

Abstract
Novel delivery strategies are necessary to effectively address glioblastoma without systemic toxicities. Triptolide is a therapy derived from the thunder god vine that has shown potent anti-proliferative and immunosuppressive properties but exhibits significant adverse systemic effects. Dendrimer-based nanomedicines have shown great potential for clinical translation of systemic therapies targeting neuroinflammation and brain tumors. Here we present a novel dendrimer-triptolide conjugate that specifically targets tumor-associated macrophages (TAMs) in glioblastoma from systemic administration and exhibits triggered release under intracellular and intratumor conditions. This targeted delivery improves phenotype switching of TAMs from pro- towards anti-tumor expression in vitro. In an orthotopic model of glioblastoma, dendrimer-triptolide achieved significantly improved amelioration of tumor burden compared to free triptolide. Notably, the triggered release mechanism of dendrimer-mediated triptolide delivery significantly reduced triptolide-associated hepatic and cardiac toxicities. These results demonstrate that dendrimers are a promising targeted delivery platform to achieve effective glioblastoma treatment by improving efficacy while reducing systemic toxicities.
Keywords: Dendrimer, triptolide, glioblastoma, tumor-associated macrophages, targeted delivery
Introduction
Glioblastoma (GBM) is the most common and aggressive form of primary brain cancer, accounting for 55% of brain cancer patients.1 More than 15,000 new cases are diagnosed each year in the United States for an annual incidence of 5.26 cases per 100,000 people.2 Current standard of care consists of surgical tumor resection followed by combination chemo- and radiotherapy, resulting in median survival of 15-20 months.3 In addition to high rates of recurrence and mortality, patients with GBM also experience substantial impacts to their cognitive function and quality of life.1 Incidence among patients over 65 is growing steadily, and these elderly patients face much poorer prognoses, with median survival of only 3-4 months.4 These poorer prognoses arise from number of factors, including less aggressive intervention and lower tolerance of treatment toxicities.5 Therefore, innovative new strategies that are less invasive and can be more well-tolerated are necessary to address the treatment challenges and rising incidence facing GBM.
Advancements in cancer therapy have shown immunotherapies as the promising next stage of cancer treatment by leveraging the body’s own tumor fighting immune response.6, 7 While these have largely focused on targeting T-cells to promote recognition of cancer antigens and enable their anti-cancer functions,8, 9 tumor-associated macrophages (TAMs) have emerged as promising therapeutic targets for cancer immunotherapy due to their roles in mediating the tumor immune response.10 GBM tumors actively recruit host macrophages and resident microglia and repolarize them into TAMs,11 which promote tumor growth, metastasis, and angiogenesis while suppressing the tumor-killing immune response.12, 13 Reprograming TAMs from their tumor-supporting state towards an anti-tumor phenotype can therefore inhibit tumor growth and bolster downstream anti-cancer immune signaling. Ablation of TAMs has also shown to exert therapeutic efficacy by reducing support of tumor growth.14, 15 TAMs-focused strategies have shown strong preclinical results, and several are undergoing clinical translation as mono- or combination therapies for synergistic effect (NCT02829723, NCT02452424, BCT01790503). However, translation of these therapies have faced hurdles, including low response rates, drug resistance, and systemic toxicities induced by broad, nonspecific immune activity.16, 17 Targeted delivery strategies that can bring these immunotherapies into solid tumors and directly to TAMs in a highly specific manner have the potential to substantially improve patient outcomes and reduce treatment-associated toxicities in GBM and other cancers.
Nonspecific activity resulting in systemic toxicities have long plagued the development of anti-cancer therapies.6 Triptolide is one such example, with promising immunosuppressive and anti-cancer properties but limited by significant systemic toxicities.18, 19 Triptolide acts as a STAT3 inhibitor to block STAT3 over activity and upregulation and suppress of tumor signaling in TAMs.20, 21 Triptolide has well established toxicity in nearly every organ system, with major toxicities observed in livers and hearts.22 These toxicities, along with poor solubility, result in a narrow therapeutic window that limits its clinical translation. Analogs aimed at improving its formulation and toxicity profile are being explored to limited success, with many analogs retaining triptolide’s toxic effects.23 Therefore, nanoparticle-mediated strategies to limit triptolide-induced toxicity and selectively deliver it to the tumor site may yield improvements to triptolide’s safety profile for clinical application.24, 25
Dendrimer-based targeting of neuroinflammation and brain tumors from systemic administration has emerged as promising strategies for addressing lack of efficacy and systemic toxicity of therapies.26, 29 We have previously shown that generation-4 hydroxyl-terminated polyamidoamine dendrimers are able to cross impaired blood brain barriers and selectively localize within activated microglia/macrophages at the site of brain injury from systemic administration.30, 36. In the context of GBM, we have shown that these dendrimers are able to penetrate solid brain tumors and specifically target TAMs in an orthotopic model of gliosarcoma from systemic administration.29, 33, 37 In addition, these dendrimers are nontoxic and scalable, facilitating clinical translation.38, 39 In this study, we present a dendrimer-triptolide conjugate for targeted systemic delivery to solid brain tumor and specifically to TAMs. We present the design and synthesis of dendrimer-triptolide conjugates using highly efficient click chemistry approach, in vitro analysis of immune reprogramming, and in vivo impacts on tumor burden and systemic toxicities in an orthotopic, immunocompetent model of glioblastoma.
Materials and Methods
Synthesis of D-triptolide conjugate
Materials
Triptolide was purchased from Chem Shuttle and was used as received. Generation-4 ethylenediamine-core polyamidoamine dendrimer (pharmaceutical grade) was purchased from Dendritech (Midland, MI, USA) as a methanolic solution. Methanol was evaporated prior to use. Azido-PEG-6-acid was purchased from Broadpharm (San Diego, CA, USA). 1-[3-(Dimethylamino)propyl]-3-ethylcarbodiimide methiodide (EDC), 4(dimethylamino)pyridine (DMAP), copper sulfate pentahydrate, hexynoic acid and sodium ascorbate were purchased from Sigma Aldrich (St. Louis, MO, USA). All other solvents were purchased from Sigma Aldrich and were used as received. The reactions were carried out in standard glassware under inert atmosphere at room temperature unless otherwise stated.
Synthesis procedures
Synthesis of triptolide-azide (Compound 2):
To a dried 100mL round bottom flask, triptolide (1g, 2.7mmoles) was taken and dissolved in 15mL of anhydrous DMF. The reaction mixture was stirred for 10 minutes until clear. Azido-PEG6-acid (1.26 g, 3.3 mmoles), EDC.HCl (0.62 g, 3.24 mmoles), and DMAP (0.32 g, 2.7 mmoles) were then added to the reaction mixture and stirred overnight. The progress of the reaction was monitored by thin layer chromatography. On completion, DMF was evaporated. The reaction mixture was diluted with 100mL of dichloromethane and washed with saturated NaHCO3 solution (20mL) three times. The organic layer was further washed with saturated NH4Cl solution (20mL) three times, followed by washing with brine solution. The organic layer was dried over anhydrous sodium sulphate, filtered, and concentrated via evaporation. The crude mixture was purified using flash column chromatography (Teledyne Combiflash). The pure compound was obtained in 5% methanol in DCM. The yield of the triptolide-PEG6-azide was 79%, and the pure compound was a transparent oil.
1H NMR: (500 MHz, DMSO) δ 4.99 (s, 1H), 4.82 (dd, J = 40.1, 17.6 Hz, 2H), 4.10 (d, J = 5.2 Hz, 1H), 3.95 (d, J = 2.9 Hz, 1H), 3.72 – 3.46 (m, 27H), 3.44 – 3.36 (m, 2H), 3.17 (d, J = 5.1 Hz, 3H), 2.65 – 2.55 (m, 3H), 2.26 – 2.07 (m, 2H), 2.02 – 1.92(s, 1H), 1.90 – 1.72 (m, 2H), 1.37 – 1.18 (m, 2H), 0.92 (s, 3H), 0.87 (d, J = 6.9 Hz, 3H), 0.75 (d, J = 6.8 Hz, 3H). (Figure S1)
13C NMR: (126 MHz, DMSO) δ 173.1, 170.4, 162.2, 123.1, 70.8, 70.2, 69.7, 69.2, 66.1, 63.3, 62.7, 60.9, 59.2, 54.9, 54.4, 50.0, 48.6, 35.0, 29.1, 27.0, 22.3, 17.4, 16.4, 13.8. (Figure S2)
HPLC: Retention time: 23.8 minutes; Purity: 96% (Figure S3)
HRMS: Theoretical: 736.37 (m/z); obtained C35H51N3NaO13: 744.33 (Na adduct) (Figure S4)
Synthesis of D-hexyne (Compound 4):
Generation 4 hydroxyl terminated PAMAM dendrimers (G4-OH) (1 g, 0.07 mmoles) in anhydrous DMF (20mL) was stirred until clear. 5-Hexynoic acid (172 mg, 1.54 mmoles) was dissolved in 2 ml of DMF and added to the stirring solution. After 10 minutes, EDC.HCl (295mg, 1.54 mmoles) and DMAP (187.88mg, 1.54 mmoles) were added and reacted for 48 hours. On completion, DMF dialysis in 1kDa MWCO was performed for 8 hours with one solvent change. Then, 30mL of D.I. water was added to the solution and dialyzed against water overnight. The solution was then lyophilized to obtain white solid at 74% yield.
1H NMR: (500 MHz, DMSO) δ 8.21 – 7.57 (m, 124H for internal amides), 4.71 (s, 50H), 4.01 (t, J = 5.5 Hz, 12–13 CH2), 3.5 – 2.1 (m, dendrimer CH2), 1.71 – 1.59 (m, 13-15 CH2). (Figure S5)
HPLC: Retention time: 13.5 minutes; Purity: 99.8% (Figure S6)
Synthesis of D-triptolide (Compound 5):
Compound 4 (2 g, 0.131 mmoles) was dissolved in 20mL of DMF by sonication. Triptolide-azide 2 (1.48g, 1.97mmoles) in DMF (5mL) was added in the reaction mixture and stirred. Copper sulfate pentahydrate (16.3mg,0.065mmoles) dissolved in 5mL water was then added dropwise. After 5 minutes, sodium ascorbate (26mg, 0.131mmoles) dissolved in 5mL water was added dropwise. The reaction mixture was stirred and heated for 24 hours at 40°C. On completion, the reaction mixture was dialyzed against DMF in 1000 MWCO. To this solution, EDTA (50μL, 0.5M) solution was added for copper removal by chelation. The DMF dialysis was followed by water dialysis overnight. The product yield was 69%. The 1H NMR indicated the formation of product with 12-13 arms of triptolide attached. The drug loading is calculated by proton integration where peaks corresponding to dendrimer and drug are compared.
1H NMR: (500 MHz, DMSO) δ 8.19 – 7.67 (m, D-internal amide H and triazole H), 4.99 (s, triptolide H), 4.82 (dd, J = 39.5, 16.6 Hz, triptolide H), 4.47 (t, J = 5.1 Hz, linker H), 4.06 – 3.86 (m, linker H), 3.80 (t, J = 5.2 Hz, linker H), 3.73 – 3.58 (m, linker H), 3.56 – 3.19 (m, triptolide H), 3.45-3.20 (m, dendrimer H), 3.17 – 3.00 (m, dendrimer H), 2.86-2.57 (m, dendrimer H), 2.45 – 2.12 (m, dendrimer H), 1.86 – 1.73 (m, linker H), 1.35-1.22 (m, triptolide H), 0.89 (s, triptolide H), 0.86 (d, J = 6.8 Hz, triptolide H), 0.74 (d, J = 6.8 Hz, triptolide H). (Figure S7)
1H NMR (500 MHz, MeOD) δ 7.84 (s, triazole H), 5.08 (s, triptolide H), 4.58-4.51 (m, triptolide H), 4.14 (m, linker H), 3.96 – 3.69 (m, linker H and triptolide H), 3.69 – 3.52 (m, linker H), 3.52-3.38 (m, dendrimer H), 3.20-2.20 (dendrimer H and triptolide H), 1.99-1.86 (m, linker H), 1.51 – 1.24 (m, triptolide H), 0.99 (s, triptolide H), 0.93 (d, triptolide H), 0.82 (d, triptolide H). (Figure S8)
HPLC: Retention time: 24.37 minutes; Purity: 99.7% (Figure S9)
Instrumentation Characterization
NMR spectroscopy
Proton and carbon nuclear magnetic resonance spectroscopy techniques were used to analyse the structures of compounds. The NMR spectra were recorded on a Bruker spectrometer (MA, USA). All the spectra were recorded at room temperatures using CDCl3, DMSO-D6 or CD3OD as solvents. The chemical shifts are reported in ppm.
Mass spectroscopy
Mass measurements were performed using high resolution mass spectrometry (HRMS, Bruker microTOF-II mass spectrometer). The measurements were performed using ESI in the positive mode. Direct flow sample introduction was carried out in acetonitrile/ water (9:1) solvent system. The spectra was recorded to show protonated molecular ion peaks [M + nH]n+ or adducts [M + nX]n+ (X = Na, K, or NH4) for empirical formula confirmation.
High Performance Liquid Chromatography (HPLC)
HPLC instrument (Waters Corporation, Milford, MA, USA) was used to analyse the purity of the drug linker and the dendrimer drug conjugates. The instrument has a 1525 binary pump, an in-line degasser AF, an autosample and two detectors (2998 photodiode array and 2457 multi λ fluorescence). The samples were run through C18 symmetry 300 column (5μm, 4.6x250mm). Waters empowers software was used for sample characterization and analysis. The traces were recorded at 210 nm. A gradient flow was used to run the samples using eluent A as water with 5% acetonitrile and B as acetonitrile. The method started with 80% A which continued for 5 minutes, then decreased to 45% A at 25 minutes, which stayed at 45% at 35 minutes and increased back to 80% A at 45 minutes maintaining a flow rate of 1 mL/min.
Hydrodynamic diameter and zeta potential distribution
Zetasizer Nano ZS (Malvern Instrument Ltd., Worchester, U.K.) instrument was used to measure the size and zeta potential of D-Triptolide conjugate using our previously established protocols.34, 35 Briefly, the conjugate was dissolved in ultrapure deionized water at a concentration of 0.2mg/mL followed by the filtration through 0.2 μm syringe filters (Pall Corporation, 0.2 μm HT Tuffryn membrane). The size was measured in triplicates via dynamic light scattering using UV transparent disposable cuvette (Dimensions: 12.5 x 12.5 x 45mm). The sample for the zeta potential distribution was prepared by dissolving the dendrimer conjugate in 10mM sodium chloride solution at the concentration of 0.2 mg/mL. The measurements were recorded in triplicates in using Malvern Zetasizer Nanoseries disposable folded capillary cells.
In vitro drug release studies
In vitro studies to estimate the release of Trip from D-Trip was carried out under plasma conditions (PBS, pH 7.4) and intracellular/intratumor conditions (citrate buffer, pH 5.5 with esterase). The studies were performed in triplicates. D-Trip conjugate was dissolved in appropriate buffers to achieve a concentration of 1 mg/mL and incubated at 37°C with constant shaking. At specified time points, the aliquots were taken out, diluted with equal volume of methanol and were stored at −20°C. A standard curve of free Trip was prepared on HPLC. The release samples were run on HPLC, and the drug release was calculated comparing to the standard curve of free Trip. The drug release values are presented as average values from three samples.
Biological studies
Materials
DMEM media, RPMI media, trypsin, fetal bovine serum (FBS), penicillin/streptomycin (P/S), L-glutamine, TRIzol, MTT reagent, and green SYBR reagent were obtained from Invitrogen (Carlsbad, CA, USA). Recombinant mouse interferon-γ (IFNγ) was obtained from R&D Systems (Minneapolis, MN, USA). Chloroform, isopropanol, DEPC water, and 200 proof ethanol were obtained from ThermoFisher (Waltham, MA, USA). Normal saline and phosphate buffered saline (PBS) were obtained from Corning Inc. (Corning, NY, USA). Sucrose and formalin solution were obtained from Sigma Aldrich (St. Louis, MO, USA).
Cell culture
BV2 murine microglia were obtained from the Children’s Hospital of Michigan’s cell culture facility. GL261 murine glioblastoma cells were obtained from the DTP/DCTD/NCI Tumor Repository (Frederick, MA, USA). Cells were incubated at 37°C and 5% CO2 atmosphere. BV2 microglia were grown in DMEM with 10% FBS and 1% P/S, while GL261 cells were grown in low glutamine RPMI with 10% FBS, 1% P/S, and 1% L-glutamine. Treatments were performed in media with 5% FBS instead of 10%.
In vitro analysis of inflammatory response
Ablation of activated microglia was assessed in IFNγ-stimulated microglia via MTT assay. Cells were preactivated with 50 ng/mL IFNγ due to its role in mediating STAT3 activation, the major immune target of triptolide.40, 41 Cells were then incubated in triptolide (Trip) or dendrimer-triptolide conjugate (D-Trip) for 24 hours at equivalent drug dosages calculated based on triptolide weight loading on the dendrimer. Treatments were replaced with fresh media and 10 μL of MTT solution (5 mg/mL MTT in PBS) was added to each well. Cells were incubated in MTT solution for 4 hours to convert to formazan, which was solubilized in DMSO and read using a Synergy MX plate reader (Biotek, Winooski, VT, USA). IC50 was calculated using GraphPad Prism analysis function [inhibitor] vs. normalized response with variable slope. For anti-proliferative activity in GL261 cells, the protocol above was followed without stimulation.
To assess impacts of treatment on inflammatory phenotype, BV2 microglia were activated with IFNγ for 3 hours at 50 ng/mL. Cells were then cotreated with IFNγ and Trip or D-Trip for 24 hours. Treatments were then removed, and cells were incubated in fresh media for 24 hours. Cells were collected in TRIzol and processed for analysis of mRNA expression according to manufacturer’s protocol. Briefly, 200 μL of chloroform was added, and samples were shaken roughly and incubated on ice for 15 minutes. Samples were then spun down for 15 min at 15,000g to separate aqueous and TRIzol fractions. The aqueous layer was collected and combined with 500 μL of isopropanol, shaken, and centrifuged for 15 minutes at 15,000g. Samples were then washed with 75 % ethanol and resuspended in 40 μL of DEPC water. Samples were nanodropped to determine RNA concentration and converted to cDNA (Applied Biosystems, Foster City, CA, USA). rt-qPCR analysis was performed on a StepOne Plus system (Applied Biosystems). PCR primers for STAT3, CD206, and SOD1 were obtained from Bio-Rad Laboratories (Hercules, CA, USA). PCR primers for TNFα (F: CCA GTG TGG GAA GCT GTC TT; R: AAG CAA AAG AGG AGG CAA CA), IL1β (F: AGC TTC AAA TCT CGA AGC AG; R: TGT CCT CAT CCT GGA AGG TC), and IL6 (F: TCC AGT TGC CTT CTT GGG AC; R: GTG TAA TTA AGC CTC CGA CTT G) were obtained from Integrated DNA Technologies (Coralville, IA, USA), three anti-tumor signals.42–44
Tumor inoculations
Animal experiments were conducted in accordance with approved protocols by the Johns Hopkins University institutional animal care and use committee. All animals were housed at Johns Hopkins University animal facilities with free access to food and water.
The GL261 syngeneic, orthotopic, immunocompetent brain tumor model in C57BL/6 mice was chosen because it is well-established for effectively evaluating immunotherapies 45 Male and female C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME, USA) 6-8 weeks in age were intracranially inoculated with GL261 murine glioblastoma cells. Mice were anesthetized with a ketamine xylazine anesthesia cocktail. A burr hole was drilled 1 mm posterior and 2 mm lateral to the midline to inject cells into the striatum. 2 μL of GL261 cell solution (100,000 cells total) was injected into the striatum at a depth of 2.5 mm by a Hamilton syringe (Hamilton Company, Reno, NV, USA) using a stereotactic frame and automated syringe pump (Stoelting Co., Wood Dale, IL, USA). The syringe was withdrawn at a rate of 0.5 mm/min to reduce backflow, and the incision was sutured together.
Evaluation of tumor burden and markers of disease progression
Five mice were randomly assigned to control, Trip, or D-Trip cohorts. Mice were administered daily with intraperitoneal injections of Trip or D-Trip at 0.5 mg/kg equivalent drug dosage starting on day 5 after tumor inoculation. This dosage was chosen because previous studies of triptolide anti-cancer efficacy in vivo have administered doses ranging from 0.42 mg/kg to 6 mg/kg.46–48 Based on our previous studies with this dendrimer platform, we chose a dose on the low end of that range. Trip was formulated in 10% DMSO in normal saline to solubilize drug. Control treatments consisted of 10% DMSO in normal saline to account for potential biological effects of DMSO. A vehicle control group of unmodified dendrimer treatment was not performed based on previous studies where G4-OH has been shown to exhibit no therapeutic/inflammatory activity.49–51 To assess disease burden, mice were monitored daily. Kyphosis scoring was performed daily by blinded graders based on the degree of hunched posture as a marker of neurodegeneration.52 A score of 0 to 3 was assigned, with 0 indicating normal posture and 3 indicating severe spinal cord curvature that does not resolve upon movement. To assess mobility, mice were recorded in open field experiments on day 10, 13, and 17 post-inoculation and analyzed with EthoVision XT video tracking software (Noldus Information Tech Inc., Leesburg, VA, USA). Distance travelled was normalized to day 10 average distances. On day 20 after inoculation (or at morbidity if mice met euthanasia criteria prior to day 20), brains were collected. Euthanasia criteria for mice include weight loss exceeding 20% of pre-surgery mass, ataxia, and lack of grooming. Tumors were dissected out and massed, and tumor burden was calculated as the tumor mass percent out of total brain mass.
Evaluation of systemic toxicities
Mice were monitored daily to assess impacts of treatment on disease progression. Injected site irritation and adverse responses were recorded when observed, including fibrosis, inflammation, and hair loss at the site of injection. No adverse responses were observed with control treatments. Organs were fixed in 4% formalin solution, followed by a sucrose gradient to remove residual formalin from tissues (10%, 20%, then 30% sucrose in PBS). Kidneys, livers, and hearts were sectioned to 4 μm thick slices and H&E stained. Stained slices were imaged on a Nikon Eclipse NiU microscope (Minato City, Tokyo, Japan). Blinded scoring of livers was performed based on globular lymphocyte infiltration, steatosis, necrosis, and subcapsular edema. Blinded scoring of hearts was performed based on fibrosis, edema, inflammation, cardiomyocyte degeneration, and vacuolation. No morphological markers of toxicity were observed in kidneys. For scoring, n=5 mice per group were evaluated, with 2 images per mouse.
Statistical analyses
Statistical analyses and plots were produced using GraphPad Prism v8.0 software (San Diego, CA, USA). Significance between IC50 of Trip and D-Trip was calculated with Student’s t-test. Significances between Trip and D-Trip in in vitro inflammatory analyses were performed using two-way ANOVAs. Comparisons between Trip and D-Trip in vivo was performed using two-way ANOVA with Bonferroni corrections for multiple comparisons for kyphosis progression and one-way ANOVAs for tumor burden. Student’s t-tests were performed for comparisons between Trip and D-Trip for morphological scoring of toxicity. Error bars presented in plots represent mean ± standard error.
Results and Discussion
Synthesis of the dendrimer-triptolide conjugate
To enable TAMs targeting, enhance water solubility, and decrease systemic toxicity, triptolide was attached to generation-4 hydroxyl-terminated PAMAM dendrimers (PAMAM-G4-OH) via covalent surface conjugation using alkyne-azide click chemistry (Figure 1). Copper (I) catalyzed alkyne-azide click (CuAAC) is a well-established click reaction which has gained tremendous popularity as a facile tool for the synthesis of dendrimer and polymer conjugates.53, 54 To enable the conjugation via CuAAC reaction on the dendrimer surface, we first modified the triptolide by attaching an azide terminating linker through enzyme sensitive ester linkages for the triggered release of the drug in the tumor microenvironment (Figure 1A). The linker was attached at the free β hydroxyl group at C14 position of triptolide (1) via an esterification reaction with azido-PEG6-acid using EDC, DMAP coupling agents to obtain triptolide-azide (2). Conjugation at this position was chosen because the β hydroxyl group at C14 in triptolide is essential for its biological activity;55 therefore, triptolide will remain inactive while attached to the dendrimer and only become active upon release in low pH environments such as intratumor and intracellular conditions. We expect conjugation through this location, along with the pH-sensitive linker and TAMs targeting property of G4-OH to significantly enhance therapeutic efficacy and reduce systemic toxicity.
Figure 1: Schematic representation of synthesis of dendrimer-triptolide conjugate.
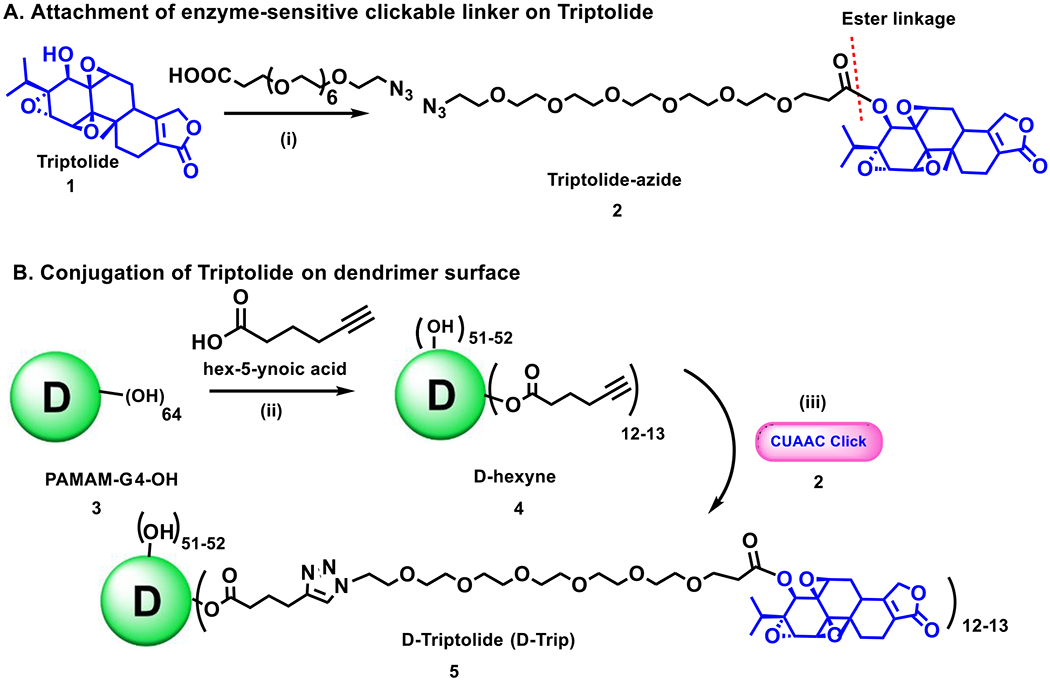
(A) synthesis of triptolide-azide containing enzyme sensitive ester linkages; (B) Covalent surface conjugation of triptolide-azide on alkyne modified dendrimer via CuAAC click reaction.
We partially modified the surface of the dendrimer to display alkyne functional groups for participation in click reaction with triptolide-azide (2) (Figure 1B). The dendrimer (3) was reacted with 5-hexynoic acid using standard esterification conditions to afford D-hexyne (4) with ~12-13 terminal alkyne functional groups. This was followed by the CuAAC reaction between D-hexyne (4) and triptolide azide (2) in the presence of catalytic amount of copper sulfate pentahydrate and sodium ascorbate to obtain D-triptolide conjugate (D-Trip) with ~12-13 molecules of the triptolide covalently linked on the dendrimer surface. The excess amount of copper was removed by chelation with EDTA to obtain pure product.
Physicochemical characterization of triptolide-linker and dendrimer-triptolide conjugate
Key challenges to translation of nanomedicines include lack of detailed physicochemical characterization for reproducible scaleup. To address these issues, we performed detailed characterization of D-Trip and intermediate compounds throughout the synthesis. We first confirmed the attachment of PEG linker on triptolide-azide. After the esterification reaction on β hydroxyl group at C14 position, the proton at C14 showed a downfield shift from δ 3.55 ppm to δ 4.99 ppm along with the disappearance of a doublet from OH proton at δ 4.68 ppm and the appearance of PEG protons (Figure 2A). In addition, the mass spectra confirmed the formation of the product showing a peak at 744.3 (m/z) corresponding to the sodium adduct of the product (Figure S4). HPLC chromatograms showed a clear shift in retention time from 13.8 minutes to 23.8 minutes after the attachment of PEG linker on triptolide (Figure 2B). The purity of triptolide-azide was ~96% (Figure S3). The number of hexyne linkers on the surface of D-hexyne was calculated using proton integration by comparing the number of internal amide protons of the dendrimer at δ8.11 – 7.70 ppm to the ester-linked methylene protons at δ 4.1 ppm, which showed the attachment of ~12-13 molecules of hexyne linkers per dendrimer. The success of the click reaction between D-hexyne and triptolide-azide to obtain D-Trip was confirmed with NMR which showed the presence of triptolide protons along with the dendrimer protons. To see the characteristic triazole proton in the NMR, we switched the solvent from DMSO-D6 to CD3OD which lead to the disappearance of internal amide protons interfering with the triazole protons and showed the presence of triazole protons at 7.8 ppm (Figure 2A). HPLC traces showed the shift in the retention time from 13.5 minutes for D-hexyne to 24.37 minutes for D-Trip (Figure 2C), and a purity of >99% for the final dendrimer-drug conjugate (Figure S9).
Figure 2. Characterization of intermediates and dendrimer-triptolide conjugate.
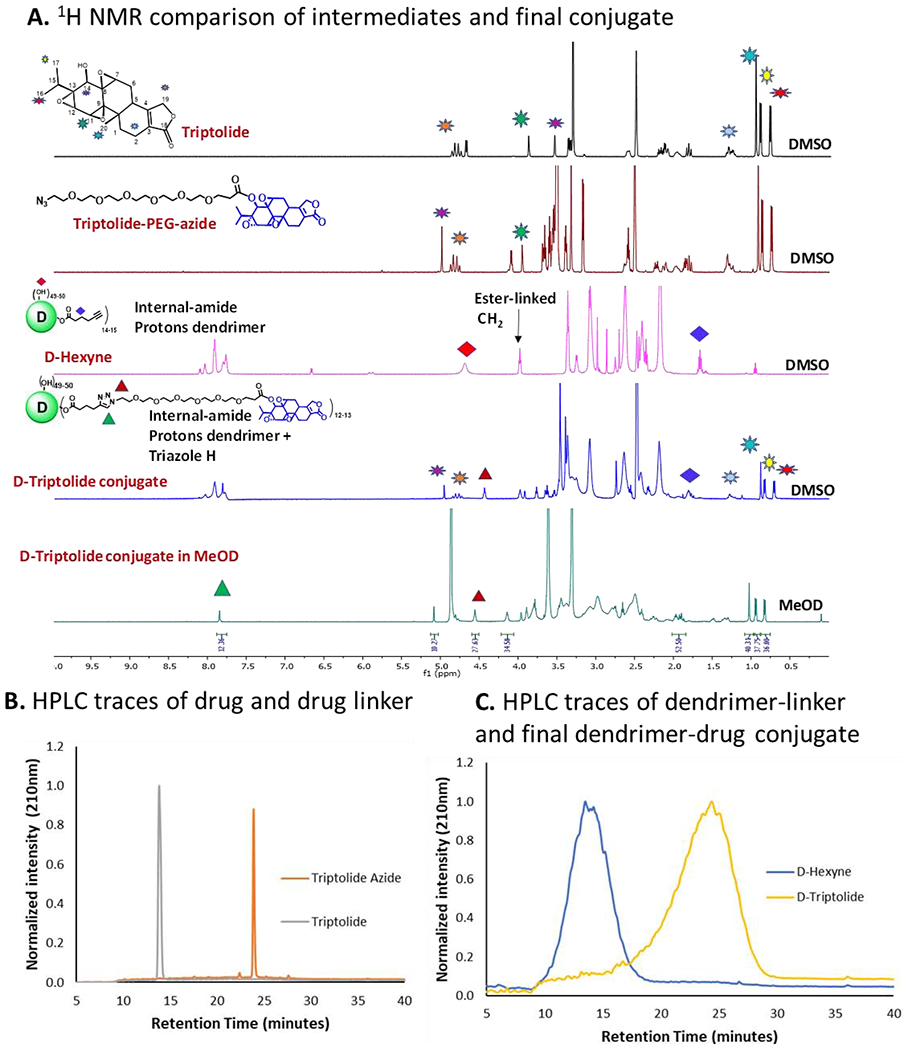
(A) 1H NMR spectra of free drug, drug linker, D-hexyne nad D-triptolide representing characteristic proton signals; (B) HPLC comparison of triptolide and triptolide-azide showing a shift in the retention time from 13.8 minutes to 23.8 minutes after the attachment of linker; and (C) HPLC traces showing a shift in retention time from D-hexyne (13.5 minutes) to D-triptolide (24.4 minutes).
The attachment of ~12-13 triptolide molecules per dendrimer corresponds to the drug loading of ~18% w/w. We have previously shown that the dendrimer-drug conjugates with up to ~20% w/w drug loading do not exhibit alterations from the biodistribution profile of the parent hydroxyl dendrimer.35, 36, 38 Zetasizer measurements of D-triptolide suggested a size distribution of 4.7±0.6 nm (Figure 3A) and a nearly neutral zeta potential distribution of +4.8±1.1 mV (Figure 3B). A small size and nearly neutral zeta potential are the key requirements for the dendrimer-drug conjugates in order to keep the inherent properties of dendrimers intact for targeting TAMs from systemic administration and for rapid renal clearance.
Figure 3. Physico-chemical properties and in vitro drug release study of D-Triptolide conjugate.
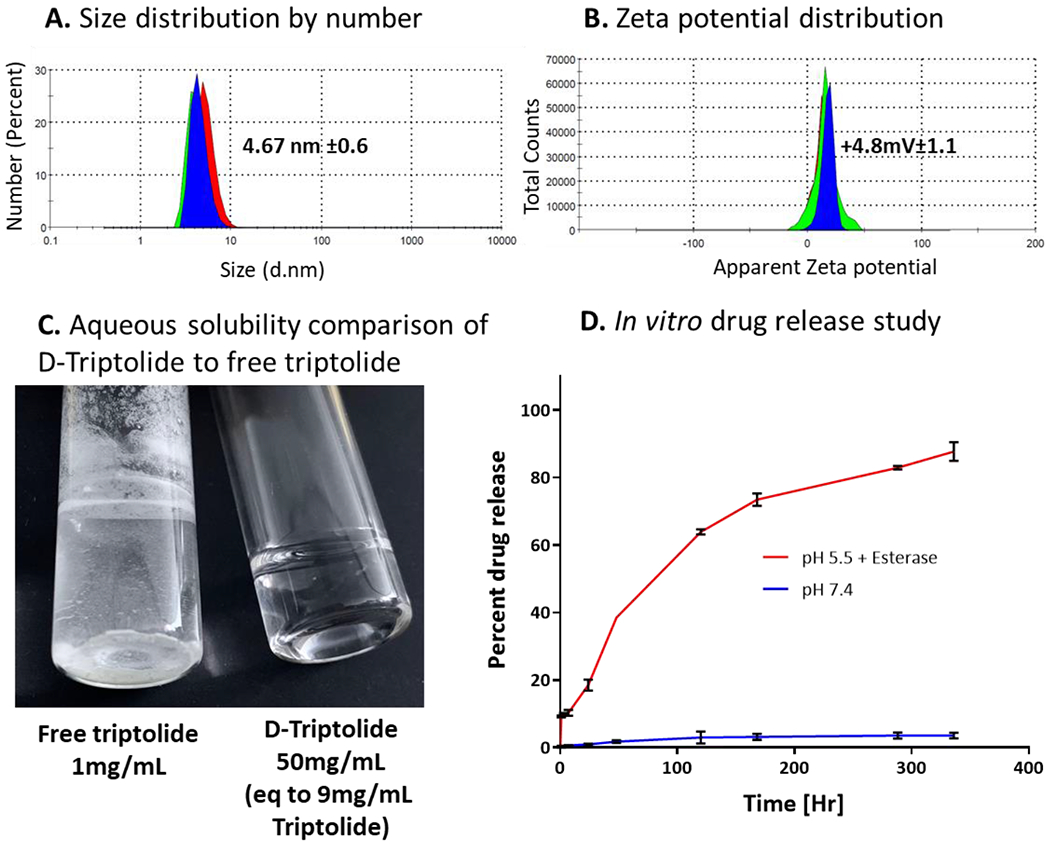
(A) Hydrodynamic diameter of D-triptolide conjugate measured in triplicates by dynamic light scattering; (B) Zeta potential distribution of D-Triptolide showing a near neutral zeta potential; (C) Aqueous solubility comparison of D-triptolide to free triptolide showing triptolide sparingly soluble in water at 1mg/mL while D-triptolide completely soluble in water at 50mg/mL (9mg/mL triptolide equivalent) demonstrating enhanced solubility of triptolide resulting from dendrimer conjugation; and (D) In vitro drug release study of D-Triptolide at extracellular plasma conditions (pH 7.4) and intracellular conditions (pH 5.5 plus esterases).
Dendrimer conjugation enhances the aqueous solubility of triptolide
The clinical development of triptolide is hindered by its toxicity and poor aqueous solubility. The conjugation of triptolide on dendrimer surface eases clinical formulation by significantly enhancing its aqueous solubility. The water solubility of triptolide is 0.017 mg/mL, while D-Trip exhibits aqueous solubility of 50 mg/mL which translates to 9 mg/mL free triptolide (18% w/w loading), suggesting >500-fold increase in the aqueous solubility of triptolide (Figure 3C).
In vitro drug release study of the dendrimer-triptolide conjugate
Triptolide was conjugated to the dendrimer via an ester linkage at its C14 position, which enables pH and esterase sensitive release. This linker design enables triggered release under intracellular and intratumor conditions, thereby limiting triptolide exposure outside the brain tumor (Figure 3D). Since β OH group at C14 position is important for the anti-cancer activity of the drug, the drug needs to be released from the conjugate to exert its effect.55 Under plasma conditions (pH 7.4), less than 1% of the triptolide load on the dendrimer was released in first 24 hours, and only ~4% was released over a period of 2 weeks. The release study at intracellular conditions were carried out at pH 5.5 in the presence of esterases to mimic endosomal conditions since dendrimers have shown to internalize into activated microglia and macrophages via fluid phase endocytotic pathways.56 At intracellular conditions, approximately 30% drug is released in first 24 hours, with more than 80% released in ~10 days.
Dendrimer-triptolide conjugate improves phenotype switching in TAMs-like microglia
Triptolide has been shown to inhibit XPB, a subunit of TFIIH, to inhibit RNA transcription and DNA repair to induce apoptosis.57 This strategy has primarily been employed to directly address tumor cell growth58, 59 but may yield therapeutic benefits when applied to TAMs as well. Macrophage ablation has been shown to be a promising strategy for reducing the immunosuppressive tumor immune environment.14 We have previously shown that systemically administered hydroxyl PAMAM dendrimers specifically localize within TAMs in orthotopic glioblastoma and reactive microglia in other CNS disease models, with negligible localization within other cell types.29, 37, 51 We attribute this selective uptake to the extravasation of dendrimers into the tumor due to the impaired blood brain tumor barrier, followed by selective internalization by TAMs via fluid-phase endocytosis.56 This enables us to deliver triptolide specifically to TAMs, with clearance from other organs through the kidney. Therefore, we examined the therapeutic efficacy of dendrimer-delivered triptolide compared to triptolide in TAMs-like IFNγ stimulated murine microglia. IFNγ was chosen as the stimulant due to its role in regulating the STAT3 pathway, the primary target of triptolide immunosuppression.40 Despite its application as an immune signal to promote anti-tumor signaling,60 IFNγ has shown conflicting roles in tumor immunity, with other reports demonstrating its tumor-promoting functions.61 Treatments were administered at equivalent drug bases. In these IFNγ stimulated microglia, both triptolide (Trip) and dendrimer-triptolide conjugate (D-Trip) exhibited dose dependent ablation (Figure 4A). D-Trip exhibited a lower IC50 of 88.1 ± 13 μg/mL compared to 140.3 ± 29 μg/mL of Trip (p = 0.022 free Trip vs. D-Trip), indicating stronger anti-proliferative activity. In vitro anti-proliferative activity in GL261 cells directly showed that Trip exerted greater effect than D-Trip, which we theorize is due to <20% of the triptolide being released from the dendrimer during the tested incubation time (Figure S10). The discrepancy in activity of D-Trip in microglia compared to tumor cells is the high degree of cellular internalization of dendrimers in these highly phagocytic cells.62
Figure 4. In vitro activity of triptolide and dendrimer-triptolide conjugate treatment.
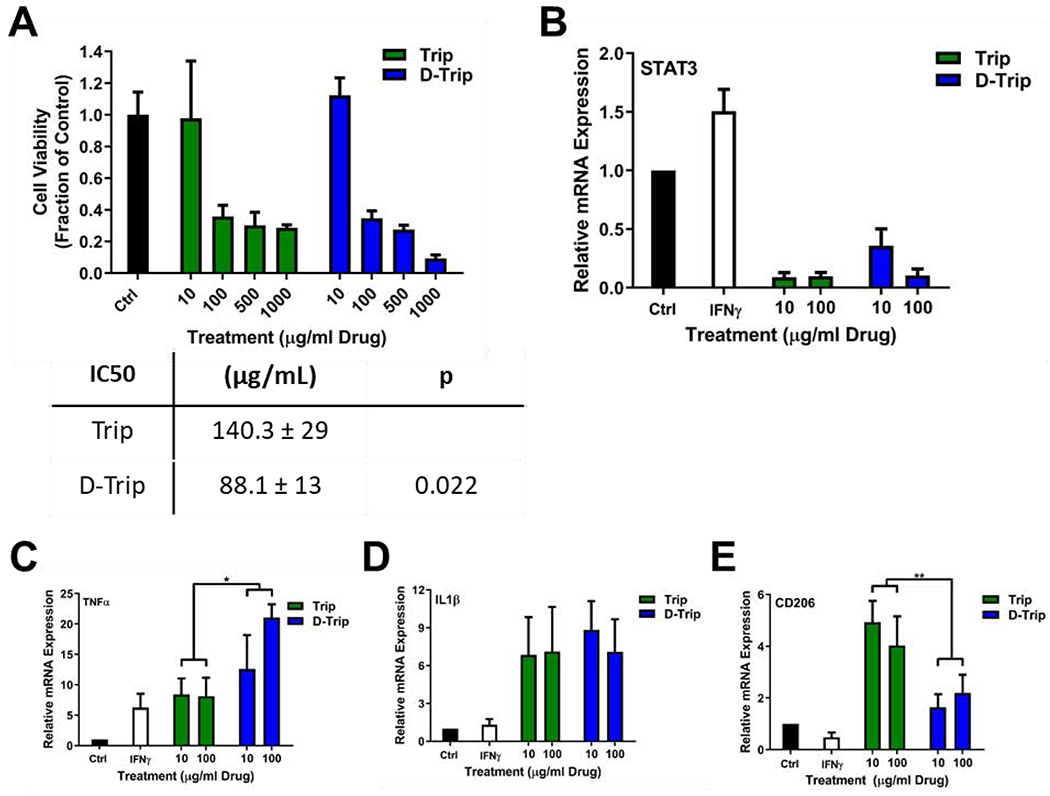
(A) Dendrimer delivery of triptolide (D-Trip) improves ablation of BV2 murine microglia compared to free triptolide (Trip) treatment. ** p < 0.01 Trip 1000 vs. D-Trip 1000. For analyses of immune modulation, BV2 cells were stimulated with 50 ng/mL of IFNγ for 3 hours, then cotreated with Trip or D-Trip for 24 hours. Cells were then incubated in fresh media for 24 hours, followed by RNA extraction for rt-qPCR analyses. (B) Both Trip and D-Trip reduce IFNγ-induced STAT3 expression to below control levels. D-Trip also significantly increases the expression of anti-tumor cytokines (C) TNFα and (D) IL1β while limiting expression of (E) protumor signal CD206. * p < 0.05, ** p < 0.01.
Incomplete ablation may result in remaining TAMs continuing to exert immunosuppressive effects. Therefore, to assess immune reprogramming through STAT3 inhibition by Trip and D-Trip, mRNA expression of immune signals was assessed in IFNγ stimulated microglia. Both Trip and D-Trip exhibited highly effective inhibition of STAT3, reducing STAT3 expression to ~10% of healthy levels with 100 μg/mL (Figure 4B), and SOD1 (Figure S11B). This resulted in upregulation of anti-tumor signals TNFα, IL1β, and IL6 (Figure 4C, D, Figure S11A). Notably, D-Trip significantly improved ~2.5-fold the increased expression of TNFα (p = 0.0335 free Trip vs. D-Trip). In the context of autoimmune diseases, triptolide has been shown to inhibit TNFα expression,63, 64 while in certain types of cancers it has been found to promote TNFα expression.22 Triptolide treatment has been shown to improve the efficacy of TNFα-based anti-cancer therapy to promote cancer apoptosis,65, 66 suggesting that the increased upregulation of TNFα by D-Trip contributes to its enhanced ablative effects. Free Trip treatment upregulated expression of anti-inflammatory cytokines CD206, while the upregulation with D-Trip treatment was clearly seen, the effect was more pronounced for free Trip (Figure 4E, p = 0.0066 free Trip vs. D-Trip) which may be due to the slower drug release rate from the dendrimer. In contrast, previous studies have established that triptolide prevents monocyte differentiation and activation into TAMs in part by inhibiting CD206 expression to reduce antiinflammatory phenotype.67, 68 Triptolide has been shown to upregulated CD206 in dendritic cells, although the mechanism behind this property is not well understood.69, 70 Further exploration is necessary to determine the differential effects of triptolide in TAMs vs. dendritic cells to determine why the observed upregulation in CD206 occurs. Nevertheless, these results indicate that triptolide treatment ablates and repolarizes TAMs-like microglia, and dendrimer delivery can improve these activities for enhanced anti-tumor immune efficacy. Based on these in vitro results, we hypothesize that in vivo TAMs targeting of dendrimers will enable significantly improved therapy for glioblastoma treatment.
Dendrimer targeted TAMs delivery of triptolide improves therapeutic efficacy
We have previously demonstrated that in vivo in orthotopic glioblastoma models, the dendrimer accumulates within the tumor in significant amounts. In addition, this localization is specifically within TAMs, with no dendrimer signals observed in cancer cells or other cell types.29, 37 To evaluate the impact of dendrimer TAMs targeting on triptolide efficacy in glioblastoma, mice were inoculated intracranially with GL261 murine glioblastoma cells to establish an orthotopic immunocompetent model of glioblastoma. Mice were administered systemically with 0.5 mg/kg Trip or D-Trip on equivalent drug dosage daily starting on day 5 after tumor inoculation. Mice were monitored daily and recorded in open field experiments to assess disease progression. Brains were collected and tumors dissected out to measure tumor mass as a percentage of total brain mass on day 20 post-inoculation (or, if earlier, when mice met criteria for euthanasia).
Trip treatment reduced tumor burden from 35% of total brain mass to 20% (Figure 5A, p = 0.0299 Ctrl vs. free Trip). D-Trip further reduced the tumor burden to ~12% of total brain mass (p = 0.0043 Ctrl vs. D-Trip, p = 0.076 free Trip vs. D-Trip) for a ~65% reduction compared to control saline treatment. While triptolide has been explored for the treatment of glioblastoma, such studies have focused on in vitro induction of apoptosis in glioma cells.71–73 In vivo, D-Trip compares favorably to other nanoparticle-mediated triptolide treatment has been explored for other cancer types. In an H22 flank model of hepatocarcinoma, systemically administered free Trip reduced tumor burden ~13%, while lipid polymer nanoparticles delivering Trip did not exhibit any reduction in tumor burden compared to untreated mice.74 Codelivery of Trip with doxorubicin in these lipid polymer nanoparticles exhibited synergistic effects, reducing tumor burden by ~70%. In an orthotopic model of hepatocarcinoma, folate-receptor targeted Trip nanoparticles decreased tumor burden by ~65%.75 In an orthotopic model of non-small cell lung cancer, Trip dosed every two days at 6 mg/kg reduced tumor size by ~25%, while Trip lipid nanoparticles reduced tumor size by ~50%.76 Co-delivery of Trip with paclitaxel in lipid nanoparticles reduced tumor burden by 80%. Compared to these nanoparticle formulations, D-Trip exhibits greater reduction in tumor burden at significantly lower doses, with efficacy rivaling combination therapy with chemotherapies or receptor-targeted nanoparticles. In addition, D-Trip is able to achieve this level of efficacy in an orthotopic brain tumor model with multiple biological barriers that flank and other tumors lack. Taken together, these results suggest that D-Trip is an effective nanoparticle delivery platform for targeting TAMs in glioblastoma for improving reduction in tumor burden. This therapy may be further enhanced through combination with chemotherapies for synergistic effects.
Figure 5. Dendrimer delivery improves therapeutic efficacy of triptolide in glioblastoma.
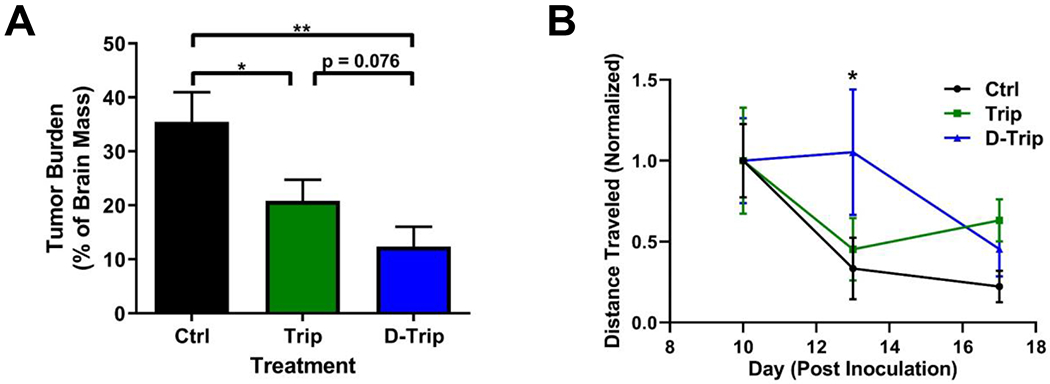
Glioblastoma tumor-bearing mice were administered intraperitoneally with 0.5 mg/kg triptolide (Trip) or dendrimer-triptolide conjugate (D-Trip) daily on day 5 after tumor inoculation. A) D-Trip treatment significantly improves reduction in tumor burden compared to control and Trip treatment group. * p < 0.05, ** p < 0.01. B) D-Trip improves mobility of treated mice as measured by open field analyses on day 13 after inoculation. * p < 0.05 Ctrl vs. D-Trip.
Mobility in open field experiments has been shown to be impacted by tumor burden as a proxy for fatigue, with partial recovery after tumor resection.77 This is of particular importance in glioblastoma, where patients often experience cognitive impairments and significant fatigue that is poorly accounted for clinically.78 To explore the impact of treatment on this parameter, mice were recorded on days 10, 13, and 17 after tumor inoculation. Glioblastoma tumor burden resulted in deterioration of mobility as the disease progressed, as measured by distance traveled in open field experiments (Figure 5B). Trip treatment did not improve this measure, but D-Trip treatment resulted in improvement in distance traveled, exhibiting a significant improvement in mobility on day 13 after tumor inoculation (p = 0.0423 Trip vs. D-Trip). These results indicate that in addition to amelioration of tumor burden, D-Trip can reduce the severity of mobility-associated markers of disease progression, albeit temporarily, associated with neurodegeneration and fatigue. Future studies focusing on more aggressive dosing regimens will be explored to determine if the temporary improvements to mobility as the disease progresses can be extended to more long-lasting benefits.
Triggered release of triptolide via dendrimers reduces triptolide-associated systemic toxicities
Off-target, nonspecific activity of triptolide is associated with significant systemic toxicities, which have limited its clinical applications.19 To assess degree of neurodegeneration, mice were scored daily for kyphosis, a measure of hunched posture associated with disease progression.52, 79 Trip treatment exacerbated neurodegeneration by inducing GBM-associated kyphosis 5 days earlier compared to untreated and D-Trip groups (Figure 6A, p < 0.0001 Ctrl vs. free Trip, p < 0.0001 free Trip vs. D-Trip). Intraperitoneal injection of Trip also induced fibrosis, hair loss, and swelling at the injection site (Figure 6B). Targeted delivery and triggered release of Trip via dendrimers significantly reduced the proportion of mice that experiences such adverse responses at the injection site (Figure 6C, p = 0.0287 free Trip vs. D-Trip).
Figure 6. Dendrimer delivery mitigates triptolide-induced toxicity.
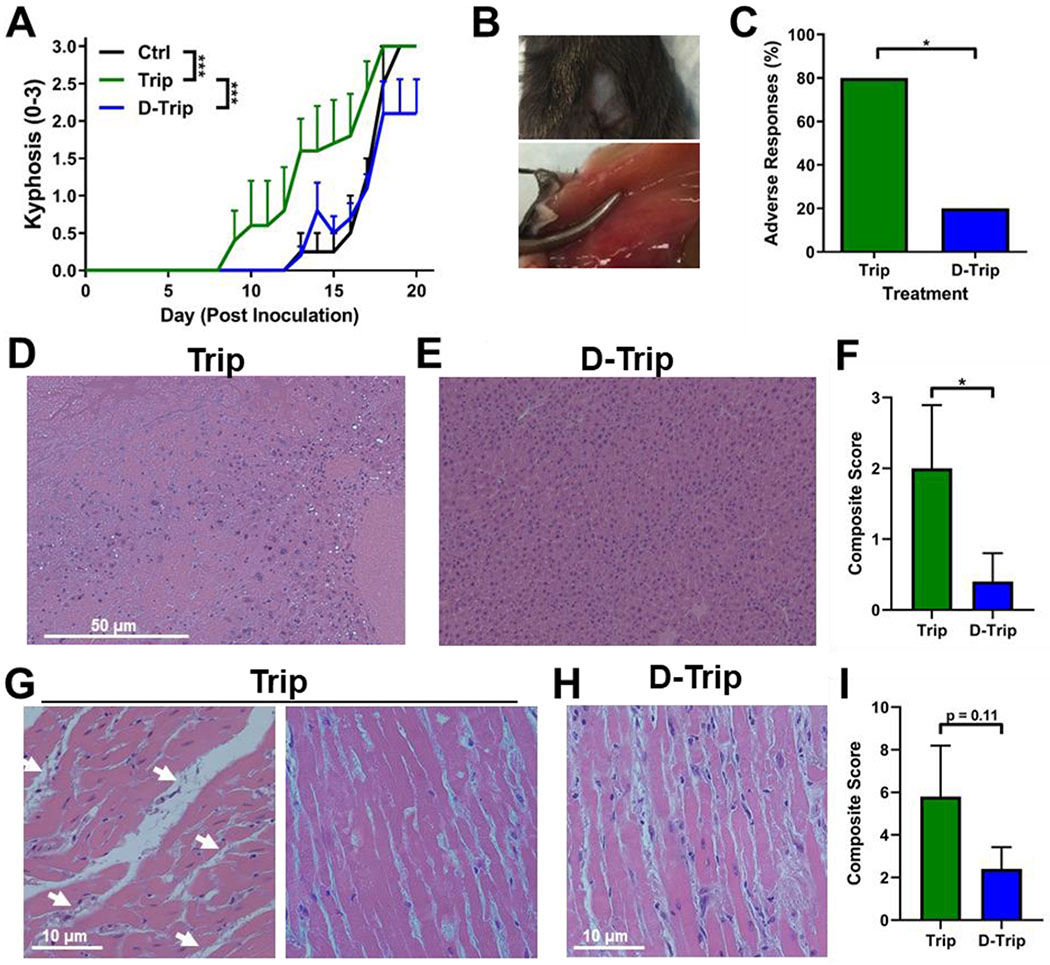
Glioblastoma tumor-bearing mice were administered intraperitoneally with 0.5 mg/kg triptolide (Trip) or dendrimer-triptolide conjugate (D-Trip) daily on day 5 after tumor inoculation. A) Treatment with Trip exacerbates progression of glioblastoma-associated kyphosis, an indicator of neurodegeneration. B) Representative images of adverse responses to Trip at the injection site. Loss of fur, fibrosis, inflammation, and swelling were observed. C) Targeted delivery by D-Trip reduces the percentage of treated animals that exhibit these adverse responses. * p < 0.05. Livers and hearts were collected for H&E staining to reveal morphological signs of triptolide-induced hepatic and cardiac toxicity. Livers of D) triptolide treated mice exhibit necrosis, edema, and steatosis while livers of E) D-Trip treated mice exhibit normal hepatic structure without morphological markers of toxicity. F) D-Trip reduces the scoring of hepatic morphological markers of toxicity compared to Trip. * p < 0.05. Hearts of G) triptolide treated mice exhibit fibrosis indicated by collagen deposits (white arrows), edema, and cardiomyocyte damage while hearts of H) D-Trip treated mice exhibit only mild edema. I) D-Trip reduces the scoring of cardiac morphological markers of toxicity compared to Trip. N=5 per group. For morphological scoring of toxicity, 2 images per mouse were evaluated.
In terms of organ toxicities, free Trip is associated with major renal, hepatic, and cardiac toxicities.22 H&E stained organ slices were imaged and graded for morphological markers of toxicity. No toxicity was observed in kidney sections (data not shown). All livers exhibited mild globular lymphocyte infiltration. Free Trip treatment induced significant morphological markers of liver degeneration, including subcapsular edema, globular lymphocyte infiltration (white arrows), regions of necrosis, and steatosis (Figure 6D). In contrast, livers of D-Trip treated mice exhibit mild globular lymphocyte infiltration but otherwise healthy hepatic structure (Figure 6E). D-Trip therefore reduced these markers of toxicity, as shown by the ~5-fold reduction in composite scoring (Figure 6F, p = 0.035). Individual subject clinical notes are denoted in Table S1. In hearts, Trip treatment induced cardiac toxicity as shown by collagen deposits between cardiomyocytes indicating fibrosis (Figure 6G, left), edema, inflammation, and loss of cardiomyocyte structure indicating necrosis (Figure 6G, right). D-Trip reduced these triptolide-associated cardiac toxicities, with only mild edema observed and sporadic vacuolation (Figure 6H). This is further shown by the ~3-fold reduction in composite clinical score (Figure 6I, p = 0.11). Individual subject scores are denoted in Table S2. This reduction in systemic toxicity is enabled by the ester linker design of Trip conjugation to the dendrimer surface, which exhibits pH and esterase sensitive cleavage for triggered release within intracellular and intratumor conditions. This is consistent with previous studies have shown that nanoparticle packaging can reduce the toxicity of Trip by inhibiting its action until reaching the target site within the tumor.75, 76, 80, 81 This targeting, along with dendrimer-conjugates being cleared rapidly and intact through kidneys, limits accumulation in systemic tissues that lead to toxicities.31, 33 Therefore, through the targeted delivery capabilities of the dendrimer, the therapeutic efficacy and systemic safety profile of free Trip can be improved for a wider therapeutic window.
Conclusion
In this study, we present a dendrimer conjugate for TAMs specific targeted delivery of triptolide in an orthotopic, immunocompetent model of glioblastoma. We show that conjugation to the dendrimer with an ester linker enables triggered intracellular and intratumor release of triptolide and enhances the aqueous solubility of triptolide by ~500 folds. Treatment in vitro exhibited STAT3 inhibition and upregulation of anti-tumor immune signaling. Systemic dendrimer delivery of triptolide significantly improved amelioration of tumor burden compared to freely administered triptolide in vivo. Dendrimer targeting and triggered release also significantly reduced triptolide-induced injection site, hepatic, and cardiac toxicities. These results suggest that dendrimer targeted delivery can significantly improve therapeutic efficacy and widen the therapeutic window of triptolide and other anti-cancer therapies that exhibit poorly tolerated systemic toxicities. Future work will explore the specific in vivo immune mechanisms responsible for these effects.
Supplementary Material
Highlights.
Dendrimer conjugation enables specific release of triptolide in intratumor conditions
Dendrimer delivery of triptolide promotes anti-tumor immune signaling in vitro
Systemic dendrimer-triptolide significantly reduces tumor burden vs. free triptolide
Dendrimer delivery ameliorates triptolide-induced toxicity
Acknowledgements
This work was funded by the Arnall Patz Distinguished Professorship endowment. We thank the Wilmer Core Grant for Vision, Research, Microscopy, and Imaging core module (Grant #EY001865) for access to the Leica CM 1905 and Zen LSM710 confocal microscope. We also would like to thank Drs. Marc Halushka and Andres Matoso for guidance with scoring morphological markers of organ toxicity.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The authors have awarded and pending patents relating to the TAMs targeting ability of hydroxyl terminated PAMAM dendrimers. RMK and SK are co-founders and have financial interests in Ashvattha Therapeutics LLC, Orpheris Inc., and RiniSight, three startups involved with the translation of dendrimer drug delivery platforms.
References
- 1.Wen PY; Kesari S The New England journal of medicine 2008, 359, (5), 492–507. [DOI] [PubMed] [Google Scholar]
- 2.Omuro A; DeAngelis LM JAMA 2013, 310, (17), 1842–1850. [DOI] [PubMed] [Google Scholar]
- 3.Adamson CD; Kanu OO; Mehta AI; Di C; Lin N; Mattox AK; Bigner DD Expert Opinion on Investigational Drugs 2009, 18, (8), 1061–1083. [DOI] [PubMed] [Google Scholar]
- 4.Iwamoto FM; Reiner AS; Panageas KS; Elkin EB; Abrey LE Annals of neurology 2008, 64, (6), 628–34. [DOI] [PubMed] [Google Scholar]
- 5.Arvold ND; Reardon DA Clin Interv Aging 2014, 9, 357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanneman M; Dranoff G Nature Reviews Cancer 2012, 12, (4), 237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blattman JN; Greenberg PD Science 2004, 305, (5681), 200. [DOI] [PubMed] [Google Scholar]
- 8.Fousek K; Ahmed N Clinical Cancer Research 2015, 21, (15), 3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyauchi JT; Tsirka SE J Neurol 2018, 265, (4), 741–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin Y; Xu J; Lan H Journal of Hematology & Oncology 2019, 12, (1), 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sevenich L Frontiers in Immunology 2018, 9, (697). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noy R; Pollard JW Immunity 2014, 41, (1), 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mantovani A; Marchesi F; Malesci A; Laghi L; Allavena P Nature reviews. Clinical oncology 2017, 14, (7), 399–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gabrusiewicz K; Hossain MB; Cortes-Santiago N; Fan X; Kaminska B; Marini FC; Fueyo J; Gomez-Manzano C Neoplasia (New York, N.Y.) 2015, 17, (4), 374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hua L; Shi J; Shultz LD; Ren G Methods Mol Biol 2018, 1784, 243–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sauter KA; Pridans C; Sehgal A; Tsai YT; Bradford BM; Raza S; Moffat L; Gow DJ; Beard PM; Mabbott NA; Smith LB; Hume DA Journal of leukocyte biology 2014, 96, (2), 265–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tap WD; Gelderblom H; Palmerini E; Desai J; Bauer S; Blay JY; Alcindor T; Ganjoo K; Martin-Broto J; Ryan CW; Thomas DM; Peterfy C; Healey JH; van de Sande M; Gelhorn HL; Shuster DE; Wang Q; Yver A; Hsu HH; Lin PS; Tong-Starksen S; Stacchiotti S; Wagner AJ Lancet (London, England) 2019, 394, (10197), 478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ziaei S; Halaby R Avicenna J Phytomed 2016, 6, (2), 149–164. [PMC free article] [PubMed] [Google Scholar]
- 19.Xi C; Peng S; Wu Z; Zhou Q; Zhou J Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 2017, 90, 531–541. [DOI] [PubMed] [Google Scholar]
- 20.Kim JH; Park B International journal of molecular medicine 2017, 40, (5), 1566–1572. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y; Shen Y; Wang S; Shen Q; Zhou X Cancer letters 2018, 415, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen S-R; Dai Y; Zhao J; Lin L; Wang Y; Wang Y Frontiers in Pharmacology 2018, 9, (104). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noel P; Von Hoff DD; Saluja AK; Velagapudi M; Borazanci E; Han H Trends in pharmacological sciences 2019, 40, (5), 327–341. [DOI] [PubMed] [Google Scholar]
- 24.Zheng W; Wang C; Ding R; Huang Y; Li Y; Lu Y International Journal of Pharmaceutics 2019, 572, 118721. [DOI] [PubMed] [Google Scholar]
- 25.Zhang YQ; Shen Y; Liao MM; Mao X; Mi GJ; You C; Guo QY; Li WJ; Wang XY; Lin N; Webster TJ Nanomedicine : nanotechnology, biology, and medicine 2019, 15, (1), 86–97. [DOI] [PubMed] [Google Scholar]
- 26.Srinageshwar B; Peruzzaro S; Andrews M; Johnson K; Hietpas A; Clark B; McGuire C; Petersen E; Kippe J; Stewart A; Lossia O; Al-Gharaibeh A; Antcliff A; Culver R; Swanson D; Dunbar G; Sharma A; Rossignol J International Journal of Molecular Sciences 2017, 18, (3), 628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lesniak WG; Mishra MK; Jyoti A; Balakrishnan B; Zhang F; Nance E; Romero R; Kannan S; Kannan RM Molecular pharmaceutics 2013, 10, (12), 4560–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma A; Sharma R; Zhang Z; Liaw K; Kambhampati SP; Porterfield JE; Lin KC; DeRidder LB; Kannan S; Kannan RM Science Advances 2020, 6, (4), eaay8514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang F; Mastorakos P; Mishra MK; Mangraviti A; Hwang L; Zhou J; Hanes J; Brem H; Olivi A; Tyler B; Kannan RM Biomaterials 2015, 52, 507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma A; Liaw K; Sharma R; Zhang Z; Kannan S; Kannan RM Theranostics 2018, 8, (20), 5529–5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mishra MK; Beaty CA; Lesniak WG; Kambhampati SP; Zhang F; Wilson MA; Blue ME; Troncoso JC; Kannan S; Johnston MV; Baumgartner WA; Kannan RM ACS Nano 2014, 8, (3), 2134–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo Y; Johnson MA; Mehrabian Z; Mishra MK; Kannan R; Miller NR; Bernstein SL PloS one 2016, 11, (4), e0154437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khoury ES; Sharma A; Ramireddy RR; Thomas AG; Alt J; Fowler A; Rais R; Tsukamoto T; Blue ME; Slusher B; Kannan S; Kannan RM Theranostics 2020, 10, (13), 5736–5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma A; Porterfield JE; Smith E; Sharma R; Kannan S; Kannan RM Journal of Controlled Release 2018, 283, 175–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharma R; Kambhampati SP; Zhang Z; Sharma A; Chen S; Duh EI; Kannan S; Tso MOM; Kannan RM Journal of Controlled Release 2020, 323, 361–375. [DOI] [PubMed] [Google Scholar]
- 36.Sharma R; Kim S-Y; Sharma A; Zhang Z; Kambhampati SP; Kannan S; Kannan RM Bioconjugate chemistry 2017, 28, (11), 2874–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liaw K; Zhang F; Mangraviti A; Kannan S; Tyler B; Kannan RM Bioeng Transl Med 2020, 5, (2), e10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma R; Sharma A; Kambhampati SP; Reddy RR; Zhang Z; Cleland JL; Kannan S; Kannan RM Bioengineering & translational medicine 2018, 3, (2), 87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobrovolskaia MA Current pharmaceutical design 2017, 23, (21), 3134–3141. [DOI] [PubMed] [Google Scholar]
- 40.Qing Y; Stark GR The Journal of biological chemistry 2004, 279, (40), 41679–85. [DOI] [PubMed] [Google Scholar]
- 41.Liu Q; Chen T; Chen G; Li N; Wang J; Ma P; Cao X Biochemical and Biophysical Research Communications 2006, 345, (3), 1122–1130. [DOI] [PubMed] [Google Scholar]
- 42.van Horssen R; Ten Hagen TL; Eggermont AM The oncologist 2006, 11, (4), 397–408. [DOI] [PubMed] [Google Scholar]
- 43.Bent R; Moll L; Grabbe S; Bros M Int J Mol Sci 2018, 19, (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fisher DT; Appenheimer MM; Evans SS Seminars in immunology 2014, 26, (1), 38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oh T; Fakurnejad S; Sayegh ET; Clark AJ; Ivan ME; Sun MZ; Safaee M; Bloch O; James CD; Parsa AT Journal of translational medicine 2014, 12, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang H; Zhu W; Su X; Wu S; Lin Y; Li J; Wang Y; Chen J; Zhou Y; Qiu P; Yan G; Zhao S; Hu J; Zhang J J Neurooncol 2012, 109, (1), 53–62. [DOI] [PubMed] [Google Scholar]
- 47.Oliveira A; Beyer G; Chugh R; Skube SJ; Majumder K; Banerjee S; Sangwan V; Li L; Dawra R; Subramanian S; Saluja A; Dudeja V Laboratory investigation; a journal of technical methods and pathology 2015, 95, (6), 648–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu J; Cheng H; Han L; Qiang Z; Zhang X; Gao W; Zhao K; Song Y Drug Des Devel Ther 2018, 12, 3199–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kannan S; Dai H; Navath RS; Balakrishnan B; Jyoti A; Janisse J; Romero R; Kannan RM Science translational medicine 2012, 4, (130), 130ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lei J; Rosenzweig JM; Mishra MK; Alshehri W; Brancusi F; McLane M; Almalki A; Bahabry R; Arif H; Rozzah R; Alyousif G; Shabi Y; Alhehaily N; Zhong W; Facciabene A; Kannan S; Kannan RM; Burd I Sci Rep 2017, 7, (1), 6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nance E; Porambo M; Zhang F; Mishra MK; Buelow M; Getzenberg R; Johnston M; Kannan RM; Fatemi A; Kannan S Journal of controlled release : official journal of the Controlled Release Society 2015, 214, 112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Au - Guyenet SJ; Au - Furrer SA; Au - Damian VM; Au - Baughan TD; Au - La Spada AR; Au - Garden GA JoVE 2010, (39), e1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma A; Mejía D; Regnaud A; Uhlig N; Li C-J; Maysinger D; Kakkar A ACS Macro Letters 2014, 3, (10), 1079–1083. [DOI] [PubMed] [Google Scholar]
- 54.Sharma R; Zhang I; Abbassi L; Rej R; Maysinger D; Roy R Polymer Chemistry 2015, 6, (9), 1436–1444. [Google Scholar]
- 55.Wang X; Matta R; Shen G; Nelin LD; Pei D; Liu Y Journal of molecular medicine (Berlin, Germany) 2006, 84, (5), 405–15. [DOI] [PubMed] [Google Scholar]
- 56.Perumal OP; Inapagolla R; Kannan S; Kannan RM Biomaterials 2008, 29, (24–25), 3469–76. [DOI] [PubMed] [Google Scholar]
- 57.Titov DV; Gilman B; He Q-L; Bhat S; Low W-K; Dang Y; Smeaton M; Demain AL; Miller PS; Kugel JF; Goodrich JA; Liu JO Nature Chemical Biology 2011, 7, (3), 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liang X; Xie R; Su J; Ye B; Wei S; Liang Z; Bai R; Chen Z; Li Z; Gao X Journal of Experimental & Clinical Cancer Research 2019, 38, (1), 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang W; He T; Chai C; Yang Y; Zheng Y; Zhou P; Qiao X; Zhang B; Liu Z; Wang J; Shi C; Lei L; Gao K; Li H; Zhong S; Yao L; Huang M-E; Lei M PloS one 2012, 7, (5), e37693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ni L; Lu J Cancer Med 2018, 7, (9), 4509–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mojic M; Takeda K; Hayakawa Y International journal of molecular sciences 2017, 19, (1), 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liaw K; Gök O; DeRidder LB; Kannan S; Kannan RM Journal of Nanoparticle Research 2018, 20, (4), 111. [Google Scholar]
- 63.Wei X; Gong J; Zhu J; Wang P; Li N; Zhu W; Li J Clinical Immunology 2008, 129, (2), 211–218. [DOI] [PubMed] [Google Scholar]
- 64.Kong X; Zhang Y; Liu C; Guo W; Li X; Su X; Wan H; Sun Y; Lin N PloS one 2013, 8, (10), e77513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee KY; Park JS; Jee YK; Rosen GD Experimental & molecular medicine 2002, 34, (6), 462–8. [DOI] [PubMed] [Google Scholar]
- 66.Chang WT; Kang JJ; Lee KY; Wei K; Anderson E; Gotmare S; Ross JA; Rosen GD The Journal of biological chemistry 2001, 276, (3), 2221–7. [DOI] [PubMed] [Google Scholar]
- 67.Li H; Li L; Mei H; Pan G; Wang X; Huang X; Wang T; Jiang Z; Zhang L; Sun L Cancer biology & therapy 2020, 21, (2), 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun L; Li H; Zhang L; Zhou H The FASEB Journal 2016, 30, (1_supplement), 1193.9–1193.9. [Google Scholar]
- 69.Chen X, Maturation and Activation of Dendritic Cells by Botanicals Used in Traditional Chinese Medicine: Role in Immune Enhancement In Dietary Components and Immune Function, Watson RR; Zibadi S; Preedy VR, Eds. Humana Press: Totowa, NJ, 2010; pp 497–514. [Google Scholar]
- 70.Zhu K-J; Shen Q-Y; Cheng H; Mao X-H; Lao L-M; Hao G-L International Immunopharmacology 2005, 5, (9), 1415–1426. [DOI] [PubMed] [Google Scholar]
- 71.Lin J; Chen LY; Lin ZX; Zhao ML The Journal of international medical research 2007, 35, (5), 637–43. [DOI] [PubMed] [Google Scholar]
- 72.Sai K; Li W-Y; Chen Y-S; Wang J; Guan S; Yang Q-Y; Guo C-C; Mou Y-G; Li W-P; Chen Z-P The American Journal of Chinese Medicine 2014, 42, (02), 485–503. [DOI] [PubMed] [Google Scholar]
- 73.Zhang H; Zhu W; Su X; Wu S; Lin Y; Li J; Wang Y; Chen J; Zhou Y; Qiu P; Yan G; Zhao S; Hu J; Zhang J Journal of Neuro-Oncology 2012, 109, (1), 53–62. [DOI] [PubMed] [Google Scholar]
- 74.Wu B; Lu S-T; Zhang L-J; Zhuo R-X; Xu H-B; Huang S-W Int J Nanomedicine 2017, 12, 1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ling D; Xia H; Park W; Hackett MJ; Song C; Na K; Hui KM; Hyeon T ACS Nano 2014, 8, (8), 8027–8039. [DOI] [PubMed] [Google Scholar]
- 76.Liu J; Cheng H; Han L; Qiang Z; Zhang X; Gao W; Zhao K; Song Y Drug Des Devel Ther 2018, 12, 3199–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Santos JC; Bever SR; Sullivan KA; Pyter LM Sci Rep 2019, 9, (1), 6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Valko PO; Siddique A; Linsenmeier C; Zaugg K; Held U; Hofer S Neuro Oncol 2015, 17, (2), 274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burkholder T; Foltz C; Karlsson E; Linton CG; Smith JM Curr Protoc Mouse Biol 2012, 2, 145–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xu L; Qiu Y; Xu H; Ao W; Lam W; Yang X Food and Chemical Toxicology 2013, 57, 371–379. [DOI] [PubMed] [Google Scholar]
- 81.Zhang C; Peng F; Liu W; Wan J; Wan C; Xu H; Lam CW; Yang X Int J Nanomedicine 2014, 9, 1049–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.