

Abstract
Background:
Next generation sequencing has facilitated the diagnosis of neurodevelopmental disorders with variable and non-specific clinical findings. Recently, a homozygous missense p.(Asp37Tyr) variant in TRAPPC2L, a core subunit of TRAPP complexes which function as tethering factors during membrane trafficking, was reported in two unrelated individuals with neurodevelopmental delay, post-infectious encephalopathy associated developmental arrest, tetraplegia, and accompanying rhabdomyolysis.
Methods:
We performed whole genome sequencing on members of an Ashkenazi Jewish (AJ) pedigree to identify the underlying genetic etiology of global developmental delay/intellectual disability in three affected siblings. To assess the effect of the identified TRAPPC2L variant, we performed biochemical and cell biological functional studies on the TRAPPC2L protein.
Results:
A rare homozygous predicted deleterious missense variant, p.(Ala2Gly), in TRAPPC2L was identified in the affected siblings and it segregated with the neurodevelopmental phenotype within the family. Using a yeast two hybrid assay and in vitro binding, we demonstrate that the p.(Ala2Gly) variant, but not the p.(Asp37Tyr) variant, disrupted the interaction between TRAPPC2L and another core TRAPP protein, TRAPPC6a. Size exclusion chromatography suggested that this variant affects the assembly of TRAPP complexes. Employing two different membrane trafficking assays using fibroblasts from one of the affected siblings, we found a delay in traffic into and out of the Golgi. Similar to the p.(Asp37Tyr) variant, the p.(Ala2Gly) variant resulted in an increase in the levels of active RAB11.
Conclusion:
Our data fill in a gap in the knowledge of TRAPP architecture with TRAPPC2L interacting with TRAPPC6a, positioning it as a putative adaptor for other TRAPP subunits. Collectively, our findings support the pathogenicity of the TRAPPC2L p.(Ala2Gly) variant.
Keywords: TRAPPC2L, neurodevelopmental disorder, genome sequencing, TRAPP complex, TRAPPC6a, RAB11, membrane trafficking, Ashkenazi Jewish
INTRODUCTION
Exome/genome sequencing has, in addition to identifying new gene-disease associations, identified the underlying genetic causes of neurodevelopmental disorders in many individuals with variable non-specific clinical manifestations. Recently, a homozygous missense p.(Asp37Tyr) variant in TRAPPC2L (MIM:610970) was reported in two unrelated individuals with neurodevelopmental delay, seizures, post-infectious encephalopathy, and episodes of rhabdomyolysis.1 TRAPPC2L is one of the core subunits of TRAPP (TRAnsport Protein Particle) complexes II and III that play an important role as tethering factors in membrane trafficking.2 Membrane trafficking is a cellular process involved in protein localization to the various intracellular compartments using membrane bound vesicles. This process requires numerous proteins and complexes including Rab family GTPases, guanine nucleotide exchange factors, Soluble NSF Attachment protein REceptors (SNAREs) and tethering factors. TRAPP (TRAnsport Protein Particle) are highly conserved tethering factor complexes in eukaryotes which regulate fusion of vesicles to membranes. These conserved multi-subunit complexes were originally identified in yeast and consist of core and modular subunits. In humans, two TRAPP complexes, TRAPP II and III, have been described. Each has a common core of subunits (TRAPPC1, TRAPPC2, TRAPPC2L, TRAPPC3, TRAPPC4, TRAPPC5 and TRAPPC6) as well as complex-specific proteins (TRAPPC8, TRAPPC9, TRAPPC10, TRAPPC11, TRAPPC12 and TRAPPC13). TRAPPC2L was initially identified based on homology to the TRAPPC2 core subunit and TRAPPC2L complexes may function in a post-Golgi compartment. Many members of the TRAPP complexes have been implicated in neurodevelopmental disorders (TRAPPC6A, TRAPPC6B, TRAPPC9, and TRAPPC12), muscular dystrophies (TRAPPC11), and skeletal dysplasias (TRAPPC2) following autosomal and X-linked recessive patterns, respectively.3–8
Here, we report three siblings from an Ashkenazi Jewish family with neurodevelopmental delay/intellectual disabilities in whom we identified a homozygous missense TRAPPC2L variant and performed functional studies to assess the effect of the variant on TRAPPC2L function using human and yeast cells.
METHODS
Patient Ascertainment and Genomic Analysis
This study was approved by the Institutional Review Board of Columbia University. Written informed consents were obtained from all participants. We performed whole genome sequencing on peripheral blood DNA samples from three affected siblings with a similar clinical presentation and clinical course and their unaffected parents. Sequencing methods and variant filtering criteria were as previously described.9 The candidate variant and segregation in the family with the neurodevelopmental phenotype were confirmed by Sanger sequencing.
Haplotype Analysis
For the haplotype analysis, variant calling was done with DeepVariant (v0.8.0), and variants from chromosome 16 (hg38) with a quality score ≥30 were used. The Bcftools (v1.9) RoH tool along with Ashkenazi Jewish population allele frequency data from Genome Aggregation Database (gnomAD) v3 release (with whole genome sequencing analyses of 1,662 Ashkenazi Jewish individuals), were used to identify the stretch of homozygosity flanking the TRAPPC2L variant of interest.10–12
Yeast experiments
Standard yeast methods were used for transformation, strain construction, and growth.13 TRAPP genes were cloned into pGADT7 and pGBKT7 plasmids (Addgene) for yeast-two-hybrid analysis. pGADT7 and pGBKT7 constructs were then transformed into AH109 yeast and Y187 yeast, respectively. Mating was performed at 30°C on YPD agar plates overnight. To assess the mating efficiency, plates were then replicated onto double drop-out (DDO) YPD agar plates lacking leucine and tryptophan. To evaluate interactions, plates were replicated on triple drop-out (TDO) YPD agar plates lacking tryptophan, and histidine. Single colonies of each diploid were then cultured in liquid selective media (DDO) for 24 hours, and serial dilutions were prepared. The diploid cells were then spotted on selective media (DDO) and (TDO) and plates were left for 3 days at 30°C.
Size exclusion chromatography
Fibroblasts were plated in two 15-cm plates. When the cell confluency reached 80–90%, the cells were washed 3 times with ice-cold PBS (10 ml each wash), then lysed in a total of 500 µL ( 250 µL for each 15-cm plate) of lysis buffer (50 mM Tris pH 7.2, 150 mM NaCl, 0.5 mM EDTA, 1 mM DTT, 1.0% Triton, and protease inhibitor cocktail, EDTA-free, Roche). The lysate was centrifuged at 16,000 g for 30 minutes at 4° C then the supernatant was transferred to a fresh tube. 5mg of total protein was loaded on a 24 mL Superose 6 Increase 10/300 GL column (GE Healthcare) and 0.5 mL fractions were collected in the wash buffer (50 mM Tris pH 7.2, 150 mM NaCl, 0.5 mM EDTA, 1mM DTT, 0.1% Triton). The fractions were loaded on two gels for western blotting probed with antibodies recognizing TRAPPC8 (monoclonal, Abcam), TRAPPC12 (polyclonal, homemade), TRAPPC2L (monoclonal, Santa cruz), or TRAPPC10 (monoclonal, Santa cruz).
Recombinant protein preparation
Bacterial cultures (500 ml) were grown to an OD600 of 0.5–0.6 at 37°C. Protein production was induced by adding 1 mM IPTG while shaking overnight at 20 °C. Cells were then pelleted at 4,000 rpm for 10 minutes and resuspended in 30ml lysis buffer (50mM Tris-HCl pH 8.0; 0.4 M NaCl; 1mM DTT, 5% glycerol, 0.5 mM AEBSF, 0.1% Triton X-100) for glutathione-S-transferase (GST) fusion proteins, or 35 ml of lysis buffer (0.3 M NaCl, 50 mM Tris-HCl pH 8.5, 10 mM β-mercaptoethanol, 5% glycerol, 0.1% Triton X-100, 0.5 mM AEBSF) for (His6)-tagged proteins. 3.5 ml of 10% Triton X-100 was added for GST fusion proteins. The cell lysates were sonicated for 2 minutes by pulsing for, 10 seconds ON and 10 seconds OFF at an amplitude setting of 25%. The resulting lysates were then cleared by centrifugation at 30,000 g for 30 minutes.
The crude extracts were incubated with either glutathione sepharose beads (Thermofisher) or Ni-NTA Agarose (Qiagen) for 1h at 4°C. The samples were then transferred into a column and washed once with 15 ml of appropriate lysis buffer and twice with 10 ml wash buffer (50mM Tris-HCl pH 8.0; 0.4 M NaCl, 5% glycerol, 1mM DTT) for GST-tagged proteins or (50 mM Tris-HCl pH 8.5, 200 mM NaCl, 5 mM β-mercaptanol, 0.5 mM EDTA pH 8.0) for His6-tagged proteins. GST-tagged proteins were eluted in 1.0 ml fractions of elution buffer (50mM Tris-HCl pH 8.0; 0.4 M NaCl, 15mM glutathione). His6-tagged proteins were eluted in 1.0 ml fractions of elution buffer (50 mM Tris pH 8.8, 200 mM NaCl, 50–250 mM imidazole).
In vitro binding assay
In vitro binding assays contained 0.5 µM of the GST fusion proteins (TRAPPC2L, TRAPPC2L A2G, or TRAPPC2L D37Y) with increasing amounts (0, 0.1, 0.2, 0.5, 1.0 µM) of the His6-tagged heterodimeric complex composed of TRAPPC3 and TRAPPC6a. Samples were made up to a total volume of 250 µl with 1x binding assay buffer (25mM Hepes pH 7.4, 115mM KOAc, 2.5 mM MgCl2, 1mM DTT)and left on ice at 4 °C overnight. The GST-tagged proteins were collected onto 20 µl of glutathione-agarose resin by incubating on a nutator for 1 hour at 4 °C. Samples were washed 3x with 250 µl of 1x binding assay buffer. Proteins were then eluted from the beads by heating to 95°C in 25 µl of 1x sample buffer for 3 minutes. Western analysis used homemade polyclonal antibody recognizing TRAPPC3.
UV–visible spectroscopy
The UV–visible spectra (200–500 nm) for TRAPPC2L and the two variants were collected using the Varian Cary 100 Bio UV–Visible Spectrophotometer. The approximate concentration of protein was calculated using the molar extinction coefficient of 15025 M−1 cm−1 at A280 for TRAPPC2L.
CD spectroscopy
CD spectra in the Far-UV region (200–280 nm) were recorded for TRAPPC2L and the two variants in a 0.2 cm cell under a constant nitrogen flow using a Jasco-815 CD spectropolarimeter. A protein concentration of 0.5 mg/ml in buffer (50mM Tris pH 8.5, 1mM DTT, 200NaCl, and 1mM EDTA) was used. The parameters used were as follows: bandwidth of 1 nm, a response time of 0.25 s, a data pitch of 0.2 nm, and a scanning speed of 20 nm/min. The spectra obtained were smoothed using the Spectra Analysis Manager program from Jasco.
Thermal denaturation
Thermal denaturation was examined by CD spectroscopy using a protein concentration of 0.5 mg/ml in buffer (50mM Tris pH 8.5, 1mM DTT, 200NaCl, and 1mM EDTA) in a 0.2 cm cell. Standard parameters were used (bandwidth of 1 nm, a response time of 0.25 s, and a data pitch of 0.2 nm). The midpoint of the unfolding transition (TM) was obtained by measuring the change in ellipticity at 222 nm with standard sensitivity using the variable temperature settings on a Jasco-815 CD spectropolarimeter. The rate used was 60°C/h with a start temperature of 25°C and an end temperature of 65°C.
Fluorescence measurements
Fluorescence of the proteins (0.1mg/ml) was measured in (50mM Tris pH 8.5, 1mM DTT, 200NaCl) using the Varian Cary Eclipse Fluorescence Spectrophotometer with a 1 cm pathlength at room temperature (25°C). The samples were excited at 280 nm and the emission spectra were recorded in the range of 290–500 nm with a scan rate of 600 nm/min and excitation and emission slits of 5 nm. The emission spectra were averaged over 10 separate scans with a 1 nm data interval.
Levels of active RAB11
Monitoring the levels of active RAB11 was performed using the RAB11 Mouse Monoclonal Antibody Kit (Neweast Biosciences) as per the manufacturer’s instructions.
RESULTS
Clinical findings
Three affected individuals in a sibship of five from a non-consanguineous family of Ashkenazi Jewish ancestry were evaluated for neurodevelopmental delay (figure 1A). Clinical findings of the individuals are given in table 1 along with two previously reported individuals with TRAPPC2L pathogenic variants. Detailed medical histories of each individual are provided in the supplemental notes. Previous genetic testing including chromosome analysis, Fragile-X testing and clinical exome sequencing were non-diagnostic.
Figure 1. Molecular genetic analysis of individuals with a TRAPPC2L variant.
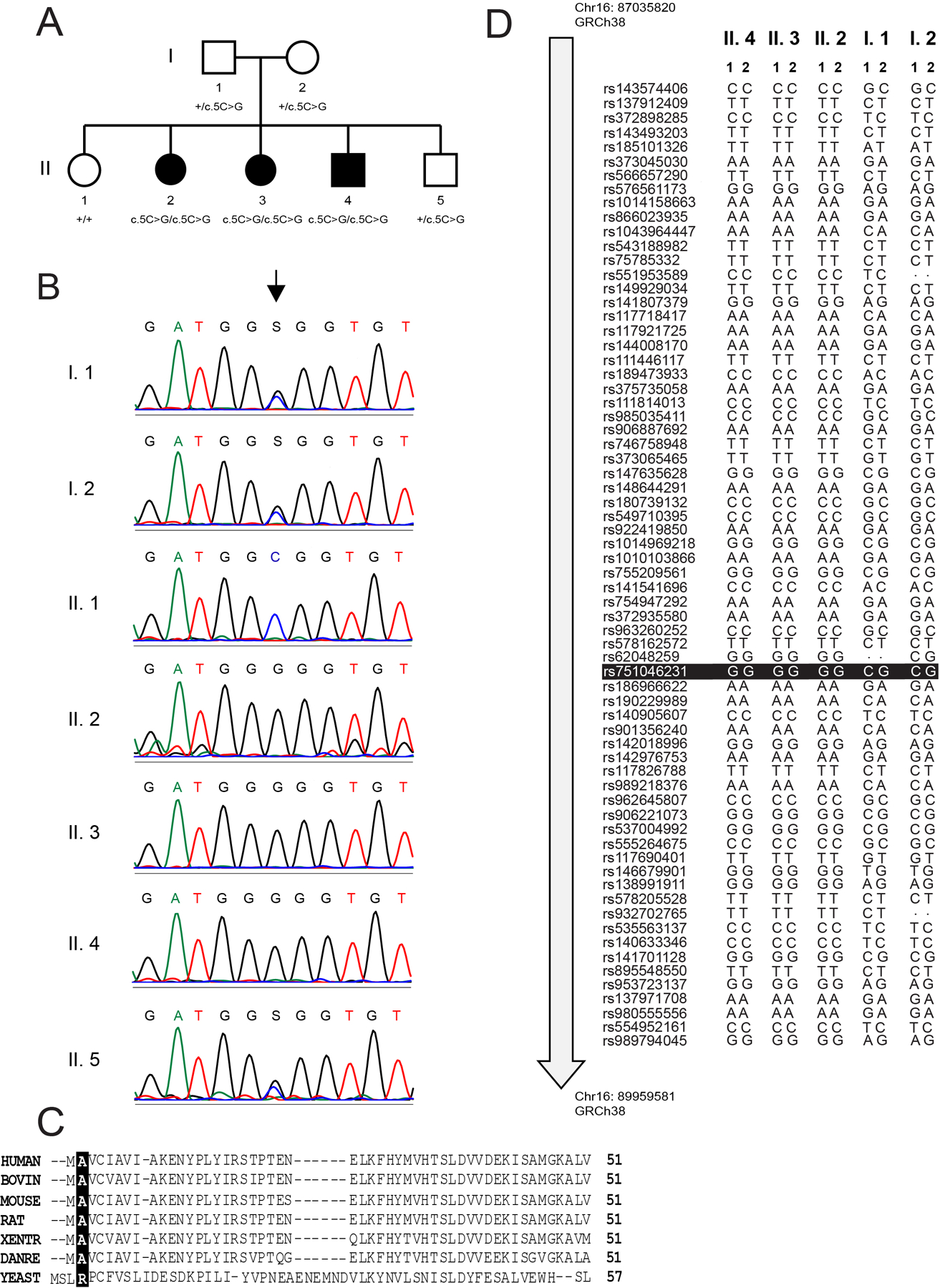
(A) A pedigree of the family presented in this study. (B) Sanger sequencing of all seven individuals in the pedigree. The arrow points to the affected nucleotide (c.5C in wild type). (C) Multiple sequence alignment of TRAPPC2L form various species indicating the highly-conserved alanine at residue 2 in the proteins. (D) Analysis of the SNPs on chromosome 16 revealed a contiguous stretch of homozygosity (Chr16:86948799–90118806, 3.17Mb, hg38) around the TRAPPC2L gene in the 3 affected individuals of this family. In this region, rare variants with at least one alternate allele at the genomic position in the parents and with allele frequency ≤ 0.01 in the Ashkenazi Jewish population (gnomAD v3), were filtered (n=68) to plot the figure. For each individual, the two alleles are represented as 1 and 2. The TRAPPC2L gene variant seen in this family is highlighted in black (Chr16:88857155-C-G,hg38; rs751046231). Dots in the figure represent genomic positions in the sample where the genotype call could not be assessed or did not cross the filtering criteria threshold.
Table 1.
Clinical findings of individuals with homozygous TRAPPC2L variants
| Individual | This Study | Milev et al., 20181 | |||
|---|---|---|---|---|---|
| II.2 | II.3 | II.4 | S1 | S2 | |
| Age / Sex | F / 38 yo | F / 36 yo | M / 33 yo | F / 3 yo | F / 16 yo |
| Ancestry | Ashkenazi Jewish | Italian | Austrian | ||
| Variant | c.5G>C:p.(Ala2Gly) | c.109G>T:p.(Asp37Tyr) | |||
| Prenatal and Perinatal | Unremarkable | Perinatal distress | Perinatal distress, preterm labor | ||
| Presenting symptom (age of onset) | Developmental delay (12 months) | Developmental delay (4 months) | Developmental delay (6 months) | Developmental delay (9 months) | Developmental regression (9 months) |
| Illness provoked regression | No | Yes | Yes | ||
| Development prior to illness | N/A | Delayed | Normal | ||
| CK during illness | Unknown | Up to 16000 | Up to 5500 | ||
| Intermittent CK levels | Unknown | Normal to 1000 | Normal range | ||
| Brain MRI before the first event & after the first event | Not performed | Normal at 2 years old | Delayed myelination at 10 months and Acute encephalopathy with posterior edema (16 months) Progressive brain atrophy (18 and 30 months) | Delayed myelination at 10 months and No follow-up MRI | |
| Developmental delay | Yes | ||||
| Motor milestones | sat independently at 12 months never crawled, walked at 2 years old | very mild motor delay, walked at 15 months old | sat at 12 months, crawled at 18 months, walked at 2–3 years old | could sit alone at 11 months but subsequently lost this ability, never achieved independent walking | could sit unsupported at 7 months but subsequently lost this ability, never achieved independent walking |
| Speech | severe expressive language delays | Non-verbal | Non-verbal | ||
| Tetraplegia | No | Yes | Yes | ||
| Dystonia | No | No | Yes | Yes | Yes |
| Seizure | No | No | No | Yes | Yes |
| Vision | No reported problems | Cerebral visual impairment | Clinical suspicion of cerebral visual impairment | ||
| Other | None | Protein losing enteropathy at 39 months old | Vitamin B12 and Folic acid deficiency, cholelithiasis, recurrent pneumonia | ||
Molecular findings
Filtering based on allele frequency (alternate allele frequency <1%) yielded homozygous missense variants in TRAPPC2L and PIEZO1 (MIM:611184) in all three affected siblings (table 2 and table S1). We assessed the PIEZO1 variant as non-causative since both the human and mice phenotypes associated with PIEZO1/piezo1 were different than the phenotype observed in our patients, leaving the ultra-rare, predicted pathogenic TRAPPC2L missense variant (NM_016209.5:c.5G>C:p.(Ala2Gly)) as the most plausible candidate that could explain the observed phenotype. The population allele frequencies and the computational prediction scores for this variant are given in table 2. Notably, this missense variant was only observed in the Ashkenazi Jewish individuals in gnomAD v2.1.1 release (http://gnomad.broadinstitute.org/) with an allele frequency of 0.0002243, and was not observed in gnomAD v3 release containing 71,702 genomes and Trans-Omics for Precision Medicine (TOPMed) Freeze 5 database containing 62,784 genomes (https://bravo.sph.umich.edu/freeze5/hg38/). The variant was confirmed by Sanger sequencing (figure 1B) and segregated with the neurodevelopmental phenotype in the family (figure 1A). The affected alanine residue is well-conserved across phyla except for yeast where an adjacent leucine may act as the hydrophobic residue (figure 1C).
Table 2.
Homozygous TRAPPC2L variants identified in affected individuals with relevant population frequencies and in silico prediction scores.
| Genomic coordinates on chr16 (hg19 hg38) | HGVS (NM_016209.5) | Population frequency (n/MAF/hom) | In silico predictions | |||||
|---|---|---|---|---|---|---|---|---|
| cDNA | Protein | gnomAD (v2.1.1; v3) | TOPMed (Freeze 5) | CADD | REVEL | PROVEAN | SIFT | |
| 88923563-C-G 88857155-C-G |
c.5C>G | p.(Ala2Gly) | 2/9.6e-6/No; 0/0/No | 0/0/No | 28.3 | 0.68 | D (−3.16) | T (0.117) |
| 88925102-G-T* 88858694-G-T |
c.109G>T | p.(Asp37Tyr) | 4/1.6e-5/No; 0/0/No | 3/0.0024/No | 29.5 | 0.981 | D (−8.75) | D (0) |
Abbreviations: MAF, minor allele frequency; No, no homozygotes were seen; D, damaging (SIFT)/deleterious (Provean); T, tolerated
TRAPPC2L variant reported in J Med Genet. 2018 Nov;55(11):753–764
CADD v1.3 scores: https://cadd.gs.washington.edu/snv
REVEL scores: https://rothsj06.u.hpc.mssm.edu/revel/revel_segments/” https://rothsj06.u.hpc.mssm.edu/revel/revel_segments/
SIFT, Provean scores for the variant were added from http://provean.jcvi.org/genome_submit_2.php?species=human
Haplotype Analysis
Haplotype analysis in TRAPPC2L and its flanking region using single nucleotide variants from the genome sequencing data revealed a stretch of homozygosity spanning 3,170,007 base-pairs in the three affected siblings, indicating a common ancestral chromosomal region carrying the variant from each parent was transmitted to the affected individuals (figure 1D). The parental SNPs were heterozygous in this region.
The TRAPPC2L missense variant p.(Ala2Gly) fails to interact with TRAPPC6a and TRAPPC12
To assess the effects of the missense p.(Ala2Gly) variant on the interactions of TRAPPC2L with other TRAPP subunits, a yeast two-hybrid assay was performed (figure 2A) using the subunits that were previously shown to interact with TRAPPC2L by this method.1 This variant disrupted the interactions with TRAPPC6a (MIM: 610396) and the TRAPP III-specific subunit TRAPPC12 (MIM: 614139). Interestingly, unlike the previously reported TRAPPC2L missense variant p.(Asp37Tyr),1 the interaction with the TRAPP II-specific subunit TRAPPC10 (MIM: 602103) was not affected. It is noteworthy that the interaction with TRAPPC6a was affected regardless of whether TRAPPC2L was in the pGADT7 or pGBKT7 vector. In contrast, an interaction with TRAPPC12 was only seen with TRAPPC2L in the pGADT7 vector.
Figure 2. TRAPPC2L p.(Ala2Gly) has reduced function compared to wild type.

(A) The TRAPP subunits indicated were cloned into either pGBKT7 or pGADT7. TRAPPC2L and the two variants p.(Ala2Gly) and p.Aspr37Tyr were also cloned into these same vectors. A yeast two-hybrid assay was performed whereby growth on DDO (double drop-out medium lacking leucine and tryptophan) indicates the presence of the indicated plasmids, while growth on TDO (triple drop-out medium lacking leucine, tryptophan and histidine) indicates an interaction. (B) A yeast strain harboring a deletion of the TRAPPC2L homologue TCA17 and an HA-tagged TRS130 gene (TRS130 is the yeast homologue of TRAPPC10) is temperature sensitive for growth at 35°C and 37°C. The growth sensitivity is rescued by wild type TCA17 and partially rescued by wild type TRAPPC2L. The two TRAPPC2L variants p.(Ala2Gly) and p.(Asp37Tyr) do not rescue the growth defect. (C) A model of a portion of the TRAPP core complex that shows TRAPPC4 (salmon colored), TRAPPC1 (yellow colored), TRAPPC3 (cyan colored) and TRAPPC6a (residues 1–83 colored in pink and residues 84–159 colored in orange). (D) The yeast two-hybrid assay was repeated using TRAPPC2L and the two variants. The second plasmid expressed TRAPPC6a residues 1–83 or residues 84–159 as indicated.
We next employed the previously established yeast system to study the functionality of the TRAPPC2L variant by using the conditionally lethal tca17Δ TRS130-HA yeast strain.1 The expression of only the wild-type TRAPPC2L, but not the p.(Ala2Gly) variant, could suppress the conditional lethality of the yeast (figure 2B), consistent with the previous report asp.(Asp37Tyr) was also incapable of supporting growth. Together with our previous data,1 we suggest that TRAPPC2L functions as an adaptor protein to aid in the assembly of TRAPP complexes most likely through an interaction with the core TRAPP protein TRAPPC6a.
TRAPPC2L binds directly to the TRAPPC6a-TRAPPC3 heterodimer.
TRAPPC2L was previously suggested to bind to the side of the TRAPP complex opposite to that of TRAPPC2.14–16 Our yeast two-hybrid assay above supports this notion. To test this hypothesis, we performed an in vitro protein binding assay to investigate the association of TRAPPC2L with TRAPPC6a. Since TRAPPC6a forms a tight complex with TRAPPC3,17,18 we purified a heterodimer of the latter two proteins for these studies and assessed binding to GST-tagged TRAPPC2L wild type or to either the p.(Ala2Gly) or the previously-reported p.(Asp37Tyr) variants (figure 3A) by western analysis using anti-TRAPPC3. As shown in figure 3B and quantified in figure 3C, while TRAPPC2L and the p.(Asp37Tyr) variant both showed a concentration-dependent increase in binding to TRAPPC6a-TRAPPC3, the p.(Ala2Gly) variant was severely impaired in its ability to bind to this heterodimer. This was not due to gross changes to the three-dimensional structure of the protein since circular dichroism spectroscopy (figure 3D), thermal denaturation temperature (figure S1A) and tyrosine fluorescence (figure S1B) were similar amongst TRAPPC2L wild type and the two missense variants. Collectively, our results suggest that the Ala2 residue, but not Asp37, is necessary for the association of TRAPPC2L with the TRAPP core.
Figure 3. TRAPPC2L p.(Ala2Gly) has a weakened interaction with TRAPPC6a.
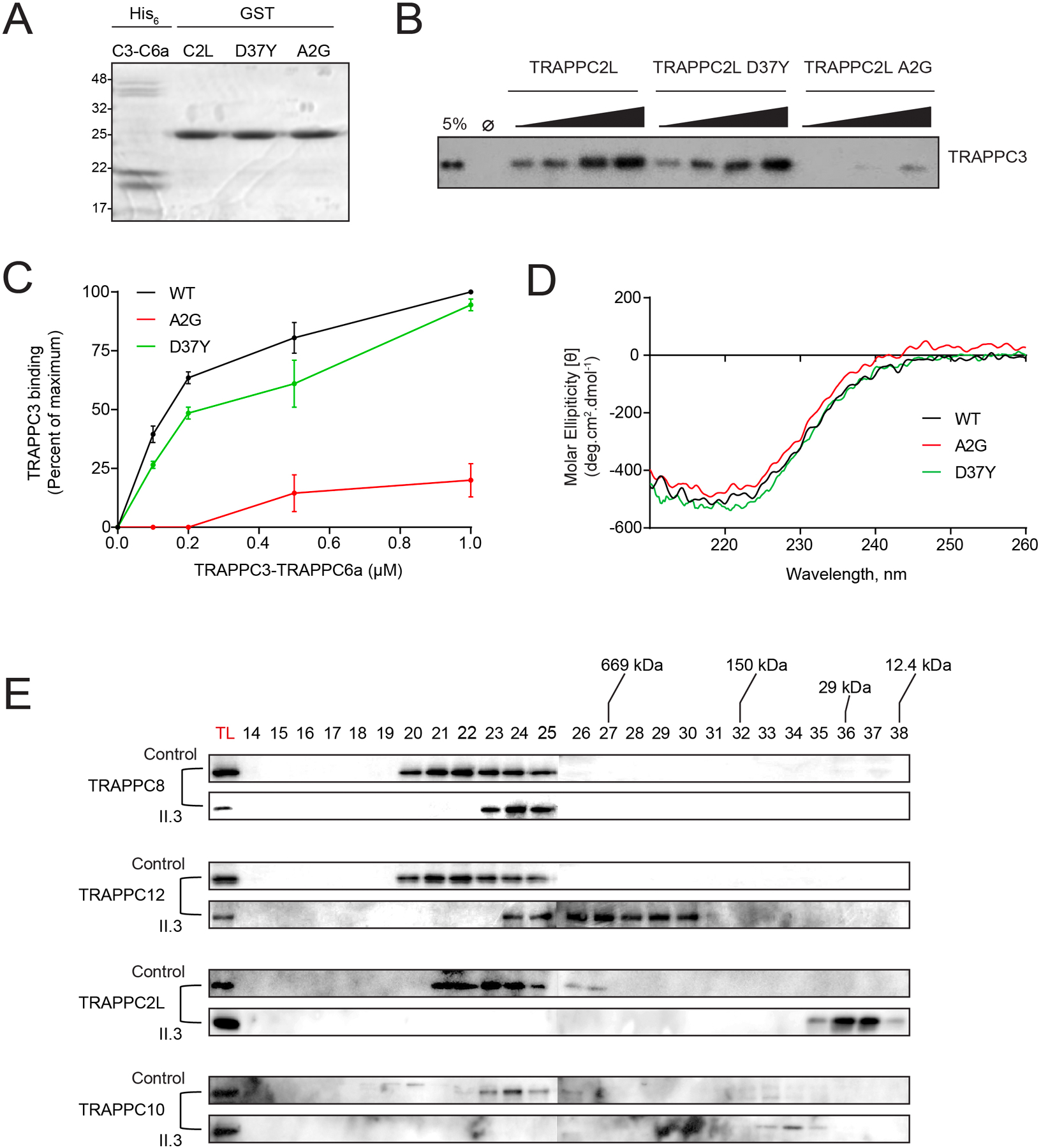
(A) an SDS-polyacrylamide gel of His-tagged recombinant TRAPPC3-TRAPPC6a heterodimer and GST-tagged TRAPPC2L or the TRAPPC2L variants p.(Ala2Gly) and p.(Asp37Tyr). Molecular size standards in kD are displayed on the left. (B) The GST-tagged proteins from panel (A) were incubated with increasing concentrations of the TRAPPC3-TRAPPC6a heterodimer. Following binding, the samples were probed by western analysis using anti-TRAPPC3 IgG. A sample representing 5% of the maximum amount of the heterodimer is shown to the left of the panel. (C) The TRAPPC3 signal from panel (B) was quantified and plotted versus the concentration of the heterodimer added to the reaction. (D) A circular dichroism curve for His-tagged recombinant TRAPPC2L or the two variants was performed. (E) Lysates from control fibroblasts (control) and fibroblasts derived from the individual harboring the p.(Ala2Gly) variant (subject) were prepared and subjected to size exclusion chromatography. Total cell lysate (TL) and fractions (indicated above the panels) from the column were analyzed by western analysis for the indicated TRAPP proteins.
We then mapped the region on TRAPPC6a that was important for this interaction. A yeast two-hybrid assay was employed by examining residues 1–83 and 84–159 individually. This effectively separates the protein into a region that is involved in interaction with the TRAPP core (1–83) and a region that is theoretically exposed and available for non-core interactions (84–159) (figure 2C). As shown in figure 2D, none of the TRAPPC2L constructs interacted with TRAPPC6a (1–83). Interestingly, while wild type TRAPPC2L and the p.(Asp37Tyr) variant interacted with TRAPPC6a (84–159), the p.(Ala2Gly) variant did not. These results further support that notion that Ala2 is critical for the interaction between TRAPPC2L and TRAPPC6a and that this interaction takes place on a region of TRAPPC6a that is not involved in its interaction with the TRAPP core subunits.
The TRAPPC2L missense variant p.(Ala2Gly) disrupts the assembly of TRAPP.
Our results thus far suggest that the p.(Ala2Gly) variant has impaired association with the TRAPP core. As an adaptor for other complex-specific proteins, this might affect the assembly of TRAPP complexes. To test this, we followed the fractionation of TRAPP complexes by size exclusion chromatography in lysates from fibroblasts derived from an affected individual with the p.(Ala2Gly) variant (II.3). We used TRAPPC8 and TRAPPC12 as markers for TRAPP III, and TRAPPC10 as a marker for TRAPP II. In all cases the proteins were shifted to a smaller molecular size fraction in lysates from fibroblasts harboring the p.(Ala2Gly) variant (figure 3E), suggesting that this variant affects the assembly or stability of TRAPP complexes.
The TRAPPC2L missense variant p.(Ala2Gly) affects membrane trafficking in fibroblasts but not autophagic flux.
Since the TRAPP complexes appear to be destabilized in the presence of the p.(Ala2Gly) variant, and these complexes are important for membrane trafficking in cells,19 we hypothesized that membrane trafficking would be affected in fibroblasts derived from the individual with the p.(Ala2Gly) variant (II.3). To test this notion, we employed the RUSH assay20 to examine trafficking between the endoplasmic reticulum (ER) and Golgi. As shown in figure 4A, there was a noticeable delay in arrival of the marker protein in the Golgi, similar to what was seen for the p.(Asp37Tyr) variant (S1). This was not due to an absence of the TRAPPC2L protein since the protein was present at a similar level as the wild type protein in fibroblasts from an unaffected individual (figure 4A inset). The delay in trafficking was rescued to near wild type levels upon expression of an RFP-tagged wild type TRAPPC2L, suggesting that the delay in membrane trafficking was due to a dysfunctional TRAPPC2L protein. We also examined VSVG-GFP ts045, a cargo whose traffic through the entire biosynthetic pathway can be followed by fluorescence microscopy and demonstrated a similar delay in traffic into and out of the Golgi in fibroblasts from both affected individuals (figure 4B). Collectively, our data suggest that the p.(Ala2Gly) variant affects assembly of TRAPP complexes resulting in membrane trafficking defects in cells.
Figure 4. Functional studies reveal a membrane trafficking defect and increased active RAB11 in fibroblasts derived from an individual with TRAPPC2L p.(Ala2Gly).
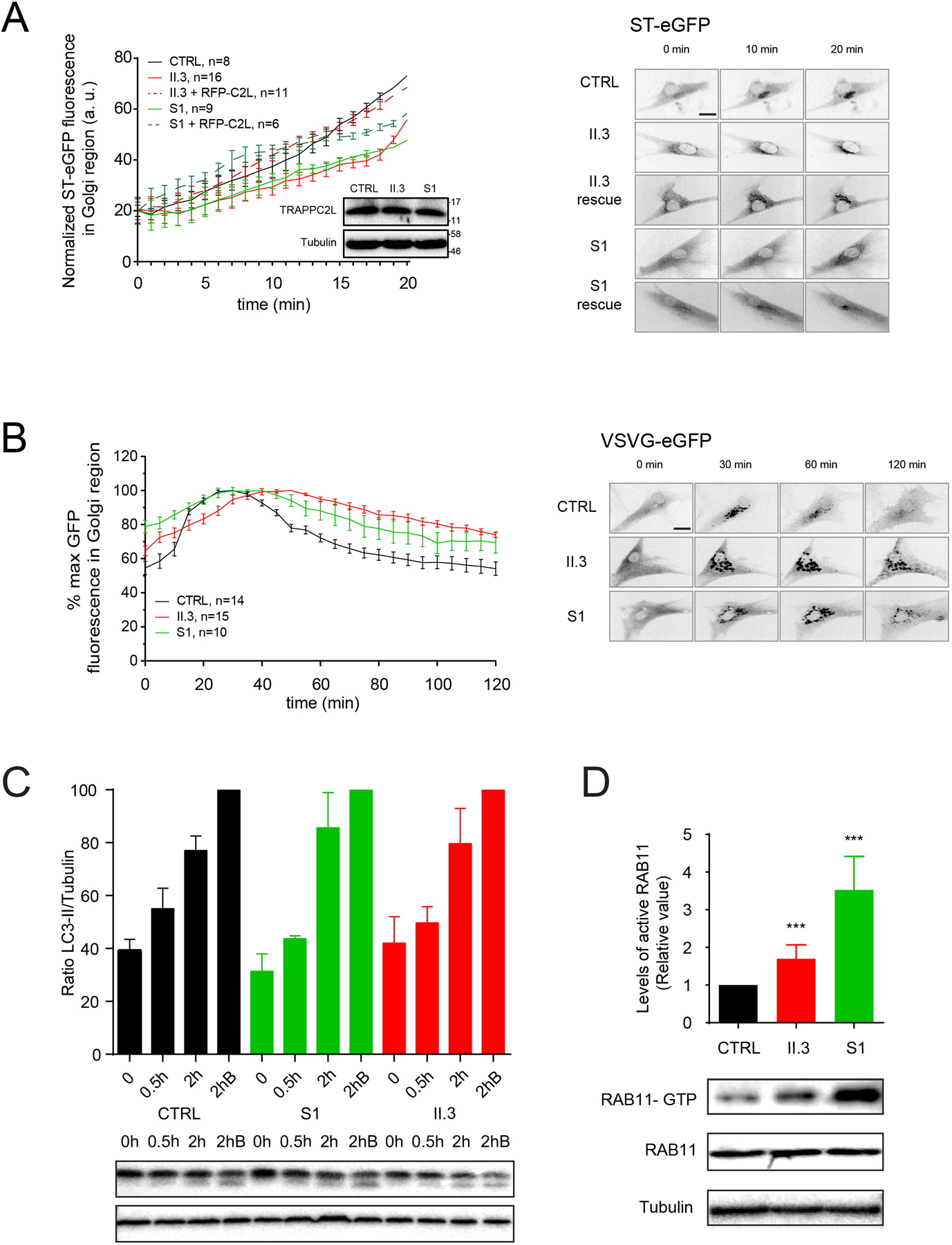
(A) The RUSH assay measuring traffic between the endoplasmic reticulum and the Golgi was performed and quantified for control (CTRL), p.(Ala2Gly) (II.3) and p.(Asp37Tyr) (S1) fibroblasts using the cargo protein ST-eGFP. The inset shows a western analysis for TRAPPC2L and tubulin from the cells that were analyzed, with molecular size standards on the right. Representative images used for the quantification are shown to the right of the graph. (B) The transport of VSVG-GFP ts045 was performed on control (CTRL), p.(Ala2Gly) (II.3) and p.(Asp37Tyr) (S1) fibroblasts, and quantified. Representative images used for the quantification are shown to the right of the graph. (C) Cells from control (CTRL) and p.(Ala2Gly) (II.3) and p.(Asp37Tyr) (S1) fibroblasts were left in nutrient-rich medium (0) .or starved for 0.5h or 2h. Some cells were starved for 2h in the presence of bafilomycin A1 which prevents formation of autolysosomes (2hB). Lysates were prepared and analyzed by western analysis for the autophagy marker LC3-II as well as tubulin. The normalized LC3-II/tubulin ratio was determined. A sample western blot is shown beneath the graph. (D) Lysates from control (CTRL), p.(Ala2Gly) (II.3) and p.(Asp37Tyr) (S1) fibroblasts were treated with IgG recognizing active (GTP-bound) RAB11. The precipitated RAB11 was then revealed by western analysis using anti-RAB11 IgG. The immunoprecipitated protein is shown I the top panel while the lysates are in the lower two panels. The signal was quantified and plotted as relative levels. *** indicates p<0.001.
Since the assembly and/or stability of TRAPP III was affected in vitro in cells harboring the p.(Ala2Gly) variant, and since TRAPP III functions in autophagy,21 we examined if autophagic flux was affected in the presence of this variant. As shown in figure 4C, there was no significant difference in the appearance of LC3-II, an autophagy marker, during starvation of the cells. This is similar to what was reported for the p.(Asp37Tyr) variant.1
The TRAPPC2L missense variant p.(Ala2Gly) affects the levels of active RAB11.
We previously demonstrated that fibroblasts derived from an individual with the p.(Asp37Tyr) variant had elevated levels of active RAB11,1 suggesting a role for TRAPP in RAB11 function. Previous reports have indicated that the yeast and Drosophila TRAPP II complexes are guanine nucleotide exchange factors for the RAB11 homologues.22–24 Given that TRAPP II is destabilized in fibroblasts harboring the p.(Ala2Gly) variant, we asked whether this might result in elevated levels of active RAB11. Using a conformation-specific antibody we found that the p.(Ala2Gly) variant resulted in a small but significant increase in active RAB11 (figure 4D). The increase, however, was not as high as that seen for the p.(Asp37Tyr) variant, suggesting that the former variant may not affect TRAPP II assembly and/or stability in vivo as severely as the latter variant.
DISCUSSION
We assessed a homozygous rare missense variant p.(Ala2Gly) in TRAPPC2L in three affected siblings with a neurodevelopmental disorder. The family is of Ashkenazi Jewish ethnicity, and sequence analysis showed that the haplotype surrounding this variant (AJ allele frequency is 0.0002243) is likely from a common ancestor. Recently, another missense homozygous variant p.(Asp37Tyr) in TRAPPC2L was reported in two unrelated individuals with post-infectious encephalopathy, spastic quadriplegia, and intermittent rhabdomyolysis. Haplotype analysis suggested a common founder for those individuals. Strengthening the evidence that the p.(Ala2Gly) and p.Asp37Tyr variants are causative of disease is the fact that non-synonymous variants of Ala2 reported in gnomAD are of low allele frequency, are predicted to be pathogenic and that neither p.Ala2Gly and p.Asp37Tyr variants have not been observed in a homozygous state in gnomAD (v2.1.1 and v3.0), TOPMed Freeze 5, and The Greater Middle East (GME) Variome Project, which is enriched for consanguineous populations. Also, the recently available Trappc2l homozygous knock-out mice (https://www.mousephenotype.org/“ https://www.mousephenotype.org/), show significant nervous system phenotypes that are in parallel to neurodevelopmental phenotype observed in thus far reported individuals.
While neurodevelopmental delay and intellectual disabilities are the common clinical findings between our family and the previously reported individuals,1 there are also differences in the clinical findings, severity and clinical course (table 1). While cerebral visual impairment, dystonia, and seizures were reported in both individuals in the previous study, dystonia (lower extremities, progressive dystonia) was observed in only one out of three affected siblings in our study and seizures have not been documented. Additionally, in contrast to the patients reported herein, the two previously reported individuals experienced post-infectious encephalopathy with rhabdomyolysis, which may have complicated the clinical spectrum and resulted in additional findings such as spastic paraplegia and rhabdomyolysis. In our family, one of the affected individuals (II.3) had viral meningitis at 5–6 weeks of life, though without any obvious acute neurologic sequelae. Neither sibling has had their serum creatine kinase levels measured nor have there been manifestations of a myopathy. Furthermore, in one of the previously reported individuals, developmental delay and delayed myelination had already been noted at 9 and 10 months of age, respectively, before the first infectious event took place at 16 months of age.1 Brain MRI of one of the affected siblings in the present study at 23 years of age was normal. The variability in clinical manifestations might be due to differences in the molecular effects of the two missense variants. Future case reports and longitudinal follow-up studies are warranted to delineate the complete clinical spectrum and natural history of the TRAPPC2L-associated neurodevelopmental disorder and to determine if infectious insults contribute to the neurological course.
TRAPPC2L is a member of the highly conserved tethering factor complexes TRAPP II and III that share a common core of subunits (TRAPPC1, TRAPPC2, TRAPPC2L, TRAPPC3, TRAPPC4, TRAPPC5, TRAPPC6) as well as complex-specific proteins (TRAPPC9 and TRAPPC10 for TRAPP II; TRAPPC8, TRAPPC11, TRAPPC12 and TRAPPC13 for TRAPP III). TRAPP complexes play an important role in membrane trafficking between the endoplasmic reticulum and Golgi (TRAPP III) and at later stages of the secretory pathway (TRAPP II), and the genes encoding both core and complex-specific subunits are associated with Mendelian disorders.3–8
Although clinically there are some differences between the individuals in the present study and those reported previously,1 at the cellular level there are a number of similarities including a TRAPPC2L-dependent delay in ER-to-Golgi traffic, no effect on autophagic flux, and a rise in the levels of active RAB11. One interesting difference is the impaired interaction between TRAPPC2L and TRAPPC6a in the presence of the p.(Ala2Gly) variant compared to the previously-reported p.(Asp37Tyr) variant. It has previously been suggested that TRAPPC2L binds to the complex close to the TRAPPC3-C6 interface.14–16 The data we now present is consistent with such a model and further delineates the region of TRAPPC6a that is important for this interaction. There are presently no homozygous or compound heterozygous TRAPPC6a variants in affected individuals that fall within the region of the protein (residues 84–159) that we now show to be important for its interaction with TRAPPC2L except for one study which identified an individual with a homozygous p.(Tyr107Asn) variant.8 That study also identified four other homozygous variants in the affected individual, making comparison to the affected individuals in the present study complicated.
Unlike TRAPPC2L p.(Ala2Gly), the p.(Asp37Tyr) variant did not affect the interactions with TRAPPC6a/TRAPPC12 subunits in the yeast two-hybrid assay,1 but rather disrupted the interaction with TRAPPC10, a TRAPP II-specific subunit. Furthermore, the p.(Ala2Gly) variant resulted in a modest increase in the levels of active RAB11 in fibroblasts from an affected individual, whereas the levels increased substantially for the p.(Asp37Tyr) variant as was shown previously.1 RAB11A GTPase is a member of the RAS oncogene superfamily, which cycles between an inactive GDP-bound form and an active GTP-bound form and regulates intracellular membrane trafficking. The active form recruits downstream effectors to membranes and is directly responsible for vesicle formation, movement, tethering and fusion. These data along with the yeast two hybrid assay, suggest that the two TRAPPC2L missense variants may result in slightly different cellular effects on membrane trafficking. It is noteworthy that both TRAPP II and III have been reported to have nucleotide exchange factor activity towards RAB11 and RAB1 in Drosophila.24 Thus, it is possible that the levels of active RAB1 are also affected in individuals with TRAPPC2L variants.
The lack of an effect on autophagic flux was surprising given the shift in TRAPPC12 seen by size exclusion chromatography, which indicates a change in TRAPP III. The subunits of this complex, including TRAPPC11 and TRAPPC12, have been implicated in autophagy.21 While a role for autophagy in other TRAPP gene mutations has not been reported, we cannot rule out a subtle defect in autophagy as being involved in the presently reported individuals, perhaps consistent with a relatively mild phenotype.
The core clinical findings of individuals with pathogenic TRAPPC2L variants show significant overlap with individuals carrying pathogenic variants in other TRAPP subunit genes (TRAPPC6A, TRAPPC6B, TRAPPC9, TRAPPC11) (reviewed in Sacher et. al, 2019)2; the majority of these individuals have neurodevelopmental delay with severe speech difficulties, microcephaly, seizures, and extrapyramidal symptoms like dystonia.3–8 A possible neurodegenerative course was also reported for some individuals with TRAPPC6B variants, and zebrafish models were consistent with the microcephaly and neuronal hyperexcitability seen in humans.5
Although a larger cohort of TRAPPC2L mutations is required to draw conclusions regarding genotype-phenotype correlations, TRAPPC2, TRAPPC11 and TRAPPC9 cohorts are the largest groups for which such correlations may be drawn. TRAPPC2 mutations have been exclusively linked to a skeletal disorder (SEDT) to the exclusion of other TRAPP proteins.2 Mutations include missense, nonsense and splicing variants. To date, no other features have been reported in such individuals including those seen in other TRAPPopathies (e.g. neurodevelopmental, microcephaly, muscular). TRAPPC9 mutations all share common features which include non-syndromic autosomal recessive intellectual disability (NS-ARID) and microcephaly.2 Some phenotypic features have been suggested to be due to impaired NF-κB signaling as TRAPPC9 was identified as a regulator of NIK, a kinase that functions in this pathway.25 Mutations in TRAPPC11 result in a spectrum of phenotypes including muscular dystrophy (LGMD2S or congenital muscular dystrophy), intellectual deficit, ocular and liver involvement as well as cerebral atrophy.2 Although this protein has been implicated in a number of different cellular processes including membrane traffic,26 protein glycosylation,27 endocytosis,28 and autophagy,21 it is unclear which impaired function results in which phenotype.
Collectively, these data suggest that the TRAPPC2L p.(Ala2Gly) variant leads to reduced interaction with the core subunit TRAPPC6a, affects membrane trafficking and support the pathogenicity of the p.(Ala2Gly) variant.
Supplementary Material
Figure S1.
A thermal denaturation curve (A) and tryptophan fluorescence emission (B) was performed for His-tagged recombinant TRAPPC2L or the two variants.
{kind=link}
Acknowledgements:
We thank the family for their contribution. This work was supported by funding from the JPB Foundation, SFARI to WKC, the Center for Mendelian Genomics (NHGRI HG009141), and by the Canadian Institutes of Health Research and the Natural Sciences and Engineering Research Council of Canada to MS. Sequencing and analysis were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 and in part by National Human Genome Research Institute grant R01 HG009141.
Footnotes
Conflict of interest: The authors declare they have no competing interests.
REFERENCES
- 1.Milev MP, Graziano C, Karall D, Kuper WFE, Al-Deri N, Cordelli DM, Haack TB, Danhauser K, Iuso A, Palombo F, Pippucci T, Prokisch H, Saint-Dic D, Seri M, Stanga D, Cenacchi G, van Gassen KLI, Zschocke J, Fauth C, Mayr JA, Sacher M, van Hasselt PM. Bi-allelic mutations in TRAPPC2L result in a neurodevelopmental disorder and have an impact on RAB11 in fibroblasts. J Med Genet 2018;55:753–764. [DOI] [PubMed] [Google Scholar]
- 2.Sacher M, Shahrzad N, Kamel H, Milev MP. TRAPPopathies: An emerging set of disorders linked to variations in the genes encoding transport protein particle (TRAPP)-associated proteins. Traffic 2019;20:5–26. [DOI] [PubMed] [Google Scholar]
- 3.Bögershausen N, Shahrzad N, Chong JX, von Kleist-Retzow J-C, Stanga D, Li Y, Bernier FP, Loucks CM, Wirth R, Puffenberger EG, Hegele RA, Schreml J, Lapointe G, Keupp K, Brett CL, Anderson R, Hahn A, Innes AM, Suchowersky O, Mets MB, Nürnberg G, McLeod DR, Thiele H, Waggoner D, Altmüller J, Boycott KM, Schoser B, Nürnberg P, Ober C, Heller R, Parboosingh JS, Wollnik B, Sacher M, Lamont RE. Recessive TRAPPC11 mutations cause a disease spectrum of limb girdle muscular dystrophy and myopathy with movement disorder and intellectual disability. Am J Hum Genet 2013;93:181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gedeon AK, Colley A, Jamieson R, Thompson EM, Rogers J, Sillence D, Tiller GE, Mulley JC, Gécz J. Identification of the gene (SEDL) causing X-linked spondyloepiphyseal dysplasia tarda. Nat Genet 1999;22:400–404. [DOI] [PubMed] [Google Scholar]
- 5.Marin-Valencia I, Novarino G, Johansen A, Rosti B, Issa MY, Musaev D, Bhat G, Scott E, Silhavy JL, Stanley V, Rosti RO, Gleeson JW, Imam FB, Zaki MS, Gleeson JG. A homozygous founder mutation in TRAPPC6B associates with a neurodevelopmental disorder characterised by microcephaly, epilepsy and autistic features. J Med Genet 2018;55:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mir A, Kaufman L, Noor A, Motazacker MM, Jamil T, Azam M, Kahrizi K, Rafiq MA, Weksberg R, Nasr T, Naeem F, Tzschach A, Kuss AW, Ishak GE, Doherty D, Ropers HH, Barkovich AJ, Najmabadi H, Ayub M, Vincent JB. Identification of mutations in TRAPPC9, which encodes the NIK- and IKK-beta-binding protein, in nonsyndromic autosomal-recessive mental retardation. Am J Hum Genet 2009;85:909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mochida GH, Mahajnah M, Hill AD, Basel-Vanagaite L, Gleason D, Hill RS, Bodell A, Crosier M, Straussberg R, Walsh CA. A truncating mutation of TRAPPC9 is associated with autosomal-recessive intellectual disability and postnatal microcephaly. Am J Hum Genet 2009;85:897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohamoud HS, Ahmed S, Jelani M, Alrayes N, Childs K, Vadgama N, Almramhi MM, Al-Aama JY, Goodbourn S, Nasir J. A missense mutation in TRAPPC6A leads to build-up of the protein, in patients with a neurodevelopmental syndrome and dysmorphic features. Sci Rep 2018;8:2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okur V, LeDuc CA, Guzman E, Valivullah ZM, Anyane-Yeboa K, Chung WK. Homozygous noncanonical splice variant in LSM1 in two siblings with multiple congenital anomalies and global developmental delay. Cold Spring Harb Mol Case Stud 2019;5. doi: 10.1101/mcs.a004101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, The Genome Aggregation Database Consortium, Neale BM, Daly MJ, MacArthur DG. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv Published Online First:28January2019. doi: 10.1101/531210 [DOI]
- 11.Narasimhan V, Danecek P, Scally A, Xue Y, Tyler-Smith C, Durbin R. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 2016;32:1749–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poplin R, Chang P-C, Alexander D, Schwartz S, Colthurst T, Ku A, Newburger D, Dijamco J, Nguyen N, Afshar PT, Gross SS, Dorfman L, McLean CY, DePristo MA. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol 2018;36:983–987. [DOI] [PubMed] [Google Scholar]
- 13.Guide to yeast genetics and molecular biology. Meth Enzymol 1991;194:1–863. [PubMed] [Google Scholar]
- 14.Scrivens PJ, Shahrzad N, Moores A, Morin A, Brunet S, Sacher M. TRAPPC2L is a novel, highly conserved TRAPP-interacting protein. Traffic 2009;10:724–736. [DOI] [PubMed] [Google Scholar]
- 15.Duarte DT, Hul S, Sacher M. A yeast two hybrid screen identifies SPATA4 as a TRAPP interactor. FEBS Lett 2011;585:2676–2681. [DOI] [PubMed] [Google Scholar]
- 16.Wang C, Gohlke U, Roske Y, Heinemann U. Crystal structure of the yeast TRAPP-associated protein Tca17. FEBS J 2014;281:4195–4206. [DOI] [PubMed] [Google Scholar]
- 17.Kümmel D, Müller JJ, Roske Y, Henke N, Heinemann U. Structure of the Bet3-Tpc6B core of TRAPP: two Tpc6 paralogs form trimeric complexes with Bet3 and Mum2. J Mol Biol 2006;361:22–32. [DOI] [PubMed] [Google Scholar]
- 18.Kim M-S, Yi M-J, Lee K-H, Wagner J, Munger C, Kim Y-G, Whiteway M, Cygler M, Oh B-H, Sacher M. Biochemical and crystallographic studies reveal a specific interaction between TRAPP subunits Trs33p and Bet3p. Traffic 2005;6:1183–1195. [DOI] [PubMed] [Google Scholar]
- 19.Kim JJ, Lipatova Z, Segev N. TRAPP complexes in secretion and autophagy. Front Cell Dev Biol 2016;4:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boncompain G, Divoux S, Gareil N, de Forges H, Lescure A, Latreche L, Mercanti V, Jollivet F, Raposo G, Perez F. Synchronization of secretory protein traffic in populations of cells. Nat Methods 2012;9:493–498. [DOI] [PubMed] [Google Scholar]
- 21.Stanga D, Zhao Q, Milev MP, Saint-Dic D, Jimenez-Mallebrera C, Sacher M. TRAPPC11 functions in autophagy by recruiting ATG2B-WIPI4/WDR45 to preautophagosomal membranes. Traffic 2019;20:325–345. [DOI] [PubMed] [Google Scholar]
- 22.Jones S, Newman C, Liu F, Segev N. The TRAPP complex is a nucleotide exchanger for Ypt1 and Ypt31/32. Mol Biol Cell 2000;11:4403–4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas LL, Fromme JC. GTPase cross talk regulates TRAPPII activation of Rab11 homologues during vesicle biogenesis. J Cell Biol 2016;215:499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riedel F, Galindo A, Muschalik N, Munro S. The two TRAPP complexes of metazoans have distinct roles and act on different Rab GTPases. J Cell Biol 2018;217:601–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu W-H, Pendergast JS, Mo X-M, Brambilla R, Bracchi-Ricard V, Li F, Walters WM, Blits B, He L, Schaal SM, Bethea JR. NIBP, a novel NIK and IKK(beta)-binding protein that enhances NF-(kappa)B activation. J Biol Chem 2005;280:29233–29241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scrivens PJ, Noueihed B, Shahrzad N, Hul S, Brunet S, Sacher M. C4orf41 and TTC-15 are mammalian TRAPP components with a role at an early stage in ER-to-Golgi trafficking. Mol Biol Cell 2011;22:2083–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeRossi C, Vacaru A, Rafiq R, Cinaroglu A, Imrie D, Nayar S, Baryshnikova A, Milev MP, Stanga D, Kadakia D, Gao N, Chu J, Freeze HH, Lehrman MA, Sacher M, Sadler KC. trappc11 is required for protein glycosylation in zebrafish and humans. Mol Biol Cell 2016;27:1220–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bassik MC, Kampmann M, Lebbink RJ, Wang S, Hein MY, Poser I, Weibezahn J, Horlbeck MA, Chen S, Mann M, Hyman AA, Leproust EM, McManus MT, Weissman JS. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell 2013;152:909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
A thermal denaturation curve (A) and tryptophan fluorescence emission (B) was performed for His-tagged recombinant TRAPPC2L or the two variants.