full complement of UCSC genome browser (https://genome.ucsc.edu/) deoxyribonuclease I hypersensitive sites (~2 million loci of variable length). Read count quantitation was performed by correcting for total count per million reads and data were log-transformed. Where appropriate, the data were further transformed by matching distributions function.
For ATAC-seq, we retained the DNAseI hypersensitive sites (DHS) with absolute read count of >−1 (log2) in control A549-MGT#1 ATAC profile replica. Read count quantitation using the 913,656 DHS included correcting for total count only in probes per million reads and for probe length, and data were log-transformed by size factor normalization and percentile normalization using “add to the 75.0 percentile” and further transformed by matching distributions. Statistical analysis of gene set enrichment was performed with standard settings in SeqMonk.
For ChIP-seq, peaks were calculated using a magnetic cell sorter (MACS) with cutoff P > 10−5 and assuming a fragment size of 300 bp. Individual ChIP-seq profiles (with replicas, wherever available) were compared to the IgG background (similar outcomes were obtained if the input DNA was used). The individual peaks lists were then merged into one containing 140,761 loci bona fide bound by either ARID1A, DOT1L, BRD2, ZMYND8, H3K27ac, H3K27me3, and RNAPII in naїve or GSK126-treated cells. Read count quantitation was performed using all reads overlapping the selected probes and correcting for total count to largest store, followed by log transformation. Additional filtering retained all probes with normalized values >2 and duplicates removed. Moreover, as some of the ChIP-seq profiles were single reads, and some were paired-end, some specific artifacts present in all the 2× PE (paired end) were removed by statistical analysis and FC of 2× PE versus 1× SR (single read) groups. In Fig. 6A, for each ChIP-seq profile, we retained the top ~75% peaks, as a measure of bona fide binding. For instance, we identified 56,540 naїve RNAPII peaks and 55,844 GSK126-treated RNAPII peaks, which were reduced to 44,932 and 44,771, respectively. These peaks were then annotated to 79,165 Ensembl transcripts for which a TSS and a TES were uniquely identified (see below).
Heatmap plots for comparing ChIP-seq profiles and density plots in Fig. 5F were generated from bigWig files using ChAsE v.1.0.11 (http://chase.cs.univie.ac.at/overview) using as parameters: exponential normalization, global stats, and binning by size. For Fig. 4A, clustering of 144,007 regions with size >150 and <5000 bp with variable extent of ZMYND8 binding and clusters I, II, and III were obtained by k-means function using the following parameters: left and right extension = 5000 bp, internal bins (with respect to the peak) = 200 bp, and number of clusters k = 3. The genomic coordinates from each cluster were exported and annotated to the HG38.
To normalize ChIP-seq data for Fig. 4E, .bam files were imported in SeqMonk, reads were extended by either 225 or 300 bp, depending on read length (150 or 76 bp), and reads were quantitated over the selected probes by standard log2, further quantitated by the size factor filter, and eventually transformed in z scores. The common regions bound by both ZMYND8 and BRD2 and regulated by TGF-b1 were identified the intensity difference data filter in SeqMonk on normalized data. The final list was the overlap between individual ChIP-seq significant loci. The Giraph plot in Fig. 6G was generated on normalized JUN ChIP-seq data, using individual MACS peak probe lists as input. Annotation was performed by querying the probe lists for the closest gene to each probe (±5000 bp, or otherwise indicated). IGV (integrative genomics viewer) of CHIP-seq data was used as a visualization tool, and autoscale or group autoscale was applied where appropriate to correct for different sequencing depth.
For DESeq2 analyses, SeqMonk calculated significance by read count quantitation using all aligned reads, and significance was called when P < 0.05 after Benjamini-Hochberg correction. Where indicated, additional stringency was applied through filtering on FC thresholds. To count reads aligning to the MGT#1-mVenus, we built a custom genome.
RNAPII ChIP-seq analysis
The GSK126-driven RNAPII traveling ratio was inspired by previous reports (48, 49) and modified and adapted to fit the data and the setting specs. Briefly, the “classic” RNAPII traveling ratio was calculated by annotating the +500 bp of each Ensembl gene retained in the analysis. Criteria for retaining a transcript were the availability of a unique TSS and TES, which resulted in 79,165 of 194,423. For the RNAPII-RNAPII + GSK126 profiles, the raw read count ranged from 0 to 9782/9760. Rather than removing the 2607/79,165 transcripts that featured “0” counts or retaining the transcripts with >50 in both profiles, 31,892/79,165 (~40%), we established a procedure that would reward transcripts with reasonable normalized counts. Therefore, we assessed the 5′ TSS and 3′ TES “RNAPII load” scores (log2 5′ TSS or 3′ TES read count + log2 whole transcript), in addition to calculating the classic traveling ratio of highly expressed transcripts (log2 5′ TSS or 3′ TES read count − log2 whole transcript). Follow-up IGV visualization was used to determine the best conditions of the analysis.
Pan-cancer analysis
The processed data and clinical data were collected from TCGA database. The discovery data consisted of 9112 tumor samples and 19,672 genes across 32 cancer types, which were obtained via the R package “TCGAbiolinks” v2.15.3. The genome was aligned against hg19 and was normalized in each sample.
Epithelial and mesenchymal gene expression scoring of human NSCLC cell lines
We used 18 EMT markers from (12). Six EPI markers consisted of CDH1, ERBB3, TJP3, CLDN7, CLDN4, and MUC1; and 12 MES markers included TWIST1, CCL11, IL1A, FN1, CDH2, TWIST2, SNAI1, SNAI2, ZEB1, ZEB2, VIM, and L1CAM. The epithelial and mesenchymal gene expression scores for each human NSCLC cell line were computed by multiplying gene expression values of 6 EPI markers and 12 MES markers, respectively, as follows.
Epithelial gene expression score = CDH1 × ERBB3 × TJP3 × CLDN7 × CLDN4 × MUC1;
Mesenchymal gene expression score = TWIST1 × CCL11 × IL1A × FN1 × CDH2 × TWIST2 × SNAI1 × SNAI2 ×ZEB1 × ZEB2 ×VIM × L1CAM.
Functional and pathway analysis
Functional and pathway analysis was performed with the R package clusterProfiler v3.10.1. In particular, we focused on the ontologies: Kyoto Encyclopedia of Genes and Genomes pathways, Biological Processes (BP), Molecular Function (MF), and Cellular Components (CC). Terms associated with an FDR ≤ 0.05 were considered significantly enriched. Where indicated, other functional analyses were conducted using Ingenuity Pathway Analysis software (QIAGEN).
EMT-MES dual scoring system defining MES and EPI patients
In each cancer type, we computed the EMT score for each patient as follows:
EMT score = [(MES1 + 1) × (MES2 + 1) × … (MES12 + 1)] / [(EPI1 + 1) × (EPI2 + 1) × … (EPI6 + 1)]
where MES1,...,12 referred the expression value of 12 MES markers and EPI1,...,6 represented the expression value of six EPI markers (see above). Then, the 10% of patients with highest EMT scores were defined as the most likely MES patients and the 10% of patients with lowest EMT scores were defined as the most likely EPI patients.
We performed the different expression analysis with DESeq2 to identify the significantly differentially expressed EMT markers between these 10% MES and EPI patients in each of the 32 cancer types. If the MES and EPI markers were significantly differentially expressed in at least 16 cancer types (>50% of 32), they were then defined as solid signature EMT markers. Finally, we obtained seven solid MES markers across cancer types: CCL11, FN1, L1CAM, SNAI2, TWIST1, TWIST2, and ZEB2. We computed a MES score as follows:
MES score = (MES1 + 1) × (MES2 + 1) × … (MES7 + 1)
where MES1,...,7 referred to the expression value of seven solid MES markers as above.
Last, we defined the 10% patients with highest MES score to be most likely MES patients and the 10% patients with lowest MES score as most likely EPI patients. The patients who were defined as MES patients by both EMT score and MES score were be the best-represented MES patients; the patients who were defined as EPI patients by both EMT score and MES score were the best-represented EPI patients.
NMF, PAM, and SAM analysis
NMF was applied to identify sample clusters. We run NMFconsensus.R for NMF analysis using the script available in the GenePattern (Broad Institute website, www.broadinstitute.org/cancer/software/genepattern/). The ZMYND8/BRD2 targets clusters I, II, and III were used as input. Particularly, each normalized gene expression value was further normalized to N(0, 1) across all samples independent of cancer types, which can adjust gene expression values to remove systematic variation between microarray experiments. This step is useful in rendering sample data comparable. ZMYND8/BRD2 clusters I, II, and III were merged as input for pan-cancer patient clustering. The NMF parameters were default, including number of clusterings to build consensus matrix = 20, error function = Euclidean, iterations = 500, and cluster K from 2 to 7.
For pan-cancer analysis, the preferred clustering result was determined by the cophenetic correlation between clusters and the average silhouette width of the consensus membership matrix. To identify the most representative samples with each cluster, silhouette widths were computed. The samples with negative silhouette widths were regarded as the outliers, which were filtered out. In the NMF consensus analysis, K = 3 (cophenetic coefficient = 0.9738) and K = 5 (cophenetic coefficient = 0.973) both exhibited good consensus. Although K = 3 displayed slightly higher cophenetic coefficient, we chose K = 5 (average silhouette width 0.86) because there was evidence for five consensus clusters, which is more in line with the literature and K = 3 versus K = 5 did not affect the overall identity of the mesenchymal cluster (C1) but rather the subdivided the other clusters. We next used a two-step process to identify subcluster-specific signature genes. First, Significance Analysis of Microarray (SAM) with cutoff FDRs set to 0 was used to identify significantly differential expression genes in each cluster defined by NMF consensus clustering. SAM function in R package “siggenes” v1.50.0 was used. Second, Prediction Analysis for Microarrays (PAM) was used to predict subcluster signature genes R package “pamr” v1.56.1 (https://CRAN.R-project.org/package=pamr). In addition, 74 patients with LUAD were clustered with NMF clustering in an analogous manner as we did for pan-cancer patients above but based on the ZMYND8/BRD2 clusters I, II, and III separately. K2 was chosen as the best class (see fig. S6).
Enrichment analysis
We performed enrichment analysis to examine the enrichment: (i) if MES and EPI patients of each cancer type was enriched in any one of the five NMF clusters; (ii) if all MES or EPI patients independent of cancer types were enriched in any one of the five NMF clusters; and (iii) if ZMYND8/BRD2 clusters I to III were enriched in any one of the five NMF clusters. Enrichments were reported as odds ratios, and their significance was evaluated using single-sided Fisher’s exact tests. P values were adjusted for multiple testing using the FDR. For enrichment analysis for (i) as example, the odds ratio was calculated as follows
where Nij is the number of MES patients of cancer type i in NMF cluster j, is the number of MES patients of cancer type i not in NMF cluster j, is the number of MES patients not from cancer type i but in NMF cluster j, and is the number of MES patients that were neither in cancer type i nor in NMF cluster j.
Independent validation of NMF clustering analysis in the ICGC cohorts
Because of limitation of gene expression availability, additional cohorts comprising nine non-TCGA projects were obtained from the data portal of the ICGC (https://dcc.icgc.org/releases/release_28/Projects) spanning the following entities: Ewing sarcoma (“BOCA-FR”), liver cancer (“LICA-FR” and “LIRI-JP”), malignant lymphoma (“MALY-DE”), ovarian cancer (“OV-AU”), pancreatic cancer (“PACA-AU” and “PACA-CA”), prostate cancer (“PRAD-CA” and “PRAD-FR”), and renal cell cancer (“RECA-EU”) patients. We used these data to first identify 112 mostly likely epithelial and mesenchymal patients with the EMT-MES dual scoring system as described above. Next, those patients were clustered with NMF clustering in an analogous manner as we did with the data from TCGA based on the 3798 signature genes for TCGA cohorts (see fig. S10).
CIBERSORT immune fraction resource
The relative fraction of 22 immune cell types within the leukocyte compartment was estimated by CISERSORT analytical tools. Recently, the CIBERSORT immune fraction for TCGA patients is available in Genomic Data Commons portal (https://gdc.cancer.gov/about-data/publications/panimmune). The major six aggregated tumor immune cell classes, including lymphocytes, macrophages, dendritic cells, mast cells, neutrophils, and eosinophils, were considered. Lymphocytes were selected as reference. Other five classes are compared to lymphocytes; significance was computed by Wilcoxon rank sum test; and P values were adjusted for multiple testing using the FDR. P < 0.05 was considered as significance.
Acknowledgments
We are grateful to A. Gardini, A. Alendar, G. Frigè, and M. Fletcher for critical reading of this manuscript; to M. van Lohuizen for support and discussions; and C. Company, M. Massaro, and G. Chandrasekaran for technical support. We are also grateful to the genomics [R. Kerkhoven, I. de Rink, and R. Kluin (NKI)] and FACS [F.v. Diepen (NKI) and H-P. Rahn (MDC)] facilities for assistance and indebted with B. Evers and R. Bernards (NKI) for sharing the kinome and epigenome sgRNA libraries and the dox-inducible KRAB-dCas9 expression system, which consist of pHR-TRE3G-KRAB-dCas9-P2A-mCherry and pHR-Tet3G, gifted by L. Gilbert and J. Weissman. We also thank S. Minucci and K. Schmidt-Ott for sharing stomach and kidney cancer cell lines, respectively. Data analyses include data generated by the TCGA Research Network (www.cancer.gov/tcga). M.J.S. and Y.D. are graduate students at Humboldt University. S.K. is a graduate student at BSIO-Charité Medical University. Funding: The G.G. laboratory acknowledges funding from MDC, Helmholtz (VH-NG-1153), KWF (NKI-2014-7208), and ERC (714922), and M.S. received funding from DFG (DFG SE 2847/2-1). Author contributions: M.S. performed most the experiments with support from S.K., M.J.S., Y.D., J.W., D.H., and G.G. M.S. and G.G. designed the experiments and analyzed data. L.L., Y.D., and G.G. performed the bioinformatics analysis. M.S. and G.G. supervised the studies. G.G. conceived the study and wrote the first draft of the manuscript. Competing interests: G.G. is an inventor on a patent application related to this work filed by Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft (no. EP18192715.3, filed 5 September 2018). The authors declare that they have no other competing interests. Data and materials availability: ChIP-seq and RNA-seq data generated in this study are made available from GEO (series GSE155117). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. CTCF (ENCFF205XEH_alignment_hg38.bw) and FOS (ENCFF338VNY.bigWig) ChIP-seq profiles were downloaded from ENCODE (www.encodeproject.org/). Additional data related to this paper may be requested from the corresponding authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabd7974/DC1
REFERENCES AND NOTES
- 1.Theveneau E., Mayor R., Neural crest delamination and migration: From epithelium-to-mesenchyme transition to collective cell migration. Dev. Biol. 366, 34–54 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Tam W.-L., Weinberg R. A., The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 19, 1438–1449 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng X., Carstens J. L., Kim J., Scheible M., Kaye J., Sugimoto H., Wu C. C., LeBleu V. S., Kalluri R., Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525–530 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Genovese G., Carugo A., Tepper J., Robinson F. S., Li L., Svelto M., Nezi L., Corti D., Minelli R., Pettazzoni P., Gutschner T., Wu C. C., Seth S., Akdemir K. C., Leo E., Amin S., Molin M. D., Ying H., Kwong L. N., Colla S., Takahashi K., Ghosh P., Giuliani V., Muller F., Dey P., Jiang S., Garvey J., Liu C. G., Zhang J., Heffernan T. P., Toniatti C., Fleming J. B., Goggins M. G., Wood L. D., Sgambato A., Agaimy A., Maitra A., Roberts C. W. M., Wang H., Viale A., DePinho R. A., Draetta G. F., Chin L., Synthetic vulnerabilities of mesenchymal subpopulations in pancreatic cancer. Nature 542, 362–366 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.David C. J., Huang Y.-H., Chen M., Su J., Zou Y., Bardeesy N., Iacobuzio-Donahue C. A., Massagué J., TGF-β Tumor Suppression through a Lethal EMT. Cell 164, 1015–1030 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heldin C.-H., Vanlandewijck M., Moustakas A., Regulation of EMT by TGFβ in cancer. FEBS Lett. 586, 1959–1970 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Yoshida T., Ozawa Y., Kimura T., Sato Y., Kuznetsov G., Xu S., Uesugi M., Agoulnik S., Taylor N., Funahashi Y., Matsui J., Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. Br. J. Cancer 110, 1497–1505 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tam W.-L., Lu H., Buikhuisen J., Soh B. S., Lim E., Reinhardt F., Wu Z. J., Krall J. A., Bierie B., Guo W., Chen X., Liu X. S., Brown M., Lim B., Weinberg R. A., Protein kinase C α is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 24, 347–364 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antony J., Tan T. Z., Kelly Z., Low J., Choolani M., Recchi C., Gabra H., Thiery J. P., Huang R. Y.-J., The GAS6-AXL signaling network is a mesenchymal (Mes) molecular subtype-specific therapeutic target for ovarian cancer. Sci. Signal. 9, ra97 (2016). [DOI] [PubMed] [Google Scholar]
- 10.McFaline-Figueroa J. L., Hill A. J., Qiu X., Jackson D., Shendure J., Trapnell C., A pooled single-cell genetic screen identifies regulatory checkpoints in the continuum of the epithelial-to-mesenchymal transition. Nat. Genet. 51, 1389–1398 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su J., Morgani S. M., David C. J., Wang Q., Er E. E., Huang Y.-H., Basnet H., Zou Y., Shu W., Soni R. K., Hendrickson R. C., Hadjantonakis A. K., Massagué J., TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 577, 566–571 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serresi M., Gargiulo G., Proost N., Siteur B., Cesaroni M., Koppens M., Xie H., Sutherland K. D., Hulsman D., Citterio E., Orkin S., Berns A., van Lohuizen M., Polycomb repressive complex 2 is a barrier to KRAS-driven inflammation and epithelial-mesenchymal transition in non-small-cell lung cancer. Cancer Cell 29, 17–31 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Shao D. D., Xue W., Krall E. B., Bhutkar A., Piccioni F., Wang X., Schinzel A. C., Sood S., Rosenbluh J., Kim J. W., Zwang Y., Roberts T. M., Root D. E., Jacks T., Hahn W. C., KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158, 171–184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brabletz T., Kalluri R., Nieto M. A., Weinberg R. A., EMT in cancer. Nat. Rev. Cancer 18, 128–134 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Bernstein B. E., Mikkelsen T. S., Xie X., Kamal M., Huebert D. J., Cuff J., Fry B., Meissner A., Wernig M., Plath K., Jaenisch R., Wagschal A., Feil R., Schreiber S. L., Lander E. S., A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Lin C., Garruss A. S., Luo Z., Guo F., Shilatifard A., The RNA Pol II elongation factor Ell3 marks enhancers in ES cells and primes future gene activation. Cell 152, 144–156 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marks H., Kalkan T., Menafra R., Denissov S., Jones K., Hofemeister H., Nichols J., Kranz A., Francis Stewart A., Smith A., Stunnenberg H. G., The transcriptional and epigenomic foundations of ground state pluripotency. Cell 149, 590–604 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chaffer C. L., Marjanovic N. D., Lee T., Bell G., Kleer C. G., Reinhardt F., D’Alessio A. C., Young R. A., Weinberg R. A., Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 154, 61–74 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kadoch C., Hargreaves D. C., Hodges C., Elias L., Ho L., Ranish J., Crabtree G. R., Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 45, 592–601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dawson M. A., The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 355, 1147–1152 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Tiwari N., Tiwari V. K., Waldmeier L., Balwierz P. J., Arnold P., Pachkov M., Meyer-Schaller N., Schübeler D., van Nimwegen E., Christofori G., Sox4 is a master regulator of epithelial-mesenchymal transition by controlling ezh2 expression and epigenetic reprogramming. Cancer Cell 23, 768–783 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Serresi M., Siteur B., Hulsman D., Company C., Schmitt M. J., Lieftink C., Morris B., Cesaroni M., Proost N., Beijersbergen R. L., van Lohuizen M., Gargiulo G., Ezh2 inhibition in Kras-driven lung cancer amplifies inflammation and associated vulnerabilities. J. Exp. Med. 215, 3115–3135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmitt M. J., Company C., Dramaretska Y., Barozzi I., Göhring A., Kertalli S., Grossman M., Naumann H., Sanchez-Bailon M. P., Hulsman D., Glass R., Squatrito M., Serresi M., Gargiulo G., Phenotypic mapping of pathological crosstalk between glioblastoma and innate immune cells by synthetic genetic tracing. Cancer Discov. 10 (2020). 10.1158/2159-8290.CD-20-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ballaré C., Lange M., Lapinaite A., Martin G. M., Morey L., Pascual G., Liefke R., Simon B., Shi Y., Gozani O., Carlomagno T., Benitah S. A., di Croce L., Phf19 links methylated Lys36 of histone H3 to regulation of Polycomb activity. Nat. Struct. Mol. Biol. 19, 1257–1265 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brien G. L., Gambero G., O’Connell D. J., Jerman E., Turner S. A., Egan C. M., Dunne E. J., Jurgens M. C., Wynne K., Piao L., Lohan A. J., Ferguson N., Shi X., Sinha K. M., Loftus B. J., Cagney G., Bracken A. P., Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat. Struct. Mol. Biol. 19, 1273–1281 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Trojer P., Cao A. R., Gao Z., Li Y., Zhang J., Xu X., Li G., Losson R., Erdjument-Bromage H., Tempst P., Farnham P. J., Reinberg D., L3MBTL2 protein acts in concert with PcG protein-mediated monoubiquitination of H2A to establish a repressive chromatin structure. Mol. Cell 42, 438–450 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wamsley J. J., Kumar M., Allison D. F., Clift S. H., Holzknecht C. M., Szymura S. J., Hoang S. A., Xu X., Moskaluk C. A., Jones D. R., Bekiranov S., Mayo M. W., Activin upregulation by NF-κB is required to maintain mesenchymal features of cancer stem-like cells in non-small cell lung cancer. Cancer Res. 75, 426–435 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsherniak A., Vazquez F., Montgomery P. G., Weir B. A., Kryukov G., Cowley G. S., Gill S., Harrington W. F., Pantel S., Krill-Burger J. M., Meyers R. M., Ali L., Goodale A., Lee Y., Jiang G., Hsiao J., Gerath W. F. J., Howell S., Merkel E., Ghandi M., Garraway L. A., Root D. E., Golub T. R., Boehm J. S., Hahn W. C., Defining a cancer dependency map. Cell 170, 564–570.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsu S. C., Gilgenast T. G., Bartman C. R., Edwards C. R., Stonestrom A. J., Huang P., Emerson D. J., Evans P., Werner M. T., Keller C. A., Giardine B., Hardison R. C., Raj A., Phillips-Cremins J. E., Blobel G. A., The BET protein BRD2 cooperates with CTCF to enforce transcriptional and architectural boundaries. Mol. Cell 66, 102–116.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoadley K. A., Yau C., Hinoue T., Wolf D. M., Lazar A. J., Drill E., Shen R., Taylor A. M., Cherniack A. D., Thorsson V., Akbani R., Bowlby R., Wong C. K., Wiznerowicz M., Sanchez-Vega F., Robertson A. G., Schneider B. G., Lawrence M. S., Noushmehr H., Malta T. M.; Cancer Genome Atlas Network, Stuart J. M., Benz C. C., Laird P. W., Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trizzino M., Barbieri E., Petracovici A., Wu S., Welsh S. A., Owens T. A., Licciulli S., Zhang R., Gardini A., The tumor suppressor ARID1A controls global transcription via pausing of RNA polymerase II. Cell Rep. 23, 3933–3945 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eferl R., Wagner E. F., AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859–868 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Gargiulo G., Cesaroni M., Serresi M., de Vries N., Hulsman D., Bruggeman S. W., Lancini C., van Lohuizen M., In vivo RNAi screen for BMI1 targets identifies TGF-β/BMP-ER stress pathways as key regulators of neural- and malignant glioma-stem cell homeostasis. Cancer Cell 23, 660–676 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Evers B., Jastrzebski K., Heijmans J. P. M., Grernrum W., Beijersbergen R. L., Bernards R., CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat. Biotechnol. 34, 631–633 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Buczkowicz P., Hoeman C., Rakopoulos P., Pajovic S., Letourneau L., Dzamba M., Morrison A., Lewis P., Bouffet E., Bartels U., Zuccaro J., Agnihotri S., Ryall S., Barszczyk M., Chornenkyy Y., Bourgey M., Bourque G., Montpetit A., Cordero F., Castelo-Branco P., Mangerel J., Tabori U., Ho K. C., Huang A., Taylor K. R., Mackay A., Bendel A. E., Nazarian J., Fangusaro J. R., Karajannis M. A., Zagzag D., Foreman N. K., Donson A., Hegert J. V., Smith A., Chan J., Lafay-Cousin L., Dunn S., Hukin J., Dunham C., Scheinemann K., Michaud J., Zelcer S., Ramsay D., Cain J., Brennan C., Souweidane M. M., Jones C., Allis C. D., Brudno M., Becher O., Hawkins C., Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 46, 451–456 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stonestrom A. J., Hsu S. C., Jahn K. S., Huang P., Keller C. A., Giardine B. M., Kadauke S., Campbell A. E., Evans P., Hardison R. C., Blobel G. A., Functions of BET proteins in erythroid gene expression. Blood 125, 2825–2834 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li N., Li Y., Lv J., Zheng X., Wen H., Shen H., Zhu G., Chen T. Y., Dhar S. S., Kan P. Y., Wang Z., Shiekhattar R., Shi X., Lan F., Chen K., Li W., Li H., Lee M. G., ZMYND8 reads the dual histone Mark H3K4me1-H3K14ac to antagonize the expression of metastasis-linked genes. Mol. Cell 63, 470–484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delgado-Benito V., Rosen D. B., Wang Q., Gazumyan A., Pai J. A., Oliveira T. Y., Sundaravinayagam D., Zhang W., Andreani M., Keller L., Kieffer-Kwon K.-R., Pękowska A., Jung S., Driesner M., Subbotin R. I., Casellas R., Chait B. T., Nussenzweig M. C., Virgilio M. D., The chromatin reader ZMYND8 regulates Igh enhancers to promote immunoglobulin class switch recombination. Mol. Cell 72, 636–649.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Surface L. E., Fields P. A., Subramanian V., Behmer R., Udeshi N., Peach S. E., Carr S. A., Jaffe J. D., Boyer L. A., H2A.Z.1 monoubiquitylation antagonizes BRD2 to maintain poised chromatin in ESCs. Cell Rep. 14, 1142–1155 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Douillet D., Sze C. C., Ryan C., Piunti A., Shah A. P., Ugarenko M., Marshall S. A., Rendleman E. J., Zha D., Helmin K. A., Zhao Z., Cao K., Morgan M. A., Singer B. D., Bartom E. T., Smith E. R., Shilatifard A., Uncoupling histone H3K4 trimethylation from developmental gene expression via an equilibrium of COMPASS, Polycomb and DNA methylation. Nat. Genet. 128, 693–611 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Voorneveld P. W., Kodach L. L., Jacobs R. J., Liv N., Zonnevylle A. C., Hoogenboom J. P., Biemond I., Verspaget H. W., Hommes D. W., de Rooij K., van Noesel C. J. M., Morreau H., van Wezel T., Offerhaus G. J. A., van den Brink G. R., Peppelenbosch M. P., Dijke P. T., Hardwick J. C. H., Loss of SMAD4 alters BMP signaling to promote colorectal cancer cell metastasis via activation of Rho and ROCK. Gastroenterology 147, 196–208.e13 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Shi H., Tao T., Abraham B. J., Durbin A. D., Zimmerman M. W., Kadoch C., Look A. T., ARID1A loss in neuroblastoma promotes the adrenergic-to-mesenchymal transition by regulating enhancer-mediated gene expression. Sci. Adv. 6, eaaz3440 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim K. H., Kim W., Howard T. P., Vazquez F., Tsherniak A., Wu J. N., Wang W., Haswell J. R., Walensky L. D., Hahn W. C., Orkin S. H., Roberts C. W. M., SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat. Med. 21, 1491–1496 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McDonald E. R. III, de Weck A., Schlabach M. R., Billy E., Mavrakis K. J., Hoffman G. R., Belur D., Castelletti D., Frias E., Gampa K., Golji J., Kao I., Li L., Megel P., Perkins T. A., Ramadan N., Ruddy D. A., Silver S. J., Sovath S., Stump M., Weber O., Widmer R., Yu J., Yu K., Yue Y., Abramowski D., Ackley E., Barrett R., Berger J., Bernard J. L., Billig R., Brachmann S. M., Buxton F., Caothien R., Caushi J. X., Chung F. S., Cortés-Cros M., deBeaumont R. S., Delaunay C., Desplat A., Duong W., Dwoske D. A., Eldridge R. S., Farsidjani A., Feng F., Feng J. J., Flemming D., Forrester W., Galli G. G., Gao Z., Gauter F., Gibaja V., Haas K., Hattenberger M., Hood T., Hurov K. E., Jagani Z., Jenal M., Johnson J. A., Jones M. D., Kapoor A., Korn J., Liu J., Liu Q., Liu S., Liu Y., Loo A. T., Macchi K. J., Martin T., Allister G. M., Meyer A., Mollé S., Pagliarini R. A., Phadke T., Repko B., Schouwey T., Shanahan F., Shen Q., Stamm C., Stephan C., Stucke V. M., Tiedt R., Varadarajan M., Venkatesan K., Vitari A. C., Wallroth M., Weiler J., Zhang J., Mickanin C., Myer V. E., Porter J. A., Lai A., Bitter H., Lees E., Keen N., Kauffmann A., Stegmeier F., Hofmann F., Schmelzle T., Sellers W. R., Project DRIVE: A compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi Screening. Cell 170, 577–592.e10 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Schick S., Rendeiro A. F., Runggatscher K., Ringler A., Boidol B., Hinkel M., Májek P., Vulliard L., Penz T., Parapatics K., Schmidl C., Menche J., Boehmelt G., Petronczki M., Müller A. C., Bock C., Kubicek S., Systematic characterization of BAF mutations provides insights into intracomplex synthetic lethalities in human cancers. Nat. Genet. 51, 1399–1410 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohm J. E., McGarvey K. M., Yu X., Cheng L., Schuebel K. E., Cope L., Mohammad H. P., Chen W., Daniel V. C., Yu W., Berman D. M., Jenuwein T., Pruitt K., Sharkis S. J., Watkins D. N., Herman J. G., Baylin S. B., A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 39, 237–242 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gargiulo G., Serresi M., Cesaroni M., Hulsman D., Van Lohuizen M., In vivo shRNA screens in solid tumors. Nat. Protoc. 9, 2880–2902 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Rahl P. B., Lin C. Y., Seila A. C., Flynn R. A., Cuine S. M., Burge C. B., Sharp P. A., Young R. A., c-Myc regulates transcriptional pause release. Cell 141, 432–445 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parsa J.-Y., Boudoukha S., Burke J., Homer C., Madhani H. D., Polymerase pausing induced by sequence-specific RNA-binding protein drives heterochromatin assembly. Genes Dev. 32, 953–964 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabd7974/DC1