

Abstract
Objective
To assess the burden of rare genetic variants and to estimate the contribution of known amyotrophic lateral sclerosis (ALS) genes in an Italian population-based cohort, we performed whole genome sequencing in 959 patients with ALS and 677 matched healthy controls.
Methods
We performed genome sequencing in a population-based cohort (Piemonte and Valle d'Aosta Registry for ALS [PARALS]). A panel of 40 ALS genes was analyzed to identify potential disease-causing genetic variants and to evaluate the gene-wide burden of rare variants among our population.
Results
A total of 959 patients with ALS were compared with 677 healthy controls from the same geographical area. Gene-wide association tests demonstrated a strong association with SOD1, whose rare variants are the second most common cause of disease after C9orf72 expansion. A lower signal was observed for TARDBP, proving that its effect on our cohort is driven by a few known causal variants. We detected rare variants in other known ALS genes that did not surpass statistical significance in gene-wise tests, thus highlighting that their contribution to disease risk in our cohort is limited.
Conclusions
We identified potential disease-causing variants in 11.9% of our patients. We identified the genes most frequently involved in our cohort and confirmed the contribution of rare variants in disease risk. Our results provide further insight into the pathologic mechanism of the disease and demonstrate the importance of genome-wide sequencing as a diagnostic tool.
Amyotrophic lateral sclerosis (ALS; OMIM 105400) is a neurologic disorder characterized by progressive degeneration of upper and lower motor neurons resulting in progressive paralysis and, ultimately, death from respiratory failure. Approximately 10% of patients with ALS report a family history, whereas the remainder of cases occur randomly.1 Since the discovery of pathogenic mutations in SOD1 in 1993,2 researchers have made great strides in delineating the genetic basis underlying ALS. More than 40 genes have been reported in association with the disease, and the genetic etiology of approximately two-thirds of familial cases is known.1,3
With few exceptions, targeted mutational screening has been performed on samples collected from patients attending ALS clinics. Such clinic-based cohorts represent a self-selected subset of the ALS population, making it challenging to determine accurate mutational rates. In contrast, the Piedmont and Valle d'Aosta Registry for ALS (PARALS) was established in 1995 to study the epidemiologic characteristics of the disease in Northern Italy.4 This comprehensive registry offers an opportunity to explore the frequency of mutations in a population-based cohort that has been in operation for over 20 years and has near-complete case ascertainment.
We analyzed the frequency and burden of mutations in 40 known ALS genes within a population-based cohort of nearly 1,000 Italian patients who underwent whole genome sequencing. Our efforts provide insight into the precise frequency of mutations in ALS genes in Northern Italy.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
Informed written consent was obtained from all participants, and the ethics committee of Azienda Ospedaliero Universitaria Città della Salute e della Scienza of Turin approved the study.
Study Participants
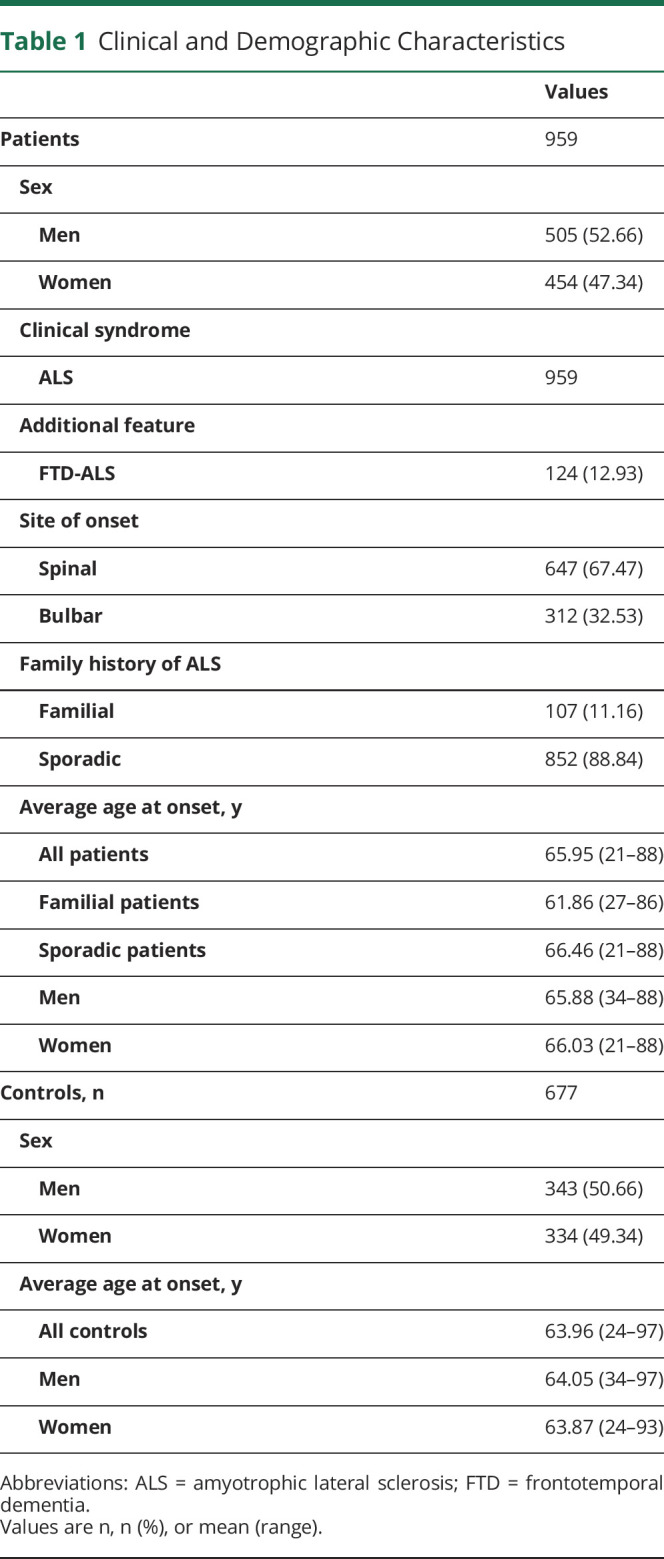
We included 959 patients who had been diagnosed with ALS and who had been enrolled in PARALS. PARALS is a prospective epidemiologic register that covers a population of 4.5 million from January 1, 1995.5 Patients are included in PARALS if they meet the diagnosis of definite, probable, or probable laboratory-supported ALS according to the revised El Escorial criteria.6 The 959 patients represent 72.9% of incident cases in the 2007–2015 period. Controls were chosen randomly from the same population and matched to the cases by sex, age (±5 years), and geographic location. Medical history was obtained for all controls and those with history of neurologic disease were excluded. Table 1 lists the clinical and demographic details of this cohort.
Table 1.
Clinical and Demographic Characteristics
Mutation Screening
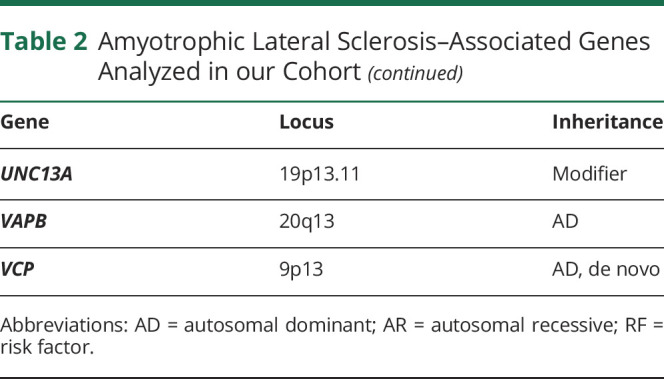
DNA was extracted from blood according to standard protocols. All participants were screened for the C9orf72 intronic expansion using a conventional repeat-primed PCR, as previously described.7 Repeat lengths of ≥30 units with the characteristic sawtooth pattern were considered to be pathogenic. Whole-genome sequencing was performed at The American Genome Center located at the Uniformed Services University on the Walter Reed National Military Medical Center campus in Bethesda, Maryland. Libraries were prepared using TruSeq DNA PCR-Free High Throughput Library Prep Kit (Illumina) as per the manufacturer's instructions, before sequencing on an Illumina HiSeqX10 sequencer using paired-end 150 base pair reads. Sequence reads were processed according to Genome Analysis Toolkit's (GATK) best practices (software.broadinstitute.org/gatk/best-practices/). Variant quality control was performed using the GATK variant quality score method with default filters. Genome Reference Consortium Human Build 38 was used as the reference. We extracted variant information from the data for 40 genes previously implicated in the pathogenesis of ALS3,8 (table 2).
Table 2.
Amyotrophic Lateral Sclerosis–Associated Genes Analyzed in our Cohort

Variant Annotation
We annotated all genotyped variants using KGGseq version 1.0 (grass.cgs.hku.hk/limx/kggseq/). We selected coding, nonsynonymous, and loss-of-function single nucleotide polymorphisms for analysis. Synonymous variants were used as a negative control.
Variant Filtering
Variants were filtered based on their minor allele frequencies (MAFs) in non-Finnish Europeans derived from the gnomAD (gnomad.broadinstitute.org/) database. Only variants with MAF <1% were retained.
Single Variant Association Testing
Firth regression was used for single variant association testing as it performs well in the study of unbalanced rare variants.9 Odds ratio (OR) estimates of the burden were derived by logistic regression using the Single Score test.
Gene Burden Analysis
To perform gene-based analysis, we further filtered rare variants based on their MAF in our cohort. As standard in gene-burden analysis, the upper ceiling for the MAF threshold was set at 5% and 1%.10 For gene burden testing, we applied different aggregation tests and set the false discovery rate (FDR) p value threshold at 0.05 and the Bonferroni-adjusted p value threshold at 1.25 × 10−2. p Values for Sequence-Based Kernel Association Test (SKAT), Combined-Multivariate Collapsing Burden Test, and Madson-Browning Burden Test were obtained using a permutation approach. Because none of these tests offers a clear advantage in terms of performance, we evaluated the concordance across the different tests to assess the importance of each gene. The analysis was repeated after removing the most influential variant from the dataset for significant genes.
All analyses were implemented in RVTEST (github.com/zhanxw/rvtests). The first 5 principal components, as calculated by flashPCA (github.com/gabraham/flashpca), and sex were included as covariates. FDR and Bonferroni-corrected significance threshold were calculated in R v.3.6.0 (r-project.org/).
Estimation of the Population Prevalence of Causative Genetic Variants
In addition to gene-based testing, we investigated the genetic structure of our ALS population by assessing the prevalence of causative mutations. We therefore applied further filtering criteria to select a high-confidence set of pathogenic variants associated with ALS. We report as patients with disease-causing mutation all the patients with ALS who carried a rare variant (MAF in European Non-Finnish population lower than 0.01%) who meet the criteria for clinical significance as recommended by the American College of Medical Genetics and the Association for Molecular Pathology for variant interpretation in Mendelian disorders11 and absent from the control population in our cohort. This highly conservative design is likely to minimize the risk of a false-positive genetic diagnosis.
Figure 1 provides a schematic visualization of the analytical workflow used in this study.
Figure 1. Experimental Workflow.

ACMG/AMP = American College of Medical Genetics and Genomics/Association for Molecular Pathology; ALS = amyotrophic lateral sclerosis; CMC = Combined-Multivariate Collapsing Burden Test; KBAC = Kernel-Based Adaptive Clustering Model; MAF = minor allele frequency; SKAT = Sequence-Based Kernel Association Test.
Data Availability
Data are available from the authors upon reasonable request.
Results
Frequency of Mutations in ALS Genes
We analyzed the sequences of 40 ALS-related genes in a population-based cohort of 959 Italian patients with ALS and 677 unaffected controls. Of the 959 patients examined, 71 carried the C9orf72 pathogenic repeat expansions, representing 7.4% of the entire group and 42.1% of the familial cases. An additional 34 patients carried causative mutations, representing a further 3.5% of the overall cohort and 13.1% of the familial cases. Figure 2 shows the mutational frequency of the genes identified in our cohort and table 3 describes the clinical features of mutation carriers.
Figure 2. Frequency of Major Amyotrophic Lateral Sclerosis (ALS) Gene Mutations in our Cohort.

(A) Relative contributions of ALS genes to the total estimate of patients harboring a causative variant. (B) Distribution of pathogenic variants among patients with sporadic ALS (sALS) and patients with ALS with a family history of the disease (fALS).
Table 3.
Pathogenic Mutations and Corresponding Clinical Features


Single Variant Analysis
After adjustment for multiple testing, only a single variant was associated with the disease. This was a c.2009T>A variant resulting in a valine to glutamic acid change at residue 670 of the neurofilament heavy gene (NEFH) (p value = 3.7 × 10−5, FDR-adjusted p value = 0.047). This variant (rs190692435) decreased the risk of ALS (OR 0.28, 95% confidence interval 0.13–0.55). Variants that are known to cause ALS did not surpass the significance threshold in our cohort, likely reflecting the fact that our sample size was insufficiently powered to detect them (table 4).
Table 4.
Results From Single-Variant Tests

Gene Burden Analysis Results
NEFH was significantly associated with the risk of ALS under the SKAT-O model at a minor allele threshold of 5% (p value = 7.7 × 10−5, FDR p value = 0.0035, figure 3). The gene-level test remained significant after removing the top variants in NEFH from the analysis.
Figure 3. Results of Rare Variant Association Testing.

Results of rare variant association testing with minor allele frequency (MAF) <5% (A) and <1% (B). Solid line: p value 0.05. Dashed line: Bonferroni-adjusted p value 0.001. All the log p values obtained by the gene-wise association tests applied in our study are reported. CMC = Combined-Multivariate Collapsing Burden Test17; Fp = burden test with rare variants upweighted using frequency controls20; Kbac = kernel-based adaptive clustering model16; Madsen-Browning = Madsen-Browning Burden Test with rare variants upweighted using inverse frequency controls19; Skat = Sequence-Based Kernel Association Test10; SkatO = Optimized Sequence-Based Kernel Association Test15; Zeggini = Zeggini-Morris Burden Test with aggregate counts of rare variants.18
The only other significant gene was SOD1 (p value under Madson-Browning analysis = 8.4 × 10−4, FDR p value = 0.036, figure 3). Based on Sanger sequence data, we had previously reported that variant to be the most prevalent in our cohort after the C9orf72 expansion.4
Repeating the gene burden analysis at a more stringent minor allele threshold of 1% found that SOD1 remained significantly associated with the risk of ALS (p value under the KBAC model = 7.0 × 10−4, FDR p value = 0.032). TARDBP was the second most enriched gene, although it did not reach statistical significance (p value under the KBAC model = 1.9 × 10−3, FDR p value = 0.084). The common founder mutation TARDBP p.A382T12 mostly drove the association signal for TARDBP (see table 4 for details).
As a negative control, we verified that supposedly neutral variation did not differ in frequency between case and control groups at MAF <5% (lowest FDR p value under SKAT-O model = 0.259) or MAF <1% (lowest FDR p value under SKAT-O model = 0.077). This null study is meant to reinforce the validity of the results obtained, excluding possible artifactual inflation or deflation of test statistics.
Discussion
We sequenced the genomes of 959 patients diagnosed with ALS and 677 neurologically healthy individuals obtained from an Italian population-based registry. We then estimated the frequency of mutations of major ALS genes in this population-based epidemiologic series. This study was an opportunity to understand the contribution of variants and genes to the genetic and phenotypic spectrum of ALS. We found that a significant portion of our cohort (n = 105 cases, 11.0%) carried mutations that are known to be pathogenic. This frequency of patients harboring a genetic mutation is consistent with previous epidemiologic studies in the same area.13 It is reasonable to assume that the application of less stringent criteria for pathogenic mutations would raise the prevalence of patients with ALS carrying at least one genetic variant, albeit at the cost of increasing false-positive results.
Our analysis demonstrates the utility of genome sequencing to assess the role of multiple genes in disease. Undertaking this type of broad analysis with traditional Sanger sequencing would be costly and time-consuming. Even if we applied restrictive criteria in the definition of causal genetic variants, we demonstrated that genome sequencing yields a broader insight into the genetic architecture of ALS than targeted sequencing or exome sequencing. Whole genome sequencing provides a complete catalog of rare variants present in patients and has the potential not only to answer specific diagnostic questions but also to uncover clinically important genetic information.
In gene-based rare variant association tests, SOD1 was the only gene to achieve significance for all of the frequency thresholds tested. Interestingly, we found low-frequency variants within the NEFH gene that lowered the risk of disease. There are few previous reports in the literature concerning protective variants in this gene in ALS. However, neurofilaments are currently regarded as a promising biomarker in ALS, possibly reflecting involvement in neuronal degeneration.14 It may be interesting to stratify NEFH blood and CSF measurements according to genotype in future studies.
The relatively small size of our cohort limited the power to detect a significant association. Furthermore, we did not consider other types of genetic mutations, such as intronic variants and structural variants that are known to be relevant to ALS. Thus, the mutational frequencies presented here are considered to be floor estimates. Nevertheless, our data allow us to conclude that coding mutations in these genes are not a common cause of sporadic disease.
In summary, we performed genome sequencing in a large Italian case–control cohort (n = 959 cases, n = 677 controls) to capture genetic variation across the ALS spectrum in our population. Our study provides evidence and an effective way to evaluate the role of low-frequency and rare variants underpinning the complex nature of ALS.
Glossary
- ALS
amyotrophic lateral sclerosis
- FDR
false discovery rate
- GATK
Genome Analysis Toolkit
- MAF
minor allele frequency
- OR
odds ratio
- PARALS
Piedmont and Valle d'Aosta Registry for ALS
- SKAT
Sequence-Based Kernel Association Test
Appendix. Authors


Acknowledgment
The authors wish to thank Yevgeniya Abramzon, BSc, Joshua Geiger, BSc, and Marya Sabir, BSc, for their assistance in preparing the DNA aliquots for the whole-genome sequencing experiments.
Study Funding
This work was supported by the Italian Ministry of Health (Ministero della Salute, Ricerca Sanitaria Finalizzata, grant RF-2016-02362405); the Progetti di Rilevante Interesse Nazionale program of the Ministry of Education, University and Research (grant 2017SNW5MB); the European Commission's Health Seventh Framework Programme (FP7/2007–2013 under grant agreement 259867); and the Joint Programme–Neurodegenerative Disease Research (Strength, ALS-Care and Brain-Mend projects), granted by Italian Ministry of Education, University and Research. This study was performed under the Department of Excellence grant of the Italian Ministry of Education, University and Research to the “Rita Levi Montalcini” Department of Neuroscience, University of Torino, Italy. This work was also supported by the Intramural Research Program of the NIH, National Institute on Aging (Z01-AG000949-02), and by the National Institute of Neurologic Disorders and Stroke (1ZIANS003154). The sponsor organizations had no role in data collection or analysis and did not participate in writing or approving the manuscript.
Disclosure
M. Grassano reports no disclosures. A. Calvo has received a research grant from Cytokinetics. C. Moglia, M. Brunetti, M. Barberis, L. Sbaiz, A. Canosa, U. Manera, R. Vasta, L. Corrado, S. D'Alfonso, L. Mazzini, S.W. Scholz, C. Dalgard, J. Ding, R.J. Gibbs, and R. Chia report no disclosures. B.J. Traynor holds European, Canadian, and American patents on the clinical testing and therapeutic intervention for the hexanucleotide repeat expansion of C9orf72. A. Chiò serves on scientific advisory boards for Mitsubishi Tanabe, Roche, and Cytokinetics, and has received a research grant from Italfarmaco. Go to Neurology.org/N for full disclosures.
References
- 1.Renton EA, Chiò A, Traynor JB. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 2014;17:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosen RD. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993;362:59–62. [DOI] [PubMed] [Google Scholar]
- 3.Chia R, Chiò A, Traynor JB. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol 2018;17:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiò A, Mora G, Calvo A, et al. Epidemiology of ALS in Italy: a 10-year prospective population-based study. Neurology 2009;72:725–731. [DOI] [PubMed] [Google Scholar]
- 5.Chiò A, Mora G, Moglia C, et al. Secular trends of amyotrophic lateral sclerosis: the Piemonte and Valle d'Aosta register. JAMA Neurol 2017;74:1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brooks RB, Miller GR, Swash M, Munsat LT; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Mot Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 7.Renton EA, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen HP, Van Broeckhoven C, van der Zee J. ALS genes in the genomic era and their implications for FTD. Trends Genet 2018;34:404–423. [DOI] [PubMed] [Google Scholar]
- 9.Ma C, Blackwell T, Boehnke M, Scott LJ. GoT2D investigators. Recommended joint and meta-analysis strategies for case-control association testing of single low-count variants. Genet Epidemiol 2013;37:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu CM, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet 2011;89:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corrado L, Ratti A, Gellera C, et al. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum Mutat 2009;30:688–694. [DOI] [PubMed] [Google Scholar]
- 13.Chiò A, Calvo A, Mazzini L, et al. Extensive genetics of ALS: a population-based study in Italy. Neurology 2012;79:1983–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steinacker P, Feneberg E, Weishaupt J, et al. Neurofilaments in the diagnosis of motoneuron diseases: a prospective study on 455 patients. J Neurol Neurosurg Psychiatry 2016;87:12–20. [DOI] [PubMed] [Google Scholar]
- 15.Lee S, Emond JM, Bamshad JM, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet 2012;91:224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu DJ, Leal SM. A novel adaptive method for the analysis of next-generation sequencing data to detect complex trait associations with rare variants due to gene main effects and interactions. PLoS Genet 2010;6:e1001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li B, Leal MS. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet 2008;83:311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morris PA, Zeggini E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol 2010;34:188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet 2009;5:e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin DY, Tang ZZ. A general framework for detecting disease associations with rare variants in sequencing studies. Am J Hum Genet 2011;89:354–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available from the authors upon reasonable request.