

Abstract
Fibroblast growth factor 1 (FGF-1) is the first FGF family member, and it induces proliferation of fibroblasts and other types of the cells. However, recent studies are uncovering unexpected functions of this molecule. Our previous study redefined this growth factor as a catabolic molecule produced in cartilage upon metabolic insult. Indeed, FGF-1 was found to repress the gene expression of cellular communication network factor 2 (CCN2), which protects and regenerates cartilage, amplifying its own production through positive feedback regulation. In the present study, we investigated the molecular mechanism of this bipartite CCN2 repression and FGF1 activation by FGF-1 in chondrocytes. Repression of CCN2 and induction of FGF1 in human chondrocytic cells were both partly abolished by valproic acid, an inhibitor of histone deacetylase 1 (HDAC1), indicating the involvement of chromatin remodeling by histone acetylation in this system. In contrast, RNA degradation analysis suggested no contribution of post-transcriptional regulation of the mRNA stability to the effects conferred by FGF-1. Suspecting a regulation by a specific transcription factor, we next sought a candidate in silico from a large dataset. As a result, we found fork head box protein A1 (FOXA1) as the transcription factor that bound to both CCN2 and FGF1 loci. Functional analysis demonstrated that FOXA1 silencing significantly attenuated the CCN2 repression and FGF1 induction caused by FGF1. These findings collectively indicate that the bipartite regulation by FGF-1 is enabled by the combination of chromatin remodeling by HDACs and transcriptional modulation by FOXA1 with unknown transcriptional coactivators of opposite functionalities.
Supplementary Information
The online version of this article (10.1007/s12079-020-00600-4) contains supplementary material, which is available to authorized users.
Keywords: FGF-1, CCN2, Osteoarthritis, Chondrocytes, Cartilage
Introduction
Fibroblast growth factor 1 (FGF-1), which was initially named acidic FGF, is the first member of the FGF family, and it comprises at least 22 members in humans (Ornitz and Itoh 2015). As the name suggests, this protein was originally discovered as an extracellular messenger that promotes the proliferation of various types of the cells, including fibroblasts. However, later studies revealed the multiple functionalities of FGF-1, some of which were unexpected based on the original name. Indeed, FGF-1 is known to play significant roles in embryonic development, wound healing, neurogenesis and angiogenesis (Ornitz and Itoh 2015). Unexpectedly, genetic removal of Fgf1 from mice revealed no apparent deficiency in development, probably due to the compensatory actions of other family members; FGF-2, in particular. Interestingly, although Fgf1-null mice showed no apparent phenotypic changes under regular conditions, these mice were found to develop hyperglycemia and insulin resistance upon high fat diet challenge (Suh et al. 2014). Subsequent studies uncovered that this growth factor acts as a metabolic hormone, which normalizes hyperglycemia by improving central glucose sensing and peripheral glucose uptake (Gasser et al. 2017). As such, FGF-1 is now believed to be involved in a variety of physiological and pathological events in human body.
From a pathological point of view, it is widely known that activating mutations in FGF receptor (FGFR) genes cause several skeletal disorders, such as achondroplasia, in humans (Ornitz and Marie 2015). Therefore, FGFR ligands are indicated to play significant roles in skeletal development and diseases. In the case of FGF-1, early studies suggested an active role of this protein in bone fracture repair (Wang et al. 2019). In this context, our recent study uncovered a novel role of FGF-1 in articular cartilage. Namely, we found that FGF-1 was produced in the articular cartilage in a rat experimental osteoarthritis (OA) model induced by a glycolysis inhibitor, monoiodoacetatic acid (El-Seoudi et al. 2017). OA is a common locomotive disorder caused by degenerative changes in synovial joints, particularly in the articular cartilage (van der Kraan and van den Berg 2012). This cartilage degeneration is usually incurred by continual mechanical overload onto the joints; thus, OA is most frequently observed weight-bearing knee and hip joints. Since OA in these joints severely impairs the quality of life of the patients, OA treatment is a critical medical issue, especially in advanced countries with a number of aged OA patients.
Regarding the pathological role of FGF-1 in OA (Fig. 1), we clarified in a previous study that FGF-1 induced the expression of its own gene and matrix metalloproteinase (MMP) -13, which destroyed cartilaginous extracellular matrix (ECM) and repressed the gene expression of cellular communication factor 2 (CCN2), as well as aggrecan and type II collagen, which are major components of the ECM (El-Seoudi et al. 2017). Among these target genes, FGF1 and CCN2 are of particular interest; FGF-1 autoactivates FGF1 to amplify its catabolic actions on cartilage, whereas CCN2 conducts regeneration of damaged articular cartilage in vitro and in vivo (Nishida et al. 2004; Abd El Kader et al. 2014), supporting energy metabolism (Maeda-Uematsu et al. 2014: Kubota et al. 2015, Akashi et al. 2020). CCN2 is the best-investigated member of the CCN family of six proteins (Perbal 2018) and is known to orchestrate extracellular signaling network via multiple interactions with growth factors (Khattab et al. 2015), cell surface receptors (Lau 2016), and ECM proteins (Kubota and Takigawa 2015). It should be noted that CCN2 overexpressed in the articular cartilage counteracts the development of age-related OA in mice (Itoh et al. 2013). Therefore, the positive and negative regulatory systems of FGF1 and CCN2, respectively, are the central machinery for FGF-1 to exert its catabolic mission in cartilage (Fig. 1). Nevertheless, how CCN2 and FGF1 are regulated by FGF-1 is still largely unknown. In this study, we investigated the molecular mechanism of the regulation of CCN2 and FGF1 by FGF-1 in chondrocytes and clarified that the same chromatin remodeler and DNA binding transcription factor are mediating the bipartite regulation of these two target genes, in collaboration with unknown transcription coactivators.
Fig. 1.
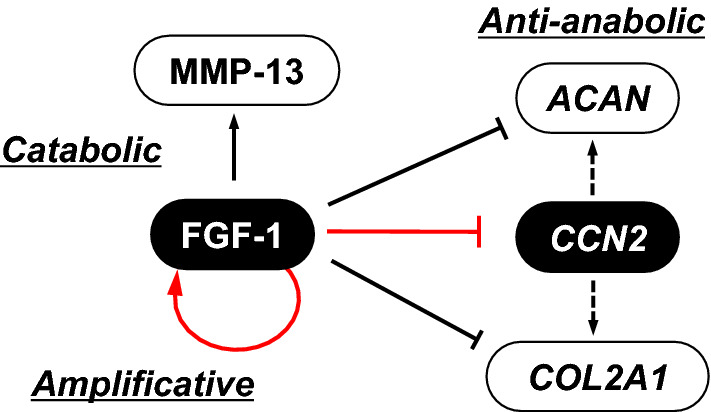
Summary of catabolic effects of fibroblast growth factor (FGF)-1 on chondrocytes. FGF-1 is involved in osteoarthritis (OA); in which, once FGF-1 is produced, the production is amplified through the auto-induction feedback loop of FGF1 expression, and activates the matrix metalloproteinase (MMP) -13 gene, which destroys the cartilaginous extracellular matrix (ECM). This could be restored by the supplemental production of cartilaginous ECM components including type II collagen (COL2A1 product) and aggrecan (ACAN product) as well as cellular communication network factor (CCN)2 by articular chondrocytes, which are strongly restrained by FGF-1 at the gene expression level. ECM components and degrading enzyme are shown as white objects, whereas extracellular signaling molecules are in black. Central regulatory actions investigated in this study are described in red
Materials and methods
Cell culture
A human chondrocytic cell line HCS-2/8 that originates from a human chondrosarcoma and retains the chondrocytic phenotype (Takigawa et al. 1989; Akashi et al. 2018) was employed as an in vitro model of human chondrocytes (Maeda-Uematsu et al. 2014; El-Seoudi et al. 2017). These cells were grown at 37 °C in a humidified atmosphere containing 5% CO2 with Dulbecco’s modified Eagle’s medium (D-MEM) supplemented with 10% fetal bovine serum (FBS).
Evaluation of the effects of valproic acid
Valproic acid (VPA), which is an inhibitor of class I and II histone deacetylases (HDACs), was purchased from Cayman Chemical (Ann Arbor, MI, USA). Before the treatment, HCS-2/8 cells were cultured in D-MEM with 10% FBS and allowed to reach confluence. In order to estimate the cytotoxicity, confluent HCS-2/8 cells in a 96-well multiplate were treated with 0, 0.125, 0.25, 0.5, 1, or 2 µM of VPA for 24 h, and their metabolic activities were evaluated after 2 h by Cell Countng Kit-8 (Dojindo, Kumamoto, Japan). For the evaluation of the effects of FGF-1 in the presence of 0, 0.125, or 0.25 µM of VPA, the cells were prepared in 6-well multiplates. In the beginning, the medium was replaced with DMEM containing 0.5% FBS together with either concentration of VPA and the cells were then incubated for 12 h. Subsequently, FGF-1 was added at a concentration of 25 ng/ml, and the incubation was continued for an additional 12 h. Thereafter, total RNA was harvested and purified as described in the next subsection.
RNA extraction and quantitative real-time reverse transcription polymerase chain reaction (RT-PCR)
Total RNA extraction and purification from the cells were performed by using Isogen (Nippongene, Tokyo, Japan) or an RNeasy kit (Qiagen, Hilden, Germany) as instructed by the manufacturers. Each RNA sample (500 ng) was reverse transcribed by avian myeloblastosis virus (AMV) reverse transcriptase (Takara, Otsu, Japan) for 30 min at 42 °C, as indicated in the manufacturer’s instructions. Quantitative real-time PCR was performed with a StepOnePlusTM Real-time PCR System (Applied Biosystems, Basel, Switzerland) by using TOYOBO SYBR Green PCR Master Mix (TOYOBO, Osaka, Japan).
The nucleotide sequences of the DNA primers used for amplification were: 5′- GCA GGC TAG AGA AGC AGA GC -3′ (sense) and 5′-ATG TCT TCA TGC TGG TGC AG -3′ (antisense) for human CCN2; 5’-ACA AGG GAC AGG AGC GAC−3’ (sense) and 5’-TCC AGC CTT TCC AGG AAC A -3’ (antisense) for human FGF1; 5’-CTA CTA CGC AGA CAC GCA GG-3’ (sense) and 5’-CCG CTC GTA GTC ATG GTG TT-3’ for human FOXA1; and 5’- GCC AAA AGG GTC ATC ATC TC -3’ (sense) and 5’- GTC TTC TGG GTG GCA GTG AT -3’ (antisense) for human GAPDH.
RNA degradation analysis
The RNA degradation profile was analyzed as previously described (Kubota et al. 2003). Briefly, HCS-2/8 at a density of 4 × 104 cells/cm2 were seeded into 6-well cell culture plates and were maintained in DMEM containing 10% FBS until the cells reached confluence. Next, the medium was changed to DMEM containing 0.5% FBS, and the cells were then incubated for 12 h. Subsequently, 10 µg/ml of actinomycin D was added to arrest de novo mRNA synthesis, in the presence of 25 ng/ml of recombinant FGF-1. At the same time, total RNA was extracted from cultures without the treatment as 0 h controls. One, two and four hours after the initiation of the treatment, total RNA was isolated as described in the last subsection. RNA degradation profiles were analyzed by quantifying the remaining mRNAs of CCN2 and FGF-1 after the addition of actinomycin D, in comparison with those of 0 h control samples. Four independent cultures were prepared for each experimental condition.
Western blotting analysis
Protein analysis by Western blotting was performed essentially as described in our previous report (Sumiyoshi et al. 2013). HCS-2/8 cells in 6-well cell culture plates were treated with 0, 25, or 50 ng/ml of FGF-1, or the siRNA cocktail against FOXA1, as described in other subsections. Protein concentrations of cell lysates were determined by Pierce™ BCA Protein Assay using bovine serum albumin (BSA) as standards (Thermo Scientific, Rockford, IL, USA). Cell lysate containing equal amounts of total proteins was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and separated proteins were transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). A semi-dry transfer apparatus (Atto Corp., Tokyo, Japan) was used for protein transfer. The membranes were incubated in an anti-FOXA1 (Cat#53,528: Cell Signaling Technology, Danvers, MA, USA) and anti-β-actin (Sigma-Aldrich; Cat# A2228) antibodies overnight at 4 °C. After extensive wash with Tris-buffered saline (TBS) buffers, the blots were incubated for 60 min at room temperature in horseradish peroxidase (HRP)-conjugated secondary antibodies. Thereafter, the membranes were washed again, and the bands were visualized with a chemiluminescence substrate. Images were captured and processed by an image analyzer (LAS-4000 mini, Fuji Film, Tokyo, Japan).
Reporter gene assay
HCS-2/8 cells were seeded into 24-well plates, maintained in DMEM containing 10% FBS, and allowed to reach subconfluence. Then, the medium was changed to DMEM with 1% FBS prior to the addition of FGF-1 at 25 ng/ml and/or plasmid DNA. A reporter gene construct with SV40 promoter-driven firefly luciferase gene containing the entire human CCN2 3’-UTR downstream, pGL3-UTRS, was used to evaluate the post-transcriptional regulation mediated by the 3’-UTR (Kubota et al. 1999). As a negative control, the parental plasmid lacking a promoter was also employed. To standardize the transfection efficiency, the herpes simplex virus TK promoter-driven Renilla luciferase construct, pRL-TK, was simultaneously introduced. The cells were transfected with the DNA in an optimized concentration of TransIT-IKO transfection reagent (Mirus Bio, Madison, WI, USA). After addition of FGF-1 and/or plasmid DNA, the medium was changed every 12 h with concurrent addition of FGF-1; incubation was continued for 48 h. Then, the cell lysate was extracted with Passive Lysis Buffer (Promega, Fitchburg, WI, USA) following the protocol provided by the manufacturer. Thereafter, the firefly and Renilla luciferase activities were measured with the Dual Luciferase system (Promega) by a luminometer (Fluroskan Ascent FL, Labsystems, Helsinki, Finland). Transfection experiments were performed with 3 independent sets of the samples, each in quadruplicate, on 3 different occasions.
Dataset analysis
The datasets of chromatin immunoprecipitation-sequencing (ChIP-seq) experiments targeting H3K4me2 (ENCSR000AMC) and transcription factors (ENCSR000BLE and ENCSR000BMO) in HepG2 cells were downloaded from the ENCODE portal (https://www.encodeproject.org/) (Sloan et al. 2016). The data was analyzed and visualized by the University of California Santa Cruz (UCSC) Genome Browser (http://genome.ucsc.edu).
Gene silencing
In order to silence FOXA1 via RNAi, we purchased a small interfering RNA (siRNA) cocktail containing 3 distinct synthetic siRNAs targeted to human FOXA1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). A non-targeting control siRNA was also obtained from Santa Cruz Biotechnology. HCS-2/8 cells were seeded into 6-well plates and were grown until the cells reach subconflucence. The cells were then transfected with 10 nM of each siRNA using the Lipofectamine® RNAiMAX Reagent (Thermo Fisher Scientific, Carlsbad, CA), according to the protocol supplied by the manufacturer, and the cells were then incubated for 36 h. Next, the medium was changed to DMEM containing 0.5% FBS together with 25 ng/ml of recombinant FGF-1 and the incubation was continued for an additional 12 h. After 48 h of gene silencing, the total cellular RNA was extracted for further analyses.
Statistical analysis
Unless otherwise specified, all of the evaluations were performed at least twice, yielding comparable results. Comparisons between two experimental groups were performed by using Student’s t-test. Statistical comparison among three groups were performed by Fisher’s least significant difference test.
Results
Effects of VPA, a HDAC1 inhibitor, on the regulation of CCN2 and FGF1 by FGF-1
In our previous study, we showed that CCN2 and FGF1 mRNA levels were drastically decreased and increased upon FGF-1 stimulation, respectively (El-Seoudi et al. 2017). As a first step to clarify the molecular mechanism of this bipartite regulation, we initially examined the involvement of chromatin remodeling therein, using an HDAC inhibitor, VPA. First, we estimated the cytotoxicity of this compound to HCS-2/8 cells and found that 0.125 µg/ml of VPA had no significant effect on the metabolic activity of these cells, whereas 0.25 µg/ml VPA showed modest reduction (< 15%) therein (Fig. S1). Therefore, we decided to treat HCS-2/8 cells with these two doses.
As clearly observed in Fig. 2a, FGF-1 dramatically repressed the steady state mRNA level of CCN2 in the absence of VPA, recapitulating our previous finding. However, in the presence of increasing concentrations of VPA, CCN2 expression without FGF-1 stimulation was strongly inhibited, suggesting that chromatin loosening by histone acetylation led to CCN2 silencing. Interestingly, VPA contrarily increased the CCN2 mRNA level repressed by FGF-1. Collectively, CCN2 repression by FGF-1 was significantly abolished by VPA in a dose-dependent manner, as clearly represented by the data in Fig. 2b
Fig. 2.

Effects of valproic acid (VPA), an histone deacetylase (HDAC) 1 inhibitor, on the regulation of CCN2 and FGF1 in chondrocytic HCS-2/8 cells. a CCN2 (left panel) and FGF1 (right panel) mRNA levels with (gray columns) or without (black columns) 25 ng/ml of FGF-1 in the presence of indicated concentrations of VPA. Relative gene expression levels versus the glyceraldehyde 3-phosphate dehydrogenase gene (GAPDH) are presented. Asterisks (*) and (**) denote a significant difference at p < 0.05 and 0.01, respectively, between the two groups. b Fold repression of CCN2 expression (left panel) and enhancement of FGF1 expression (right panel) in the presence of VPA of indicated concentrations. Data represent mean values from 3 independent cell cultures with error bars representing standard deviations. Asterisks (*), (**) and (***) denote significant differences at p < 0.05, 0.01 and 0.001, respectively, against the control
In contrast, VPA did not show a prominent effect on FGF1 mRNA levels in the absence or presence of FGF-1 at the lower concentration. However, the higher concentration of VPA markedly attenuated the autoinduction of FGF1, while the basal level of FGF1 expression without exogenous FGF-1 was unchanged (Fig. 2a). As such, autoinduction of FGF1 was also repressed significantly by 0.25 µg/ml of VPA (Fig. 2b). These results indicate that chromatin remodeling is involved in both CCN2 repression and FGF1 induction by FGF-1 and that both events depend on HDAC activity.
Evaluation of possible post-transcriptional regulation of CCN2 and FGF1 mRNA by FGF-1
Steady-state mRNA levels are determined under the balance of nascent transcription and mRNA degradation. Therefore, the observed decrease in CCN2 mRNA and increase in FGF1 mRNA might result from accelerated CCN2 mRNA degradation and FGF1 mRNA stabilization, respectively. To examine these possibilities, we arrested nascent mRNA synthesis by actinomycin D and determined the fate of CCN2 and FGF1 mRNAs in HCS-2/8 cells in the presence or absence of exogenous FGF-1. In our previous report, we found relatively rapid degradation of CCN2 mRNA in the same cells under regular conditions (Kubota et al. 2003), which was also confirmed in this study (Fig. 3a). Surprisingly, in the presence of FGF-1, CCN2 mRNA was found to be stabilized. Indeed, no appreciable CCN2 mRNA decay was observed 4 h after transcription, which counteracted the resultant decrease in the steady-state mRNA level caused by FGF-1 (Fig. 2a). Also, to our surprise, FGF-1 mRNA was found to be unusually stable in HCS-2/8 cells, regardless of FGF-1 treatment. These data indicate that CCN2 and FGF1 mRNAs are not dominantly regulated by FGF-1 at a post-transcriptional stage.
Fig. 3.

Post-transcriptional effects of FGF-1 on CCN2 and FGF1 mRNAs. a RNA degradation profile of CCN2 (left panel) and FGF1 (right panel) mRNAs in the absence (dotted lines) or presence (solid lines) of 25 ng/ml FGF-1. Relative levels of remaining mRNAs after the indicated hours of actinomycin D treatment are plotted with error bars of standard deviations. Data were computed from the results obtained with 8 independent cultures for each condition. Asterisks (**), (***) and (****) indicate significant difference at p < 0.01, p < 0.001 and p = 0.0001, respectively, between the two groups at the same time point. b Structure of the plasmid reporting the post-transcriptional regulatory functions of CCN2 3’-UTR. This plasmid, pGL3-UTRS (shown at the top), produces firefly luciferase (F luc) mRNA with the entire 3’-UTR of CCN2 mRNA (at the bottom). SV40p and pA represent the SV40 promoter and polyadenylation signal with a t-splice site, respectively. The cis-repressive elements involved in the CCN2 3’-UTR are also noted. c Expression of the firefly luciferase gene with the CCN2 3’-UTR in the absence or presence of exogenous FGF-1. NC represents the control experiments with a vector containing no promoter (pGL3∆P). Data are shown as firefly luciferase activities standardized by the internal control (Renilla luciferase activities from pRL-TK). Each value represents the average and standard deviation of the data obtained from 3 experiments performed on separate occasions, in which 3 independent sets of samples with 4 quadruplicate cultures were evaluated. Asterisks (*) denote significant differences at p < 0.05 between the two groups, FGF-1(−) and FGF-1(+)
It is generally recognized that stability of mRNA is usually determined by the cis-elements built in their 3’-UTR. In fact, the 3’-UTR of human CCN2 mRNA contains a number of post-transcriptional repressive elements, including a cis-acting element for structure-anchored repression (CAESAR) (Kubota et al. 2000; Leask and Abraham 2006) and various miRNA targets (Ohgawara et al. 2009; Kubota and Takigawa 2015). Thus, we evaluated the possible contribution of these cis-repressive elements to the CCN2 repression by FGF-1 with a reporter plasmid (Fig. 3b) that expresses a firefly luciferase mRNA with the 3’-UTR of human CCN2. The reporter gene assay showed, however, that this CCN2 3’-UTR containing luciferase gene did not respond to FGF-1 (Fig. 3c), indicating that the UTR-mediated post-transcriptional regulation did not contribute to the observed repression of CCN2 expression by FGF-1.
Identification of FOXA1 as an FGF-1 inducible transcription factor that binds to both CCN2 and FGF1 loci
The results shown in Figs. 2 and 3 suggest that the effects of FGF-1 on CCN2 and FGF1 are the outcomes of collaborative transcriptional regulation of chromatin remodelers and transcription factors that recognize CCN2 and FGF1 loci. As a next step, we tried to find a transcription factor candidate which actually binds to these loci. For this objective, we analyzed datasets deposited in the ENCODE portal site. By utilizing chromatin immunoprecipitation (ChIP) sequencing datasets, histone modifications representing chromatin status and transcription factors binding profiles of any locus in particular cell(s) can be analyzed. Unfortunately, no such data in chondrocytes were available to date. Instead, we analyzed data from another human cell line, in which both CCN2 and FGF1 loci are active in transcription, as observed in HCS-2/8 cells. As illustrated in Fig. 4a, these loci are characterized by histone H3 di-methylation on Lys 4 (H3K4me2), representing euchromatin that is open for transcription. Therefore, in these cells, a number of transcription factors are accessible to these loci, and thus transcription factor–ChIP datasets indicated the binding of a number of different transcription factors therein. Among them, we found FOXA1 bound to both loci, FGF1 and CCN2 (Fig. 4a, b). Most interestingly, this transcription factor binds to 3 distinct areas within the 5 kb-long proximal promoter area of human CCN2. In our previous study, we evaluated the FGF1 responsiveness of a CCN2 proximal promoter of < 1 kb in length (El-Seoudi et al. 2017), in which only one FOXA1 binding site was involved. Consistent with its molecular structure, this short proximal promoter fragment showed a significant, but modest, response to FGF-1. Next, we evaluated the effect of exogenous FGF-1 on FOXA1 expression. As observed in Fig. 4c, the gene expression and production of FOXA1 was enhanced by FGF-1 stimulation in HCS-2/8. Therefore, we chose FOXA1 as a candidate that might regulate both CCN2 and FGF1 in an opposite manner upon FGF-1 stimulation, and forwarded this candidate to subsequent analysis.
Fig. 4.

Binding of FOXA1 to CCN2 and FGF1 loci and its induction by FGF-1. a, b Preferential binding of FOXA1 to the FGF1 (a) and CCN2 (b) loci. Analysis of the chromatin immunoprecipitation-sequencing (ChIP-seq) datasets in the ENCODE portal site revealed strong binding signals of FOXA1 to the regions characterized by H3K4me2 histone modification (shown in pink). Transcripts from the illustrated region are also summarized (shown in dark blue; boxes and lines represent exons and introns, respectively). c Induction of FOXA1 by exogenous FGF-1 in HCS-2/8 cells. Relative gene expression levels were computed against those of GAPDH. Mean values from 3 independent cell cultures are shown with error bars representing standard deviations. Asterisks (**) indicate a statistically significant difference at p < 0.01 between the two groups (left). Dose-dependent enhancement of FOXA1 protein production by FGF-1 was confirmed by Western blotting. Positions of molecular weight markers (in kDa) are indicated at the left. Signals of β-actin are also shown as an internal control (right)
FOXA1-dependence of the bipartite regulation of CCN2 and FGF1 by FGF-1
In order to clarify whether FOXA1 mediated the regulation of CCN2 and FGF1, we utilized an RNAi strategy to silence FOXA1 and evaluated its effects on each mRNA level modified by exogenous FGF-1. As shown in Fig. 5a, transfection of a synthetic siRNA cocktail into HCS-2/8 cells significantly silenced FOXA1 expression in those cells, compared to that with control siRNAs. Efficient silencing of FOXA1 production was also confirmed at a protein level (Fig. 5b). Under the same conditions, the cells were exposed to FGF-1 in the medium, and the CCN2 and FGF1 mRNA levels were compared. As expected, silencing of FOXA1 significantly de-repressed the CCN2 expression repressed by FGF-1, whereas it repressed the enhanced FGF1 expression (Fig. 5c). However, the effects of FOXA1 silencing were not striking, which is probably due to the limited level of FOXA1 silencing. This single transcription factor was shown to mediate the regulation of these two genes by FGF-1, at least in part, in chondrocytes.
Fig. 5.

Reversion of the effects of FGF-1 on CCN2 and FGF1 expression by FOXA1 silencing. a FOXA1 silencing by an siRNA cocktail. Relative FOXA1 mRNA levels in HCS-2/8 cells were quantified after the transfection with non-targeting control (siCtrl) or siRNAs against FOXA1 (siFOXA1). b Confirmation of FOXA1 silencing by Western blotting. Positions of molecular weight markers (in kDa) are indicated at the left. Signals of β-actin are also shown as an internal control. c Effects of FOXA1 silencing on the FGF-1-modulated expression of CCN2 (left panel) and FGF1 (right panel). All of the cells were treated with FGF-1 after the respective siRNA transfection. CCN2 expression was de-repressed, and FGF1 induction was suppressed by siFOXA1. Values were standardized against the mRNA levels of GAPDH and displayed with standard deviations (error bars). Asterisks (*) and (***) indicate statistically significant differences at p < 0.05 and p < 0.001, respectively, between the two groups
Discussion
In the present study, we investigated the molecular mechanism of the central regulatory system of FGF-1-mediated cartilage degeneration. Consequently, we found a specific transcription factor, FOXA1, mediates both CCN2 transcriptional repression and FGF1 induction by FGF-1, which is dependent on chromatin remodeling complexes, HDACs. Additionally, possible involvement of post-transcriptional regulation in these regulatory processes was experimentally addressed, and this possibility was ruled out.
HDACs are enzyme complexes that catalyze the deacetylation of acetylated lysine residues in histones (Khan and Haqqi 2018). Human HDACs consist of HDAC 1 to 11, classified as classes I, II, and III HDACs and sirtuin (SIRT) 1 to 7, classified as class III. By the removal of the acetyl groups by HDACs, histones redeem the positive charge on the lysine residues and DNA binding ability, which results in chromatin condensation. The requirement of HDAC I or HDAC II activity for the process of CCN2 and FGF1 regulation, which conducts the FGF-1-induced cartilage degeneration, is consistent with the fact that HDACs are critical factors in OA development. Indeed, HDACs 1, 2 and 7 were found upregulated in OA chondrocytes, and more interestingly, HDAC gene silencing or enzymatic inhibition could protect from the degeneration of cartilaginous ECM in vitro and showed protective effects in animal OA models in vivo (Khan and Haqqi 2018). Therefore, HDAC inhibitors are currently considered for potential OA therapeutics. In this context, our study adds a novel mechanistic view of the anti-OA actions of HDAC inhibitors through CCN2 and FGF1.
FOXA1 is a member of a huge transcription factor family with 50 members, which are classified into 19 subgroups, FOXA to FOXS (Lam et al. 2013). These members are structurally characterized by the retention of fork head DNA binding domains, and thus are believed to possess the ability to directly bind to DNA. The first subgroup of this family, FOXA, comprises three members, FOXA1, A2 and A3. These transcription factors regulate the temporo-spatial expression of a number of genes during development and at adult stages (Lam et al. 2013). In relation to cartilage biology, FOXA1 and A2 are produced in notochordal cells during vertebral development, regulating Shh expression, and thus contribute to intervertebral disc formation (Nakamichi and Asahara 2020). More importantly, FoxA members including FoxA1 were found to be critically involved in the regulation of chondrocyte hypertrophy in mice (Ionescu et al. 2012). This notion was further confirmed by a recent study with deer antler chondrocytes, which constructs and reconstructs the antlers of deer, revealed that FoxA1, A2 and A3 are all required for the chondrocytes to proceed to hypertrophy properly, under the regulation of desert hedgehog (Dhh) (Ma et al. 2020). Mechanistically, FOXA members are shown to compete with SOX9 that is an inhibitor of hypertrophy (Tan et al. 2018). It should be noted that hypertrophic changes are frequently observed in osteoarthritic articular chondrocytes (van der Kraan and van den Berg 2012). FGF-1, which induces FOXA1, is a mediator of osteoarthritic changes in articular cartilage in a rat model (El-Seoudi et al. 2017). Therefore, FOXA members could also play critical roles in the OA pathogenesis.
In this study, we elucidated the involvement of FOXA1 in the regulation of CCN2 and FGF1 by the signal evoked by FGF-1. However, no information is available on the cell surface receptors and intracellular signaling molecule that actually transmit the FGF signal to FOXA1. Since FGF receptor 3 is demonstrated on chondrocytes and emits signals to control the chondrocytic phenotype (Wang et al. 2019), it is the most plausible receptor that may accept FGF-1 and initiate a signaling cascade towards FOXA1. Subsequent studies in the future may clarify the signaling molecules involved in this regulation.
These FOXA members exert highly context-dependent effects on target gene expression. For example, FOXA1 upregulates CDKN1B encoding p27, while it downregulates SLUG in cancer cells (Lam et al. 2013). Similarly, FOXA1 may regulate CCN2 and FGF1 in opposite directions, depending upon the nano-environment around each locus. Such a bipartite regulation is supposed to be performed under the collaboration of transcriptional co-activators and co-repressors, which are conditionally recruited to the target loci (Fig. 6).
Fig. 6.
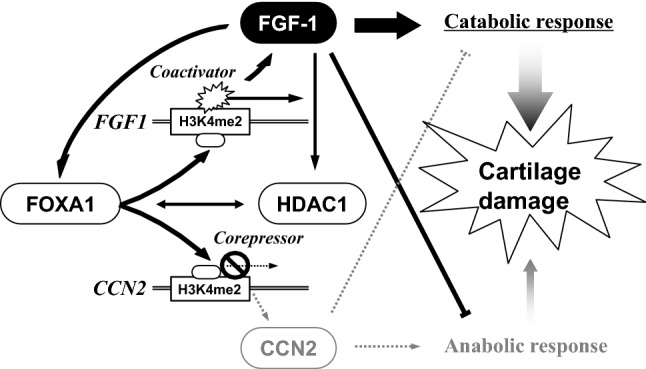
Schematic representation of the molecular mechanism of the bipartite regulation by FGF-1. Chromatin remodeling by histone methylation and acetylation, in which HDAC1 plays a significant role, is required for this regulation. FGF-1 induces FOXA1, which acts as a pioneer factor to open the chromatin structure with H3K4me2 regions in CCN2 and FGF1 loci. Thereafter, FOXA1 binds to the CCN2 silencer and FGF1 enhancer to facilitate the access of the coactivator and corepressor, respectively. In consequence, amplified FGF-1 signal incurs catabolic and anti-anabolic responses of chondrocytes, leading to OA development
FOXA1 is not only a DNA binding transcriptional factor, but also a pioneer factor that is able to actively modify chromatin structure. In fact, this factor induces H3K4 methylation and DNA demethylation to open the chromatin structure, allowing the access of other transcription factors represented by steroid hormone receptors (Lam et al. 2013). FOXA1 is likely opening the cis-elements in the H3K4me2-marked loci in CCN2 and FGF1 for co-activators and co-repressors, which are, however, still unidentified, and thus further investigation is needed to verify this hypothesis.
Considering the HDAC-dependence of the CCN2 and FGF1 regulation by FGF-1, it is suspected that the pioneer actions of FOXA1 are exerted under collaboration with HDACs. Consistent with this idea, HDAC7 was reported to form a complex with FOXA1, together with estrogen receptors, to repress Reprimo, which encodes a cell cycle inhibitor (Malik et al. 2010). If similar machinery is utilized for the regulation of CCN2 and FGF1, FOXA1 and HDAACs may synergistically work to yield maximal effects, which may account for the limited effect of partial FOXA1 silencing alone on the CCN2 and FGF1 expression in the presence of FGF-1. Alternatively, these findings indicate the utility of anti-FOXA1 molecules as a novel therapeutic tool for OA treatment in combination with HDAC inhibitors.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank Ms. Yoshiko Miyake for her secretarial assistance. This study was supported by JSPS KAKENHI Grant Numbers JP18J10073, JP19H03817, JP19K22716 and Research Grant from Koyanagi Foundation.
Compliance with ethical standards
Conflict of interest
The authors have no conflict of interests to declare.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Abd El Kader T, Kubota S, Nishida T, Hattori T, Aoyama E, Janune D, Hara ES, Ono M, Tabata Y, Kuboki T, Takigawa M. The regenerative effects of CCN2 independent modules on chondrocytes in vitro and osteoarthritis models in vivo. Bone. 2014;59:180–188. doi: 10.1016/j.bone.2013.11.010. [DOI] [PubMed] [Google Scholar]
- Akashi S, Nishida T, El-Seoudi A, Takigawa M, Iida S, Kubota S. Metabolic regulation of the CCN family genes by glycolysis in chondrocytes. J Cell Commun Signal. 2018;12:245–252. doi: 10.1007/s12079-017-0420-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi S, Nishida T, Mizukawa T, Kawata K, Takigawa M, Iida S, Kubota S (2020) Regulation of cellular communication factor 2 (CCN2) in breast cancer cells via the cell-type dependent interplay between CCN2 and glycolysis. J Oral Biosci (in press) [DOI] [PubMed]
- El-Seoudi A, Abd El Kader T, Nishida T, Eguchi T, Aoyama E, Takigawa M, Kubota S. Catabolic effects of FGF-1 on chondrocytes and its possible role in osteoarthritis. J Cell Commun Signal. 2017;11:255–263. doi: 10.1007/s12079-017-0384-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser E, Moutos CP, Downes M, Evans RM. FGF1: a new weapon to control type 2 diabetes mellitus. Nat Rev Endocrinol. 2017;13:599–609. doi: 10.1038/nrendo.2017.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionescu A, Kozhemyakina E, Nicolae C, Kaestner KH, Olsen BR, Lassar AB. FoxA family members are crucial regulators of the hypertrophic chondrocyte differentiation program. Dev Cell. 2012;22:927–939. doi: 10.1016/j.devcel.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh S, Hattori T, Tomita N, Aoyama E, Yutani Y, Yamashiro T, Takigawa M. CCN family member 2/connective tissue growth factor (CCN2/CTGF) has anti-aging effects that protect articular cartilage from age-related degenerative changes. PLoS ONE. 2013;8:e71156. doi: 10.1371/journal.pone.0071156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NM, Haqqi TM. Epigenetics in osteoarthritis: potential of HDAC inhibitors as therapeutics. Pharmacol Res. 2018;128:73–79. doi: 10.1016/j.phrs.2017.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khattab HM, Aoyama E, Kubota S, Takigawa M. Physical interaction of CCN2 with diverse growth factors involved in chondrocyte differentiation during endochondral ossification. J Cell Commun Signal. 2015;9:247–254. doi: 10.1007/s12079-015-0290-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota S, Takigawa M. Cellular and molecular actions of CCN2/CTGF and its role under physiological and pathological conditions. Clin Sci (Lond) 2015;128:181–196. doi: 10.1042/CS20140264. [DOI] [PubMed] [Google Scholar]
- Kubota S, Hattori T, Nakanishi T, Takigawa M. Involvement of cis-acting repressive element(s) in the 3’-untranslated region of human connective tissue growth factor gene. FEBS Lett. 1999;450:84–88. doi: 10.1016/S0014-5793(99)00480-9. [DOI] [PubMed] [Google Scholar]
- Kubota S, Kondo S, Eguchi T, Hattori T, Nakanishi T, Pomerantz RJ, Takigawa M. Identification of an RNA element that confers post-transcriptional repression of connective tissue growth factor/hypertrophic chondrocyte specific 24 (ctgf/hcs24) gene: similarities to retroviral RNA-protein interactions. Oncogene. 2000;19:4773–4786. doi: 10.1038/sj.onc.1203835. [DOI] [PubMed] [Google Scholar]
- Kubota S, Moritani NH, Kawaki H, Mimura H, Minato M, Takigawa M. Transcriptional induction of connective tissue growth factor/hypertrophic chondrocyte-specific 24 gene by dexamethasone in human chondrocytic cells. Bone. 2003;33:694–702. doi: 10.1016/S8756-3282(03)00227-8. [DOI] [PubMed] [Google Scholar]
- Kubota S, Maeda-Uematsu A, Nishida T, Takigawa M. New functional aspects of CCN2 revealed by trans-omic approaches. J Oral Biosci. 2015;57:37–43. doi: 10.1016/j.job.2014.09.002. [DOI] [Google Scholar]
- Lam EW, Brosens JJ, Gomes AR, Koo CY. Forkhead box proteins: tuning forks for transcriptional harmony. Nat Rev Cancer. 2013;13:482–495. doi: 10.1038/nrc3539. [DOI] [PubMed] [Google Scholar]
- Lau LF. Cell surface receptors for CCN proteins. J Cell Commun Signal. 2016;10:121–127. doi: 10.1007/s12079-016-0324-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A, Abraham DJ. All in the CCN family: essential matricellular signaling modulators emerge from the bunker. J Cell Sci. 2006;119:4803–4810. doi: 10.1242/jcs.03270. [DOI] [PubMed] [Google Scholar]
- Ma L, Duan CC, Yang ZQ, Ding JL, Liu S, Yue ZP, Guo B. Novel insights into Dhh signaling in antler chondrocyte proliferation and differentiation: involvement of Foxa. J Cell Physiol. 2020;235:6023–6031. doi: 10.1002/jcp.29528. [DOI] [PubMed] [Google Scholar]
- Maeda-Uematsu A, Kubota S, Kawaki H, Kawata K, Miyake Y, Hattori T, Nishida T, Moritani N, Lyons KM, Iida S, Takigawa M. CCN2 as a novel molecule supporting energy metabolism of chondrocytes. J Cell Biochem. 2014;115:854–865. doi: 10.1002/jcb.24728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, Jiang S, Garee JP, Verdin E, Lee AV, O’Malley BW, Zhang M, Belaguli NS, Oesterreich S. Histone deacetylase 7 and FoxA1 in estrogen-mediated repression of RPRM. Mol Cell Biol. 2010;30:399–412. doi: 10.1128/MCB.00907-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamichi R, Asahara H. The transcription factors regulating intervertebral disc development. JOR Spine. 2020;3:e1081. doi: 10.1002/jsp2.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida T, Kubota S, Kojima S, Kuboki T, Nakao K, Kushibiki T, Tabata Y, Takigawa M. Regeneration of defects in articular cartilage in rat knee joints by CCN2 (connective tissue growth factor) J Bone Miner Res. 2004;19:1308–1319. doi: 10.1359/JBMR.040322. [DOI] [PubMed] [Google Scholar]
- Ohgawara T, Kubota S, Kawaki H, Kondo S, Eguchi T, Kurio N, Aoyama E, Sasaki A, Takigawa M. Regulation of chondrocytic phenotype by micro RNA 18a: involvement of Ccn2/Ctgf as a major target gene. FEBS Lett. 2009;583:1006–1010. doi: 10.1016/j.febslet.2009.02.025. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol. 2015;4:215–266. doi: 10.1002/wdev.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz DM, Marie PJ. Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. 2015;29:1463–1486. doi: 10.1101/gad.266551.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perbal B. The concept of the CCN protein family revisited: a centralized coordination network. J Cell Commun Signal. 2018;12:3–12. doi: 10.1007/s12079-018-0455-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan CA, Chan ET, Davidson JM, Malladi VS, Strattan JS, Hitz BC, Gabdank I, Narayanan AK, Ho M, Lee BT, Rowe LD, Dreszer TR, Roe G, Podduturi NR, Tanaka F, Hong EL, Cherry JM. ENCODE data at the ENCODE portal. Nucleic Acids Res. 2016;44:D726–D732. doi: 10.1093/nar/gkv1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh JM, Jonker JW, Ahmadian M, Goetz R, Lackey D, Osborn O, Huang Z, Liu W, Yoshihara E, van Dijk TH, Havinga R, Fan W, Yin YQ, Yu RT, Liddle C, Atkins AR, Olefsky JM, Mohammadi M, Downes M, Evans RM. Endocrinization of FGF1 produces a neomorphic and potent insulin sensitizer. Nature. 2014;513:436–439. doi: 10.1038/nature13540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumiyoshi K, Kubota S, Ohgawara T, Kawata K, Abd El Kader T, Nishida T, Ikeda N, Shimo T, Yamashiro T, Takigawa M. Novel role of miR-181a in cartilage metabolism. J Cell Biochem. 2013;114:2094–2100. doi: 10.1002/jcb.24556. [DOI] [PubMed] [Google Scholar]
- Takigawa M, Tajima K, Pan HO, Enomoto M, Kinoshita A, Suzuki F, Takano Y, Mori Y. Establishment of a clonal human chondrosarcoma cell line with cartilage phenotypes. Cancer Res. 1989;49:3996–4002. [PubMed] [Google Scholar]
- Tan Z, Niu B, Tsang KY, Melhado IG, Ohba S, He X, Huang Y, Wang C, McMahon AP, Jauch R, Chan D, Zhang MQ, Cheah KSE. Synergistic co-regulation and competition by a SOX9-GLI-FOXA phasic transcriptional network coordinate chondrocyte differentiation transitions. PLoS Genet. 2018;14:e1007346. doi: 10.1371/journal.pgen.1007346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kraan PM, van den Berg WB. Chondrocyte hypertrophy and osteoarthritis: role in initiation and progression of cartilage degeneration? Osteoarthr Cartil. 2012;20:223–232. doi: 10.1016/j.joca.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu S, Li J, Yi Z. The role of the fibroblast growth factor family in bone-related diseases. Chem Biol Drug Des. 2019;94:1740–1749. doi: 10.1111/cbdd.13588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.