

Abstract
Immune responses against polyethylene glycol (PEG) can lead to the rapid clearance of PEGylated drugs and are associated with increased risk of serious adverse events such as infusion reactions and anaphylaxis. Although select PEGylated therapeutics can induce anti-PEG antibodies (APA), there is currently no readily deployable strategy to mitigate their negative effects. Given the large number of PEGylated therapeutics that are either FDA-approved or in clinical development, methods that suppress APA induction to ensure the safety and efficacy of PEGylated drugs in patients would be a valuable clinical tool. We previously showed that infusion of high molecular weight (MW) free PEG can safely and effectively restore the circulation of PEG liposomes in animals with high pre-existing titers of APA, without stimulating additional APA production. Here, we explored the effectiveness of prophylaxis with free PEG or tolerogenic PEGylated liposomes in reducing APA induced by subsequently administered PEGylated liposomes. Surprisingly, we found that a single administration of free PEG alone was capable of markedly reducing the APA response to PEG-liposomes for ~2 months; the effectiveness was comparable to, and frequently exceeded, interventions with different tolerogenic PEG-liposomes. These results support further investigation of free PEG prophylaxis as a strategy to ameliorate the APA response to sensitizing PEGylated therapeutics.
Keywords: polyethylene glycol antibodies, anti-PEG antibodies, PEGylation, polyethylene glycol, anti-drug antibodies, drug delivery, accelerated blood clearance, nanoparticles, tolerogenesis
Introduction
Polyethylene glycol (PEG) is a polymer with an extensive history of use in the pharmaceutical industry. The hydrophilicity of PEG increases the aqueous solubility of hydrophobic drugs, improving stability and reducing aggregation [1–5]. As PEG chains are highly flexible, they form a conformational cloud that can sterically hinder the adsorption of opsonic proteins and other blood components, diminish interactions with the immune system, increase particle size, and reduce enzymatic degradation [2, 6]. This results in a ‘stealth’ effect that prolongs the systemic pharmacokinetic (PK) profiles of various PEGylated therapeutics, thereby reducing the frequency of administration, improving the quality of life of patients, and lowering clinical costs [3, 4]. At least 17 pharmaceutical products on the market are PEGylated, with several more currently under evaluation in clinical trials [2].
Unfortunately, repeated administration of PEGylated nanoparticles and some PEGylated proteins can induce the formation of anti-PEG antibodies (APA) [7–10]. Circulating APA can quickly clear subsequent doses of PEGylated therapeutics from the blood, a process commonly referred to as accelerated blood clearance (ABC) [7, 11–19], which renders treatment with PEGylated therapeutics ineffective. Clinical studies of pegloticase and pegaspargase have shown that ~40% of patients develop high-titer APA responses that directly result in treatment failure [20, 21]. Furthermore, patients with APA are at substantially greater risks of serious adverse events (SAEs) including anaphylaxis [20, 22, 23]. For instance, in a phase III trial of pegnivacogin, a PEGylated RNA aptamer, SAEs were correlated with high titers of APA, and ultimately contributed to early termination of the study [22, 23].
We recently began to explore the use of high molecular weight (MW) free PEG to saturate APA and restore a time window for the safe and efficacious use of PEGylated therapeutics [24]. We found that administering 40 kDa free PEG to mice with high APA titers shortly before injecting PEG-liposomes restored the circulation of those PEG-liposomes to the same level as mice without APA for at least 48 hours. This represents a marked improvement over mice with high APA titers that did not receive free PEG, which eliminated all PEG-liposomes within 3 hours of injection [24]. Surprisingly, the repeated weekly administration of free PEG did not result in increasing APA titers, detectable immune complex deposition in the kidneys or liver, changes to complete blood counts, or altered AST, ALT, creatinine, or BUN values compared to mice dosed weekly with saline [24]. These results strongly underscore the use of high MW free PEG as a potentially safe and efficacious intervention restoring the use of select PEGylated therapeutics in patients with high APA titers.
However, it remains unclear whether free PEG can prevent the induction of APA in response to PEGylated therapeutics. Notably, PEG is a thymus independent antigen (i.e. anti-PEG immunity does not require T cell activation), with B cells in the marginal zone of the spleen being the critical site for initial generation of APA [7, 8]. Thus, blocking stimulation of PEG-specific B-cells in the spleen represents a strategy for lessening the production of APA. The co-administration or encapsulation of cytotoxic or immunosuppressive agents along with antigen has previously been shown to reduce antibody responses to both T-dependent and T-independent antigens [25]. For instance, conjugating rapamycin to PEGylated proteins reduced APA responses in mice by ~90% compared to administering the PEGylated protein alone [26]. Ongoing clinical trials are investigating whether co-administering PEGylated nanoparticles containing rapamycin with PEGylated proteins can reduce the APA response [27]. Similarly, Siglec-engaging tolerance-inducing antigenic liposomes (STALs) have been shown to utilize B cells’ suppressive CD22 function to suppress specific naïve and memory B-cells, thus reducing subsequent antibody response to antigen [28]. Finally, Ishida et al. have demonstrated that the encapsulation of doxorubicin in PEGylated liposomes hinders subsequent APA responses [9]. In this work, we compared the effectiveness of prophylaxis with free PEG to these various tolerogenic nanoparticle formulations in suppressing the APA response to subsequently administered PEG-liposomes.
Methods
Thin film hydration and extrusion to produce liposomes.
The challenge dose used to sensitize mice against PEG consisted of empty PEGylated liposomes with a 39:56:5 molar ratio composition of 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), cholesterol, and methoxy-PEG 2000 DSPE, respectively. These were produced by thin film hydration and extrusion. Briefly, each was dissolved in 2:1 chloroform:methanol, added to a glass tube, and mixed. Using a slow stream of nitrogen gas, the solvent was evaporated while rotating the tube, leaving a thin film. The film was desiccated overnight under vacuum to ensure complete evaporation. Warm PBS (>55 °C) was added to completely cover the film and sonicated for 15 rounds of 30 seconds each. The liposomes were extruded through a 400 nm and 100 nm filter using an Extruder Kit (Avanti Polar Lipids).
PEG-rapamycin (PEG-rapa) liposomes were also produced using thin-film hydration and extrusion with a composition of 83:9:5:3 molar ratio of phosphatidylcholine, cholesterol, methoxy-PEG 2000, and rapamycin, respectively. The PEG-rapa liposomes were extruded with 400 nm and 100 nm filters. Information on the doses of each PEGylated liposome can be found in Supplementary table 1.
Particle size and surface charge analysis.
The particle size and surface charge of liposomes were confirmed using a Zetasizer dynamic light scattering (DLS) device (Malvern Instruments, Malvern, UK). The liposome solutions were diluted to a total lipid concentration of ~10 nM in PBS before DLS analysis.
Evaluation of tolerogenic treatments in vivo.
Tolerogenic nanoparticles were administered to mice either before or after challenge with a dose of PEGylated liposomes (challenge dose: 0.1 μmol/kg). The following doses of particles were tested: PL-dox (5 μmol/kg), PL-rapa (5 μmol/kg), STALS (200 μL of a 0.05 mM solution), and PL-control (200 μL of a 0.05 mM solution). Pre-treatment with free PEG was with a dose of 5.5 mg/kg of PEG 40 kDa, and was given 30 min prior to the challenge dose of PEG-liposomes into the opposite tail vein (Fig 1A). It is important to note that the in vivo studies conducted to generate data for months 2 and 3 of the experiment shown in Figure 1D were only conducted one time, with 4 animals per treatment group. Future studies should repeat this long-term dosing strategy to assess the external reproducibility of these results. All other experiments in this work were repeated a minimum of two times.
Figure 1: Free PEG and tolerogenic nanoparticles reduce titer of APA induced by PEGylated liposomes.
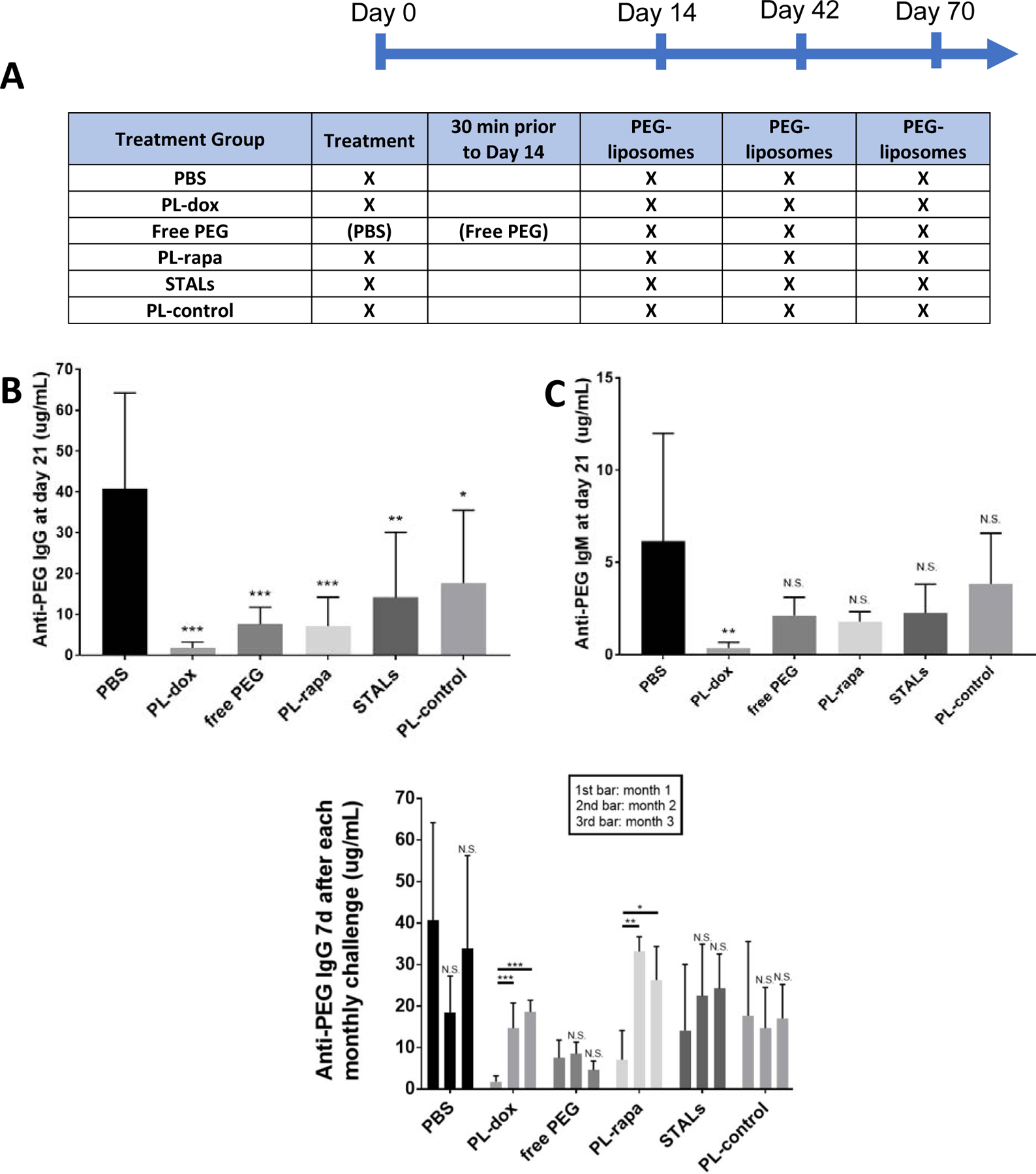
Mice were given an i.v. injection of tolerogenic nanoparticles or PBS on Day 0, prior to i.v. injection of empty PEG-liposomes and NP-Ficoll (a control T-independent antigen) on Day 14. Animals in the free PEG group received PBS at Day 0, and a dose of 40 kDa free PEG 30 minutes prior to the dosing of PEG-liposomes on day 14. PEG-liposomes were also dosed on Day 42 and Day 70. At Day 21, 49 and 77, blood was collected, and the concentrations of anti-PEG IgG and IgM were measured in plasma. (A) Scheme of experimental design, where X indicates specific interventions given. (B) Comparison of anti-PEG IgG produced in mice treated with saline, free PEG, or various tolerogenic particles at Day 21. (C) Comparison of anti-PEG IgM produced in mice treated with saline, free PEG, or various tolerogenic particles at Day 21. Statistical comparisons across groups were T tests and were Bonferroni-adjusted for multiple comparisons. (D) Anti-PEG IgG response 7 days after each of the three PEG-liposome challenges: the first bar is anti-PEG IgG 1 week after first PEG-liposome infusion (Day 21), the second bar is anti-PEG IgG 1 week after second PEG-liposome infusion (Day 49), and the third bar is anti-PEG IgG one week after the third PEG-liposome infusion (Day 77). The experiments in panels B and C were conducted twice, with 4 mice per group, per experiment, for a total of 8 animals per condition. The long-duration studies in panel D (2nd and 3rd challenges) were conducted once, with n=4 per treatment group except for the PBS control group, which had 8 mice. Comparisons across group averages were conducted using unpaired t-tests, with *,**, and *** representing p<0.05, 0.01, and 0.001, respectively.
APA quantification via competition ELISA.
APA were quantified using a competitive ELISA, as previously described [14]. Briefly, blood was collected into EDTA vacutainer tubes. Blood was centrifuged at 2,000 rcf at 4° C for 15 minutes to generate plasma. ELISA plates (Corning Costar 3695) were coated overnight at 4° C with 50 μg/ml DSPE-PEG 5 kDa. Plates were blocked with 5% (w/v) milk in PBS. Monoclonal mouse anti-PEG IgG (Silverlake Research, CH2076) or IgM (IBMS Sinica, AGP4) were used to generate a standard curve. Plasma and standards were incubated in 1% milk in PBS overnight at 4° C. Replicates of plasma samples were also competitively incubated with free PEG 8 kDa to allow determination of non-PEG-specific background signal (e.g., from other antibodies). Secondary antibodies against mouse IgG (ThermoFisher Supraclonal, SA5–10226), and IgM were diluted 1:10,000 in 1% milk and incubated overnight at 4° C. Plates were developed using TMB, quenched with 2N H2SO4, and absorbance was read at 450nm (signal) and 570nm (background) using a plate reader. Typical lower limits of quantitation for the assay were in the range of 5 to 10 ng/mL, prior to accounting for plasma dilution.
Flow cytometry.
The phenotype of splenic B cells was analyzed by flow cytometry. In brief, 5 days after vaccination with PEGylated liposomes (0.1 μmole lipid / kg bodyweight, i.v.), the spleen was harvested and digested with collagenase A and DNase at 37°C for 60 min. After lysis of red blood cells, cells were resuspended in 5 mL of PBS. Approximately 2×106 cells were stained with fluorescently labeled antibodies for surface marker expression analysis at room temperature for 15 mins. After surface marker staining, the cells were washed twice with FACS buffer for 5 mins. The cells were then incubated with fluorescent PEGylated nanoparticles for 30 mins at 4°C. After staining, cells were washed and analyzed on an 18-color flow cytometer (BD LSR II). Data were analyzed with FlowJo 8.6 software (TreeStar).
Clinical blood chemistry.
Complete blood counts (CBCs) were performed on the IDEXX Procyte Dx hematology analyzer utilizing murine species-specific parameters. Untreated control data was combined across three separate studies of male and female PBS-treated BALB/c mice of various ages (n=18). CBC data for treatment groups was from a single study (n=4 per group). CBC data for mice treated with free PEG was from a separate experiment using a different group of mice.
Results
Free PEG and tolerogenic nanoparticles reduce the amount of APA produced after PEG challenge
The classic animal model for PEG-sensitization is generated by i.v. administration of empty PEGylated liposomes to naïve mice; this has been shown by multiple labs to lead to the rapid production (beginning at day 3) of both IgG and IgM APA that plateaus ~7 days later, consistent with prior data from other T-independent antigens such as bacterial polysaccharides that directly result in class switching and memory B cell production [7–9, 14, 15, 18, 19, 29, 30]. This is notably different than the more common T-dependent antigen response that results in an initial IgM response followed by somatic hypermutation, creating high affinity IgGs weeks later. We first assessed whether pre-administration of free PEG could reduce induction of APA against PEG-liposomes. Surprisingly, despite the lack of any tolerogenic or cytotoxic active pharmaceutical ingredients, pre-administration of a relatively low dose of high MW (40 kDa) free PEG (i.e. 5.5 mg/kg, or ~100-fold lower than the amount required to saturate high titer APA [24]) led to a 6-fold reduction in the concentration of anti-PEG IgG (from 41 ± 24 μg/ml to 8 ± 4 μg/ml, p < 0.001), and a 3-fold reduction in anti-PEG IgM (from 6± 6 μg/ml to 2 ± 1 μg/ml, p > 0.05) vs. PBS control 7 days following dosing of empty PEG-liposomes (Figure 1B and C). High MW PEG was equally effective at limiting APA stimulation compare to lower MW PEG, and additional increases or decreases in the amount of free PEG administered did not appear to further improve the effectiveness of free PEG prophylaxis (Supplementary Figure 1).
To put these results in perspective, we also assessed different liposome-based interventions containing various cytotoxic or tolerogenic agents, including PEGylated liposomal (PL) doxorubicin (PL-dox), PEGylated liposomal rapamycin (PL-rapa), siglec-engaging tolerance-inducing antigenic liposomes (STALs), and empty PEGylated liposomes (PL-control). Notably, all of these liposomal interventions included PEG to improve the specificity of induced tolerance against PEG-binding B cells. PL-dox led to the most extensive reduction in APA, reducing average anti-PEG IgG and IgM concentrations 7 days after PEG-liposome dosing to ~2 ± 1 μg/ml and 0.4 ± 0.3 μg/ml, respectively (p < 0.001, Figure 1B and C). PL-rapa also reduced induced APA to ~7 ± 7 μg/ml anti-PEG IgG (p < 0.001) and ~2 ± 1 μg/ml anti-PEG IgM (p < 0.05) (Figure 1). In contrast, STALs induced only a modest suppression of induced APA response (14 ± 16 μg/ml IgG (p < 0.01) and 2 ± 2 μg/ml IgM (p > 0.05). Thus, at the doses tested here, free PEG suppressed induced APA with potency comparable to PL-rapa but was slightly outperformed by PLD (dose information in Supplementary table 1).
We next evaluated the duration of induced tolerance by quantifying APA titers one week following each of two additional rounds of PEG-liposomes, administered on Day 42 and Day 70 (Figure 1D). Although mice treated with PL-dox initially had the lowest IgG and IgM APA response after the first challenge with PEG-liposomes (Figure 1B and C), it only had minor tolerogenic effect against later rounds of PEG-liposome, with anti-PEG IgG levels reaching 15 ± 6 μg/ml and 19 ± 3 μg/ml one week after the second and third round of PEG-liposome infusion, respectively. These levels are ~8- and ~10- fold greater than anti-PEG IgG titers detected one week following the first PEG-liposome dosing (Figure 1D). A similar trend was observed for mice treated with PL-rapa: anti-PEG IgG levels increased from 7 ± 4 μg/ml one week following initial PEG-liposome dosing to 33 ± 4 μg/ml and 26 ± 8 μg/ml one week following the second and third rounds of PEG-liposome dosing, respectively (Figure 1D). Surprisingly, mice treated with a single infusion of high MW free PEG alone exhibited a durable suppression of the APA response. The amount of anti-PEG IgG detected one week following the second (9 ± 3 μg/ml) and third (5 ± 2 μg/ml) round of PEG-liposomes was not significantly different from the anti-PEG IgG detected one week following the first round of PEG-liposomes dosing (8 ± 4 μg/ml, p > 0.05), despite only dosing free PEG once immediately prior to the first PEG-liposome dose. Mice that were initially treated with STALs exhibited little evidence of a dampened APA response, with comparable levels of anti-PEG IgG detected one week after the second (23 ± 13 μg/ml, p>0.05) and third (24 ± 8 μg/ml, p>0.05) round of PEG-liposomes.
To gain a better mechanistic understanding of how free PEG may interact with anti-PEG B cells, we next measured the relative number of PEG-binding splenic B cells after treatment with free PEG, PEG liposomes, PL-dox, or saline. We hypothesized that PL-dox, due to its cytotoxic payload, may lead to a reduction in PEG-binding B cells in the spleen compared to saline control, whereas pre-treatment with free PEG may not reduce B cell numbers relative to saline control. In good agreement with our expectations, PEG-sensitized mice had greater number of PEG-specific B cells in the marginal zone of the spleen, whereas PL-dox treatment resulted in an appreciable but non-statistically significant decrease in PEG-binding B cells in the spleen (Supplementary Figure 2). Interestingly, we found that mice treated with free PEG followed by PEG-liposomes had comparable number of PEG-binding B cells in the spleen compared to mice treated with PEGylated liposomes alone, despite having much lower APA titers in circulation (Supplementary Figure 2). This suggests that, mechanistically, free PEG does not limit induction of APA by reducing the number of PEG-binding B cells. Instead, it appears free PEG most likely bind temporarily saturates the B cell receptors (BCRs) on anti-PEG B-cells, thereby preventing subsequently infused PEG-liposomes from accessing and activating BCRs.
Pre-existing APA, including those elicited by tolerogenic PEG-liposomes, reduce the magnitude of APA induction by later doses of PEG-liposomes
Empty PEG-liposomes (PL-control) dosed 14 days prior reduced APA titers generated by a subsequent dose of PEG-liposomes by ~50%, to a similar extent as STALs (Figure 1B–C). Prior work has shown that empty PEG-liposomes potently elicit an APA response, and are largely completely eliminated by Day 7 post-dosing. This led us to speculate that the apparent tolerogenic effects we observed may be partially attributed to the rapid hepatic clearance of the subsequent dose of PEG-liposomes, which in turn limits the activation of APA+ B cells in the spleen. In support of this hypothesis, we previously found that substantial APA titers can quickly clear PEG-liposomes from the circulation into the liver within 3 hours [24]. Further, it has been shown that patients with higher pre-existing titers of anti-influenza antibodies produce less of an antibody response to influenza vaccination [31]. The dose of tolerogenic PEG-liposomes administered was in the range of ~1–5 μmole lipids/kg bodyweight, and the dose of each additional challenge with PL-control was 0.1 μmole lipids/kg bodyweight. To test our hypothesis, we administered either saline or passively immunized naïve mice with APA IgG, followed by dosing empty PEG-liposomes (0.1 μmole lipid/kg bodyweight) one day later. Finally, we measured the amount of IgG and IgM APA one week later, mirroring the same sampling schedule as the study as in Figure 1. Strikingly, we found that pre-existing APA IgG from passive immunization markedly reduced the APA titers produced in response to empty PEG-liposomes (Figure 2A), reducing the average serum anti-PEG IgG from ~41 ± 24 μg/mL in the saline control to ~3 ± 0.4 μg/mL APA IgG in pre-treated mice after 7 days (p<0.01).
Figure 2: The presence of APA at time of PEG-liposome dosing reduces induction of APA.
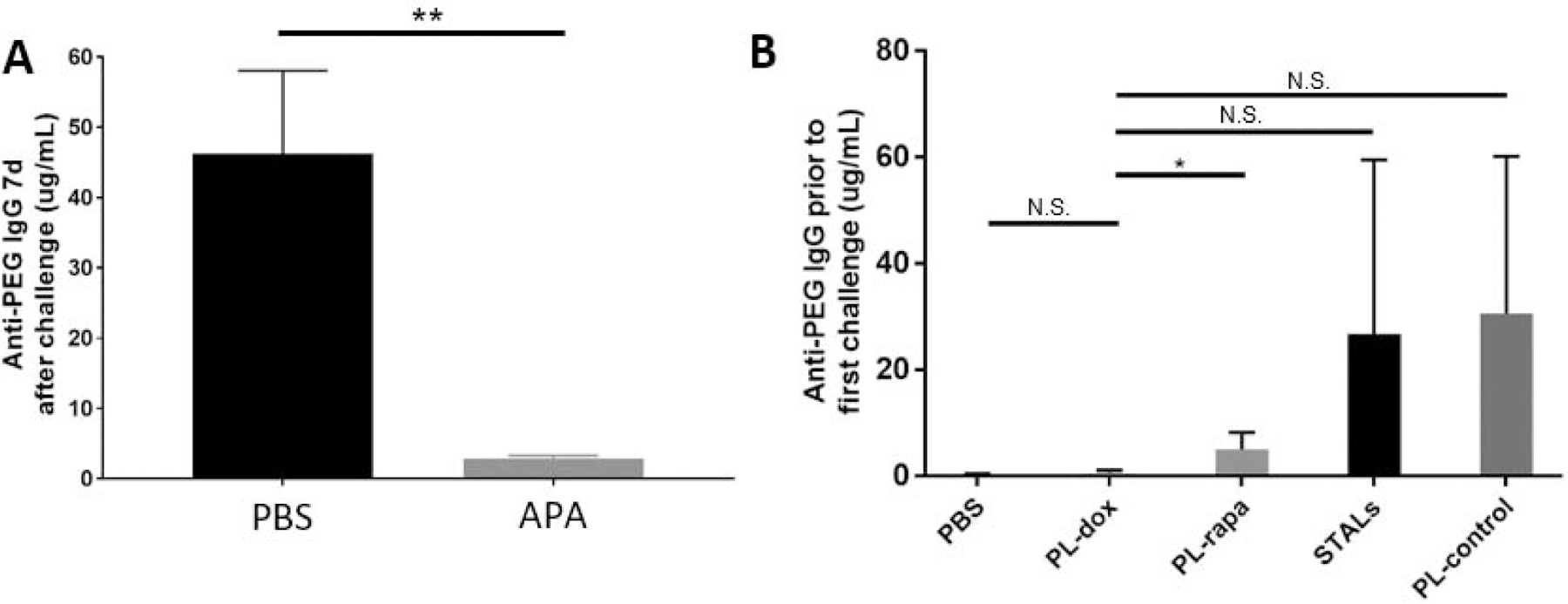
(A) Naïve mice were administered monoclonal anti-PEG IgG (“APA”; n=4) or PBS (“PBS”; n=8) 24 h prior to infusion of PEG-liposomes. Then, 7 days later, blood was collected to quantify APA. (B) Naïve mice (n=4) were treated at Day 0 and blood was collected at Day 7 for the measurement of APA induced by the various treatments. Comparisons across group averages were conducted using unpaired t-tests, with *,**, and *** representing p<0.05, 0.01, and 0.001, respectively. NS; no significant difference.
These results explain why the administration of PEG-liposomes alone can dampen the APA response to subsequent doses of PEG-liposome: the circulating anti-PEG antibodies clear the challenge dose from circulation before it has time to stimulate anti-PEG B cells. Importantly, this suggests that if the various tolerogenic PEG-liposome interventions actually induce appreciable APA production, the lack of APA stimulation by empty PEG-liposomes administered 2 weeks later could be caused by rapid hepatic clearance that limits access to PEG-specific B cells in the spleen. The end result would falsely appear to be a tolerogenic effect, with the mechanism mistakenly attributed to the tolerogenic APIs exerting specific effects on the splenic APA B-cell populations. To further investigate this effect, we conducted ELISAs on plasma samples taken one week following the initial tolerogenic PEG-liposomes but prior to dosing with PL-control. We found that, with the lone exception of cytotoxic PL-dox (which induced only ~300 ng/mL APA), all tolerogenic PEG-liposome formulations actually induced substantial APA production, in some cases reaching very high titers of APA (~15–30 μg/ml IgG APA) (Figure 2B).
Treatment of previously sensitized mice with tolerogenic nanoparticles does not prevent future sensitization
The previous experiments modeled the possible prophylactic treatment of patients with no appreciable pre-existing APA. We next investigated whether the same interventions could also reduce APA production in previously PEG-sensitized mice. We hypothesized that the administration of tolerogenic nanoparticles would kill or anergize the existing APA B cells, thus leading to marked reduction of APA stimulation by subsequently-dosed empty PEG-liposomes. For these studies, we first sensitized naïve mice with empty PEG-liposomes, then administered various tolerogenic interventions six weeks later, followed again by dosing with empty PEG-liposomes two weeks later, and finally measured the titers of anti-PEG IgG and IgM one week after the last dose of PL-control. Much to our surprise, we found that the various tolerogenic interventions, including PL-dox, did not reduce APA following dosing with the empty PEG-liposomes compared to saline control (Figure 3A). The limited effectiveness of the various tolerogenic interventions is consistent with the hypothesis that the pre-existing APA (induced by the initial sensitization 6 weeks prior) quickly cleared the PEG-liposomal interventions to the liver, thus minimizing their ability to access and exert tolerogenic effects on anti-PEG B cells in the spleen.
Figure 3: The administration of tolerogenic particles to mice previously sensitized to secrete APA is not effective at reducing APA titers.
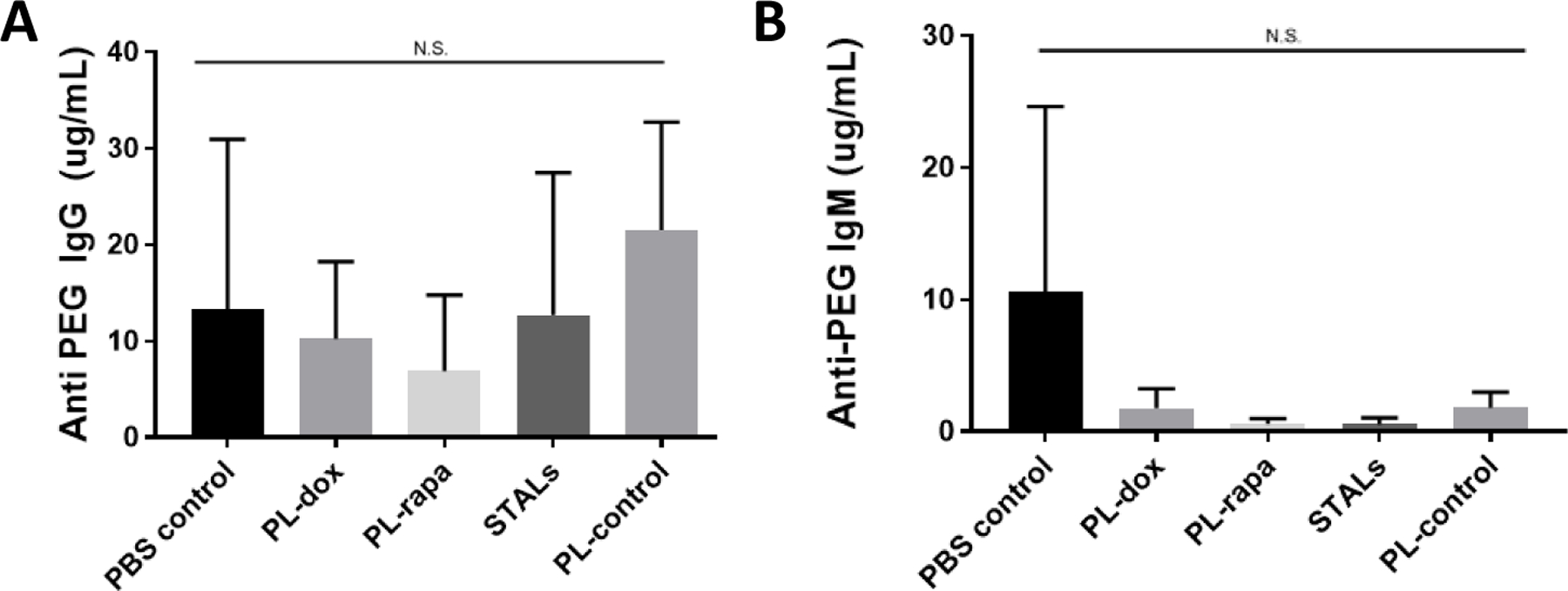
Naïve mice were first sensitized by administration of an i.v. injection of PEGylated liposomes (0.1 μmole lipid/kg) at day 0. Six weeks later, mice were treated with the indicated tolerogenic particles. Two weeks later i.e. Week 8 [28], mice were infused with empty PEG-liposomes. Blood was collected one week after i.e. Week 9, and (A) IgG and (B) IgM APA levels were measured. There were no significant differences measured across any of the groups studied compared to saline control, per ANOVA. This experiment was conducted once with n=4 mice per treatment group.
Tolerogenesis does not cause broad B cell toxicity
There is concern that some of the tolerogenic approaches, especially cytotoxic liposomes, although capable of suppressing anti-PEG B cells, might negatively affect the function of unrelated B cells. To investigate whether the treatments’ inhibitory activity was specific to anti-PEG B cells, we simultaneously measured antibody responses to a separate model T-independent antigen, NP-Ficoll. For the in vivo study shown in Figure 1, when the mice were given a dose of PL-control at day 14, they also received NP-Ficoll antigen. If these tolerogenic treatments were specific to PEG-binding B cells, the anti- Ficoll response should not be impacted relative to saline control mice. Indeed, we found no statistically significant difference in NP-Ficoll antibody titers among all treatment group (Figure 4A). This suggests that the observed tolerogenic effects were specific to APA B cells and not other B cells, presumably due to the presence of PEG in all of the interventions evaluated. Importantly, this suggests that it is indeed possible to achieve antigen-specific attenuation of anti-PEG B cells, while leaving other T-independent responses (e.g., against capsular bacteria) intact.
Figure 4: Tolerogenic nanoparticles have few apparent off-target effects.
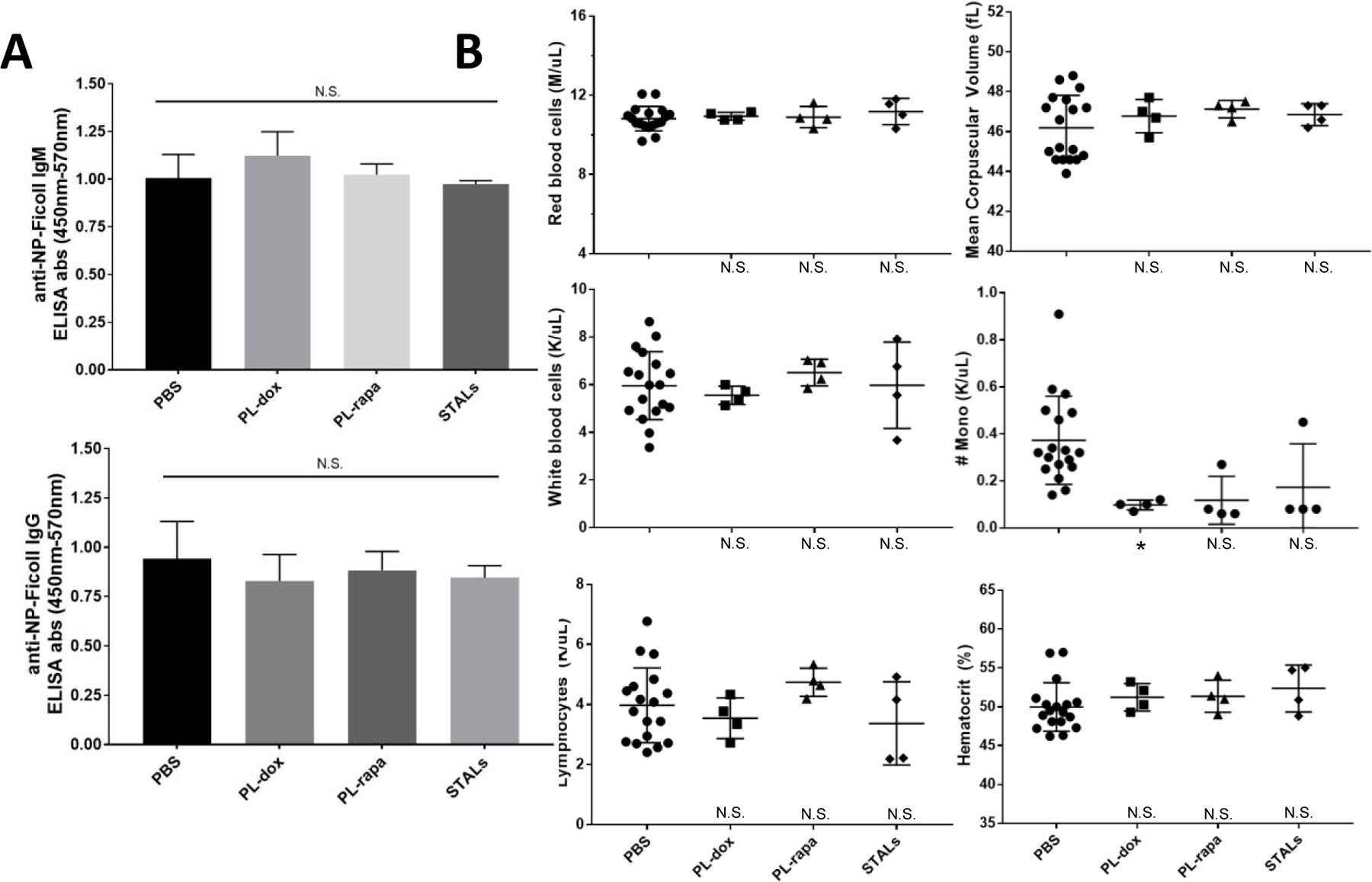
Mice were given an IV injection of tolerogenic nanoparticles two weeks prior to infusion with empty PEGylated liposomes and NP-Ficoll. One week later, on Day 21, all mice were bled, and anti-Ficoll IgM and IgG measured. (B) Comparison of complete blood count results in mice treated with various tolerogenic nanoparticles. CBC blood samples were taken 7 days after treatment with each particle. Groups with significantly different CBC measurements are designated by: p<0.05, *; p<0.01, **; p<0.001, **. Group comparisons to PBS controls are marked near the X-axis. This experiment was conducted once, with n=4 in all groups in panel A, and the indicated numbers of mice per group in panel B (each data point is one mouse). Comparisons across groups were conducted using unpaired t-tests relative to PBS. In panel C, statistical results relative to PBS are indicated near the X-axis.
Although the antibody response to NP-Ficoll was maintained, this does not rule out the possibility of toxic effects to other cell types in circulation, particularly since some of the interventions involved cytotoxic small molecules (e.g. doxorubicin, rapamycin). For example, macrophages, dendritic cells, and other leukocytes might phagocytose the PEG-liposomes, leading to a decline in white blood cell counts despite maintaining a comparable anti-Ficoll response. We performed complete blood counts on mice treated with PL-dox, PL-rapa, STALs, or PBS. At the dosages tested here, we found no statistically significant difference for most groups in red blood cell count, hemoglobin, hematocrit, mean corpuscular volume, white blood cell count, or lymphocyte count across treatment groups (Figure 4B). All treatments led to a measurable decrease in absolute monocyte count (Figure 4B).
Discussion
Several studies of PEGylated drugs that are currently in clinical use have demonstrated that, in up to ~40% of patients, APA can result in ineffective or adverse responses to therapy [20, 21, 32]. Unfortunately, there is currently no FDA-approved or readily employable strategy to manage APA, other than to simply discontinue treatment. To tackle this challenge, we recently explored the pre-infusion of high MW free PEG as a safe, efficacious, and easily translatable solution to restore the use of PEGylated drugs in patients with substantial APA titers [24]. Despite the promising results in vivo, we believe preventing or eliminating induction of high APA titers in patients receiving these PEGylated drugs is a superior strategy. This led us to compare the prophylactic effectiveness of pre-treatment with free PEG in minimizing APA induction by PEG-liposomes to a variety of tolerogenic liposomal formulations. Surprisingly, despite the simplicity of this intervention and the complete lack of any cytotoxic or immunosuppressive agents, pre-treatment with free PEG offered potent and durable reduction in APA induction, reducing systemic APA IgG (the dominant APA Ig in humans) one week following dosing with empty PEG-liposomes by ~5-fold for at least 2 months in mice. In contrast, PL-dox and PL-rapa mediated comparable suppression of APA only to the first dose of PEG-liposomes, with their effects lost by the second and third challenge. None of the interventions evaluated in this study affected IgM or IgG responses to the model TI antigen NP-Ficoll, underscoring the antigen-specificity to PEG. These results, considered together with our earlier findings of safety and effectiveness in overcoming high APA titers [24], underscore the potential use of high MW free PEG as a promising strategy to enable the safe use of sensitizing PEGylated drugs in patients who otherwise must currently discontinue therapy if they develop APA.
There are a number of other strategies under pursuit to reduce APA induced by PEGylated therapeutics. One advanced candidate is a current Phase 2 study evaluating the use of rapamycin-loaded PLGA particles to reduce anti-drug antibodies (including APA) when co-administered with a PEGylated protein (Selecta Biosciences, NCT03905512) [27]. This combination therapy is apparently well tolerated, but a small number of patients appeared to still lose response to the therapy. There is also an active Phase 4 clinical study combining methotrexate (an immunosuppressive agent) with pegloticase (Horizon Pharma, NCT03635957). These strategies are similar in that they combine a small molecule immunosuppressive drug with an immunogenic PEGylated protein drug, causing temporary systemic immunosuppression and decreasing the likelihood of B and T cell activation [33–38]. Methotrexate and rapamycin have both been found to be a successful means of overcoming anti-drug antibodies, including in rheumatoid arthritis (preventing TNF inhibitors), viral gene therapy (preventing antibodies to the viral vectors), and hemophilia replacement therapy (preventing Ab responses to Factor IX), among others [39–41].
There are multiple avenues of ongoing preclinical efforts to overcome APA at this time. These include an elegant approach based on reducible chemical conjugation of rapamycin to PEG-uricase, which showed a 35-fold reduction in anti-drug antibodies (combined anti-uricase and anti-PEG Ab reduction) [26]. There are also ongoing efforts to develop new PEG variants, such as modifying uricase with poly(oligoethylene glycol) methacrylate (POEGMA), which has a bottlebrush conformation containing very short PEG sidechains of only two to three OEG monomer repeats that exhibit reduced binding by pre-existing APA [42]. Although it is clear that POEGMA is not readily bound by APA in human plasma samples, it remains to be determined whether it can cause the induction of APA [43]. Another tolerogenic strategy has used a DNA-protein polymeric nanocomplex to mimic the inherent tolerogenic properties of chromatin to induce tolerance to therapeutic proteins [44]. Other approaches have aimed to leverage the tolerogenic potential of red blood cells in order to counteract inflammatory responses against therapeutic proteins [45]. Compared to these approaches that involve the creation and development of new chemical entities that require extensive preclinical development efforts and costs, we believe the primary advantage of intervening with free PEG is its simplicity and lower hurdles to clinical evaluation. Specifically, the manufacturing of USP-grade PEG has been established and is readily available. The safety of IV infusion of 40 kDa free PEG has also been established in other animal models, including rats and, notably, primates [46].
Surprisingly, pre-treatment with free PEG was nearly as effective as cytotoxic PL-dox and more effective than other PEG-liposomal interventions in reducing the APA response. We hypothesize that this is because free PEG can saturate B cell receptors (BCRs) on the relatively limited number of APA-secreting B cells in naïve mice, thus greatly attenuating the APA response to subsequently dosed PEGylated drugs. Free PEG chains, when not anchored to a protein or nanoparticle, are likely rather inefficient at crosslinking multiple B cell receptors on the surface of APA-specific B cells, especially at high ratios of free PEG to APA-BCR (i.e. virtually all free PEG molecules would have at most one anti-PEG BCR bound, in contrast to multiple BCR binding to the same PEG molecule). Since the crosslinking of multiple BCRs is a necessary first step to activation of antigen-specific B-cells, the infusion of free PEG could significantly lower induction of APA compared to injection of PEGylated particles/proteins. What was also very surprising was the very long duration of this suppressive effect: a single infusion of free PEG was able to attenuate APA response to at least 3 injections of PEG-liposomes administered at monthly intervals. We speculate that this may be attributed to the fact that, once bound to the BCR on an anti-PEG B cells in the spleen or bone marrow, 40 kDa free PEG is not able to readily diffuse away; any PEG molecule that became unbound from the BCR would likely quickly rebind the BCR, thus enabling prolonged inhibition of APA induction. It should be noted that the long-lasting suppression of APA induction by a single infusion of free PEG was not anticipated, and should be interpreted with caution until externally validated, ideally in a different animal model. Interestingly, high MW free PEG (40 kDa) appeared to be no more effective than smaller PEG (10 kDa) in attenuating the APA response to PEGylated liposomes (Supplementary Figure 1).
STALs function by drawing together the BCR and CD22. Previously, the ratio of antigen to CD22 ligand was shown to be a critical parameter for the successful induction of tolerance [28]; optimal formulations have a 20- to 50-fold excess of CD22 ligand compared to antigen. Since the stealth formulation of STALs necessitates the use of 5% PEG, the STALs used in the experiments here actually had an excess of antigen compared to CD22 ligand. Thus, it is quite likely that STALs did not engage the appropriate amount of CD22 relative to BCR, leading to poor suppression of BCR signaling. While we considered the possibility of using less PEG on the STALs for this study, this would negate their stealth properties and, therefore, complicate the experiments with additional variables.
Whereas the tolerogenic liposomes such as PL-dox, PL-rapa, and STALs were administered 14 days prior to the challenge with PEG-liposomes, free PEG was administered just 30 min prior to challenge. This was due to our hypothesis of differing mechanisms of action between the two strategies. For tolerogenic PEG-liposomes, prior work has shown that they are taken up by B cells, whereupon the encapsulated small molecules act to either cause apoptosis or to lead to anergy [28]. Our prior studies of STAL-mediated induction of tolerance found that administration 2 weeks prior to antigenic challenge led to optimal attenuation of the humoral response [28]. In contrast, we hypothesized that free PEG acts by temporarily saturating BCRs on APA+ B-cells in the spleen, such that PEG-liposomes would not encounter freely available BCRs on APA+ B-cells to stimulate. Pharmacokinetic studies of free PEG have shown that PEG 40 kDa is eliminated with a terminal half-life of about 15 hours in mice [47]; thus, it is highly unlikely that there is sufficient quantities of free PEG in the blood to competitively inhibit PEG-liposomes from encountering APA+ B-cells in the blood. Smaller PEGs are eliminated even more quickly; PEG 6 kDa has a terminal half-life in the range of just 18 minutes, and 20 kDa has a half-life of ~3 hours [47]. Free PEG’s apparent ability to block induction of APA by PEG-liposomes over many weeks indicates that it would also be effective if administered 14 days in advance of PEG-liposome dosing. In clinical practice, it is actually far preferable to administer an intervention (e.g. free PEG) either together with- or just prior to-dosing of the PEGylated therapies, rather than to require patients to have to separately visit the clinic two weeks prior to their normal infusion in order to receive the necessary tolerogenic intervention. Thus, the current dosing regimen of introducing free PEG may actually be, as it could be given in the same visit to the clinic.
We previously found that up to ~70% of the population possesses a detectable level of pre-existing anti-PEG IgM or IgG, highlighting the potential for rapid immune activation after treatment with immunogenic PEGylated therapeutics [48]. Our prior modeling studies with a physiologically-based pharmacokinetic model suggest that low APA titers are unlikely to substantially impact the pharmacokinetics and/or biodistribution of PEGylated drugs, leading to clearance of only a small fraction of the administered dose [14]. As a result, we believe it is possible that a successful prophylactic therapy will not need to completely eliminate APA. This is supported by the fact that most PEGylated drugs do not suffer from APA-mediated ABC despite the high general fraction of the population possessing low titers of APA [14].
With at least 15 PEGylated therapeutics currently in clinical trials, we anticipate that the clinical implications of antibody responses against PEG will become increasingly difficult to manage, particularly given the limited recognition of APA among physicians [49]. As it becomes more common for patients to be prescribed multiple PEGylated therapeutics concurrently, the incidence of cross anti-drug antibody interactions will inevitably rise. For instance, a patient may experience APA stimulation in response to one PEGylated therapeutic, producing APA titers substantially exceeding what would typically be stimulated by a second, less immunogenic, PEGylated therapy, and consequently rendering both PEG-drugs ineffective. These realities underscore the need for a safe and inexpensive method to address PEG-sensitivity and enable the effective use of PEGylated drugs, restoring treatment options among patients with substantial APA titers who currently have no choice but to discontinue therapy. Our work to date suggests that intervening with high molecular weight free PEG prior to the administration of a PEGylated therapeutic may be a promising and readily translatable option, either as a prophylactic to minimize the likelihood and/or intensity of APA stimulation or as a direct intervention to overcome high titers of pre-existing APA. Further studies to optimize the dose, timing, and characteristics of this strategy, as well as studies to rigorously investigate its safety in larger animal models, would be key next steps for the translation of this work.
Supplementary Material
Funding:
This work was supported by a National Science Foundation Graduate Research Fellowship (DGE-1650116; MDM), The David and Lucile Packard Foundation (2013-39274, SKL), National Institutes of Health (R01 HL141934; SKL), and Eshelman Institute of Innovation (SKL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: McSweeney and Lai are inventors on Intellectual Property (IP) related to this research. This IP has not been licensed.
References
- 1.Ivens IA, et al. , PEGylated Biopharmaceuticals: Current Experience and Considerations for Nonclinical Development. Toxicologic Pathology, 2015. 43(7): p. 959–983. [DOI] [PubMed] [Google Scholar]
- 2.Swierczewska M, Lee KC, and Lee S, What is the future of PEGylated therapies? Expert Opin Emerg Drugs, 2015. 20(4): p. 531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gefen T, et al. , The impact of PEGylation on protein immunogenicity. Int Immunopharmacol, 2013. 15(2): p. 254–9. [DOI] [PubMed] [Google Scholar]
- 4.Veronese FM and Mero A, The impact of PEGylation on biological therapies. BioDrugs, 2008. 22(5): p. 315–29. [DOI] [PubMed] [Google Scholar]
- 5.Veronese FM and Pasut G, PEGylation, successful approach to drug delivery. Drug Discov Today, 2005. 10(21): p. 1451–8. [DOI] [PubMed] [Google Scholar]
- 6.Jevsevar S, Kunstelj M, and Porekar VG, PEGylation of therapeutic proteins. Biotechnol J, 2010. 5(1): p. 113–28. [DOI] [PubMed] [Google Scholar]
- 7.Ishida T, et al. , Spleen plays an important role in the induction of accelerated blood clearance of PEGylated liposomes. J Control Release, 2006. 115(3): p. 243–50. [DOI] [PubMed] [Google Scholar]
- 8.Ishida T, et al. , PEGylated liposomes elicit an anti-PEG IgM response in a T cell-independent manner. J Control Release, 2007. 122(3): p. 349–55. [DOI] [PubMed] [Google Scholar]
- 9.Ishida T, et al. , Accelerated blood clearance of PEGylated liposomes upon repeated injections: effect of doxorubicin-encapsulation and high-dose first injection. J Control Release, 2006. 115(3): p. 251–8. [DOI] [PubMed] [Google Scholar]
- 10.Mohamed M, et al. , PEGylated liposomes: immunological responses. Sci Technol Adv Mater, 2019. 20(1): p. 710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grenier P, et al. , Anti-polyethylene glycol antibodies alter the protein corona deposited on nanoparticles and the physiological pathways regulating their fate in vivo. J Control Release, 2018. 287: p. 121–131. [DOI] [PubMed] [Google Scholar]
- 12.Li B, et al. , Revealing the Immunogenic Risk of Polymers. Angewandte Chemie International Edition, 2018. 0(0). [DOI] [PubMed] [Google Scholar]
- 13.Hsieh YC, et al. , Pre-existing anti-polyethylene glycol antibody reduces the therapeutic efficacy and pharmacokinetics of PEGylated liposomes. Theranostics, 2018. 8(11): p. 3164–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McSweeney MD, et al. , A minimal physiologically based pharmacokinetic model that predicts anti-PEG IgG-mediated clearance of PEGylated drugs in human and mouse. Journal of Controlled Release, 2018. 284: p. 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fix SM, et al. , Accelerated Clearance of Ultrasound Contrast Agents Containing Polyethylene Glycol is Associated with the Generation of Anti-Polyethylene Glycol Antibodies. Ultrasound Med Biol, 2018. 44(6): p. 1266–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang P, et al. , Anti-PEG antibodies in the clinic: Current issues and beyond PEGylation. J Control Release, 2016. 244(Pt B): p. 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng TL, et al. , Accelerated clearance of polyethylene glycol-modified proteins by anti-polyethylene glycol IgM. Bioconjug Chem, 1999. 10(3): p. 520–8. [DOI] [PubMed] [Google Scholar]
- 18.Ichihara M, et al. , Anti-PEG IgM Response against PEGylated Liposomes in Mice and Rats. Pharmaceutics, 2011. 3(1): p. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mima Y, et al. , Anti-PEG IgM Is a Major Contributor to the Accelerated Blood Clearance of Polyethylene Glycol-Conjugated Protein. Mol Pharm, 2015. 12(7): p. 2429–35. [DOI] [PubMed] [Google Scholar]
- 20.Hershfield MS, et al. , Induced and pre-existing anti-polyethylene glycol antibody in a trial of every 3-week dosing of pegloticase for refractory gout, including in organ transplant recipients. Arthritis Research & Therapy, 2014. 16(2): p. R63–R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Armstrong JK, et al. , Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer, 2007. 110(1): p. 103–11. [DOI] [PubMed] [Google Scholar]
- 22.Ganson NJ, et al. , Pre-existing anti-polyethylene glycol antibody linked to first-exposure allergic reactions to pegnivacogin, a PEGylated RNA aptamer. J Allergy Clin Immunol, 2016. 137(5): p. 1610–1613.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Povsic TJ, et al. , Pre-existing anti-PEG antibodies are associated with severe immediate allergic reactions to pegnivacogin, a PEGylated aptamer. Journal of Allergy and Clinical Immunology, 2016. 138(6): p. 1712–1715. [DOI] [PubMed] [Google Scholar]
- 24.McSweeney MD, et al. , Overcoming anti-PEG antibody mediated accelerated blood clearance of PEGylated liposomes by pre-infusion with high molecular weight free PEG. J Control Release, 2019. 311–312: p. 138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kontos S, Grimm AJ, and Hubbell JA, Engineering antigen-specific immunological tolerance. Curr Opin Immunol, 2015. 35: p. 80–8. [DOI] [PubMed] [Google Scholar]
- 26.Zhang P, et al. , Proactively Reducing Anti-Drug Antibodies via Immunomodulatory Bioconjugation. Angewandte Chemie International Edition, 2019. 58(8): p. 2433–2436. [DOI] [PubMed] [Google Scholar]
- 27.Earl Sands AK, Johnston Lloyd, and Kishimoto Takashi K., Mitigation of Anti-Drug Antibodies Against a Pegylated Uricase in Patients with Hyperuricemia Results in Enhanced Control of Serum Uric Acid. American College of Rheumatology Meeting Abstracts, 2018. W.86. [Google Scholar]
- 28.Macauley MS, et al. , Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. The Journal of clinical investigation, 2013. 123(7): p. 3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Defrance T, Taillardet M, and Genestier L, T cell-independent B cell memory. Curr Opin Immunol, 2011. 23(3): p. 330–6. [DOI] [PubMed] [Google Scholar]
- 30.Obukhanych TV and Nussenzweig MC, T-independent type II immune responses generate memory B cells. The Journal of experimental medicine, 2006. 203(2): p. 305–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng J, et al. , Antibody quantity versus quality after influenza vaccination. Vaccine, 2009. 27(45): p. 6358–6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipsky PE, et al. , Pegloticase immunogenicity: the relationship between efficacy and antibody development in patients treated for refractory chronic gout. Arthritis Res Ther, 2014. 16(2): p. R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan ESL and Cronstein BN, Molecular action of methotrexate in inflammatory diseases. Arthritis Research & Therapy, 2002. 4(4): p. 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian H and Cronstein BN, Understanding the mechanisms of action of methotrexate: implications for the treatment of rheumatoid arthritis. Bull NYU Hosp Jt Dis, 2007. 65(3): p. 168–73. [PubMed] [Google Scholar]
- 35.Weinblatt ME, Methotrexate in rheumatoid arthritis: a quarter century of development. Transactions of the American Clinical and Climatological Association, 2013. 124: p. 16–25. [PMC free article] [PubMed] [Google Scholar]
- 36.Kishimoto TK and Maldonado RA, Nanoparticles for the Induction of Antigen-Specific Immunological Tolerance. Frontiers in immunology, 2018. 9: p. 230–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li J, Kim SG, and Blenis J, Rapamycin: one drug, many effects. Cell metabolism, 2014. 19(3): p. 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomson AW, Turnquist HR, and Raimondi G, Immunoregulatory functions of mTOR inhibition. Nature reviews. Immunology, 2009. 9(5): p. 324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meliani A, et al. , Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nature communications, 2018. 9(1): p. 4098–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biswas M, et al. , Combination therapy for inhibitor reversal in haemophilia A using monoclonal anti-CD20 and rapamycin. Thromb Haemost, 2017. 117(1): p. 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cronstein BN, Methotrexate BAFFles anti-drug antibodies. Nature Reviews Rheumatology, 2018. 14(9): p. 505–506. [DOI] [PubMed] [Google Scholar]
- 42.Joh DY, et al. , Architectural Modification of Conformal PEG-Bottlebrush Coatings Minimizes Anti-PEG Antigenicity While Preserving Stealth Properties. Adv Healthc Mater, 2019. 8(8): p. e1801177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qi Y, et al. , A brush-polymer conjugate of exendin-4 reduces blood glucose for up to five days and eliminates poly(ethylene glycol) antigenicity. Nat Biomed Eng, 2016. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li B, et al. , A Chromatin-Mimetic Nanomedicine for Therapeutic Tolerance Induction. ACS Nano, 2018. 12(12): p. 12004–12014. [DOI] [PubMed] [Google Scholar]
- 45.Kontos S, et al. , Engineering antigens for in situ erythrocyte binding induces T-cell deletion. Proceedings of the National Academy of Sciences, 2013. 110(1): p. E60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.FDA, Summary Basis for Regulatory Action for REBINYN, (N9-GP, GlycoPEGylated rFIX). 2017.
- 47.Yamaoka T, Tabata Y, and Ikada Y, Distribution and tissue uptake of poly(ethylene glycol) with different molecular weights after intravenous administration to mice. J Pharm Sci, 1994. 83(4): p. 601–6. [DOI] [PubMed] [Google Scholar]
- 48.Yang Q, et al. , Analysis of Pre-existing IgG and IgM Antibodies against Polyethylene Glycol (PEG) in the General Population. Anal Chem, 2016. 88(23): p. 11804–11812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McSweeney MD, et al. , Physician Awareness of Immune Responses to Polyethylene Glycol-Drug Conjugates. Clinical and Translational Science, 2018. 11(2): p. 162–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.