

Summary
Rho is a general transcription termination factor playing essential roles in RNA polymerase (RNAP) recycling, gene regulation, and genomic stability in most bacteria. Textbook models of transcription termination postulate that hexameric Rho loads onto RNA prior to contacting RNAP and then translocates along the transcript in pursuit of the moving RNAP to pull RNA from it. Here, we report the cryo-EM structures of two termination process intermediates. Prior to interacting with RNA, Rho forms a specific “pre-termination complex” (PTC) with RNAP and elongation factors NusA and NusG, which stabilize the PTC. RNA exiting RNAP interacts with NusA before entering the central channel of Rho from the distal C-terminal side of the ring. We map the principal interactions in the PTC and demonstrate their critical role in termination. Our results overturn the traditional termination models and support a mechanism in which the formation of a persistent PTC is a prerequisite for termination.
One Sentence Summary:
An integrated structure-function approach elucidates the pathway leading to factor-dependent transcription termination.
Introduction
RNA helicase Rho is a multitasking transcription termination factor, almost universally present in bacteria (Boudvillain et al., 2013; Grylak-Mielnicka et al., 2016; Svetlov and Nudler, 2020). In E. coli, Rho separates transcription units (Roberts, 1969), controls global gene expression (Sedlyarova et al., 2016), silences toxic “foreign” genes (Cardinale et al., 2008), prevents detrimental transcription–replication collisions (Dutta et al., 2011) and R-loops (Leela et al., 2013), and curbs pervasive antisense transcription (Magan et al., 2019; Peters et al., 2012; Sedlyarova et al., 2017).
Structural and biochemical studies propose that hexameric Rho interacts with nascent RNA in two consecutive steps. First, the “primary binding site” of each monomer interacts with a pair of pyrimidines of an unstructured and preferably cytosine-rich RNA segment known as the Rho-utilization transcript (RUT) (Hart and Roberts, 1991; Richardson and Richardson, 1996; Skordalakes and Berger, 2003). RNA is then enclosed in the central cavity of the ring, which comprises the “secondary binding site”. This process activates ATP binding and hydrolysis, enabling Rho to thread RNA through its central pore (Boudvillain et al., 2013; Skordalakes and Berger, 2003).
In spite of decades of genetic, biochemical, and biophysical studies, some of the basic principles of Rho-dependent termination remain unresolved. Traditionally, it was assumed that Rho loading onto RNA precedes its interaction with RNAP and that Rho contacts RNAP only transiently, at the moment of the actual termination event. Although biochemical evidence argued to the contrary (Epshtein et al., 2010), the “classical” model has persisted (Koslover et al., 2012; Mitra et al., 2017; Ray-Soni et al., 2016).
To clarify the mechanism of Rho-dependent termination, we determined two cryo-EM structures of the pre-termination complex (PTC) on its path to termination. Probing RNAP–Rho interactions in the PTC demonstrates their critical importance for the termination process. A combination of structural, genetic, and biochemical data presented here illuminates the principal steps leading to Rho-dependent termination, most of which are substantially different from those postulated by the traditional model.
Results and Discussion
Preparation and Structure Determination of the PTC from E. coli
Two general transcription elongation factors, NusA and NusG, have been implicated in the process of Rho-dependent termination in vitro and in vivo (Burns et al., 1998; Cardinale et al., 2008; Nehrke and Platt, 1994; Schmidt and Chamberlin, 1984; Sullivan and Gottesman, 1992). Given that both, NusA and NusG, crosslink to RNAP and Rho in vivo, even in the presence of the antibiotic bicyclomycin (BCM) that specifically inhibits Rho helicase activity (Table S1), we reasoned that both factors should be part of a stable Rho–RNAP complex. Indeed, we were able to reconstitute such a quaternary PTC from individually purified E. coli proteins (Figure 1). To assemble the active transcription elongation complex (EC), we modified the nucleic-acid scaffold previously used to determine the cryo-EM structure of the E. coli EC (Kang et al., 2017) by shortening the RNA to 18 nucleotides or extending it with the partial RUT sequence at the 5’ end (Figure 1A). The EC–NusA–NusG–Rho complexes assembled on these RNA scaffolds, PTC18 and PTC60, were stable enough to withstand size-exclusion chromatography and appeared highly homogeneous and monodispersed (Figures 1B and 1C), allowing us to use single-particle cryo-EM to determine their structures. The map of PTC60 reached a nominal resolution of 3.1 Å. In contrast, the “overall” map of PTC18 reached a nominal resolution of only 7.9 Å. Focused classification and refinement of the EC and Rho regions separately allowed us to generate a “composite” map of PTC18, in which the resolution of the EC density was improved to 4.0 Å while the Rho density remained at a resolution of 7.9 Å (Figures 2, 3, S1 – S4 and Table S2).
Figure 1. Formation and Isolation of the Pre-Termination Complexes (PTCs).
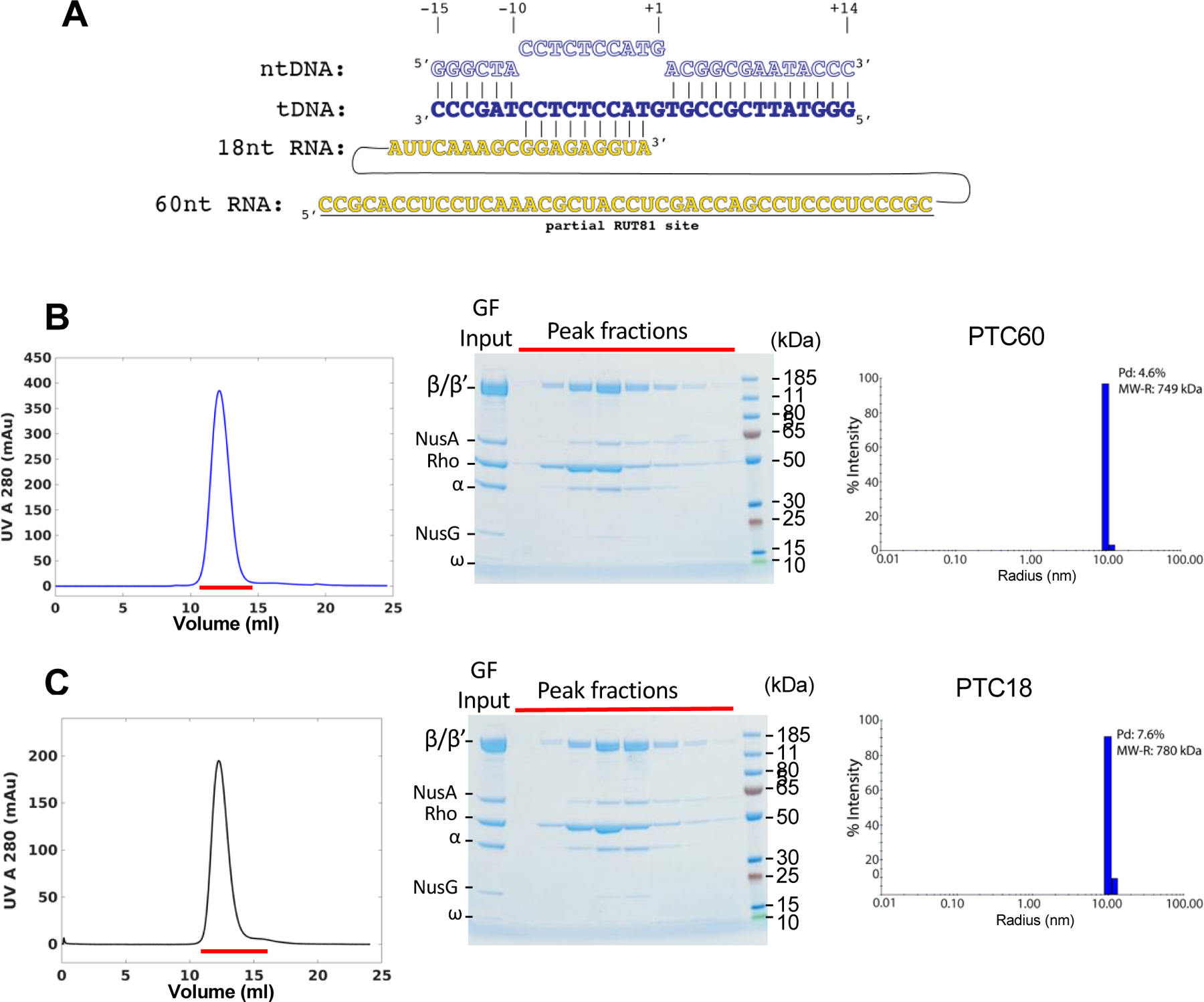
(A) Schematic of the nucleic acid scaffold used to assemble the tertiary elongation complexes (EC18 and EC60). ntDNA - nontemplate DNA strand; tDNA - template DNA strand; nt - nucleotide; partial RUT81 – the first 40 nucleotides of canonical 81-nucleotides Rho-utilization site.
(B) Size-exclusion chromatography (SEC) of PTC60 (left panel). Red line indicates the peak fractions that were analyzed by SDS-PAGE (middle panel). Right panel shows the dynamic light scattering (DLS) analysis of the peak fraction from the SEC purification of PTC60 (%Pd, the polydispersity statistics; MW-R, estimated molecular weight).
(C) SEC of PTC18 (left panel). Red line indicates the peak fractions that were analyzed by SDS-PAGE (middle panel). Right panel shows DLS of the peak fraction from the SEC purification of PTC18.
Figure 2. Overview of the Cryo-EM Structure of the PTC60.
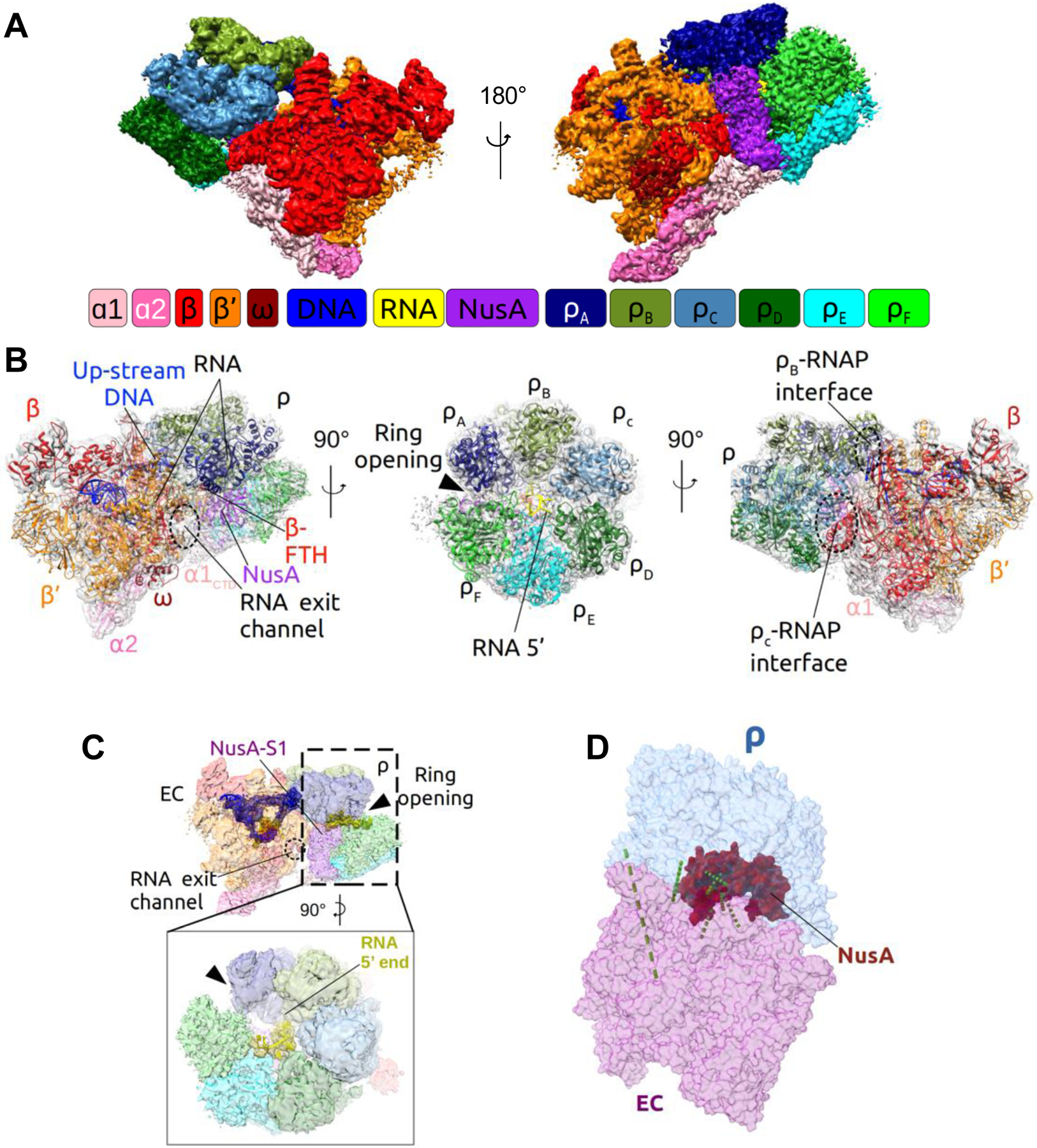
(A) Surface view of the cryo-EM map of PTC60 at 3.1-Å nominal resolution. The diagram represents the color coding that is maintained in all figures, unless otherwise indicated. RNAP core subunits: ɑ1 - light pink, ɑ2 - pink, β - red, β’ - orange, ω - dark red; Rho subunits: ρA - navy, ρB - drab, ρC - light blue, ρD - dark green, ρE - cyan, ρF - green; NusA - purple; DNA - blue, RNA - yellow.
(B) Structural model of PTC60 with the density map shown as transparent gray surface and individual components denoted and shown in ribbon representation.
(C) Cryo-EM map of PTC60 with the model of the nucleic acid scaffold. The densities for DNA and RNA are shown in blue and yellow mesh representation, respectively, and the remaining map as transparent surface.
(D) Model of PTC60 with identified in-vivo crosslinks between the different modules (see Table S1 for details). RNAP is shown in purple, Rho in light blue, NusA in red, and in-vivo crosslinks as green dashed lines.
Figure 3. Major Protein-protein Interactions of Rho in the PTC and Their Functional Validation.
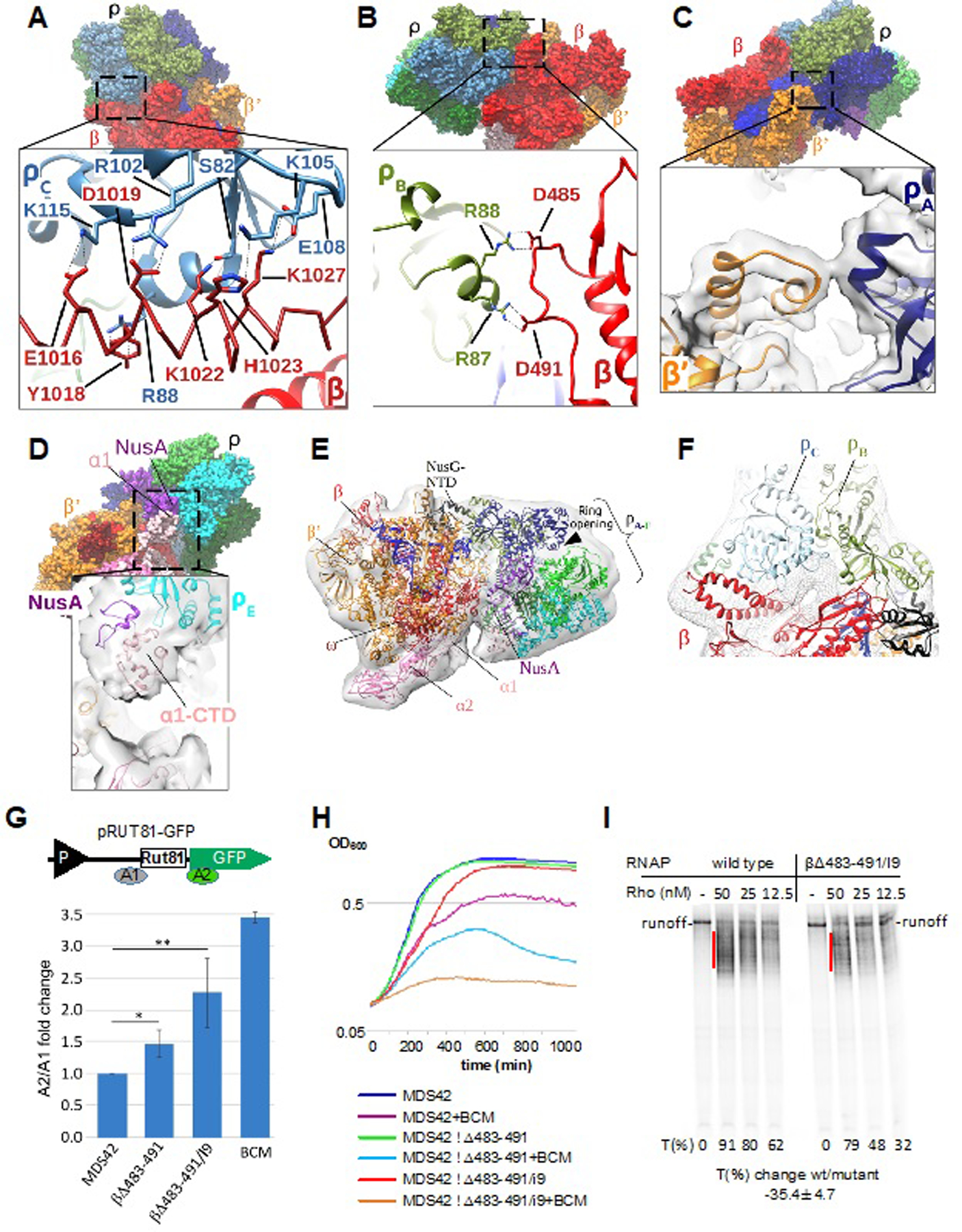
(A) Interaction of Rho subunit C (ρC) with residues of the RNAP β I9 domain in PTC60. Upper panel: overview of PTC60. Lower panel: magnified view of the boxed region. The RNAP β I9 domain is colored red with α-helix residues 1012–1038 shown as a Cα model, and ρC is colored light blue. Interacting residues are labeled and shown as sticks. Black dashed lines denote H-bonds, salt bridges and cation–π interactions.
(B) Interaction of Rho subunit B (ρB) with the RNAP β subunit (residues 483–491) in PTC60. Upper panel: overview of PTC60. Lower panel: magnified view of the boxed region. β is colored red and ρB is colored drab. Interacting residues are labeled and shown as sticks. Black dashed lines indicate H-bonds and salt bridges.
(C) Interaction of Rho subunit A (ρA) with RNAP β’ subunit (residues 283–287) in PTC60. Upper panel: overview of PTC60. Lower panel: magnified view of the boxed region. β’ is colored orange, ρA is colored navy, and the map is shown as transparent surface.
(D) Interactions of ρB with the RNAP α1-CTD in PTC60. Upper panel: overview of PTC60. Lower panel: magnified view of the boxed region. The map is shown as transparent surface.
(E) PTC18 overall map at 7.9-Å nominal resolution with fitted model. The map is shown as gray transparent surface. The PTC18 structure model is shown in ribbon representation, and DNA and RNA are shown as blue and yellow ladders, respectively.
(F) Interactions of ρB and ρC with RNAP β in PTC18. The β subunit is colored red, ρB and ρC are colored drab and light blue, respectively, and the map is shown as gray mesh surface. Red dash lines denote the inter-protein crosslinks βLys115–ρLys115 and βLys1027–ρLys105.
(G) Mutations of RNAP residues that interact with Rho in the PTC compromise Rho-dependent termination in vivo. Upper panel: Schematic diagram of the plasmid-based Rho-dependent termination reporter(Sedlyarova et al., 2017) used to test the effect of RNAP chromosomal mutations. P (black triangle) indicates the constitutive promoter, and Rut81 is the 81-nt long canonical Rho-utilization site. Colored bars A1 and A2 indicate the locations of qRT-PCR amplicons. Lower panel: fold changes in Rho-dependent termination for E. coli MDS42 strains containing different mutations in the RNAP β subunit. BCM is bicyclomycin (5 µg/ml), which was used as positive control. qRT-PCR data are shown in fold changes of the A2/A1 amplicon signal; data from three independent experiments are presented as the means ± SEM; **P < 0.01; *P < 0.05.
(H) Δβ483–491 and Δβ483–491/I9 are progressively more sensitive to bicyclomycin (BCM) than the parent MDS42 strain. Bioscreen C™-generated automated growth curves for MDS42 and mutant strains in LB with or without BCM (50 µg/ml) are shown.
(I) Δβ483–491/I9 RNAP is less susceptible to Rho-dependent termination in vitro. The radiogram shows a representative runoff assay used to assess the efficiency of Rho-dependent termination. The initial radiolabeled EC20 was chased in the absence or presence of the indicated amounts of Rho. The termination efficiency (%T) was estimated as the ratio between the signals within the termination zones (red lines) and the runoff. The average value of the termination change was calculated from three independent experiments using 12.5 nM Rho.
The PTC Is Held Together by Specific Protein–protein Interactions
The PTC60 map revealed defined protein–protein interactions between RNAP and Rho. Two major points of contact with Rho occur at the rear face of RNAP (with respect to the direction of transcription) (Figures 3A and 3B). Rho interacts with RNAP in a hexameric open-ring conformation, using at least two of its monomeric N-terminal domains (NTDs). The most prominent interface is established between the α-helical insertion I9 domain (residues 939–1038) of the RNAP β subunit (rpoB) and the basic shallow pocket formed by the α4 helix (residues 82–90), the loop region between β4-β5 and the β5 strand of Rho-ring subunit C (ρC) (Figure 3A). Well-defined interactions are the ionic and hydrogen bonds βGlu1016–ρCLys115, βAsp1019–ρCArg102, βLys1022–ρCSer82, and βLys1027–ρCGlu108, and the cation–π interactions βTyr1018–ρCArg88 and βHis1023–ρCLys105. Another important interaction occurs between a loop region of β (residues 483–491) and the α4 helix (residues 82–90) of ρB (Figure 3B). Here, βAsp485 makes contacts with ρBArg88, and βAsp491 interacts with ρBArg87. There may be two more interactions between RNAP and Rho, but density for these was more ambiguous. A further density linkage indicates an interaction between a loop region of the RNAP β’ subunit (rpoC) (residues 283–287) and helix α4 (residues 82–90) in ρA (Figure 3C). At a lower contour level, density extends from one of the RNAP α subunits toward ρE (Figure 3D). This density linkage may account for a previous finding that implicated the C-terminal domain (CTD) of RNAP α in Rho-dependent termination (Kainz and Gourse, 1998).
The RNAP module of PTC60 remains largely unchanged when compared to the previously reported cryo-EM structure of the E. coli EC (PDB: 6ALH) (Kang et al., 2017), with a root-mean-square deviation (RMSD) of 1.454 Å between 2,615 corresponding Cα atoms, except for some significant changes in a region that interacts with Rho and the flexible β’ I3 (β’948–1126) (Table S3, Figures S5A and S5B). It follows that the formation of PTC should not affect the basic properties of the EC, such as its stability and processivity, supporting previous biochemical results (Epshtein et al., 2010).
Traditionally, it has been assumed that Rho loading onto RNA precedes its interaction with RNAP. To unambiguously confirm that the PTC forms without any RNA tethering, we determined the structure of PTC18. The shape of the PTC18 overall map is very similar to that of the PTC60 map (Figure S2), demonstrating that Rho interacts with the EC without Rho interacting with RNA. However, the lower resolution of the PTC18 map suggests higher structural variability in this PTC. We therefore used multibody refinement to analyze the heterogeneity in the PTC18 and PTC60 datasets. Movies illustrating the variability in the particle populations as motions show similar motions for both PTCs (Movies S1 and S2), indicating that the structural variability is not due to differences in how Rho interacts with the EC. Focused classification improved the resolution of the EC density in the PTC18 map to 4.0 Å, but did not improve the density for Rho, indicating that most of the structural variability in PTC18 derives from heterogeneity in the Rho hexamer. Since the Rho density reached much higher resolution in the PTC60 map, interactions of Rho with RNA appear to greatly stabilize the conformation of the hexamer.
By placing atomic models of the components into the PTC18 overall map and then refining them against the composite map, we were able to create a model for PTC18 that allowed us to assess the interactions between the components (Figure 3E). The map clearly shows the protein–protein interfaces between Rho and the EC, with Rho again binding at the rear face of RNAP. Importantly, the same points of contact between RNAP and the Rho ring observed in PTC60 are also seen in PTC18 (Figure 3F), confirming that the PTC is held together primarily by protein–protein interactions.
To further map the interacting surfaces between Rho and the EC, and to validate the PTC18 model, we used lysine-specific crosslinking coupled with mass spectrometry (XLMS). The high-abundance crosslinking sites confirm the PTC18 model (Table S4 and Figure 4A). The structure of PTC18, in which the RNA scaffold is short and lacks the Rho-binding RUT sequence, implies that the interactions between Rho and RNAP are likely established very early during elongation and even before the nascent RNA could have reached the Rho ring.
Figure 4. Dynamic Interactions of NusG in the PTC.
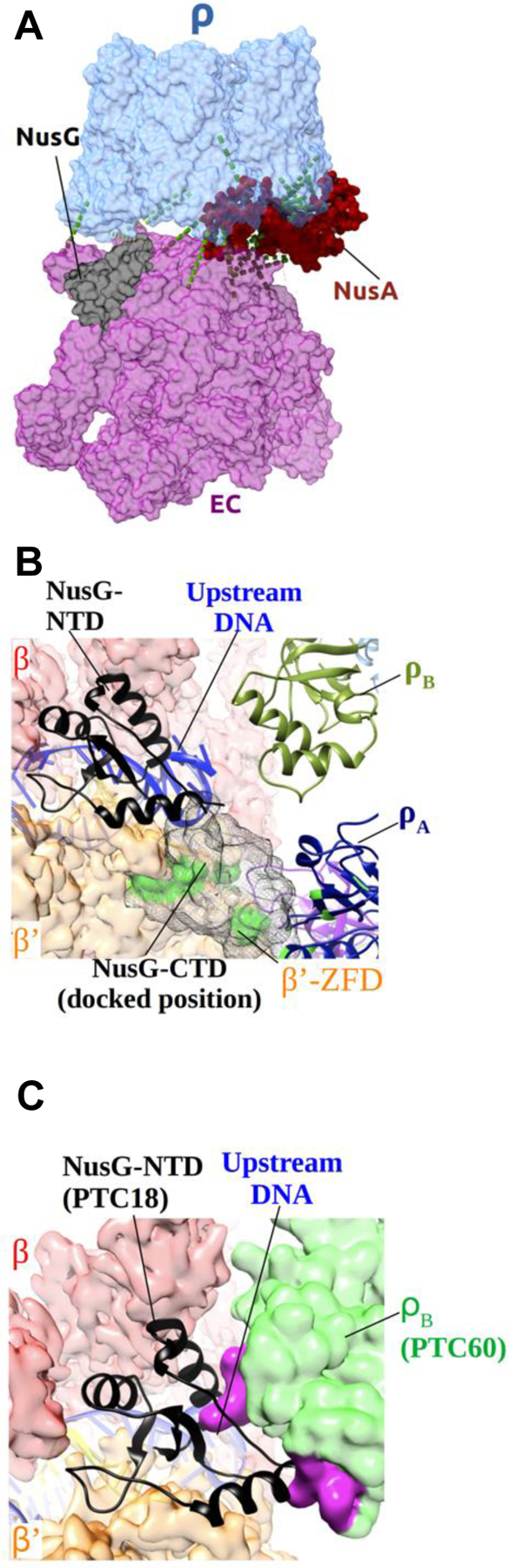
(A) Identified in-vitro crosslinks between PTC18 modules (see Table S4 for details). RNAP is colored purple; Rho is colored light blue; NusA is colored red; NusG is colored black. Green dashed lines represent the most abundant in-vitro crosslinks.
(B) Close-up view of NusG-NTD in PTC18. RNAP β and β’ subunits are shown as transparent red and orange surfaces, respectively. NusG-NTD is shown in black cartoon representation. ρB and ρA are shown as drab and navy ribbon diagrams, respectively. NusG-CTD, docked in a position consistent with in-vitro crosslinking sites (green spots), is shown as dark gray mesh surfaces.
(C) Close-up view of NusG-NTD and the upstream DNA in PTC18, also showing the modeled position of ρB of PTC60. The NusG-restrained upstream DNA is not compatible with the position of ρB in PTC60 as it would cause steric clashes. Rho subunit B in the position it adopts in PTC60 is shown as green transparent surface, and the residues that would cause steric clashes as solid magenta surface. The RNAP β subunit is shown as red transparent surface, the β’ subunit as orange transparent surface, NusG-NTD in black cartoon representation, and the upstream DNA in blue cartoon representation.
RNAP–Rho Interactions in the PTC Are Important for Termination
Based on our structural findings, we designed two E. coli chromosomal rpoB deletion mutants that should progressively weaken the Rho–RNAP interactions without significantly affecting the basic function of RNAP: Δβ483–491 and a double deletion of the β483–491 and I9 (β939–1038) regions (Δβ483–491/I9). To minimize distortion of the RNAP structure, we replaced the deleted residues with glycine linkers. To construct these mutant cells, we used E. coli strain MDS42, which can tolerate severely compromised Rho-dependent termination (Cardinale et al., 2008). To evaluate the effect of these mutations, we measured Rho-dependent termination efficiencies in vivo using a plasmid-based system we established previously (Sedlyarova et al., 2016) (Figure 3G, upper panel). We hypothesized that PTC formation is crucial for Rho-dependent termination (Epshtein et al., 2010), so when Rho–RNAP interactions are weakened, the efficiency of termination should be reduced. Two pairs of primers were designed to amplify mRNA regions flanking the canonical Rho-dependent terminator, RUT81 (Ciampi, 2006; Sedlyarova et al., 2017), which was under the control of a constitutive promoter. The quantitative RT-PCR signal from the segment downstream to the termination site normalized to the upstream signal increased, respectively, by approximately 1.5- and 2-fold in the single- and double-deletion mutant cells, when compared to wild-type cells (Figure 3G, lower panel). The Δβ483–491/I9 double deletion showed an even stronger anti-termination effect of ~ 2.5-fold (Figure S6) for the native chromosomal Rho/NusG-dependent terminator downstream of the dsbB ORF (Dar and Sorek, 2018). Accordingly, MDS42 Δβ483–491 and MDS42 Δβ483–491/I9 cells became progressively more sensitive to BCM (Figure 3H). Indeed, as Δβ483–491/I9 and BCM acted on different steps in the Rho-mediated termination mechanism, the former weakens the interactions in the PTC, whereas the latter inhibits Rho catalytic activity, the effects from the mutants and the antibiotic are expected to be additive/synergistic, and not epistatic. The purified Δβ483–491/I9 mutant enzyme also exhibited compromised Rho-dependent termination in a single-round transcriptional assay (Figure 3I). These results demonstrate that Rho–RNAP interactions in the PTC are crucial for Rho-dependent termination.
NusG Interactions in the PTC
The PTC18 overall and composite maps showed clear density for the NusG-NTD (Figures 3E and S4C). The NusG-NTD binds to the upstream face of the EC cleft. The location and orientation are consistent with those in a cryo-EM structure of the E. coli EC–NusG complex (Kang et al., 2018) (Figure S5C). The NusG-NTD bridges across the β and β’ subunits on top of the upstream duplex DNA (Figure 4B). The density for the NusG-CTD was ambiguous. However, we identified several inter-protein crosslinks between the NusG-CTD and RNAP, and the NusG-CTD and Rho (Table S4 and Figure 4B), which indicate that the NusG-CTD extends toward the zinc-finger domain (ZFD) of RNAP β’ and interacts with at least one of the six Rho subunits. This NusG-mediated dual bridging of Rho and RNAP should further stabilize the early-stage PTC and facilitate its formation in vivo. It also rationalizes previous observations that the NusG-CTD mediates Rho interaction with EC (Li et al., 1993; Mooney et al., 2009). Notably, despite its better resolution, the PTC60 map did not resolve NusG. We argue that this is due to a conformational change of Rho in PTC60: as the NusG-NTD constrains the path of the upstream duplex DNA, the upstream DNA would clash with the location of one of the Rho monomers in PTC60 (Figure 4C). Thus, when the nascent transcript is loaded onto the Rho ring, NusG is pushed off the upstream DNA and either becomes more mobile in the complex or is released completely.
Recent structural studies of the binary Rho–NusG-CTD complex proposed that the principal role of NusG is to stimulate Rho-ring closure (Lawson et al., 2018). However, Rho remains an open ring in the PTC in the presence of NusG (Figure 3E and S2B). Moreover, the relative orientation of Rho and NusG seen in the PTC (Figure 2B and 3E) rules out the possibility that the NusG-CTD could reach Rho from its CTD side. Indeed, we do not detect a NusG-CTD–Rho-CTD interaction in the PTC by XLMS (Tables S1 and S4) in vivo and in vitro. We thus conclude that the main role of NusG is to stabilize Rho–RNAP interactions in the PTC and, possibly, also to help RNAP to adopt the non-backtracked conformation (Bar-Nahum et al., 2005; Turtola and Belogurov, 2016) that is compatible with Rho-dependent termination (Dutta et al., 2008).
NusA Interactions in the PTC
We observed clear density for the first 200 residues of NusA in the PTC60 map (Figures 2A and S4B); the more flexible NusA-CTD was not resolved. The NusA-NTD interacts with Rho and RNAP (Figure 5A), thereby providing an additional anchor for Rho in the PTC. In the PTC60 map, we observe density for the mobile flap tip helix (FTH) of RNAP β interacting with an α-helix bundle formed by NusA α-helix 1 (residues 1–10), α-helix 2 (residues 19–39) and α-helix 4 (residues 103–132) (Figure 5B). This observation is consistent with previous findings describing NusA-NTD–RNAP interactions (Guo et al., 2018; Krupp et al., 2019; Yang et al., 2009; You et al., 2019). On the other hand, NusA also uses a loop-helix region (residues 77–96) to interact with ρE (Figure 5C). This dual interaction of the NusA-NTD should further stabilize the PTC.
Figure 5. Principle Interactions of NusA in the PTC.
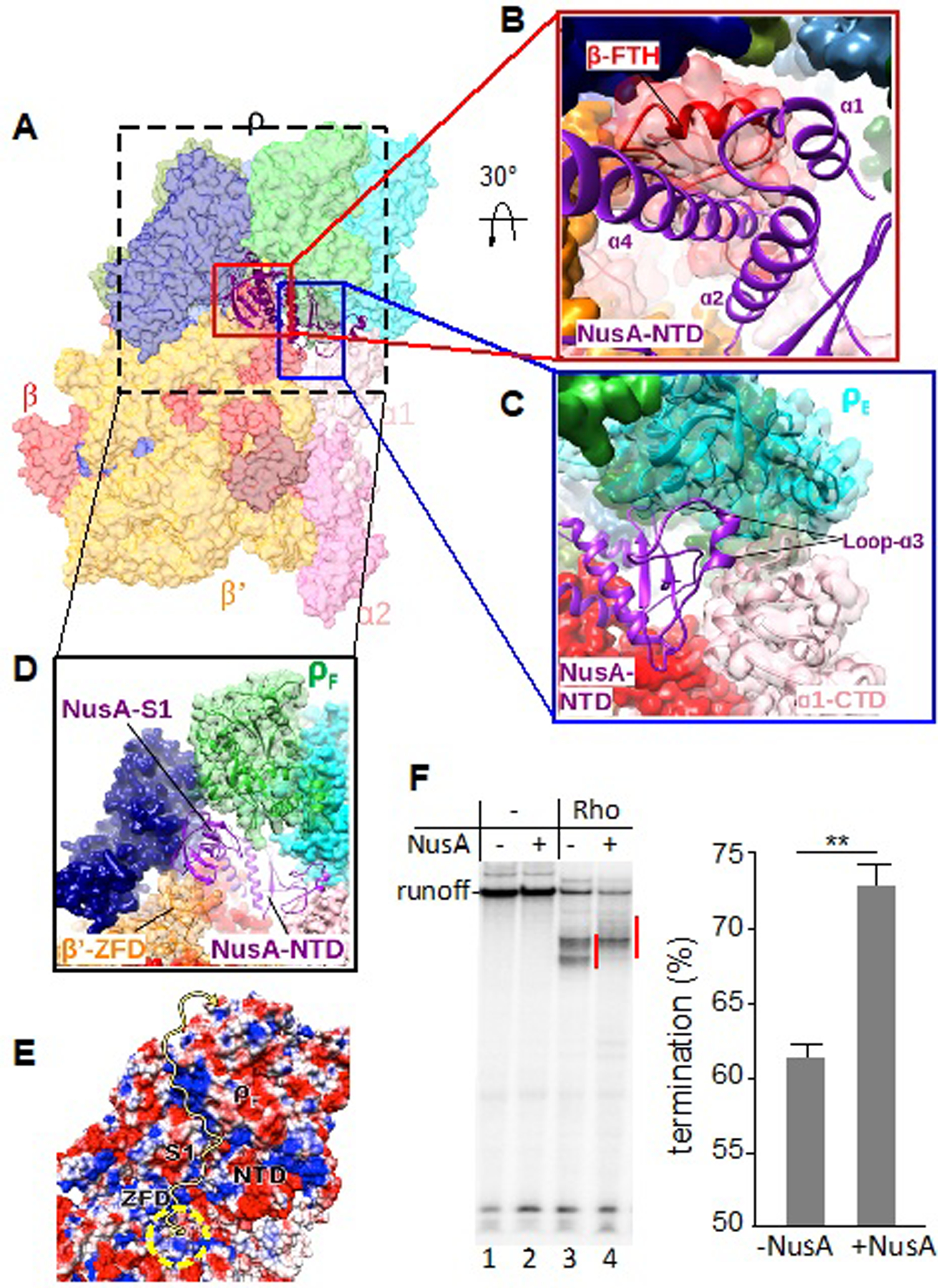
(A) Overview of PTC60 focusing on NusA interactions. The PTC60 model is color-coded as in Fig. 1 and shown in transparent surface representation, except for NusA, which is shown in ribbon representation.
(B) Close-up view of the interaction of NusA-NTD (purple) with RNAP β-FTH (red). RNAP β is shown both as transparent surface and cartoon representation to show secondary-structure elements (red). The secondary-structure elements of NusA-NTD that interact with β-FTH are labeled.
(C) Close-up view of the interaction of NusA-NTD (purple) with ρE (cyan) and ɑ1-CTD (light pink). ρE is shown both as transparent surface and cartoon representation to show secondary-structure elements. The interacting region of NusA-NTD is shown in cartoon representation and α1-CTD as transparent surface and cartoon representation in light pink.
(D) Close-up view of the interaction of the NusA S1 domain (purple) with the RNAP β’ zinc-finger domain (β’-ZFD) (orange) and ρF (green). β’ and ρF are shown as transparent surface and cartoon representation to show secondary-structure elements. e, Electrostatic potential of the surface formed by NusA S1 and NTD, β’-ZFD and ρF. The yellow arrow indicates the possible path for the nascent transcript. The yellow dashed circle indicates the RNA-exit channel of RNAP.
(F) Effect of NusA on Rho-dependent termination in vitro. Left panel: representative runoff assay used to assess the location and efficiency of Rho-dependent termination sites. The initial radiolabeled EC20 was chased in the absence (lanes 1, 2) or presence (lanes 3, 4) of Rho and/or NusA. Termination efficiency was estimated as the ratio between the signals within the termination zones (marked by red lines) and runoff. Right panel: NusA stimulates Rho-dependent termination in vitro. Data from three independent experiments are presented as the means ± SEM; **P < 0.01.
The low-resolution density map revealed extra density near the N terminus of NusA (residues 63–93) that fits one copy of the RNAP α-CTD (Figure 5C), supporting previous findings that suggested an interaction between the RNAP α-CTD and the NusA-NTD (Guo et al., 2018; Liu et al., 1996; Schweimer et al., 2011). In addition, the density for the NusA S1 domain was evident next to the β’-ZFD (Figure 5D). Here, the S1 domain of NusA bridges the RNA exit channel with ρF, essentially serving as an extension of the RNA exit channel. Together with the β’-ZFD and ρF, it forms a positively charged path for the nascent RNA (Figure 5E), directing the transcript into the central channel of the Rho ring.
Rho-dependent Termination with RUT Provided in trans
PTC formation implies that Rho does not need to load onto RUT for the purpose of “catching up” to the moving EC (Rho has been already pre-bound to RNAP). Therefore, we reasoned that RUT could activate Rho and promote termination of the PTC regardless of whether it was naturally tethered to the complex in cis (cisRUT) or provided in trans (transRUT). Indeed, adding synthesized “canonical” transRUT to PTCs stalled at positions +29 or +44 resulted in robust termination (Figure 6A). Control RNA of a similar size added in trans (Figure 6B, lane 5), or transRUT without Rho (Figure S7), did not induce termination. As in the case of cisRUT, NusG strongly stimulated transRUT-mediated termination (Figure 6C, lanes 7, 8), indicating that the latter process reflects the natural mechanism of Rho-dependent termination. PTCs stalled at positions +18 or +22 were resistant to transRUT-mediated termination (Figure 6A), probably because the nascent RNA in these complexes was too short to be reached by Rho. Remarkably, termination occurred with PTC29, even though most of its nascent RNA remained inside RNAP, forming the 9-bp RNA:DNA hybrid (Nudler et al., 1997) in the main channel and occupying ~10 nt of the RNA-exit channel (Kang et al., 2018; Kang et al., 2017). To reach this RNA, Rho must obtain access to the RNA-exit channel, as predicted by the allosteric model (Epshtein et al., 2010). Moreover, for such a short RNA to enter the central channel of Rho from its CTD side, which is opposite to that forming the interface with RNAP and RUT (Figures 6B and 6C), Rho has to eventually turn and/or invade the RNAP. This transition likely occurs after the allosteric change in RNAP that leads to its inactivation and the destabilization of the PTC (Epshtein et al., 2010).
Figure 6. Rho-mediated Termination with RUT Acting in trans.
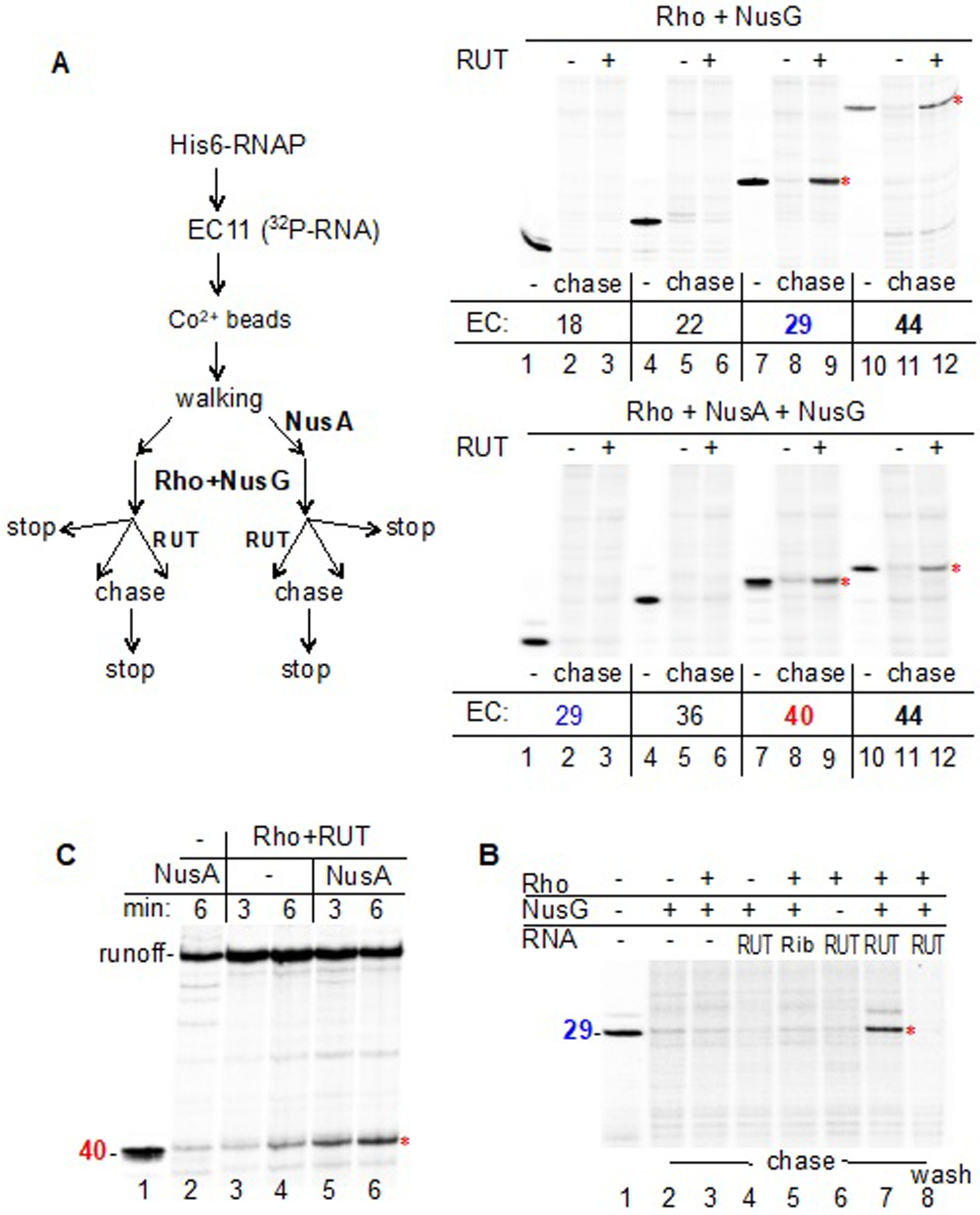
(A) Termination of promoter-proximal complexes by Rho and transRUT. The schematic (left panel) depicts the general workflow used for the experiments (right panels). The initial EC11 immobilized on Co2+ beads was “walked” to the indicated positions (from +18 to +44), followed by the addition of NusA (lower panel), NusG, and Rho. Each reaction mixture was then divided into three equal parts. One aliquot was mixed with the stoppage buffer (lanes 1, 4, 7, 10); a second aliquot was mixed with NTPs (chase) (lanes 2, 5, 8, 11); and a third aliquot was mixed with RUT RNA for 2 min followed by the addition of NTPs (chase) (lanes 3, 6, 9, 12). The earliest PTCs to be terminated without and with NusA, namely those containing EC29 and EC40, respectively, are marked in blue and red.
(B) EC29 is susceptible to termination by Rho+NusG+transRUT. Robust termination can be detected in the presence of NusG and transRUT (lane 7). An unrelated RNA of similar size (TPP riboswitch – “Rib”) does not support termination (lane 5). After completion of the chase reaction, the beads were washed to remove the dissociated RNA products (lane 8), confirming the termination event.
(C) NusA promotes termination. Rho+transRUT terminates transcription by EC40 more efficiently in the presence of NusA (lanes 5,6) than in the absence of NusA (lanes 3,4).
Role of NusA in Rho-dependent Termination
Genetic and genomic studies indicate that NusA, like NusG, facilitates Rho-dependent termination in vivo (Cardinale et al., 2008; Saxena and Gowrishankar, 2011; Ward and Gottesman, 1981), although one recent study argues to the contrary (Qayyum et al., 2016).
The structure of the PTC reveals specific interactions between NusA and Rho that contribute to PTC stability (Figure 5), supporting earlier biochemical evidence (Schmidt and Chamberlin, 1984). NusA serves as an extension of the RNA-exit channel in the PTC (Figure 5D); the nascent RNA passes along the RNA-binding domains of NusA prior to contacting Rho. This explains why NusA shifts the termination window downstream in the runoff assay (Figure 5F), and shifts the onset of Rho-dependent termination in the transRUT assay (Figure 6A); without NusA, PTC29 readily terminates with Rho+transRUT (Figure 6A, top panel), but remains resistant to termination in the presence of NusA (bottom panel). When nascent RNA reaches a length of 40 nt, the PTC+NusA complex becomes susceptible to termination by Rho+transRUT (bottom panel). Notably, this shift in the onset of termination does not imply that NusA compromises Rho activity. On the contrary, the stimulating effect of NusA on Rho-dependent termination becomes apparent in the absence of NusG (Figure 6C); NusG masks this effect as it, itself, promotes termination (Figure 6B). Likewise, NusA stimulates Rho-dependent termination in a conventional runoff assay, as measured by the ratio between termination and readthrough bands, even though it shifts the bulk of the termination zone downstream (Figure 5F) (Burns et al., 1998).
These biochemical and structural data support the earlier in-vivo findings that established NusA as a cofactor of Rho (Cardinale et al., 2008; Saxena and Gowrishankar, 2011) that, along with NusG, stabilizes Rho–RNAP interactions in the PTC, thereby promoting termination.
Revised Model of Rho-dependent Termination
The prevailing “textbook” model of Rho-dependent termination postulates the following sequence of events leading to EC dissociation: (i) Rho loads onto the ribosome-free RUT, (ii) translocates along RNA in pursuit of the moving EC, (iii) catches up with the paused EC and pulls RNA from it, or “pushes” it forward, thereby causing termination (Jocelyn E. Krebs, 2018). None of these steps implies an active role of RNAP in the termination process. Here, we provide structural, biochemical, and genetic data supporting the alternative model (Epshtein et al., 2010). In contrast to the traditional model, the process begins with the direct binding of Rho to RNAP, NusA and NusG, prior to contacting the nascent RNA. Such a pre-termination complex (PTC) serves two functions: it positions Rho in a proper orientation relative to the RNA-exit channel (Figure 7A), and it elevates the local concentration of Rho, allowing it to sample the growing transcript via linear scanning using its Q and R loop regions in its central channel (Figures 7B and 7C). Molecular crowding, a vast number of potential RNA decoys, and numerous RNA-binding proteins should confound the 3D diffusional search for the loading site by Rho in vivo. Consistently, the mutations in RNAP predicted by the PTC structure to weaken Rho–RNAP interaction compromise termination in vivo and in vitro (Figures 3G and 3I). Likewise, NusA and NusG, which stabilize Rho–RNAP interactions in the PTC (Figures 4 and 5), promote termination in vitro (Figures 5F and 6B) (Burns et al., 1998; Kainz and Gourse, 1998) and in vivo (Cardinale et al., 2008; Saxena and Gowrishankar, 2011).
Figure 7. The Allosteric Model of Rho-dependent Transcription Termination.
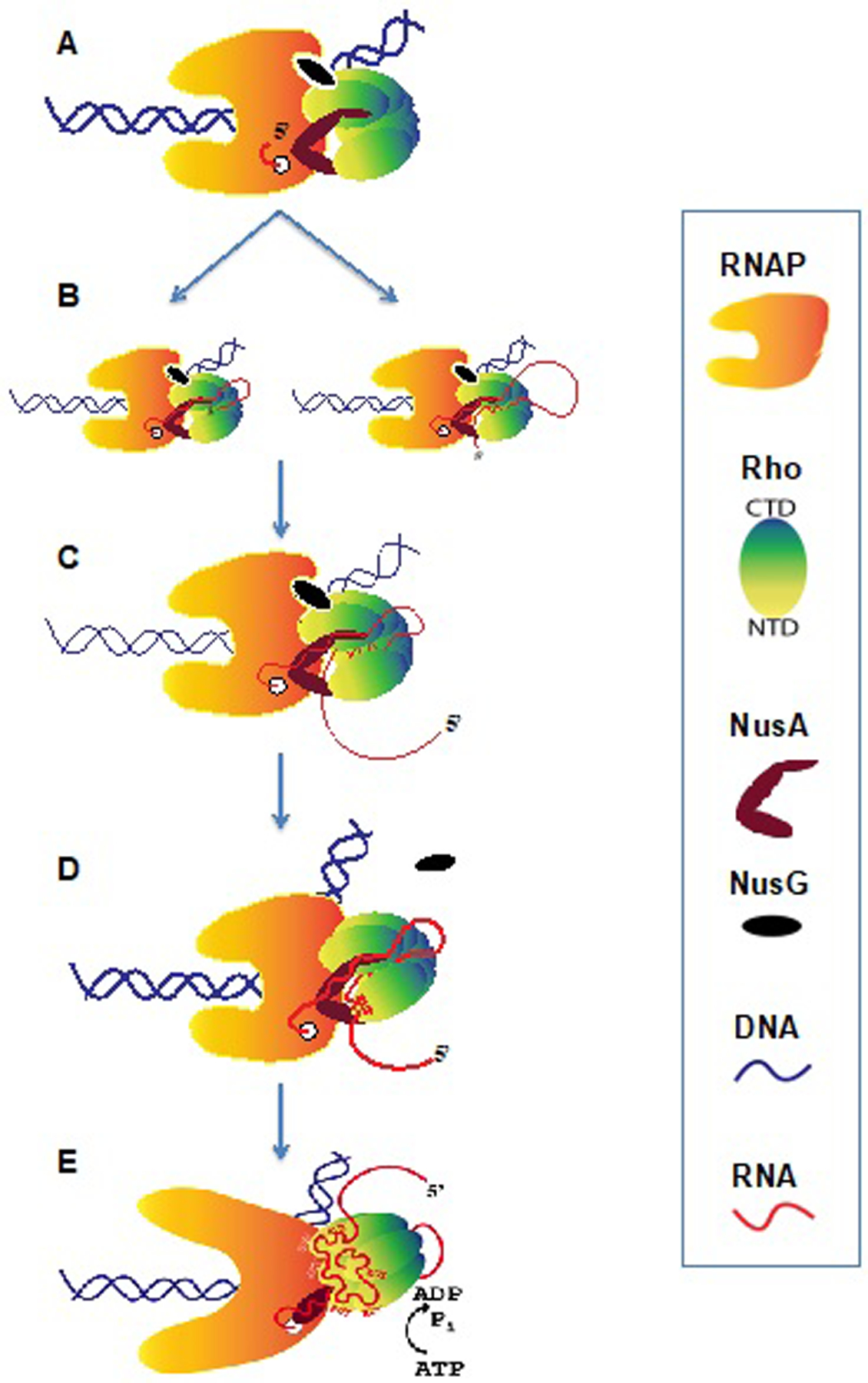
(A) A stable PTC forms at an early stage of transcription elongation with the direct binding of Rho to RNAP, NusA and NusG. These interactions do not require RNA.
(B) Nascent RNA is directed by NusA and loaded into the central channel of Rho from the distal C-terminal face either by lateral loading (via looping) or 5’ terminal entry.
(C) Rho samples the RNA sequence during transcription elongation.
(D) When RUT RNA emerges, the activation of Rho begins. RUT is loaded onto the Rho-NTDs. The PTC undergoes a conformation change causing NusG displacement to accommodate the movement of the upstream DNA that is induced by shifting Rho. The PTC is prone to termination at this stage.
(E) The complete RUT RNA (in cis or in trans) activates Rho, which allosterically inactivates and destabilizes the EC without pulling RNA or pushing RNAP forward(Epshtein et al., 2010). The PTC has transitioned to the termination complex.
Based on structural models of Rho–RNA binary complexes (Skordalakes and Berger, 2003; Thomsen and Berger, 2009), it has been proposed that Rho-NTD loads onto RUT first, followed by RNA interaction with the P-, Q- and R- loops of Rho-CTD in the central channel, eventually triggering the Rho-ring closure and ATP-driven Rho-translocase activity. However, this order of RNA-loading steps cannot explain transRUT-mediated termination (Figure 6A). Indeed, the nascent transcripts in EC29 (without NusA) or EC40 (with NusA) appear to be too short to sequentially engage with Rho-NTDs and then with the Rho central channel. Our observation that RUT RNA can localize in the Rho channel prior to interacting with Rho-NTDs (Figure 2C) indicates that the nascent RNA emerging from RNAP first occupies the “secondary” binding site by entering the Rho ring from its C-terminal side. While RNA continues threading through the central channel, the emerging RUT will be presented to the “primary” binding sites located in the Rho-NTDs (Figure 7D). Once the Rho-NTDs are fully loaded with RUT RNA, it may trigger ring closure. Note that this “reverse” order of RNA loading does not contradict previous structural models of Rho–RNA binary complexes, only their interpretations.
Rho has recently been established as a global regulator of gene expression in bacteria (Sedlyarova et al., 2016). Therefore, the PTC concept described here have important implications for gene regulation. Various factors, including ribosomes, are likely to control Rho-dependent termination by affecting protein–protein interactions between Rho and the EC, as well as influencing the conformational state of RNAP and its responsiveness to Rho. The present work provides the framework for understanding these regulatory mechanisms.
Formation of a persistent PTC implies that Rho can function not only as a dedicated termination factor, but also as a general elongation factor. Indeed, we previously showed that catalytically inactive Rho strongly influences transcriptional pausing in vitro (Epshtein et al., 2010). The PTC structure suggests that Rho can modulate pausing by acting as both an allosteric effector and RNA chaperon, akin to NusG and NusA.
The protein scaffold holding the EC together is conserved among all multi-subunit RNAPs, arguing that the structural principles of transcription termination revealed here for bacteria should be applicable to eukaryotic systems as well.
Limitations
Although our results demonstrate that the functionally important interactions between RNAP and Rho occur prior to Rho loading onto RNA, they do not rule out the possibility of additional pathways that may occur in vivo.
STAR*Methods
Resource Availability
Lead Contact
Further information and requests for reagents and resources should be directed to and will be fulfilled by the Lead Contact, Dr. Evgeny Nudler (evgeny.nudler@nyulangone.org).
Materials Availability
Plasmids generated in this study are available upon request from the Lead Contact with a completed Material Transfer Agreement.
Data and Code Availability
The cryo-EM maps included in this study have been deposited in the Electron Microscopy Data Bank with accession codes: EMD-22114 and EMD-22115. The atomic coordinates have been deposited in the Protein Data Bank with accession codes: 6XAS and 6XAV.
Method Details
Protein Expression and Purification
To purify wild-type E. coli RNAP, E. coli strain BL21 (DE3) was transformed with pVS10 (Svetlov and Artsimovitch, 2015), and recombinant protein expression was auto-induced (Studier, 2005). After 16 h at 30°C, cells were harvested by centrifugation (4,000g for 10 min at room temperature) and pellets were stored at −80°C. Cell pellets were resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol, 0.5 mM β-mercaptoethanol, 1M NaCl) supplemented with complete, EDTA-free protease inhibitor cocktail tablets (Roche Applied Science) and lysed using sonication (5-s pulses with 10-s intervals for 10 min on ice). The cell lysate was clarified by centrifugation (30,000g for 40 min at 4°C) to remove insoluble debris. The supernatant was applied to a HisTrap column (GE Healthcare) equilibrated in HisTrap Buffer A (50 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol, 0.5 mM β-mercaptoethanol, 500 mM NaCl, 10 mM imidazole). The column was washed with 20 column volumes (CV) of HisTrap Buffer A. Protein was eluted with HisTrap Buffer B (50 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol, 0.5 mM β-mercaptoethanol, 250 mM NaCl, 250 mM imidazole). Fractions containing recombinant RNAP eluted from the HisTrap column were diluted 5 times in Hep A buffer (50 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol, 20 mM NaCl) and applied to a Heparin column (GE Healthcare) equilibrated in Hep A buffer. Protein was eluted using a linear NaCl gradient (0.02 to 1.5 M NaCl) in Hep B buffer (50 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol). The RNAP-containing peak fractions were pooled, diluted 4 times in Hep A buffer and applied to a Mono Q column (GE Healthcare). RNAP was eluted using a linear NaCl gradient (0.15 to 0.5 M) in Hep B buffer. Finally, RNAP was purified over a Superose 6 Increase size-exclusion chromatography (SEC) column (GE Healthcare) that was equilibrated in 20 mM Tris-HCl, pH 7.5, 2 mM MgCl2, 150 mM KCl, 1 mM dithiothreitol, and the RNAP-containing peak fractions were collected, flash-frozen in liquid nitrogen and stored at −80°C. RNAP mutants were purified using the same protocol.
The open reading frame of the full-length E. coli Rho protein was cloned into the pET21b vector. The plasmid was used to transform E. coli strain BL21 (DE3), and recombinant protein expression was auto-induced. After 16 h at 30°C, cells were harvested by centrifugation (4,000g for 10 min at room temperature) and pellets were stored at −80°C. Cell pellets were resuspended in lysis buffer (50 mM Tris-HCl, pH 7.5, 10% (v/v) glycerol, 50 mM KCl) supplemented with complete, EDTA-free protease inhibitor cocktail tablets (Roche Applied Science) and lysed using sonication (5-s pulses with 10-s intervals for 10 min on ice). The cell lysate was clarified by centrifugation (30,000g for 40 min at 4°C) to remove insoluble debris. The supernatant was applied to an SP column (GE Healthcare) equilibrated in Buffer A (50 mM Tris-HCl, pH 7.5, 10% (v/v) glycerol, 50 mM KCl). The column was washed with 3 CV of Buffer A. Protein was eluted with Buffer B (50 mM Tris-HCl, pH 8.0, 10% (v/v) glycerol, 1 M KCl). Fractions containing recombinant Rho protein were pooled and diluted 5 times with Hep A buffer and applied to a Mono Q column (GE Healthcare) equilibrated in Hep A buffer. Protein was eluted with a linear NaCl gradient (0.02 to 0.5 M) in Hep B buffer. Finally, Rho protein was purified over a Superose 6 Increase SEC column (GE Healthcare) that was equilibrated in 10 mM Tris-HCl, pH 8.0, 300 mM NaCl, 1 mM dithiothreitol, and the peak fractions containing Rho protein were collected, flash frozen in liquid nitrogen and stored at −80°C.
The open reading frame of the full-length E. coli NusA protein was cloned into the pSUMO vector, which has 6XHis tag conjugated with a N-terminal SUMO tag. The plasmid was used to transform E. coli strain BL21 (DE3), and recombinant 6XHis-SUMO-NusA was expressed as soluble protein by inducing with 0.5 mM Isopropyl β-D-thiogalactoside (Sigma) for 3 h at 37°C upon the culture reaching OD600=0.4–0.6. Cells were harvested by centrifugation (4,000g for 10 min at room temperature) and pellets were stored at −80°C. Cell pellets were resuspended in lysis buffer (50 mM Tris-HCl, pH 7.5, 10% (v/v) glycerol, 50 mM KCl) supplemented with complete, EDTA-free protease inhibitor cocktail tablets (Roche Applied Science) and lysed using sonication (5-s pulses with 10-s intervals for 10 min on ice). The cell lysate was clarified by centrifugation (30,000g for 40 min at 4°C) to remove insoluble debris. The supernatant was applied to a HisTrap column (GE Healthcare) equilibrated in HisTrap Buffer A (50 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol, 0.5 mM β-mercaptoethanol, 500 mM NaCl, 10 mM imidazole). The column was washed with 20 column volumes (CV) of HisTrap Buffer A. Protein was eluted with HisTrap Buffer B (50 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol, 0.5 mM β-mercaptoethanol, 250 mM NaCl, 250 mM imidazole). Fractions containing recombinant 6XHis-SUMO-NusA eluted from the HisTrap column were subjected to 6XHis-SUMO tag cleavage using SUMO protease (Invitrogen) in dialysis buffer (50 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol, 0.5 mM β-mercaptoethanol, 200 mM NaCl) at 4°C for 16 h. After 6XHis-SUMO tag cleavage, protein mixture was applied to a HisTrap column (GE Healthcare) equilibrated in HisTrap Buffer A (50 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol, 0.5 mM β-mercaptoethanol, 500 mM NaCl, 10 mM imidazole). Flow-through containing non-tagged NusA was pooled and applied to a Heparin column (GE Healthcare) equilibrated in Hep A buffer. Protein was eluted using a linear NaCl gradient (0.02 to 1.5 M NaCl) in Hep B buffer (50 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol). The NusA-containing peak fractions were pooled, diluted 4 times in Hep A buffer and applied to a Mono Q column (GE Healthcare). NusA was eluted using a linear NaCl gradient (0.15 to 0.5 M) in Hep B buffer. Finally, NusA was purified over a Superose 6 Increase size-exclusion chromatography (SEC) column (GE Healthcare) that was equilibrated in 20 mM Tris-HCl, pH 7.5, 2 mM MgCl2, 150 mM KCl, 1 mM dithiothreitol, and the NusA-containing peak fractions were collected, flash-frozen in liquid nitrogen and stored at −80°C.
The open reading frame of the full-length E. coli NusG protein was cloned into the pSUMO vector. The plasmid was used to transform E. coli strain BL21 (DE3), and recombinant protein expression was auto-induced. After 16 h at 30°C, cells were harvested by centrifugation (4,000g for 10 min at room temperature) and pellets were stored at −80°C. Cell pellets were resuspended in lysis buffer (50 mM Tris-HCl, pH 7.5, 10% (v/v) glycerol, 50 mM KCl) supplemented with complete, EDTA-free protease inhibitor cocktail tablets (Roche Applied Science) and lysed using sonication (5-s pulses with 10-s intervals for 10 min on ice). The cell lysate was clarified by centrifugation (30,000g for 40 min at 4°C) to remove insoluble debris. The supernatant was applied to a HisTrap column (GE Healthcare) equilibrated in HisTrap Buffer A (50 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol, 0.5 mM β-mercaptoethanol, 500 mM NaCl, 10 mM imidazole). The column was washed with 20 column volumes (CV) of HisTrap Buffer A. Protein was eluted with HisTrap Buffer B (50 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol, 0.5 mM β-mercaptoethanol, 250 mM NaCl, 250 mM imidazole). Fractions containing recombinant 6XHis-SUMO-NusG eluted from the HisTrap column were subjected to 6XHis-SUMO tag cleavage using SUMO protease (Invitrogen) in dialysis buffer (50 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol, 0.5 mM β-mercaptoethanol, 200 mM NaCl) at 4°C for 16 h. After 6XHis-SUMO tag cleavage, protein mixture was applied to a HisTrap column (GE Healthcare) equilibrated in HisTrap Buffer A (50 mM Tris-HCl, pH 8.0, 5% (v/v) glycerol, 0.5 mM β-mercaptoethanol, 500 mM NaCl, 10 mM imidazole). Flow-through containing non-tagged NusG was pooled, diluted 4 times in Hep A buffer and applied to a Mono Q column (GE Healthcare). NusG was eluted using a linear NaCl gradient (0.15 to 0.5 M) in Hep B buffer. Finally, NusG was purified over a Superose 6 Increase size-exclusion chromatography (SEC) column (GE Healthcare) that was equilibrated in 20 mM Tris-HCl, pH 7.5, 2 mM MgCl2, 150 mM KCl, 1 mM dithiothreitol, and the NusG-containing peak fractions were collected, flash-frozen in liquid nitrogen and stored at −80°C.
Nucleic-acid Scaffold Preparation
Synthetic DNA and RNA oligonucleotides were obtained from Integrated DNA Technologies (IDT). The nucleic acids were dissolved in RNase-free deionized water at a concentration of 1 mM. To assemble the scaffold, template DNA and RNA were mixed at a 1:1 ratio, annealed by incubation at 95°C for 2 min, 75°C for 2 min, 45°C for 5 min, and then decreasing the temperature by 5°C every 2 min until reaching 25°C. The annealed template DNA:RNA hybrid was stored at −20°C until use.
Preparation of the PTC for Cryo-EM
Purified E. coli RNAP was mixed with template DNA:RNA hybrid at a molar ratio of 1:1.3 and incubated for 30 min at 30°C. Non-template DNA was added at a molar ratio of 3:1 and incubated for 20 min. To remove excess nucleic acid, the complexes were run over a Superose 6 Increase SEC column (GE Healthcare) that was equilibrated in 20 mM Tris-HCl, pH 8.0, 2 mM MgCl2, 50 mM KCl, 1 mM dithiothreitol. The peak fractions containing the elongation complex (EC) were pooled and sequentially mixed with NusG at a molar ratio of 1:3 and NusA at a molar ratio of 1:2, followed by SEC over a Superose 6 Increase column (GE Healthcare) that was equilibrated in 20 mM Tris-HCl, pH 8.0, 2 mM MgCl2, 50 mM KCl, 1 mM dithiothreitol to remove excess NusG and NusA. Purified EC–NusG–NusA complexes were then mixed with purified Rho at a molar ratio of 1:8.
To stabilize and further purify the PTC complex, freshly formed PTC was purified and crosslinked using the GraFix method (Kastner et al., 2008). The gradient solution contained 20 mM HEPES-KOH, pH 8.0, 50 mM KCl, 2 mM MgCl2, 10–30% (v/v) glycerol and 0–0.08% (v/v) gutaraldehyde. The samples were centrifuged at 36,000 rpm for 18 h at 4°C, using a Beckman-Coulter SW41 Ti swinging-bucket rotor. Fractions containing the PTC were pooled and dialyzed against buffer without glycerol and glutaraldehyde.
Cryo-EM Grid Preparation
UltrAuFoil (Quantifoil) R-1.2/1.3 Au 300 mesh grids were glow-discharged for 1 min. After applying 3.5 μl of sample, grids were blotted for 0.5–1 s with a blotting force of 0 and vitrified in liquid ethane using a Vitrobot Mark IV (FEI) with 100% humidity at 22°C.
Cryo-EM Data Acquisition and Processing
Grids were loaded into a Titan Krios electron microscope (FEI) operated at 300 kV and equipped with a Gatan K3 Summit direct electron detector (PTC60 dataset) or a Gatan K2 Summit direct electron detector (PTC18 dataset). Images of PTC60 were recorded in super-resolution mode with a pixel size of 0.539 Å and a defocus range of 0.8 – 1.8 µm, using a total dose of 50 electrons/Å2 fractionated over 50 frames. Images of PTC18 were recorded in counting mode with a pixel size of 1.048 Å and a defocus range of 1.0 – 2.5 µm, using a total dose of 68 electrons/Å2 fractionated over 50 frames.
Collected micrographs were drift-corrected and dose-weighted in MotionCor2 (Zheng et al., 2017), and the contrast transfer function (CTF) parameters were estimated using CTFFIND4 (Rohou and Grigorieff, 2015). Approximately 10,000 particles were manually picked and subjected to 2D classification in RELION-3 (Zivanov et al., 2018), which was used for all subsequent image processing. Projection averages of the most populated 10 classes were used as templates for automated particle picking in RELION-3 (Scheres, 2015). Picked particles were manually inspected, then subjected to two subsequent rounds of 2D classification. Poorly populated classes were removed after each round, resulting in datasets of 2,865,384 particles for PTC60 and 1,182,362 particles for PTC18. To better separate images of fully-assembled PTC from those of EC and Rho ring by themselves, the particle images were subjected to supervised 3D classification with alignment, giving as reference models the EC–Rho complex (initial model obtained in RELION-3), EC (PDB: 6ALF) and Rho hexamer (PDB: 1PVO). Classes representing EC and Rho by themselves were removed, resulting in datasets of 551,397 PTC60 particles and 176,340 PTC18 particles.
The PTC60 particles were 3D autorefined and subjected to 3D classification with alignment into 8 classes using the refined map and alignment angles. The best class that showed the highest resolution both in the EC and Rho sub-regions contained 82,394 particles (15% of the starting dataset), which were autorefined, post-processed and subjected to two cycles of CTF refinement and particle polishing in RELION-3, yielding the final density map at a nominal resolution of 3.1 Å. Local resolution calculations were performed using RELION-3. Multibody refinement was performed continuing on from the final 3D autorefinement, defining the EC density as Body 1 and the Rho–NusA density as Body 2. The corresponding masks were generated using the consensus map low-pass filtered to 30-Å resolution to define the boundary with the solvent region and the atomic models to define the boundaries between the two bodies. Soft-edges with a width of 8 Å were applied to the boundaries of the masks, resulting in a slight overlap of the two bodies. Motions corresponding to the first three eigenvectors are shown in Supplemental Movie S1.
The PTC18 particles were 3D autorefined and subjected to 3D classification with alignment into 8 classes using the refined map and alignment angles The class that showed the clearest features for the EC and Rho sub-regions contained 15,681 particles (12% of the starting dataset), which were autorefined, post-processed and subjected to two cycles of CTF refinement and particle polishing in RELION-3, yielding the “overall” density map at a nominal resolution of 7.9 Å. Multibody refinement was performed continuing on from the final 3D auto-refinement as described for PTC60. Motions corresponding to the first three eigenvectors are shown in Supplemental Movie S2. The 176,340 particles after 3D autorefinement were also used for focused 3D classifications with alignment into 8 classes for the EC–NusG density and for the Rho–NusA density. For the EC–NusG density, the two classes with the highest-resolved features were combined and subjected to a second round of focused 3D classifications with alignment into 6 classes. The two classes with the highest-resolved features were combined and the 50,610 particles (29% of the starting dataset) were autorefined, post-processed and subjected to two cycles of CTF refinement and particle polishing in RELION-3, yielding the final EC–NusG density map at a nominal resolution of 4.0 Å. For the Rho–NusA density, the class showing the clearest features contained 35,760 particles (20% of the starting dataset), which were autorefined, post-processed and subjected to two cycles of CTF refinement and particle polishing in RELION-3, yielding the final Rho–NusA density map at a nominal resolution of 7.9 Å. The final EC–NusG and Rho–NusA maps were fitted into the overall PTC18 map to generate the “composite” map using the fit-in-map function in Chimera. Local resolution calculations were performed using RELION-3.
Model Building and Refinement
To build an initial model for PTC60, the atomic models of EC (PDB: 6ALH; (Kang et al., 2017), Rho (PDB: 1PVO; (Skordalakes and Berger, 2003), and the NusA NTD and S1 domain (PDB: 5LM7; (Said et al., 2017) were fit into the cryo-EM map using Chimera (Pettersen et al., 2004). The same structures plus the atomic model of NusG-NTD (PDB: 6C6U; (Kang et al., 2018) were fit into the cryo-EM map of PTC18. These initial models were real-space refined in PHENIX (Adams et al., 2010). The subunits in RNAP, the 6 monomers in the Rho ring, NusA-NTD, S1 and the nucleic acids were first refined as rigid bodies and were subsequently refined with secondary-structure restraints. Residues at the 5’ end of the single-stranded RNA were built de novo in Coot (Emsley and Cowtan, 2004), and real-space refined with the previously refined model. To build the model for PTC18, we first placed the atomic models of the components into the overall map to obtain the global architecture of PTC18 and then refined the models against the composite map. Both models were then visually inspected, and outliers and problematic regions were fixed manually in Coot. The final refinement statistics are summarized in Table S2.
RNAP Mutant Strains and Plasmid Construction
E. coli strains with polypeptide loop/domain deletions were constructed by using the lambda Red recombineering method together with CRISPR-Cas9 counterselection (Reisch and Prather, 2015). Oligonucleotides (~80-mers) used for recombineering were designed to target the lagging strand of replicating DNA with upstream and downstream homology to the area of deletion. To bridge the gap between the points of the deletion, sequences encoding for one to three glycine residues were introduced in place of the deleted regions. Three phosphorothioated bases were used at the 5’end of the oligos to reduce its degradation rate in vivo. pKDsg (pSg-xxx) derivatives used for counterselection were constructed by circular polymerase extension cloning using primers with overlapping 20-bp protospacer sequences that corresponded to the fragment adjacent to appropriate PAM site (5’-NGG-3’) in the deleted parental sequences.
E. coli MDS42 strains were first transformed with the pCas9cr4 plasmid and subsequently transformed with the sgRNA encoding plasmid (pSg-xxx). Cells that possessed both plasmids were grown in Super Optimal Broth with spectimomycin (Sp, 50 mg/l) and chloramphenicol (Cm, 30 mg/l) at 30°C. When OD600 reached ~0.5, lambda Red was induced with 1.2% (w/v) L-arabinose, and cells were grown for another 20 min. Then, an oligonucleotide for recombineering was electroporated into the cells. After 2 h of recovery, the cells were plated on Luria broth (LB) with Sp, Cm and anhydrotetracycline (aTc, 100 ng/l) and incubated overnight at 30°C to select for survivors of the CRISPR/Cas9 selection. Colonies were screened with specific primers and the corresponding chromosome regions were verified by sequencing.
To eliminate the pSg-xxx plasmid, cells were incubated in LB for 12 h at 37°C and streaked on LB plates. Individual colonies were selected and assessed for the loss of Sp resistance. The next pSg-xxx plasmid was used in a subsequent iteration to make another deletion. The pKDsg-15a plasmid was used to cure the pCas9cr plasmid that targeted the p15a origin of replication of pCas9cr. Upon transformation of pKDsg-15a into cells that contained pCas9cr, the cells were recovered in SOC (Super Optimal broth with Catabolite repression) for 2 h at 30°C, then aTc (100 ng/l) was added and incubated for an additional 2 h before plating on LB with Sp and aTc. The pKDsg-15a plasmid was cured by growth at 37°C.
To create plasmid pVS10-ΔRpoB483–491/I9, a fragment of the rpoB gene with two deletions (Δβ 483–491 and I9) was amplified from genomic DNA of the SP1176 strain and was inserted into the BbvCI and SbfI sites of the pVS10 plasmid to replace the corresponding region of the wild-type gene.
mRNA Purification, Reverse Transcription and qRT-PCR
The pVE-RUT81-GFP plasmid was transformed into E. coli strain MDS42(Cardinale et al., 2008) and corresponding mutant strains that carried chromosomal RNAP mutations. Single colonies from each of the strains were inoculated from fresh LB plates into 3 ml of LB medium and grown overnight at 30°C with 50 mg/ml kanamycin (Km) with shaking. The next day (~20 h), 30 ml of fresh LB with 50 mg/ml Km were inoculated with 0.3 ml of the overnight cultures and grown in 250-ml flasks at 30°C with shaking to an OD600 of ~0.3. For the chromosome-based Rho-dependent terminator reporter (dsbB-RhoT), wild-type E. coli MDS 42 strain and the corresponding mutant strain that carried the chromosomal RNAP mutation were grown in LB (without Km).
10-ml aliquots were taken from each culture and transferred into 15-ml Falcon tubes before centrifugation at 5,000g for 5 min at 4°C. The supernatant was discarded and mRNA was purified using the MasterPure Complete DNA and RNA purification kit (Lucigen) according to the manufacturer’s instructions, except that DNAse I treatment was conducted for 30 min at 37°C and was supplemented with 500 units of ExoIII and 25 units of Sau3AI (both from New England Biolabs). RNA was re-dissolved in 50 ml TE buffer (10 mM Tris-HCl, pH 8.0, 0.1 mM EDTA) to a concentration of 1–2 mg/ml and the volume was adjusted to 500 ng/ml with TE buffer.
cDNA was produced from 1 μg RNA using QuantiTect reverse transcription kit (Qiagen) according to the manufacturer’s instructions. For strand specificity, 0.7 μM primers of pVE_B reverse, GFP_3 reverse, dsbB D1 reverse and dsbB D2 reverse were used instead of the random primers provided in the kit.
qPCR was performed using a QuantStudio 7 Flex real-time qPCR machine (Applied biosystems) from 5 ng DNA in one well (20 μl per well) in triplicates using pVE_B forward/reverse, GFP_3 forward/reverse, dsbB D1 forward/reverse and dsbB D2 forward/reverse primer pairs (5 ng/ml each) at an annealing temperature of 60°C. The sequences of the target region of the pVE-RUT81-GFP plasmid used for qPCR and the primers are shown in Table S5. The pVE-RUT81-GFP plasmid is a derivative of the pMW-3110 plasmid (Sedlyarova et al., 2017) with the 5’ UTR of the GFP gene replaced as shown in Table S5.
In-vitro Transcription Termination Assay
Elongation complexes were formed with 75 nM T7A1-Trpt1 DNA (Table S5) and 100 nM wild-type and mutant RNAP core with an equimolar concentration of σ70 in 100 μl of TB50 buffer (40 mM Tris-HCl, pH 8.0, 10 mM MgCl2, 50 mM NaCl, 0.003% (v/v) Igepal-60, 5 mM β-mercaptoethanol with 40 unit of RNasin (Promega)). Transcription was initiated with 10 μM AUC primer and 25 μM of ATP and GTP for 5 min at 22°C. 1 μl of CTP [ɑ−32P] 3000Ci/nmol, (0.33 nM) was added for 5 min at 22°C. Where indicated NusA (100 nM) was added and incubated for 5 min at 22°C (Figure 5F). Rho (50, 25, and 12.5 nM) and NusG (1 μM) were added with the NTPs and the reactions were chased with 1 mM ATP and 200 μM of the other NTPs at 37°C for 5 min. The termination efficiency of the mutant RNAP (RpoB Δ483–491/I9) was compared with that of wild-type RNAP. Reactions were terminated by adding 2X STOP buffer and were heated for 5 min at 95°C and resolved on a 6% urea-PAGE for 20 min at 50 W, and dried and exposed to a phosphor imager screen. Note: For the titration of Rho with RNAP (Figure 3I), 100 nM wild-type RNAP and 100 nM NusA were added before the final chase along with NTPs, NusG, Rho, and NTPs.
Rho-dependent Transcription Termination “in trans”
Template 1 (Table S5) was produced by PCR-directed mutagenesis (Nudler et al., 1995) using Phusion DNA polymerase (New England Biolabs) and synthetic DNA oligos (IDT). It has the identical T7A1 promoter and initial transcribed sequence up to position +10 (counting from the +1 start of the transcription) followed by a modified sequence as shown in Table S5. The template DNA was purified from a 2% agarose gel using a Qiagen Gel Extraction Kit according to the manufacturer’s instructions and diluted in TE buffer to a concentration ~1 pmol/μl. His6-tagged RNAP, NusA, NusG and Rho were purified as described previously. Rut81 synthetic RNA oligo was purchased from IDT and diluted in TE buffer up to 1 μM concentration.
5 to 10 pmol RNAP were mixed with 2X molar excess of DNA in 20 μl of TB50 (40 mM Tris-HCl, pH 8.0, 10 mM MgCl2, 50 mM NaCl, 0.003% (v/v) Igepal-60) for 5 min at 37°C, followed by addition of ApUpC (10 μM), GTP and ATP (25 μM each) for 5 min. Next, 15 μl TB50-equilibrated Ni-NTA-agarose beads (Qiagen) were added and the slurry was shaken for 5 min at room temperature in the presence of 1.5 mg/ml heparin. The sample was washed twice with 1 ml of TB1000 (as TB50 but with 1 M NaCl), incubated for 1 min at room temperature and washed 3 times with 1 ml of TB100 (as TB50 but with 100 mM NaCl). To produce EC14, ATP (5 μM) and 2 μl of CTP [ɑ−32P] 3000Ci/nmol (0.33 nM) were added for 5 min at room temperature followed by CTP (5 μM) for another 2 min. Beads were washed 4 times with 1 ml of TB100. EC14 was walked to position 29, 40, or 50. The walking reaction was performed at room temperature as described previously (Nudler et al., 1994) using limited NTP sets (5 μM). Samples of the corresponding ECs were divided into equal aliquots and NusG (up to 1 μM) or/and NusA (up to 1 μM) or/and RUT81 RNA (up to 0.1 μM, Table S5) were added for 5 min at 22° C as indicated in each figure. To measure Rho-dependent termination, the samples were mixed with either 100 nM Rho hexamer pre-mixed with ATP (up to 1 mM final concentration) or mock solution containing ATP and TB100 instead of Rho. After incubation at 22°C for the indicated time, 10-μL aliquots were withdrawn and chased with 100 μM CTP, UTP, GTP and 1 mM ATP for 5 min at 37°C. The reactions were stopped by the addition of an equal volume of Stop Buffer (SB) (1X TBE (Tris/Borate/EDTA), 20 mM EDTA; 8 M urea, 0.025% (v/v) xylenthianol, 0.025% (w/v) bromophenol blue) and the RNA products were separated by 15% sequencing PAGE. To measure the termination efficiency, the beads were not chased with NTPs but instead were washed once with 1 ml of TB1000 and once with 1 ml of TB100. The volume was readjusted to 10 μl and the samples were quenched by the addition of an equal volume of SB. The gels were exposed to a phosphor-screen and the screen was scanned using an Amersham Typhoon Scanner (GE Healthcare). The data were analyzed using Image Quant software (GE Healthcare).
In-vivo and in-vitro XLMS (Covalent Crosslink Mapping by Mass Spectrometry) and Data Analysis
Strain construction:
rpoC:10XHis and rho:FLAG3. strains were constructed by introducing coding sequences for 10X H and DYKDDDDKDYKDDDDKDYKDDDDK after codons 1407 (rpoC) and 419 (rho), respectively, into parental E. coli strain MG1655 by means of lambda Red-mediated gene replacement (Datsenko and Wanner, 2000). Successful construction was confirmed in each case by genomic sequencing and whole-cell enumerative proteomics.
In-vitro crosslinking:
Crosslinkers BS3 and DSS (Thermo Fisher Scientific) were dissolved in LC-MS grade H20 and oxygen-depleted anhydrous DMSO (ZerO2, Millipore Sigma), respectively, at a concentration of 50 mM. Crosslinker was added to the target complex prepared in NHS-ester non-reactive buffer (50 mM HEPES, pH 7.5, 500 mM NaCl, 2 mM MgSO4, 1 mM TCEP) to a final concentration of 100–500 µM. Reactions were performed at 25°C in disposable inert cuvettes (UVette, Eppendorf), and monitored by continuous looped dynamic light scattering measurements of polydispersity (Pd<10%; DynaPro NanoStar, Wyatt) (Meyer et al., 2015). Crosslinking was quenched after 30 min by addition of Tris-HCl, pH 8.0 to a final concentration of 5 mM.
In-vivo crosslinking:
Cells were grown in 0.5X Terrific Broth (Thermo Fisher Scientific) at 37°C with agitation (250 rpm). When OD600 reached 0.5, the culture was supplemented with 350 mM DSS in DMSO (ZerO2, Millipore Sigma) to a final concentration of 2 mM. The reaction was quenched after 45 min by addition of Tris-HCl, pH 8.0 to a final concentration of 5 mM. Cells were harvested by centrifugation at a 6000 g for 5 min at 4°C, and processed immediately or stored at −80°C. When needed, bicyclomycin (Takeda Schering-Plough Animal Health K.K.) was added to a final concentration of 100 µg/L at an OD600 of 0.5, and cells were incubated an additional 2 h before addition of the crosslinker.
Affinity purification of His10-tagged complexes:
Cells were suspended in lysis buffer (50 mM HEPES, pH 7.5, 500 mM NaCl, 2 mM MgSO4, 5 mM ZnSO4, 1 mM TCEP, 1X ProBlock Gold Bacterial 2D, Gold Bio) and lysed by the combined action of lysonase (Sigma Millipore) and ultrasonication. A cell-free extract was prepared by centrifugation at 29,500 g for 45 min at 4°C, and His10-tagged proteins were purified using His Mag Sepharose Ni (GE Healthsciences), according to the manufacturer’s protocol.
Affinity purification of 3XFLAG-tagged complexes:
Cells were suspended in lysis buffer (50 mM HEPES, pH 7.5, 125 mM NaCl, 1 mM TCEP, 1X ProBlock Gold Bacterial 2D, Gold Bio) and lysed by the combined action of lysonase (Sigma Millipore) and ultrasonication. Cell-free extract was prepared by centrifugation at 29,500 g for 45 min at 4°C, and His10-tagged proteins were purified using Pierce Anti-DYKDDDDK Magnetic Agarose (Thermo Fisher Scientific), according to the manufacturer’s protocol.
Mass spectrometry and data analysis:
Samples were dialyzed against 100 mM ammonium bicarbonate, reduced with 50 mM tris(2-carboxyethyl)phosphine (TCEP) for 10 min at 60°C and alkylated with 50 mM iodoacetamide for 60 min at 25°C in the dark. Digestion was carried out overnight at 37°C with 125 ng/µl sequencing grade modified trypsin (Promega) in 30 mM ammonium bicarbonate. The reaction mixture was supplemented with trifluoroacetic acid to a final concentration of 0.1%. The resulting peptides were passed though C18 Spin Tips (Thermo Scientific) before elution with 40 μL of 80% (v/v) acetonitrile (ACN) in 0.1% (v/v) trifluoroacetic acid (TFA). Eluted peptides were dried and resuspended in 20 μl 0.1% (v/v) formic acid for MS analysis. Peptides were analyzed in an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific) coupled to an EASY-nLC (Thermo Scientific) liquid chromatography system, with a 2 μm, 500 mm EASY-Spray column. The peptides were eluted over a 120-min linear gradient from 96% Buffer A (water) to 40% Buffer B (ACN), then continued to 98% Buffer B over 20 min with a flow rate of 250 nl/min. Each full MS scan (R = 60,000) was followed by 20 data-dependent MS2 (R = 15,000) with high-energy collisional dissociation and an isolation window of 2.0 m/z. The normalized collision energy was set to 35. Precursors of charge state 2 and 3 were collected for MS2 scans in enumerative mode, precursors of charge state 4–6 were collected for MS2 scans in crosslink discovery mode (both were performed for each sample); monoisotopic precursor selection was enabled and the dynamic exclusion window was set to 30.0 s. raw files obtained in the enumerative mode were analyzed using the pFind3 software (Chi et al., 2018) in open search mode, using the entire MG1655 proteome as the search space (Uniprot UP000000625). fasta sequences of identified proteins formed the search space for crosslink discovery by pLink2 (Chen et al., 2019); protein modifications inferred by pFind3 and comprising >0.5% of total were included as the variable modifications in pLink2 search parameters. pLink2 results were filtered for FDR (<5%), e-value (<1.0E−3), score (<1.0E−2), and abundance (PSMs≥5).
Data and Software Availability
Structural models of PTC60 and PTC18 have been deposited in the PDB with accession numbers 6XAS and 6XAV, respectively. The cryo-EM density maps for PTC60 and PTC18 have also been deposited in the Electron Microscopy Data Bank under accession number 22114 and 22115, respectively. All other data are included within the manuscript and its Supporting Information.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli: BL21(DE3) Tuner | Novagen | Cat#70623 |
| E. coli: MDS42 | Laboratory strains | N/A |
| E. coli: MDS42-rpoBΔ483–491 | This study | N/A |
| E. coli: MDS42-rpoBΔ483–491/I9 | This study | N/A |
| E. coli: MG1665-rpoC:10XHis | This study | N/A |
| E. coli: MG1665-rho: FLAG3 | This study | N/A |
| E. coli: MG1655 | Laboratory strains | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| EDTA-free protease inhibitor cocktail | Roche Applied Science | Cat#118361700001 |
| HiPrep Heparin Fast Flow | GE Healthcare | Cat#28-9365-49 |
| HisTrap Crude FF | GE Healthcare | Cat#17-5286-01 |
| Mono Q 10/100 GL | GE Healthcare | Cat#17-5167-01 |
| Superose 6 Increase 10/300 GL | GE Healthcare | Cat#29-0915-96 |
| HiPrep SP XL 16/10 | GE Healthcare | Cat#28-9365-40 |
| Nuclease-free water (not DEPC treated) | Ambion | Cat# 4387936 |
| 8% glutaraldehyde aqueous Solution | Electron Microscopy Sciences | Cat#16019 |
| SUMO protease | Invitrogen | Cat#12588-018 |
| bis(sulfosuccinimidyl)suberate (BS3) | Thermo Scientific | Cat#21580 |
| Phusion High-Fidelity DNA polymerase | New England Biolabs | Cat#M0530 |
| RNasin Ribonuclease Inhibitor | Promega | Cat#N2115 |
| MasterPure Complete DNA and RNA purification kit | Lucigen | Cat#MC85200 |
| QuantiTect reverse transcription kit | Qiagen | Cat#205311 |
| disuccinimidyl suberate (DSS) | Thermo Scientific | Cat#21555 |
| bicyclomycin | Takeda Schering-Plough Animal Health K.K. | Laboratory stock |
| Deposited Data | ||
| Cryo-EM Structure of E. coli Rho-dependent Transcription Pre-termination Complex | This study | PDB: 6XAS |
| Cryo-EM Structure of E. coli Rho-dependent Transcription Pre-termination Complex with bound NusG | This study | PDB: 6XAV |
| Cryo-EM Structure of E. coli Rho-dependent Transcription Pre-termination Complex, 3.1-Å resolution map | This study | EMDB: EMD-22114 |
| Cryo-EM Structure of E. coli Rho-dependent Transcription Pre-termination Complex with bound NusG, 7.9-Å resolution map | This study | EMDB: EMD-22115 |
| Oligonucleotides | ||
| EC non-template DNA GGGCTACCTCTCCATGACGGCGAATACCC |
IDT | N/A |
| EC template DNA GGGTATTCGCCGTGTACCTCTCCTAGCCC |
IDT | N/A |
| 18nt RNA AUUCAAAGCGGAGAGGUA |
IDT | N/A |
| 60nt RNA CCGCACCUCCUCAAACGCUACCUCGACCAGCCUCCCUCCCGCAUUCAAAGCGGAGAGGUA |
IDT | N/A |
| RUT81 RNA canonical RUT CCCUCAACGACCCCUUCCUUCUCCCCAUCGCUACCUCAUAUCCGCACCUCCUCAAACGCUACCUCGACCAGCCUCCCUCCC |
IDT | N/A |
| pVE_B forward qRT-PCR primer ACAGCCAACCTAATCGACAC |
IDT | N/A |
| pVE_B reverse qRT-PCR primer AAACGACGGCCAGTGAAT |
IDT | N/A |
| GFP_3 forward qRT-PCR primer GGTGATGTTAATGGGCACAAAT |
IDT | N/A |
| GFP_3 reverse qRT-PCR primer CAAAGTAGTGACAAGTGTTGGC |
IDT | N/A |
| dsaB D1 forward qRT-PCR primer ATG GTT CGT TTC CCG GAA TG |
IDT | N/A |
| dsaB D1 reverse qRT-PCR primer GCG GCA TTT CCA GAC CTA AA |
IDT | N/A |
| dsaB D2 forward qRT-PCR primer GTA TAA CGT GGT GAA AGC ATG G |
IDT | N/A |
| dsaB D2 reverse qRT-PCR primer GGG CAG AGG AAC ACT CTA TTT |
IDT | N/A |
| Template 1 DNA double strand DNA template used in RUT in trans assay tccagatcccgaaaatttatcaaaaagagtattgacttaaagtctaacctataggatacttacagccATCGAGAGGGCCACGGCGAACAGCCAACCTAATCGACACC GGGGTCCGGGATCTGGATCTGGATCGCGAATTCCAGGCCTGCTGGTAATCTTTGGATCCCCGGGTACCGAGCTCGAATTCACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAACCCTGGCG |
This study | N/A |
| T7A1-Trpt1 DNA template double strand DNA template used in in-vitro transcription termination assay tccagatcccgaaaatttatcaaaaagagtattgacttaaagtctaacctataggatacttacagccATCGAGAGGGACACGGCGAAGGCGGCATTTTAACTTTCTTTAATGAAGCCGGAAAAATCCTAAATTCATTTAATATTTATCTTTTTACCGTTTCGCTTACCCCGGTCGAACGTCAACTTACGTCATTTTTCCGCCCAACAGTAATATAATCAAACAAATTAATCCCGCAACATAACACCAGTAAAATCAATAATTTTCTCTAAGTCACTTATTCCTCAGGTAATTGTTAATATATCCAGAATGTTCCTCAAAATATATTTTCCCTCTATCTTCTCGTTGCGCTTAATTTGACTAATTCTCATTAGCGACTAATTTTAATGAGTGTCGACACACAACACTCATATTAATGAAACAATGCAACGCAACGGGAGAAATAACATGGCCGAACATCGTGGTGGTTCAGGAAATTTCGCCGAAGACCGTGAGAAGGCATCCGACGCAGGCCGTAAAGGCGGTCAGCATAGCGGCGGTAATTTTAAAAATGATCCGCAACGCGCATCTGAAGCGGGTAAAAAAGGCGGTCAACAAAGCGGTGGTAATAAATCAGGCAAATCCTG |
This study | N/A |
| Recombinant DNA | ||
| pVS 10 wild-type E. coli RNAP overexpression plasmid |
Svetlov and Artsimovitch, 2015 | N/A |
| pET21b-Rho Rho overexpression plasmid, no tag |
This study | N/A |
| pSUMO-NusA NusA overexpreesion plasmid, 6XHis-SUMO tag |
This study | N/A |
| pSUMO-NusG NusG overexpreesion plasmid, 6XHis-SUMO tag |
This study | N/A |
| pVE-RUT81-GFP plasmid used to measure the in-vivo Rho-dependent transcription termination |
This study | N/A |
| pVS10-ΔRpoB483–491/I9 RNAP-Δβ483–491/I9 mutant overexpression plasmid |
This study | N/A |
| Software and Algorithms | ||
| Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera |
| COOT | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| CTFFIND4 | Rohou and Grigorieff, 2015 | http://grigoriefflab.janelia.org/ctffind4 |
| MotionCor2 v1.2.1 | Zheng et al., 2017 | http://msg.ucsf.edu/em/software/motioncor2.html |
| Relion 3.0 | Zivanov et al., 2018 | https://github.com/3dem/relion |
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org/ |
| MolProbity | Chen et al., 2010 | http://molprobity.biochem.duke.edu/ |
| pLink2 | http://pfind.ict.ac.cn/software/pLink/ | http://pfind.ict.ac.cn/software/pLink/ |
| Other | ||
| UltrAuFoil R-1.2/1.3 Au 300 mesh grids | Electron Microscopy Sciences | Cat#Q350AR13A |
Acknowledgments
We thank William Rice, Bing Wang, David Stokes and Zheng Liu for helping with sample screening and data collection at NYU Langone Health’s Cryo-EM Laboratory. We thank Zhiheng Yu, Doreen Matthies, Rui Yan and Xiaowei Zhao at the HHMI Janelia Cryo-EM Facility for help in microscope operation and data collection. We thank the BigPurple HPC core at NYU Langone Health for computer access. This work was supported by the Russian Science Foundation Grant 17-74-30030 (A.M.), NIH grant R01 GM126891, Blavatnik Family Foundation, and by the Howard Hughes Medical Institute (E.N.).
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Nahum G, Epshtein V, Ruckenstein AE, Rafikov R, Mustaev A, and Nudler E (2005). A ratchet mechanism of transcription elongation and its control. Cell 120, 183–193. [DOI] [PubMed] [Google Scholar]
- Boudvillain M, Figueroa-Bossi N, and Bossi L (2013). Terminator still moving forward: expanding roles for Rho factor. Current opinion in microbiology 16, 118–124. [DOI] [PubMed] [Google Scholar]
- Burns CM, Richardson LV, and Richardson JP (1998). Combinatorial effects of NusA and NusG on transcription elongation and Rho-dependent termination in Escherichia coli. Journal of molecular biology 278, 307–316. [DOI] [PubMed] [Google Scholar]
- Cardinale CJ, Washburn RS, Tadigotla VR, Brown LM, Gottesman ME, and Nudler E (2008). Termination factor Rho and its cofactors NusA and NusG silence foreign DNA in E. coli. Science 320, 935–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZL, Meng JM, Cao Y, Yin JL, Fang RQ, Fan SB, Liu C, Zeng WF, Ding YH, Tan D, et al. (2019). A high-speed search engine pLink 2 with systematic evaluation for proteome-scale identification of cross-linked peptides. Nature communications 10, 3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H, Liu C, Yang H, Zeng WF, Wu L, Zhou WJ, Wang RM, Niu XN, Ding YH, Zhang Y, et al. (2018). Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine. Nature biotechnology. [DOI] [PubMed]
- Ciampi MS (2006). Rho-dependent terminators and transcription termination. Microbiology 152, 2515–2528. [DOI] [PubMed] [Google Scholar]
- Dar D, and Sorek R (2018). High-resolution RNA 3’-ends mapping of bacterial Rho-dependent transcripts. Nucleic acids research 46, 6797–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, and Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proceedings of the National Academy of Sciences of the United States of America 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta D, Chalissery J, and Sen R (2008). Transcription termination factor rho prefers catalytically active elongation complexes for releasing RNA. The Journal of biological chemistry 283, 20243–20251. [DOI] [PubMed] [Google Scholar]
- Dutta D, Shatalin K, Epshtein V, Gottesman ME, and Nudler E (2011). Linking RNA polymerase backtracking to genome instability in E. coli. Cell 146, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Epshtein V, Dutta D, Wade J, and Nudler E (2010). An allosteric mechanism of Rho-dependent transcription termination. Nature 463, 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grylak-Mielnicka A, Bidnenko V, Bardowski J, and Bidnenko E (2016). Transcription termination factor Rho: a hub linking diverse physiological processes in bacteria. Microbiology 162, 433–447. [DOI] [PubMed] [Google Scholar]
- Guo X, Myasnikov AG, Chen J, Crucifix C, Papai G, Takacs M, Schultz P, and Weixlbaumer A (2018). Structural Basis for NusA Stabilized Transcriptional Pausing. Molecular cell 69, 816–827 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart CM, and Roberts JW (1991). Rho-dependent transcription termination. Characterization of the requirement for cytidine in the nascent transcript. The Journal of biological chemistry 266, 24140–24148. [PubMed] [Google Scholar]
- Krebs Jocelyn E., E.S.G., Kilpatrick Stephen T. (2018). “How Does Rho Factor Work?” In LEWIN’S GENES XII (Burlington: JONES & BARTLETT LEARNING; ), pp. 1744–1750. [Google Scholar]
- Kainz M, and Gourse RL (1998). The C-terminal domain of the alpha subunit of Escherichia coli RNA polymerase is required for efficient rho-dependent transcription termination. Journal of molecular biology 284, 1379–1390. [DOI] [PubMed] [Google Scholar]
- Kang JY, Mooney RA, Nedialkov Y, Saba J, Mishanina TV, Artsimovitch I, Landick R, and Darst SA (2018). Structural Basis for Transcript Elongation Control by NusG Family Universal Regulators. Cell 173, 1650–1662 e1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JY, Olinares PD, Chen J, Campbell EA, Mustaev A, Chait BT, Gottesman ME, and Darst SA (2017). Structural basis of transcription arrest by coliphage HK022 Nun in an Escherichia coli RNA polymerase elongation complex. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner B, Fischer N, Golas MM, Sander B, Dube P, Boehringer D, Hartmuth K, Deckert J, Hauer F, Wolf E, et al. (2008). GraFix: sample preparation for single-particle electron cryomicroscopy. Nature methods 5, 53–55. [DOI] [PubMed] [Google Scholar]
- Koslover DJ, Fazal FM, Mooney RA, Landick R, and Block SM (2012). Binding and translocation of termination factor rho studied at the single-molecule level. Journal of molecular biology 423, 664–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp F, Said N, Huang YH, Loll B, Burger J, Mielke T, Spahn CMT, and Wahl MC (2019). Structural Basis for the Action of an All-Purpose Transcription Anti-termination Factor. Molecular cell 74, 143–157 e145. [DOI] [PubMed] [Google Scholar]
- Lawson MR, Ma W, Bellecourt MJ, Artsimovitch I, Martin A, Landick R, Schulten K, and Berger JM (2018). Mechanism for the Regulated Control of Bacterial Transcription Termination by a Universal Adaptor Protein. Molecular cell 71, 911–922 e914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leela JK, Syeda AH, Anupama K, and Gowrishankar J (2013). Rho-dependent transcription termination is essential to prevent excessive genome-wide R-loops in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America 110, 258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Mason SW, and Greenblatt J (1993). Elongation factor NusG interacts with termination factor rho to regulate termination and antitermination of transcription. Genes & development 7, 161–172. [DOI] [PubMed] [Google Scholar]
- Liu K, Zhang Y, Severinov K, Das A, and Hanna MM (1996). Role of Escherichia coli RNA polymerase alpha subunit in modulation of pausing, termination and anti-termination by the transcription elongation factor NusA. The EMBO journal 15, 150–161. [PMC free article] [PubMed] [Google Scholar]
- Magan A, Amman F, El-Isa F, Hartl N, Shamovsky I, Nudler E, Schroeder R, and Sedlyarova N (2019). iRAPs curb antisense transcription in E. coli. Nucleic acids research 47, 10894–10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer A, Dierks K, Hussein R, Brillet K, Brognaro H, and Betzel C (2015). Systematic analysis of protein-detergent complexes applying dynamic light scattering to optimize solutions for crystallization trials. Acta crystallographica Section F, Structural biology communications 71, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra P, Ghosh G, Hafeezunnisa M, and Sen R (2017). Rho Protein: Roles and Mechanisms. Annual review of microbiology 71, 687–709. [DOI] [PubMed] [Google Scholar]
- Mooney RA, Schweimer K, Rosch P, Gottesman M, and Landick R (2009). Two structurally independent domains of E. coli NusG create regulatory plasticity via distinct interactions with RNA polymerase and regulators. Journal of molecular biology 391, 341–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehrke KW, and Platt T (1994). A quaternary transcription termination complex. Reciprocal stabilization by Rho factor and NusG protein. Journal of molecular biology 243, 830–839. [DOI] [PubMed] [Google Scholar]
- Nudler E, Goldfarb A, and Kashlev M (1994). Discontinuous mechanism of transcription elongation. Science 265, 793–796. [DOI] [PubMed] [Google Scholar]
- Nudler E, Kashlev M, Nikiforov V, and Goldfarb A (1995). Coupling between transcription termination and RNA polymerase inchworming. Cell 81, 351–357. [DOI] [PubMed] [Google Scholar]
- Nudler E, Mustaev A, Lukhtanov E, and Goldfarb A (1997). The RNA-DNA hybrid maintains the register of transcription by preventing backtracking of RNA polymerase. Cell 89, 33–41. [DOI] [PubMed] [Google Scholar]
- Peters JM, Mooney RA, Grass JA, Jessen ED, Tran F, and Landick R (2012). Rho and NusG suppress pervasive antisense transcription in Escherichia coli. Genes & development 26, 2621–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Qayyum MZ, Dey D, and Sen R (2016). Transcription Elongation Factor NusA Is a General Antagonist of Rho-dependent Termination in Escherichia coli. The Journal of biological chemistry 291, 8090–8108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray-Soni A, Bellecourt MJ, and Landick R (2016). Mechanisms of Bacterial Transcription Termination: All Good Things Must End. Annual review of biochemistry 85, 319–347. [DOI] [PubMed] [Google Scholar]
- Reisch CR, and Prather KL (2015). The no-SCAR (Scarless Cas9 Assisted Recombineering) system for genome editing in Escherichia coli. Scientific reports 5, 15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson LV, and Richardson JP (1996). Rho-dependent termination of transcription is governed primarily by the upstream Rho utilization (rut) sequences of a terminator. The Journal of biological chemistry 271, 21597–21603. [DOI] [PubMed] [Google Scholar]
- Roberts JW (1969). Termination factor for RNA synthesis. Nature 224, 1168–1174. [DOI] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. Journal of structural biology 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said N, Krupp F, Anedchenko E, Santos KF, Dybkov O, Huang YH, Lee CT, Loll B, Behrmann E, Burger J, et al. (2017). Structural basis for lambdaN-dependent processive transcription antitermination. Nature microbiology 2, 17062. [DOI] [PubMed] [Google Scholar]
- Saxena S, and Gowrishankar J (2011). Compromised factor-dependent transcription termination in a nusA mutant of Escherichia coli: spectrum of termination efficiencies generated by perturbations of Rho, NusG, NusA, and H-NS family proteins. J Bacteriol 193, 3842–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2015). Semi-automated selection of cryo-EM particles in RELION-1.3. Journal of structural biology 189, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MC, and Chamberlin MJ (1984). Binding of rho factor to Escherichia coli RNA polymerase mediated by nusA protein. The Journal of biological chemistry 259, 15000–15002. [PubMed] [Google Scholar]
- Schweimer K, Prasch S, Sujatha PS, Bubunenko M, Gottesman ME, and Rosch P (2011). NusA interaction with the alpha subunit of E. coli RNA polymerase is via the UP element site and releases autoinhibition. Structure 19, 945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlyarova N, Rescheneder P, Magan A, Popitsch N, Rziha N, Bilusic I, Epshtein V, Zimmermann B, Lybecker M, Sedlyarov V, et al. (2017). Natural RNA Polymerase Aptamers Regulate Transcription in E. coli. Molecular cell 67, 30–43 e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlyarova N, Shamovsky I, Bharati BK, Epshtein V, Chen J, Gottesman S, Schroeder R, and Nudler E (2016). sRNA-Mediated Control of Transcription Termination in E. coli. Cell 167, 111–121 e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skordalakes E, and Berger JM (2003). Structure of the Rho transcription terminator: mechanism of mRNA recognition and helicase loading. Cell 114, 135–146. [DOI] [PubMed] [Google Scholar]
- Studier FW (2005). Protein production by auto-induction in high density shaking cultures. Protein expression and purification 41, 207–234. [DOI] [PubMed] [Google Scholar]
- Sullivan SL, and Gottesman ME (1992). Requirement for E. coli NusG protein in factor-dependent transcription termination. Cell 68, 989–994. [DOI] [PubMed] [Google Scholar]
- Svetlov V, and Artsimovitch I (2015). Purification of bacterial RNA polymerase: tools and protocols. Methods in molecular biology 1276, 13–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svetlov V, and Nudler E (2020). Towards the unified principles of transcription termination. The EMBO journal 39, e104112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen ND, and Berger JM (2009). Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell 139, 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turtola M, and Belogurov GA (2016). NusG inhibits RNA polymerase backtracking by stabilizing the minimal transcription bubble. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward DF, and Gottesman ME (1981). The nus mutations affect transcription termination in Escherichia coli. Nature 292, 212–215. [DOI] [PubMed] [Google Scholar]
- Yang X, Molimau S, Doherty GP, Johnston EB, Marles-Wright J, Rothnagel R, Hankamer B, Lewis RJ, and Lewis PJ (2009). The structure of bacterial RNA polymerase in complex with the essential transcription elongation factor NusA. EMBO Rep 10, 997–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You L, Shi J, Shen L, Li L, Fang C, Yu C, Cheng W, Feng Y, and Zhang Y (2019). Structural basis for transcription antitermination at bacterial intrinsic terminator. Nature communications 10, 3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM maps included in this study have been deposited in the Electron Microscopy Data Bank with accession codes: EMD-22114 and EMD-22115. The atomic coordinates have been deposited in the Protein Data Bank with accession codes: 6XAS and 6XAV.
Structural models of PTC60 and PTC18 have been deposited in the PDB with accession numbers 6XAS and 6XAV, respectively. The cryo-EM density maps for PTC60 and PTC18 have also been deposited in the Electron Microscopy Data Bank under accession number 22114 and 22115, respectively. All other data are included within the manuscript and its Supporting Information.