

Plasma cells shape the mesenchymal identity of ovarian cancers through exosome-mediated miR-330-3p/JAM2 axis.
Abstract
Ovarian cancer represents a highly lethal disease that poses a substantial burden for females, with four main molecular subtypes carrying distinct clinical outcomes. Here, we demonstrated that plasma cells, a subset of antibody-producing B cells, were enriched in the mesenchymal subtype of high-grade serous ovarian cancers (HGSCs). Plasma cell abundance correlated with the density of mesenchymal cells in clinical specimens of HGSCs. Coculture of nonmesenchymal ovarian cancer cells and plasma cells induced a mesenchymal phenotype of tumor cells in vitro and in vivo. Phenotypic switch was mediated by the transfer of plasma cell–derived exosomes containing miR-330-3p into nonmesenchymal ovarian cancer cells. Exosome-derived miR-330-3p increased expression of junctional adhesion molecule B in a noncanonical fashion. Depletion of plasma cells by bortezomib reversed the mesenchymal characteristics of ovarian cancer and inhibited in vivo tumor growth. Collectively, our work suggests targeting plasma cells may be a novel approach for ovarian cancer therapy.
INTRODUCTION
Ovarian cancer ranks among the most lethal malignancies for women, displaying substantial heterogeneity in tumor biology and clinical outcome (1, 2). Genomic changes in cancer cells stratify patients into different subgroups with distinct prognosis and response to therapies (3). As cancer tissues are composed of both cancer cells and nonneoplastic cells (4, 5), the functions of these nonneoplastic cells are less well studied. The success of oncoimmunology has prompted the interrogation of infiltrating immune cells to predict clinical outcome and response to treatment (6).
Large-scale transcriptional profiling of patient specimens has led to the pioneering work of molecular subtyping for ovarian cancer (7). Later, it has become well recognized that ovarian cancer, primarily high-grade serous ovarian cancer (HGSC), could be categorized into four distinct transcriptional subtypes: immunoreactive, differentiated, proliferative, and mesenchymal subtypes, among which the mesenchymal subtype has a relatively poor overall survival, confirmed by RNA sequencing data for about 500 patients with serous ovarian cancer by The Cancer Genome Atlas (TCGA) network (3). However, rare reports could be found on whether different subtypes of ovarian cancer exhibit distinct patterns of immune infiltration (8, 9), which is critical to the design of precision medicine for this deadly disease.
On the basis of this background, we hypothesized that deconvolution of the transcriptomic signatures from whole-tumor specimens can distinguish tumor and immune cell contributions and yield insights into subtype-specific immunologic responses in ovarian cancer. A recently described gene expression deconvolution algorithm (CIBERSORT) estimates the relative proportions of 22 distinct functional subsets of immune cells (10, 11). Using this method, we quantified the immune infiltration of the four subtypes of HGSC, the most common pathological subtype of ovarian cancer, and found a significant correlation with subtypes. In particular, using this integrated computational analysis together with further functional experiments, we identified an immune-associated cellular, molecular, and clinical network that highlights the defining role of plasma cells in the mesenchymal identity of HGSCs.
RESULTS
Plasma cells are enriched in the mesenchymal subtype of HGSCs
To investigate the subtype-specific immune infiltration pattern in ovarian cancer, we applied CIBERSORT to bulk gene expression profiles (GEPs) of ovarian cancer in the publicly available Bonome dataset (12) to infer the proportions of 22 subsets of immune cells in the four subtypes. The most prevalent immune cells were CD8+ T cells, plasma cells, M2 macrophages, and follicular helper T cells in the ovarian cancer microenvironment (Fig. 1A). For external validation, we further interrogated the abundance of plasma cells in the mesenchymal-subtype HGSCs in two other training cohorts (both excluding non-HGSC patients): Mateescu dataset (13) and Tothill dataset (table S1) (7). It was found that the plasma cell abundance was significantly higher in the mesenchymal subtype compared with that in other three subtypes in both Mateescu dataset and Tothill dataset (Fig. 1B). Moreover, in both Bonome dataset and Mateescu dataset, the mesenchymal-subtype patients were prone to have higher plasma cell abundance compared with nonmesenchymal-subtype patients (fig. S1A). Further analysis indicated that M1 macrophages were enriched in the immunoreactive subtype (P < 0.001), CD8+ T cells in the differentiated subtype (the most abundant although not significant), and plasma cells in the mesenchymal subtype of HGSCs (P < 0.001) (Fig. 1C).
Fig. 1. Plasma cells enrich in mesenchymal-subtype ovarian cancer compared with other subtypes.
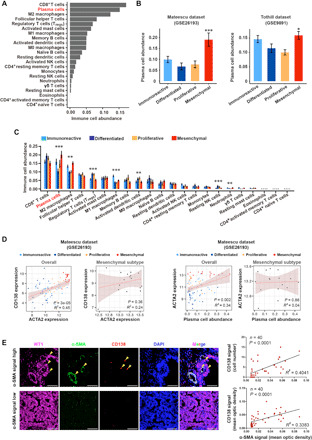
(A) Computational biology analysis revealed the total content of different immune cells in ovarian cancer (Bonome dataset, n = 182). Tregs, regulatory T cells; NK, natural killer. (B) Plasma cell abundance in four different molecular subtypes of ovarian cancer in Mateescu’s cohort and Tothill’s cohort, as calculated by the CIBERSORT algorithm (Mateescu dataset, n = 79; Tothill dataset, n = 260). P values were calculated by the Wilcoxon rank sum tests. (C) Boxplot showing the abundance of the 22 subsets of immune cells for each subtype of ovarian cancer (Bonome dataset). Data are presented as mean ± SEM. Kruskal-Wallis test, ***P < 0.001 and **P < 0.01. (D) Correlation analysis for ACTA2 with CD138 (left) and plasma cell abundance (right) in all patients and the mesenchymal-subtype patients, respectively. (E) Immunofluorescent staining of WT1, CD138, and α-SMA in 40 independent clinical ovarian cancer specimens. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar, 50 μm. Images were captured by confocal fluorescence microscopy, and the signal intensity as protein expression level was quantified by ImageJ software. Correlation analysis was performed for the expression of CD138 protein (or CD138+ cell number) and α-SMA protein in ovarian cancer specimens. Coefficient of determination (R2) and statistical significance levels were determined by linear regression with linear model method. Shaded area indicates 95% confidence interval. Data are shown as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
As the mesenchymal subtype is associated with the worst prognosis (14), we were particularly interested in the functional role of plasma cells in the maintenance of the mesenchymal identity of HGSCs. It was observed that in the Mateescu dataset, actin alpha 2 (ACTA2) expression, the gene that encodes α–smooth muscle actin (α-SMA), was significantly correlated with CD138 expression (P = 3 × 10−5, R2 = 0.45) and plasma cell abundance (P = 0.002, R2 = 0.34) in all molecular subtypes, while no significant correlation between ACTA2 expression and CD138 expression as well as plasma cell abundance was observed in merely the mesenchymal subtype (Fig. 1D). Similar results were also noted in the Bonome dataset (fig. S1B and table S1). As direct clinical validation, we performed immunohistochemical labeling on a clinical ovarian cancer cohort (patient characteristics described in table S2) with α-SMA, a marker for cancer-associated fibroblasts (15), CD138, a marker for human plasma cells, and Wilms’ tumor gene 1 (WT1), a marker to tumor cells (16, 17). Mesenchymal tumors displayed high levels of both α-SMA and CD138 compared with nonmesenchymal tumors (Fig. 1E). The number of CD138+-infiltrated plasma cells positively correlated with the number of mesenchymal cells in 40 ovarian cancer patient samples using immunofluorescent analysis (Fig. 1E). The number of immunoglobulin κ constant (IGKC)–positive cells, another plasma cell marker, was also positively correlated with the number of mesenchymal cells in ovarian cancer patient samples using immunofluorescent analysis (fig. S1C). Further flow cytometry analysis combined with immunostaining assay also revealed that the abundance of plasma cells correlated with the expression of α-SMA in fresh ovarian cancer patient samples (fig. S1, D and E). In 16 patients, the abundance of plasma cells varied significantly in the examined ovarian cancer tissues (fig. S1D), and plasma cell abundance was well correlated with ACTA2 gene expression (P = 0.0034, R2 = 0.46; fig. S1E). Gene set enrichment analysis (GSEA) in the TCGA ovarian cancer cohort confirmed that mesenchymal genes were highly enriched for ovarian cancer samples with high plasma cell abundance (fig. S1F). Thus, plasma cells are enriched in the mesenchymal subtype of HGSCs.
Plasma cells secrete exosomes to induce a mesenchymal phenotype of ovarian cancer cells
The infiltration of plasma cells into mesenchymal tumors could be either a cause or an effect. In other words, plasma cells could contribute to the acquisition of a mesenchymal signature or mesenchymal tumor cells could attract plasma cells. To distinguish between these possibilities, we investigated whether plasma cells could directly induce nonmesenchymal ovarian cancer cells to undergo a phenotypic switch to a mesenchymal state. We isolated plasma cells from ovarian cancer patient blood samples (Fig. 2A) and then cocultured them with either the nonmesenchymal human ovarian cancer cell lines, COV318 and OVCAR-3, or the mesenchymal human ovarian cancer cell lines, SKOV-3 and COV504, which have been previously described (18, 19). Plasma cells induced a spindle-like morphology in nonmesenchymal human ovarian cancer cell lines including COV318 and OVCAR-3 cells while did not induce any changes in mesenchymal human ovarian cancer cell lines including SKOV-3 and COV504 (Fig. 2B). We further stained different ovarian cancer cells cocultured with plasma cells with phalloidin to visualize F-actin. It was also found that after coculture with plasma cells, while the cell area of COV318 and OVCAR-3 cells did not change much, there was a significant increase in cell perimeter and elongation index and decrease in circularity of both cells compared with control. However, no significantly changes were observed in the mesenchymal cells including SKOV-3 and COV504 (Fig. 2B). Loss of the epithelial marker, E-cadherin, was confirmed by immunoblotting, while the mesenchymal marker vimentin increased in the two cell lines upon coculture with plasma cells (Fig. 2C). Plasma cells induced a loss of E-cadherin mRNA levels and gain in vimentin, fibronectin, and Twist mRNA (fig. S2A). These results were confirmed by immunofluorescence (fig. S2B).
Fig. 2. Treatment of ovarian cancer cells with plasma cells induces a mesenchymal phenotype.
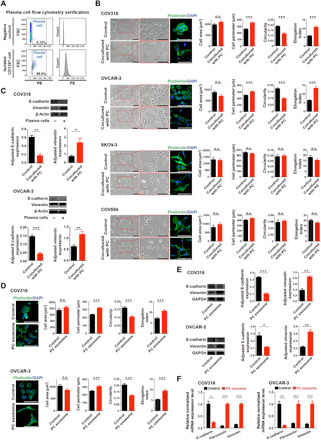
(A) Plasma cells that are marked by CD138 antibody were isolated from ovarian cancer patient blood by magnetic bead sorting and quantitatively verified by flow cytometry. PE, phycoerythrin; FSC, forward scatter. (B) Phase-contrast micrographs of COV318, OVCAR-3, SKOV-3, and COV504 cells treated with control or cocultured with plasma cells (PC) for 48 hours. Scale bar, 100 μm. Phalloidin staining was shown to quantify the difference between control or cocultured groups. The quantifications include cell area, perimeter, circularity, and elongation index (mean ± SEM of no less than 40 cells per group). n.s., not significant. (C) Western blotting analysis of epithelial-mesenchymal transition (EMT) markers in COV318 and OVCAR-3 cells cocultured with plasma cells compared with control for 48 hours. ImageJ software was used to quantify protein expression levels (n = 3 for each group). (D) Phalloidin staining of COV318 and OVCAR-3 cells was shown to quantify the difference between control and plasma cell exosome–treated groups for 48 hours. Scale bar, 100 μm. The quantifications include cell area, perimeter, circularity, and elongation index (mean ± SEM of no less than 40 cells per group). (E) Western blotting analysis of EMT markers in COV318 and OVCAR-3 cells treated with plasma cell exosomes compared with control for 48 hours (n = 3 for each group). ImageJ software was used to quantify protein expression levels. GAPDH, glyceraldehyde phosphate dehydrogenase. (F) mRNA levels of EMT markers in COV318 and OVCAR-3 cells treated with plasma cell exosomes compared with control for 48 hours (n = 3 for each group). In (B) to (F), statistical significance was determined by a two-tailed, unpaired Student’s t test. Data are shown as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Our results suggest that plasma cells provide cell-to-cell signaling to alter the state of tumor cells. To determine the components in the conditioned media of plasma cells that mediated this phenotypic switch in ovarian cancer, we considered a potential role for exosomes. It was found that after treatment with plasma cell exosomes, although the cell area of COV318 cells did not change much, there was a significant increase in cell perimeter and elongation index and decrease in circularity of COV318 cells compared with control. Similar changes were also observed in OVCAR-3 cells after treatment with plasma cell exosomes (Fig. 2D). Furthermore, treatment with plasma cell exosomes led to decreased E-cadherin and increased vimentin expression on both protein (Fig. 2E) and mRNA (Fig. 2F) levels in both COV318 and OVCAR-3 cells. COV318 and OVCAR-3 cells incubated with plasma cell–derived exosomes exhibited enhanced migratory capacity measured by wound healing and Transwell chamber assays compared with control cells (fig. S2, C and D). These results support a role of plasma cell–derived exosomes in modulating ovarian tumor cell phenotypes.
Exosomal microRNA profiling identifies plasma cell–derived microRNA-330-3p (miR-330-3p) as a key regulator of ovarian cancer mesenchymal identity
To determine the downstream mechanisms by which plasma cell–derived exosomes induce mesenchymal features in ovarian cancer cells, we cocultured PKH67-labeled primary plasma cells with ovarian cancer cell lines for 24 hours and observed the transfer of membrane vesicles from plasma cells to ovarian cancer cells (Fig. 3, A and B). To determine whether exosomes released from plasma cells mediate cross-talk with ovarian cancer cells, we purified exosomes from the supernatant of plasma cells and excluded contamination by other vesicle types and protein aggregates. Purified exosomes were confirmed by their high expression of exosome markers CD63 and CD81 (Fig. 3C). Scanning electron microscopy of the exosomes showed typical rounded particles ranging from 50 to 200 nm in diameter (Fig. 3D). These exosomes were further characterized using a NanoSight NS300 system (Fig. 3E). NanoSight analysis indicated the size distributions of exosome particles released by plasma cells, especially in extracellular vesicles (EVs) of ~110 nm in size (Fig. 3E). Exosome uptake was observed 1 hour after treatment, and exosomes accumulated in recipient cells over time, with transfer blocked, as expected, following incubation at 4°C or preincubation of exosomes with proteinase K (Fig. 3, F and G). Temperature dependence and involvement of specific proteins point toward an active exosome uptake mechanism in the interaction between plasma cells and ovarian cancer cells. These observations implicated exosomes released by plasma cells and internalized by ovarian cancer cells as a key driver of the mesenchymal identity of ovarian cancer.
Fig. 3. Plasma cell exosomal miR-330-3p could be transferred to ovarian cancer.
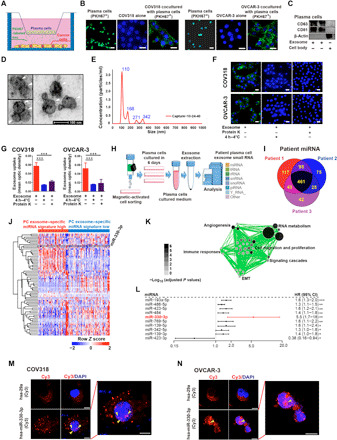
(A) Schematic diagram of plasma cells and ovarian cancer cells cocultured in six-well plates. (B) Ovarian cancer cells were cocultured in the absence or presence of primary PKH67-labeled plasma cells (green). Nuclei were stained with DAPI (blue) (n = 3 for each group). Scale bar, 20 μm. (C) Western blot analysis for CD63, CD81, and β-actin in plasma cell exosomes (n = 3 for each group). (D) Electron micrograph of plasma cell exosomes shows the morphological size (50 to 200 nm). Scale bar, 100 nm. (E) Size distribution of exosomes was measured using NanoSight analysis. (F and G) Immunofluorescent images of PKH67 abrogation in ovarian cancer cells with respective treatment. Scale bar, 20 μm. Statistical chart was plotted (n = 3 for each group). (H) Scheme chart for small RNA sequencing in plasma cell exosomes in patients with ovarian cancer. tRNA, transfer RNA; rRNA, ribosomal RNA; snRNA, small nuclear RNA; snoRNA, small nucleolar RNA; piRNA, Piwi-interacting RNA.(I) Venn diagram for overlapped miRNAs identified in ovarian cancer plasma cell exosomes. (J) Heatmap for unsupervised hierarchical clustering of GSE73582 dataset using plasma cell exosome–specific miRNA panel as classifiers. (K) Cellular programs enriched by GSEA for plasma cell exosome–specific miRNAs represented using Enrichment Map. (L) The univariate regression analyses of the identified top miRNAs associated with patient survival, OC179 (GSE73581), n = 179. CI, confidence interval. (M and N) Fluorescence diagram shows subcellular localization of miR-330-3p (yellow arrowheads) (n = 3 to 4 for each group). Scale bar, 20 μm. In (G), data are shown as mean ± SEM. All statistical significance was determined by a two-tailed, unpaired Student’s t test. *P < 0.05; **P < 0.01; ***P < 0.001.
Exosomes transport a wide variety of payloads that are delivered to recipient cells. Therefore, we profiled specific RNAs shuttled via plasma cell–derived exosomes from three patients with ovarian cancer using small RNA sequencing analysis (Fig. 3H). A total of 461 microRNAs (miRNAs) overlapped in the three ovarian carcinoma patient–derived plasma cell exosomes (Fig. 3I). To determine whether the plasma cell exosome–specific miRNAs were clinically important, we derived a miRNA signature to classify patients with ovarian cancer. Using an unsupervised hierarchical clustering, we identified two clusters that formed the most robust classification from plasma cell exosome–specific miRNAs in an ovarian cancer dataset (Fig. 3J) (20). GSEA (21) coupled to Enrichment Map visualization identified RNA metabolism, angiogenesis, cell migration, and proliferation among the differentially enriched biological pathways between the two groups (Fig. 3K). Among the top miRNAs correlated with survival of patients with ovarian cancer, the hazard ratio (HR) of miR-330-3p ranked the highest using univariate regression analysis (HR = 5.5) (Fig. 3L). To validate the expression pattern of miR-330-3p across different immune cell types and ovarian cancer cells in vitro, we measured the expression levels of miR-330-3p in human T cells, B cells, plasma cells, neutrophils, natural killer cells, dendritic cells, macrophages, and human COV318 ovarian cancer cells. miR-330-3p was uniquely overexpressed in plasma cells compared with other immune cell types and COV318 ovarian cancer cells (fig. S7A). Thus, we investigated the functional importance of exosomal miR-330-3p derived from plasma cells.
If miR-330-3p functions in ovarian carcinoma cells, then its subcellular location in ovarian cells upon transfer from plasma cell exosomes would be expected to determine its function. After transfection of both COV318 and OVCAR-3 cells with Cy3-labeled miR-330-3p, Cy3-miR-330-3p was detected in the nucleus of the two cell lines. No signal of a control miRNA, Cy3-miR-29a, was detected in the nucleus of the two cell lines (Fig. 3, M and N). We further evaluated the functional role of plasma cell exosome–containing miR-330-3p in shaping the mesenchymal identity of ovarian cancer cells. It was demonstrated that in COV318 cells transfected with miR-330-3p mimic, while the cell area of COV318 cells did not significantly change, there was a remarkable increase in cell perimeter and elongation index and decrease in circularity of COV318 cells compared with control. Similar changes were also observed in OVCAR-3 cells after transfection with miR-330-3p mimic (fig. S3A). We further found that in both COV318 and OVCAR-3 cells treated with plasma cell exosomes and miR-330-3p inhibitor, the cell area did not remarkably change. However, it was noted that there were a notable decrease in cell perimeter and elongation index and increase in circularity of both ovarian cancer cells compared with cells treated with plasma cell exosomes and control inhibitor (fig. S3B). Epithelial-mesenchymal transition (EMT) markers E-cadherin and vimentin were examined in COV318 cells treated with control plasma cell exosomes, control inhibitor–transfected plasma cell exosomes and miR-330-3p inhibitor–transfected plasma cell exosomes. Inhibition of miR-330-3p within plasma cell–derived exosomes induced up-regulation of E-cadherin and down-regulation of vimentin in COV318 and OVCAR-3 cells (fig. S3C). On the mRNA levels, we also noted similar changes in both cells (fig. S3D). Immunofluorescent analysis further supported the EMT-inducing role of exosome-contained miR-330-3p for ovarian cancer cells, demonstrating increased E-cadherin and decreased fibronectin, and vimentin following inhibition of miR-330-3p while treated with plasma cell exosomes (fig. S3E). Supporting a key role for miR-330-3p in mediating plasma cell–induced EMT, miR-330-3p inhibitor–transfected plasma cell exosomes do not promote ovarian cancer cell migration compared with control plasma cell exosomes, measured by wound healing (fig. S3F) and Transwell chamber migration (fig. S3G). These data suggest that exosomal miR-330-3p derived from plasma cells could enter the nuclei of ovarian cancer cells and induce a mesenchymal program.
miR-330-3p enhances junctional adhesion molecule 2 transcription via enhancer-induced gene activation mechanisms
Our preliminary analysis showed that the predicted canonical targets of miR-330-3p did not have any correlation with the prognosis of patients with ovarian cancer (fig. S4A). As previous reports have demonstrated that nuclear miRNAs could enhance gene expression through noncanonical mechanisms (22, 23), we next investigated the potential up-regulated target genes of nuclear miR-330-3p essential for the mesenchymal identity of ovarian cancer. A computational biology strategy was used to identify the commonly changed gene expression based on the two criteria: (i) genes up-regulated in the mesenchymal-subtype ovarian cancer tissues compared with the nonmesenchymal-subtype tissues in the TCGA dataset and (ii) genes up-regulated in both OVCAR-3 and COV318 cells after miR-330-3p mimic transfection compared with control (Fig. 4A). Among the six overlapping genes, junctional adhesion molecule 2 (JAM2) was the most significantly correlated with the prognosis of patients with ovarian cancer (fig. S4B); thus, we further characterized the functional role of JAM2 in ovarian cancer (Fig. 4B). JAM2 mRNA levels were decreased in COV318 and OVCAR-3 cells cocultured with miR-330-3p inhibitor–transfected plasma cells compared with control inhibitor–transfected plasma cells (Fig. 4C). Inhibition of miR-330-3p within plasma cells decreased JAM2 expression in both ovarian cancer cell lines compared to treatment with a nontargeting inhibitor (Fig. 4D). Conversely, exosomes derived from miR-330-3p mimic–transfected plasma cells induced up-regulation of JAM2 expression and a promesenchymal mRNA program in COV318 and OVCAR-3 cells (Fig. 4E). Likewise, JAM2 protein levels trended higher in COV318 and OVCAR-3 cells treated with exosomes derived from miR-330-3p mimic–transfected plasma cells compared with control (Fig. 4F). E-cadherin protein levels were decreased in COV318 and OVCAR-3 cells treated with exosomes derived from miR-330-3p mimic–transfected plasma cells compared with control, while protein levels of vimentin were significantly increased in the experimental group (Fig. 4F). Migration of both COV318 and OVCAR-3 cells was enhanced by treatment with exosomes derived from plasma cells transfected with a miR-330-3p mimic compared with control, as revealed by wound healing analysis (Fig. 4G).
Fig. 4. miR-330-3p targets junctional adhesion molecule 2 for the maintenance of mesenchymal identity of ovarian cancer.
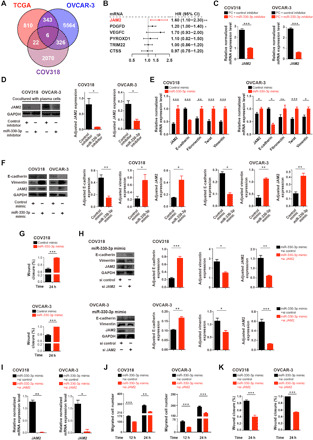
(A) Venn diagram showing the most possible up-regulated genes targeted by plasma cell exosome–containing mir-330-3p. (B) Univariate regression analysis of the six overlapped target genes associated with ovarian cancer patient survival (Bonome dataset, n = 182). (C) mRNA level of JAM2 in COV318 and OVCAR-3 cells with respective treatment (n = 3 to 4 for each group). (D) Western blotting analysis of JAM2 protein levels in COV318 and OVCAR-3 cells with respective treatment (n = 3 for each group). (E) mRNA levels of JAM2 and EMT markers in miR-330-3p mimic–transfected or control COV318 and OVCAR-3 cells (n = 3 to 4 for each group). (F) Western blotting analysis of JAM2 and EMT markers in miR-330-3p mimic–transfected or control COV318 and OVCAR-3 cells (n = 3 for each group). (G) Wound healing analysis to assess the migration ability of COV318 and OVCAR-3 cells with respective treatment (n = 3 to 5 for each group). (H) Western blotting analysis of JAM2 and EMT markers in COV318 and OVCAR-3 cells with respective treatment (n = 3 for each group). si, small interfering. (I) mRNA levels of JAM2 in COV318 and OVCAR-3 cells with respective treatment (n = 3 to 4 for each group). (J) Transwell chamber analysis to assess the migration ability of COV318 and OVCAR-3 cells with respective treatment (n = 3 to 5 for each group). (K) Wound healing analysis to assess the migration ability of COV318 and OVCAR-3 cells with respective treatment (n = 3 to 5 for each group). Data are shown as mean ± SEM. All statistical significance was determined by a two-tailed, unpaired Student’s t test. *P < 0.05; **P < 0.01; ***P < 0.001.
We next examined the functional role of miR-330-3p/JAM2 in driving the mesenchymal phenotype and invasion of ovarian cancer cells in vitro. In miR-330-3p mimic–transfected ovarian cancer cells, treatment with JAM2 small interfering RNA (siRNA) induced up-regulation of E-cadherin and down-regulation of vimentin and JAM2 in both COV318 and OVCAR-3 cells compared with a nontargeting control (Fig. 4H). On the mRNA level, treatment with JAM2 siRNA reduced JAM2 expression in both COV318 and OVCAR-3 cells, compared with nontargeting controls (Fig. 4I). JAM2 knockdown impaired migration of both COV318 and OVCAR-3 ovarian cancer cells, as measured by both Transwell chamber analysis (Fig. 4J) and wound healing analysis (Fig. 4K). Therefore, the miR-330-3p/JAM2 axis might be involved in the maintenance of the mesenchymal identity of ovarian cancer in vitro.
We further explored the noncanonical mechanisms underlying nuclear miR-330-3p–mediated overexpression of JAM2. miRNAs often negatively regulate mRNAs in the cytoplasm by binding to the 3′ untranslated region (3′UTR) of target genes to degrade mRNA or inhibit translation (24). However, miRNAs are also present in the nucleus, which can activate gene expression by binding to enhancer elements (23, 25). Given the nuclear localization of miR-330-3p, we investigated its potential regulatory mechanisms for JAM2 expression. We noticed that the sequence of miR-330-3p and the human JAM2 promoter region shared a remarkable complementary match (fig. S4C). To investigate whether sequence-specific interactions between miR-330-3p and the JAM2 promoter are required to up-regulate promoter activity, we introduced mutation or deletion into the synthetic miR-330-3p mimic. Mutation or deletion in the synthetic miR-330-3p mimic abrogated promoter activity enhancement for JAM2 validated by immunoblot (fig. S4D) and quantitative polymerase chain reaction (qPCR) analysis (fig. S4E). We further investigated whether miR-330-3p could bind with the specific promoter region of JAM2 (fig. S4F). It was verified that elevated RNA polymerase II (Pol II) occupancy by chromatin immunoprecipitation (ChIP)–qPCR analysis on the JAM2 promoter was observed when ovarian cancer cells were transfected with miR-330-3p mimic, whereas no significant difference was observed in the RNA Pol II occupancy when transfected with mutated or deleted miR-330-3p mimic compared with control (fig. S4G). We also treated COV318 and OVCAR-3 cells transfected with miR-330-3p mimic with JQ1, a small-molecule bromodomain inhibitor that abolishes bromodomain-containing protein 4 binding (26), which decreased JAM2 expression levels. In contrast, JAM2 expression levels were essentially unchanged in COV318 and OVCAR-3 cell lines treated with JQ1 and control mimic (fig. S4H). These results prove the computational prediction and suggest that miR-330-3p binds to the JAM2 promoter in a highly sequence-specific manner.
miR-330-3p/JAM2 axis is vital for the mesenchymal identity of ovarian cancer in vivo
To determine the functional importance of the miR-330-3p/JAM2 axis in plasma cell–ovarian cancer cell interactions, we examined the effects of the mesenchymal identity of ovarian cancer in both ovarian cancer subcutaneous and peritoneal metastasis mouse models. Subcutaneous tumors derived from the murine epithelial ovarian cancer cell line ID8 following treatment with plasma cell–derived exosomes were significantly larger and displayed more rapid growth (Fig. 5, A and B) and more peritoneal metastasis (Fig. 5C) compared with the control group. ID8 cells subcutaneously implanted together with plasma cell–derived exosomes led to decreased expression of E-cadherin and increased expression of Ki67 and vimentin by immunohistochemistry, consistent with a molecular signature for mesenchymal identity and cellular proliferation (fig. S5A). To further characterize the indispensability of plasma cells in driving the mesenchymal phenotype transition of ovarian cancer, we treated subcutaneous ID8 ovarian cancer cells with bortezomib (BTZ), a well-established plasma cell–depleting agent (27–29). We first validated the inhibition specificity of BTZ for plasma cells. BTZ treatment reduced plasma cell infiltration in the tumor, as expected, while no significant changes were noted in the contents of other immune cells, including B cells, T cells, or myeloid-derived suppressor cells (MDSCs) (fig. S6, A to D). Moreover, it was found that while BTZ induced apoptosis of a small fraction of ID8 ovarian cancer cells, it caused massive apoptosis in plasma cells, indicating that plasma cells are far more sensitive to BTZ treatment compared with ovarian cancer cells, and BTZ is a relatively specific plasma cell–depletion agent (fig. S6E). In vivo, we observed that BTZ treatment decreased the size and growth rate of subcutaneous tumors compared with the control group (Fig. 5, D and E) and inhibited peritoneal metastasis (Fig. 5F). ID8 cells treated with BTZ displayed elevated expression of E-cadherin and reduced expression of Ki67 and vimentin (fig. S5B). In contrast, ID8 cells treated locally with anti-CD3 (T cell depletion), anti-CD19 (B cell depletion), or anti–granulocyte receptor-1 (MDSC depletion) antibodies displayed no significant alterations of in vivo growth or alterations of EMT markers. Collectively, these results support a critical role of plasma cells in epithelial ovarian cancer cell growth in vivo.
Fig. 5. The importance of miR-330-3p/JAM2 axis for the mesenchymal identity of ovarian cancer in vivo.
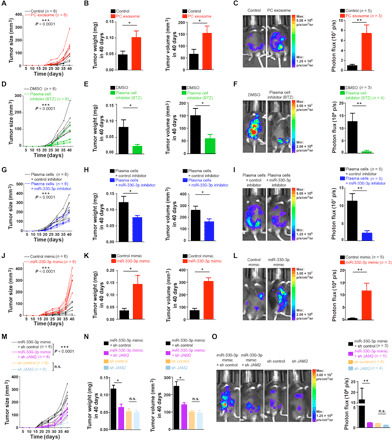
(A, D, G, J, and M) A total of 5 × 106 ID8 cells with respective treatment were subcutaneously injected into C57 mice together with plasma cell–derived exosomes and control (n = 6 for each group). Growth curve was plotted. (B, E, H, K, and N) Tumor weight and volume of ID8 cells in each group were measured at indicated time. (C, F, I, L, and O) In vivo bioluminescent imaging of tumor growth in each group was performed in mice 30 days after injection (n = 3 to 5 for each group). Data are shown as mean ± SEM. In (A), (D), (G), (J), and (M), P value out of two-way repeated measures analysis of variance (ANOVA). Otherwise, statistical significance was determined by a two-tailed, unpaired Student’s t test. *P < 0.05; **P < 0.01. ***P < 0.001. DMSO, dimethyl sulfoxide; sh, small hairpin.
We next characterized the vital role of miR-330-3p/JAM2 axis in vivo. It was demonstrated that ID8 cells inoculated with plasma cells transfected with miR-330-3p inhibitor displayed reduced tumor growth (Fig. 5, G and H) and decreased peritoneal metastasis (Fig. 5I) compared with ID8 cells inoculated with plasma cells transfected with control inhibitor. Treatment of ID8 cells with miR-330-3p inhibitor–treated plasma cells elevated expression of E-cadherin and reduced expression of Ki67 and vimentin (fig. S5C). Conversely, ID8 cells transfected with miR-330-3p mimic grew larger tumors (Fig. 5, J and K) and developed more peritoneal metastasis (Fig. 5L) than the control group. Treatment of ID8 cells with miR-330-3p mimic reduced expression of E-cadherin and increased expression of Ki67 and vimentin (fig. S5D). To demonstrate specificity of the effects of miR-330-3p, we performed rescue experiments to elucidate the functional importance of miR-330-3p/JAM2 axis (fig. S5, E and F). Tumors formed by ID8 cells transfected with miR-330-3p mimic were smaller after JAM2 knockdown (Fig. 5, M and N) and had weaker peritoneal metastasis capacity (Fig. 5O) compared with control. Tumors formed by ID8 cells transfected with miR-330-3p mimic showed increased expression of E-cadherin and decreased expression Ki67 and vimentin after JAM2 knockdown compared with control (fig. S5G). Therefore, in vivo epithelial ovarian tumor growth is accelerated by miR-330-3p/JAM2 induced by plasma cell infiltration.
miR-330-3p/JAM2 axis in the cross-talk between plasma cells and ovarian cancer cells informs prognosis
To explore the clinical importance of the miR-330-3p/JAM2 axis in the cross-talk between plasma cells and HGSC cells, we examined whether the plasma cell gene signature curated from the CIBERSORT algorithm informed ovarian cancer patient prognosis. Using the Tothill ovarian cancer dataset, patients were categorized on the basis of transcriptional profiles into two subgroups: plasma cell–high and plasma cell–low groups (Fig. 6, A and B). The overall survival for plasma cell–high patients with HGSC was significantly worse than plasma cell–low patients in the Tothill dataset (GSE9891) (P = 0.036; Fig. 6C). Similar results were also observed in the Mateescu dataset (P = 0.02; Fig. 6D), although in the subset of the mesenchymal subtype, no significant difference was observed between plasma cell–high and plasma cell–low patients with HGSC (P = 0.71; Fig. 6D). As miRNAs contained in plasma cell exosomes classify patients with ovarian cancer into two clusters, we explored whether these two subgroups have different prognoses. Progression-free survival of plasma cell exosome miRNA signature–high patients was worse than plasma cell exosome miRNA signature–low patients in both the OC133 dataset (P = 0.02; Fig. 6E) and the OC179 dataset (P = 0.004; Fig. 6E). These findings support the clinical significance of plasma cells and plasma cell exosomal miRNAs in patients with HGSC.
Fig. 6. Clinical significance of miR-330-3p/JAM2 axis in the cross-talk between plasma cells and ovarian cancer cells.
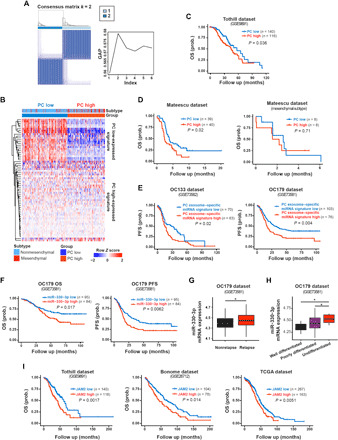
(A) Unsupervised classification of plasma gene signature shows the optimal classification using two clusters (left), as supported by the gap statistic (right). (B) Heatmap for unsupervised hierarchical clustering of the Tothill dataset using plasma cell gene signature as classifiers. (C) Survival difference between the plasma cell–high subgroup and the plasma cell–low subgroup in the Tothill dataset. OS, overall survival. (D) Survival difference between the plasma cell–high subgroup and the plasma cell–low subgroup in all patients and the mesenchymal-subtype patients in the Mateescu dataset. (E) Differential progression-free survival (PFS) between plasma cell exosome–specific miRNA signature–high group and plasma cell exosome–specific miRNA signature–low group of patients with ovarian cancer in OC133 and OC179 datasets. (F) Differential overall survival and progression survival between miR-330-3p–high group and miR-330-3p–low group of patients with ovarian cancer in the OC179 dataset. (G) Expression levels of miR-330-3p in patients with ovarian cancer with relapse and without relapse. (H) Expression levels of miR-330-3p in patients with ovarian cancer with distinct differentiation grades. (I) Differential overall survival between JAM2-high group and JAM2-low group of patients with ovarian cancer in the Tothill, Bonome, and TCGA datasets. Data are shown as mean ± SEM. In (C), (D), (E), (F), and (I), survival difference was calculated by log-rank test; in (G) and (H), P value was calculated using the two-tailed Student’s t test. **P < 0.01.
We further assessed whether miR-330-3p and its target, JAM2, inform the prognosis of patients with ovarian cancer. The overall survival (P = 0.017) and the progression-free survival (P = 0.0062) of miR-330-3p–high patients were significantly worse than those of miR-330-3p–low patients in the OC179 dataset (Fig. 6F). In addition, miR-330-3p expression was higher in patients with ovarian cancer with relapse compared with those without relapse (Fig. 6G) and higher in poorly differentiated and undifferentiated ovarian carcinoma patients compared with well-differentiated patients (Fig. 6H). Expression of miR-330-3p was also higher in the mesenchymal subtype of ovarian cancer compared with nonmesenchymal subtypes (fig. S7B). We next explored whether miR-330-3p was also differentially expressed in subgroups of ovarian cancer samples with different immune cell–subtype infiltrates. miR-330-3p expression levels were elevated in plasma cell–enriched (P = 0.03) and γδ T cell–enriched (P = 0.01) ovarian cancer samples compared with plasma cell–rare and γδ T cell–rare ovarian cancer samples, while no significant difference was observed on the basis of enrichment for other immune components (fig. S8). Overall survival was consistently worse in JAM2-high patients compared with JAM2-low patients in the Tothill dataset (P = 0.0017), Bonome dataset (P = 0.014), and TCGA dataset (P = 0.0051) (Fig. 6I). These results demonstrate that the miR-330-3p/JAM2 axis is associated with poor ovarian cancer patient prognosis.
DISCUSSION
On the basis of the deconvolution of bulk gene expression data of ovarian cancer, we unraveled distinct patterns of immune infiltration between tumors. Our comprehensive analysis of the immune microenvironment in ovarian cancer revealed subtype-specific immune infiltration patterns that might shed light on new immunotherapeutic strategies for this deadly disease. In particular, the mesenchymal-subtype ovarian cancer is characteristically enriched with plasma cell infiltration, which is prognostically important.
The prognostic roles of B cell/plasma cell infiltration in different types of cancer appear to be context dependent. Plasma cell recruitment to the tumor microenvironment was associated with better prognosis (30), whereas other studies showed that B cell infiltration was correlated with poorer prognosis (31). Berntsson et al. (30) found that a higher density of plasma cells correlated significantly with an improved overall survival in patients with colorectal cancer. However, the clinical significance of B cells and plasma cells in ovarian cancer remains obscure. Lundgren and colleagues (32) reported that high CD20 and CD138 expression levels correlated significantly with high tumor grade and high CD138 expression were associated with a significantly reduced overall survival as well as ovarian cancer–specific survival. However, another group showed that plasma cells were associated with the most robust, prognostically favorable CD8+ tumor-infiltrating lymphocyte responses in HGSCs (33). To clarify the exact role of plasma cells in ovarian cancer, we performed integrated analysis in large-scale HGSC gene expression data and found that plasma cell infiltration is enriched in the mesenchymal subtype, among which the prognosis was the worst. Further clinical sample validation indicated that plasma cell abundance was correlated with the content of mesenchymal cells, giving additional support to the possible functional role of plasma cells in shaping the mesenchymal phenotype of HGSCs.
Exosomes are small membrane vesicles formed by the inward budding of late endosomes (34). They are released into the extracellular environment upon fusion with the plasma membrane under both physiological and pathological conditions. Exosomes contain a variety of functional proteins, mRNAs, and miRNAs that facilitate these structures to operate as signaling platforms for short-range or long-range delivery of information to other cells (35). Exosomes constitute an important component of the tumor microenvironment that serves as a messenger between different cells. Existing literature has characterized the importance of exosomes in remodeling the tumor niche and maintained the hallmarks of ovarian cancer. Zhou et al. (36) demonstrated that exosomes released from tumor-associated macrophages transfer miRNAs that induce a T regulatory/T helper 17 cell imbalance in epithelial ovarian cancer. Dorayappan et al. (37) found that hypoxic ovarian cancer cell–derived exosomes were potent in augmenting metastasis/chemotherapy resistance in ovarian cancer and may serve as a novel mechanism for tumor metastasis and chemoresistance and an effective intervention for improving clinical outcomes. In this study, we found that plasma cells secreted exosomes to shape the mesenchymal identity of HGSCs both in vitro and in vivo. Small RNA sequencing revealed that these exosomes secreted by plasma cells in the tumor microenvironment contained miR-330-3p that was transmitted to the ovarian cancer cells to induce the mesenchymal phenotype. Thus, exosomes might be an important messenger between plasma cells and HGSCs and essential for the mesenchymal phenotype.
miR-330-3p has been implicated as a critical factor in tumor development and progression, but whether it acts as a tumor suppressor or an oncogene is still controversial. Yao et al. (38) reported that miR-330-3p inhibits the proliferation of melanoma cells by negatively regulating the expression of Targeting protein for Xklp2, which was pro-proliferative in melanoma cells. However, another group demonstrated that miR-330-3p was overexpressed in non–small cell lung cancer tissues and brain metastasis tissues (39). Currently, the functional role of miR-330-3p in ovarian cancer has not been elucidated. Our study showed that miR-330-3p derived from plasma cell exosomes functions as a key driving force in determining the mesenchymal identity of ovarian cancer and could serve as a strong poor prognosis indicator for patients with ovarian cancer. It is traditionally considered that miRNAs usually negatively regulate gene expression by binding to the 3′UTR of target genes to degrade mRNA or inhibit translation (40). However, recent evidence shows that apart from functioning in the cytoplasm, miRNAs are also present in the nucleus, acting unconventionally to activate gene transcription by targeting enhancers (23, 25). While we did not find any correlation between canonical miR-330-3p targets and ovarian cancer patient prognosis, we observed that after transmitted from the plasma cells, miR-330-3p could enter the nucleus of ovarian cancer cells and directly interact with the promoter region of JAM2, enhancing its gene expression in a noncanonical fashion. Overexpressed JAM2 is important for the mesenchymal identity of HGSCs. Our study gave further support to the notion that miRNAs could activate gene transcription apart from inducing gene repression. In addition, the noncanonical miR-330-3p/JAM2 axis newly identified by our study offers new therapeutic vulnerabilities for HGSCs. JAM2 is a member of JAM, which are a family of adhesion molecules belonging to the immunoglobulin superfamily that localize to the tight junctions. JAM protein family members are type 1 transmembrane glycoproteins consisting of two immunoglobulin-like domains, one transmembrane domain, and one cytoplasmic tail. Jam proteins function through either homo- or heterodimerization with Jam family members or other proteins, such as integrins (41). Previous report showed that JAM2high hematopoietic stem cells are characterized by enhanced potential for T lymphopoiesis (42). In the cancer context, while not frequently reported, JAM2 was demonstrated to promote invasiveness of melanoma (43) and breast cancer (44). We presented that the noncanonical miR-330-3p/JAM2 axis maintains the mesenchymal phenotype of HGSCs and that disruption of this axis could potently impair ovarian cancer growth and invasiveness both in vitro and in vivo. Thus, this novel noncanonical miR-330-3p/JAM2 axis could potentially serve as an efficient therapeutic target for HGSCs.
Collectively, our study used integrated systems biology analysis and found specific immune infiltration patterns among different molecular subtypes of HGSCs. We found that enrichment of plasma cells is a specific feature of the mesenchymal-subtype HGSCs. Further in vivo and in vitro experiments unraveled an exosome-derived miR-330-3p/JAM2 axis that mediated induction of core EMT programs in ovarian cancer, which extrinsically controlled ovarian cancer metastasis and affected patient outcome. Therefore, therapeutic targeting the plasma cell–ovarian cancer cell interactions by blocking the noncanonical exosomal miR-330-3p/JAM2 axis might be an efficient treatment option for patients with HGSC.
METHODS
Reagents, cell culture, and cell transfection
The antibodies used in this study are listed as follows: For flow cytometry: phycoerythrin (PE) anti-human CD138 (1:20; Invitrogen, 12-1389-41), PE anti-mouse CD138 (1:50; BD Pharmingen, 561070), Peridinin-Chlorophyll-Protein Complex (PerCP)/Cy5.5 anti-mouse CD45 (1:400; BioLegend, 103132), PE anti-mouse CD45 (1:50; BD Pharmingen, 561087), allophycocyamin (APC) anti-mouse/human CD11b (1:400; BioLegend, 101212), APC anti-mouse/human CD45R/B220 (1:400; BioLegend, 103211), PE anti-mouse Gr-1 (1:50; BD Pharmingen, 561084); and for immunofluorescent staining and Western blot: CD138 (1:200; BioLegend, 352302), CD81 (1:500; Invitrogen, #10630D), CD63 (1:500; Invitrogen, #10628D), α-SMA (1:1000; Novus, NB300-978SS), IGKC (1:200; Bioss Antibodies, bs-3800R), E-cadherin (1:1000; Abcam, ab76055), vimentin (1:1000; Abcam, ab8978), fibronectin (1:200; MXB-bio, RAB-0071), JAM2 (1:1000; Abcam, ab139645), Ki67 (1:200; ZSGB-BIO, ZA-0502), and WT1 (1:1000; Abcam, ab89901). Secondary antibodies used include Alexa Fluor 488 goat anti-rabbit (1:1000; Invitrogen, A-11008), Alexa Fluor 555 goat anti-rabbit (1:1000; Invitrogen, A-21428), Alexa Fluor 555 goat anti-mouse (1:1000; Invitrogen, 1837985), Alexa Fluor 488 goat anti-mouse (1:1000; Invitrogen, A11001), Alexa Fluor 488 donkey anti-goat (1:1000; Invitrogen, A-11055), Alexa Fluor 555 donkey anti-mouse (1:1000; Invitrogen, A-31570), Alexa Fluor 647 goat anti-rabbit (1:1000; Invitrogen, A-21244), peroxidase-labeled affinity-purified goat anti-mouse (1:5000; KPL, 0741806), and peroxidase-labeled affinity-purified goat anti-rabbit (1:5000; KPL, 0741506).
All cell lines were maintained at 37°C and 5% CO2. The human ovarian cancer cell lines OVCAR-3, COV318, and SKOV-3 were cultured in RPMI 1640 containing 10% fetal bovine serum (FBS) and penicillin-streptomycin (100 U/ml) (both from Gibco-BRL, Grand Island, NY, USA). The human ovarian cancer cell line COV504 was cultured in Dulbecco’s modified Eagle medium (DMEM) containing 10% FBS and penicillin-streptomycin (100 U/ml) (both from Gibco-BRL, Grand Island, NY, USA). The mouse ovarian cancer cell line ID8 was cultured in DMEM supplemented with 10% FBS, 2 mM l-glutamine, 1 mM sodium pyruvate, and penicillin-streptomycin (100 U/ml) (both from Gibco-BRL, Grand Island, NY, USA). ID8 cells were transduced with a lentiviral vector containing a luciferase reporter (GenBank no: MF693179.1) together with the puromycin resistance gene (ID8-Luc) for establishment of peritoneal metastasis model. Primary lymphocytes and plasma cells were cultured in RPMI 1640/DMEM containing 10% FBS and penicillin-streptomycin (100 U/ml) no more than 6 days. Cell lines were tested for authenticity in 2019 using short tandem repeat genotyping. They were also tested negative for mycoplasma.
In each plasma cell–tumor cell coculture experiment, plasma cells (1 × 106) and ovarian cancer cells (1 × 105) were cocultured in six-well plates for 48 hours. In exosome-involving experiments, 5 × 104 ovarian cancer cells were treated with culture medium with concentrations of about 6.0 × 105 exosomes/ml for each group for 48 hours. A total of 6.0 × 105 exosomes derived from about 200 ml of 1 × 106 plasma cell culture media. The number was calculated on the basis of 3.0 × 102 particles (30 to 200 nm) each 100 μl of plasma cell culture media directly from NanoSight result. For cell transfection experiment, siRNA, miRNA mimic, and miRNA inhibitor were transfected into cells using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions; small hairpin RNAs (shRNAs) were transfected into cells using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions.
For cell transfection experiment, siRNA, miRNA mimic, and miRNA inhibitor manufactured by RiboBio (Guangzhou, China) were transfected into cells using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. Transfection efficiency was guaranteed by fluorescent probe cotransfected. In all experiments involving siRNA, miRNA mimic, and miRNA, the concentrations were 50 to 60 nM equally in both experimental and control groups. As for shRNA lentiviral or pLenti-CBh-3FLAG-luc2-tCMV-mNeonGreen-F2A-Puro (ID8 luciferase reporter lentivirus) infection experiment, the virus titer used was based on the following formula: virus (μl) = cell number × multiplicity of infection/virus (stock solution) titer × 103. Final concentration of polybrene (5 μg/ml) was used to facilitate infection. In 72 hours, puromycin (4 μg/ml) was added to culture medium for cell screening. The screening process lasted for 2 weeks, and culture medium was replaced with puromycin (4 μg/ml) every day.
Other important reagents are as follows: PKH67 (Sigma-Aldrich), Matrigel (Corning, 356234), phalloidin (Abcam, ab176753), and RNA Pol II (5 μg per test; BioLegend, 920102). The reagent BTZ was purchased from Sigma-Aldrich.
Quantification of cell morphology
Ovarian cells in each experiment were fixed with 4% paraformaldehyde and stained with fluorescein isothiocyanate–phalloidin (Abcam) and 4′,6-diamidino-2-phenylindole. Individual cells were manually outlined using Adobe Photoshop CS6 software. Area, perimeter, circularity, and elongation index were quantified for no less than 40 cells per substrate using ImageJ software. Circularity was defined as 4π(cell area)/(cell perimeter)2. Elongation index was defined as (cell perimeter)2/(4π(cell area)) as reported previously (45).
Public datasets
We used four different mRNA datasets comprising together a total of 989 patients for this study, including TCGA cohort (n = 483) (3), Bonome cohort (GSE26712, n = 185) (12), Tothill cohort (GSE9891, n = 242) (7), and Mateescu cohort (GSE26193, n = 79) (13). Only patients with HGSC in these datasets were included in our analysis. In addition, two independent miRNA datasets were analyzed in this study, among which the OV179 cohort includes 179 patients with ovarian cancer (GSE73581) (20) and the OV133 cohort comprises 133 patients with ovarian cancer (GSE73582) (20). TCGA data was downloaded from Firebrowse (http://firebrowse.org/) (46). For the other cohorts, gene expression data together with clinical profiles were downloaded from Gene Expression Omnibus (GEO) directly in their processed form using R package GEOquery (version 1.0.7) (47). For each dataset, the expression profiles were collapsed from probe sets to genes, followed by z-normalization across all samples. Only patients with complete survival information were used for survival analyses. The molecular subtyping information for the Bonome, Tothill, TCGA, and Mateescu datasets were retrieved from the study of Verhaak et al. (48).
Profiling of infiltrating immune cells
To dissect immune cell infiltration heterogeneity, we used CIBERSORT (10), a popular algorithm for characterizing cell composition from bulk-tumor GEPs. More specifically, normalized gene expression datasets were uploaded to the CIBERSORT web portal (http://cibersort.stanford.edu/) for analysis, using the default signature matrix at 1000 permutations. For each sample, CIBERSORT inferred the relative proportions of 22 types of infiltrating immune cells (all P values of <0.05), where P values were empirically calculated using Monte Carlo sampling indicating the statistical confidence for deconvolution of bulk tumor GEPs into a mixture of cell type–specific GEPs.
Identification of plasma cell gene signature subgroups
The z-score normalized microarray data from the Tothill cohort was used. Plasma signature genes were retrieved from CIBERSORT. Next, we clustered these samples using hierarchical clustering with agglomerative average linkage. Consensus clustering (48) was applied to assess the clustering stability, with 1000 iterations and a 0.98 subsampling ratio. To determine the optimal cluster number, we computed the gap statistic (49) for k = 1 to 10, and a peak was found at k = 2.
Identification of plasma cell exosomal miRNA gene signature subgroups
The OV133 cohort was first normalized followed by z-score transformation. The overlapped miRNAs among OV133, OV179, and plasma cell exosome small RNA sequencing data were selected. The k means clustering algorithm (51) was used to classify the OV133 samples into two groups. To build a classifier, the overlapped miRNAs were used to train a classifier by prediction analysis for microarrays (PAMs) (52). We classified the OV179 cohort using the PAM classifier.
Gene set enrichment analysis
GSEA (21) was performed using GSEA software (www.broadinstitute.org/gsea) with 1000 permutations. Phenotype used for the analysis was log2 fold change between GEPs of samples with high and low plasma cell abundance (mean-dichotomized). Gene sets used were obtained from MSigDB (Molecular Signatures Database; C2 and C5 databases, version 6). False discovery rate–adjusted P < 0.05 was used to select statistically significant gene sets.
RNA sequencing and data analysis
Whole-transcriptome sequencing libraries were constructed as described previously (50) and were sequenced on the Illumina HiSeq platform (Novogene, China). Briefly, a total amount of 3 μg of RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using the NEBNext UltraTM RNA Library Prep Kit for Illumina (New England Biolabs, USA) following the manufacturer’s recommendations, and index codes were added to attribute sequences to each sample. Briefly, mRNA was purified from total RNA using poly-T oligo–attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5×). First-strand complementary DNA (cDNA) was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase [ribonuclease H− (RNase H−)]. Second-strand cDNA synthesis was subsequently performed using DNA Pol I and RNase H. Remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of 3′ ends of DNA fragments, NEBNext Adaptor with hairpin loop structure were ligated to prepare for hybridization. To select cDNA fragments of preferentially 250 to 300 base pairs (bp) in length, the library fragments were purified with the AMPure XP system (Beckman Coulter, Beverly, USA). Then, 3 μl of USER Enzyme (New England Biolabs, USA) was used with size-selected, adapter-ligated cDNA at 37°C for 15 min followed by 5 min at 95°C before PCR. Then, PCR was performed with Phusion High-Fidelity DNA Polymerase, Universal PCR primers, and Index (X) Primer. Last, PCR products were purified (AMPure XP system), and library quality was assessed on the Agilent Bioanalyzer 2100 system. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina HiSeq platform, and 125/150-bp paired-end reads were generated (51). Here, a total of 6-gauge clean reads were generated from whole-transcriptome sequencing for each group. For quality control: Raw data of Fastq format were first processed through in-house perl scripts. In this step, clean data were obtained by removing reads containing adapter and low-quality reads from raw data. Meanwhile, Q20, Q30, and GC content, the clean data were calculated (52). All the downstream analyses were based on the clean data with high quality. For reads mapping to the reference genome: Reference genome and gene model annotation files were downloaded from genome website directly. Index of the reference genome was built using STAR, and paired-end clean reads were aligned to the reference genome GRCh38 using STAR (v2.5.1b) (53). STAR used the method of Maximal Mappable Prefix, which can generate a precise mapping result for junction reads. Quantification of gene expression level: HTSeq v0.6.0 was used to count the reads numbers mapped to each gene. Then, FPKM of each gene was calculated on the basis of the length of the gene and reads count mapped to this gene. FPKM, expected number of fragments per kilobase of transcript sequence per millions base pairs sequenced, considers the effect of sequencing depth and gene length for the read count at the same time and is currently the most commonly used method for estimating gene expression levels.
Patients and clinical specimens
The tumor specimens of patients with ovarian cancer were collected from West China Second Hospital, Sichuan University. The freshly resected tumor samples of patients with ovarian cancer for flow cytometry analysis were also obtained from West China Second Hospital, Sichuan University. All of these samples were examined by experienced pathologists who confirmed the diagnosis of disease samples. Blood samples used for isolation of plasma cells derived from three patients with HGSC. Exosomes subjected to miRNA sequencing were also obtained from plasma cells isolated from these patients with HGSC. We combined the exosomes for further functional validation experiments. This study was approved by the Institutional Ethics Committee of Sichuan University. Informed consents were obtained from all patients before analysis.
Cell sorting method
Plasma cell–sorting progress was divided into two steps: First, total lymphocytes were isolated from patient blood using a lymphocyte separation kit (DKW-KLSH-0400), which was based on the density gradient centrifugation method; second, plasma cells were isolated using magnetic separation (CD138 MicroBeads: MACS, 130-051-301) from the lymphocytes obtained in the former step. The concentration of CD138 MicroBeads used was 20 μl of beads/1 × 107 cells per 40 μl of buffer. Other immune cells were sorted and enriched as described previously using magnetic separation as well (49).
Immunoblotting, immunohistochemistry, and immunofluorescent assays
Immunoblotting was performed as previously described (34, 50). Briefly, for immunoblotting, whole-cell lysates of ovarian cancer cells were prepared using radioimmunoprecipitation assay lysis buffer (C1053), 1 mM phenylmethylsulfonyl fluoride (P0100), and 1 mM EDTA. Protein lysates were resolved on SDS–polyacrylamide gel electrophoresis (P0012A) and blotted on Amersham Hybond P 0.45 polyvinylidene difluoride. Membranes were blocked for 1 hour in tris-buffered saline (TBS) containing 0.1% (v/v) Tween 20 with 20% (w/v) nonfat dry milk powder (blocking solution). Primary antibodies were incubated at 4°C in blocking solution. Secondary peroxidase-labeled affinity-purified antibodies (KPL) were diluted 1:5000 in blocking solution and incubated for 1 hour at room temperature. Membranes were washed in 0.1% TBS–Tween 20, and immune complexes were detected using the Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, 32106).
For immunohistochemistry analysis, 4-μm-thick paraffin sections were deparaffinized and rehydrated, and antigens were retrieved in antigen unmasking solution, citric acid based (Vector Laboratories, H-3300), using a pressure boiler for 10 min under a slightly boiling state. Slides were treated with a 3% solution of hydrogen peroxidase in methanol to block the endogenous peroxidase activity and then washed in a phosphate buffer solution before immunoperoxidase staining. Slides were then incubated at 5% goat serum blocked for 2 hours and then 4°C overnight with primary antibody. Tissue sections were then washed three times with phosphate-buffered saline (PBS) and next incubated with biotinylated antibody immunoglobulin and then stained with streptavidin labeled with peroxidase; the signal was developed by using 3,3′-diaminobenzine chromogen as substrate. After chromogen development, slides were counterstained with hematoxylin, washed, dehydrated with alcohol, and mounted with coverslips using a permanent mounting medium (Absin, abs9177).
For immunofluorescent analysis, before antibody incubation, 4-μm-thick paraffin section samples were deparaffinized and rehydrated, and antigens were retrieved in antigen unmasking solution, citric acid based (Vector Laboratories H-3300), using a pressure boiler for 10 min under a slightly boiling state. For formaldehyde-fixated cell climbing tablet samples, PBST (0.3% Triton X-100 with PBS) incubation was processed for 30 min, and then, the paraffin section samples or cell climbing tablet samples were incubated at 5% goat serum for 2 hours and then 4°C overnight with primary antibody, then washed three times, and next incubated with secondary antibody. Last, samples were washed and mounted with coverslips with a permanent mounting medium. Photos were required by using an Olympus IX73 fluorescence microscope and a Leica SP5 confocal fluorescence microscope. We have defined those clinical specimens with a mean optic density of α-SMA over 0.05 as the mesenchymal subtype and otherwise as the nonmesenchymal subtype.
Flow cytometry analysis
Quantification of different immune cells was performed using a BD FACSAria III and a BD LSRFortessa as described previously (54). Antibody combination strategies for different kinds of immune cells could directly be seen in the figures. The antibody concentrations were used according to the manufacturer’s instructions.
Quantitative polymerase chain reaction
The mRNA levels of each gene were measured via qPCR. Following procedures previously described (50), RNA was isolated using a total RNA isolation kit, including an on-column deoxyribonuclease treatment (Norgen). cDNA synthesis was carried out using the SuperScript III Reverse Transcriptase Kit using a mixture of oligo(dT) and random hexamers for priming (Life Technologies). qPCR was conducted with Fast SYBR Green Master Mix (Applied Biosystems), and fluorescence was monitored using a 7900HT Fast real-time instrument (Applied Biosystems). Data were analyzed using the ΔΔCt method. The endogenous control transcripts were used for normalization. Statistical significance was determined using a one-tailed Student’s t test. The sequences of the primers used for all qPCR assays are in table S3.
Transwell chamber and wound healing analysis
Transwell 24-well chambers (Corning) were applied for in vitro cell migration assay as described previously (55). Ten contiguous fields of each sample were assessed to obtain a representative number of cells that had migrated across the membrane. The cell number was calculated by Countstar Automated Cell Counter machine and adjusted by diluting with cell culture medium. As for the wound healing analysis, wounds were scratched in confluent cells using a pipette tip, and the cells were then rinsed with medium to remove free-floating cells and debris. Serum-free medium was subsequently added, and culture plates were incubated at 37°C for 2 days. Wound healing was observed at 0 and 24 hours within the scrape line, and representative scrape lines for each cell line were photographed.
Mice subcutaneous and peritoneal metastatic tumor model
The C57 mice used for subcutaneous tumor inoculation were obtained from Beijing Vital River Laboratory Animal Technology Co. Ltd. Animal studies were reviewed and approved by the Institutional Ethics Committee of Sichuan University. For subcutaneous model, mice were injected with ID8 cells subcutaneously, and tumors were allowed to establish for 40 days. For the peritoneal metastatic tumor model, ID8-luc cells were intraperitoneally injected, and mice were examined for Luc expression using d-luciferin (100 mg/kg; Invitrogen, Life Technologies). Images were captured with the PerkinElmer IVIS Lumina III instrument to assess tumor development every week. Image analyses were carried out with Living Image Software–IVIS Lumina Series software.
Exosome extraction and treatment
Exosomes were purified by differential centrifugation processes as described previously (56). Plasma cell culture media were subsequently subjected to sequential centrifugation steps at 300g for 10 min and 2000g for 10 min. This resulting supernatant was then subjected to ultracentrifugation (Thermo Fisher Scientific Sorvall WX 100+) sequentially for 30 and 70 min. The first 30 min was to remove cell debris, and a pellet was recovered after 70 min of the resulting supernatant subjected to ultracentrifugation. The pellet was resuspended in PBS and subsequently ultracentrifuged at 100,000g for another 70 min. Purified exosomes were then analyzed and used for experimental procedures. For in vitro experiments, tumor cells were treated with exosomes (10 μg/ml).
Electron microscopy and NanoSight analysis
Exosomes were examined with a transmission electron microscope after isolation and loading. The samples were dropped in the carbon support membrane with a copper net and placed for 20 min. Then, excess liquid was absorbed using filter paper. The carbon support membrane was then dripped with 2% phosphotungstic acid and laid up for 5 min. Excess liquid was absorbed using filter paper, and sample was visualized with a Tecnai G2 transmission electron microscope. A NanoSight NS300 instrument equipped with NTA 3.0 analytical software was used to quantify the size distribution of exosomes.
miRNA sequencing
Total RNA from exosomes was then subjected to miRNA sequencing. Library preparation and miRNA sequencing were performed by RiboBio (Guangzhou, China). Only small RNAs ranging from 18 to 30 nt were used for library preparation. PCR amplification products were sequenced using the Illumina HiSeq 2500 platform. The 3′ adapters were removed by Cutadapt (54). The adapter-trimmed reads were remapped to human genome hg38 using Burrows-Wheeler Aligner (BWA) aligner (51). miRBase version 21 (57) (www.mirbase.org/) was used for annotation.
Chromatin immunoprecipitation
For ChIP assay, formaldehyde was added to cell culture medium to a final concentration of 1%. After 10 min of incubation at room temperature, glycine was added to a concentration of 125 mM to stop cross-linking reaction. Cells were quickly washed with cold PBS in a culture dish twice and then added to 1 ml of PBS mixed with protease inhibitors. Then, they were pelleted by centrifugation at 800g for 5 min at 4°C. Cell pellet was resuspended in 1 ml of lysis buffer A [50 mM Hepes-KOH (pH7.5), 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, and 0.25% Triton X-100] with protease inhibitors and then was rotated modestly for 10 min at 4°C. Then, they were pelleted by centrifugation at 800g for 5 min at 4°C. Resulting nuclear pellet was resuspended in 300 μl of lysis buffer B [10 mM tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA (pH 8.0), and 0.1% Na-deoxycholate] with protease inhibitors, which was next put on ice for 30 min. Resulting suspension was sonicated in ultrasound to achieve DNA fragments of 200 to 500 bp. Ultrasonic product (25 μl) was saved as an input, whereas another 250 μl was added to 555 μl of dilution buffer [0.01% SDS, 1.1% Triton X-100,1.2 mM EDTA, 16.7 mM tris-HCl (pH 8.1), and 167 mM NaCl] with protease inhibitors combined with 50 μl of magnetic beads coupled anti-mouse immunoglobulin G. The mixture was rotated modestly for 30 min at 4°C. Then, Magnetic Separation Rack was used to absorb magnetic beads for 2 min. The suspension was then equally divided into IP (Immunoprecipitation) group and negative group, and the original magnetic beads were discarded. Add 5 μg of antibody into the IP group tube rotated for a night at 4°C. The next day, 50 μl of magnetic beads was added into two group tubes that next rotated for 1 hour at 4°C. Magnetic Separation Rack was used to absorb magnetic beads for 2 min. The suspension was discarded, and the beads were washed with the following solution of 500 μl, respectively, each time rotated for 5 min at 4°C: (i) Low salt [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM tris-HCl (pH 8.1), and 150 mM NaCl], wash once; (ii) high salt [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM tris-HCl (pH 8.1), and 500 mM NaCl], wash once; (iii) LiCl [0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, and 10 mM tris-HCl (pH8.1)]; and (iv) TE [10 mM tris-HCl and 1 mM EDTA (pH 8.0)], wash twice.
Then, when it comes to the elution procedure, the One-Day Chromatin Immunoprecipitation Kit (Millipore) was performed according to the manufacturer’s instructions. The final step adopted a standard phenol/chloroform/isoamyl alcohol procedure to purify DNA.
Statistical analysis
Kaplan-Meier curves were generated to show survival significance calculated on the basis of log-rank tests. Continuous variables were presented as mean and SEM. Wilcoxon rank sum tests were used for comparisons of different groups in all public cohorts. Two-sided Student’s t tests were used to assess differences in all experiments. For any test, a P value of <0.05 was considered to be significant. Statistical significance is shown as *P < 0.05, **P < 0.01, and ***P < 0.001. Statistical analyses were performed using R (version 3.4.3, www.r-project.org).
Acknowledgments
Funding: This work was supported by grants from the National Natural Science Foundation of China (grant nos. 81822034, 81821002, and 81773119), the National Key Research and Development Program of China (2017YFA0106800, 2018YFA0109200, and 2017YFA0106500), the Sichuan Science-Technology International Cooperation Project (grant no. 2019YFH0144), the Distinguished Young Scientists Program of Sichuan Province (2019JDJQ0029), the Direct Scientific Research Grants from West China Second Hospital, Sichuan University (grant nos. KS021 and K1907), the 135 Program for Excellent Scholars at West China Hospital, Sichuan University (ZYYC20019), the Graduate Student’s Research and Innovation Fund of Sichuan University (grant no. 2018YJSY117), a grant from the Research Grants Council of the Hong Kong Special Administrative Region, China (project no. CityU 11103718), and a grant from the Guangdong Basic and Applied Basic Research Foundation (2019B030302012). The work was also supported by NIH grants (CA197718, CA154130, CA169117, CA171652, CA217065, NS087913, and NS089272). Author contributions: Z.Y., Y.W., J.N.R., and S.Z. designed the experiments, analyzed the data, and prepared the manuscript with contributions from all authors. Z.Y., L.Z., and L.X. performed the experiments. W.W., X.W., and R.C.G. performed database analyses. S.Z. and L.X. provided clinical samples. R.C.G. helped in the revision of the research. R.C.G., Y.W., S.Z., and J.N.R. provided scientific input and helped edit the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All high-throughput sequencing data in this study have been deposited in the GEO database under accession number GSE158317. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabb0737/DC1
REFERENCES AND NOTES
- 1.Bowtell D. D., Bohm S., Ahmed A. A., Aspuria P. J., Bast R. C. Jr., Beral V., Berek J. S., Birrer M. J., Blagden S., Bookman M. A., Brenton J. D., Chiappinelli K. B., Martins F. C., Coukos G., Drapkin R., Edmondson R., Fotopoulou C., Gabra H., Galon J., Gourley C., Heong V., Huntsman D. G., Iwanicki M., Karlan B. Y., Kaye A., Lengyel E., Levine D. A., Lu K. H., McNeish I. A., Menon U., Narod S. A., Nelson B. H., Nephew K. P., Pharoah P., Powell D. J. Jr., Ramos P., Romero I. L., Scott C. L., Sood A. K., Stronach E. A., Balkwill F. R., Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 15, 668–679 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raab M., Kobayashi N. F., Becker S., Kurunci-Csacsko E., Kramer A., Strebhardt K., Sanhaji M., Boosting the apoptotic response of high-grade serous ovarian cancers with CCNE1 amplification to paclitaxel in vitro by targeting APC/C and the pro-survival protein MCL-1. Int. J. Cancer 146, 1086–1098 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research Network , Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang Y. A., Chen Y. F., Bao Y., Mahara S., Yatim S., Oguz G., Lee P. L., Feng M., Cai Y., Tan E. Y., Fong S. S., Yang Z. H., Lan P., Wu X. J., Yu Q., Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-β2 to promote chemoresistance in colorectal cancer. Proc. Natl. Acad. Sci. U.S.A. 115, E5990–E5999 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shang S., Ji X., Zhang L., Chen J., Li C., Shi R., Xiang W., Kang X., Zhang D., Yang F., Dai R., Chen P., Chen S., Chen Y., Li Y., Miao H., Macrophage ABHD5 suppresses NFκB-Dependent matrix metalloproteinase expression and cancer metastasis. Cancer Res. 79, 5513–5526 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Wei Y., Zhao Q., Gao Z., Lao X. M., Lin W. M., Chen D. P., Mu M., Huang C. X., Liu Z. Y., Li B., Zheng L., Kuang D. M., The local immune landscape determines tumor PD-L1 heterogeneity and sensitivity to therapy. J. Clin. Invest. 129, 3347–3360 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tothill R. W., Tinker A. V., George J., Brown R., Fox S. B., Lade S., Johnson D. S., Trivett M. K., Etemadmoghadam D., Locandro B., Traficante N., Fereday S., Hung J. A., Chiew Y. E., Haviv I., Gertig D., DeFazio A., Bowtell D. D., Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 14, 5198–5208 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Zhang A. W., McPherson A., Milne K., Kroeger D. R., Hamilton P. T., Miranda A., Funnell T., Little N., de Souza C. P. E., Laan S., LeDoux S., Cochrane D. R., Lim J. L. P., Yang W., Roth A., Smith M. A., Ho J., Tse K., Zeng T., Shlafman I., Mayo M. R., Moore R., Failmezger H., Heindl A., Wang Y. K., Bashashati A., Grewal D. S., Brown S. D., Lai D., Wan A. N. C., Nielsen C. B., Huebner C., Tessier-Cloutier B., Anglesio M. S., Bouchard-Côté A., Yuan Y., Wasserman W. W., Gilks C. B., Karnezis A. N., Aparicio S., McAlpine J. N., Huntsman D. G., Holt R. A., Nelson B. H., Shah S. P., Interfaces of malignant and immunologic clonal dynamics in ovarian cancer. Cell 173, 1755–1769.e22 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Ray M., Ruffalo M. M., Bar-Joseph Z., Construction of integrated microRNA and mRNA immune cell signatures to predict survival of patients with breast and ovarian cancer. Genes Chromosom. Cancer 58, 34–42 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Newman A. M., Liu C. L., Green M. R., Gentles A. J., Feng W., Xu Y., Hoang C. D., Diehn M., Alizadeh A. A., Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu R., Hu R., Zeng Y., Zhang W., Zhou H. H., Tumour immune cell infiltration and survival after platinum-based chemotherapy in high-grade serous ovarian cancer subtypes: A gene expression-based computational study. EBioMedicine 51, 102602 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonome T., Levine D. A., Shih J., Randonovich M., Pise-Masison C. A., Bogomolniy F., Ozbun L., Brady J., Barrett J. C., Boyd J., Birrer M. J., A gene signature predicting for survival in suboptimally debulked patients with ovarian cancer. Cancer Res. 68, 5478–5486 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mateescu B., Batista L., Cardon M., Gruosso T., de Feraudy Y., Mariani O., Nicolas A., Meyniel J. P., Cottu P., Sastre-Garau X., Mechta-Grigoriou F., miR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat. Med. 17, 1627–1635 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Zhao L., Wang W., Xu L., Yi T., Zhao X., Wei Y., Vermeulen L., Goel A., Zhou S., Wang X., Integrative network biology analysis identifies miR-508-3p as the determinant for the mesenchymal identity and a strong prognostic biomarker of ovarian cancer. Oncogene 38, 2305–2319 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalluri R., The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Yeong J., Lim J. C. T., Lee B., Li H., Chia N., Ong C. C. H., Lye W. K., Putti T. C., Dent R., Lim E., Thike A. A., Tan P. H., Iqbal J., High densities of tumor-associated plasma cells predict improved prognosis in triple negative breast cancer. Front. Immunol. 9, 1209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lohr M., Edlund K., Botling J., Hammad S., Hellwig B., Othman A., Berglund A., Lambe M., Holmberg L., Ekman S., Bergqvist M., Ponten F., Cadenas C., Marchan R., Hengstler J. G., Rahnenfuhrer J., Micke P., The prognostic relevance of tumour-infiltrating plasma cells and immunoglobulin kappa C indicates an important role of the humoral immune response in non-small cell lung cancer. Cancer Lett. 333, 222–228 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Domcke S., Sinha R., Levine D. A., Sander C., Schultz N., Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 4, 2126 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan T. Z., Miow Q. H., Huang R. Y., Wong M. K., Ye J., Lau J. A., Wu M. C., Bin Abdul Hadi L. H., Soong R., Choolani M., Davidson B., Nesland J. M., Wang L. Z., Matsumura N., Mandai M., Konishi I., Goh B. C., Chang J. T., Thiery J. P., Mori S., Functional genomics identifies five distinct molecular subtypes with clinical relevance and pathways for growth control in epithelial ovarian cancer. EMBO Mol. Med. 5, 1051–1066 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bagnoli M., Canevari S., Califano D., Losito S., Maio M. D., Raspagliesi F., Carcangiu M. L., Toffoli G., Cecchin E., Sorio R., Canzonieri V., Russo D., Scognamiglio G., Chiappetta G., Baldassarre G., Lorusso D., Scambia G., Zannoni G. F., Savarese A., Carosi M., Scollo P., Breda E., Murgia V., Perrone F., Pignata S., De Cecco L., Mezzanzanica D.; Multicentre Italian Trials in Overian cancer (MITO) translational group , Development and validation of a microRNA-based signature (MiROvaR) to predict early relapse or progression of epithelial ovarian cancer: A cohort study. Lancet Oncol. 17, 1137–1146 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H., Fan J., Zhao Y., Zhang X., Dai B., Zhan J., Yin Z., Nie X., Fu X. D., Chen C., Wang D. W., Nuclear miR-320 mediates diabetes-induced cardiac dysfunction by activating transcription of fatty acid metabolic genes to cause lipotoxicity in the heart. Circ. Res. 125, 1106–1120 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao M., Li J., Li W., Wang Y., Wu F., Xi Y., Zhang L., Ding C., Luo H., Li Y., Peng L., Zhao L., Peng S., Xiao Y., Dong S., Cao J., Yu W., MicroRNAs activate gene transcription epigenetically as an enhancer trigger. RNA Biol. 14, 1326–1334 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jonas S., Izaurralde E., Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 16, 421–433 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Suzuki H. I., Young R. A., Sharp P. A., Super-enhancer-mediated RNA processing revealed by integrative microrna network analysis. Cell 168, 1000–1014.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ren C., Zhang G., Han F., Fu S., Cao Y., Zhang F., Zhang Q., Meslamani J., Xu Y., Ji D., Cao L., Zhou Q., Cheung K. L., Sharma R., Babault N., Yi Z., Zhang W., Walsh M. J., Zeng L., Zhou M. M., Spatially constrained tandem bromodomain inhibition bolsters sustained repression of BRD4 transcriptional activity for TNBC cell growth. Proc. Natl. Acad. Sci. U.S.A. 115, 7949–7954 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alexander T., Sarfert R., Klotsche J., Kuhl A. A., Rubbert-Roth A., Lorenz H. M., Rech J., Hoyer B. F., Cheng Q., Waka A., Taddeo A., Wiesener M., Schett G., Burmester G. R., Radbruch A., Hiepe F., Voll R. E., The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann. Rheum. Dis. 74, 1474–1478 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hiepe F., Radbruch A., Plasma cells as an innovative target in autoimmune disease with renal manifestations. Nat. Rev. Nephrol. 12, 232–240 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Taylor E. B., Barati M. T., Powell D. W., Turbeville H. R., Ryan M. J., Plasma cell depletion attenuates hypertension in an experimental model of autoimmune disease. Hypertension 71, 719–728 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berntsson J., Nodin B., Eberhard J., Micke P., Jirstrom K., Prognostic impact of tumour-infiltrating B cells and plasma cells in colorectal cancer. Int. J. Cancer 139, 1129–1139 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Wei Y., Lao X. M., Xiao X., Wang X. Y., Wu Z. J., Zeng Q. H., Wu C. Y., Wu R. Q., Chen Z. X., Zheng L., Li B., Kuang D. M., Plasma cell polarization to the immunoglobulin G phenotype in hepatocellular carcinomas involves epigenetic alterations and promotes hepatoma progression in mice. Gastroenterology 156, 1890–1904.e16 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Lundgren S., Berntsson J., Nodin B., Micke P., Jirstrom K., Prognostic impact of tumour-associated B cells and plasma cells in epithelial ovarian cancer. J. Ovarian Res. 9, 21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kroeger D. R., Milne K., Nelson B. H., Tumor-infiltrating plasma cells are associated with tertiary lymphoid structures, cytolytic t-cell responses, and superior prognosis in ovarian cancer. Clin. Cancer Res. 22, 3005–3015 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Zhao L., Huang S., Mei S., Yang Z., Xu L., Zhou N., Yang Q., Shen Q., Wang W., Le X., Lau W. B., Lau B., Wang X., Yi T., Zhao X., Wei Y., Warner M., Gustafsson J. A., Zhou S., Pharmacological activation of estrogen receptor beta augments innate immunity to suppress cancer metastasis. Proc. Natl. Acad. Sci. U.S.A. 115, E3673–E3681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tran N., Cancer exosomes as miRNA factories. Trends Cancer 2, 329–331 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Zhou J., Li X., Wu X., Zhang T., Zhu Q., Wang X., Wang H., Wang K., Lin Y., Wang X., Exosomes released from tumor-associated macrophages transfer miRNAs that induce a Treg/Th17 cell imbalance in epithelial ovarian cancer. Cancer Immunol. Res. 6, 1578–1592 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Dorayappan K. D. P., Wanner R., Wallbillich J. J., Saini U., Zingarelli R., Suarez A. A., Cohn D. E., Selvendiran K., Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: A novel mechanism linking STAT3/Rab proteins. Oncogene 37, 3806–3821 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao Y., Zuo J., Wei Y., Targeting of TRX2 by miR-330-3p in melanoma inhibits proliferation. Biomed. Pharmacother. 107, 1020–1029 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Liu X., Shi H., Liu B., Li J., Liu Y., Yu B., miR-330-3p controls cell proliferation by targeting early growth response 2 in non-small-cell lung cancer. Acta Biochim. Biophys. Sin. 47, 431–440 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Fabian M. R., Sonenberg N., Filipowicz W., Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 79, 351–379 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Lauko A., Mu Z., Gutmann D. H., Naik U. P., Lathia J. D., Junctional adhesion molecules in cancer: A paradigm for the diverse functions of cell-cell interactions in tumor progression. Cancer Res. 80, 4878–4885 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Radulovic V., van der Garde M., Koide S., Sigurdsson V., Lang S., Kaneko S., Miharada K., Junctional adhesion molecule 2 represents a subset of hematopoietic stem cells with enhanced potential for T lymphopoiesis. Cell Rep. 27, 2826–2836.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Arcangeli M. L., Frontera V., Bardin F., Thomassin J., Chetaille B., Adams S., Adams R. H., Aurrand-Lions M., The junctional adhesion molecule-B regulates JAM-C-dependent melanoma cell metastasis. FEBS Lett. 586, 4046–4051 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Costa A., Kieffer Y., Scholer-Dahirel A., Pelon F., Bourachot B., Cardon M., Sirven P., Magagna I., Fuhrmann L., Bernard C., Bonneau C., Kondratova M., Kuperstein I., Zinovyev A., Givel A. M., Parrini M. C., Soumelis V., Vincent-Salomon A., Mechta-Grigoriou F., Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 33, 463–479.e10 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Loye A. M., Kinser E. R., Bensouda S., Shayan M., Davis R., Wang R., Chen Z., Schwarz U. D., Schroers J., Kyriakides T. R., Regulation of mesenchymal stem cell differentiation by nanopatterning of bulk metallic glass. Sci. Rep. 8, 8758 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deng M., Bragelmann J., Kryukov I., Saraiva-Agostinho N., Perner S., FirebrowseR: An R client to the Broad Institute’s firehose pipeline. Database , baw160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davis S., Meltzer P. S., GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 23, 1846–1847 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Verhaak R. G., Tamayo P., Yang J. Y., Hubbard D., Zhang H., Creighton C. J., Fereday S., Lawrence M., Carter S. L., Mermel C. H., Kostic A. D., Etemadmoghadam D., Saksena G., Cibulskis K., Duraisamy S., Levanon K., Sougnez C., Tsherniak A., Gomez S., Onofrio R., Gabriel S., Chin L., Zhang N., Spellman P. T., Zhang Y., Akbani R., Hoadley K. A., Kahn A., Köbel M., Huntsman D., Soslow R. A., Defazio A., Birrer M. J., Gray J. W., Weinstein J. N., Bowtell D. D., Drapkin R., Mesirov J. P., Getz G., Levine D. A., Meyerson M.; Cancer Genome Atlas Research Network , Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Invest. 123, 517–525 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peters C. E., Woodside S. M., Eaves A. C., Isolation of subsets of immune cells. Methods Mol. Biol. 302, 95–116 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Zhao L., Wang W., Huang S., Yang Z., Xu L., Yang Q., Zhou X., Wang J., Shen Q., Wang C., Le X., Feng M., Zhou N., Lau W. B., Lau B., Yao S., Yi T., Wang X., Zhao X., Wei Y., Zhou S., The RNA binding protein SORBS2 suppresses metastatic colonization of ovarian cancer by stabilizing tumor-suppressive immunomodulatory transcripts. Genome Biol. 19, 35 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo J., Han X., You Y., Huang W., Zhan J., IgA dysfunctions induced by the early-lifetime disruption of gut microbitoa result in metabolic syndrome in mice. Research Square, 10.21203/rs.3.rs-40336/v1, (2020). [DOI]
- 52.Mao J., Tian Y., Wang C., Jiang K., Li R., Yao Y., Zhang R., Sun D., Liang R., Gao Z., Wang Q., Wang L., CBX2 regulates proliferation and apoptosis via the phosphorylation of YAP in hepatocellular carcinoma. J. Cancer 10, 2706–2719 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang S., Zhou N., Zhao L., Gimple R. C., Ahn Y. H., Zhang P., Wang W., Shao B., Yang J., Zhang Q., Zhao S., Jiang X., Chen Z., Zeng Y., Hu H., Gustafsson J., Zhou S., Pharmacological activation of estrogen receptor beta overcomes tumor resistance to immune checkpoint blockade therapy. iScience 23, 101458 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao L., Ji G., Le X., Wang C., Xu L., Feng M., Zhang Y., Yang H., Xuan Y., Yang Y., Lei L., Yang Q., Lau W. B., Lau B., Chen Y., Deng X., Yao S., Yi T., Zhao X., Wei Y., Zhou S., Long noncoding RNA LINC00092 acts in cancer-associated fibroblasts to drive glycolysis and progression of ovarian cancer. Cancer Res. 77, 1369–1382 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Paggetti J., Haderk F., Seiffert M., Janji B., Distler U., Ammerlaan W., Kim Y. J., Adam J., Lichter P., Solary E., Berchem G., Moussay E., Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 126, 1106–1117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kozomara A., Birgaoanu M., Griffiths-Jones S., miRBase: From microRNA sequences to function. Nucleic Acids Res. 47, D155–D162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/9/eabb0737/DC1