

Abstract
Fibrosis is perpetuated by an autocrine, pro-adhesive signaling loop maintained by the synthetic and contractile abilities of myofibroblasts and the stiff, highly-crosslinked extracellular matrix. Transcriptional complexes that are exquisitely responsive to mechanotransduction include the co-activator YAP1, which regulates the expression of members of the CCN family of matricellular proteins such as CCN2 and CCN1. Although selective YAP1 inhibitors exist, the effect of these inhibitors on profibrotic gene expression in fibroblasts is largely unknown, and is the subject of our current study. Herein, we use genome-wide expression profiling, real-time polymerase chain reaction and Western blot analyses, cell migration and collagen gel contraction assays to assess the ability of a selective YAP inhibitor verteporfin (VP) to block fibrogenic activities in dermal fibroblasts from healthy individual human controls and those from isolated from fibrotic lesions of patients with diffuse cutaneous systemic sclerosis (dcSSc). In control fibroblasts, VP selectively reduced expression of fibrogenic genes and also blocked the ability of TGFbeta to induce actin stress fibers in dermal fibroblasts. VP also reduced the persistent profibrotic phenotype of dermal fibroblasts cultured from fibrotic lesions of patients with dcSSc. Our results are consistent with the notion that, in the future, YAP1 inhibitors may represent a novel, valuable method of treating fibrosis as seen in dcSSc.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12079-020-00596-x.
Keywords: YAP1, Scleroderma, Verteporfin, CCN2, Connective tissue growth factor, CTGF, Fibrosis, Mechanotransduction, Myofibroblasr
Introduction
There is presently no broadly effective treatment for fibrosis, the excess production and restructuring of the extracellular matrix (ECM), which is estimated to contribute to approximately 45% of deaths in the developed world (Borthwick et al. 2013). Novel therapeutic options are desperately needed. Initially, fibrotic conditions, including scleroderma which is characterized by progressive fibrosis of the skin and internal organs (Allanore et al. 2015). were thought to occur due to a persistent wound repair program that failed to terminate (Bainbridge 2013). For example, it was believed that persistent, excessive canonical (ALK5/Smad) transforming growth factor beta (TGFβ) signaling was necessary and sufficient to explain the fibrotic phenotype. We now know that is overly simplistic. Specifically, a non-canonical proadhesive (FAK/integrin/TAK) signaling pathway that responds to increased mechanical tension, as well as signaling molecules such as TGFβ, is responsible for activating fibroblasts, resulting in their differentiation into myofibroblasts (Tomasek et al. 2002; Schulz et al. 2018; Hinz et al. 2019). Myofibroblasts are characterized by the presence of actin stress fibers, allowing for contraction of the ECM (Tomasek et al. 2002; Schulz et al. 2018; Hinz et al. 2019). In fibrotic conditions, including scleroderma, myofibroblasts are persistent, as they stably differentiate from a fibroblast-derived progenitor-like cell, often called a fibroadipogenic precursor (Shi-wen et al. 2012; Xu et al. 2009; Tsang et al. 2020). It is the persistence of this cell type and the presence of highly-crosslinked, collagen-rich scar tissue that produces an autocrine proadhesive signaling loop that perpetuates the fibrotic phenotype, including that of lesional SSc fibroblasts (Schulz et al. 2018; Parapuram et al. 2011).
Many proteins are involved in transmitting signals that mediate mechanical tension from the ECM to cells, including connective tissue growth factor (CTGF;CCN2) (Leask 2020). CCN2, a matricellular protein, is a non-structural ECM component that interacts with signaling molecules, constituents of the ECM, and cell surface receptors, including integrins, to regulate a variety of cellular processes including adhesion, migration and proliferation (Leask 2020). Over-expression of CCN2 has been observed in various fibrotic disorders; CCN2 is required for differentiation of fibroblast-derived progenitor cells into myofibroblasts, therefore targeting fibroblast-specific expression of CCN2 is considered a bona fide antifibrotic therapeutic approach (Tsang et al. 2020; Leask 2020; Makino et al. 2017; Liu et al. 2013). Intriguingly, fibroblast-specific deletion of CCN2 had no significant impact on normal cutaneous wound repair (Liu et al. 2014).
These observations contribute to the long-standing hypothesis that targeting the expression of CCN2 may be a useful antifibrotic approach. We showed that Yes-associated-protein (YAP), a transcriptional co-activator, is upstream of CCN2, operating through a non-Smad TGFbeta response element in the CCN2 promoter (Leask et al. 2003; Peidl et al. 2019; Chaqour 2020). YAP activity is exquisitely sensitive to high mechanical stress, which causes translocation of YAP into the nucleus, enabling YAP to promote gene transcription (Piersma et al. 2015; Toyama et al. 2018). Constitutive nuclear localization of YAP is a feature of lesional SSc dermal fibroblasts (Zanconato et al. 2016; Toyama et al. 2018). Verteporfin (VP), a drug currently used to treat macular degeneration, suppresses aberrant growth of a range of tumor cell lines via a intriguing mechanism linked to the specific suppression of YAP (Wang et al. 2015; Feng et al. 2016; Mulder et al. 2019; Li et al. 2019; Pellosi et al. 2019). Previously, we showed that VP reduced TGFβ1-induced CCN2 mRNA and protein expression in dermal fibroblasts from healthy humans (Peidl et al. 2019); however, the overall effect of VP on dermal fibroblasts from healthy individuals and the persistent fibrotic phenotype of dermal fibroblasts isolated from individuals with SSc has not been evaluated.
Methods
Cell culture
Human neonatal dermal fibroblasts (HDF) (CRL-2094, American Type Culture Collection) were cultured in high glucose Dulbecco’s modified eagle medium (DMEM), 10% fetal bovine serum (FBS) and 1% antibiotic-antimycotic (Thermo-Fisher), 37 °C, 5% CO2. Cells were cultured to 70% confluency and incubated overnight in the same media with 0.5% FBS and treated with either dimethyl sulfoxide (DMSO) (Sigma) or the selective YAP inhibitor verteporfin (VP) (0.2 µg/mL; Sigma; Liu-Chittenden et al. 2012) for an additional 6 h (for RNA extraction), or 24 h (for protein extraction). Cells were used at passage 8–10.
Patients were diagnosed as having SSc according to the 2013 American College of Rheumatology/European League Against Rheumatism SSc classification criteria (van den Hoogen et al. 2013). Samples were obtained under ethical approval (Royal Free Hospital, NHS Health Research Authority, NRES Committee London, Hampstead, Research Ethics Committee London Centre, reference number 6398). Cells were used for experiments at passage 3–5. Primary cultures of 3 SSc and 3 normal skin fibroblasts were derived from 4-mm punch biopsies sampled from the involved anterior forearm skin of patients with diffuse subset SSc less than 2 years since the first non-Raynaud’s phenomenon symptoms, and from the normal forearm skin in healthy controls, after written informed consent using a study specific consent form and information sheet. Biopsy material was minced and cultured in DMEM with 20% serum. Cultures were inspected daily by light microscopy, and when a fibroblast outgrowth was detected, cells were washed with Dulbecco’s phosphate buffered saline, and contaminating epithelial cells were detached by Trypsin/EDTA. Fibroblasts were further cultured, and then subcultured in T75 tissue culture flasks. Passaged cells were maintained in DMEM supplemented with 10% FCS and penicillin/streptomycin solution, incubated at 37 °C with 5% CO2 in T75 flasks and used for experiment at passages 3–5.
RNA isolation and analysis
RNAs were isolated using TRIzol (ThermoFisher) according to the manufacturer’s instructions. RNA was re-dissolved in 30µL of DNAase and RNAase free water, and RNA concentrations were determined (Nanodrop). RNA was transcribed into cDNA9 qScript cDNA supermix (VWR cat#CA101414-112) in a BioRad T100 Thermal Cycler (5 min at 25 °C, 30 min at 42 °C, 5 min at 85 °C, after which the samples were held at 4 °C) and then subjected to quantitative reverse transcriptase Polymerase Chain Reaction (q-RT-PCR) using SYBR green fastmix (VWR) each containing a different primer set. Cycles were: 95 °C for 2 min, 40 times at 95 °C for 10 s and 60 °C for 30 s. To obtain the melt curve, the temperature was raised to 95 °C for 15 s 65 °C for 5 s, and then 95 °C in 0.2 °C increments. Each experiment was conducted in triplicate. The resultant data were then analyzed using the △△Ct method with β-actin as the internal control. At least three separate experiments were conducted, and the average fold-change was analyzed using an unpaired t-test, as previously described Guo et al. 2011; Murphy-Marshman et al. 2017: Quesnel et al. 2019).
Primers used were: CCN2 (fwd: GAGGAGTGGCTGTGTGACG; rev: TCTTCCAGTCGGTAAGCCGC); COL1A1 (fwd: CCCCGAGGCTCTGAAGGTC; rev: GGAGCACCATTGGCACCTTT); COL1A2 (fwd: CCCCCGGTCCTGCTGGAAGT; rev: GCCAGGGGGACCAACTGCAC); TGFB1 (fwd: GCCGTGGAGGGGAAATTGAG; rev: TGAACCCGTTGATGTCCACTT)); YAP (fwd: GAACTCGGCTTCAGGTCCTC; rev: AGGGTCAAGCCTTGGGTCTA); LRP1 (fwd: GAGTCTGCTTCGTGTGCCTA; rev: CAGTCATTGTCATTGTCGCATCT); ITGA4 (fwd: TCATTTCTTCCATGCTTCCTCC; rev: TTTGGCATTGGCATTGTGTACC); ITGA5 (fwd: TGCCGAGTTCACCAAGACTG; rev: TGCAATCTGCTCCTGAGTGG); ITGA11 (fwd: CTGTGGCCAGGGTTCACG; rev: TGTAGCCAAAGAAGGCGGTC); ITGB1 (fwd: GCCGCGCGGAAAAGATGAAT; rev: CACAATTTGGCCCTGCTTGTA); ITGB3 (fwd: TTGGAGACACGGTGAGCTTC; rev: GCCCACGGGCTTTATGGTAA); TGFBR1 (fwd: CCTCGAGATAGGCCGTTTGT; rev: GCAATGGTAAACCAGTAGTTGGA); EGFR (fwd: GGCACTTTTGAAGATCATTTTCTC; rev: CTGTGTTGAGGGCAATGAG); PDGFRB (fwd: GTGGTGATCTCAGCCATCCT; rev.: CCGACATAAGGGCTTGCTT); and ACTB (fwd: CCTCGCCTTTGCCGATCC; rev: CGCGGCGATATCATCATCG).
Microarrays were conducted, essentially as previously described (Guo et al. 2011; Tsang et al. 2020), at the London Regional Genomics Centre using Affymetrix GeneChip Mouse Gene 2.0 ST arrays (Affymetrix, Santa Clara, CA). DAVID Functional Annotation Software version 6.7 (https://david.ncifcrf.gov) was employed for cluster analysis. Experiments were performed twice; the complete data set after average values was calculated (fold change > 1.7; N = 2 and cluster analysis) are shown in Supplemental Data. Cluster analysis is shown in Supplemental Table I whereas the complete data set is shown in Supplemental Table II.
Protein isolation and analysis
Cells were lysed using radioimmunoprecipitation assay (RIPA) buffer combined with a protease inhibitor/ethylenediaminetetraacetic acid (EDTA) (Sigma). Protein concentrations [bicinchoninic acid (BCA)] were determined (ThermoFisher) and equal amounts of protein (50 µg) were subjected to SDS/PAGE, transferred to a nitrocellulose membrane (iBlot, ThermoFisher). Membranes were blocked in BLOTTO (5% milk, tris-buffered saline with tween 20 (TBST), pH 7.6) 1 hr. Membranes were placed diluted primary antibody in BLOTTO (16 h, 4 °C). The primary antibodies were: anti-Collagen type I 1:3000 (Cat Nu: AB758; Lot: 2659197 Millipore), α-SMA (Clone 1A4; REF M0851; Lot 20019176 Dako,1:000), integrin beta1 (Cell signaling cat#34971S; 1:350), CCN2 (L-20) 1:1000 (Cat Nu: SC-14939; Lot B115; Santa Cruz Biotechnology, and anti-PDGFRβ (ab32570;1:600). Appropriate horseradish peroxidase-conjugated (HRP) secondary antibodies diluted in BLOTTO (RT, 1 h; Jackson ImmunoResearch 1:5000 for non-SSc samples) were added, membranes were washed in BLOTTO and blots developed using chemiluminescence (Thermo Fisher, cat#34080, 5 min) and X-ray film. Membranes were then washed with 1× TBST and stripped with stripping buffer (ThermoFisher, cat#46430) for 10 min. The membranes were then blocked in BLOTTO, and the same protocol repeated with β-actin (Sigma cat#A1978, 1:8000), and HRP-conjugated secondary anti-mouse antibody (Jackson ImmunoResearch cat#715-036-150, 1:5000). Gels involving dcSSc fibroblasts, using cells from 9 healthy individuals and 9 with SSc, were run (20 µg/sample) on 4–12% Nupage gels (Invitrogen) and processed in parallel using an anti-GAPDH antibody (6C5; Cat Nu: ab8245 Abcam 1:50.000) or appropriate horseradish peroxidase-conjugated (HRP) secondary antibodies ( Dako Polyclonal Rabbit Anti-Goat Immunoglobulins/HRP Cat No.P 0160 1:1000 for CCN2, 1:10,000 for collagen type I; Cell Signalling anti-mouse IgG/HRP-linked antibody Lot:31; Cat Nu: 7076S; 1:1000 Cell Signalling anti-rabbit IgG/HRP-linked antibody Lot:25; Cat Nu: 7074S; 1:1000). Experiments were repeated thrice. Bands were detected using ECL and X-ray film (Amersham, Buckinghamshire, UK) and quantified using densitometry relative to β-actin or GAPDH (ImageJ or Biospectrum AC Imaging; UVP, Cambridge, UK), and relative expression assessed (unpaired t-test).
Immunofluorescence analysis of actin stress fibers
A 24-well plate with glass slide inserts was coated with 500 µL of collagen type I overnight (10 µg/mL) (Invitrogen Cat#A1064401). HDFs were then plated (8000 cells/well) and allowed to attach overnight. Cells were then serum starved and treated VP or DMSO ± TGFβ for 24 h, administering TGFβ1 30 min after VP. Cells were then serum starved and treated VP or DMSO ± TGFβ1 for 24 h, administering TGFβ1 30 min after VP. Cells were fixed in 4% paraformaldehyde (Sigma), washed in PBS two times for 2 min each, and blocked for 45 min in a solution containing 0.1% TritonX-100 in PBS and 10% donkey serum (Jackson Immunoresearch, West Grove, PA, USA). Cells were then incubated with rhodamine-conjugated phalloidin (ThermoFisher #R415) diluted 1:1000 in blocking solution for 45 min in the dark. After washing in PBS two times for 2 min, the glass slides were mounted on coverslips using DAPI fluorescent mounting medium (Vectashield #H-1200). Fluorescence images were acquired with a Zeiss Imager M1 fluorescence microscope (Toronto, ON, Canada), using 360 nm to visualize DAPI in blue and 540 nm to visualize rhodamine-phalloidin in red. Images were overlaid using Northern Eclipse software (Empix, Mississauga, ON, Canada).
Cell migration assay
Type I collagen (Calbiochem cat# 341635) (10 ug/mL in DPBS) was plated in 12-well plates. Fibroblasts were cultured until confluent and then passaged onto the collagen-treated plates; each well had 1 mL of high glucose DMEM media supplemented with 10% FBS, 1% antimycotic-antibiotic. Cells were serum-starved in low glucose DMEM media supplemented with 0.5% FBS and 1% anti-antimycotic–antibiotic overnight before a scratch was made along the middle of each well using a 1 mL tip. Cells were detached and removed by washing with DPBS thrice, and then low glucose DMEM media supplemented with 0.5% FBS and 1% antimycotic-antibiotic was added. Mitomycin C (5 µg/mL) (Sigma cat# M4287) was added to each well, in the presence of absence of TGFbeta1, VP or DMSO, as described above. Three images of the scratch width were taken with a microscope (100× magnification) at 0 h and 48 h at the same location. Percentage wound closure relative to the initial wound, which was taken to represent 100%, was calculated for each treatment. Mean ± SD (N = 3) was calculated.
Collagen gel contraction assay
Experiments were performed essentially as described (Xu et al. 2009). Wells 24-well tissue culture plates were pre-coated with BSA. Trypsinized fibroblasts, used at passage three, were suspended in MCDB medium (Sigma) and collagen solution [one part 0.2 M HEPES, pH 8.0, four parts 3 mg/mL collagen (Nutragen, Inamed) and five parts of 2× MCDB] to a final concentration of 80,000 cells and 1.2 mg collagen per ml. One ml of the suspension was then added to each well to polymerize for ~ 1 h. Gels were detached by addition of 1 ml MCDB medium. Twenty-four hours later, gel contraction was quantified measuring changes in weight.
Results
As an initial approach to assessing whether YAP inhibition, for example by VP, might be used as an antifibrotic strategy, potentially in SSc, we treated dermal fibroblasts for 24 h in the presence or absence of VP and conducted genome-wide expression profiling on RNAs extracted from these cells. Cluster analysis revealed that genes whose expression was reduced by VP were those specifically involved with fibrogenic responses (Supplemental Table I). The entire list of mRNAs whose expression was reduced greater or equal to 1.7-fold is shown in Supplemental Table II. Genes affected included those involved with cell adhesion, extracellular matrix, cell migration, collagen and matrix metalloproteinases. Real time PCR verified that VP reduced CCN2 mRNA expression, consistent with prior data that CCN2 is a YAP target (Fig. 1) (Leask et al. 2003). We subsequently used quantitative PCR to confirm our microarray data that VP treatment reduced expression of matrix and matrix-associated genes, specifically COL1A1, COL1A2 (Fig. 1a) and TGFB1 (Fig. 1). Conversely, VP did not appreciably decrease YAP expression (Fig. 1a). VP also reduced mRNA expression of the integrins ITGA1, ITGA4, ITGA5, ITGB1 and ITGB3 (Fig. 1a) and the signaling receptors LRP1, TGFBR1, EGFR1 and PDGFRB (Fig. 1a). Given their role in the fibrotic phenotype (Bonner 2004; Liu et al. 2009; Leask 2013), we confirmed by Western blot analysis that VP reduced expression of ITGβ1 and PDGFRβ protein in dermal fibroblasts (Fig. 1b).
Fig. 1.

Verteporfin (VP) reduces profibrotic gene expression in human dermal fibroblasts. As described in methods, human dermal fibroblasts were cultured in the presence or absence of verteporfin (VP) (0.2 µg/mL) for 6 h (for RNA analysis) or 24 h (for protein analysis). (a) RNA analysis. RT-qPCR was used to determined mRNA expression of CCN2, COL1A1, COL1A2, TGFB1, YAP, LRP1, TGFBR1, EGFR, PDGFRB, ITGA5, ITGA11, ITGB1 and ITGB3. [N = 5, except for PDGFRB (N = 4) and YAP (N = 3)]. Averages ± SD (unpaired t test, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, **** p ≤ 0.0001) are shown. b Protein analysis. Western Blot assay was utilized to determine protein levels of PDGFRβ and integrin beta1 (ITGβ1). Optical densitometry quantification using ImageJ was used to calculate relative protein expression compared to their respective β-actin controls. N = 3 [average ± SD. (*p ≤ 0.05, unpaired student’s t-test)]
We then extended these observations by investigating the phenotypic effects of VP on cultured human dermal fibroblasts. Given the role of integrins, and specifically mechanotransduction via FAK, in myofibroblast differentiation (Thannickal et al. 2003; Shi-wen et al. 2012: Lagares et al. 2012), and since YAP mediates mechanotransductive responses, we assessed if VP impaired the ability of exogenously added TGFβ1 to induce myofibroblast differentiation, as monitored by rhodamine phalloidin staining of actin stress fibers, in dermal fibroblasts. As anticipated, exposure of fibroblasts to TGFβ1 readily induced formation of actin stress fibers, characteristic of myofibroblasts; however, this action was blocked by VP (Fig. 2).
Fig. 2.

TGFbeta1-induced myofibroblast differentiation in human dermal fibroblasts is inhibited by VP. Human dermal fibroblasts were plated at approximately 8000 cells/well in a 24-well plate, precoated with 10 µg/mL collagen on glass slides. Cells were treated with either DMSO or VP and ± TGFbeta1 in triplicate for 24 h. Cells were then fixed with 4% paraformaldehyde and actin stress fibers detected by staining fixed cells with rhodamine-phalloidin, as described in methods. Representative images are shown. Dermal fibroblasts treated with TGFbeta1 alone differentiated into myofibroblasts, as indicated by formation of actin stress fibers, the key hallmark of myofibroblasts, visualized in red; a 30 min pretreatment with VP prior to addition of TGFβ1 blocked this effect (Bar, 100 µm)
To extend these data, we used western blot analysis to assess if VP reduced the persistent fibrotic phenotype of lesional SSc fibroblasts. To do this, we treated dermal fibroblasts from healthy control individuals and fibrotic areas of patients with dcSSc with or without TGFβ1 and with or without VP. Addition of VP reduced expression of CCN2, type I collagen and α-SMA in control and scleroderma fibroblasts both in the presence and absence of added TGFβ1 (Fig. 3).
Fig. 3.

VP reduced the overexpression of alpha-SMA, CCN2 and type I collagen in lesional SSc dermal fibroblasts. Healthy control and scleroderma dermal fibroblasts, each from three individuals, were grown in a monolayer and cultured in the presence of absence of TGFbeta1 (TGFβ) (4 ng/mL) and/or the presence of DMSO or VP (0.1 or 1.0 µg/mL, as indicated, *p < 0.05, N = 3, Mann U Whitney). VP reduced TGFβ1-induced alpha-SMA, CCN2 and type I collagen expression and basal type I collagen expression in healthy dermal fibroblasts and the basal overexpression of alpha-SMA, CCN2 and type I collagen characteristic of SSc fibroblasts. Please note that TGFbeta1 induction of protein expression and protein overexpression in SSc fibroblasts was statistically significant (p < 0.05) (not shown on figure)
To provide a functional context for these observations, we used a standard scratch wound assay to show that VP could reduce migration of healthy fibroblasts treated with TGFβ1 and SSc fibroblasts both basally and treated with TGFβ1 (Fig. 4). Moreover, VP reduced the enhanced ability of lesional SSc dermal fibroblasts to contract a collagen gel matrix (Fig. 5).
Fig. 4.

VP reduced the enhanced migratory ability of lesional SSc dermal fibroblasts. Healthy (NF) and SSc (SScF) dermal fibroblasts were grown in a monolayer and then subjected to a scratch wound assay in the presence of absence of TGFβ1 (TGFβ) (4 ng/mL) and/or the presence of DMSO or VP (Vert or Verteporfin, 0.2 µg/mL), as indicated, and compared with untreated controls. Percent of the original wound size was calculated, and mean ± SD calculated. analyzed with unpaired student’s t-test presented as mean. Representative images of scratch and wound closure from (n = 3) independent repetitions are shown (*p ≤ 0.05, unpaired t test)
Fig. 5.
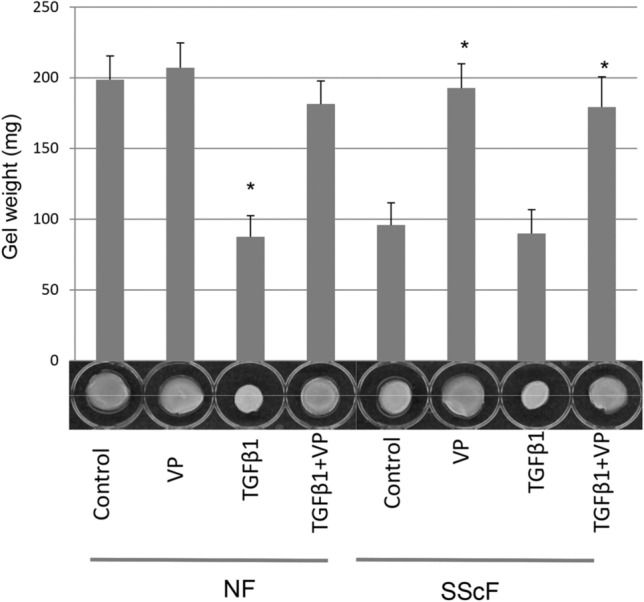
VP reduced the enhanced contractile ability of lesional SSc dermal fibroblasts. Floating gel contraction analysis reveals that VP (0.2 µg/mL) reduced TGFβ1 (4 ng/mL)-induced contraction of healthy dermal fibroblasts and the enhanced basal contractile ability of SSc dermal fibroblasts. The ability of fibroblasts to contract a floating collagen gel matrix, in the presence or absence to TGFβ1 or VP (as indicated), was assessed over 24 h, and compared with untreated controls. Contraction was assessed photographically and by measuring the weight of contracted gels. Experiments were performed in from cells from three different individuals. Means ± SD are indicated (*p ≤ 0.05; Student’s t-test), relative to untreated healthy control fibroblasts
Thus, VP, an inhibitor of the mechanosensitive transcription cofactor YAP, can block profibrotic gene expression in dermal fibroblasts and the persistent fibrotic phenotype of SSc fibroblasts (Fig. 6). Our results are consistent with the hypothesis that targeting YAP inhibition may be a useful therapeutic approach to treat SSc.
Fig. 6.
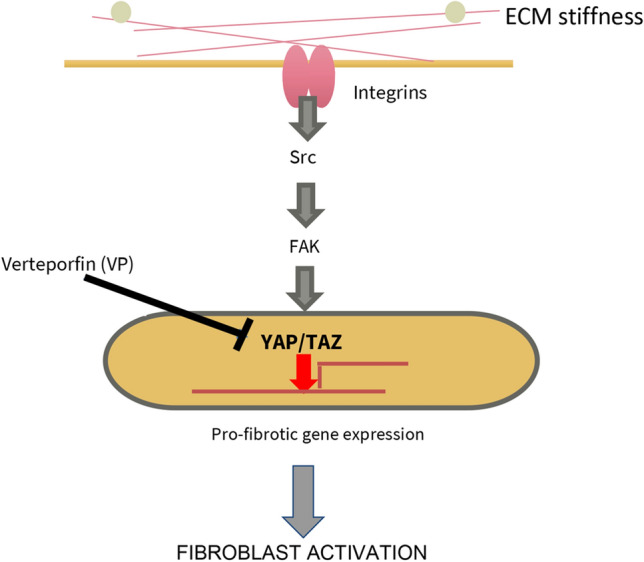
Summary of our results. The YAP inhibitor VP selectively blocks profibrotic gene expression in fibroblasts
Discussion
The key effector cell of fibrosis, the myofibroblast, is irreversibly differentiated from fibroblasts, likely through a progenitor cell intermediate, and continuously secretes pro-fibrotic proteins and ECM components that stiffen the ECM and stimulates further differentiation of local cells into myofibroblasts (Tomasek et al. 2002; Hinz et al. 2019). This positive proadhesive signaling feedback loop perpetuates pathological fibrosis; blocking this loop should therefore provide a large therapeutic benefit.
ECM stiffness seen in fibrotic disease can influence gene transcription in a process known as mechanotransduction. Pro-fibrotic ECM components bind to mechanosensitive integrin receptors and translate mechanical tension on the cell surface into nuclear signaling to alter gene expression and drive myofibroblast differentiation (Hinz et al. 2019). A nuclear effector of this pathway is YAP/TAZ, a heterodimer that activates TEAD (Piersma et al. 2015; Gibault et al. 2016).
Visudyne© (VP for injection) is an approved drug currently used to treat macular degeneration. The drug is injected intravenously and photoactivated in the eye. Interestingly, the non-photoinduced molecular form of VP has been shown to act as a selective YAP inhibitor. with no significant cytotoxicity below 2 µM (Gibault et al. 2016: Wang et al. 2015; Feng et al. 2016). VP acts by blocking translocation of YAP into the nucleus (Wang et al. 2015). If VP can disrupt pathological fibrosis through inhibition of YAP, then cross-purposing this drug could treat fibrosis. In this report, we show that VP, when applied to fibroblasts, selectively reduces expression of profibrotic genes, including type I collagen, integrin beta 1 and CCN2. VP also blocked TGFβ1-induced myofibroblast differentiation of dermal fibroblasts, as well as the persistent fibrotic phenotype of lesional SSc fibroblasts, as visualized by enhanced contractile and migratory ability and overexpression of alpha-SMA, collagen type I and CCN2 protein. Since TGFβ1 was not able to rescue the inhibitory effects of VP, the effects of VP on fibroblasts were not caused by loss of TGFβ1 expression. Our data are consistent with the notion that TGFbeta acts to promote fibrogenic gene expression through the non-canonical pro-adhesive signaling pathway (Liu et al. 2007); however, it remains unclear if VP could block the effects of TGFbeta if it were applied after TGFbeta addition.
One limitation of our study is that we have only used cultured fibroblasts, and the effects of VP on other SSc cell types including epithelial, endothelial and immune cells has not been explored, nor has the interaction among all these cell types. However, as there is no established in vivo model that recapitulates all features of SSc, we feel justified, in our initial proof-of-concept study, in focusing on the effects of VP on the final effector cell of SSc fibrosis, the fibroblast.
In conclusion, our results are consistent with the notion that further evaluation of drugs targeting YAP as a therapeutic approach to treat patients with SSc is warranted.
Supplementary Material
Below is the link to the electronic supplementary material.
Acknowledgements
AL was funded by the Canadian Institute of Health Research (MOP-77603 and MOP-119410), the Natural Sciences and Engineering Council of Canada (CPG- 146479) and The Arthritis Society (SOG-17-0039). RJS was funded by Versus Arthritis. We thank David Carter (London Regional Genomics Center) for expert assistance with microarray analysis.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, et al. Systemic sclerosis. Nat Rev Dis Primers. 2015;2015(1):15002. doi: 10.1038/nrdp.2015.2. [DOI] [PubMed] [Google Scholar]
- Bainbridge P. Wound healing and the role of fibroblasts. J Wound Care. 2013;22:407–12. doi: 10.12968/jowc.2013.22.8.407. [DOI] [PubMed] [Google Scholar]
- Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15:255–73. doi: 10.1016/j.cytogfr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Borthwick LA, Wynn TA, Fisher AJ. Cytokine-mediated tissue fibrosis. Biochim Biophys Acta. 2013;1832:1049–1060. doi: 10.1016/j.bbadis.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaqour B. Caught between a “Rho” and a hard place: Are CCN1/CYR61 and CCN2/CTGF the arbiters of microvascular stiffness? J Cell Commun Signal. 2020;14:21–29. doi: 10.1007/s12079-019-00529-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Gou J, Jia J, Yi T, Cui T, Li Z. Verteporfin, a suppressor of YAP–TEAD complex, presents promising antitumor properties on ovarian cancer. Onco Targets Ther. 2016;9:5371–5381. doi: 10.2147/OTT.S102733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibault F, Corvaisier M, Bailly F, Huet G, Melnyk P, Cotelle P. Non-photoinduced biological properties of verteporfin. Curr Med Chem. 2016;23:1171–1184. doi: 10.2174/0929867323666160316125048. [DOI] [PubMed] [Google Scholar]
- Guo F, Carter DE, Leask A. Mechanical tension increases CCN2/CTGF expression and proliferation in gingival fibroblasts via a TGFβ-dependent mechanism. PLoS One. 2011;6:e19756. doi: 10.1371/journal.pone.0019756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, McCulloch CA, Coelho NM. Mechanical regulation of myofibroblast phenoconversion and collagen contraction. Exp Cell Res. 2019;379:119–128. doi: 10.1016/j.yexcr.2019.03.027. [DOI] [PubMed] [Google Scholar]
- Lagares D, Busnadiego O, García-Fernández RA, Kapoor M, Liu S, Carter DE, et al. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum. 2012;64:1653–1664. doi: 10.1002/art.33482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A. Integrin beta1: a mechanosignaling sensor essential for connective tissue deposition by fibroblasts. Adv Wound Care. 2013;2:160–166. doi: 10.1089/wound.2012.0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A. Conjunction junction, what’s the function? CCN proteins as targets in fibrosis and cancers. Am J Physiol Cell Physiol. 2020;318(6):C1046–C1054. doi: 10.1152/ajpcell.00028.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A, Holmes A, Black CM, Abraham DJ. Connective tissue growth factor gene regulation. Requirements for its induction by transforming growth factor-beta 2 in fibroblasts. J Biol Chem. 2003;278:13008–13015. doi: 10.1074/jbc.M210366200. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang S, Wei X, Zhang S, Song Z, Chen X, et al. Role of inhibitor of yes-associated protein 1 in triple-negative breast cancer with taxol-based chemoresistance. Cancer Sci. 2019;110:561–567. doi: 10.1111/cas.13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Xu SW, Kennedy L, Pala D, Chen Y, Eastwood M, Carter DE, Black CM, Abraham DJ, Leask A. FAK is required for TGFbeta-induced JNK phosphorylation in fibroblasts: implications for acquisition of a matrix-remodeling phenotype. Mol Biol Cell. 2007;18:2169–2178. doi: 10.1091/mbc.e06-12-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Kapoor M, Denton CP, Abraham DJ, Leask A. Loss of beta1 integrin in mouse fibroblasts results in resistance to skin scleroderma in a mouse model. Arthritis Rheum. 2009;60:2817–21. doi: 10.1002/art.24801. [DOI] [PubMed] [Google Scholar]
- Liu S, Parapuram SK, Leask A. Brief report: Fibrosis caused by Loss of pten expression in mouse fibroblasts is crucially dependent on CCN2. Arthritis Rheum. 2013;65:2940–2944. doi: 10.1002/art.38121. [DOI] [PubMed] [Google Scholar]
- Liu S, Thompson K, Leask A. CCN2 expression by fibroblasts is not required for cutaneous tissue repair. Wound Repair Regen. 2014;22:119–124. doi: 10.1111/wrr.12131. [DOI] [PubMed] [Google Scholar]
- Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300–1305. doi: 10.1101/gad.192856.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino K, Makino T, Stawski L, Lipson KE, Leask A, Trojanowska M. Anti-connective tissue growth factor (CTGF/CCN2) monoclonal antibody attenuates skin fibrosis in mice models of systemic sclerosis. Arthritis Res Ther. 2017;19:134. doi: 10.1186/s13075-017-1356-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder CL, Eijkenboom LL, Beerendonk CCM, Braat DDM, Peek R. Enhancing the safety of ovarian cortex autotransplantation: cancer cells are purged completely from human ovarian tissue fragments by pharmacological inhibition of YAP/TAZ oncoproteins. Hum Reprod. 2019;34(3):506–518. doi: 10.1093/humrep/dey384. [DOI] [PubMed] [Google Scholar]
- Murphy-Marshman H, Quesnel K, Shi-Wen X, Barnfield R, Kelly J, Peidl A, et al. Antioxidants and NOX1/NOX4 inhibition blocks TGFβ1-induced CCN2 and α-SMA expression in dermal and gingival fibroblasts. PLoS One. 2017;12:e0186740. doi: 10.1371/journal.pone.0186740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parapuram SK, Shi-wen X, Elliott C, Welch ID, Jones H, Baron M, Denton CP, Abraham DJ, Leask A. Loss of PTEN expression by dermal fibroblasts causes skin fibrosis. J Investig Dermatol. 2011;131:1996–2003. doi: 10.1038/jid.2011.156. [DOI] [PubMed] [Google Scholar]
- Peidl A, Perbal B, Leask A. Yin/Yang expression of CCN family members: transforming growth factor beta 1, via ALK5/FAK/MEK, induces CCN1 and CCN2, yet suppresses CCN3, expression in human dermal fibroblasts. PLoS One. 2019;14:e0218178. doi: 10.1371/journal.pone.0218178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellosi DS, Paula LB, de Melo MT, Tedesco AC. Targeted and synergic glioblastoma treatment: multifunctional nanoparticle delivering verteporfin as adjuvant therapy for temozolomide chemotherapy. Mol Pharm. 2019;16:1009–1024. doi: 10.1021/acs.molpharmaceut.8b01001. [DOI] [PubMed] [Google Scholar]
- Piersma B, Bank RA, Boersema M. Signaling in fibrosis: TGF-β, WNT, and YAP/TAZ converge. Front Med. 2015;2:1–14. doi: 10.3389/fmed.2015.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesnel K, Shiwen X, Hutchenreuther J, Xiao Z, Liu S, Peidl A, et al. CCN1 expression by fibroblasts is required for bleomycin-induced skin fibrosis. Matrix Biol Plus. 2019;3:100009. doi: 10.1016/j.mbplus.2019.100009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JN, Plomann JM, Sengle G, Gullberg D, Krieg T, Eckes B. New developments on skin fibrosis - essential signals emanating from the extracellular matrix for the control of myofibroblasts. Matrix Biol. 2018;68–69:522–532. doi: 10.1016/j.matbio.2018.01.025. [DOI] [PubMed] [Google Scholar]
- Shi-wen X, Thompson K, Khan K, Liu S, Murphy-Marshman H, Baron M, Denton CP, Leask A, Abraham DJ. Focal adhesion kinase and reactive oxygen species contribute to the persistent fibrotic phenotype of lesional scleroderma fibroblasts. Rheumatology. 2012;51:2146–2154. doi: 10.1093/rheumatology/kes234. [DOI] [PubMed] [Google Scholar]
- Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, et al. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechanoregulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–336. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- Toyama T, Looney AP, Baker BM, Stawski L, Haines P, Simms R, et al. Therapeutic targeting of TAZ and YAP by dimethyl fumarate in systemic sclerosis fibrosis. J Investig Dermatol. 2018;138:78–88. doi: 10.1016/j.jid.2017.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang M, Quesnel K, Vincent K, Hutchenreuther J, Postovit LM, Leask A. Insights into fibroblast plasticity: cellular communication network 2 is required for activation of cancer-associated fibroblasts in a murine model of melanoma. Am J Pathol. 2020;190:206–221. doi: 10.1016/j.ajpath.2019.09.006. [DOI] [PubMed] [Google Scholar]
- van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A et al (2013) Classification criteria forsystemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum 65:2737-2774 [DOI] [PMC free article] [PubMed]
- Wang C, Zhu X, Feng W, Yu Y, Jeong K, Guo W, Lu Y, Mills GB. Verteporfin inhibits YAP function through up-regulating 14-3-3σ sequestering YAP in the cytoplasm. Am J Cancer Res. 2015;6:27–37. [PMC free article] [PubMed] [Google Scholar]
- Xu SW, Liu S, Eastwood M, Sonnylal S, Denton CP, Abraham DJ, Leask A. Rac inhibition reverses the phenotype of fibrotic fibroblasts. PLoS One. 2009;4:e7438. doi: 10.1371/annotation/654a1794-3ca1-45ac-bbf6-20ae9d33c016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Battilana G, Cordenonsi M, Piccolo S. YAP/TAZ as therapeutic targets in cancer. Curr Opin Pharmacol. 2016;29:26–33. doi: 10.1016/j.coph.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.