

SUMMARY
Crop productivity depends on activity of meristems that produce optimized plant architectures, including that of the maize ear. A comprehensive understanding of development requires insight into the full diversity of cell types and developmental domains, and the gene networks required to specify them. Until now, these were identified primarily by morphology and insights from classical genetics, that are limited by genetic redundancy and pleiotropy. Here, we investigated the transcriptional profiles of 12,525 single cells from developing maize ears. The resulting developmental atlas provides a single-cell RNA sequencing (scRNA-seq) map of an inflorescence. We validated our results by mRNA in situ hybridization and by Fluorescence-Activated Cell Sorting (FACS) RNA-seq, and show how these data may facilitate genetic studies by predicting genetic redundancy, integrating transcriptional networks, and identifying candidate genes associated with crop yield traits.
Keywords: Maize ear, meristem, scRNA-seq, developmental networks, GWAS, trait genes
In Brief
Xu et al. construct and validate a single-cell transcriptomic atlas of developing maize ears. Their single cell gene co-expression networks will facilitate developmental genetics studies by predicting genetic redundancy and revealing transcriptional regulatory networks. Their results also inform maize breeding by identifying candidate trait-associated genes.
Graphical Abstract
INTRODUCTION
Plant architecture is initiated by meristems made up of pluripotent stem cells and their descendants that are organized in distinct cell types and domains with specific functions. In developing maize ears, a series of meristems build inflorescence architecture, including spikelet pair meristems (SPMs) formed from the inflorescence meristem (IM) and spikelet meristems (SMs) made from the branching of SPMs (Irish, 1997). Mutant studies have identified key cell type or domain-specific regulators that orchestrate inflorescence architecture by specifying different developmental domains (Vollbrecht and Schmidt, 2009). For example, the homeodomain transcription factor encoded by KNOTTED1 (KN1) is critical for meristem establishment and maintenance, and is expressed throughout shoot meristems (Jackson et al., 1994). The production of axillary meristems to elaborate branching architecture depends on expression of a basic helix-loop-helix transcription factor encoded by BARREN STALK1 (BA1), expressed specifically in the adaxial meristem periphery where axillary meristems initiate (Gallavotti et al., 2004). Another transcription factor, BRANCHED SILKLESS1 (BD1), is expressed at the boundary of meristems and glumes to control spikelet architecture by promoting meristem determinacy (Chuck et al., 2002), whereas RAMOSA genes, such as RA1 and RA3, are expressed in an arc of cells at the base of meristems, to impose determinacy on spikelet branches (Satoh-Nagasawa et al., 2006). Many of these key regulators have reshaped inflorescence architecture during evolution or domestication, and their discovery was enabled by the availability of mutants that block specific aspects of development. However, such insights are limited by genetic redundancy and pleiotropy, thus, a high-resolution expression atlas of specific cell types and domains is needed to gain further insights into the gene networks that control development.
Single-cell RNA sequencing (scRNA-seq) offers the opportunity to assay gene expression with high resolution, and to construct developmental maps of complex organs or organisms (Kulkarni et al., 2019, Potter, 2018). Recently, the 10x Genomics Chromium scRNA-seq platform has been used extensively to identify cell type or domain markers in Arabidopsis roots (Rich-Griffin et al., 2020), however the application of this technology to shoot tissues has been limited. As well as providing expression information, scRNA-seq data can be integrated with other genomic datasets, such as ChIP-seq identification of targets of transcription factors, or surveys of chromatin status. Such datasets have been generated from developing maize inflorescences (Bolduc et al., 2012, Eveland et al., 2014, Pautler et al., 2015), however single-cell data has not yet been integrated.
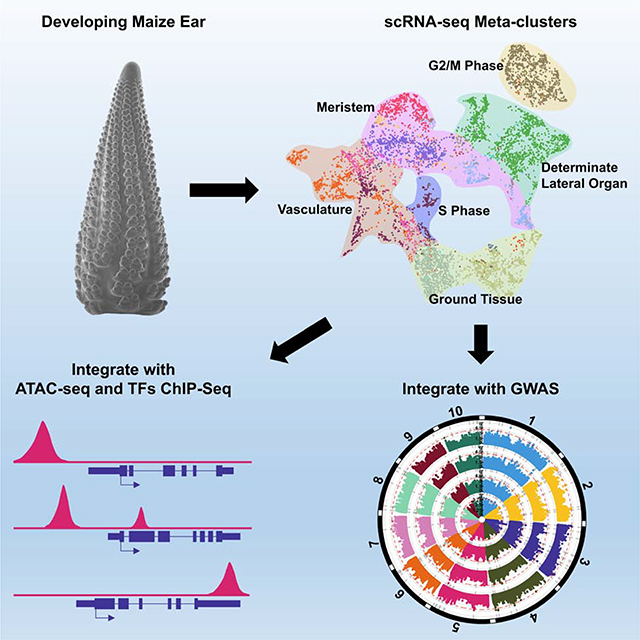
Productivity of maize depends on development of the inflorescences, in particular the seed-bearing ear. Genome-Wide Association Studies have identified candidate genes associated with yield-related traits (Liu et al., 2020) that can guide breeding or trait engineering. Regulatory genes functioning in early stages of ear development show significant association with ear yield traits (Vollbrecht and Schmidt, 2009, Bommert et al., 2013, Je et al., 2016, Liu et al., 2020), yet it remains challenging to identify and validate such regulators on a genome-wide scale. To fill these gaps, we optimized protocol using 10x Genomics scRNA-seq technology to generate a high-resolution transcriptome atlas of the developing maize ear inflorescence. We illustrate how this data can enhance maize genetics by predicting genetic redundancy, build transcriptional regulatory networks at cell-type resolution, and identify candidate loci associated with ear yield traits.
RESULTS
Construction of a Single-Cell Transcriptome Atlas of the Developing Maize Ear
To generate a single-cell atlas from developing maize ears, we used the 5–10mm stage, where major developmental and architectural decisions, including meristem initiation, maintenance and determinacy, organ specification and differentiation of vascular and ground tissues, are being made (Irish, 1997, Vollbrecht and Schmidt, 2009). We first optimized a cell wall digestion method, taking into account the different composition of grass cell walls (Ortiz-Ramírez et al., 2018), that allowed us to isolate ear protoplasts within ~45 minutes (see STAR Methods). However, developing ear protoplasts were fragile, and we removed small debris and organelles from broken cells (Figure S1A) by filtration followed by FACS (see STAR Methods; Figure 1A) before loading into the 10x Genomics Chromium Controller. Then, scRNA-seq libraries were generated and sequenced on the Illumina platform (Table S1). In total, we profiled 12,525 individual cells from three independent replicates (Table S1), and detected expression from 28,899 genes using maize V3 reference, comparable to the number detected by bulk RNA-seq of the same tissue (Eveland et al., 2014, Pautler et al., 2015).
Figure 1: Isolation of Maize Ear Protoplasts to Construct a Single-Cell Transcriptomic Atlas.

(A) Experimental design, the first panel shows a scanning electron microscope image of a 5–10mm developing ear (scale bar = 2mm), second panel image of ear protoplasts, scale bar = 50μm. (B) MetaNeighbor identifies 12 reproducible meta-clusters (left color blocks) across three biological replicates (top color blocks) of single-cell RNA-seq datasets. (C) 12 meta-clusters displayed by an integrated Uniform Manifold Approximation and Projection (UMAP) plot in two dimensions, with each dot representing a cell. (D-N) UMAP plots of marker genes predicting the identities of meta-clusters, with color scale indicating normalized expression level. (D) KN1, meristem, all meta-clusters except 3 and 6; (E) BD1, meristem boundary, meta-cluster 9; (F) BA1, adaxial meristem periphery, meta-cluster 11; (G) RA3, meristem base, meta-cluster 10; (H) ZmHDZIV8, epidermis, meta-clusters 6 and part of 3; (I) ZmYAB14, determinate lateral organ, meta-cluster 3; (J) ZmTMO5, xylem, meta-cluster 4; (K) ZmAPL, phloem, meta-cluster 5; (L) ZmSHR1, bundle sheath, meta-cluster 12; (M) GRMZM2G345700 (2G345700), cortex, meta-cluster 1; (N) ZmNAC122, pith, meta-cluster 8. (O) Sketches of longitudinal section of a spikelet meristem (left panel) and transverse section of vascular bundle (right panel) showing cell/domain identities in scRNA-seq meta-clusters.
Technical variation and sparse data in scRNA-seq make it challenging to identify reproducible clusters (groups of cells) that represent homogenous cell types across technical replicates (Crow et al., 2018). We used MetaNeighbor to ask how well the identity of cells in a given cluster of one replicate can be predicted based on their similarity to a cluster from another replicate (Crow et al., 2018), reported as the average area under the receiver operating characteristic curve (AUROC). All cluster pairs with AUROCs > 0.9 in both directions, across at least two replicates, were used to merge and identify 12 replicable cell identity clusters (hereafter referred to as meta-clusters) (Figure 1B). As some genes may be affected by protoplasting (the process to isolate protoplasts), we used mRNA-seq to compare total ear protoplasts with freshly dissected, intact developing ear tissue, and identified 713 protoplasting-responsive genes (FDR < 0.05, |log2FC| > 2, Table S1). After excluding these genes, > 97% of the highly variable genes used for generating clusters were unchanged, as were the meta-clusters (Figures S1B and S1C). Thus, protoplasting-responsive genes did not affect clustering, as reported previously (Ma et al., 2020).
Prediction and Validation of Meta-Cluster Identities
We visualized meta-clusters using a Uniform Manifold Approximation and Projection (UMAP) plot, where we could track the distribution of genes of interest (Figure 1C). Next, to predict identities for each meta-cluster, we compiled a list of known or predicted inflorescence development marker genes, whose expression patterns have been studied in maize or Arabidopsis (Table S1). Among them, 74 are functionally characterized by their mutant phenotype in maize. Importantly, we detected the expression of 73 of these genes, and each had enriched expression in one or more of the 12 meta-clusters (Figures 1D–1N and S2; Table S1). For example, to identify meristem cell types, we used KN1, which is expressed throughout the meristem as well as the developing stem and vascular tissues, but strictly excluded from the epidermis and determinate lateral organ (Jackson et al., 1994). KN1 was expressed in multiple (10 out of 12) meta-clusters, as expected (Figure 1D). Ear architecture is governed by branching events that are controlled by genes expressed in different meristem domains. To identify meta-clusters representing these domains, we searched for expression of characterized marker genes, including BD1, which is expressed at the boundary of spikelet meristems (Figures 1E and 1O) (Chuck et al., 2002), and found it to be uniquely expressed in meta-cluster 9, identifying this as a meristem boundary meta-cluster (Figure 1E). Another well-characterized marker, BA1, is expressed in a distinct adaxial meristem periphery domain (Figures 1F and 1O) (Gallavotti et al., 2004), and was expressed in meta-cluster 11 (Figure 1F). A third cellular domain that controls branching is marked by an arc of expression of RAMOSA genes at the meristem base (Figures 1G and 1O) that partially overlaps with BA1 (Satoh-Nagasawa et al., 2006, Vollbrecht and Schmidt, 2009). Correspondingly, we found expression of RAMOSA genes in meta-cluster 11 (Satoh-Nagasawa et al., 2006), similar to BA1, as well as in meta-cluster 10 (Figure 1G). As such, we could identify three of the KN1 expressing meta-clusters as distinct branching domains.
KN1 expression is excluded from the meristem epidermis and determinate lateral organs (Jackson et al., 1994). We found epidermis marker gene ZmHOMEODOMAIN LEUCINE ZIPPER IV8 (ZmHDZIV8) (Javelle et al., 2011) was highly enriched in the two meta-clusters, 3 and 6, that did not express KN1 (Figure 1H), while determinate lateral organ marker gene ZmYABBY14 (ZmYAB14) (Strable et al., 2017) was expressed throughout meta-cluster 3 (Figure 1I), suggesting that meta-cluster 3 was a determinate lateral organ meta-cluster while meta-cluster 6 corresponded to meristem epidermis cells (Figure 1O). Consistently, meta-cluster 3 was significantly enriched for organ initiation genes from a maize shoot laser capture microdissection (LCM) study (q < 0.001) (Table S1) (Knauer et al., 2019). Using additional markers, we identified four distinct sub-clusters of meta-cluster 3 (Figure S2A), including determinate lateral organ epidermis domain (marker = LIPID TRANSFER PROTEIN1 (LTP1)) (Takacs et al., 2012), and non-epidermis domain (marker = ZmRIBULOSE BISPHOSPHATE CARBOXYLASE SMALL SUBUNIT 1A (ZmRBCS1A)), as well as adaxial domain (marker = DROOPING LEAF2/ZmYABBY7 (DRL2/ZmYAB7)) and abaxial domain (marker = Homolog of Arabidopsis AUXIN RESPONSE FACTOR3/4 (ZmARF3/4-LIKE1)) (Chitwood et al., 2007).
KN1 is also expressed in developing vascular tissues (Jackson et al., 1994). To identify distinct vascular meta-clusters, we used maize homologs of Arabidopsis genes (De Rybel et al., 2016). For example, xylem marker ZmTARGET OF MONOPTEROS 5 (ZmTMO5), and its paralogs ZmTMO5-LIKE1 and 2, were expressed in meta-cluster 4 (Figures 1J, 1O and S2B). We also found a sub-cluster of predicted maturing xylem cells in meta-cluster 4 (Figure S2B), using homologs of Arabidopsis marker genes for secondary cell walls and tracheary elements, including ZmMYB DOMAIN PROTEIN 46 (ZmMYB46) (Zhong et al., 2007) and ZmXYLEM CYSTEINE PEPTIDASE 2 (ZmXCP2) (Funk et al., 2002). In contrast, we found that meta-cluster 5 represented phloem cells, as shown by specific expression of a maize homolog of Arabidopsis ALTERED PHLOEM DEVELOPMENT (Figures 1K and 1O) (De Rybel et al., 2016). Phloem tissues include distinctive sieve element and companion cells, which were reflected in sub-clusters marked by maize homologs of Arabidopsis protophloem sieve element marker PHLOEM EARLY DOF 1 (PEAR1)/PEAR2 (Miyashima et al., 2019) and companion cell marker PHLOEM PROTEIN 2-LIKE A1 (Figure S2C) (Guo et al., 2018). Therefore, meta-clusters 4 and 5 correspond to xylem and phloem cells, respectively, and we found significant enrichment of genes in these meta-clusters with vascular markers in a maize LCM study (q < 0.001) (Table S1) (Knauer et al., 2019). In addition to meta-cluster 4 and 5, cells in meta-cluster 12 also expressed vascular marker genes (q < 0.001) (Table S1) (Knauer et al., 2019), including bundle sheath markers, such as ZmSHORT-ROOT (ZmSHR1) (Figures 1L and 1O) (Chang et al., 2012), thus meta-cluster 12 was predicted to be bundle sheath.
The remainder of the developing ear corresponds to ground tissue, including the outer cortex and inner pith tissues (Figures 1M–1O and S2D). A maize homolog, GRMZM2G345700, of the most unique Arabidopsis root cortex marker, AT1G62510, a bifunctional lipid-transfer / 2S albumin superfamily gene (Denyer et al., 2019), had restricted expression in meta-cluster 1 (Figure 1M), as did stem cortex marker homolog ZmNITRATE TRANSPORTER 1/PEPTIDE TRANSPORTER FAMILY 6.4-LIKE 2 (ZmNPF6.4-LIKE2) (Figure S2D) (Tong et al., 2016). We thus predicted this meta-cluster to be cortex. In contrast, we predicted that meta-cluster 8 was comprised of pith cells, by specific expression of homologs of sorghum or Arabidopsis markers, such as ZmNO APICAL MERISTEM DOMAIN CONTAINING (NAC) TRANSCRIPTION FACTOR 122 (ZmNAC122), GRMZM2G430849, a homolog of Sobic.006G147400 (Figure 1N) (Fujimoto et al., 2018), or GRMZM2G039074, a homolog of AT2G3830, an MYB transcriptional regulator (Figure S2D) (Schürholz et al., 2018). Finally, meta-clusters 2 and 7 were highly enriched for expression of cell cycle genes, such as ZmCYCLINB1;2 (ZmCYCB1;2) and ZmHISTONE2A12 (ZmHIS2A12), indicating that these two meta-clusters contained dividing cells at different phases of the cell cycle (Figure S2D); similar cell cycle clusters are found in root scRNA-seq studies (Denyer et al., 2019, Rich-Griffin et al., 2020). We calculated the percentage of cells in each meta-cluster (Figure S2E). 23% of cells were from meristem domains (meta-clusters 6, 9, 10 and 11), 21% from ground tissues (meta-clusters 1 and 8), 20% from vascular tissues (meta-clusters 4, 5 and 12) and 19% from determinate lateral organ tissues (meta-cluster 3). In summary, using maize inflorescence development markers and homologs of markers from other plants, we predicted the cell or domain identities of all 12 meta-clusters (Figure 1C), and in several cases sub-divided them into more specific cell types or developmental stages.
To validate our predicted meta-cluster identities, we first used differential expression (DE) analysis to identify marker genes with AUROCs ≥ 0.7 in at least one replicate, and identified 813 candidate markers (Table S1). The top markers of each meta-cluster were further selected based on the percentage of cells expressing the marker, and showed highly enriched expression, as expected (Figure 2A). We prioritized a set of these markers by predicted developmental roles and validated them using in situ hybridization (Figures 2B–2M; Table S1). For example, marker genes for meta-cluster 9, GRMZM2G004528, annotated as ZmMYO-INOSITOL PHOSPHATE SYNTHASE2 (ZmMIPS2), predicted to act in auxin signaling and transport (Chen and Xiong, 2010), and GRMZM2G097989, annotated as ZmGLUTATHIONE TRANSFERASE 41 (ZmGST41), involved in meristem size control (Horváth et al., 2019), showed specific expression in the meristem boundary, similar to BD1 (Figures 2B and 2C) (Chuck et al., 2002). ZmGST41 was also a DE marker for meta-cluster 12, and consistently showed vascular trace expression (Figure 2C, arrow). Markers of a second meristem meta-cluster, 11, included GRMZM2G038284, which was expressed in the adaxial meristem periphery similar to BA1 (Figure 2D), and encodes a homolog of Arabidopsis DROUGHT INDUCED19, of interest because maize ear development is especially sensitive to drought stress (Nuccio et al., 2015). Two additional markers of meta-cluster 11, GRMZM2G034152, which encodes a ZmPOLYAMINE OXIDASE 1 (ZmPAO1) (Figure 2E), and GRMZM2G430522, a homolog of Arabidopsis CUP-SHAPED COTYLEDON 3 (ZmCUC3-LIKE) (Figure S3A) also showed restricted expression at the adaxial meristem periphery. Meta-clusters 3 and 6 were predicted to have an epidermal identity, and specific expression was observed as expected for markers such as EF517601.1_FG016, annotated as MALE FLOWER SPECIFIC 18 (Figure 2F), and GRMZM2G126397, a ZmPHOSPHOLIPID TRANSFER PROTEIN3 (ZmPLTP3) gene (Figure 2G). Moving away from the meristem, marker genes for meta-cluster 3, predicted to be determinate lateral organ, included GRMZM2G019686, annotated as ZmFLOWERING PROMOTING FACTOR 1 (ZmFPF1) (Figure 2H), and GRMZM2G075255, annotated as ZmECERIFERUM1 (ZmCER1) (Figure 2I), and showed expected expression pattern.
Figure 2: Validation of scRNA-seq by mRNA in situ and FACS RNA-seq.

(A) The top two marker genes of each meta-cluster are shown in dot plots with circle size indicating the percentage of cells expressing the marker and color representing Z_scored expression value. (B-M) mRNA in situ of meta-clusters marker genes validates the predicted identities: (B) ZmMIPS2, meristem boundary; (C) ZmGST41, meristem boundary (meta-cluster 9) and bundle sheath (meta-cluster 12, red arrow); (D-E) ZmDI19 (D) and ZmPAO1 (E), adaxial meristem periphery; (F-G) MFS18 (F) and ZmPLTP3 (G), meristem epidermis (meta-cluster 6) and determinate lateral organ (meta-cluster 3) epidermis; (H-I) ZmFPF1 (H) and ZmCER1 (I), determinate lateral organ; (J) ZmTMO5-LIKE3, xylem (red arrow indicates xylem vessels); (K) ZmZNF30, phloem. (L) ZmCYCB2–4, cell cycle G2/M phase; (M) ZmHIS2A, cell cycle S phase. Scale bar = 100μm. (N) Collection of RFP protoplasts from pZmYAB14-TagRFPt reporter line using FACS. Scale bar = 100μm. Three biological replicates are collected for FACS RNA-seq. One biological replicate is collected for FACS ATAC-seq. (O) Log2(fold change(FC)) of determinate lateral organ domain enriched markers, ZmYAB genes, and depleted marker, KN1, between RFP and total control protoplasts (Control) in FACS RNA-seq. (P) Volcano plot with 1-sided test positions the hits of enriched markers from pZmYAB14-TagRFPt FACS RNA-seq (red dots) on the ranked list of scRNA-seq differentially expressed (DE) genes from meta-cluster 3 (black circles). X-axis indicates the mean log2(FC) of DE genes between meta-cluster 3 and all other meta-clusters. Y-axis indicates corresponding −log10(p-value). (Q) pZmYAB14-TagRFPt FACS RNA-seq and scRNA-seq meta-cluster 3 have concordant differential gene expression patterns with Area Under the Receiver Operating Characteristics (AUROC) score = 0.8 (indicated by curved line; dashed line indicates the null (AUROC score = 0.5)). Axes indicates the true and false positive rate, the proportion of pZmYAB14-TagRFPt FACS RNA-seq enriched markers that do or do not match to scRNA-seq meta-cluster 3 enriched markers, respectively. (R) meta-cluster 3 DE genes are enriched in open chromatin in pZmYAB14-TagRFPt FACS sorted cells, see text for details.
We also identified candidate vascular markers in meta-clusters 4 and 5. Predicted xylem markers included ZmTARGET OF MONOPTEROS5-LIKE3 (ZmTMO5-LIKE3), GRMZM2G176141 (Figure 2J), ZmTRANSMEMBRANE AMINO ACID TRANSPORTER FAMILY PROTEIN (ZmTMAAT), GRMZM2G109865 (Figure S3B), and ZmWALLS ARE THIN 1 (ZmWAT1), GRMZM2G007953 (Figure S3C), and all showed specific expression identified by the distinctive cell walls of xylem vessels. Interestingly, some xylem markers were also expressed in meristem tips, mostly enriched in central zone (Figures S3D–S3F). We also confirmed the specific expression of meta-cluster 5 (phloem) marker, GRMZM2G116079, which encodes Zinc Finger Protein 30 (ZmZNF30), whose Arabidopsis homolog, AT3G15680, is predicted to be involved in RNA regulation (Figure 2K) (Gipson et al., 2020). Lastly, candidate markers from meta-clusters 2 and 7, predicted to be dividing cells, included several cyclin and histone encoding genes, such as ZmCYCLINB2–4 (ZmCYCB2–4), GRMZM2G061287 (Figure 2L), and ZmHISTONE2A (ZmHIS2A), GRMZM2G305046 (Figure 2M), and had punctate expression, as expected.
FACS RNA-seq has been used to validate scRNA-seq data in Arabidopsis roots (Rich-Griffin et al., 2020). Few marker lines are available in maize, but one, pZmYAB14-TagRFPt (Je et al., 2016), is specifically expressed in determinate lateral organ (Figure 2N). We introgressed this reporter into a bd1;Tunicate (bd1;Tu) double mutant background, which produces highly proliferative ears, to generate large amounts of ear tissue. We made protoplasts from this tissue, and used FACS to sort RFP positive cells, followed by RNA-seq to identify lateral organ domain-specific genes (Figure 2N; Table S2). We found highly enriched expression of ZmYAB14 and other YAB genes (Strable et al., 2017) in the FACS sorted cells, while negative control markers, such as KN1 (Jackson et al., 1994) were significantly depleted (Figure 2O), as expected. We identified 2,040 differentially expressed genes (FDR < 0.05) (Table S2), and as we expected the majority were differentially expressed (AUROC score 0.8) in scRNA-seq meta-cluster 3, with predicted lateral organ identity, validating our scRNA-seq data (Figures 2P and 2Q).
We also used FACS sorted pZmYAB14-TagRFPt expressing cells in an ATAC-seq experiment to investigate how chromatin accessibility changes during differentiation of determinate lateral organs in the ear. Genome-wide analysis of accessible chromatin regions (ACRs) found that 31% mapped within 10kb upstream of the transcription start site (TSS), and 18% localized to transcription termination sites (TTS), Untranslated Regions (UTRs), exons or introns, comparable to whole ear tissue ATAC-seq results (Figure S3G; Table S2) (Ricci et al., 2019). 60% of all maize genes had ACRs in pZmYAB14-TagRFPt FACS sorted cells (Figure 2R; Table S2), and this value was significantly enriched for DE genes from scRNA-seq meta-cluster 3 (71%; p < 0.001, chi-squared test, Figure 2R; Table S2). As expected, several ZmYAB genes that were significantly enriched in scRNA-seq meta-cluster 3 had accessible chromatin, (Figure S3H), and we validated the expression of two meta-cluster 3 marker genes with ACRs by in situ hybridization (Figures S3I and S3J), including GRMZM2G026556, a homolog of Arabidopsis BLADE ON PETIOLE2, which controls lateral organ fate (Ha et al., 2007), and GRMZM2G004012, homolog of Arabidopsis PLANTACYANIN, that plays a role in the development of reproductive organs (Dong et al., 2005). To gain further insight into these data, we subtracted the determinate lateral organ specific ATAC-seq peaks from whole developing ear ATAC-seq peaks (Ricci et al., 2019) to predict ACRs specific to indeterminate meristem tissues, as well as vascular and ground tissues (Figure S3K, Table S2). scRNA-seq marker genes corresponding to these domains (Figure S3K; Table S1) were significantly enriched with these ACR associated genes (p < 0.001, chi-squared test; Figure S3K; Table S2). Thus, the integration of ATAC-seq and scRNA-seq may provide insights into chromatin accessibility and its effect on gene expression in specific cell types or developmental contexts.
scRNA-seq Networks Predict Redundancy
Gene redundancy often masks the phenotype of single-gene knockouts (Lloyd and Meinke, 2012), however, distinguishing redundant from non-redundant paralogs can be challenging. In a recent study of the maize branching mutant ramosa3 (ra3), we identified a ra3 enhancer as its’ paralog, ZmTREHALOSE PHOSPHATE PHOSPHATASE 4 (ZmTPP4) (Claeys et al., 2019). Among 12 maize ZmTPP genes, two of them, ZmTPP4 and ZmTPP12, are upregulated in ra3 mutants (Eveland et al., 2014), a common predictor of compensating redundant paralogs (Rodriguez-Leal et al., 2019). However, CRISPR knockouts of ZmTPP12 do not affect ear development, nor do they enhance ra3 (Claeys et al., 2019). Neither of the two paralogs is more similar in sequence to RA3, so to ask why ZmTPP4, and not ZmTPP12, acts as a redundant compensator, we queried their co-expression. In an aggregate network across 89 maize bulk tissue RNA-seq datasets (Lee et al., 2020), RA3 and its two paralogs had similar co-expression scores (Figure S4A). In contrast, ZmTPP4 was highly co-expressed with RA3 in our single-cell data, similar to a RA3-RA1 positive control (Satoh-Nagasawa et al., 2006), whereas ZmTPP12 was not (Figure S4B). Thus, functional redundancy in maize ear branching could be predicted by co-expression in scRNA-seq, but not in bulk tissue RNA-seq networks. To test this idea further, we identified a small gene family of VASCULAR PLANT ONE-ZINC-FINGER (ZmVOZ) genes, whose homologs regulate flowering time in Arabidopsis (Yasui et al., 2012). Two of them, ZmVOZ4 and ZmVOZ5 exhibited highly similar co-expression within our scRNA-seq data (Spearman correlation 0.88, Figures S4C–S4H), and we identified a common set of high confidence genes that showed consistent co-expression with both genes (FDR < 0.05, Table S3). To ask if these paralogs acted redundantly, we made CRISPR/Cas9 knockouts of all ZmVOZ members, including ZmVOZ1 and 2 that were not detected in our scRNA-seq dataset, possibly due to their low expression in ears. As predicted, single Zmvoz mutants had no obvious phenotype, but Zmvoz1,2,4,5 quadruple mutants were severely delayed in the floral transition, reminiscent of voz1,2 double mutants in Arabidopsis (Figure 3A) (Yasui et al., 2012). Therefore, these two examples highlight the utility of maize ear scRNA-seq data in predicting genetic redundancy.
Figure 3: scRNA-seq Can Predict Genetic Redundancy and Aid in Predicting Transcriptional Regulatory Networks.

(A) Maize plant with CRISPR/cas9 knockout of four ZmVOZ paralogs fails to transition to flowering, as shown by two month old shoot apex (left bottom panel, scale bar = 100μm), and a 6 month old plant that lacks ears or tassel. (B) Directly modulated transcriptional targets of KN1 are significantly co-expressed with KN1 at the single-cell level; all maize genes are used as control (p < 0.01, one-way ANOVA with Tukey’s HSD). (C-D) Expression of TF translational fusion lines, ZmHDZIV6-YFP (C, merge of YFP channel and bright field) and ZmM16-YFP (D, merge of YFP and DAPI channels), used for two biological replicates of ChIP-seq. Scale bar = 100μm. (E-F) Expected motifs are significantly over-represented in bound peaks of ZmHDZIV6 (E) or ZmM16 (F). (G, J) ZmHDZIV6 candidate modulated targets, ZmNIP1A (G) and ZmPROPEP1 (J) are highly co-expressed with ZmHDZIV6 in scRNA-seq (Jaccard index = 0.155 for both targets). (H, K) ZmHDZIV6 bound peaks in ZmNIPA1 (H) and ZmPROPEP1 (K). (I, L) ZmNIP1A (I) and ZmPROPEP1 (L) are specifically expressed in the epidermis, by mRNA in situ. Scale bar = 100μm.
Using scRNA-seq to Build Transcriptional Regulatory Networks
We next asked if scRNA-seq might aid in building transcriptional regulatory networks, given that directly modulated targets of a transcription factor (TF) should be co-expressed in the same cell types. We used our scRNA-seq data to calculate co-expression of KN1 with its published directly modulated targets (Table S3) (Bolduc et al., 2012), and found that it was significantly higher than expected compared to a control using all maize genes (p < 0.01), supporting our hypothesis (Figure 3B). Thus, we next generated two additional ChIP-seq datasets, for ZmHOMEODOMAIN LEUCINE ZIPPER IV6 (ZmHDZIV6) (Javelle et al., 2011), which was uniquely expressed in the epidermis (Figures 3C and S4I), and ZmMADS16 (ZmM16) (Bartlett et al., 2015), which was expressed in specific floral organ (Figure 3D). Biological replicates for each TF ChIP-seq had significant overlap (Figure S4J; Table S3), and we identified 907 high confidence peaks for ZmHDZIV6, and 1,155 for ZmM16 (Table S3). ~60% of these peaks mapped to gene regions, with a preference for promoters (Figures S4K and S4L), similar to other maize ChIP-seq studies (Bolduc et al., 2012). Members of the homeodomain leucine zipper IV family bind a GCATTAAATGC consensus sequence (Nakamura et al., 2006), and we found a similar sequence in motif analysis of ZmHDZIV6 bound peaks (Figure 3E). Similarly, motif analysis of ZmM16 bound peaks found an expected MADS binding motif, CC(A/T)6GG (Figure 3F) (Aerts et al., 2018). We were thus confident in our ZmHDZIV6 and ZmM16 bound target predictions (Table S3).
Modulated TF targets are often inferred by comparison of ChIP-seq bound targets and expression changes in mutant RNA-seq. However, since ZmHDZIV6 and ZmM16 are members of large gene families and mutant RNA-seq of them was not available. We therefore asked if we could predict modulated targets based on scRNA-seq co-expression with each TF (Table S3). We thereby identified 79 and 55 candidate modulated targets for ZmHDZIV6 and ZmM16 respectively, using a Jaccard index co-expression cutoff of ≥ 0.05 (Table S3). Among the predicted modulated targets of ZmHDZIV6, we identified five additional members of the ZmHDZIV family (Figures S4M and S4N; Table S3), some of which are similarly expressed in the maize SAM epidermis (Javelle et al., 2011) and might form transcriptional cascades to regulate epidermal differentiation. We validated the epidermal expression of additional candidate modulated targets, including ZmNOD26-LIKE MEMBRANE INTRINSIC PROTEIN1 (ZmNIP1A, GRMZM2G041980) and ZmPRECURSOR ELICITOR PEPTIDE1 (ZmPROPEP1, GRMZM5G899080) (Figures 3G–3L). Homologs of these genes in Arabidopsis function as transporters (Liu et al., 2009), or in pathogen defense (Huffaker et al., 2011), suggesting similar roles in the maize ear epidermis. Similarly, among the 55 co-expressed ChIP-seq targets identified for ZmM16, we found additional members of the MADS family, such as ZmSEP3/ZmMADS7 and ZmAGAMOUS-LIKE 8 (ZmAGL8) (Figures S4O and S4P; Table S3), indicating that these genes might act downstream of ZmM16 to form gene regulatory networks controlling inflorescence development, analogous to MADS networks in Arabidopsis (Chen et al., 2018).
scRNA-seq Identifies Genes Associated with Maize Yield Traits
Maize ear morphology is associated with yield traits (Je et al., 2016, Liu et al., 2020). To ask if the cell- or domain-specific genes identified in our scRNA-seq overlapped with candidate regulators of maize yield, we used a targeted GWAS approach, by comparing our scRNA-seq marker genes from meristem, determinate lateral organ and vascular meta-clusters against a GWAS panel of 281 maize lines phenotyped for ear morphology traits related to yield (Figures 4A–4D; Table S3) (Rice et al., 2020). Using SNPs in or within 2kb of scRNA-seq marker genes, we found the meta-cluster 3 marker gene ZmYABBY9 (ZmYAB9) had two significant SNPs (at 5% FDR) for cob weight (CW) (Figure 4B). We also found two significant SNPs (at 10% FDR) associated with ear diameter (ED), with minor additive effects (Figures 4C and 4D). One was associated with GRMZM2G361210, a marker of meristem branching related meta-clusters 9, 10, and 11 (Figure 4C), that encodes a C2H2-type zinc finger transcription factor related to RAMOSA1 (RA1), a major player in maize domestication that controls branching and grain yield traits (Sigmon and Vollbrecht, 2010). A second significant SNP for ear diameter (ED) was associated with ZmTMO5, GRMZM2G043854 (Figure 4D), a xylem meta-cluster marker (Figure 1J), whose Arabidopsis homolog controls periclinal cell divisions during vascular development (De Rybel et al., 2016).
Figure 4: scRNA-seq Marker Genes Are Associated with Maize Ear Traits.

(A) Diagrams of 9 different ear traits measured for GWAS analysis: Ear Length (EL), Seed Set Length (SSL), Ear Rank Number (EKN), Ear Diameter (ED), Cob Diameter (CD), Ear Row Number (ERN), Ear Weight (EW), Cob Weight (CW), Kernel Weight (20 Seeds) (KW). (B-D) Targeted GWAS using SNPs in or within 2kb of genes reveals that scRNA-seq marker gene ZmYAB9, has significant SNPs for ear weight (B), and two marker genes GRMZM2G361210 (2G361210) (C) and ZmTMO5 (D) have significant SNPs for ear diameter. ** FDR threshold of 0.05, * FDR threshold of 0.1. Y-axis indicates the –log10(p-value) (Table S3). (E-G) Lambda values of scRNA-seq marker genes (red lines) are greater than two standard deviations from mean lambda values of 1,000 random gene sets (histogram distributions) for ear diameter (2kb partition, E, 200kb partition, F), and for seed set length trait (200kb partition, G). Lambda values are reported in Table S3. (H-I) Distributions of SNP heritability (ℎ2) using 2kb (H) or 200kb (I) partitions; ℎ2 values for scRNA-seq marker genes (purple dots) for the given traits (*) are greater than the top 5% permuted ℎ2 values (red bars) using 1000 random subsets of maize genes (grey violin plots). ℎ2 values are reported in Table S3.
We also conducted lambda analysis (Parvathaneni et al., 2020) to ask if scRNA-seq marker SNPs were more significantly associated with yield traits compared to a random subset of maize genes (Table S3; see STAR Methods). Using SNPs in or within 2kb of genes (2kb partition), we indeed found that ear diameter (ED) was significantly associated (Figures 4A and 4E; Table S3), suggesting that scRNA-seq marker genes preferentially control this trait (Figure 4E). Given that natural variation in distal regulatory elements also controls maize domestication and yield traits (Liu et al., 2020), we also considered SNPs within 200kb of gene coding regions (200kb partition), and found again that ear diameter (ED) was significantly associated (Figure 4F), as was an additional yield related trait, seed set length (SSL) (Figures 4A and 4G).
Although lambda analysis detects significance for a target set of markers, it does not quantify the level of trait variability that is explained by those markers. Therefore, we next estimated narrow-sense heritability (ℎ2) for scRNA-seq markers, compared to a distribution of ℎ2 estimates from random subsets of markers (Table S3; see STAR Methods). We found that SNP heritability for scRNA-seq markers was consistently greater than the 95th percentile of ℎ2 estimates from random markers for ear diameter (ED), cob weight (CW), and seed set length (SSL) traits (Figures 4A, 4H–4I), validating our targeted GWAS and lambda analysis. Similar findings were obtained for ear length (EL) in the 2kb partition (Figure 4H), as well as for ear row number (ERN) and ear rank number (EKN) in the 200kb partition (Figure 4I).
To ask if scRNA-seq data was required for these insights, we also calculated a list of “whole ear” specific genes from bulk tissue RNA-seq data (Table S3) (Walley et al., 2016), and found a low overlap with our scRNA-seq markers (4%, Table S3), indicating that the bulk tissue RNA-seq lacked cell or developmental domain specific information, as expected. We performed GWAS analysis using the whole ear specific genes, and they were also enriched for association with ear morphology traits, however in most cases for different traits and genes (Table S3). For instance, this analysis identified ZmYAB9, that was also identified using scRNA-seq markers (Figure 4B). However, the ear diameter (ED) trait was associated with different genes in scRNA-seq (Figures 4C–4D) and bulk tissue RNA-seq (Table S3) datasets. Furthermore, in heritability (ℎ2) analysis at a 200kb partition, no traits were associated with whole ear specific genes (Table S3), whereas four of them were significantly associated with scRNA-seq marker genes (Figure 4I). In summary, scRNA-seq markers revealed associations with multiple ear traits that were not found using bulk RNA-seq data, suggesting a unique application of this approach in identifying candidates to improve crop yield traits.
DISCUSSION
Development requires programmed cell- or domain-specific expression of regulatory and effector genes that together orchestrate stereotypical patterns of morphogenesis. The maize ear has a complex morphology with multiple indeterminate meristem and determinate organ types, and optimization of morphology is important for maize yield. To identify spatial regulators of ear development, we performed scRNA-seq of ~12,500 single cells from developing ears, and predicted 12 meta-clusters that were identified using known markers. As expected, many meta-clusters were meristem-associated, including for discrete domains that control branching, and we also found distinct vasculature meta-clusters, including xylem, phloem, and bundle sheath. We identified meta-clusters from determinate lateral organs and ground tissues, and in several cases, sub-clusters could be identified. Our method was sensitive enough to detect most maize genes, though we failed to detect the expression of CLV3 and WUS orthologs, possibly due to their low expression, or to a relatively low representation of central zone and organizing center cells in our experiments using whole developing ears. Another possibility is that these cell types were not recovered with our current protocol, and further improvements or profiling of cells from more finely dissected meristem tissues may address this issue. It is also intriguing that some xylem markers were expressed in the tips of meristems. In Arabidopsis, class III HD-ZIP and KANADI genes, which are expressed in xylem and phloem, respectively, are expressed in complementary patterns in adaxial and abaxial sides of lateral organs to specify their polarity (Emery et al., 2003). This polarity is pre-patterned by their correponding central and peripheral expression in the shoot meristem (Caggiano et al., 2017). Thus, our finding of maize ear xylem markers expressed in the meristem tip suggests that additional vascular genes specify a pre-pattern in meristems.
We validated our scRNA-seq results by mRNA in situ hybridization and by comparing to a FACS RNA-seq dataset, thus constructing a robust single-cell transcriptome atlas of a developing inflorescence. We also provided three applications showing how this atlas can enhance functional studies. First, we highlight how the cellular resolution of scRNA-seq data can accurately predict redundancy, a major obstacle in genetic analyses. We could predict redundancy in a family of maize TPPs that control inflorescence branching, and identified a family of redundant maize VOZ genes that produced a delayed floral transition phenotype through multiplex CRISPR/Cas9 mutagenesis. Flowering time is a major target of maize breeding (Liu et al., 2020), thus our findings provide candidates to fine tune flowering for crop improvement. In some cases, redundant paralogs may show only partial co-expression, as observed for SQUAMOSA PROMOTER BINDING (SBP)-box transcription factors UNBRANCHED2 (UB2) and UB3 which control initiation of lateral organ (Chuck et al., 2014, Du et al., 2020), or AINTEGUMENTA (ANT) and ANT-LIKE6, whose expression overlaps partially to redundantly regulate floral organ patterning and growth (Krizek, 2009). Therefore, care should be taken in use of scRNA-seq data in discerning such partially overlapping expression patterns.
In a second example, we hypothesized that the resolution of scRNA-seq could be combined with ChIP-seq to predict directly modulated targets of TFs. We created ChIP-seq datasets for two TFs that are likely to act redundantly, precluding the use of single mutants to find their modulated targets. We also integrated scRNA-seq with FACS ATAC-seq to provide evidence of spatially-regulated accessible chromatin. The limited availability of maize reporter lines for FACS may be overcome in the future by application of single-cell ATAC-seq (Rich-Griffin et al., 2020). Lastly, we hypothesized that scRNA-seq marker genes with spatially restricted expression in developing ears are enriched for regulators of ear morphology traits important for crop yields. Indeed, the scRNA-seq marker genes were significantly associated with ear morphology trait SNPs in a GWAS panel. These marker candidates could be selected in breeding programs or genetically modified to test their effects on yield.
In summary, scRNA-seq allowed valuable insights into maize ear development. The atlas can inform developmental genetics studies and breeding, and the methods we developed can be applied to studies of other complex shoot systems. As more plant scRNA-seq datasets are generated, a cross-species (eg. between maize and Arabidopsis) or cross-tissue (eg. between shoot and root) comparative analysis at single-cell resolution will inform how gene signatures were selected during evolution to shape the diverse morphologies that are critical to reproductive success and agricultural production.
STAR⋆METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Jackson (jacksond@cshl.edu).
Materials Availability
Requests for materials should be directed to Lead Contact, David Jackson (jacksond@cshl.edu). Requests for transgenic plant materials will require a Materials Transfer Agreement (MTA).
Data and Code Availability
All raw high-throughput sequencing data have been deposited in NCBI’s Sequence Read Archive (SRA). SRA IDs are listed in KEY RESOURCES TABLE. This study used codes from published software described in QUANTIFICATION AND STATISTICAL ANALYSIS.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GFP antibody (GFP-Trap magnetic agarose) | ChromoTek | Cat#gtma-20; RRID: AB_2631358 |
| Bacterial and Virus Strains | ||
| Agrobacterium | N/A | EHA101 |
| E.coli | N/A | DH5α |
| Biological Samples | ||
| Zea mays B73 | Maize Genetics COOP Stock Center | N/A |
| Zea mays pYAB14-TagRFPt reporter line | Je et al., 2016 | N/A |
| Zea mays ZmM16-YFP translational fusion line | Bartlett et al., 2015 | N/A |
| Zea mays ZmHDZIV6-YFP translational fusion line | This paper | N/A |
| Zea mays CRISPR/Cas9 knock out mutants of ZmVOZs | This paper | N/A |
| Zea mays branched silkless1;Tunicate (bd1;Tu) mutants | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Mannitol | Sigma-Aldrich | Cat# M4125 |
| Bovine serum albumin | Sigma-Aldrich | Cat#A7907-50G |
| T7 RNA polymerase | Sigma-Aldrich | 10881775001 |
| Calcofluor white stain | Sigma-Aldrich | Cat#18909 |
| Toluidine blue | Sigma-Aldrich | Cat#T3260 |
| CelLytic™ PN Isolation/Extraction Kit | Sigma-Aldrich | Cat#CELLYTPN1 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat#15714-s |
| Glutaraldehyde | Electron Microscopy Sciences | Cat#16537-16 |
| Cacodylate buffer | Electron Microscopy Sciences | Cat#11652 |
| LR white resin | Electron Microscopy Sciences | Cat#905072 |
| Cellulase RS | Onozuka | N/A |
| Cellulase R-10 | Onozuka | N/A |
| Macerozyme R-10 | Onozuka | N/A |
| Pectolyase Y-23 | Duchefa Biochem. | Cat#P8OO4.0001 |
| Trypan blue | Thermo Fisher Scientific | Cat#15250061 |
| Paraplast | McCormick Scientific | Cat#39503002 |
| ProbeOn Plus™ Slides | Fisher Scientific | Cat#22-230-900 |
| NBT/BCIP Ready-to-Use Tablets | Roche | 11697471001 |
| Critical Commercial Assays | ||
| Chromium i7 Multiplex Kit | 10X Genomics | Cat# PN-120262 |
| Chromium Single Cell 3’ Library & Gel Bead Kit v2 | 10X Genomics | Cat#PN-120237 |
| Chromium Single Cell A Chip Kit v2 | 10X Genomics | Cat#PN-1000009 |
| Dynabeads™ MyOne™ Silane Beads | Thermo Fisher Scientific | Cat#37002D |
| Arcturus PicoPure RNA Isolation Kit | Thermo Fisher Scientific | KIT0204 |
| RNA Bioanalyzer kit | Agilent | Cat#5067-1513 |
| DNA High Sensitivity Bioanalyzer kit | Agilent | Cat#5067-4626 |
| SMART-Seq™ v4 Ultra™ Low Input RNA Kit | Takara Bio USA, Inc. | Cat#634890 |
| Nextera XT DNA Library Prep Kit | Illumina | Cat#FC-131-1024 |
| AMPure XP Beads | Beckman Coulter | Cat#A63880 |
| KAPA Library Quantification Kits | Roche | Cat#KK4824 |
| NEXTflex ChIP-seq Kit | PerkinElmer Applied Genomics | NOVA-5143-02 |
| Deposited Data | ||
| B73 whole ear scRNA-seq_replicate 1 | This paper | PRJNA646989 |
| B73 whole ear scRNA-seq_replicate 2 | This paper | PRJNA646996 |
| B73 whole ear scRNA-seq_replicate 3 | This paper | PRJNA647001 |
| Protoplasting-response bulk RNAseq | This paper | PRJNA647196 |
| pZmYAB14-TagRFPt FACS RNAseq | This paper | PRJNA647195 |
| pZmYAB14-TagRFPt FACS ATACseq | This paper | PRJNA647197 |
| ZmHDZIV6-YFP ChIP-seq | This paper | PRJNA647198 |
| ZmM16-YFP ChIP-seq | This paper | PRJNA647200 |
| Experimental Models: Cell Lines | ||
| N/A | N/A | N/A |
| Experimental Models: Organisms/Strains | ||
| N/A | N/A | N/A |
| Oligonucleotides | ||
| See Table S4 for primers or sgRNAs sequences for mRNA in-situ, ZmVOZs crispr and genotyping, ZmHDZIV6-YFP transgene, and bd1 genotyping. | N/A | N/A |
| Recombinant DNA | ||
| pGW-Cas9 vector | Wang et al., 2014 | Addgene Plasmid # 50661; RRID: Addgene_50661 |
| pTF101 Gateway-compatible vector | Je et al., 2016 | NA |
| Software and Algorithms | ||
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR/wiki |
| R | R Core Team, 2013 | https://www.r-project.org/ |
| EmptyDrops | Lun et al., 2019 | https://rdrr.io/github/MarioniLab/DropletUtils/man/emptyDrops.html |
| DoubletFinder | McGinnis et al., 2019 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| Scater | McCarthy et al., 2017 | https://github.com/Alanocallaghan/scater |
| Scran | Lun et al., 2016 | https://github.com/MarioniLab/scran |
| Rsvd package | Erichson et al., 2019 | https://github.com/erichson/rSVD |
| Cccd package | Marchette, 2015 | https://github.com/cran/cccd |
| InfoMap | Csardi and Nepusz, 2006 | https://github.com/mapequation/infomap |
| MetaNeighbor | Crow et al., 2018 | https://github.com/maggiecrow/MetaNeighbor |
| UMAP package | Konopka, 2020 | https://github.com/tkonopka/umap |
| EGAD | Ballouz et al., 2017 | https://github.com/sarbal/EGAD |
| Trimmomatic - version 0.36 | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| edgeR | Robinson et al., 2010 | https://bioconductor.riken.jp/packages/devel/bioc/html/edgeR.html |
| elprep | Herzeel et al., 2015 | https://github.com/ExaScience/elprep |
| BWA-MEM | Li and Durbin, 2009 | https://github.com/lh3/bwa |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| LDAK software v5.0 | Speed et al., 2012 | http://www.ldak.org |
| Other | ||
| Cell Strainer | pluriStrainer | Cat#43-50030-50 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All analyses were performed with Zea mays (Maize). Maize plants were grown in the summer field (June – October) of Uplands Farm Agricultural Station at Cold Spring Harbor, New York or in the greenhouse with 16hr daytime, 26–28°C, and 8hr night, 22–24°C (Wu et al., 2020). Reference B73 inbred plants were used for single-cell experiments. The pYAB14-TagRFPt reporter line (Je et al., 2016) and ZmM16-YFP translational fusion line (Bartlett et al., 2015) were obtained from previous studies. The ZmHDZIV6-YFP translational fusion line was constructed using ZmHDZIV6 native promoter and coding sequence as previously described in the pTF101 Gateway-compatible vector (Je et al., 2016), primer sequences are listed in Table S4. The pYAB14-TagRFPt reporter line and ZmHDZIV6-YFP translational fusion line were introgressed into a proliferative cauliflower-like double mutant line, bd1;Tu, to generate a large amount of ear meristem tissue for FACS RNA-seq and ChIP-seq respectively. Primer sequences for genotyping bd1 are listed in Table S4. Tu genotyping was performed as previously described (Han et al., 2012).
CRISPR/Cas9 was used to knockout ZmVOZs genes following Agrobacterium-mediated transformation of Hi-II embryos (Je et al., 2016). Guide RNAs (sgRNAs) were designed based on B73 V3 reference genome, one pair targeting ZmVOZ1 and ZmVOZ2, and a second pair targeting ZmVOZ4 and ZmVOZ5, Table S4. The sgRNAs were introduced by Gateway Recombination into pGW-Cas9 vector (Addgene plasmid # 50661; RRID: Addgene_50661) (Wang et al., 2014) and transferred to Agrobacterium (EHA101) for maize transformation. 29 plants from 8 transformation events were obtained and analyzed by PCR amplification and Sanger sequencing to identify mutations in the targeted regions. The Cas9 transgene was segregated away by crossing with B73 to recover stable mutant alleles. Primer sequences and PCR assays for genotyping are listed in Table S4.
METHOD DETAILS
Protoplast preparation and 10x Genomics library construction and sequencing
For the three biological replicates of wild type (B73) whole ear samples, protoplasts were prepared as previously described but without L-cysteine pretreatment (Ortiz-Ramírez et al., 2018). 5–10mm developing ears were dissected into protoplast washing buffer and diced with a razor blade to 0.5–1mm pieces, then washed three times with protoplast washing buffer before adding enzyme solution. Tissues were protoplasted for ~ 45min at room temperature with gentle shaking. The mixture was filtered through a 30μm cell strainer (pluriStrainer, 43–50030-50) then collected by centrifugation at 500g for 3 mins at 4°C. The supernatant was gently removed without disturbing the protoplast pellet, which was washed by gentle resuspension in protoplast washing buffer. Protoplasts were then filtered as before and purified by FACS sorting (FACSAria II SORP with 100-micron setup, purity precision, yield mask at 32, purity mask at 32, plates voltage at 2,500, voltage centering at 20, sheath pressure at 20, and target gap at 12). Protoplasts were sorted into 1×PBS with 0.1% BSA and 0.4M mannitol, and pelleted at 400g for 2 mins at 4°C. The supernatant was carefully removed, leaving 20–40μl to gently resuspend the pellet. The protoplasts were stained with trypan blue (Thermo Fisher Scientific, 15250061) to check concentration and viability with a hemocytometer under a light microscope, and good quality protoplasts with viability ≥ 70% were immediately loaded into the 10x Genomics Chromium System using V2 chemistry kits. scRNA-seq libraries were sequenced by Illumina short reads with ~ 400M paired end reads per library (read1 = 28bp, read2 = 56bp) (Table S1). Raw sequencing data were deposited in NCBI’s Sequence Read Archive (SRA). SRA IDs are listed in Table S1.
Anatomy and confocal microscopy
For anatomy, wild type (B73) developing ears from fresh plants were hand dissected and fixed in 4% paraformaldehyde (Electron Microscopy Sciences, 15714-s) (Jackson et al., 1994). The fixed ear tissue was dehydrated through a graded alcohol series (50%, 70%, 85%, 95%, and 100%) and a histoclear series, then embedded in paraplast (McCormick Scientific, 39503002) (Jackson et al., 1994). 5μm sections were cut using a Leica microtome, then mounted on ProbeOn Plus™ Slides (Fisher Scientific, 22–230-900) and rehydrated and stained with Calcofluor white stain (Sigma-Aldrich, 18909). Images were taken on a ZEISS LSM 710 confocal microscope using DAPI channel.
For vascular bundle anatomy, wild type (B73) developing ears were fixed overnight at 4°C in 0.1 M cacodylate buffer pH 7.4 (Electron Microscopy Sciences, 11652) with 4% paraformaldehyde (Electron Microscopy Sciences, 15714-s) and 1% glutaraldehyde (Electron Microscopy Sciences, 16537–16), washed three times with 0.1M cacodylate buffer and then dehydrated in a graded ethanol series (50%, 60%, 70%, 80%, 90%, 95%, two times 100%). The dehydrated ear samples were transferred to LR white resin / ethanol in 1:3, 1:1, and 3:1 ratios, and twice overnight in 100% LR white resin (Electron Microscopy Sciences, 905072). Tissues in LR white resin were then polymerized in a 60 °C oven for 48 h, and 0.5–1μm thick sections were cut using 45 Diamond DiATOME Histo Knife on a Reichert Ultracut E microtome. Sections were collected and stained by toluidine blue (Sigma-Aldrich, T3260). Images were taken by Nikon DS-Ri2 microscope.
pYAB14-TagRFPt, ZmM16-YFP, and ZmHDZIV6-YFP lines were imaged using ZEISS LSM 710 or 780 confocal microscopes. The DAPI channel was used for capturing autofluorescence (blue color) in Figures 2N and 3D.
mRNA in situ hybridization
mRNA in situs were conducted as previously described (Jackson et al., 1994). Briefly, wild type (B73) developing ears were freshly collected, fixed in 4% paraformaldehyde (Electron Microscopy Sciences, 15714-s) (Jackson et al., 1994). The fixed ear tissue was dehydrated through a graded alcohol series (50%, 70%, 85%, 95%, and 100%) and a histoclear series, then embedded in paraplast (McCormick Scientific, 39503002) (Jackson et al., 1994). 10μm sections were cut using a Leica microtome, then mounted on ProbeOn Plus™ Slides (Fisher Scientific, 22–230-900). To prepare probes for marker genes, we added T7 promoter sequences GAGTAATACGACTCACTATAGGGAGA into reverse primers used to amplify gene-specific PCR products from cDNA templates. Then probes were synthesized by in vitro transcription using T7 RNA Polymerase (Sigma-Aldrich, 10881775001). Primer sequences for all genes were listed in Table S4. Probes were then applied on tissue sections and incubated at 50°C overnight. To detect the hybridization signal, we applied freshly dissolved NBT/BCIP Ready-to-Use Tablets (Roche, 11697471001) in the alkaline phosphatase reaction solution. Images were taken using a Nikon DS-Ri2 DIC microscope.
FACS and bulk RNA-seq, ATAC-seq library preparation
For three biological replicates of bulk RNA-seq to identify protoplasting-responsive genes, wild type B73 background ear tissue was used. RNA was isolated from protoplasted and equivalent non-protoplasted tissue to compare side by side. For three biological replicates of pYAB14-TagRFPt FACS RNA-seq, ear tissue of pYAB14-TagRFPt/bd1;Tu and bd1;Tu negative control plants were collected and digested as described earlier. Protoplasts were gently washed, filtered, and resuspended as before. bd1;Tu negative control protoplasts were first loaded into FACSAria II SORP to set up the gate for identifying autofluorescence signals. pYAB14-TagRFPt/bd1;Tu protoplasts were then loaded using the same settings. RFP cells were collected based on specific signals from the mStrawberry channel, and examined under a ZEISS LSM 710 confocal microscope. RNA for RFP positive protoplast samples and control samples (total protoplasts without sorting) was extracted using Arcturus PicoPure RNA Isolation Kit (Thermo Fisher Scientific, KIT0204). RNA was examined by a RNA Bioanalyzer kit (Agilent, 5067–1513). RNA-seq libraries were built using SMART-Seq™ v4 Ultra™ Low Input RNA Kit (Takara Bio USA, Inc., 634890) and Nextera XT DNA Library Prep Kit (Illumina, FC-131–1024). Library quality and size was examined by a DNA High Sensitivity Bioanalyzer chip (Agilent, 5067–4626), and quantified using the KAPA Library Quantification Kit (Roche, KK4824) before Illumina sequencing. Raw sequencing data were deposited into NCBI’s Sequence Read Archive (SRA). SRA IDs are listed in Tables S1–S2. For one biological replicate of pYAB14-TagRFPt FACS ATAC-seq, RFP protoplasts were collected as described above, and nuclei isolated for library construction and sequencing as previously (Lu et al., 2017). Raw sequencing data were deposited in NCBI’s Sequence Read Archive (SRA). SRA ID is listed in Table S2.
ChIP-seq library preparation
ChIP experiments were conducted as previously described (Pautler et al., 2015) with some modifications. Briefly, two biological replicates of freshly harvested ear tissues of ZmHDZIV6-YFP/bd1;Tu and ZmM16-YFP were immediately cross-linked in buffer containing 1% formaldehyde, 10mM HEPES-NaOH PH7.4, 0.4 M sucrose, 1 mM EDTA, and 1 mM PMSF, for 20min under vacuum. Glycine was then added to a concentration of 0.1 M to for another 5 min under vacuum. Nuclei extraction was conducted using CelLytic™ PN Isolation/Extraction Kit (Sigma-Aldrich, CELLYTPN1). For immunoprecipitation, we used high-affinity GFP-Trap magnetic agarose (ChromoTek, gtma-20; RRID: AB_2631358). For building ChIP-seq libraries, we used NEXTflex ChIP-seq Kit (PerkinElmer Applied Genomics, NOVA-5143–02) with AMPure XP beads (Beckman Coulter, A63880). ChIP-seq libraries were quantified by KAPA Library Quantification Kits (Roche, KK4824) before Illumina sequencing. Raw sequencing data were deposited in NCBI’s Sequence Read Archive (SRA). SRA IDs are listed in Table S3.
QUANTIFICATION AND STATISTICAL ANALYSIS
scRNA-seq analysis, clustering, and selection of marker genes
Sequencing reads of three biological replicates of wild type (B73) whole ear scRNA-seq samples were aligned to the maize v3 reference genome using STARsolo v2.7.0.f (Dobin et al., 2013). We updated the v3.31 GTF annotation file by adding four maize CLAVATA3/EMBRYO SURROUNDING REGION-RELATED (ZmCLE) genes, including ZmCLE7 (GRMZM2G372364), ZmCLE14 (AC191109.3_FG001), ZmCLE25 (GRMZM2G525788), and ZmCLEug-2 (GRMZM2G054501) (Table S4; MaizeGDB), before using it to build a STAR genome index with default parameters (Dobin et al., 2013). All downstream processing was performed in R (R Core Team, 2013), with each dataset analyzed separately. In brief, we removed droplets that lacked a protoplast using EmptyDrops with a minimum threshold of 800 UMIs (Lun et al., 2019), and removed probable doublets using DoubletFinder (McGinnis et al., 2019). Expression data was log2 normalized using scater (McCarthy et al., 2017). We identified highly variable genes using the trendVar function in scran (Lun et al., 2016), selecting genes at FDR<0.05, and we used the rsvd package (Erichson et al., 2019) to calculate approximate principal components for all cells after subsetting to highly variable genes. We then generated a nearest neighbor graph for cells with the cccd package (Marchette, 2015) using the Euclidean distance across the top 20 PCs, with k=100. To find clusters, we used the InfoMap algorithm implemented in the igraph R package (Csardi and Nepusz, 2006), resampling 100 times (Rosvall and Bergstrom, 2008). MetaNeighbor analysis was performed as previously described (Crow et al., 2018). We used the umap package to generate embeddings for visualization (Konopka, 2020). Within in each UMAP, every dot represents a cell, and the color scale indicates the normalized expression level by adding a constant 75% of each cell’s expression value to nearest neighbors for clear visualization. Differential expression statistics were calculated with an AUROC test on log counts using the auc multifunc function from EGAD (Ballouz et al., 2017). Differential expression genes with AUROC scores of ≥ 0.7 in at least one replicate were considered as meta-cluster marker genes (Table S1).
FACS and bulk RNA-seq, ATAC-seq analysis
Three biological replicates of FACS and bulk RNA-seq analyses were performed as previously (Wang et al., 2020) with some modifications. Bulk RNA-seq datasets for extracting ear tissue specific genes were download from a previous study (Walley et al., 2016). Raw sequencing reads were firstly trimmed with Trimmomatic (Bolger et al., 2014), and then mapped with STAR (Dobin et al., 2013) using the same updated maize V3 reference as scRNA-seq analysis. edgeR (Robinson et al., 2010) was used to perform differential expression analysis. We calculated similarity between scRNA-seq and FACS RNA-seq data using the auroc_analytic function in EGAD (Ballouz et al., 2017), with ranked scRNA-seq meta-cluster 3 p-values as the “scores” and FACS RNA-seq DE genes (log2 FC > 0 and FDR < 0.05, Table S2) as the “labels”. Whole ear ATAC-seq datasets were download from a previous study (Ricci et al., 2019). One biological replicate of FACS ATAC-seq analysis was performed as previously (Ricci et al., 2019), using the same reference as scRNA-seq analysis.
ChIP-seq analysis
Two biological replicates of ChIP-seq reads for both ZmHDZIV6-YFP and ZmM16-YFP datasets were trimmed using sickle (https://github.com/najoshi/sickle). Duplicated reads were further removed by elprep (Herzeel et al., 2015), before aligning to the same updated maize V3 reference used for scRNA-seq analysis with BWA-MEM (Li and Durbin, 2009). Alignment reads were filtered for those above a MAPping Quality (MAPQ) threshold above 40. Peak calling, peak annotation, and motif enrichment were performed with HOMER (Heinz et al., 2010), with peak calling parameters as the following: -F = 8: Fold enrichment threshold of IP tag count over input tag count, -L = 2: Fold enrichment of a putative peak’s tag count over surrounding (local) genomic region, and -LP = 0.0001: p-value cutoff for local fold enrichment to a peak to be considered. High confidence peaks between two biological replicates were determined by finding midpoints of peaks positioned within 300bp of each other (Pautler et al., 2015).
scRNA-seq co-expression analysis
89 maize bulk tissue RNA-seq datasets were used for calculating co-expression values as previously described (Lee et al., 2020). Briefly, co-expression networks were constructed using Spearman’s correlation. Three gene pairs including RA3-ZmTPP4, RA3-ZmTPP12, and ZmTPP4-ZmTPP12 were then extracted from the datasets. The aggregated co-expression value of each gene pair was calculated and reported in Figure S4. For scRNA-seq co-expression to predict RA3-ZmTPPs redundancy, ZmVOZ co-expression, and TF ChIP-Seq directly modulated targets, the average Jaccard index was calculated for B73 whole ear datasets, using genes expressed in >1% of cells. Genes with at least 1 UMI were assigned a value of 1, and all others were assigned a value of 0. The Jaccard index was calculated for each ear dataset separately and then averaged. To calculate significance (Figure S4) we took a non-parametric approach with the null hypothesis that there is no replicable co-expression across batches, calculated by convolving the uniform distributions obtained for each batch after ranking. To compare overlaps in co-expression between two genes, we reranked and assessed similarity against the null using the same approach. FDRs were calculated by dividing cumulative nulls by cumulative empiricals.
Integration of GWAS with scRNA-seq or ear tissue bulk RNA-seq
To integrate GWAS with scRNA-seq, we firstly selected unique scRNA-seq marker genes from ear meristem, determinate lateral organ, and vasculature meta-clusters (3, 4, 5, 6, 9, 10, 11, 12) using two different cutoffs. The stringent cutoff was AUROC ≥ 0.7 across all replicates, which gave 68 scRNA-seq marker genes (Table S3). The less stringent cutoff was AUROC ≥ 0.7 in at least one replicate, which gave 241 scRNA-seq marker genes (Table S3). For targeted GWAS analysis: we used the best linear unbiased predictors (BLUPs) from nine ear phenotypes to perform a targeted GWAS using the unified mixed linear model. BLUPs estimation, GWAS model details, and genomic marker filtering procedures are described in (Rice et al., 2020). The SNPs in or within 2kb of scRNA-seq marker genes from lists of both stringent and less stringent cutoffs were used (Table S3).
For lambda analysis, the procedure has been previously described (Parvathaneni et al., 2020). Briefly, lambda is a ratio of the FDR adjusted p-values for a given set of markers when included with the full marker set compared to when considered on their own (equation below).
We looked at the significance of 99th percentile (q = 99) of FDR adjusted p-values subset of markers (SNPs in the region of genes of interest). We performed 1,000 replicates (random subsets) and estimated the lambda distributions for each trait. Lambda values of 241 unique markers from scRNA-seq meta-clusters (3, 4, 5, 6, 9, 10, 11, 12) with less stringent cutoff (Table S3) were compared against random subsets lambda distributions to determine significance. Traits with λ ≥ mean ± 2SD were considered to be biologically significant.
For SNP Heritability analysis, we estimated narrow-sense heritability (ℎ2) from the subsets of the scRNA-seq SNPs considered in the lambda analysis using the LDAK software v5.0 (Speed et al., 2012). Thus, the resulting estimate of ℎ2 provides an estimate of the additive genetic variance explained by genes of interest. To determine if the resulting heritability for a given trait was greater than chance, the heritability for 1000 permutations using a random subset of maize genes was estimated. For a given permutation, genes with at least one SNPs within the genic region were randomly selected. Enough genes were selected to ensure the total number in a permuted subset was ± 5 compared to the target set. A target set was declared significant for a given trait if its heritability was greater than the top 5% of permuted values.
To integrate GWAS with ear tissue bulk RNA-seq, we firstly extracted ear tissue specific genes from a previous study (Table S3) (Walley et al., 2016). Then we used these genes to perform same analyses, including targeted GWAS, lambda, and SNP Heritability as mentioned above for scRNA-seq (Table S3).
Supplementary Material
Table S4: Primers, sgRNAs sequences and ZmCLE genes’ annotation. Related to all Figures.
Highlights.
scRNA-seq of developing maize ears reveals major cell types and developmental markers
scRNA-seq co-expression networks predict genetic redundancy
Integration of scRNA-seq and ChIP-seq/ ATAC-seq helps build transcriptional networks
Integration of scRNA-seq and GWAS identifies candidate maize yield associated genes
ACKNOWLEDGMENTS
We thank Drs. Kenneth Birnbaum, Carlos Ortiz Ramírez, and Bruno Guillotin for advice on scRNA-seq and FACS RNA-seq experiments, and Drs. Bert De Rybel and Joyce Chery for discussions on vascular biology. We thank support from Dr. Jon Preall and the CSHL single-cell core facility, flow cytometry facility, microscopy facility, NGS facility, and farm management team. We thank Richelle Chen for assisting bioinformatic analyses, and Micheal Okoro for assisting field work and sample preparation. We acknowledge funding support from NSF (IOS-1833182, IOS-1445025, IOS- 1027445, IOS-1934388, IOS-1755141). R.J.S. acknowledges support from NSF (IOS-1856627). J.G. acknowledges support from R01 LM012736 and R01 MH113005. M.C. acknowledges support from K99 MH120050. B.H. acknowledges support from The Robertson Research Fund, CSHL School for Biological Sciences. We thank Drs. Munenori Kitagawa and Penelope Lindsay for their feedback on the manuscript.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aerts N, de Bruijn S, van Mourik H, Angenent GC, and van Dijk ADJ (2018). Comparative analysis of binding patterns of MADS-domain proteins in Arabidopsis thaliana. BMC plant biology 18, 131–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballouz S, Weber M, Pavlidis P, and Gillis J (2017). EGAD: ultra-fast functional analysis of gene networks. Bioinformatics 33, 612–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett ME, Williams SK, Taylor Z, DeBlasio S, Goldshmidt A, Hall DH, Schmidt RJ, Jackson DP, and Whipple CJ (2015). The Maize PI/GLO Ortholog Zmm16/sterile tassel silky ear1 Interacts with the Zygomorphy and Sex Determination Pathways in Flower Development. Plant Cell 27, 3081–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc N, Yilmaz A, Mejia-Guerra MK, Morohashi K, O’Connor D, Grotewold E, and Hake S (2012). Unraveling the KNOTTED1 regulatory network in maize meristems. Genes & development 26, 1685–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bommert P, Nagasawa NS, and Jackson D (2013). Quantitative variation in maize kernel row number is controlled by the FASCIATED EAR2 locus. Nature genetics 45, 334–337. [DOI] [PubMed] [Google Scholar]
- Caggiano MP, Yu X, Bhatia N, Larsson A, Ram H, Ohno CK, Sappl P, Meyerowitz EM, Jönsson H, and Heisler MG (2017). Cell type boundaries organize plant development. Elife 6, e27421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y-M, Liu W-Y, Shih AC-C, Shen M-N, Lu C-H, Lu M-YJ, Yang H-W, Wang T-Y, Chen SCC, Chen SM, et al. (2012). Characterizing regulatory and functional differentiation between maize mesophyll and bundle sheath cells by transcriptomic analysis. Plant physiology 160, 165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Yan W, Fu L-Y, and Kaufmann K (2018). Architecture of gene regulatory networks controlling flower development in Arabidopsis thaliana. Nature Communications 9, 4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, and Xiong L (2010). myo-Inositol-1-phosphate synthase is required for polar auxin transport and organ development. The Journal of biological chemistry 285, 24238–24247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitwood DH, Guo M, Nogueira FTS, and Timmermans MCP (2007). Establishing leaf polarity: the role of small RNAs and positional signals in the shoot apex. Development 134, 813. [DOI] [PubMed] [Google Scholar]
- Chuck G, Muszynski M, Kellogg E, Hake S, and Schmidt RJ (2002). The control of spikelet meristem identity by the branched silkless1 gene in maize. Science 298, 1238–1241. [DOI] [PubMed] [Google Scholar]
- Chuck G, Brown PJ, Meeley R, and Hake S (2014). Maize SBP-box transcription factors unbranched2 and unbranched3 affect yield traits by regulating the rate of lateral primordia initiation. Proc Natl Acad Sci U S A 111, 18775–18780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claeys H, Vi SL, Xu X, Satoh-Nagasawa N, Eveland AL, Goldshmidt A, Feil R, Beggs GA, Sakai H, Brennan RG, et al. (2019). Control of meristem determinacy by trehalose 6-phosphate phosphatases is uncoupled from enzymatic activity. Nature Plants 5, 352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow M, Paul A, Ballouz S, Huang ZJ, and Gillis J (2018). Characterizing the replicability of cell types defined by single cell RNA-sequencing data using MetaNeighbor. Nature communications 9, 884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csardi G, Nepusz T (2006). “The igraph software package for complex network research.” InterJournal, Complex Systems, 1695 http://igraph.org. [Google Scholar]
- De Rybel B, Mähönen AP, Helariutta Y, and Weijers D (2016). Plant vascular development: from early specification to differentiation. Nature Reviews Molecular Cell Biology 17, 30–40. [DOI] [PubMed] [Google Scholar]
- Denyer T, Ma X, Klesen S, Scacchi E, Nieselt K, and Timmermans MC (2019). Spatiotemporal developmental trajectories in the Arabidopsis root revealed using high-throughput single-cell RNA sequencing. Developmental cell 48, 840–852. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Kim ST, and Lord EM (2005). Plantacyanin plays a role in reproduction in Arabidopsis. Plant physiology 138, 778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Liu L, Peng Y, Li M, Li Y, Liu D, Li X, and Zhang Z (2020). UNBRANCHED3 Expression and Inflorescence Development is Mediated by UNBRANCHED2 and the Distal Enhancer, KRN4, in Maize. PLoS Genet 16, e1008764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery JF, Floyd SK, Alvarez J, Eshed Y, Hawker NP, Izhaki A, Baum SF, and Bowman JL (2003). Radial patterning of Arabidopsis shoots by class III HD-ZIP and KANADI genes. Curr Biol 13, 1768–1774. [DOI] [PubMed] [Google Scholar]
- Erichson NB, Voronin S, Brunton SL, Kutz JN (2019). Randomized Matrix Decompositions Using R. Journal of Statistical Software 89, 1–48. doi: 10.18637/jss.v089.i11. [DOI] [Google Scholar]
- Eveland AL, Goldshmidt A, Pautler M, Morohashi K, Liseron-Monfils C, Lewis MW, Kumari S, Hiraga S, Yang F, Unger-Wallace E, et al. (2014). Regulatory modules controlling maize inflorescence architecture. Genome Res 24, 431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Sazuka T, Oda Y, Kawahigashi H, Wu J, Takanashi H, Ohnishi T, Yoneda JI, Ishimori M, Kajiya-Kanegae H, et al. (2018). Transcriptional switch for programmed cell death in pith parenchyma of sorghum stems. Proc Natl Acad Sci U S A 115, E8783–e8792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk V, Kositsup B, Zhao C, and Beers EP (2002). The Arabidopsis xylem peptidase XCP1 is a tracheary element vacuolar protein that may be a papain ortholog. Plant physiology 128, 84–94. [PMC free article] [PubMed] [Google Scholar]
- Gallavotti A, Zhao Q, Kyozuka J, Meeley RB, Ritter MK, Doebley JF, Pe ME, and Schmidt RJ (2004). The role of barren stalk1 in the architecture of maize. Nature 432, 630. [DOI] [PubMed] [Google Scholar]
- Gipson AB, Giloteaux L, Hanson MR, and Bentolila S (2020). Arabidopsis RanBP2-Type Zinc Finger Proteins Related to Chloroplast RNA Editing Factor OZ1. Plants (Basel) 9, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P, Zheng Y, Peng D, Liu L, Dai L, Chen C, and Wang B (2018). Identification and expression characterization of the Phloem Protein 2 (PP2) genes in ramie (Boehmeria nivea L. Gaudich). Scientific Reports 8, 10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha CM, Jun JH, Nam HG, and Fletcher JC (2007). BLADE-ON-PETIOLE 1 and 2 control Arabidopsis lateral organ fate through regulation of LOB domain and adaxial-abaxial polarity genes. Plant Cell 19, 1809–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J-J, Jackson D, and Martienssen R (2012). Pod corn is caused by rearrangement at the Tunicate1 locus. The Plant cell 24, 2733–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Molecular Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzeel C, Costanza P, Decap D, Fostier J, and Reumers J (2015). elPrep: High-Performance Preparation of Sequence Alignment/Map Files for Variant Calling. PLoS One 10, e0132868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horváth E, Bela K, Holinka B, Riyazuddin R, Gallé Á, Hajnal Á, Hurton Á, Fehér A, and Csiszár J (2019). The Arabidopsis glutathione transferases, AtGSTF8 and AtGSTU19 are involved in the maintenance of root redox homeostasis affecting meristem size and salt stress sensitivity. Plant Science 283, 366–374. [DOI] [PubMed] [Google Scholar]
- Huffaker A, Dafoe NJ, and Schmelz EA (2011). ZmPep1, an ortholog of Arabidopsis elicitor peptide 1, regulates maize innate immunity and enhances disease resistance. Plant Physiol 155, 1325–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish EE (1997). Class II Tassel Seed Mutations Provide Evidence for Multiple Types of Inflorescence Meristems in Maize (Poaceae). American Journal of Botany 84, 1502–1515. [PubMed] [Google Scholar]
- Jackson D, Veit B, and Hake S (1994). Expression of maize KNOTTED1 related homeobox genes in the shoot apical meristem predicts patterns of morphogenesis in the vegetative shoot. Development 120, 405–413. [Google Scholar]
- Javelle M, Klein-Cosson C, Vernoud V, Boltz V, Maher C, Timmermans M, Depège-Fargeix N, and Rogowsky PM (2011). Genome-Wide Characterization of the HD-ZIP IV Transcription Factor Family in Maize: Preferential Expression in the Epidermis. Plant Physiology 157, 790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Je BI, Gruel J, Lee YK, Bommert P, Arevalo ED, Eveland AL, Wu Q, Goldshmidt A, Meeley R, Bartlett M, et al. (2016). Signaling from maize organ primordia via FASCIATED EAR3 regulates stem cell proliferation and yield traits. Nature genetics 48, 785–791. [DOI] [PubMed] [Google Scholar]
- Knauer S, Javelle M, Li L, Li X, Ma X, Wimalanathan K, Kumari S, Johnston R, Leiboff S, Meeley R, et al. (2019). A high-resolution gene expression atlas links dedicated meristem genes to key architectural traits. Genome Res 29, 1962–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka T (2020). umap: Uniform Manifold Approximation and Projection. R package version 0.2.6.0. https://CRAN.R-project.org/package=umap. [Google Scholar]
- Krizek B (2009). AINTEGUMENTA and AINTEGUMENTA-LIKE6 act redundantly to regulate Arabidopsis floral growth and patterning. Plant Physiol 150, 1916–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A, Anderson AG, Merullo DP, and Konopka G (2019). Beyond bulk: a review of single cell transcriptomics methodologies and applications. Curr Opin Biotechnol 58, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Shah M, Ballouz S, Crow M, and Gillis J (2020). CoCoCoNet: conserved and comparative co-expression across a diverse set of species. Nucleic acids research 48, W566–w571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Fernie AR, and Yan J (2020). The Past, Present, and Future of Maize Improvement: Domestication, Genomics, and Functional Genomic Routes toward Crop Enhancement. Plant Communications 1, 100010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Wang H, Zhang Z, Wu J, Feng Y, and Zhu Z (2009). Divergence in function and expression of the NOD26-like intrinsic proteins in plants. BMC Genomics 10, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd J, and Meinke D (2012). A comprehensive dataset of genes with a loss-of-function mutant phenotype in Arabidopsis. Plant physiology 158, 1115–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Hofmeister BT, Vollmers C, DuBois RM, and Schmitz RJ (2017). Combining ATAC-seq with nuclei sorting for discovery of cis-regulatory regions in plant genomes. Nucleic acids research 45, e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun AT, McCarthy DJ, and Marioni JC (2016). A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research 5, 2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun ATL, Riesenfeld S, Andrews T, Dao TP, Gomes T, Marioni JC, and participants in the 1st Human Cell Atlas J (2019). EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biology 20, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Denyer T, and Timmermans MC (2020). PscB: A Browser to Explore Plant Single Cell RNA-Sequencing Datasets. Plant Physiology, pp.00250.02020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchette DJ (2015). cccd: Class Cover Catch Digraphs. R package version 1.5. https://CRAN.R-project.org/package=cccd. [Google Scholar]
- McCarthy DJ, Campbell KR, Lun AT, and Wills QF (2017). Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 33, 1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Systems 8, 329–337.e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashima S, Roszak P, Sevilem I, Toyokura K, Blob B, Heo J. o., Mellor N, Help-Rinta-Rahko H, Otero S, Smet W, et al. (2019). Mobile PEAR transcription factors integrate positional cues to prime cambial growth. Nature 565, 490–494. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Katsumata H, Abe M, Yabe N, Komeda Y, Yamamoto KT, and Takahashi T (2006). Characterization of the Class IV Homeodomain-Leucine Zipper Gene Family in Arabidopsis. Plant physiology 141, 1363–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuccio ML, Wu J, Mowers R, Zhou HP, Meghji M, Primavesi LF, Paul MJ, Chen X, Gao Y, Haque E, et al. (2015). Expression of trehalose-6-phosphate phosphatase in maize ears improves yield in well-watered and drought conditions. Nat Biotechnol 33, 862–869. [DOI] [PubMed] [Google Scholar]
- Ortiz-Ramírez C, Arevalo ED, Xu X, Jackson DP, and Birnbaum KD (2018). An Efficient Cell Sorting Protocol for Maize Protoplasts. Current protocols in plant biology 3, e20072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvathaneni RK, Bertolini E, Shamimuzzaman M, Vera DL, Lung P-Y, Rice BR, Zhang J, Brown PJ, Lipka AE, Bass HW, et al. (2020). The regulatory landscape of early maize inflorescence development. Genome Biology 21, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pautler M, Eveland AL, LaRue T, Yang F, Weeks R, Je BI, Meeley R, Komatsu M, Vollbrecht E, and Sakai H (2015). FASCIATED EAR4 encodes a bZIP transcription factor that regulates shoot meristem size in maize. The Plant Cell 27, 104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter SS (2018). Single-cell RNA sequencing for the study of development, physiology and disease. Nature reviews Nephrology 14, 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: URL http://www.R-project.org/. [Google Scholar]
- Ricci WA, Lu Z, Ji L, Marand AP, Ethridge CL, Murphy NG, Noshay JM, Galli M, Mejía-Guerra MK, Colomé-Tatché M, et al. (2019). Widespread long-range cis-regulatory elements in the maize genome. Nature Plants 5, 1237–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice BR, Fernandes SB, and Lipka AE (2020). Multi-Trait Genome-wide Association Studies Reveal Loci Associated with Maize Inflorescence and Leaf Architecture. Plant & cell physiology 61, 1427–1437. [DOI] [PubMed] [Google Scholar]
- Rich-Griffin C, Stechemesser A, Finch J, Lucas E, Ott S, and Schäfer P (2020). Single-Cell Transcriptomics: A High-Resolution Avenue for Plant Functional Genomics. Trends in Plant Science 25, 186–197. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England) 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Leal D, Xu C, Kwon C-T, Soyars C, Demesa-Arevalo E, Man J, Liu L, Lemmon ZH, Jones DS, Van Eck J, et al. (2019). Evolution of buffering in a genetic circuit controlling plant stem cell proliferation. Nature genetics 51, 786–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosvall M, and Bergstrom CT (2008). Maps of random walks on complex networks reveal community structure. Proceedings of the National Academy of Sciences 105, 1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh-Nagasawa N, Nagasawa N, Malcomber S, Sakai H, and Jackson D (2006). A trehalose metabolic enzyme controls inflorescence architecture in maize. Nature 441, 227–230. [DOI] [PubMed] [Google Scholar]
- Schürholz A-K, López-Salmerón V, Li Z, Forner J, Wenzl C, Gaillochet C, Augustin S, Barro AV, Fuchs M, Gebert M, et al. (2018). A Comprehensive Toolkit for Inducible, Cell Type-Specific Gene Expression in Arabidopsis. Plant physiology 178, 40–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigmon B, and Vollbrecht E (2010). Evidence of selection at the ramosa1 locus during maize domestication. Molecular ecology 19, 1296–1311. [DOI] [PubMed] [Google Scholar]
- Speed D, Hemani G, Johnson MR, and Balding DJ (2012). Improved heritability estimation from genome-wide SNPs. American journal of human genetics 91, 1011–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strable J, Wallace JG, Unger-Wallace E, Briggs S, Bradbury PJ, Buckler ES, and Vollbrecht E (2017). Maize YABBY Genes drooping leaf1 and drooping leaf2 Regulate Plant Architecture. Plant Cell 29, 1622–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takacs EM, Li J, Du C, Ponnala L, Janick-Buckner D, Yu J, Muehlbauer GJ, Schnable PS, Timmermans MC, Sun Q, et al. (2012). Ontogeny of the maize shoot apical meristem. Plant Cell 24, 3219–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong W, Imai A, Tabata R, Shigenobu S, Yamaguchi K, Yamada M, Hasebe M, Sawa S, Motose H, and Takahashi T (2016). Polyamine Resistance Is Increased by Mutations in a Nitrate Transporter Gene NRT1.3 (AtNPF6.4) in Arabidopsis thaliana. Frontiers in Plant Science 7, 834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollbrecht E, and Schmidt RJ (2009). Development of the Inflorescences In Handbook of Maize: Its Biology, Bennetzen JL, and Hake SC, eds. (New York, NY: Springer; ), pp. 13–40. [Google Scholar]
- Walley JW, Sartor RC, Shen Z, Schmitz RJ, Wu KJ, Urich MA, Nery JR, Smith LG, Schnable JC, Ecker JR, et al. (2016). Integration of omic networks in a developmental atlas of maize. Science 353, 814–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lu Z, delaBastide M, Van Buren P, Wang X, Ghiban C, Regulski M, Drenkow J, Xu X, Ortiz-Ramirez C, et al. (2020). Management, Analyses, and Distribution of the MaizeCODE Data on the Cloud. Front Plant Sci 11, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, and Lander ES (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Xu F, Liu L, Char SN, Ding Y, Je BI, Schmelz E, Yang B and Jackson D (2020). The maize heterotrimeric G protein β subunit controls shoot meristem development and immune responses. Proceedings of the National Academy of Sciences 117, 1799–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui Y, Mukougawa K, Uemoto M, Yokofuji A, Suzuri R, Nishitani A, and Kohchi T (2012). The phytochrome-interacting vascular plant one-zinc finger1 and VOZ2 redundantly regulate flowering in Arabidopsis. Plant Cell 24, 3248–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong R, Richardson EA, and Ye Z-H (2007). The MYB46 transcription factor is a direct target of SND1 and regulates secondary wall biosynthesis in Arabidopsis. The Plant cell 19, 2776–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S4: Primers, sgRNAs sequences and ZmCLE genes’ annotation. Related to all Figures.
Data Availability Statement
All raw high-throughput sequencing data have been deposited in NCBI’s Sequence Read Archive (SRA). SRA IDs are listed in KEY RESOURCES TABLE. This study used codes from published software described in QUANTIFICATION AND STATISTICAL ANALYSIS.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GFP antibody (GFP-Trap magnetic agarose) | ChromoTek | Cat#gtma-20; RRID: AB_2631358 |
| Bacterial and Virus Strains | ||
| Agrobacterium | N/A | EHA101 |
| E.coli | N/A | DH5α |
| Biological Samples | ||
| Zea mays B73 | Maize Genetics COOP Stock Center | N/A |
| Zea mays pYAB14-TagRFPt reporter line | Je et al., 2016 | N/A |
| Zea mays ZmM16-YFP translational fusion line | Bartlett et al., 2015 | N/A |
| Zea mays ZmHDZIV6-YFP translational fusion line | This paper | N/A |
| Zea mays CRISPR/Cas9 knock out mutants of ZmVOZs | This paper | N/A |
| Zea mays branched silkless1;Tunicate (bd1;Tu) mutants | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Mannitol | Sigma-Aldrich | Cat# M4125 |
| Bovine serum albumin | Sigma-Aldrich | Cat#A7907-50G |
| T7 RNA polymerase | Sigma-Aldrich | 10881775001 |
| Calcofluor white stain | Sigma-Aldrich | Cat#18909 |
| Toluidine blue | Sigma-Aldrich | Cat#T3260 |
| CelLytic™ PN Isolation/Extraction Kit | Sigma-Aldrich | Cat#CELLYTPN1 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat#15714-s |
| Glutaraldehyde | Electron Microscopy Sciences | Cat#16537-16 |
| Cacodylate buffer | Electron Microscopy Sciences | Cat#11652 |
| LR white resin | Electron Microscopy Sciences | Cat#905072 |
| Cellulase RS | Onozuka | N/A |
| Cellulase R-10 | Onozuka | N/A |
| Macerozyme R-10 | Onozuka | N/A |
| Pectolyase Y-23 | Duchefa Biochem. | Cat#P8OO4.0001 |
| Trypan blue | Thermo Fisher Scientific | Cat#15250061 |
| Paraplast | McCormick Scientific | Cat#39503002 |
| ProbeOn Plus™ Slides | Fisher Scientific | Cat#22-230-900 |
| NBT/BCIP Ready-to-Use Tablets | Roche | 11697471001 |
| Critical Commercial Assays | ||
| Chromium i7 Multiplex Kit | 10X Genomics | Cat# PN-120262 |
| Chromium Single Cell 3’ Library & Gel Bead Kit v2 | 10X Genomics | Cat#PN-120237 |
| Chromium Single Cell A Chip Kit v2 | 10X Genomics | Cat#PN-1000009 |
| Dynabeads™ MyOne™ Silane Beads | Thermo Fisher Scientific | Cat#37002D |
| Arcturus PicoPure RNA Isolation Kit | Thermo Fisher Scientific | KIT0204 |
| RNA Bioanalyzer kit | Agilent | Cat#5067-1513 |
| DNA High Sensitivity Bioanalyzer kit | Agilent | Cat#5067-4626 |
| SMART-Seq™ v4 Ultra™ Low Input RNA Kit | Takara Bio USA, Inc. | Cat#634890 |
| Nextera XT DNA Library Prep Kit | Illumina | Cat#FC-131-1024 |
| AMPure XP Beads | Beckman Coulter | Cat#A63880 |
| KAPA Library Quantification Kits | Roche | Cat#KK4824 |
| NEXTflex ChIP-seq Kit | PerkinElmer Applied Genomics | NOVA-5143-02 |
| Deposited Data | ||
| B73 whole ear scRNA-seq_replicate 1 | This paper | PRJNA646989 |
| B73 whole ear scRNA-seq_replicate 2 | This paper | PRJNA646996 |
| B73 whole ear scRNA-seq_replicate 3 | This paper | PRJNA647001 |
| Protoplasting-response bulk RNAseq | This paper | PRJNA647196 |
| pZmYAB14-TagRFPt FACS RNAseq | This paper | PRJNA647195 |
| pZmYAB14-TagRFPt FACS ATACseq | This paper | PRJNA647197 |
| ZmHDZIV6-YFP ChIP-seq | This paper | PRJNA647198 |
| ZmM16-YFP ChIP-seq | This paper | PRJNA647200 |
| Experimental Models: Cell Lines | ||
| N/A | N/A | N/A |
| Experimental Models: Organisms/Strains | ||
| N/A | N/A | N/A |
| Oligonucleotides | ||
| See Table S4 for primers or sgRNAs sequences for mRNA in-situ, ZmVOZs crispr and genotyping, ZmHDZIV6-YFP transgene, and bd1 genotyping. | N/A | N/A |
| Recombinant DNA | ||
| pGW-Cas9 vector | Wang et al., 2014 | Addgene Plasmid # 50661; RRID: Addgene_50661 |
| pTF101 Gateway-compatible vector | Je et al., 2016 | NA |
| Software and Algorithms | ||
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR/wiki |
| R | R Core Team, 2013 | https://www.r-project.org/ |
| EmptyDrops | Lun et al., 2019 | https://rdrr.io/github/MarioniLab/DropletUtils/man/emptyDrops.html |
| DoubletFinder | McGinnis et al., 2019 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| Scater | McCarthy et al., 2017 | https://github.com/Alanocallaghan/scater |
| Scran | Lun et al., 2016 | https://github.com/MarioniLab/scran |
| Rsvd package | Erichson et al., 2019 | https://github.com/erichson/rSVD |
| Cccd package | Marchette, 2015 | https://github.com/cran/cccd |
| InfoMap | Csardi and Nepusz, 2006 | https://github.com/mapequation/infomap |
| MetaNeighbor | Crow et al., 2018 | https://github.com/maggiecrow/MetaNeighbor |
| UMAP package | Konopka, 2020 | https://github.com/tkonopka/umap |
| EGAD | Ballouz et al., 2017 | https://github.com/sarbal/EGAD |
| Trimmomatic - version 0.36 | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| edgeR | Robinson et al., 2010 | https://bioconductor.riken.jp/packages/devel/bioc/html/edgeR.html |
| elprep | Herzeel et al., 2015 | https://github.com/ExaScience/elprep |
| BWA-MEM | Li and Durbin, 2009 | https://github.com/lh3/bwa |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| LDAK software v5.0 | Speed et al., 2012 | http://www.ldak.org |
| Other | ||
| Cell Strainer | pluriStrainer | Cat#43-50030-50 |