

Abstract
Precise regulation of circulating glucose is crucial for human health and ensures a sufficient supply to the brain, which relies almost exclusively on glucose for metabolic energy. Glucose homeostasis is coordinated by hormone-secreting endocrine cells in the pancreas, as well as glucose utilization and production in peripheral metabolic tissues including the liver, muscle, and adipose tissue. Glucose-regulatory tissues receive dense innervation from sympathetic, parasympathetic, and sensory fibers. In this review, we summarize the functions of peripheral nerves in glucose regulation and metabolism. Dynamic changes in peripheral innervation have also been observed in animal models of obesity and diabetes. Together, these studies highlight the importance of peripheral nerves as a new therapeutic target for metabolic disorders.
Keywords: autonomic, sensory, pancreatic islets, hormone secretion, brown fat, white fat, liver, diabetes, obesity
Neural regulation of blood glucose
Glucose homeostasis is the tight maintenance of blood glucose levels (within 4–6 mM) in response to internal changes or external events such as feeding, fasting, stress, or exercise. Both hyper- and hypo-glycemia have life-threatening consequences, with the global prevalence of chronic hyperglycemia or diabetes being approximately one in eleven among adults [1]. Further, diabetes brings increased risks for cardiac dysfunction, renal failure, and blindness [2]. A better understanding of diverse mechanisms by which the body regulates blood glucose levels has significant implications for human health.
Multiple organs contribute to the regulation of blood glucose levels. One key locus is the pancreatic islets of Langerhans, which secrete the hormones insulin and glucagon to exert opposing actions on blood glucose levels. Insulin signals to peripheral tissues such as the liver, skeletal muscles, and fat depots, to promote glucose uptake from the bloodstream. Conversely, glucagon signals to liver and muscle to elevate circulating glucose levels by breakdown of glycogen (glycogenolysis) or promotes de novo glucose synthesis from simpler precursors (gluconeogenesis) in liver and kidneys. The central nervous system (CNS) plays a key role in coordinating the glucose-regulatory activity of peripheral organs. Hypothalamic regions control islet hormone secretion and whole-body glucose homeostasis via the autonomic nervous system, which consists of the anatomically and functionally distinct branches of the sympathetic and parasympathetic nervous systems [3]. Autonomic nerves directly innervate islets, liver, and adipose tissue, to control hormone secretion, glucose production, and metabolism through release of neurotransmitters and neuropeptides. Further, sensory nerves relay information about metabolic cues from peripheral tissues to the brain and locally release neuropeptides to influence glucose homeostasis. In this review, we focus on the peripheral nervous system and functions of autonomic and sensory innervation of islets, adipose tissue, and liver, in glycemic control. Further, we discuss emerging evidence that suggests that changes in peripheral innervation might contribute to metabolic dysfunction.
Islet innervation
Pancreatic islets are micro-organs consisting of distinct endocrine cell types including insulin-secreting β-cells, glucagon-secreting α-cells, somatostatin-secreting δ-cells, ε-cells that secrete ghrelin, and pancreatic polypeptide (pp)-secreting cells, as well as vascular cells, immune cells, fibroblasts, nerves and glia. Recent studies have revealed distinct inter-species variability in islet organization [4]. Murine islets have a segregated architecture with β-cells found at the core, and α-cells and other endocrine cells located at the periphery or mantle, whereas in human islets, endocrine cells are largely randomly dispersed [5]. Pancreatic islets are richly innervated by sympathetic and parasympathetic fibers, as well as by nociceptive sensory fibers [6] (Figure 1A). While islet-intrinsic mechanisms governing endocrine cell development and functions have received much attention, the role of extrinsic factors, specifically innervation, remain under-appreciated.
Figure 1: Peripheral nerves modulate islet hormone secretion and development.
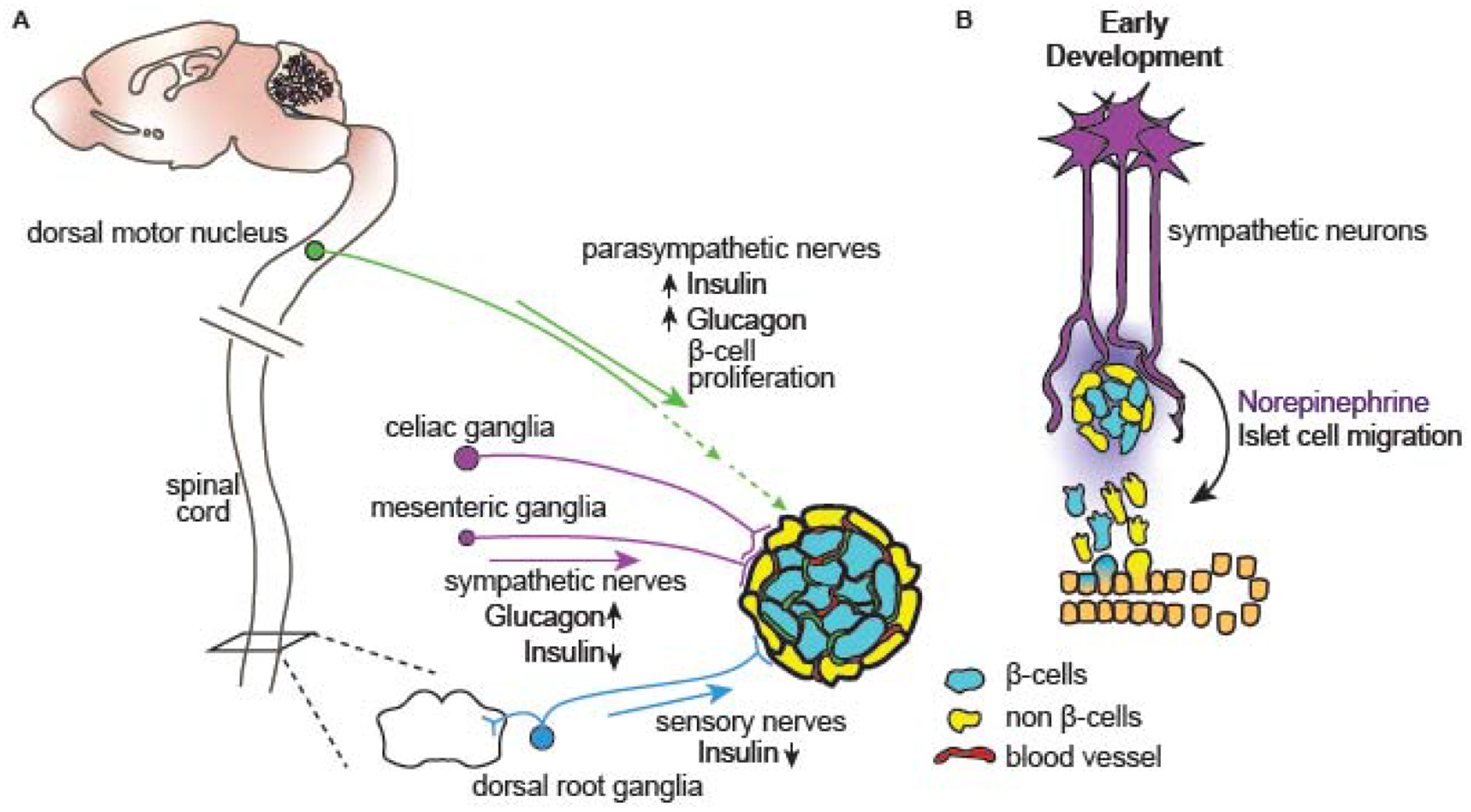
(A) Pancreatic islets of Langerhans are innervated by sympathetic, parasympathetic, and sensory fibers originating from celiac and mesenteric ganglia, dorsal motor vagal nuclei, and dorsal root ganglia, respectively. Sympathetic nerves act to limit insulin secretion and promote glucagon secretion, whereas parasympathetic nerves stimulate secretion of insulin and glucagon, and also influence β-cell proliferation during obesity. Sensory nerves inhibit insulin release. (B) During embryonic development, sympathetic innervation promotes endocrine cell migration from the ductal epithelium and governs islet organization through the release of the neurotransmitter norepinephrine.
Sympathetic nerves
Sympathetic nerves arrive at the pancreas via splanchnic nerves originating from prevertebral celiac and superior mesenteric ganglia [6, 7]. In mice, sympathetic innervation is localized largely at the islet periphery, with nerves making direct contacts with glucagon-secreting α-cells (Figure 1A) [5]. In human islets, sympathetic nerves primarily contact contractile vascular cells, specifically, pericytes and smooth muscle cells, within the islet [5, 8]. These findings suggest that sympathetic innervation influences islet hormone secretion via distinct mechanisms in mice versus humans, with nerve-mediated modulation of hormone release in human islets likely to be indirect. However, other studies, using high-resolution 3-dimensional imaging, argue for species similarities, with mouse islets exhibiting abundant sympathetic nerve-vascular contacts within the islet core similar to that in humans [9].
Defining specific effects of sympathetic innervation on peripheral organ function has been challenging due to wide-spread distribution of sympathetic nerves and difficulties in manipulating projections to a single target tissue. The impact of innervation on islet function has been traditionally investigated through a combination of surgical, electrophysiological, and pharmacological approaches in rodents. Early studies indicated that sympathetic innervation acts to rapidly elevate blood glucose levels. Electrical stimulation of splanchnic nerves inhibits glucose-stimulated insulin secretion (GSIS) and increases glucagon secretion in islets [7]. Sympathetic activity is also critical for the body’s response to hypoglycemia, also known as the counter-regulatory response. Prolonged hypoglycemia, as during fasting, enhances sympathetic activity, which in turn stimulates glucagon release from α-cells to enhance circulating glucose levels [10]. The effect of sympathetic innervation on other islet hormones is less well-characterized, but sympathetic activity reduces secretion of somatostatin and pancreatic polypeptide [7], suggesting that sympathetic nerves have a more expansive role in regulation of blood glucose than currently appreciated.
Effects of sympathetic innervation on islet hormone secretion are largely mediated by the neurotransmitter norepinephrine (NE), acting via α- or β-adrenergic receptors [7]. Electrical activation of sympathetic nerves induces NE release [11], and exogenous NE treatment or adrenergic receptor agonists recapitulate effects of electrical activation of sympathetic nerves on inhibiting GSIS [12, 13]. Conversely, adrenergic receptor antagonists prevent the ability of sympathetic activity to inhibit insulin secretion [14]. The inhibitory effects of sympathetic nerves on insulin secretion are largely mediated by α-adrenergic receptors [7]. In particular, pharmacological and genetic studies in mice support a critical role for the Adra2a receptor in mediating NE-dependent suppression of insulin secretion [15]. Adra2a is the most abundant adrenergic receptor in adult mouse and human islets [15]. Adra2a receptor-specific agonists elicit a profound decrease in islet insulin secretion [13], and animals with overexpression of Adra2a, specifically in β-cells, are glucose-intolerant [16]. In contrast, Adra2a knockout mice have enhanced insulin secretion and improved glucose tolerance [17]. Notably, in humans, a single-nucleotide polymorphism in Adra2a that results in protein over-expression leads to reduced insulin secretion and increased pre-disposition to type 2 diabetes [18]. These findings highlight the potential advantages of Adra2a antagonists in enhancing insulin secretion and treating hyperglycemia in diabetes. In contrast to α-adrenergic receptors, NE acting via islet β-adrenergic receptors are capable of enhancing insulin secretion [7], and activation of both α- and β-adrenergic receptors promotes glucagon secretion [7]. Thus, the precise effects of NE on islet hormone secretion relies on adrenergic receptor abundance and activity. Sympathetic nerves also influence islet function via the neuropeptides galanin and Neuropeptide Y (NPY) [7]. Like NE, Galanin and NPY inhibit insulin secretion and enhance glucagon secretion [19, 20], suggesting these neuropeptides augment neurotransmitter effects.
While the role of sympathetic nerves in acutely regulating islet hormone secretion is well-appreciated, an emergent concept is the potential role for innervation in islet development and maturation. In embryonic mice, ablation of sympathetic nerves results in perturbed islet morphology, with a loss of β-cell-cell contacts and intermingling of α- and β cells at the core [21]. Together, these findings suggest that sympathetic nerves provide key organizational cue(s) during islet formation (Figure 1B). Mice lacking sympathetic nerves are also glucose intolerant later in life [21]. The early effects of sympathetic innervation on islet architecture are mediated, in part, via signaling through β-adrenergic receptors, since developmental blockade of β-adrenergic signaling using propranolol recapitulated the effects of nerve ablation on islet architecture [21]. Together, these findings suggest that sympathetic innervation and NE exert distinct effects on developing versus mature islets (Figure 1B), likely by engaging separate adrenergic receptors.
Autonomic dysfunction is a component of diabetic neuropathy, where damage to peripheral nerves occurs as a long-term consequence of hyperglycemia [22]. However, an early and selective loss of sympathetic innervation to islets occurs in humans with type 1 diabetes and in animal models of the disease, prior to the onset of hyperglycemia [23, 24]. The mechanisms underlying this early and selective loss of islet sympathetic nerves remain unclear. One possibility could involve immune cell infiltration of islets, since the denervation is only observed in animal models of immune-mediated diabetes [25]. The nerve loss could also occur by mechanisms analogous to axon pruning during development, since deletion of the p75 neurotrophin receptor, an essential mediator of the process, also inhibited the loss of innervation in diabetic mice [26, 27]. Further, impaired synaptic transmission in sympathetic ganglia, where the cell bodies reside, is another early event observed in in streptozotocin-induced diabetes, a model of type 1 diabetes [28]. Elevated glucose results in inactivation of nicotinic Acetylcholine Receptors (nAChRs) in sympathetic ganglia, impairing their ability to receive input from pre-ganglionic projections. Together, these studies suggest that sympathetic neuropathy could be an early contributor to pathogenesis in diabetes.
Major questions that remain to be addressed include elucidating the signaling pathways that mediate acute effects of sympathetic neurotransmitters/neuropeptides on hormone secretion, and identity of nerve-derived factors and pathways that underlie effects of innervation on islet development. Notably, β-adrenergic receptor antagonists (β-blockers) are commonly used to treat cardiovascular disease and hypertension, including in pregnant women [29]. Therefore, inhibition of adrenergic signaling during fetal development could have lasting impacts on glycemic control. Finally, neurons and β-cells have distinct developmental origins, but share remarkable similarities in electrical properties, ion channel composition, and exocytosis machinery involved in regulated secretion [30]. Recent studies suggest that islet endocrine cells are capable of secreting several neurotransmitters, including dopamine (see Box 1) [4]. The contributions of islet-intrinsic neurotransmitters to islet function, as well as their mechanisms of action relative to neurotransmission, remain to be elucidated.
Box 1. Neurotransmitter secretion by islet cells.
Recent studies highlight that innervating fibers are not the only source of neurotransmitters in pancreatic islets, but that neurotransmitters are also produced and secreted from islet endocrine cells [4]. Among islet neurotransmitters, dopamine (DA) is the best characterized [31]. DA is synthesized in β-cells and packaged into insulin secretory granules via the vesicular monoamine transporter 2 (VMAT2) [32]. DA then acts in an autocrine fashion to suppress insulin secretion by binding to dopamine receptors on the surface of β-cells [33, 34]. DA is also a biological precursor for NE, raising the possibility that islet cells may secrete NE, although it remains unclear whether Dopamine Beta Hydroxylase (DBH), the enzyme needed to convert DA to NE is expressed in islets [35].
While pancreatic parasympathetic fibers are cholinergic, a recent study suggested that, in human islets, glucagon-producing α-cells are the predominant source of Acetylcholine (ACh). In human islets, parasympathetic fibers are sparser compared to murine islets [5], and the cholinergic enzymes, vesicular acetylcholine transporter vAChT and choline acetyltransferase (ChAT) are detected in α-cells [36]. Notably, human α-cells secrete ACh in response to lowered glucose levels, which has been suggested to act as a paracrine signal to potentiate β-cell function [36]. Serotonin is another neurotransmitter affecting islet function; serotonin regulates β-cell proliferation in rodents and zebrafish [37], although the source of serotonin remains unclear. Serotonin signaling may play a specific role during pregnancy when there is a compensatory expansion in β-cell mass to counter the mild insulin resistance that typically develops during this period [38]. During pregnancy, a dramatic rise in serotonin-synthesizing enzymes, serotonin production, and signaling machinery occurs in rodent islets [38]. Notably, inhibition of the serotonin receptor, 5-hydroxytryptamine receptor-2b (Htr2b), blocks the increase in β-cell mass and causes glucose intolerance in pregnant mice.
The classical inhibitory neurotransmitter, Gamma-amino butyric acid (GABA), has also been suggested to play an important role in modulating islet hormone secretion [39]. GABA is secreted from β-cells, and acts in an autocrine fashion to promote insulin secretion while inhibiting glucagon secretion [39]. Other studies have suggested that GABA contributes to maintenance of islet mass and protects β-cells from apoptosis [40]. Intriguingly, two recent studies reported that GABA or GABA receptor activation by the anti-malarial drug, artemisinin, induces trans-differentiation of α-to-β cells in islets in several species [41, 42]. However, these findings have since been challenged by rigorous lineage tracing of mature α-cells that revealed no effects on their conversion either by long-term GABA or artemisinin treatment [43, 44].
In future studies, it will be of interest to determine the spatial and temporal regulation of endogenous neurotransmitter secretion from islet cells, and to dissect their relative contribution to islet function compared to neurotransmitter release from nerve terminals.
Parasympathetic nerves
Parasympathetic innervation to the pancreas originates from the dorsal motor nucleus of the vagus [7]. Preganglionic vagal nerves project to intrapancreatic ganglia, from which post-ganglionic projections directly innervate islets. Parasympathetic nerves densely innervate mouse islets, contacting both α- and β-cells, in contrast to human islets where the innervation is sparser and follows the blood vessels (Figure 1A) [5]. Stimulation of vagal nerves increases both circulating glucagon and insulin in mammals [7]. Parasympathetic nerves are specifically critical for the body’s response to acute changes in blood glucose [7]. Following a meal, there is an early increase in circulating insulin (<10 minutes in humans) in anticipation of an impending rise in blood glucose levels, termed the “cephalic phase” of insulin secretion [7]. Cephalic insulin secretion is blunted by blockade of parasympathetic nerve activity [45]. Parasympathetic activity also contributes to the counter-regulatory response during hypoglycemia, when vagal efferent nerve activity is increased, which in turn, stimulates glucagon secretion to elicit enhanced circulating glucose [10].
Acetylcholine (ACh) is the primary neurotransmitter secreted by parasympathetic nerves, and islets express all five subtypes of muscarinic ACh receptors (AChRs) [46]. Pharmacological inhibition or deletion of M3 AChRs results in impaired insulin secretion and glucose intolerance in mice, suggesting that it is the most relevant receptor for mediating the effects of cholinergic signaling on insulin secretion [47]. Pancreatic parasympathetic nerves also secrete the neuropeptides vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase-activating polypeptide (PACAP), to augment the effects of ACh in promoting islet hormone secretion [48]. Gene deletion of VIP or PACAP receptors in mice or their pharmacological blockade results in defects in GSIS and impairs glucose tolerance [49, 50].
In addition to regulating hormone secretion, parasympathetic innervation regulates β-cell proliferation. Early studies suggested that vagal hyper-activation, resulting from injury or lesions to the ventromedial hypothalamus, resulted in pancreatic hyperplasia and β-cell proliferation in adult rats [51]. Conversely, vagotomy prevents an increase in β-cell proliferation that occurs to compensate for insulin resistance in mouse models of obesity [52].
Sensory nerves
Afferent sensory nerves leave the pancreas via the splanchnic nerves and relay information from visceral tissues to the CNS [53]. Neuronal tracing indicates that cell bodies of pancreatic sensory nerves lie within the dorsal root ganglia (DRG) and nodose ganglion [54]. Sensory nerves in mouse islets contact the periphery where non-β-cells are found and occasionally penetrate the islet interior (Figure 1A) [8]. The role of sensory nerves in regulating islet hormone secretion remain poorly defined, relative to autonomic innervation. A large proportion of sensory nerves express the non-selective cation channel transient receptor potential vanilloid 1 (TRPV1), and are sensitive to capsaicin [55]. Capsaicin-mediated ablation of sensory nerves at the whole-body level, and specifically in the pancreas, results in enhanced GSIS and improved glucose tolerance without influencing insulin sensitivity or β-cell mass in mice [56]. Thus, sensory innervation appears to inhibit insulin secretion in response to elevated glucose. Interestingly, the effects of sensory denervation are only observed in male mice [56], suggesting sex-specific effects of innervation on glucose homeostasis. Studies in animal models of diabetes suggest that sensory denervation also has beneficial effects on glucose metabolism in pathological conditions. Capsaicin-mediated ablation of sensory nerves prevents progression of diabetes in Zucker Diabetic Fatty (ZDF) rats, a model for type 2 diabetes [57], and limits islet inflammation and β-cell stress in non-obese diabetic (NOD) mice, a model for type 1 diabetes [58].
The neuropeptides calcitonin gene-related polypeptide (CGRP) and substance P (SP), present in sensory nerve terminals in the pancreas, may mediate inhibitory effects of sensory innervation on insulin secretion. CGRP treatment inhibits GSIS in isolated islets [59]. Further, mice with global deletion of α-CGRP, one of two CGRP isoforms in mice, exhibit improved glucose tolerance, insulin sensitivity, and are protected from diet-induced obesity [60]. The effects of SP are less clear, with reports of both inhibitory and stimulatory effects on insulin secretion in isolated islets [7].
Adipose innervation
Adipose tissue is broadly distributed throughout the body as either white adipose tissue (WAT) or brown adipose tissue (BAT), and serves as both an energy storage and endocrine organ. WAT serves primarily as an energy reservoir by storing triglycerides that can be broken down by the process of lipolysis and released during times of need [61]. WAT also dynamically communicates with other peripheral tissues by releasing adipokines to control energy balance and glucose homeostasis [62]. In contrast, BAT has a large capacity for glucose and lipid clearance, which it metabolizes to generate heat in a process called non-shivering adaptive thermogenesis [63]. Similar to WAT, activated BAT releases endocrine factors called batokines to influence glycemic control and insulin sensitivity [64]. In mice, genetic or pharmacological activation of BAT protects from a gain in body fat and metabolic dysfunction caused by high fat feeding [65], whereas BAT dysfunction or ablation elicits insulin resistance and obesity [66]. Notably, WAT adipocytes can be induced to acquire a brown-like phenotype, or de novo adipogenesis can occur in BAT, in response to chronic cold exposure or other stimuli, in a process called “browning/beiging” [65]. The identification of functional BAT and beige fat in humans [67] has raised interest in their potential as anti-obesity targets.
Adipose tissue is densely innervated by sympathetic fibers, which play a key role in adipose tissue-mediated lipolysis, thermogenesis, and glycemic control (Figure 2) [68, 69]. Sensory nerves are key for relaying information to the CNS regarding the adiposity of fat depots [70–72], and for secretion of neuropeptides that locally impact adipocyte function [72] (Figure 2). Parasympathetic nerves are barely detected in adipose tissue, and their role in adipose functions have been proposed to be minor [68]. Attaining more comprehensive knowledge of how peripheral innervation governs adipose function is critical for understanding the role of fat depots in glucose homeostasis, and for designing successful therapies to improve glucose homeostasis and combat obesity.
Figure 2: Adipose tissue innervation regulates lipolysis, thermogenesis, and glucose homeostasis.
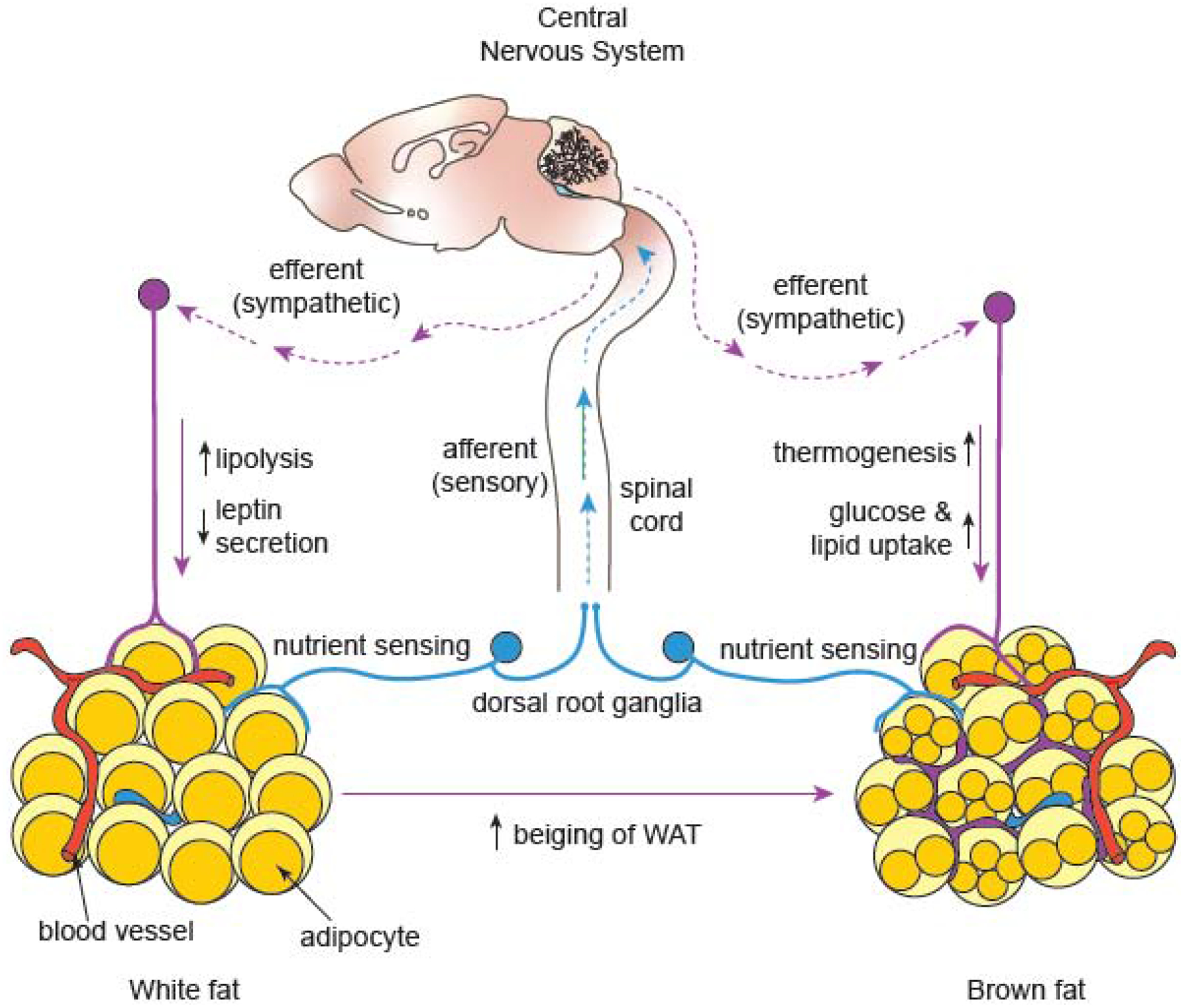
White adipose tissue (WAT) serves as a storage depot for energy-rich triglycerides that are broken down in times of need (lipolysis), whereas brown adipose tissue (BAT) burns glucose and fat to generate heat (thermogenesis). Sympathetic nerves stimulate WAT lipolysis through the release of norepinephrine. Leptin, a WAT-derived hormone, acts on the central nervous system (CNS) to promote lipolysis, which is mediated by sympathetic efferents to WAT. Sympathetic activity also suppresses leptin release as part of a negative feedback loop. In BAT, sympathetic innervation is a key regulator of thermogenesis and promotes the uptake of excess glucose and lipids from the bloodstream. Sympathetic nerves are involved in the conversion of white adipocytes to brown or “beiging”. Sensory nerves relay information about metabolic cues from adipose tissue to the CNS, which in turn, controls sympathetic output to regulate WAT lipolysis and BAT thermogenesis.
Sympathetic innervation
Initially, the degree of sympathetic innervation of WAT, as assessed using conventional methods of immunostaining in thin slices of fixed tissue, was unclear [61]. However, recent advances in tissue clearing and whole organ imaging reveal dense networks of sympathetic fibers in WAT, with nerve terminals in close apposition to over 90% of adipocytes in mice (Figure 2) [73]. Innervation is observed in both the WAT parenchyma and vasculature in several species [69]. Decades of work from several laboratories provide evidence that sympathetic activity enhances lipolysis and fat mobilization [61]. Optogenetic stimulation of sympathetic nerves in WAT indicate that sympathetic activation is sufficient to promote increased lipolysis and deplete fat depots in mice [74]. Conversely, ablation of sympathetic nerves blocks lipolysis, increases fat pad size, and adipocyte proliferation and differentiation [75]. Nerve-derived effects are primarily due to NE acting on adipose β-adrenergic receptors. Mice with deletion of all three β-adrenergic receptors (Adrb1, Adrb2, and Adrb3) exhibit massive obesity in response to a high-fat diet [76].
WAT also exerts endocrine control over blood glucose through secretion of adipokines, most importantly leptin. Circulating leptin acts on neurons in the hypothalamus to inhibit food intake, deplete fat mass, and promote glucose metabolism in the periphery [77]. The sympathetic nervous system mediates some of leptin’s actions, as leptin-sensitive neuronal circuits in the CNS converge on efferent sympathetic nerves to promote lipolysis in white adipocytes [74]. Notably, leptin synthesis and secretion itself is under the control of sympathetic activity, where sympathetic nerves act via β-adrenergic receptors on adipocytes to lower circulating leptin in the fasted state, as a mechanism to couple leptin levels to nutrient availability [78]. Sympathetic neurite density in WAT is also reduced in both mouse and human tissues under pathophysiological conditions of obesity and diabetes [79].
Similar to WAT, sympathetic fibers envelop BAT vasculature and penetrate into the parenchyma to contact adipocytes [80]. However, BAT is more densely innervated by sympathetic nerves than WAT [68]. Enriched innervation of BAT may stem in part from the secretion of tissue-specific growth factors. For example, brown adipocytes selectively express a novel endoplasmic reticulum (ER)-membrane-bound protein, Calsyntenin3b (Clstn3b), which controls the secretion of the growth factor S100b to promote sympathetic innervation of BAT and thermogenesis [81]. PRDM16, a master transcriptional co-regulator in brown adipocytes, promotes brown fat gene expression but suppresses white fat-selective genes, and is also a key regulator of sympathetic innervation of BAT [65]. Sympathetic innervation may not only be dictated by adipocyte-derived factors, but also by monocytes or macrophages that either infiltrate the tissue or are tissue-resident [82–84]. In a recent study, T lymphocytes were found to enhance sympathetic innervation of BAT, in part by driving expression of the TGFβ1 growth factor in adipocytes [85], suggesting interactions between immune, adipose, and nervous systems that regulate metabolism.
The sympathetic nervous system is a central regulator of BAT-mediated thermogenesis in response to cold exposure [63]. Sympathetic nerve-derived NE acts via adrenergic receptors on brown adipocytes to generate heat by triggering biochemical events in mitochondria that uncouple oxidative phosphorylation from ATP synthesis with the help of a specialized mitochondrial protein, Uncoupling Protein 1 (UCP1) [86]. Denervation of BAT leads to loss of UCP1 expression, decreased mitochondrial function, and glucose uptake in animals exposed to cold or on a high-fat diet [65, 87]. Conversely, activation of β-adrenergic receptors, in particular, Adrb3, is sufficient to stimulate BAT thermogenesis and glucose uptake in rodents and humans [88].
Emerging evidence suggests that, in addition to promoting thermogenesis, sympathetic innervation of BAT influences whole-body metabolism by enabling import of excess glucose and lipids from the bloodstream [63]. In mouse models of diabetes and obesity, BAT activity corrects hyperglycemia and hyperlipidemia [65]. BAT is also a source of factors that seem to have endocrine functions by acting on peripheral tissues including the pancreas, liver, or bone to promote insulin secretion, improve glucose tolerance and insulin sensitivity [64]. Knowledge of BAT innervation is largely based on rodent studies. However, similar to rodents, human BAT responds to cold or adrenergic stimulation, and BAT activation correlates with improved glucose tolerance and insulin sensitivity [89].
An important function of sympathetic activity is the development of beige fat, which are white adipocytes that can be triggered to express UCP1, increase mitochondrial activity, and become thermogenic under certain conditions such as cold exposure [90]. The appearance of beige fat is positively correlated with the degree of sympathetic innervation, and UCP1-positive beige adipocytes are specifically localized in areas of dense sympathetic innervation [91]. Notably, sympathectomy or deletion of β-adrenergic receptors in mice prevents beiging in response to cold [73], while treatment with β-adrenergic agonists is sufficient to induce beiging [92], pointing to the necessity and sufficiency of sympathetic activity in the process. Since browning of WAT promotes energy expenditure, there is much interest in generation of beige fat as a therapeutic target in obesity and diabetes.
Sensory innervation
Both CGRP- and SP-positive sensory nerves innervate WAT and BAT in rodents in patterns similar to sympathetic nerves [93], but with the cell bodies residing in the DRG [94] (Figure 2). In contrast to sympathectomy, sensory denervation of WAT increases fat pad mass by hypertrophy rather than proliferation [93], suggesting that sympathetic and sensory nerves influence white adipocytes by distinct mechanisms. A key function for sensory nerves is to detect metabolic signals in adipose tissue and relay the information to the brain [70–72], which in turn controls the sympathetic drive to ultimately affect metabolic homeostasis [72]. Viral tracing of sensory nerves innervating adipose tissue revealed labeling in several hypothalamic and brain stem regions, especially regions involved in the control of sympathetic outflow [94], suggesting that the sensory innervation may be part of a sensory-CNS-sympathetic circuit [72]. Selective denervation of sensory fibers in WAT reduced sympathetic activity in not only WAT, but also BAT, and suppressed BAT-mediated thermogenesis in response to cold exposure [71]. Together, these studies support an essential role for sensory nerves in WAT to relay signals to the CNS to drive sympathetic output to not only control lipid mobilization in WAT, but also to regulate thermogenesis in distant fat depots in BAT (Figure 2). Sensory nerve terminals may be responding to WAT-derived leptin, since nerves express the leptin receptor and are activated by direct leptin injections into WAT in Siberian hamsters [95]. Leptin has been proposed to signal directly through afferent sensory nerves that relay information to the CNS [72, 95]. Localized denervation of sensory nerves by injecting capsaicin directly into intercapsular BAT, the largest BAT depot, impaired thermogenesis, suggesting a similar role in surveillance of adiposity [94].
There are several key open questions regarding functions of sensory innervation in adipose tissue, including the role of sensory innervation in beiging, identity of adipose-derived factors that stimulate sensory nerve terminals, and functions of neuropeptides, CGRP and SP.
Liver innervation in regulation of glucose homeostasis
The liver is a key organ in regulating whole-body metabolism, and disruptions in liver function have major consequences for glucose and lipid homeostasis [96]. The liver is the principal site of glucose storage. During fasting, the liver elevates circulating glucose levels by breakdown of glycogen or de novo glucose synthesis from simpler precursors [96]. Conversely, following a meal, the liver responds by shutting down hepatic glucose production, suppressing glycogenolysis and synthesizing triglycerides for storage [96]. In humans, the influence of peripheral innervation in regulation of hepatic glucose metabolism was initially thought to be limited, since severing of all hepatic nerves during liver transplants did not elicit significant effects on whole-body blood glucose regulation [97]. However, long-term, increased incidence of diabetes, dyslipidemia, and obesity have been reported in liver transplant recipients [98], suggesting a role for the nervous system, in particular autonomic nerves, in contributing to liver regulation of metabolism.
The liver receives both sympathetic and parasympathetic innervation (Figure 3) [99]. Sympathetic innervation arrives at the liver via the celiac and superior mesenteric ganglia, which in turn receive input from pre-ganglionic neurons in the intermediolateral column in the spinal cord and higher brain regions in the brainstem and hypothalamus [99]. According to the few developmental studies to date on liver innervation in both human and mouse tissues, sympathetic innervation starts in the liver during late embryonic development, with the density of innervation progressively increasing postnatally [100]. Parasympathetic innervation originates in the vagal dorsal motor nucleus from which pre-ganglionic nerves project to the liver [99]. In mammals, a common feature in liver innervation is that autonomic nerves are largely found surrounding the hepatic artery, portal vein, and bile ducts [99]. However, there are species differences in the extent of innervation in liver parenchyma, with rodents having reduced innervation relative to humans [99]. In rodents, nerve-mediated modulation of metabolic functions is predominantly indirect, either via the vasculature or by regulating gap junction signaling between parenchymal cells [99, 101].
Figure 3: Liver innervation in the regulation of glucose homeostasis.
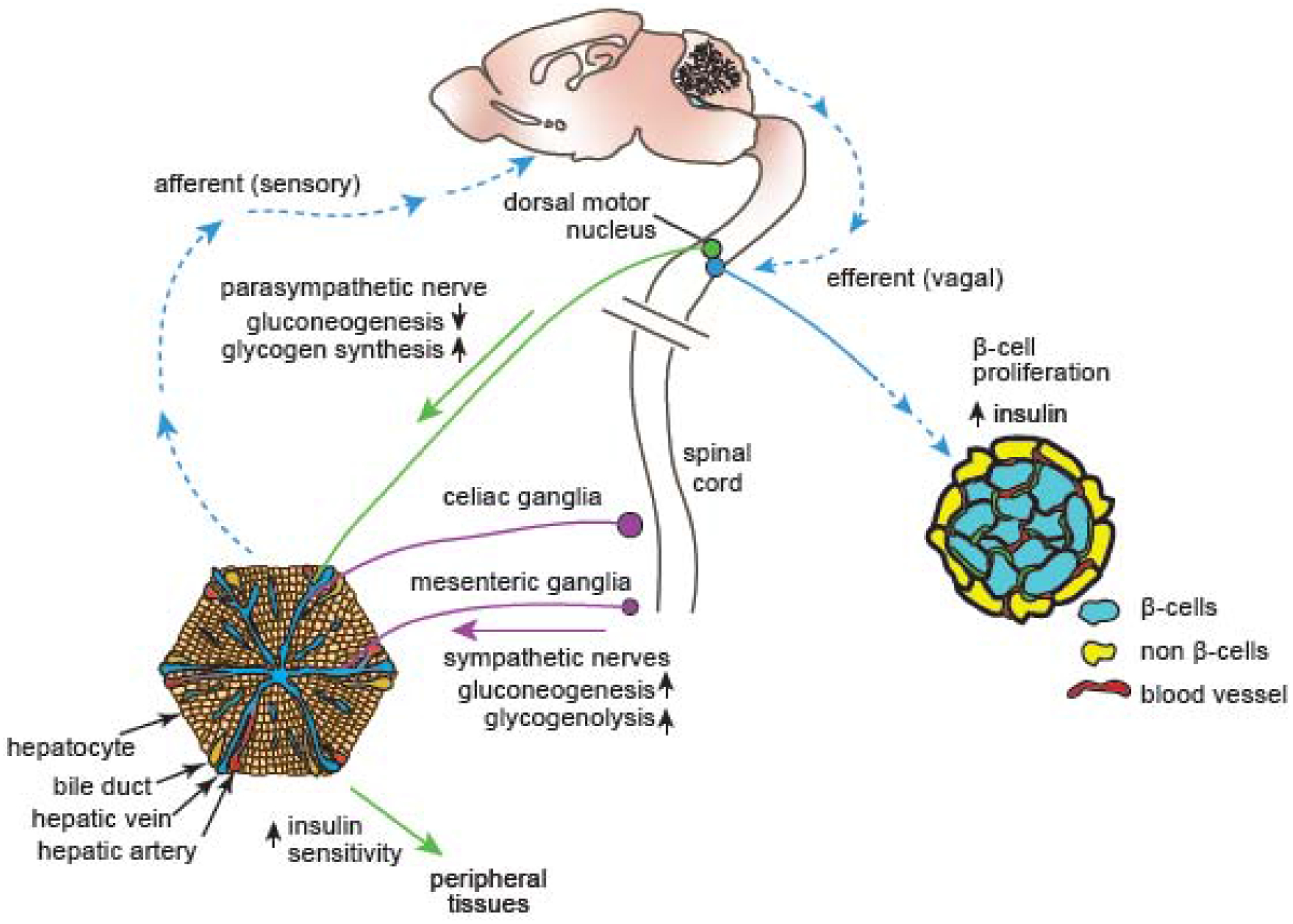
The liver receives sympathetic and parasympathetic innervation. Sympathetic nerves elevate blood glucose levels by promoting glucose production (gluconeogenesis) and the breakdown of glycogen into glucose (glycogenolysis). Conversely, parasympathetic nerves lower blood glucose levels by promoting the storage of glucose as glycogen and inhibiting gluconeogenesis. Hepatic parasympathetic innervation also modulates the insulin responsiveness of several peripheral tissues. A liver-brain-pancreas neuronal relay, consisting of afferent sensory nerves, the central nervous system, and efferent vagal nerves, promotes β-cell proliferation and enhanced insulin secretion as a compensatory response during insulin-resistant conditions of obesity and diabetes.
In early studies, sympathetic activity in the liver was found to rapidly increase circulating glucose by promoting glycogen breakdown to glucose (glycogenolysis) and suppressing glycogen production [102]. Sympathetic control of glycogenolysis proceeds primarily via activation of α-adrenergic receptors, particularly Adra1b, since its deletion resulted in excessive accumulation of glycogen [103]. Sympathetic nerves may also influence hepatic glucose production through release of neuropeptides [104]. Galanin augments the liver’s response to NE in elevating circulating glucose levels [104], while NPY increases glucose uptake by the liver, and opposes hepatic glucose release in response to glucagon and norepinephrine [104].
In contrast to sympathetic innervation, parasympathetic innervation promotes glycogen synthesis and suppresses glucose production [105]. In addition to directly regulating glucose availability, the liver also indirectly regulates blood glucose levels by modulating the insulin sensitivity of other organs [97]. Parasympathetic denervation or cholinergic blockade of the liver reduces insulin sensitivity globally, and diminishes glucose uptake in peripheral tissues [106].
In insulin-resistant states such as in obesity or type 2 diabetes, the liver communicates to the pancreas via systemic factors and neuronal signaling to promote β-cell proliferation to compensate for insulin resistance [107]. In mouse models of insulin resistance or obesity, the liver secretes factors such as serpin, a protease inhibitor, or hepatocyte growth factor (HGF) to enhance β-cell mass [108, 109]. Further, afferent splanchnic nerves carry sensory information from the liver to the brain, which in turn communicates via vagal efferent nerves to the pancreas to elicit enhanced β-cell proliferation or insulin secretion (Figure 3) [110]. This neuronal relay mechanism is activated by extracellular signal-regulated kinase (ERK) signaling in the liver [110], although how ERK signaling activates sensory fibers innervating the liver, and the identity of the vagal-derived factors that promote the pancreatic effects remain to be determined. Nevertheless, these results highlight the potential of developing therapeutic approaches based on innervation in ameliorating aberrant glucose homeostasis in obesity and type 2 diabetes, and perhaps even promoting β-cell regeneration after their loss in type 1 diabetes. The liver is also the only major internal organ that shows significant regenerative potential in adult mammals [111]. Intriguingly, liver regeneration appears to exert beneficial effects on islet function, with increases in islet mass, β-cell proliferation, and insulin secretion observed after partial hepatectomy [112]. Thus, neuronal signaling in the liver-pancreas axis could contribute to restoring metabolic homeostasis following injury or disease.
Concluding Remarks and Future Perspectives
The findings of crosstalk between nerves and peripheral metabolic tissues open up a fertile area for detailed studies that have broad translational implications. Modulation of innervation in glucose-regulatory peripheral tissues presents an emerging avenue for treatment of metabolic dysfunction. To date, much of the information on nerve effects on individual tissues has been gleaned from surgical or genetic ablation of nerves or pharmacological manipulations that come with widespread and non-specific effects. Most previous studies have also focused on the role of neural activity in regulating glucose homeostasis. However, recent studies highlight the considerable structural plasticity of adult peripheral nerves, specifically under conditions of obesity or diabetes [9, 73, 79, 113, 114]. The molecular mechanisms underlying the dynamic regulation of innervation in pathological conditions are poorly understood. Notably, it remains to be determined whether nerve remodeling that occurs in pathological situations is a contributing factor to the pathogenesis or a “bystander effect”. Exciting areas for future research include: gaining a more comprehensive understanding of the anatomical, molecular, and cellular basis of neural connections in glucose-regulatory tissues; clarifying how innervation is established during development and maintained during adult life; identifying the cell types that are directly targeted by nerves; and elucidating nerve-derived factors, in addition to neurotransmitters, that mediate effects on glucose regulation (see Outstanding Questions). In addition to tissues covered in this review, nerves also impact the ability of bone and muscle to regulate glucose homeostasis, either through influencing release of bone-derived hormones that augment insulin secretion [115], or via insulin-independent mechanisms of glucose uptake in skeletal muscle [116]. Thus, the general role of peripheral nerves in promoting inter-organ communication in regulating glucose homeostasis and the mechanistic underpinnings warrant further studies. Further, recent advances in whole organ imaging, tissue clearing, and 3D reconstructions make it possible to comprehensively map peripheral innervation in specific target tissues and reveal regional differences in innervation. Peripheral nerves do not make classical synaptic contacts with targets as in the CNS [117]. Instead, nerve terminals are located 100–200 μm from target cells, and neurotransmitter release occurs from varicosities along the axonal shaft [117]. Advances in serial section electron microscopy have the potential to provide high-resolution insight into cell types that are directly connected by peripheral nerves and morphology of the nerve-target contacts. Further, technological advances in single cell sequencing, optogenetics, and chemical genetic tools offer new opportunities to explore the molecular profiles of specific neuronal sub-types projecting to end-organs and associated circuits. Together, these, and other approaches will allow for more targeted and localized manipulation of interactions between local nerve fibers and target cell types to advance the knowledge of the role of peripheral innervation in metabolic health and whole-body energy balance.
Outstanding Questions.
How is innervation of glucose-regulatory tissues established during development and how is it maintained during adulthood?
Which cell types in metabolic tissues are directly contacted by peripheral nerves, and what morphological specializations and synaptic machinery underlie the nerve-target cell connectivity?
Other than the classical neurotransmitters and neuropeptides, what nerve-derived factors act on peripheral cell types to elicit metabolic outcomes?
Do peripheral nerves play a broader role than currently appreciated in mediating crosstalk between glucose-regulatory organs? If so, what are the underlying mechanisms of inter-organ communication?
To date, little is known about heterogeneity of sympathetic neurons. Do molecularly distinct sympathetic neuron populations control diverse functional effects in islets, adipose tissue, and liver? If so, what are the identities of these neuronal sub-types?
Peripheral nerves innervating metabolic tissues remodel their connections and arborization patterns in pathological conditions such as diabetes and obesity. Do these changes in innervation contribute to metabolic dysfunction? What molecular mechanisms underlie nerve plasticity in disease states?
Highlights.
Metabolic tissues such as pancreatic islets, adipose tissue, and liver receive extensive innervation from peripheral sympathetic, parasympathetic, and sensory nerves.
Peripheral nerves regulate hormone secretion, energy expenditure, and systemic glucose homeostasis through the release of neurotransmitters and neuropeptides.
Structural plasticity of innervation in glucose-regulatory tissues is observed in pathophysiological conditions including obesity and diabetes.
Defining the anatomical, cellular, and molecular bases of nerve-metabolic target tissue crosstalk may allow for specific modulation of peripheral nerves in the development of therapeutic avenues for metabolic disorders.
Acknowledgements:
We thank Jessica Houtz for helpful comments. We apologize to authors whose work could not be cited due to space limitations. The authors’ work is supported by a NIH F32 fellowship award to E.E. Lin (DK11648), NIH R01 awards (NS114478 and NS107342) to R. Kuruvilla.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: The authors declare no competing interests.
References:
- 1.Guariguata L et al. (2014) Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res Clin Pract 103 (2), 137–49. [DOI] [PubMed] [Google Scholar]
- 2.Hogan P et al. (2003) Economic costs of diabetes in the US in 2002. Diabetes Care 26 (3), 917–32. [DOI] [PubMed] [Google Scholar]
- 3.Bentsen MA et al. (2019) Revisiting How the Brain Senses Glucose-And Why. Cell Metab 29 (1), 11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caicedo A (2013) Paracrine and autocrine interactions in the human islet: more than meets the eye. Semin Cell Dev Biol 24 (1), 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodriguez-Diaz R et al. (2011) Innervation patterns of autonomic axons in the human endocrine pancreas. Cell Metab 14 (1), 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woods SC and Porte D Jr. (1974) Neural control of the endocrine pancreas. Physiol Rev 54 (3), 596–619. [DOI] [PubMed] [Google Scholar]
- 7.Ahren B (2000) Autonomic regulation of islet hormone secretion--implications for health and disease. Diabetologia 43 (4), 393–410. [DOI] [PubMed] [Google Scholar]
- 8.Chien HJ et al. (2019) Human pancreatic afferent and efferent nerves: mapping and 3-D illustration of exocrine, endocrine, and adipose innervation. Am J Physiol Gastrointest Liver Physiol 317 (5), G694–G706. [DOI] [PubMed] [Google Scholar]
- 9.Chiu YC et al. (2012) 3-D imaging and illustration of the perfusive mouse islet sympathetic innervation and its remodelling in injury. Diabetologia 55 (12), 3252–61. [DOI] [PubMed] [Google Scholar]
- 10.Taborsky GJ Jr. et al. (2002) Autonomic mechanism and defects in the glucagon response to insulin-induced hypoglycaemia. Diabetes Nutr Metab 15 (5), 318–22; discussion 322–3. [PubMed] [Google Scholar]
- 11.Ahren B et al. (1987) Sympathetic nerve stimulation versus pancreatic norepinephrine infusion in the dog: 1). Effects on basal release of insulin and glucagon. Endocrinology 121 (1), 323–31. [DOI] [PubMed] [Google Scholar]
- 12.Porte D Jr. and Williams RH (1966) Inhibition of insulin release by norepinephrine in man. Science 152 (3726), 1248–50. [DOI] [PubMed] [Google Scholar]
- 13.Skoglund G et al. (1988) Selective alpha 2-adrenoceptor activation by clonidine: effects on 45Ca2+ efflux and insulin secretion from isolated rat islets. Acta Physiol Scand 132 (3), 289–96. [DOI] [PubMed] [Google Scholar]
- 14.Kurose T et al. (1990) Mechanism of sympathetic neural regulation of insulin, somatostatin, and glucagon secretion. Am J Physiol 258 (1 Pt 1), E220–7. [DOI] [PubMed] [Google Scholar]
- 15.Fagerholm V et al. (2011) alpha2-adrenoceptor regulation of blood glucose homeostasis. Basic Clin Pharmacol Toxicol 108 (6), 365–70. [DOI] [PubMed] [Google Scholar]
- 16.Devedjian JC et al. (2000) Transgenic mice overexpressing alpha2A-adrenoceptors in pancreatic beta-cells show altered regulation of glucose homeostasis. Diabetologia 43 (7), 899–906. [DOI] [PubMed] [Google Scholar]
- 17.Peterhoff M et al. (2003) Inhibition of insulin secretion via distinct signaling pathways in alpha2-adrenoceptor knockout mice. Eur J Endocrinol 149 (4), 343–50. [DOI] [PubMed] [Google Scholar]
- 18.Rosengren AH et al. (2010) Overexpression of alpha2A-adrenergic receptors contributes to type 2 diabetes. Science 327 (5962), 217–20. [DOI] [PubMed] [Google Scholar]
- 19.Ahren B et al. (2004) Loss-of-function mutation of the galanin gene is associated with perturbed islet function in mice. Endocrinology 145 (7), 3190–6. [DOI] [PubMed] [Google Scholar]
- 20.Imai Y et al. (2007) Insulin secretion is increased in pancreatic islets of neuropeptide Y-deficient mice. Endocrinology 148 (12), 5716–23. [DOI] [PubMed] [Google Scholar]
- 21.Borden P et al. (2013) Sympathetic innervation during development is necessary for pancreatic islet architecture and functional maturation. Cell Rep 4 (2), 287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bodman MA and Varacallo M (2020) Peripheral Diabetic Neuropathy. In StatPearls. [PubMed] [Google Scholar]
- 23.Mei Q et al. (2002) Early, selective, and marked loss of sympathetic nerves from the islets of BioBreeder diabetic rats. Diabetes 51 (10), 2997–3002. [DOI] [PubMed] [Google Scholar]
- 24.Persson-Sjogren S et al. (2005) Remodeling of the innervation of pancreatic islets accompanies insulitis preceding onset of diabetes in the NOD mouse. J Neuroimmunol 158 (1–2), 128–37. [DOI] [PubMed] [Google Scholar]
- 25.Mundinger TO and Taborsky GJ Jr. (2016) Early sympathetic islet neuropathy in autoimmune diabetes: lessons learned and opportunities for investigation. Diabetologia 59 (10), 2058–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taborsky GJ Jr. et al. (2014) The p75 neurotrophin receptor is required for the major loss of sympathetic nerves from islets under autoimmune attack. Diabetes 63 (7), 2369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh KK et al. (2008) Developmental axon pruning mediated by BDNF-p75NTR-dependent axon degeneration. Nat Neurosci 11 (6), 649–58. [DOI] [PubMed] [Google Scholar]
- 28.Campanucci V et al. (2010) Diabetes depresses synaptic transmission in sympathetic ganglia by inactivating nAChRs through a conserved intracellular cysteine residue. Neuron 66 (6), 827–34. [DOI] [PubMed] [Google Scholar]
- 29.Parati G and Esler M (2012) The human sympathetic nervous system: its relevance in hypertension and heart failure. Eur Heart J 33 (9), 1058–66. [DOI] [PubMed] [Google Scholar]
- 30.Arntfield ME and van der Kooy D (2011) beta-Cell evolution: How the pancreas borrowed from the brain: The shared toolbox of genes expressed by neural and pancreatic endocrine cells may reflect their evolutionary relationship. Bioessays 33 (8), 582–7. [DOI] [PubMed] [Google Scholar]
- 31.Ustione A et al. (2013) Minireview: Dopaminergic regulation of insulin secretion from the pancreatic islet. Mol Endocrinol 27 (8), 1198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raffo A et al. (2008) Role of vesicular monoamine transporter type 2 in rodent insulin secretion and glucose metabolism revealed by its specific antagonist tetrabenazine. J Endocrinol 198 (1), 41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rubi B et al. (2005) Dopamine D2-like receptors are expressed in pancreatic beta cells and mediate inhibition of insulin secretion. J Biol Chem 280 (44), 36824–32. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Tornadu I et al. (2010) Disruption of the dopamine d2 receptor impairs insulin secretion and causes glucose intolerance. Endocrinology 151 (4), 1441–50. [DOI] [PubMed] [Google Scholar]
- 35.Mitok KA et al. (2018) Islet proteomics reveals genetic variation in dopamine production resulting in altered insulin secretion. J Biol Chem 293 (16), 5860–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez-Diaz R et al. (2011) Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nat Med 17 (7), 888–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang G et al. (2015) First quantitative high-throughput screen in zebrafish identifies novel pathways for increasing pancreatic beta-cell mass. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim H et al. (2010) Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med 16 (7), 804–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Q et al. (2019) GABAergic regulation of pancreatic islet cells: Physiology and antidiabetic effects. J Cell Physiol. [DOI] [PubMed] [Google Scholar]
- 40.Soltani N et al. (2011) GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc Natl Acad Sci U S A 108 (28), 11692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ben-Othman N et al. (2017) Long-Term GABA Administration Induces Alpha Cell-Mediated Beta-like Cell Neogenesis. Cell 168 (1–2), 73–85 e11. [DOI] [PubMed] [Google Scholar]
- 42.Li J et al. (2017) Artemisinins Target GABAA Receptor Signaling and Impair alpha Cell Identity. Cell 168 (1–2), 86–100 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ackermann AM et al. (2018) GABA and Artesunate Do Not Induce Pancreatic alpha-to-beta Cell Transdifferentiation In Vivo. Cell Metab 28 (5), 787–792 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van der Meulen T et al. (2018) Artemether Does Not Turn alpha Cells into beta Cells. Cell Metab 27 (1), 218–225 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strubbe JH (1992) Parasympathetic involvement in rapid meal-associated conditioned insulin secretion in the rat. Am J Physiol 263 (3 Pt 2), R615–8. [DOI] [PubMed] [Google Scholar]
- 46.Iismaa TP et al. (2000) Quantitative and functional characterization of muscarinic receptor subtypes in insulin-secreting cell lines and rat pancreatic islets. Diabetes 49 (3), 392–8. [DOI] [PubMed] [Google Scholar]
- 47.Gautam D et al. (2006) A critical role for beta cell M3 muscarinic acetylcholine receptors in regulating insulin release and blood glucose homeostasis in vivo. Cell Metab 3 (6), 449–61. [DOI] [PubMed] [Google Scholar]
- 48.Moody TW et al. (2011) VIP and PACAP: recent insights into their functions/roles in physiology and disease from molecular and genetic studies. Curr Opin Endocrinol Diabetes Obes 18 (1), 61–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsutsumi M et al. (2002) A potent and highly selective VPAC2 agonist enhances glucose-induced insulin release and glucose disposal: a potential therapy for type 2 diabetes. Diabetes 51 (5), 1453–60. [DOI] [PubMed] [Google Scholar]
- 50.Jamen F et al. (2000) PAC1 receptor-deficient mice display impaired insulinotropic response to glucose and reduced glucose tolerance. J Clin Invest 105 (9), 1307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kiba T et al. (1996) Ventromedial hypothalamic lesion-induced vagal hyperactivity stimulates rat pancreatic cell proliferation. Gastroenterology 110 (3), 885–93. [DOI] [PubMed] [Google Scholar]
- 52.Edvell A and Lindstrom P (1998) Vagotomy in young obese hyperglycemic mice: effects on syndrome development and islet proliferation. Am J Physiol 274 (6), E1034–9. [DOI] [PubMed] [Google Scholar]
- 53.Brunicardi FC et al. (1995) Neural regulation of the endocrine pancreas. Int J Pancreatol 18 (3), 177–95. [DOI] [PubMed] [Google Scholar]
- 54.Fasanella KE et al. (2008) Distribution and neurochemical identification of pancreatic afferents in the mouse. J Comp Neurol 509 (1), 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caterina MJ et al. (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389 (6653), 816–24. [DOI] [PubMed] [Google Scholar]
- 56.Bou Karam J et al. (2018) TRPV1 neurons regulate beta-cell function in a sex-dependent manner. Mol Metab 18, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gram DX et al. (2007) Capsaicin-sensitive sensory fibers in the islets of Langerhans contribute to defective insulin secretion in Zucker diabetic rat, an animal model for some aspects of human type 2 diabetes. Eur J Neurosci 25 (1), 213–23. [DOI] [PubMed] [Google Scholar]
- 58.Razavi R et al. (2006) TRPV1+ sensory neurons control beta cell stress and islet inflammation in autoimmune diabetes. Cell 127 (6), 1123–35. [DOI] [PubMed] [Google Scholar]
- 59.Pettersson M and Ahren B (1990) Calcitonin gene-related peptide inhibits insulin secretion studies on ion fluxes and cyclic AMP in isolated rat islets. Diabetes Res 15 (1), 9–14. [PubMed] [Google Scholar]
- 60.Walker CS et al. (2010) Mice lacking the neuropeptide alpha-calcitonin gene-related peptide are protected against diet-induced obesity. Endocrinology 151 (9), 4257–69. [DOI] [PubMed] [Google Scholar]
- 61.Bartness TJ et al. (2014) Neural innervation of white adipose tissue and the control of lipolysis. Front Neuroendocrinol 35 (4), 473–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scheja L and Heeren J (2019) The endocrine function of adipose tissues in health and cardiometabolic disease. Nat Rev Endocrinol 15 (9), 507–524. [DOI] [PubMed] [Google Scholar]
- 63.Cannon B and Nedergaard J (2004) Brown adipose tissue: function and physiological significance. Physiol Rev 84 (1), 277–359. [DOI] [PubMed] [Google Scholar]
- 64.Villarroya F et al. (2017) Brown adipose tissue as a secretory organ. Nat Rev Endocrinol 13 (1), 26–35. [DOI] [PubMed] [Google Scholar]
- 65.Wang W and Seale P (2016) Control of brown and beige fat development. Nat Rev Mol Cell Biol 17 (11), 691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lowell BB et al. (1993) Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature 366 (6457), 740–2. [DOI] [PubMed] [Google Scholar]
- 67.Nedergaard J et al. (2007) Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab 293 (2), E444–52. [DOI] [PubMed] [Google Scholar]
- 68.Bartness TJ et al. (2010) Sympathetic and sensory innervation of brown adipose tissue. Int J Obes (Lond) 34 Suppl 1, S36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bartness TJ et al. (2010) Sensory and sympathetic nervous system control of white adipose tissue lipolysis. Mol Cell Endocrinol 318 (1–2), 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Garretson JT et al. (2016) Lipolysis sensation by white fat afferent nerves triggers brown fat thermogenesis. Mol Metab 5 (8), 626–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nguyen NLT et al. (2018) Sensory denervation of inguinal white fat modifies sympathetic outflow to white and brown fat in Siberian hamsters. Physiol Behav 190, 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guilherme A et al. (2019) Molecular pathways linking adipose innervation to insulin action in obesity and diabetes mellitus. Nat Rev Endocrinol 15 (4), 207–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang H et al. (2017) Dense Intra-adipose Sympathetic Arborizations Are Essential for Cold-Induced Beiging of Mouse White Adipose Tissue. Cell Metab 26 (4), 686–692 e3. [DOI] [PubMed] [Google Scholar]
- 74.Zeng W et al. (2015) Sympathetic neuro-adipose connections mediate leptin-driven lipolysis. Cell 163 (1), 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harris RBS (2018) Denervation as a tool for testing sympathetic control of white adipose tissue. Physiol Behav 190, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bachman ES et al. (2002) betaAR signaling required for diet-induced thermogenesis and obesity resistance. Science 297 (5582), 843–5. [DOI] [PubMed] [Google Scholar]
- 77.Friedman JM and Halaas JL (1998) Leptin and the regulation of body weight in mammals. Nature 395 (6704), 763–70. [DOI] [PubMed] [Google Scholar]
- 78.Caron A et al. (2018) Leptin and brain-adipose crosstalks. Nat Rev Neurosci 19 (3), 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blaszkiewicz M et al. (2019) Neuropathy and neural plasticity in the subcutaneous white adipose depot. PLoS One 14 (9), e0221766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blaszkiewicz M et al. (2019) The Importance of Peripheral Nerves in Adipose Tissue for the Regulation of Energy Balance. Biology (Basel) 8 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zeng X et al. (2019) Innervation of thermogenic adipose tissue via a calsyntenin 3beta-S100b axis. Nature 569 (7755), 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wolf Y et al. (2017) Brown-adipose-tissue macrophages control tissue innervation and homeostatic energy expenditure. Nat Immunol 18 (6), 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pirzgalska RM et al. (2017) Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat Med 23 (11), 1309–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Villarroya F et al. (2018) Toward an Understanding of How Immune Cells Control Brown and Beige Adipobiology. Cell Metab 27 (5), 954–961. [DOI] [PubMed] [Google Scholar]
- 85.Hu B et al. (2020) gammadelta T cells and adipocyte IL-17RC control fat innervation and thermogenesis. Nature 578 (7796), 610–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhao J et al. (1998) Thermogenesis is beta3- but not beta1-adrenergically mediated in rat brown fat cells, even after cold acclimation. Am J Physiol 275 (6), R2002–11. [DOI] [PubMed] [Google Scholar]
- 87.Rothwell NJ and Stock MJ (1984) Effects of denervating brown adipose tissue on the responses to cold, hyperphagia and noradrenaline treatment in the rat. J Physiol 355, 457–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cypess AM et al. (2015) Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab 21 (1), 33–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chondronikola M et al. (2014) Brown adipose tissue improves whole-body glucose homeostasis and insulin sensitivity in humans. Diabetes 63 (12), 4089–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Giordano A et al. (2014) White, brown and pink adipocytes: the extraordinary plasticity of the adipose organ. Eur J Endocrinol 170 (5), R159–71. [DOI] [PubMed] [Google Scholar]
- 91.Chi J et al. (2018) Three-Dimensional Adipose Tissue Imaging Reveals Regional Variation in Beige Fat Biogenesis and PRDM16-Dependent Sympathetic Neurite Density. Cell Metab 27 (1), 226–236 e3. [DOI] [PubMed] [Google Scholar]
- 92.Himms-Hagen J et al. (1994) Effect of CL-316,243, a thermogenic beta 3-agonist, on energy balance and brown and white adipose tissues in rats. Am J Physiol 266 (4 Pt 2), R1371–82. [DOI] [PubMed] [Google Scholar]
- 93.Shi H et al. (2005) Sensory or sympathetic white adipose tissue denervation differentially affects depot growth and cellularity. Am J Physiol Regul Integr Comp Physiol 288 (4), R1028–37. [DOI] [PubMed] [Google Scholar]
- 94.Vaughan CH and Bartness TJ (2012) Anterograde transneuronal viral tract tracing reveals central sensory circuits from brown fat and sensory denervation alters its thermogenic responses. Am J Physiol Regul Integr Comp Physiol 302 (9), R1049–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Murphy KT et al. (2013) Leptin-sensitive sensory nerves innervate white fat. Am J Physiol Endocrinol Metab 304 (12), E1338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lin HV and Accili D (2011) Hormonal regulation of hepatic glucose production in health and disease. Cell Metab 14 (1), 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mizuno K and Ueno Y (2017) Autonomic Nervous System and the Liver. Hepatol Res 47 (2), 160–165. [DOI] [PubMed] [Google Scholar]
- 98.Laryea M et al. (2007) Metabolic syndrome in liver transplant recipients: prevalence and association with major vascular events. Liver Transpl 13 (8), 1109–14. [DOI] [PubMed] [Google Scholar]
- 99.McCuskey RS (2004) Anatomy of efferent hepatic nerves. Anat Rec A Discov Mol Cell Evol Biol 280 (1), 821–6. [DOI] [PubMed] [Google Scholar]
- 100.Delalande JM et al. (2004) Hepatic nervous system development. Anat Rec A Discov Mol Cell Evol Biol 280 (1), 848–53. [DOI] [PubMed] [Google Scholar]
- 101.Seseke FG et al. (1992) Signal propagation via gap junctions, a key step in the regulation of liver metabolism by the sympathetic hepatic nerves. FEBS Lett 301 (3), 265–70. [DOI] [PubMed] [Google Scholar]
- 102.Shimazu T (1996) Innervation of the liver and glucoregulation: roles of the hypothalamus and autonomic nerves. Nutrition 12 (1), 65–6. [DOI] [PubMed] [Google Scholar]
- 103.Burcelin R et al. (2004) Impaired glucose homeostasis in mice lacking the alpha1b-adrenergic receptor subtype. J Biol Chem 279 (2), 1108–15. [DOI] [PubMed] [Google Scholar]
- 104.Mundinger TO and Taborsky GJ Jr. (2000) Differential action of hepatic sympathetic neuropeptides: metabolic action of galanin, vascular action of NPY. Am J Physiol Endocrinol Metab 278 (3), E390–7. [DOI] [PubMed] [Google Scholar]
- 105.Shimazu T (1967) Glycogen synthetase activity in liver: regulation by the autonomic nerves. Science 156 (3779), 1256–7. [DOI] [PubMed] [Google Scholar]
- 106.Lautt WW et al. (2001) Hepatic parasympathetic (HISS) control of insulin sensitivity determined by feeding and fasting. Am J Physiol Gastrointest Liver Physiol 281 (1), G29–36. [DOI] [PubMed] [Google Scholar]
- 107.Imai J (2018) Regulation of compensatory beta-cell proliferation by inter-organ networks from the liver to pancreatic beta-cells. Endocr J 65 (7), 677–684. [DOI] [PubMed] [Google Scholar]
- 108.El Ouaamari A et al. (2016) SerpinB1 Promotes Pancreatic beta Cell Proliferation. Cell Metab 23 (1), 194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Araujo TG et al. (2012) Hepatocyte growth factor plays a key role in insulin resistance-associated compensatory mechanisms. Endocrinology 153 (12), 5760–9. [DOI] [PubMed] [Google Scholar]
- 110.Imai J et al. (2008) Regulation of pancreatic beta cell mass by neuronal signals from the liver. Science 322 (5905), 1250–4. [DOI] [PubMed] [Google Scholar]
- 111.Ozaki M (2020) Cellular and molecular mechanisms of liver regeneration: Proliferation, growth, death and protection of hepatocytes. Semin Cell Dev Biol 100, 62–73. [DOI] [PubMed] [Google Scholar]
- 112.Moreau F et al. (2014) Early effects of liver regeneration on endocrine pancreas: in vivo change in islet morphology and in vitro assessment of systemic effects on beta-cell function and viability in the rat model of two-thirds hepatectomy. Horm Metab Res 46 (13), 921–6. [DOI] [PubMed] [Google Scholar]
- 113.Mundinger TO et al. (2016) Human Type 1 Diabetes Is Characterized by an Early, Marked, Sustained, and Islet-Selective Loss of Sympathetic Nerves. Diabetes 65 (8), 2322–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang P et al. (2020) A leptin-BDNF pathway regulating sympathetic innervation of adipose tissue. Nature 583 (7818), 839–844. [DOI] [PubMed] [Google Scholar]
- 115.Clemens TL and Karsenty G (2011) The osteoblast: an insulin target cell controlling glucose homeostasis. J Bone Miner Res 26 (4), 677–80. [DOI] [PubMed] [Google Scholar]
- 116.Nonogaki K (2000) New insights into sympathetic regulation of glucose and fat metabolism. Diabetologia 43 (5), 533–49. [DOI] [PubMed] [Google Scholar]
- 117.Smolen AJ (1988) Morphology of synapses in the autonomic nervous system. J Electron Microsc Tech 10 (2), 187–204. [DOI] [PubMed] [Google Scholar]