

Abstract
Objective
Astrocytes actively participate in energy metabolism in the brain, and astrocytic aerobic glycolysis disorder is associated with the pathology of Alzheimer's disease (AD). GLP-1 has been shown to improve cognition in AD; however, the mechanism remains unclear. The objectives of this study were to assess GLP-1's glycolytic regulation effects in AD and reveal its neuroprotective mechanisms.
Methods
The Morris water maze test was used to evaluate the effects of liraglutide (an analog of GLP-1) on the cognition of 4-month-old 5FAD mice, and a proteomic analysis and Western blotting were used to assess the proteomic profile changes. We constructed an astrocytic model of AD by treating primary astrocytes with Aβ1-42. The levels of NAD+ and lactate were examined, and the oxidative levels were assessed by a Seahorse examination. Astrocyte-neuron co-culture was performed to evaluate the effects of GLP-1 on astrocytes’ neuronal support.
Results
GLP-1 improved cognition in 4-month-old 5FAD mice by enhancing aerobic glycolysis and reducing oxidative phosphorylation (OXPHOS) levels and oxidative stress in the brain. GLP-1 also alleviated Aβ-induced glycolysis declines in astrocytes, which resulted in reduced OXPHOS levels and reactive oxygen species (ROS) production. The mechanism involved the activation of the PI3K/Akt pathway by GLP-1. Elevation in astrocytic glycolysis improved astrocyte cells’ support of neurons and promoted neuronal survival and axon growth.
Conclusions
Taken together, we revealed GLP-1's capacity to regulate astrocytic glycolysis, providing mechanistic insight into one of its neuroprotective roles in AD and support for the feasibility of energy regulation treatments for AD.
Keywords: Alzheimer's disease, Astrocyte, Aerobic glycolysis, GLP-1
Abbreviations: AD, Alzheimer's disease; OXPHOS, oxidative phosphorylation; GLP-1, glucagon-like peptide-1; WT, wild-type; MWM, Morris water maze; TEM, transmission electron microscopy; ELISA, enzyme-linked immunosorbent assay; HIF-1a, hypoxia-inducible factor-1 alpha; TBS, tris-buffered saline; DAB, diaminobenzidine; PSD, post-synaptic density; HK, hexokinase; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; PKM, pyruvate kinase; PDH, pyruvate dehydrogenase; LDHA, lactate dehydrogenase A
Graphical abstract
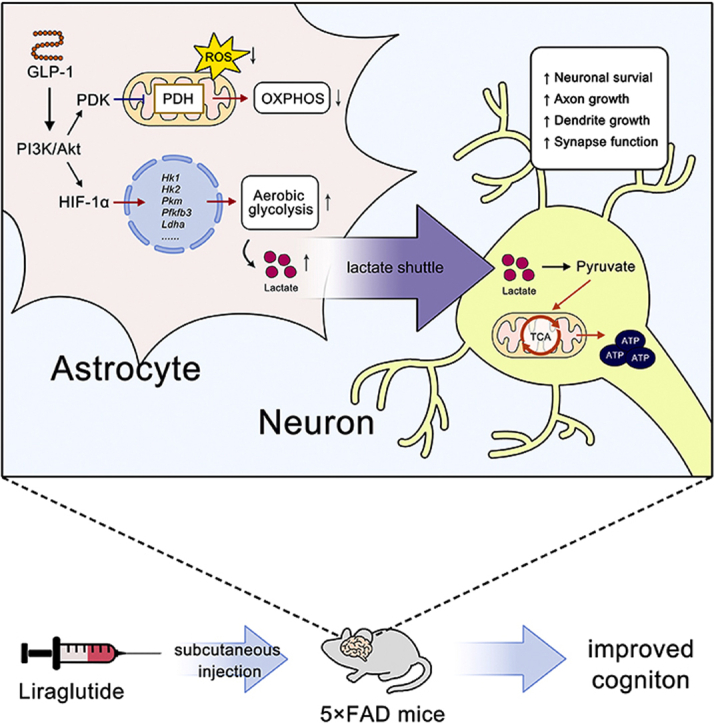
Highlights
-
•
GLP-1 mediates a metabolic shift from oxidative phosphorylation to aerobic glycolysis in Alzheimer's disease.
-
•
GLP-1's mechanism of action involves activation of the PI3K/Akt pathway.
-
•
GLP-1 enhances the supportive ability of astrocytes to neurons by promoting aerobic glycolysis.
1. Introduction
Alzheimer's disease (AD) is characterized by overproduction of beta amyloid (Aβ) peptides, leading to the formation of neurofibrillary tangles in the brain, and Aβ toxicity is considered a major cause of AD. However, in recent years, brain energy deficiency and metabolic alterations have attracted attention to the etiology of AD. It has been suggested that AD is a bioenergetic disease, and dysregulated glucose metabolism has been observed in the prodromal and progressive stages of AD [[1], [2], [3]].
The brain uses glucose as a primary source of energy, and its breakdown to produce energy in the form of ATP occurs via two complementary mechanisms, namely glycolysis and oxidative phosphorylation (OXPHOS). Astrocytes and neurons apply different methods for energy generation. Specifically, astrocytes use glycolysis as the main mechanism of energy production, whereas in neurons, the predominant mode of energy production is OXPHOS [4]. Astrocytes rely primarily on glycolytic metabolism and tend to upregulate aerobic glycolysis when they encounter energy deficiency [5]. Using glycolysis for energy production under sufficient oxygen supply conditions is termed aerobic glycolysis, which is essential for memory formation and consolidation [6]. In the central nervous system, aerobic glycolysis may represent a protective mechanism activated by nerve cells under adverse environmental conditions. In an energy crisis, astrocytes can upregulate glycolysis and increase the production of lactate, which can be transported to neurons as an energy substrate and alleviate energy deficiency in neurons [7].
Failure to elevate glycolytic levels has been observed in progressive AD and is associated with the severity of AD pathology and the expression of AD symptoms [3]. As demonstrated by Harris and Zhang's studies, higher lactate levels correlate with better cognitive performance in transgenic mice with AD and a lactate deficit-aggravated energy deficiency in neurons [2,8]. Clinical research has also shown that a reduction in glycolytic flux in regions with prominent neurofibrillary and amyloid pathology correlate with disease progression and lower cognitive performance [7,9,10]. The failure of this adaptive response or the suppression of glycolytic enzymes result in greater vulnerability of astrocytes to Aβ toxicity and acceleration of cell death [11,12]. In addition, the elevation of OXPHOS levels has been found in brain tissue affected by AD, which promotes the production of reactive oxygen species (ROS) and induces further cellular damage [13].
GLP-1 is a glycoprotein excreted by gut entero-endocrine cells and is known to exert neuroprotective functions in neurodegenerative diseases [14,15]. GLP-1 is involved in cellular energy metabolism, and studies have shown that GLP-1 treatment prevents brain glucose metabolism decline in Alzheimer's disease [16,17]; however, the mechanism has not been thoroughly investigated. GLP-1 receptor (GLP-1R) activation was found to augment glycolytic levels in β cells, which is associated with increased glucose uptake and ATP production [18]. GLP-1 also shifts substrate utilization from oxygen-consuming fatty acid metabolism toward oxygen-sparing glycolysis and glucose oxidation in coronary artery smooth muscle cells, protecting them from myocardial ischemic injury [19]. However, the influence of GLP-1 on astrocyte glycolysis has rarely been investigated.
In this study, we found that GLP-1 improved cognition and altered proteomic profiles in 5 × FAD mice, particularly the expression of glucose metabolism-related proteins. We then assessed glycolysis and OXPHOS levels in animal and astrocyte models of AD and found that GLP-1 improved astrocytes' support of neurons by upregulating aerobic glycolysis and inhibiting OXPHOS. The mechanism of GLP-1's glycolytic regulation is associated with activation of the PI3K/Akt pathway. This study highlights the energy regulation capacity of GLP-1 and provides support for the feasibility of energy regulation in the treatment of AD.
2. Materials and methods
2.1. Animals and drug administration
Male 5 × FAD mice were purchased from the Jackson Laboratory (stock no. 034848-JAX, Bar Harbor, ME, USA). The mice were kept in cohorts of 5 animals per cage in a well-ventilated and pathogen-free environment with ad libitum access to food and water. All mice were maintained under standard laboratory conditions with 40%–60% humidity and a temperature of 20 ± 3 °C. At 4 months old, the mice were randomly assigned to either an AD group or an AD + liraglutide group; 4-month-old wild-type littermates were used as controls, referred to as the wild-type (WT) group. A total of 15 mice were assigned to each group.
Liraglutide (Lira) is a slightly altered version of GLP-1 with an Arg34Lys substitution and the addition of a fatty-acid side-chain at Lys26. The compound was administered once daily to ensure continuous exposure to circulating GLP-1 [20]. The mice in the AD + Lira group received subcutaneous injections of liraglutide (25 nmol/kg body weight, once daily) for 8 weeks [21], and the mice in the two other groups were injected with an equivalent amount of 0.9% saline. All operations were conducted in accordance with the ARRIVE guidelines, the Institutional Animal Care and Use Committee of Fujian Medical University (no. FJMU IACUC 2018-034), and the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals (NIH eighth edition, revised 2011).
2.2. Morris water maze (MWM) tests
The MWM tests were performed according to a previously reported protocol [22]. Briefly, we used a circular pool 120 cm in diameter equipped with a transparent platform fixed 1 cm below the water level. The water was made opaque with a non-toxic white paint. During the test, the pool was curtained with spatial cues arranged at fixed positions. In the acquisition phase, the mice were allowed to swim and search freely for 60 s. The mice that voluntarily found the platform were permitted to remain on the platform for 10 s, while the mice who failed to find the platform were gently guided to it and retained there for 10 s. Each mouse was released from different quadrants in each trial, and each mouse underwent four trials per day for five consecutive days. The platform was removed on day 6, and each mouse was released from the quadrant opposite to previous location of the platform and given 60 s to search in the water. After each test, the mice were gently dried with a towel. Swimming speeds, escape latencies, swimming tracks, and times crossing the target spot were recorded and analyzed using SAMRT 2.0 (Pan Lab, Barcelona, Spain). For the MWM tests, 10 mice were included in each group. The test performers and data analyzers were blinded to the mice groups.
Following the MWM tests, the mice were euthanized with 50 mg/kg of pentobarbital sodium administered intraperitoneally. For Nissl staining, immunohistology, and transmission electron microscopy (TEM), the mice were perfused via the cardiac apex with 0.9% saline followed by 4% paraformaldehyde. The cortices and hippocampi were then dissected and fixed with formaldehyde or 3% buffered glutaraldehyde (for TEM). For proteomic analysis, enzyme-linked immunosorbent assays (ELISA), Western blotting, and evaluation of lactate and NAD+/NADH levels, the cortices were dissected on ice and then stored at −80 °C.
2.3. Nissl staining
The specimens (n = 3/group) were embedded in paraffin and then cut using a microtome (RM2016, Leica, Germany) to obtain 4-μm-thick sections. The sections were dewaxed and dehydrated with xylene, a graded series of alcohol, and distilled water. The samples were treated with a Nissl staining solution (C0117, Beyotime Biotechnology, Jiangsu, China) for 5 min, washed, dehydrated again, and mounted with neutral balsam.
2.4. Transmission electron microscopy
The CA1 region of the hippocampus (n = 3/group) was dissected and fixed with 3% buffered glutaraldehyde followed by 1% osmium tetroxide. The samples were dehydrated in ascending concentrations of ethanol and embedded in Epon 812. The synapse structures were observed under a transmission electron microscope (Philips, Amsterdam, Netherlands). Photos of 5 sections were obtained, yielding at least 50–100 synapses per mouse for morphometric analysis using Image-Pro Plus 6.0 software. Synapse features, including post-synaptic density (PSD), synaptic cleft width, and synaptic interface curvature were measured using previously described methods [23].
2.5. Proteomic analysis
2.5.1. Protein extraction and normalization
Approximately 100 mg of cortex tissue from each mouse (n = 3/group) was homogenized in liquid nitrogen and 1 mL of lysis buffer (8 M of urea, 0.1% SDS, and 1 mM of PMSF). The lysates were sonicated and centrifuged at 12,000 rpm for 20 min at 4 °C. The supernatants were collected, combined with 4 volume equivalents of chilled acetone, and precipitated at −20 °C overnight. The resulting lysates were centrifuged again (12,000 rpm for 20 min at 4 °C), the pellets were discarded, and 200 μL of lysis buffer was added prior to a final sonication and round of centrifugation (12,000 × g for 20 min at 4 °C) before the supernatant was collected. The concentration of proteins was determined by the Bradford method using BSA as a standard.
2.5.2. Protein digestion and peptide iTRAQ labeling
Proteins (200 μg) were reduced with 5 mM of TCEP for 1 h at 37 °C, followed by alkylation with 10 mM of MMTS for 30 min in the dark. The samples were then added to a 10 kDa molecular weight cutoff ultrafiltration centrifugal tube (OD010C34, PALL) and centrifuged for 20 min at 12,000 rpm. The samples were prepared by washing the membranes twice with 200 μL of 8 M urea by centrifugation for 15 min at 12,000 rpm, followed by three washes with 100 μL of dissolution buffer (4381664, AB Sciex) to remove urea. Dissolution buffer (50 μL) supplemented with 4 μg of trypsin (V5280, Promega) was added to the samples in the centrifugal device, and digestion proceeded overnight at 37 °C. The following day, 2 μg of trypsin was added to the samples and digestion proceeded for an additional 4 h at 37 °C. Peptides were collected by elution from the membranes via centrifugation for 20 min at 12,000 rpm, and 50 μL of dissolution buffer was added to collect a second elution sample. The peptide elution fractions were pooled and labeled with 8-plex iTRAQ according to the manufacturer's instructions. The labeled peptides were then dried by vacuum centrifugation.
2.5.3. High-pH RPLC fractionation and RPLC-MS analysis
An Agilent 1260 Infinity HPLC system (Agilent, Santa Clara, CA, USA) coupled with a chromatographic column (Waters XBridge C18 column, 1.7 μm, 150 × 2.1 mm, Waters, Milford, MA, USA) was used for high-pH RPLC fractionation. The column was equilibrated with 95% of mobile phase A (20 mM of ammonium formate with a pH of 10) and 5% of mobile phase B (80% acetonitrile) for 30 min. The dried samples were reconstituted in 200 μL of mobile phase A and 100 μL fractions were injected and eluted at a flow rate of 0.2 mL/minus with the following gradients: 5–10% buffer B (5–10 min), 10–37% buffer B (10–60 min), and 37–95% buffer B (60–85 min). A total of 48 fractions were collected, merged into 18 pooled fractions, and then lyophilized.
Prior to the RPLC-MS analysis, the 18 pooled fractions were reconstituted into solutions consisting of 5% acetonitrile and 0.1% formic acid, centrifuged at 13,500 rpm for 20 min at 4 °C to remove particulate matter, and the supernatants were carefully extracted for analysis. An Easy-nLC 1200 system (Thermo Fisher Scientific, San Jose, CA, USA) coupled with MS (Thermo Fisher Scientific Orbitrap Fusion Lumos) was used for the RPLC-MS analysis. The temperature of the column (75 μm i.d. × 150 mm, packed with Acclaim PepMap RSLC C18, 2 μm, 100 5 μm i.d. × 150 mm, packed with Acclaim PepMap RSLC C18, 2 μm, 100 Å, NanoViper, 164943; Thermo Fisher Scientific, San Jose, CA, USA) was maintained at 40 °C. Mobile phase A consisted of 0.1% formic acid in water, and mobile phase B consisted of 100% acetonitrile with 0.1% formic acid. Sample peptides were loaded in the column and eluted for 1 h at a flow rate of 300 nL/min over a 75 min period using 5–28% buffer B over 43 min, 28–38% buffer B over 8 min, and 38–100% buffer B over 2 min and retained at 100% buffer B for 7 min. MS1 was operated with a full scan range of 350–1550 m/z and the automatic gain control (AGC) was set to 4 × 105. MS/MS spectra were acquired with high-energy collision dissociation (HCD) at 35% normalized collision energy (CE) and the AGC target was set to 1 × 105.
2.5.4. Protein identification and quantification
MASCOT search algorithms were used for protein identification and quantification. All the spectral data were compared with the UniProt proteome mouse database. The search parameters were set as follows: the enzyme was set to trypsin with up to 2 missed cleavage sites, the MS1 mass tolerance was set to 10 ppm, and the MS2 to 0.02 Da; carbamidomethyl cysteine was set as a fixed modification and oxidation of methionine and N-terminal acetylation as variable modifications. Other modifications included iTRAQ-8plex modification from the quantification method (as defined in Mascot). All the proteins were identified by at least 6 unique peptide sequences. A false discovery rate (FDR) of less than 1% was required for all of the reported proteins.
2.6. Cell cultures
Astrocyte cultures were generated from cerebral cortices of neonatal Sprague–Dawley rat pups (P0–P3 purchased from Beijing Vital River Laboratory Animal Technology). The cortices were dissected, the meninges were carefully removed, and tissues were digested with 0.25% trypsin for 15 min at 37 °C. The dissociated cells were seeded into 25 cm2 flasks. The cells were cultured in DME/F-12 1:1 (SH30023.01B, HyClone) supplemented with 10% fetal bovine serum (16140071, Gibco), 2 mM of glutamine (C0212, Beyotime Biotechnology), 100 unit/mL of penicillin, and 100 g/mL of streptomycin (C0222, Beyotime Biotechnology). The cultures were maintained at 37 °C in a humidified 5% CO2 atmosphere. The culture medium was changed every 3 days. On days 12–14, confluent cultures were shaken at 37 °C and 200 rpm overnight to minimize microglial contamination. The purified astrocytes were reseeded at a density of 2.5 × 104 cells/cm2 into 96- or 6-well plates for subsequent experiments.
Co-culture of neurons and astrocytes was carried out as previously described [24]. The astrocytes were plated on the top side of poly-d-lysine-coated polycarbonate membranes with Transwell inserts (Corning) with astrocyte medium for 48 h. The culture medium was then replaced with neuron culture medium for 24 h and the astrocytes were treated with drugs before combining the cultures. Neurons were obtained from E18 embryos of Sprague Dawley rats. The cortices were dissected as previously mentioned and digested with prewarmed papain dissociation solution at 37 °C for 20 min. The solution was removed, and the tissues were washed three times with neuron culture medium, neurobasal medium (21103049, Gibco) containing 2% B27, 50 mg/mL of streptomycin, 50 U/mL of penicillin, and 0.5 mmol/L of glutamine, followed by carefully collecting the cell supernatant. After filtering through a 70-mm mesh, the cells were seeded on poly-d-lysine-coated 6-well plates. For Western blotting and immunofluorescence analysis, 1 × 10ˆ6 and 5 × 10ˆ4 cells per well were seeded, respectively. Neurons were allowed to adhere for 4 h at 37 °C in 5% CO2 before exchanging the medium. The medium was then halved every 3 days. At 3 days in vitro (DIV), neuron and astrocyte cultures were combined and co-cultured for 10 days before further testing.
2.7. ELISA
Cortices (n = 3/group) were rinsed once with ice-cold PBS and homogenized in PBS (9 mL PBS to 1 mg of tissue). They were centrifuged for 5 min at 5,000×g and the supernatants were extracted and processed using a mouse ROS ELISA kit (ml009876-C, Shanghai Enzyme-linked Biotechnology Co., Ltd., China) according to the manufacturer's instructions.
2.8. Immunofluorescence analysis
For immunofluorescence analysis, cell cultures were washed twice with PBS, fixed with 4% paraformaldehyde for 15 min, washed three additional times with PBS, permeabilized with 0.1% Triton X-100 in PBS for 5 min at 37 °C, and blocked for 2 h with 4% albumin. The cells were then incubated overnight with either anti-beta III tubulin (1:400, ab78078, Abcam), anti–HIF–1a (NB100105, Novus), or anti-GFAP (#80788, CST) antibodies at 4 °C, washed with PBS, and incubated with a goat anti-mouse secondary antibody conjugated with either a goat anti-rabbit secondary antibody (Alexa Fluor 488, ab150077 Abcam), goat anti-mouse secondary antibody (Alexa Fluor 488, ab150113, Abcam) or goat anti-rabbit secondary antibody (Alexa Fluor 594, ab150080, Abcam) for 1 h at room temperature. Cell nuclei were stained with 0.2 μg/mL of 4,6-diamino-2-phenylindole (DAPI, D9542, Sigma–Aldrich).
ImageJ software with the Sholl analysis plugin was used to quantify the neurite length, the number of neurites from soma, the number of secondary branches, and the number of neurite intersections according to a previous protocol [25]. The analyzers were blinded to the neuron groups.
2.9. Apoptosis analysis
Hoechst dye (33258) and propidium iodide (PI) labeling (C1056, Beyotime Biotechnology) were performed to distinguish between apoptotic and necrotic neurons according to the manufacturer's instructions. In each case, at least four microscopic fields were photographed randomly, with three wells were used for each group.
2.10. Aβ1-42 preparation
Soluble oligomers of Aβ1-42 (synthesized by China Peptides Co., Ltd.) were prepared as previously described [26]. Aβ was dissolved in hexafluoroisopropanol and dried in a speed-vacuum centrifuge. Before use, the aliquots were dissolved in dimethyl sulfoxide (DMSO) and then diluted in PBS to a final concentration of 400 μM/mL. The solution was allowed to oligomerize for 24 h at 4 °C.
2.11. Measurements of intracellular and mitochondrial ROS
Intracellular ROS levels were determined using a reactive oxygen species assay kit (S0033, Beyotime Biotechnology). After drug treatment, the culture medium was aspirated and the cells were washed three times with warm PBS and incubated with DCFH-DA (1:1000) at 37 °C for 20 min. The cells were then washed twice with serum-free culture medium and observed under a fluorescence microscope (CKX41, Olympus, Japan).
Mitochondrial ROS levels were detected with MitoSOX Red Mitochondrial Superoxide Indicator (M36008, Invitrogen), and mitochondria were marked with Mito-Tracker Green (C1048, Beyotime Biotechnology). The cells were washed, incubated in serum-free culture medium with 100 nM Mito-Tracker Green at 37 °C for 30 min, then washed twice with PBS, and incubated with 5 μM of MitoSOX Red Mitochondrial Superoxide Indicator for 10 min at 37 °C. The cells were washed again with prewarmed PBS and observed using a confocal microscope (Leica SP5, Germany).
2.12. ATP assay
The ATP content was determined using an Enhanced ATP assay kit (S0027, Beyotime Biotechnology) according to the manufacturer's instructions (n = 5/group). Cortex tissues or cells were homogenized, and lysates were centrifuged for 5 min at 4 °C and 12,000×g. The supernatant was added to a 96-well plate and mixed. The luminescence signals were collected with a luminometer (SpectraMax iD3, Molecular Devices). Total ATP levels were calculated using the standard curve and then normalized using the protein concentrations.
2.13. Western blotting
Cortex proteins (n = 3/group) or astrocyte proteins (n = 3 wells/group) were extracted using RIPA buffer (P0013C, Beyotime Biotechnology) supplemented with a protease and phosphatase inhibitor cocktail (Cell Signaling Technology, Danvers, MA, USA). Protein (20 μg) was separated on 8–10% (w/v) sodium dodecyl sulfate polyacrylamide gels and transferred onto polyvinylidene difluoride membranes using Trans-Blot Turbo (Bio-Rad, Singapore). After blocking with a 5% (w/v) non-fat dry milk solution in 0.1% TBS/Tween-20 for 2 h at room temperature, the membranes were incubated overnight at 4 °C with primary antibodies (rabbit anti-PSD95, #2507, CST; rabbit anti-Synaptophysin, #5467, CST; rabbit anti-PDK2, ab68164, Abcam; rabbit anti-hexokinase I, CST, #2024; rabbit anti-hexokinase II, #2867, CST; rabbit anti-PKM1/2, #3190, CST; rabbit anti-PKM2, #4053, CST; rabbit anti-PFKFB3, M02382, Bostor Biological Technology Co., Ltd.; rabbit anti-LDHA, ab101562, Abcam; rabbit anti-PDH, #3205, CST; rabbit anti-anti-pyruvate dehydrogenase E1-alpha subunit (phospho S293) antibody, ab177461, Abcam; rabbit anti-Akt, #9272,CST; rabbit anti-phospho-Akt (Ser473), #4060, CST; rabbit anti-β-actin, #4970, CST; rabbit anti-GAPDH, #5174, CST; rabbit anti-PI3 kinase p85, #4228, CST; rabbit anti-phospho-PI3 kinase p85 (Tyr458)/p55 (Tyr199), #4228,CST; rabbit anti-EAAT1, ab181036; rabbit anti-EAAT2, ab205247; and rabbit anti-GS, #80636). The membranes were washed 3 times with 0.1% TBS/Tween-20 and then incubated with horseradish peroxidase-coupled anti-rabbit or anti-mouse IgG secondary antibody (#7074 and #7076, CST) for 1 h at room temperature. ImageJ analysis software was used to quantify the bands’ density.
2.14. RT-qPCR
RNA was extracted from astrocytes using an Easy Spin Plus kit (RN28, Aidlab Biotechnologies, China) according to the manufacturer's instructions. RNA (1 mg) was reverse-transcribed using a PrimeScript RT reagent kit (RR037A, TaKaRa, Beijing, China) and an Arktik Thermal Cycler (AKC481201135, Thermo Fisher Scientific). Primers (DNA sequence 5′-3′) were synthesized by Sangon Biotech, China (Table 1). cDNA was amplified on a LightCycler 96 real-time PCR instrument (Roche). The thermal cycling program was as follows: denaturing and polymerase hot-start activating step at 95 °C for 30 s, followed by 40 repeated cycles at 95 °C for 5 s and 60 °C for 20 s. Melting curves were constructed by increasing the final temperature from 60 °C to 95 °C. The reaction consisted of 2 μL of cDNA, 0.8 μL of each amplification primer (10 μmol/mL), 6.4 μL of nucleic acid-free water, and 10.0 μL of TB Green Premix Ex Taq II (RR820A, TaKaRa). The samples were assayed in triplicate. Data were analyzed using the comparative cycle threshold (Ct) method (ΔΔCt), and β-actin was used as an internal control. These steps were conducted in accordance with the latest publication of quantitative real-time PCR experiment (MIQE) guidelines [27].
Table 1.
Primer sequences for RT-qPCR.
| Gene | Primer sequence | |
|---|---|---|
| Hk1 | Forward | 5′-CCGCAGCAGAGGGAGGAAGG-3′ |
| Reverse | 5′-AGAGTGGCAGGGCAGGGATTAG-3′ | |
| Hk2 | Forward | 5′-AGACCAGAGCATCCTCCTCAAGTG-3′ |
| Reverse | 5′-TCATTCACCACGGCAACCACATC-3′ | |
| Pkm | Forward | 5′-GTGCCGCCTGGACATTGACTC-3′ |
| Reverse | 5′-ATTCAGCCGAGCCACATTCATCC-3′ | |
| Pfkfb3 | Forward | 5′-TGCCATACCTGAAATGCCCACTTC-3′ |
| Reverse | 5′-CCGATGCGTGCTCACTGATTCTAC-3′ | |
| Ldha | Forward | 5′-TCGCACCTTGTAGCCGTTATTGG-3′ |
| Reverse | 5′-AACTGCCCTCCCGCTCTTCTC-3′ | |
| β-actin | Forward | 5′-GGAGATTACTGCCCTGGCTCCTA-3′ |
| Reverse | 5′-GACTCATCGTACTCCTGCTTGCTG-3′ |
2.15. Measurements of the oxygen consumption rate
The oxygen consumption rate (OCR) was measured using a Seahorse XFe24 analyzer (Agilent Technologies) according to the manufacturer's instructions. Astrocytes were plated at 8,000 cells/well in a 24-well Seahorse XF Microplate (100777-004, Agilent Technologies) with culture medium and incubated at 37 °C and 5% CO2 for 24 h. After drug treatment, the culture medium was replaced with Agilent Seahorse XF Base Medium (103335-100, Agilent Technologies) supplemented with 1 mM of sodium pyruvate, 2 mM of glutamine, and 1 of mM d-glucose. The cells were cultured at 37 °C in a non-CO2 incubator for 1 h. Three basal (OCR) measurements were recorded (3 min each) before adding oligomycin (0.25 mg/mL), FCCP (0.5 mM) and rotenone (0.5 mM). The effect on mitochondrial oxygen consumption was measured 3 times for 3 min each.
2.16. Lactate assay
Lactate contents were measured using an l-Lactate assay kit (ab65330, Abcam) according to the manufacturer's instructions. Tissues (10 mg, n = 3/group) or cells (10ˆ6 cells, n = 3 wells/group) were washed twice with PBS, homogenized with lactate assay buffer, and centrifuged for 5 min at 4 °C and 12,000 rpm. The resulting supernatants were collected. For tissues, deproteinization was performed using a deproteinizing sample preparation kit (K823-200, BioVision). Reaction reagents were prepared as instructed and mixed with 50 μL of the samples, incubated at room temperature for 30 min protected from light, and measured at OD 570 nm.
2.17. NAD+ and NADH measurements
NAD+ and NADH contents of cortices and astrocytes were measured using a NAD+/NADH assay kit with WST-8 (S0175, Beyotime Biotechnology). Ten milligrams of tissue or 1 x 10ˆ6 cells were washed twice with cold PBS, then homogenized with NADH/NAD extraction buffer. The samples were centrifuged at 12,000×g for 10 min, and the supernatant was extracted. To detect the total NAD (NADtotal), the supernatant was bathed at 60 °C for 30 min, while to detect the total NADH, the supernatant was maintained at 4 °C. The working solution was prepared as instructed, and 20 μL of supernatant was added to 90 μL of working solution, followed by incubation at 37 °C and shielded from light for 10 min. Then 10 μL of color developing agent was added. The mixture was incubated at 37 °C in the dark for another 30 min, at which point the absorbance was measured at 450 nm. The NAD + level and NAD+/NADH ratio were calculated as follows:
| NAD+ = NADtotal - NADH |
| NAD+/NADH = (NADtotal - NADH)/NADH |
2.18. Glutamate uptake assay
The assay was carried out as described by Shaimaa Mahmoud with slight modifications [28]. Astrocytes were seeded in a 24-well plate, and after treatments, the cells were washed 2 times with Hank's Balanced Salt Solution (HBSS) containing calcium and magnesium but without phenol (14025134, Gibco). The astrocytes were then incubated with 200 μM of glutamate in HBSS for 4 h at 37 °C with a 5% CO2 incubator. The glutamate concentration in the culture supernatant was measured using a glutamate colorimetric assay kit (MAK004, Sigma–Aldrich) according to the manufacturer's instructions. Glutamate uptake by the astrocytes was measured by subtracting the amount of glutamate measured in the medium from the amount initially added to the cells and normalized to the cell protein concentrations.
2.19. Statistical analyses
The results are reported as mean ± SEM. Comparisons among different groups were carried out using one-way ANOVA following Bonferroni's post hoc test for multiple group comparisons. IBM SPSS Statistics 24.0 (IBM Corp., Armonk, NY, USA) and GraphPad Prism 7.0 software were used for analyses. Differences were considered statistically significant at P < 0.05.
3. Results
3.1. GLP-1 improved spatial cognition in the 5 × FAD mice
5 × FAD mice are transgenic mice transfected with five FAD mutations (APP K670N/M671L [Swedish] + I716V [Florida] + V717I [London] and PS1 M146L + L286V) and develop cerebral amyloid plaques and gliosis at 2 months of age [23]. The MWM test is reliable for evaluating spatial learning ability in rodents [22]. As shown by the MWM tests, the 4-month-old 5 × FAD mice exhibited impaired spatial cognition, with longer escape latencies and fewer platform crosses. After 8 weeks of liraglutide treatment, the time required for platform searching shortened, increasing the chances of the 5 × FAD mice finding the target platform, indicating improved cognition (Figure 1).
Figure 1.

GLP-1 improved the spatial cognition of the 5 × FAD mice. (A–C) Cognition evaluation of the mice by the MWM, including escape latencies (A) and crossing the target quadrant during the probe trial (B) and representative swimming tracks of the different groups of mice in the probe trial (C). n = 10/group for MWM tests. (D) Representative images of cortex Nissl staining of the different groups. Scale bars: 500 μm; enlarged images are shown in the lower column, scale bars: 200 μm; n = 3/group. (E) Protein expression of PSD95 and SYN in the cortices of the mice by Western blotting. n = 3/group. (F) Representative images of the morphology of synapses in the cortex region under TEM. The red arrowheads indicate synapses. Scale bars: 1 μm; n = 3/group. Data are presented as means ± SEM. ∗P < 0.05, ∗∗P < 0.01, WT vs AD group; #P < 0.05, ##P < 0.01, AD vs AD + Lira group.
The number of neurons and structures of synapses were evaluated using Nissl staining and TEM. As shown in Figure 1D, liraglutide treatment increased the number of surviving neurons in the cortical and hippocampal regions, and neurons in this group exhibited a more regular synapse structure and thicker post-synaptic density (PSD) (Figure 1F). The expression levels of synapse marker proteins PSD95 and SYN were also elevated (Figure 1E). These results suggested a protective effect of GLP-1 on neuronal survival and function in the 5 × FAD mice.
We next performed an iTRAQ-based proteomic analysis to compare the three groups' proteomic profiles and investigate the molecular mechanisms of GLP-1's neuroprotective effects.
3.2. GLP-1 altered brain proteomic profiles in the 5 × FAD mice
In the proteomic analysis, a total of 7,013 proteins were identified in the cortices of the mice from the 3 groups. Among them, 300 proteins were differentially expressed between the WT and AD groups (P < 0.05), 275 proteins were differentially expressed between the AD and AD + Lira groups (P < 0.05; Figure 2C and Figure 2D), and a set of 49 proteins significantly changed in a three group comparison (P < 0.05; Figure 2C). A KEGG enrichment analysis was used to enrich pathways of differentially expressed proteins between the groups. The top 10 enriched pathways are listed in Figure 2A and Figure 2B. The metabolic pathway was prominently changed in the AD brain and also altered by GLP-1 administration. Upon further examination, the expression of several bioenergetic proteins, such as PDK2, NDUFB2, ACAA2, and GSTK1, decreased in the AD group and was upregulated by GLP-1 administration (Figure 2E). PDK2 is a regulator of the glucose metabolic phenotype, and depending on the phosphorylation status of PDH, glucose metabolism can be directed toward glycolysis or OXPHOS. ACAA2 is involved in mitochondrial fatty acid-β oxidation [29], whereas GSTK1 is critical for maintaining mitochondrial homeostasis [30]. Aside from the aforementioned proteins, the expression level of 14 other proteins involved in protein transport, proteolysis, protein glycosylation, and cell proliferation partially reversed in the presence of GLP-1. Other effected cellular structures, such as the plasma membrane, cytoskeleton, dendritic spine, and nucleoplasm, are shown in Figure 2F.
Figure 2.

GLP-1 altered brain proteomic profiles in the 5 × FAD mice. (A–B) Volcano plots of protein expression changes in the mice cortices with the top 10 KEGG enriched pathways of differentially expressed proteins (DEPs, P < 0.05). The significance of the KEGG pathway is indicated by the red intensity. (C) Venn diagram of DEPs enriched in the cortices. Differences were considered statistically significant at P < 0.05. (D) Hierarchical clustering heat map of DEPs among the three groups; the color of each section is proportional to the significance of the changes in proteins; red indicates upregulation and blue indicates downregulation. (E) Changes in cortex protein levels of ACCA2, GSTK1, NDUFB2, and PDK2 in the three groups. The protein levels were quantified and analyzed by one-way ANOVA. (F) Protein–protein interaction network analysis of DEPs. Nodes represent DEPs and the node color indicates an increase (red) to decrease (green) of the protein interaction counts. The line thickness indicates the interaction confidence. Data are presented as means ± SEM. n = 3/group. ∗P < 0.05, WT vs AD group; #P < 0.05, ##P < 0.01, AD vs AD + Lira group.
3.3. GLP-1 improved aerobic glycolysis and alleviated oxidative stress in the cortices of the 5 × FAD mice
We evaluated glycolysis and OXPHOS levels in the three groups and found that glycolysis was suppressed in the AD brains, whereas OXPHOS was upregulated. The expression levels of enzymes that are critical for glycolysis and OXPHOS are shown in Figure 3. Hypoxia-inducible factor-1 alpha (HIF-1 a) has been shown to upregulate glycolysis enzymes. Hexokinase (HK) phosphorylates glucose to form glucose-6-phosphate, which is then acted upon by 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) and pyruvate kinase (PKM) to form pyruvate, which is then converted into lactate by lactate dehydrogenase A (LDHA) [31,32]. The cellular content of PKM1/2, PKM2, and PFKFB3 decreased in the AD brains (Figure 3A), indicating a decline in glycolysis. However, OXPHOS levels were elevated in the AD brains as shown by reduced PDK levels and a higher degree of PDH phosphorylation (Figure 3E). Nevertheless, even with enhanced OXPHOS, ATP production declined and ROS levels increased (Figure 3H and Figure 3F), concurrent with a reduction in GSH content (Figure 3G), indicating an exacerbated redox state. Treatment with GLP-1 increased the expression of glycolytic enzymes, relieved radical OXPHOS levels, reduced oxidative stress, and increased ATP production.
Figure 3.

GLP-1 improved aerobic glycolysis and alleviated oxidative stress in the cortices of the 5 × FAD mice. (A–D) Evaluation of cortex glycolytic levels in the three groups, including the relative expression of glycolytic enzymes by Western blotting (A); glycolytic products of lactates (B) and NAD+ (C and D). (E–H) Evaluation of cortex oxidative phosphorylation levels in the three groups, including the relative expression of PDK2 and PDH (E); contents of ROS (F), GSH (G), and ATP (H) in the cortices. (I) Relative expression of cortex PI3K and Akt by Western blotting. Data are presented as means ± SEM. n = 3/group. ∗∗P < 0.01, ∗∗∗P < 0.001, WT vs AD group; #P < 0.05, ###P < 0.001, AD vs AD + Lira group.
The PI3K/Akt pathway is deeply involved in energetic processes, and reduced Akt phosphorylation is known to be associated with impaired cognition in AD [33]. We found that GLP-1 activated the PI3K/Akt pathway, which was suppressed in the AD mice (Figure 3I), and we hypothesize that this may contribute to the mechanism of GLP-1 action in glycolytic modulation.
3.4. GLP-1 improved aerobic glycolysis and alleviated oxidative stress in astrocyte cells
Astrocytes mainly use glycolysis for ATP production and represent the major source of glycolytic influx in the brain. Numerous studies have indicated that malfunctioning astrocytes contribute to the pathology of AD. To evaluate the effect of GLP-1 on astrocytes, we constructed a cellular model of AD by treating astrocytes with soluble oligomers of Aβ1-42.
Consistent with findings in animal experiments, GLP-1 alleviated Aβ-induced glycolytic suppression as demonstrated by the increased expression of glycolytic enzymes such as PKM2 and HK2 (Figure 4A) and enhanced glycolytic products (Figure 4B, Figure 4C, and Figure 4D). GLP-1 also increased Akt phosphorylation levels in Aβ-treated astrocytes (Figure 4E). However, when the PI3K/Akt pathway was inhibited by LY294002, a potent inhibitor of PI3K, the stimulation of glycolysis by GLP-1 was reduced. The expression of PDK2 and HIF-1α was reduced by LY294002 (Figure 4E and Figure 4I), and the transcription of Ldha, Pkm, and hexokinase II was suppressed (Figure 4J). The cellular content of lactate and NAD+ was also reduced upon PI3K/Akt pathway inhibition (Figure 4F, Figure 4G, and Figure 4H).
Figure 4.

GLP-1 improved aerobic glycolysis and alleviated oxidative stress via the PI3K/Akt pathway in astrocytes in vitro. Astrocytes were treated with 5 μM of Aβ1-42 for 24 h or incubated with 100 nM of GLP-1 for 2 h before treatment with Aβ1-42 according to the group. In the Aβ+GLP-1 + LY group, astrocytes were pretreated with 10 μM of LY294002, a potent PI3K inhibitor, 1 h prior to incubation with GLP-1 and Aβ1-42. (A–D) Evaluation of glycolytic levels in the three groups, including the relative expression of glycolytic enzymes by Western blotting (A); glycolytic products of lactates (B) and NAD+ (C and D). (E) Relative expression of cortex PI3K, Akt, PDK2, and HIF-1α by Western blotting. (F–H) Evaluation of glycolytic levels, including glycolytic products of lactates (F) and NAD+ (G and H). (I) Representative immunofluorescence images of HIF-1α and GFAP in the three groups. Scale bars: 25 μm. (J) Transcriptional level of glycolytic enzymes, including Hk1, Hk2, Pkm, Pfkmb3, and Ldha. (F) Evaluation of oxidative phosphorylation levels. The basal oxygen consumption rate (OCR), maximal OCR, and mitochondrial ATP production were calculated by adding oligomycin, FCCP, and rotenone/antimycin A during the Seahorse examination process. Data are presented as means ± SEM. n = 3–5 wells/group. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, control vs Aβ group; #P < 0.05, ##P < 0.01, ###P < 0.001, Aβ vs Aβ + GLP-1 group; &P < 0.05, &&P < 0.01, &&&P < 0.001, Aβ + GLP-1 vs Aβ + GLP-1 + LY group.
In contrast, OXPHOS was affected in the opposite manner. Using Seahorse examination, OXPHOS levels in the different groups were assessed by real-time measurement of the oxygen consumption rate (OCR). Oligomycin, an ATP synthase inhibitor; cyanide p-trifluoromethoxy-phenyl-hydrazone (FCCP), a H+ ionophore; and rotenone/antimycin A, an electron-transport chain inhibitor, were added to calculate the basal or maximal OCR. As shown in Figure 4K, the basal and maximal OCR was elevated in the Aβ group, along with increased mitochondrial ATP production, suggesting activation of mitochondrial OXPHOS levels when glycolytic influx was deficient and the energy supply was low. GLP-1 reduced the activation of OXPHOS and alleviated pressure on the mitochondria. However, when the PI3K/Akt pathway was inhibited, OXPHOS levels rebounded. These results revealed the role of the PI3K/Akt pathway in GLP-1's mechanism of glycolytic and OXPHOS regulation.
Astrocytes took up glutamate in the synaptic cleft, which was turned into glutamine by glutamine synthetase (GS) and then delivered to neurons, or glutamate could be metabolized into a-ketoglutarate and inter-TCA. We found that Aβ treatment resulted in a decline in astrocytic glutamate uptake, with a decrease in EAAT2, a type of glutamate transporter, and GS expression; however, GLP-1 failed to improve the astrocytes’ glutamate uptake ability, although it upregulated GS expression (Figure 5).
Figure 5.

GLP-1 did not enhance glutamate uptake in Aβ-treated astrocytes in vitro. Astrocytes were treated with 5 μM of Aβ1-42 for 24 h or incubated with 100 nM of GLP-1 for 2 h before treatment with Aβ1-42according to the groups. (A–D) Evaluation of EAAT1, EAAT2, and GS expression by Western blotting. (E) Glutamate uptake by astrocytes in the different groups. Data are presented as means ± SEM. n = 3–5 wells/group. ∗∗P < 0.01, ∗∗∗P < 0.001, control vs Aβ group; ##P < 0.01.
3.5. GLP-1 improved the supportive role of astrocytes to neurons by elevating aerobic glycolysis in the astrocytes
Astrocytes support neuronal survival and function by providing lactate as an energy substrate to neurons and maintaining the redox balance. We added 2-deoxyglucose (2-DG) to evaluate its influence on neuronal survival when the action of GLP-1 in astrocytic glycolysis was inhibited, as shown by decreased lactate and NAD + levels (Figure 6A-Figure 6C). 2-DG was transported across the membrane and rapidly phosphorylated to form 2-deoxyglucose-6-phosphate, which could not be metabolized further by glycolytic enzymes and thus impeded the glycolysis process. GLP-1's ability to alleviate OXPHOS activation was hampered in the presence of 2-DG as shown by reduced phosphorylation of PDH (Figure 6D) and higher OCR values (Figure 6J-M). In agreement with this, the total and mitochondrial ROS production increased (Figure 6F, Figure 6G, and Figure 6H) and GSH levels declined (Figure 6I), indicating the deterioration of a balanced cellular redox state in the astrocytes.
Figure 6.

OXPHOS level and oxidative stress of astrocytes increased after glycolysis inhibition. Astrocytes were treated with 5 μM of Aβ1-42 for 24 h or incubated with 100 nM of GLP-1 for 2 h before treatment with Aβ1-42 according to the groups. In the Aβ + GLP-1 + 2-DG group, astrocytes were pretreated with 10 mM of 2-DG 1 h prior to incubation with GLP-1 and Aβ1-42. (A–C) Glycolytic products of lactate (A) and NAD+ (B and C). (D) Relative expression of PDK2 and PDH by Western blotting. (E) ATP production of astrocytes in the different groups. (F–H) Fluorescence images of total and mitochondrial ROS in the different groups. Mitochondria were marked by MitoTracker Green; total and mitochondrial ROS were marked by DCFH-DA and MitoSox, respectively. For DCFH-DA, scale bars: 100 μm; for MitoTracker Green and MitoSox, scale bars: 10 μm. (I) GSH content in the different groups. (J–M) The basal oxygen consumption rate (OCR), maximal OCR, and mitochondrial ATP production in the different groups. Data are presented as means ± SEM. n = 3–5 wells/group. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, control vs Aβ group; #P < 0.05, ##P < 0.01, ###P < 0.001, Aβ vs Aβ + GLP-1 group; &P < 0.05, &&P < 0.01, &&&P < 0.001, Aβ + GLP-1 vs Aβ + GLP-1 + LY group.
To assess GLP-1's influence on the astrocyte's neuronal supportive ability, we performed astrocyte-neuron co-culture experiments in Transwell. Aβ administration to the astrocytes reduced the viability of neurons in the co-cultures, whereas GLP-1 increased the astrocytes' ability to support neurons under the same experimental conditions (Figure 7A and Figure 7B). Axon length and dendrite growth of neurons were promoted by GLP-1 administration, whereas the inhibition of astrocytic glycolysis by 2-DG eliminated GLP-1's beneficial effects (Figure 7C-H). The expression of PSD95 and SYN was also increased by GLP-1 and reduced by 2-DG (Figure 7I-K). These results highlight the importance of astrocytic glycolysis in neuron growth and implicate that GLP-1 improved astrocyte's supportive ability via enhancing aerobic glycolysis.
Figure 7.

GLP-1 improved astrocytes' supportive ability to neurons by elevating aerobic glycolysis in the astrocytes. Neurons were co-cultured with astrocytes that were previously treated with Aβ1-42, GLP-1, or 2-DG. (A–B) Neurons were stained with Hoechst 33342 and PI to assess cellular survival. Scale bars: 75 μm. (C–H) Evaluation of the neuritic complexity of neurons. Fluorescent images of cortical neurons (C). Sholl analysis of neurite intersections (B; control vs Aβ group, P < 0.01; Aβ vs Aβ + GLP-1 group, P < 0.001; Aβ + GLP-1 vs Aβ + GLP-1 + 2-DG group; P < 0.001), neurite length (E), number of secondary branches (F), number of neurites from soma (G), and axon length (H) were evaluated by ImageJ software with the Sholl analysis plugin. More than 100 cells per group were analyzed. Scale bars: 25 μm. (I–K) Relative expression of PSD95 and SYN in cortical neurons by Western blotting. Data are presented as means ± SEM. n = 3 wells/group. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, control vs Aβ group; #P < 0.05, ##P < 0.01, ###P < 0.001, Aβ vs Aβ + GLP-1 group; &P < 0.05, &&P < 0.01, &&&P < 0.001, Aβ + GLP-1 vs Aβ + GLP-1 + 2-DG group.
4. Discussion
In this study, we found that GLP-1 mediated a glucose metabolic shift in the AD animal and astrocyte cell models. Enhanced glycolytic influx was accompanied by increased production of glycolytic products such as lactate and NAD+, whereas OXPHOS was suppressed and mitochondrial ROS levels were reduced. GLP-1's effect on metabolic phenotype regulation was associated with activation of the PI3K/Akt pathway. The shift from OXPHOS to aerobic glycolysis improved the astrocyte cells' ability to support neurons as shown by increased cellular viability and robust dendrite and axon growth. This study verified GLP-1's action in shifting the cellular glucose metabolic phenotype not previously demonstrated in astrocytes and revealed a new mechanism of GLP-1 action in improving cognition in AD.
In our study, GLP-1 mediated a shift from OXPHOS to glycolysis in the AD animal and cellular models. This effect could represent one mechanism of GLP-1's neuroprotective role. GLP-1 has been found to upregulate cellular energy expenditure via several bioenergetic pathways [34] and is capable of shifting the energy metabolic phenotype toward glycolysis under ischemia and hypoxia conditions [35]. This mechanism helps cells cope with an energy crisis in an aerobic manner when the oxygen supply is scarce, such as in myocardial ischemic injury, improve cellular resilience to adverse conditions, and enhance cellular survival [19].
Brain energetic deficits have been outlined in neurodegenerative disorders of aging (NDAs), particularly AD and Parkinson's disease, in which the failure in glucose uptake and ATP production has been a driving force in disease progression. Lactate generated from astrocytic glycolysis is a vital alternative fuel for neurons when faced with energy challenges, and glycolytic enhancement was found to be promising in NDAs treatment [36]. Upregulation of HIF-1α was found in the prodromal state of AD, increasing the expression of several glycolytic enzymes, and is one mechanism by which astrocytes counteract the toxicity of Aβ [37,38]. However, the accumulation of Aβ and tau inhibited glycolysis, and this compensatory effect is lost in advanced AD [39], which was also proved in this study, resulting in an energy-deficient state and exacerbating oxidative stress. By improving astrocytic glycolysis, GLP-1 increased lactate production and mitigated energy crisis.
GLP-1's action in enhancing aerobic glycolysis in AD is connected with the PI3K/Akt pathway, which is deeply involved in energy metabolism and cellular survival [40]. GLP-1 has been reported to activate the PI3K/Akt pathway in β cells and neural cells, which repress FOXO, an inhibitor of PDK, and induced a decrease in OXPHOS [41,42]. In accordance with previous studies, we found that GLP-1 increased Akt phosphorylation, leading to a higher degree of PDH phosphorylation, which downregulates OXPHOS and reduces oxidative stress. In addition, the phosphorylation of Akt leads to the activation of the mTOR–HIF–1α pathway, inducing the expression of glycolytic enzymes and enhancing glycolysis [18,43]. In a recent study, Sung et al. found that metabolic reprogramming mediated by the mTOR–HIF–1α pathway was essential for microglial activation, highlighting the importance of aerobic glycolysis in glial neural functions [44]. In this study, we reported the participation of the PI3K/Akt pathway in the mechanism of GLP-1-mediated metabolic regulation, which has not been reported in an AD astrocyte model.
An elevation in aerobic glycolysis was also found to be associated with better astrocyte support of neurons as exemplified by increased viability and improved neurite growth. It has been postulated that aerobic glycolysis exerts its protective effect by providing lactate to energy-deficient neurons, and abundant evidence has shown the beneficial effects of lactate on stressed neuronal cells. Lactate supplementation has been shown to reduce brain lesions in hypoxic-ischemic encephalopathy [45] and elevate hippocampal lactate concentration, improving neurogenesis and alleviating depression syndromes [46]. It has also been reported that exercise improves cognition by producing lactate, which crosses the blood–brain barrier and promotes BDNF production [47].
Although some reports have indicated the elevated glycolytic capacity of neurons during electrical activation, lactate produced through astrocyte glycolysis might not be indispensable for neuronal activities. In fact, chronic glycolysis enhancement might be detrimental and unsustainable for neurons, as shown in Herrero-Mendez's study, in which glycolysis augmented through upregulation of Pfkfb3 expression resulted in oxidative stress and apoptotic death [48,49]. We contend that in AD, when glucose metabolism is deterred and mitochondrial function is reduced, lactate shuttling from astrocyte glycolysis becomes increasingly important for neuronal survival by providing an alternate energy substrate for neurons without increasing oxidative stress. GLP-1 can partly rescue the glycolytic function of astrocytes and increase lactate flux, which helps alleviate an energy crisis in neurons. Other energy-enhancing therapy involving triple GLP-1/GIP/glucagon receptor agonist was also found to be beneficial in mitigating neural damage in AD, which suggested that GLP-1 combined with other energy-modulating agents or conventional anti-AD drugs such as memantine should be feasible in AD treatment [50].
In our study, the lactate level in the brain of the AD mice did not vary among the WT and AD + Lira groups, whereas Aβ-treated astrocytes showed a significant decrease in lactate content. This dichotomy might have been caused by astrocyte-neuron interactions. Lactate and NAD + can be taken in by neurons and enter the TCA cycle, so that the levels of these species in the brain are affected by the metabolic levels of both. Richard et al. also reported that although brain glycolytic activity was altered in different courses of AD, the lactate content in the cortex remained roughly unchanged [8].
Improved balance of the cellular redox state is another mechanism of GLP-1's neuroprotective role. Promotion of glycolysis improved the antioxidant ability of astrocytes and reduced the production of ROS. Astrocytes are considered a major line of defense against oxidative injuries, and GSH is a key component in astrocytic detoxification processes [49]. Neurons are intrinsically OXPHOS dependent yet have low antioxidant capacity, and the production of GSH by astrocytes is an important protection against oxidative stress in neurons [51].
5. Conclusions
In summary, this study revealed that GLP-1's neuroprotective mechanism is associated with its ability to promote aerobic glycolysis and alleviate OXPHOS activation and connected with the activation of the PI3K/Akt pathway. This study suggests that modulation of the energetic phenotype is effective at slowing down the progress of AD and sheds light on the possibility of treating AD via energy modulation therapy or combination therapy with other anti-AD drugs.
Author contributions
Jiaping Zheng: Conceptualized and wrote the original draft. Yunzhen Xie and Li Wu: Writing, review, and editing. Lingjia Ren and Liqin Qi: Data curation. Xiaodong Pan and Jianxing Zhou: Resources. Libin Liu: Project administration. Zhou Chen: Supervision.
Acknowledgments
This study was supported by grants from the Startup Fund for Scientific Research of Fujian Medical University (no. 80001044), the National Natural Science Foundation of China (no. 81800712), the Science and Technology External Cooperation Program of Fujian (no. 2018I0007), and the Science and Technology Innovation Joint Fund Project of Fujian Province (no. 2017Y9018).
The authors thank Professor Dongsheng Cai from the Department of Molecular Pharmacology, Albert Einstein College of Medicine, Bronx, NY 10461, USA, and Ling Lin and Junjin Lin from the Public Technology Service Center (Fujian Medical University, Fuzhou, China) for their assistance.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2021.101180.
Contributor Information
Zhou Chen, Email: chenzhou@fjmu.edu.cn.
Libin Liu, Email: libinliu@fjmu.edu.cn.
Conflict of interest
None declared.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Figs1.

References
- 1.Butterfield D.A., Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nature Reviews Neuroscience. 2019;20(3):148–160. doi: 10.1038/s41583-019-0132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang M., Cheng X., Dang R., Zhang W., Zhang J., Yao Z. Lactate deficit in an alzheimer disease mouse model: the relationship with neuronal damage. Journal of Neuropathology & Experimental Neurology. 2018;77(12):1163–1176. doi: 10.1093/jnen/nly102. [DOI] [PubMed] [Google Scholar]
- 3.An Y., Varma V.R., Varma S., Casanova R., Dammer E., Pletnikova O. Evidence for brain glucose dysregulation in Alzheimer's disease. Alzheimers Dement. 2018;14(3):318–329. doi: 10.1016/j.jalz.2017.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolaños J.P., Almeida A., Moncada S. Glycolysis: a bioenergetic or a survival pathway? Trends in Biochemical Sciences. 2010;35(3):145–149. doi: 10.1016/j.tibs.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 5.Demetrius L.A., Simon D.K. The inverse association of cancer and Alzheimer's: a bioenergetic mechanism. Journal of The Royal Society Interface. 2013;10(82):20130006. doi: 10.1098/rsif.2013.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alberini C.M., Cruz E., Descalzi G., Bessières B., Gao V. Astrocyte glycogen and lactate: new insights into learning and memory mechanisms. Glia. 2018;66(6):1244–1262. doi: 10.1002/glia.23250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atlante A., de Bari L., Bobba A., Amadoro G. A disease with a sweet tooth: exploring the Warburg effect in Alzheimer's disease. Biogerontology. 2017;18(3):301–319. doi: 10.1007/s10522-017-9692-x. [DOI] [PubMed] [Google Scholar]
- 8.Harris R.A., Tindale L., Lone A., Singh O., Macauley S.L., Stanley M. Aerobic glycolysis in the frontal cortex correlates with memory performance in wild-type mice but not the APP/PS1 mouse model of cerebral amyloidosis. Journal of Neuroscience. 2016;36(6):1871–1878. doi: 10.1523/JNEUROSCI.3131-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashraf A., Fan Z., Brooks D.J., Edison P. Cortical hypermetabolism in MCI subjects: a compensatory mechanism? European Journal of Nuclear Medicine and Molecular Imaging. 2015;42(3):447–458. doi: 10.1007/s00259-014-2919-z. [DOI] [PubMed] [Google Scholar]
- 10.Adriaanse S.M., van Dijk K.R., Ossenkoppele R., Reuter M., Tolboom N., Zwan M.D. The effect of amyloid pathology and glucose metabolism on cortical volume loss over time in Alzheimer's disease. European Journal of Nuclear Medicine and Molecular Imaging. 2014;41(6):1190–1198. doi: 10.1007/s00259-014-2704-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bobba A., Amadoro G., La Piana G., Calissano P., Atlante A. Glycolytic enzyme upregulation and numbness of mitochondrial activity characterize the early phase of apoptosis in cerebellar granule cells. Apoptosis. 2015;20(1):10–28. doi: 10.1007/s10495-014-1049-1. [DOI] [PubMed] [Google Scholar]
- 12.Fu W., Shi D., Westaway D., Jhamandas J.H. Bioenergetic mechanisms in astrocytes may contribute to amyloid plaque deposition and toxicity. Journal of Biological Chemistry. 2015;290(20):12504–12513. doi: 10.1074/jbc.M114.618157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Demetrius L.A., Magistretti P.J., Pellerin L. Alzheimer's disease: the amyloid hypothesis and the Inverse Warburg effect. Frontiers in Physiology. 2014;5:522. doi: 10.3389/fphys.2014.00522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Athauda D., Foltynie T. Protective effects of the GLP-1 mimetic exendin-4 in Parkinson's disease. Neuropharmacology. 2018;136(Pt B):260–270. doi: 10.1016/j.neuropharm.2017.09.023. [DOI] [PubMed] [Google Scholar]
- 15.Hölscher C. Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer's and Parkinson's disease models. Neuropharmacology. 2018;136(Pt B):251–259. doi: 10.1016/j.neuropharm.2018.01.040. [DOI] [PubMed] [Google Scholar]
- 16.Drucker D.J. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metabolism. 2018;27(4):740–756. doi: 10.1016/j.cmet.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 17.Gejl M., Gjedde A., Egefjord L., Møller A., Hansen S.B., Vang K. In alzheimer's disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial. Frontiers in Aging Neuroscience. 2016;8:108. doi: 10.3389/fnagi.2016.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlessi R., Chen Y., Rowlands J., Cruzat V.F., Keane K.N., Egan L. GLP-1 receptor signalling promotes β-cell glucose metabolism via mTOR-dependent HIF-1α activation. Scientific Reports. 2017;7(1):2661. doi: 10.1038/s41598-017-02838-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siraj M.A., Mundil D., Beca S., Momen A., Shikatani E.A., Afroze T. Cardioprotective GLP-1 metabolite prevents ischemic cardiac injury by inhibiting mitochondrial trifunctional protein-α. Journal of Clinical Investigation. 2020;130(3):1392–1404. doi: 10.1172/JCI99934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lund A., Knop F.K., Vilsbøll T. Glucagon-like peptide-1 receptor agonists for the treatment of type 2 diabetes: differences and similarities. European Journal of Internal Medicine. 2014;25(5):407–414. doi: 10.1016/j.ejim.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 21.Qi L., Chen Z., Wang Y., Liu X., Liu X., Ke L. Subcutaneous liraglutide ameliorates methylglyoxal-induced Alzheimer-like tau pathology and cognitive impairment by modulating tau hyperphosphorylation and glycogen synthase kinase-3β. American Journal of Translational Research. 2017;9(2):247–260. [PMC free article] [PubMed] [Google Scholar]
- 22.Vorhees C.V., Williams M.T. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nature Protocols. 2006;1(2):848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Y., Huang L., Zheng W., An J., Zhan Z., Wang L. Recurrent nonsevere hypoglycemia exacerbates imbalance of mitochondrial homeostasis leading to synapse injury and cognitive deficit in diabetes. American Journal of Physiology. Endocrinology and Metabolism. 2018;315(5):973–e986. doi: 10.1152/ajpendo.00133.2018. [DOI] [PubMed] [Google Scholar]
- 24.Kuszczyk M.A., Sanchez S., Pankiewicz J., Kim J., Duszczyk M., Guridi M. Blocking the interaction between apolipoprotein E and Aβ reduces intraneuronal accumulation of Aβ and inhibits synaptic degeneration. American Journal Of Pathology. 2013;182(5):1750–1768. doi: 10.1016/j.ajpath.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoon G., Kim Y.K., Song J. Glucagon-like peptide-1 suppresses neuroinflammation and improves neural structure. Pharmacological Research. 2020;152:104615. doi: 10.1016/j.phrs.2019.104615. [DOI] [PubMed] [Google Scholar]
- 26.Baik S.H., Kang S., Lee W., Choi H., Chung S., Kim J.I. A breakdown in metabolic reprogramming causes microglia dysfunction in alzheimer's disease. Cell Metabolism. 2019;30(3):493–507. doi: 10.1016/j.cmet.2019.06.005. e496. [DOI] [PubMed] [Google Scholar]
- 27.Plain K.M., Marsh I.B., Waldron A.M., Galea F., Whittington A.M., Saunders V.F. High-throughput direct fecal PCR assay for detection of Mycobacterium avium subsp. paratuberculosis in sheep and cattle. Journal of Clinical Microbiology. 2014;52(3):745–757. doi: 10.1128/JCM.03233-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mahmoud S., Gharagozloo M., Simard C., Amrani A., Gris D. NLRX1 enhances glutamate uptake and inhibits glutamate release by astrocytes. Cells. 2019;8(5) doi: 10.3390/cells8050400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma S., Khare P., Kumar A., Chunduri V., Kumar A., Kapoor P. Anthocyanin-biofortified colored wheat prevents high fat diet-induced alterations in mice. Nutrigenomics Studies. 2020;64(13) doi: 10.1002/mnfr.201900999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sasagawa S., Nishimura Y., Okabe S., Murakami S., Ashikawa Y., Yuge M. Downregulation of GSTK1 is a common mechanism underlying hypertrophic cardiomyopathy. Frontiers in Pharmacology. 2016;7:162. doi: 10.3389/fphar.2016.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang W., Zheng Y., Xia Y., Ji H., Chen X., Guo F. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nature Cell Biology. 2012;14(12):1295–1304. doi: 10.1038/ncb2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. The International Journal of Biochemistry & Cell Biology. 2011;43(7):969–980. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 33.Griffith C.M., Macklin L.N., Cai Y., Sharp A.A., Yan X.X., Reagan L.P. Impaired glucose tolerance and reduced plasma insulin precede decreased AKT phosphorylation and GLUT3 translocation in the Hippocampus of old 3xTg-AD mice. Journal of Alzheimers Disease. 2019;68(2):809–837. doi: 10.3233/JAD-180707. [DOI] [PubMed] [Google Scholar]
- 34.Zhou J.Y., Poudel A., Welchko R., Mekala N., Chandramani-Shivalingappa P., Rosca M.G. Liraglutide improves insulin sensitivity in high fat diet induced diabetic mice through multiple pathways. European Journal of Pharmacology. 2019;861:172594. doi: 10.1016/j.ejphar.2019.172594. [DOI] [PubMed] [Google Scholar]
- 35.Alburquerque-Béjar J.J., Barba I., Inserte J., Miró-Casas E., Ruiz-Meana M., Poncelas M. Combination therapy with remote ischaemic conditioning and insulin or exenatide enhances infarct size limitation in pigs. Cardiovascular Research. 2015;107(2):246–254. doi: 10.1093/cvr/cvv171. [DOI] [PubMed] [Google Scholar]
- 36.Cunnane S.C., Trushina E., Morland C., Prigione A., Casadesus G., Andrews Z.B. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nature Reviews Drug Discovery. 2020;19(9):609–633. doi: 10.1038/s41573-020-0072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Gijsel-Bonnello M., Baranger K., Benech P., Rivera S., Khrestchatisky M., de Reggi M. Metabolic changes and inflammation in cultured astrocytes from the 5xFAD mouse model of Alzheimer's disease: alleviation by pantethine. PloS One. 2017;12(4) doi: 10.1371/journal.pone.0175369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ashok B.S., Ajith T.A., Sivanesan S. Hypoxia-inducible factors as neuroprotective agent in Alzheimer's disease. Clinical and Experimental Pharmacology and Physiology. 2017;44(3):327–334. doi: 10.1111/1440-1681.12717. [DOI] [PubMed] [Google Scholar]
- 39.Velliquette R.A., O'Connor T., Vassar R. Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer's disease pathogenesis. Journal of Neuroscience. 2005;25(47):10874–10883. doi: 10.1523/JNEUROSCI.2350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dibble C.C., Cantley L.C. Regulation of mTORC1 by PI3K signaling. Trends in Cell Biology. 2015;25(9):545–555. doi: 10.1016/j.tcb.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park S.H., Park J.H., Shim H.M., Na A.Y., Bae K.C., Lim J.G. Protection of pancreatic β-cells against glucotoxicity by short-term treatment with GLP-1. Biochemical and Biophysical Research Communications. 2015;459(4):561–567. doi: 10.1016/j.bbrc.2015.02.139. [DOI] [PubMed] [Google Scholar]
- 42.Jha M.K., Lee I.K., Suk K. Metabolic reprogramming by the pyruvate dehydrogenase kinase-lactic acid axis: linking metabolism and diverse neuropathophysiologies. Neuroscience & Biobehavioral Reviews. 2016;68:1–19. doi: 10.1016/j.neubiorev.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 43.Cheng S.C., Quintin J., Cramer R.A., Shepardson K.M., Saeed S., Kumar V. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345(6204):1250684. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roumes H., Dumont U., Sanchez S., Mazuel L., Blanc J., Raffard G. Neuroprotective role of lactate in rat neonatal hypoxia-ischemia. Journal of Cerebral Blood Flow and Metabolism. 2020 doi: 10.1177/0271678X20908355. 271678x20908355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roumes H., Dumont U., Sanchez S., Mazuel L., Blanc J., Raffard G. Neuroprotective role of lactate in rat neonatal hypoxia-ischemia. Journal of Cerebral Blood Flow and Metabolism. 2020 doi: 10.1177/0271678X20908355. 271678x20908355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carrard A., Elsayed M., Margineanu M., Boury-Jamot B., Fragnière L., Meylan E.M. Peripheral administration of lactate produces antidepressant-like effects. Molecular Psychiatry. 2018;23(2):392–399. doi: 10.1038/mp.2016.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El Hayek L., Khalifeh M., Zibara V., Abi Assaad R. Lactate mediates the effects of exercise on learning and memory through SIRT1-dependent activation of hippocampal brain-derived neurotrophic factor (BDNF) Journal of Neuroscience. 2019;39(13):2369–2382. doi: 10.1523/JNEUROSCI.1661-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herrero-Mendez A., Almeida A., Fernández E., Maestre C., Moncada S., Bolaños J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nature Cell Biology. 2009;11(6):747–752. doi: 10.1038/ncb1881. [DOI] [PubMed] [Google Scholar]
- 49.Dringen R., Brandmann M., Hohnholt M.C., Blumrich E.M. Glutathione-dependent detoxification processes in astrocytes. Neurochemical Research. 2015;40(12):2570–2582. doi: 10.1007/s11064-014-1481-1. [DOI] [PubMed] [Google Scholar]
- 50.Camins A., Ettcheto M., Busquets O., Manzine P.R., Castro-Torres R.D., Beas-Zarate C. Triple GLP-1/GIP/glucagon receptor agonists, a potential novel treatment strategy in Alzheimer's disease. Expert Opinion on Investigational Drugs. 2019;28(1):93–97. doi: 10.1080/13543784.2019.1552677. [DOI] [PubMed] [Google Scholar]
- 51.Bolaños J.P. Bioenergetics and redox adaptations of astrocytes to neuronal activity. Journal of Neurochemistry. 2016;139(Suppl 2):115–125. doi: 10.1111/jnc.13486. (Suppl 2) [DOI] [PMC free article] [PubMed] [Google Scholar]