

Abstract
Introduction
As clinical trials move toward earlier intervention, we sought to redefine the β-amyloid (Aβ)-PET threshold based on the lowest point in a baseline distribution that robustly predicts future Aβ accumulation and cognitive decline in 3 independent samples of clinically normal individuals.
Methods
Sequential Aβ cutoffs were tested to identify the lowest cutoff associated with future change in cognition (Preclinical Alzheimer Cognitive Composite [PACC]) and Aβ-PET in clinically normal participants from the Harvard Aging Brain Study (n = 342), Australian Imaging, Biomarker and Lifestyle study of aging (n = 157), and Alzheimer's Disease Neuroimaging Initiative (n = 356).
Results
Within samples, cutoffs derived from future Aβ-PET accumulation and PACC decline converged on the same inflection point, beyond which trajectories diverged from normal. Across samples, optimal cutoffs fell within a short range (Centiloid 15–18.5).
Discussion
These optimized thresholds can help to inform future research and clinical trials targeting early Aβ. Threshold convergence raises the possibility of contemporaneous early changes in Aβ and cognition.
Classification of Evidence
This study provides Class II evidence that among clinically normal individuals a specific Aβ-PET threshold is predictive of cognitive decline.
Due to the largely disappointing results of antiamyloid therapies in patients with mild cognitive impairment (MCI) and Alzheimer disease (AD), many clinical trials are pivoting to intervention earlier in the AD pathologic trajectory to slow or prevent disease progression.1,2 Markers of β-amyloid (Aβ) can be used to target individuals earlier in the disease process3,4 through the use of PET radioligands for Aβ such as 11C-Pittsburgh compound B (PiB),5 18F-florbetapir (FBP),6 and 18F-NAV4694.7
Elevated, or abnormal, levels of Aβ-PET are most often defined on the basis of a threshold derived from cross-sectional Aβ-PET8–13 or from combined PET-neuropathology studies.14 Some evidence indicates that current thresholds might be too high,10,15 leading to the misclassification of some subthreshold individuals as Aβ−. Recent neuropathologic studies in participants with PET before death suggest that far lower PET thresholds can be achieved.14 However, without confirmation provided by autopsy on an individual basis, low PET signal is difficult to differentiate from noise, raising the possibility of introducing greater false positives.
Thus, our aim was to determine how low thresholds can go in the Aβ-PET signal while remaining useful as a predictor of whether individuals are on an AD trajectory, signaled by continued Aβ accumulation or cognitive decline. To evaluate reliability, we examined longitudinal data from 3 independent samples. We hypothesize that longitudinal cognitive and PET outcomes will provide justification for shifting to a threshold lower than that possible with cross-sectional bimodality-based approaches, thus potentially increasing potential benefit from anti-Aβ therapy and increasing eligibility for trials.
Methods
Subjects
Clinically normal (CN) participants (Clinical Dementia Rating score = 0; Mini-Mental State Examination [MMSE] score ≥24) were included from 3 independent samples: the Harvard Aging Brain Study (HABS), the Australian Imaging, Biomarker and Lifestyle (AIBL) study of aging, and the Alzheimer's Disease Neuroimaging Initiative (ADNI). All studies included participants with a baseline Clinical Dementia Rating score of 0 and excluded participants with clinical depression, neurologic disorders, and head trauma.
HABS Sample
HABS is focused on differentiating normal aging from AD and consequently recruited only individuals initially considered CN. Detailed inclusion criteria have been published previously.16 For the primary analysis of baseline PiB predicting cognitive change, all HABS participants (n = 342) were included. Cognitive assessments were conducted annually, with a median follow-up length of 4.21 (SD = 2.34) years. Analyses relating to PiB accumulation focused on the subset with at least 2 PiB-PET scans (n = 214), 141 with 3 scans, and 39 with 4 scans. The median follow-up time was 2.82 (2.15) years.
AIBL Sample
AIBL recruited individuals across the AD continuum and enriched its CN cohort for family history and memory complaints to ensure a high proportion of Aβ+ adults.17 Detailed inclusion criteria for AIBL have been published previously.17 One hundred fifty-seven CN participants were included from AIBL. Participants underwent cognitive testing approximately every 18 months, with a median cognitive follow-up of 7.48 (1.97) years. All participants had at least 1 cognitive follow-up visit; 123 had at least 2 PiB-PET scans, 87 had at least 3, and 54 had 4 PiB-PET scans, with a median PET follow-up of 3.06 (1.83) years.
ADNI Sample
ADNI is a multisite study conducted across 63 sites in the United States and Canada, enrolling individuals across the AD continuum. In contrast to HABS and AIBL, the ADNI CN cohort was required not to have memory complaints at baseline but also was allowed a more lenient MMSE score of ≥24 (HABS and AIBL used an MMSE score ≥25). Detailed inclusion criteria for ADNI have been published previously.18 Aβ-PET scanning with FBP was introduced to ADNI at a later stage, and only longitudinal cognitive data collected prospectively from the first FBP-PET scan were included. Participants were restricted to those with a CN diagnosis at first PET scan (n = 356). All participants had at least 1 cognitive follow-up visit (median cognitive follow-up 2.97 [2.33] years); 284 had at least 2 scans, 124 had 3 scans, and 7 had 4 scans, with a median PET follow-up of 2.05 (1.60) years.
Standard Protocol Approvals, Registrations, and Patient Consents
We conducted the procedures for this study under the Partners Human Research Committee, the Institutional Review Board for the Massachusetts General Hospital and Brigham and Women's Hospital. All participants provided written consent.
Cognition
Cognition was assessed with the Preclinical Alzheimer Cognitive Composite (PACC) 5 version19,20 of the PACC,21 which was honed to detect Aβ-related cognitive decline. As previously reported,20,22 the PACC5 was computed within each sample as the averaged z scores of 5 tests, with z scores standardized using the baseline mean and SD within each cohort. Of these 5 tests, 2 were overlapping across all samples: the MMSE and Logical Memory Delayed Recall. As a measure of executive function, Digit Symbol Substitution was used in HABS and AIBL, while ADNI used Trail Making Test Part B. As a measure of word recall, HABS used the Free and Cued Selective Reminding Test, AIBL used California Verbal Learning Test (second edition) ,and ADNI used Alzheimer's Disease Assessment Scale–Cognitive Battery Word Recall. For semantic memory, each study used versions of Categories, with HABS using Animals/Vegetables/Fruit, AIBL using Animals/Names, and ADNI using Animals.
All studies show similar distributions of baseline and slopes for these study-specific composites.20,22
PET
HABS PET
PiB-PET acquisition parameters for HABS have been published previously.16,23 In brief, PET images were acquired on a Siemens (Munich, Germany) ECAT EXACT HR+ scanner with a 60-minute dynamic acquisition starting directly after injection. Distribution volume ratios (DVRs) were calculated via Logan plotting with a cerebellar gray reference tissue. At each time point, PET data were rigidly coregistered to the individual's closest magnetization-prepared rapid gradient echo with SPM12 (Wellcome Department of Cognitive Neurology, Function Imaging Laboratory, London). Cortical regions of interest (ROIs) were defined from the Desikan-Killiany atlas24 via FreeSurfer version 6.0. Frontal, lateral temporal, and retrosplenial (FLR) regions were averaged into a global aggregate, as previously reported.8,20,23 PET data were not partial volume corrected.
AIBL PET
Detailed acquisition parameters have been published previously.9 PiB-PET images were processed in-house with a modified version of the HABS pipeline to account for the lack of full dynamic data. Fifty- to 70-minute frames were summed and coregistered to their MRI at each time point. Subject-specific cortical parcellations from FreeSurfer were unavailable, so instead each individual's PET was normalized to Montreal Neurological Institute space, and the same HABS FLR ROI was selected from a customized probabilistic template space atlas25 based on the Desikan-Killiany atlas. The standardized uptake value ratio (SUVR) was computed with FLR referenced to cerebellar gray.
ADNI PET
Detailed acquisition parameters were published previously.26 FBP-PET images were processed with the same pipeline used to process AIBL data, but the FBP SUVR was normalized to whole cerebellum.
Centiloid Scale
Due to our interests in identifying thresholds within the uncertain range between clearly Aβ− and Aβ+ positive, we prioritized using in-house methods that would maximize measurement accuracy within each sample. To compare between samples and to provide generalizable estimates, we then converted each sample to the Centiloid (CL) scale.27 Notably, the CL transformation is expected to reduce but not completely eliminate between-sample differences in acquisition and processing.28 CL values should therefore be considered approximate.
As recommended27 and published previously,20,25 we ran the GAAIN dataset using our in-house pipeline and ROIs to compute the linear transformation between the in-house and CL scales and then applied the CL transformation. An additional linear transformation was required to transform the ADNI data to translate from FBP SUVR to PiB SUVR using the GAAIN dataset, similar to previous recommendations.29 Two additional steps were used to transform the HABS data to CL due to differences in acquisition time from the CL method and the expression of PiB as a DVR.25 The resulting equations were as follows: HABS: CL = 143.06 × HABS DVR − 145.60; AIBL: CL = 96.94 × AIBL SUVR − 105.20; ADNI: CL = 180.20 × ADNI FBP SUVR − 179.70.
Statistical Analysis
All analyses were conducted in R version 3.6.0. For sample descriptives and comparison with optimal thresholds, thresholds were generated within each sample at baseline by fitting the data to a gaussian mixture model (GMM).8,20,23,30 In all samples, a bimodal 2-class model fit best, exhibiting the highest Bayesian information criterion (BIC) and significantly higher likelihood than 1-class or 3-class models in bootstrapped (n = 1,000) likelihood ratio tests. Mixed-effects models were used to account for missing data and loss to follow-up.
The first objective was to identify a cognitively derived PET cutoff separately in each sample, at the lowest point in baseline Aβ-PET distribution that predicted subsequent cognitive decline. This has been classified Class II evidence that among CN individuals a specific Aβ-PET threshold is predictive of cognitive decline. To do so, we conducted a series of 2-level linear mixture models to predict PACC5 performance over time iterating through a range of possible cutoffs (c) of Aβ-PET (i.e., 1.1–1.8 by 0.01), adjusting for sex, age at baseline, APOE ε4 status, and years of education. The first level of the model was as follows:
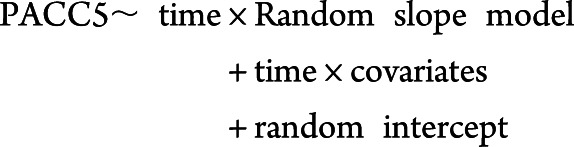 |
The second level was as follows:
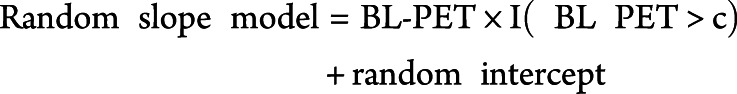 |
The nested random slope term accounts for change in PACC5 over time as a function of an threshold indicator I (baseline PET > c) that assigns a 0 or 1 depending on whether individuals fell above or below c and the continuous effect of tracer retention (baseline PET) above the cutoff. The rationale for this nested slope model was based on the assumption that below the optimal minimal cutoff the PACC5 should not relate to Aβ-PET tracer retention, whereas above the cutoff the magnitude of PACC5 decline is expected to track with the magnitude of Aβ-PET retention.12 The random intercept embedded within the nested random slope model represents an average measure of the baseline PET for all individuals in the sample regardless of the threshold value c, with random effects added to allow variations between individuals. The Akaike information criterion (AIC)/BIC was used to select the optimal cognitively derived 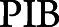 cutoff by comparing model fits under different baseline thresholds.31,32 Both are reported but identified the same cutoffs in all models. Sensitivity analysis was carried out with model expansion in both the PACC model and the PET model below to ensure that the optimal models were selected.
cutoff by comparing model fits under different baseline thresholds.31,32 Both are reported but identified the same cutoffs in all models. Sensitivity analysis was carried out with model expansion in both the PACC model and the PET model below to ensure that the optimal models were selected.
We also aimed to identify an accumulation-derived cutoff based on subsequent Aβ-PET accumulation. Here, we iterated through a series of linear mixed-effects model of the effect of time, Aβ-PET cutoff, the same set of covariates used above, and the random intercept on Aβ-PET with a range of possible cutoffs c:
The rationale for this model was based on previous evidence33–35 that at low Aβ levels roughly equivalent proportions of negative and positive PET changes are observed and are presumed to largely reflect fluctuations in nonspecific binding of the tracer, but as baseline PET increases, the observed changes become predominately positive and reflect contributions from both nonspecific fluctuations and Aβ accumulation. We sought to identify the threshold beyond which changes are predominately positive and thus maximize the difference between Aβ+/− groups. To do so, we iteratively compared models with different baseline Aβ-PET thresholds and selected the model that maximized the group differences in change36 as reflected by the greatest effect size (standardized β) for the Aβ group × time term. This selected criterion differs from that used for the PACC model. Goodness of fit was still tested with AIC/BIC, but due to the greater dynamic range in DVR/SUVR compared with change over time, overall model fit is dominated by the between-participant differences in DVR/SUVR (the main effect of Aβ group) rather than within-participant change (time × Aβ group). To identify a cutoff based on optimal change over time, we selected the cutoff associated with the maximal effect size for the Aβ group × time term.
To further describe the additional individuals who would be identified as Aβ+ with the optimal cutoffs, participants were assigned into low (below optimal cutoff), intermediate (between low and GMM cutoffs), and high (above GMM cutoff) groups within each sample and then pooled into 1 large sample (n = 855). These groups were then compared on age, sex, APOE ε4 status, Aβ slope (CL/year), and PACC5 score slope (SD/year) with independent-sample t tests and χ2 tests.
Finally, the optimal threshold was used to conduct power analyses to project the sample size needed to detect Aβ-PET accumulation and PACC score decline over a 5-year clinical trial. In the pooled sample, 5 possible CL range groups were selected with the lower bound for inclusion set at the optimal CL threshold and the CL upper bound varying from 35, 50, 70, and 100 to the maximum observed in our samples (140). Within each group, we computed the sample size needed to detect different possible percent changes (10%–100%) in Aβ-PET accumulation and PACC score decline over 5 years with 80% power. Power to detect Aβ-PET accumulation was based on estimates from a linear mixed-effect model of CL ∼ time + random intercept. For PACC score decline, estimates were based on a linear mixed effect of PACC score ∼ time + random intercept + random slope.
Data Availability
All data for ADNI and PET data for AIBL are publicly available online at ida.loni.usc.edu. AIBL cognitive data and HABS data are available on request.
Results
Sample Differences
Table 1 displays descriptive statistics for the HABS, AIBL, and ADNI samples, dichotomized within sample using GMM into Aβ+/− groups. Between samples, AIBL had a numerically higher proportion of APOE ε4 carriers due to their enriched recruitment strategy,17 although it did not reach statistical significance compared with HABS (MDifference = 0.07, p = 0.15, 95% confidence interval [CI] −0.03 to 0.17) or ADNI (MD = 0.08, p = 0.08, 95% CI 0.01–0.18). ADNI participants were older (MD = 2.91, p < 0 .001, 95% CI 1.83–3.98) and more highly educated than HABS participants (MD = 0.70, p < 0 .001, 95% CI 0.28–1.11) and older than AIBL participants (MD = 2.17, p < 0 .001, 95% CI 0.92–3.43), although HABS and AIBL participants did not differ by age. AIBL participants had a longer cognitive follow-up than HABS (MD = 2.99, p < 0 .001, 95% CI 2.57–3.41) and ADNI (MD = 3.31, p < 0 .001, 95% CI 2.89–3.73) participants.
Table 1.
Sample Descriptives for HABS, AIBL, and ADNI

Overall, each sample exhibited typical bimodal Aβ-PET distributions (figure 1) that mapped onto a similar range on the CL scale, but some small differences are noted. The AIBL sample had a greater SUVR range than HABS and ADNI participants. Notably, the GMM in AIBL also provided a lower CL threshold for positivity (CLGMM = 19.0) than in HABS (CLGMM = 24.6) or ADNI (CLGMM = 25.7). In ADNI, a larger variance was observed in the Aβ2 group, but this variance translated into more negative CL values rather than a higher GMM threshold. The proportion of Aβ+ participants based on the GMM threshold varied by sample, with a lower proportion in HABS (22.5%) than in AIBL (32.5%, χ2 = 5.61, p = 0.02) and ADNI (29.0% χ2 = 4.08, p = 0.04). The varied mixing proportions may contribute to slight differences in how the distributions transform to the CL scale.
Figure 1. Bimodal Gaussian Distribution of PET Signal in HABS, AIBL, and ADNI.

A–C) Histograms of the baseline (BL) PET data in each sample are plotted and fitted to a bimodal gaussian distribution using gaussian mixture model (GMM). Data are plotted on the tracer for each sample and processing-specific scale on the bottom x-axis and transformed to the Centiloid (CL) scale on the top x-axis. The GMM threshold (purple line) was identified within each sample as the point at which an individual had an equal probability of being in the lower β-amyloid (Aβ)− and higher Aβ+ gaussian. ADNI = Alzheimer's Disease Neuroimaging Initiative; AIBL = Australian Imaging, Biomarker and Lifestyle; DVR = distribution volume ratio; HABS = Harvard Aging Brain Study; PiB = Pittsburgh compound B; SUVR = standardized uptake value ratio.
Cognitively Derived Cutoff
We detected similar optimal cutoffs associated with cognitive decline across the 3 samples: HABS at a PiB DVR of 1.14 (CL = 17.5), AIBL at a PiB SUVR of 1.24 (CL = 15.0), and ADNI at an FBP SUVR of 1.1 (CL = 18.5) (figure 2). The optimal cutoff was selected from the model with absolute lowest AIC/BIC in HABS, and the lower of 2 thresholds associated with similar AIC/BIC in AIBL and ADNI (table 2). Across samples, the optimal cutoff fell 0.04 to 0.05 SUVR/DVR below their respective GMM cutoff. As shown in figure 1, the cutoff corresponds to an approximate inflection point in the baseline Aβ-PET distribution beyond which cognitive slopes begin to negatively deviate from zero.
Figure 2. Optimal Cutoffs Within HABS, AIBL, and ADNI Based on Longitudinal Cognitive Outcomes.

For each sample, the Akaike information criterion (AIC) demonstrating the model fit for a range of possible cutoffs are shown (A, C, E). In Harvard Aging Brain Study (HABS) (A), an optimal cutoff of Pittsburgh compound B (PiB) distribution volume ratio (DVR) 1.14/Centiloid (CL) 17.5 is derived from a clear lowest AIC (least information loss). In Australian Imaging, Biomarker and Lifestyle (AIBL), similarly low AIC was achieved twice, and the lower cutoff at a PiB standardized uptake value ratio (SUVR) 1.24/CL 15.0 was selected. Likewise, in Alzheimer's Disease Neuroimaging Initiative (ADNI) an optimal cutoff of 18F-florbetapir (FBP) SUVR 1.1/CL 18.5 was selected as the lower cutoff from 2 similarly well-fitting models with nearly equivalent AIC. Notably, across samples, the optimal cutoffs based on cognitive decline fit in a tight CL range of 15.0 to 18.5. (B, D, F) Preclinical Alzheimer Cognitive Composite 5 version (PACC5) score slope over time is plotted as a function of baseline (BL) PET within each sample using a loess curve to demonstrate the shift in trajectories of change with increasing baseline β-amyloid (Aβ)-PET tracer retention. PACC5 score slope for each participant was extracted from the slope of the linear regression of PACC5 score over time. Data are unadjusted for covariates. In each sample, the magnitude of tracer retention is unrelated to PACC score slope until an inflection point is reached, beyond which greater levels of Aβ-tracer retention are associated with increasing rates of cognitive decline on the PACC5. The inflection point corresponds to the optimal cutoff derived from the iterative models of PACC5 score over time.
Table 2.
Model Fit and Parameter Estimates at Optimal and GMM cutoffs in HABS, AIBL, and ADNI

Accumulation-Derived Cutoff
Analyses of Aβ-PET accumulation–derived optimal thresholds converged on the same threshold as with those derived from cognitive outcomes in HABS (PiB DVR 1.14/CL = 17.5), and AIBL (PiB SUVR1.24/CL = 15.0) and nearly converged in ADNI (FBP SUVR 1.09/CL = 16.7). Across samples, the standardized β criterion provided clear cutoffs for maximizing group differences in accumulation (figure 3). As shown in figure 3, these cutoffs corresponded to an inflection point in baseline Aβ-PET distribution beyond which PET slopes shift from a roughly equal mix of negative and positive PET slopes to predominately positive slopes. In contrast, the GMM cutoff had lower standardized β values (table 2) and failed to identify several individuals with low baseline but high accumulation as Aβ+.
Figure 3. Optimal Cutoffs Within HABS, AIBL, and ADNI Based on Longitudinal Aβ-PET Accumulation.

For each sample, the standardized β for the cutoff × time interaction term from iterative models across a range of possible cutoffs is shown (A, C, E). The optimal cutoff based on Aβ-PET accumulation was set at the cutoff that gave the highest standardized β in each sample. In Harvard Aging Brain Study (HABS) and Australian Imaging, Biomarker and Lifestyle (AIBL), the β-amyloid (Aβ) accumulation–derived optimal cutoff is identical to the cognitively derived optimal cutoff: (A) HABS: Pittsburgh compound B (PiB) distribution volume ratio (DVR) 1.14/Centiloid (CL) 17.5; (C) AIBL: PiB standardized uptake value ratio (SUVR) 1.24/CL 15.0. In Alzheimer's Disease Neuroimaging Initiative (ADNI), the Aβ accumulation–derived optimal cutoff (18F-florbetapir [FBP] SUVR 1.09, CL 16.7) was very slightly lower than the cognitively derived cutoff (FBP SUVR 1.1, CL 18.5). (B, D, F) DVR/SUVR slope over time is plotted as a function of baseline DVR/SUVR within each sample using a loess curve to demonstrate the shift in trajectories of change as function of baseline Aβ-PiB tracer retention. DVR/SUVR slope for each participant was extracted from the slope of the linear regression of DVR/SUVR over time. Data are unadjusted for covariates. In each sample, the slopes below the optimal cutoff consist of a roughly equal proportion of both negative and positive slopes, presumed to reflect random fluctuations in signal noise. Increasing baseline DVR/SUVR is associated with a small negative trend in this range suggestive of regression to the mean. The optimal cutoff appears to mark a shift toward more positive slopes presumed to reflect Aβ accumulation.
Participants Between the Optimal and GMM Cutoffs
A total of 54 individuals were identified with intermediate Aβ between the optimal and GMM cutoffs across samples (table 3). This group was also intermediate between the low Aβ and high Aβ group in terms of age (MD_low_int = 2.36, p = 0.01, 95% CI 0.52–4.20; MD_int_high = −1.58, p = 0.10, 95% CI −3.49 to 0.33), number of APOE ε4 carriers (MD_low_int = 0.113, p = 0.07, 95% CI −0.03 to 0.25; MD_int_high = −0.21, p = 0.01, 95% CI −0.36 to −0.06), and PACC5 score slope (MD_low_int = −0.07, p = 0.05, 95% CI −0.001 to 0.12; MD_int_high = −0.17, p = 0.01, 95% CI −0.31 to −0.03). Notably, the intermediate Aβ group had CL slopes comparable to those of the high Aβ group and higher CL slopes than the low Aβ group (MD_low_int = 2.26, p < 0 .001, 95% CI 1.09–3.43; MD_int_high = −0.64, p = 0.40, 95% CI −0.86 to 2.14).
Table 3.
Descriptives for Participants Between Optimal and GMM Cutoffs Pooled Across Samples

Power to Detect Reduced Aβ-PET Accumulation and PACC Score Decline
Power analyses (figure 4) demonstrated that a CL in the 18 to 35 range would require the smallest sample size to detect reduced Aβ-PET accumulation (n = 64 for 20% reduction over 5 years). In contrast, trials with a primary cognitive outcome would exhibit optimal power using a wider CL range of 18 to 70. Targeting individuals in the 18 to 50 CL range for an early intervention trial would provide a balance between minimizing sample size needed to detect PACC score decline while maintaining high power to detect reduced Aβ-PET accumulation (n = 886).
Figure 4. Sample Sizes Needed to Detect Aβ-PET Accumulation and PACC Score Decline.

(A and B) Sample size needed per arm to detect varying levels of change over 5 years with 80% power in (A) β-amyloid (Aβ)-PET accumulation and (B) Preclinical Alzheimer Cognitive Composite (PACC) score decline for groups with different ranges of Aβ burden measured in Centiloid (CL) at baseline. Power analyses were estimated from a combined Harvard Aging Brain Study (HABS), Alzheimer's Disease Neuroimaging Initiative (ADNI), and Australian Imaging, Biomarker and Lifestyle (AIBL) dataset measured in CL. As shown in the red line, an early intervention trial targeting individuals in the low 18 to 35 CL range would require the smallest sample to detect Aβ-PET accumulation because individuals in this range typically exhibit the highest rates of accumulation. However, detecting PACC decline in the low 18 to 35 CL range would necessitate very large sample sizes. Targeting individuals in the 18 to 50 CL range would minimize the sample size needed to detect PACC score decline while maximizing power to detect reduced Aβ-PET accumulation.
Discussion
As the field moves toward intervening earlier in the course of the disease, it is important to ascertain the lowest point in the AD-related biomarker cascade that can robustly identify individuals who are embarking on the AD pathologic trajectory and effectively exclude those unlikely to benefit from intervention. Here, we focused on Aβ-PET markers because they are currently the primary method for recruiting at-risk CN individuals in many early intervention trials.1,37 Independently deriving baseline Aβ cutoffs for PET on the basis of the prediction of both future cognitive decline and Aβ accumulation converged on the same or very nearly the same point in the distribution across 3 independent samples. Furthermore, the 3 samples indicated an optimal threshold in the short CL range of 15.0 to 18.5. In each sample, the optimal cutoff fell at an inflection point beyond which Aβ-PET and cognitive trajectories diverged from normal. This point was consistently below the GMM-derived cutoff that relies primarily on the bimodality of the Aβ distribution but was higher than cutoffs previously reported and based on autopsy data.14 Individuals in the intermediate range between the optimal and GMM cutoffs were older than those in the lowest PET tracer uptake range (below the optimal cutoff) and exhibited Aβ accumulation comparable to that displayed in those with the highest PET tracer uptake (above the GMM cutoff). The lower optimal cutoff allows excellent power to detect reductions in Aβ accumulation and sufficient power to detect slowing of cognitive decline in preclinical individuals with low to moderate Aβ burden. Consequently, this threshold may prove useful for research and clinical trials targeting individuals in the early stages of amyloidosis.
Within each sample, the optimal cutoff derived from cognitive outcomes converged on the identical or nearly identical cutoff derived from Aβ accumulation. This may suggest a more contemporaneous onset of change in Aβ and cognition than would be expected from the amyloid cascade hypothesis,38,39 which posits a cognitive lag behind Aβ accumulation. It is important to note, however, that these optimal thresholds remain thresholds of detection and not necessarily initial onset of Aβ. Undoubtedly, toxic Aβ species exist, both soluble and insoluble isoforms, below these lower PET-detection thresholds, because PET tracers primarily have been found to bind to aggregated fibrillar Aβ.40 The converging cutoffs based on Aβ-PET accumulation and cognitive decline may instead reflect the lowest point in the PET distribution where signal exceeds noise and is therefore likely to predict future outcomes. While additional studies are needed to probe the possibility of contemporaneous changes in Aβ and cognition, the present findings add to mounting evidence that even very low levels of Aβ-PET signal are associated with subtle cognitive changes35,41,42 in advance of more severe decline observed at higher levels of Aβ pathology. Furthermore, while the observed PACC changes are subtle, it has recently been demonstrated in the same 3 cohorts that subtle decline over 3 years is predictive of future progression to MCI.22 While the present study was focused earlier in the AD spectrum, it is noted that individuals with high baseline Aβ and declining cognition were the most likely to progress to MCI.
The optimal thresholds remained in a short CL range despite the use of different tracers (PiB and FBP) across the 3 unique cohorts. Although FBP is known to exhibit greater nonspecific binding than PiB and consequently requires a more conservative whole cerebellum reference region,6 both the GMM and optimal thresholds remained in a similar CL range. FBP translated primarily into a wider Aβ range for ADNI, with CL values extending further into the negatives. Furthermore, despite the differences in sample populations, the robustness of the pattern of Aβ accumulation and cognitive decline across samples yielded highly similar thresholds. Exclusive recruitment of CN adults in HABS resulted in a lower proportion of Aβ+ participants but did not hinder detection of a lower optimal threshold. The multisite ADNI exhibited greater variance in measurement over time, leading to a more modest relationship between Aβ and cognitive decline, but still arrived at a comparable threshold. In AIBL, enrichment for family history and memory complaints led to fewer individuals with low to moderate Aβ and a greater dynamic range in the PiB SUVR distribution. Despite the resulting slight discrepancy in the translation of AIBL to the CL scale, the observed 15 CL optimal threshold remained very close to 16.7 to 18.5 CL observed in HABS and ADNI.
Much attention in recent years has been paid to the question of whether existing thresholds are too high.10,43 Our CL range of 15 to 18.5 in HABS, ADNI, and AIBL was closely corroborated by a fourth PiB sample (Mayo Clinic Study of Aging) that demonstrated reliable worsening of PiB accumulation beyond a CL 19.15 In each of our 3 samples, the optimal cutoff fell approximately 0.04 to 0.05 DVR/SUVR below their respective GMM cutoffs (CL 20–26) and below the CL 24 to 35 range most often implicated in studies using similar approaches.29,44–46
These thresholds do, however, sit slightly higher than the CL 12 cutoff that was recently suggested by autopsy and antemortem PET data from La Joie et al.14 Due to the lag between PET and autopsy, it is likely that if PET were acquired closer to death, Aβ accumulation in the intervening years would result in a higher CL threshold. However, a recent combined CSF-PET study47 found a CL 12 threshold based on CSF Aβ42 levels. Thus, it may instead be that autopsy and CSF allow earlier detection of Aβ than is possible with PET due to their sensitivity to earlier forms of Aβ, including diffuse plaques and soluble Aβ oligomers. Our findings (figures 2 and 3) suggest that a CL of 12 is likely to introduce false positives in PET. A CL in the 15 to 18.5 range therefore provides a good compromise, minimizing false positives that could weaken outcomes and introduce ethical considerations while increasing inclusion of those at higher risk for future deleterious changes along the AD trajectory.
The 15 to 18.5 CL range as a lower bound may thus be a useful target threshold for clinical trials, particularly those aiming to get closer to primary prevention. As posited by the amyloid cascade hypothesis38 and supported with recent evidence,48,49 Aβ accumulates very gradually over 2 decades or more34 and leads to neocortical tau proliferation, neurodegeneration, and cognitive decline. The success of some anti-Aβ therapies may depend on very early intervention at low to moderate Aβ levels, before individuals exhibit neocortical tau and neurodegeneration.1,37 Our analyses demonstrated that when focusing on individuals with low to moderate Aβ (18–50 CL), clinical trials will be well powered to detect changes in Aβ accumulation. Furthermore, despite the subtle rate of change in cognition in this earlier stage of preclinical AD, our power analyses indicate that cognition may be still be a useful outcome measure with larger sample sizes or longer follow-up. The exact threshold chosen for a given trial will depend on the specific goals of the study and the length of follow-up available to detect change; there are design tradeoffs with targeting earlier vs later stages of preclinical AD and potential sample size considerations with increased heterogeneity in baseline Aβ burden.
By modeling possible cutoffs separately, we limit our ability to estimate uncertainty in the cutoff. However, by using 3 independent sample with different tracers and scanners, we were able to demonstrate a short CL range (15–18.5) in which an optimal cutoff likely falls. Researchers should be cautious in selecting a single cutoff within this range because linear transformations to CL cannot be expected to completely account for all differences in scanner, acquisition, processing, tracer, and sample.25,28 Evidence suggests that linear transformation to a common scale may be particularly problematic at the low levels of tracer retention around which the threshold falls.25 Furthermore, while the CL recommendations27 for translating in-house pipelines and ROIs to the CL scale were followed, the additional transformations likely confer some additional error to the CL estimation. As a result, we elected to select a more conservative 18CL in our power analyses to minimize the inclusion of false positives due to measurement error. Clinical trials aimed at later AD stages should continue using more conservative existing thresholds to minimize false positives further.
In addition, ADNI had a higher proportion of individuals with only 2 PET scans, which reduces our ability to more robustly estimate Aβ change. While ≥3 scans is optimal for measuring change, we opted to use all data presently available. As these studies continue to gather larger samples with ≥3 scans available, it is possible that more refined PET change measures will result in even lower optimal thresholds. Participants in the HABS, AIBL, and ADNI samples are predominantly White, highly educated individuals with low cardiovascular risk, and future studies with more representative samples are needed. Finally, testing specific aspects of episodic memory such as free recall and more targeted regional composites50–52 may be even more sensitive to early detection.
Across 3 independent samples of CN older adults, we found evidence that lower thresholds for Aβ-PET in the CL range of 15.0 to 18.5 are optimally predictive of future Aβ accumulation and cognitive decline. This range appears to correspond to an inflection point in the Aβ-PET distribution beyond which Aβ and cognitive trajectories diverge from normal. Such thresholds may be useful in future studies and clinical trials using Aβ-PET imaging to detect and track the earliest stages of AD. Thresholds derived from future changes in Aβ and cognition converged across 3 samples, and future studies are needed to determine whether this phenomenon suggests that early changes in Aβ and cognition are contemporaneous or if this convergence reflects signal-to-noise properties of the PET tracers.
Glossary
- Aβ
β-amyloid
- AD
Alzheimer disease
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- AIBL
Australian Imaging, Biomarker and Lifestyle
- AIC
Akaike information criterion
- BIC
Bayesian information criterion
- CI
confidence interval
- CL
Centiloid
- CN
clinically normal
- DVR
distribution volume ratio
- FBP
18F-florbetapir
- FLR
frontal, lateral temporal, and retrosplenial
- GMM
gaussian mixture model
- HABS
Harvard Aging Brain Study
- MCI
mild cognitive impairment
- MMSE
Mini-Mental State Examination
- PACC
Preclinical Alzheimer Cognitive Composite
- PiB
Pittsburgh compound B
- ROI
region of interest
- SUVR
standardized uptake value ratio
Appendix. Authors

Footnotes
Class of Evidence: NPub.org/coe
Study Funding
This work was supported with funding from the NIH, including P01 AG036694 (Sperling and Johnson), P50 AG005134 (Sperling, Johnson), and K24 AG035007 (Sperling). This research was carried out in part at the Athinoula A. Martinos Center for Biomedical Imaging at the Massachusetts General Hospital using resources provided by the Center for Functional Neuroimaging Technologies, P41EB015896, a P41 Biotechnology Resource Grant supported by the National Institute of Biomedical Imaging and Bioengineering, NIH. This work also involved the use of instrumentation supported by the NIH Shared Instrumentation Grant Program and/or High-End Instrumentation Grant Program, specifically grants S10RR021110, S10RR023401, and S10RR023043. Data collection and sharing for this project were funded by the ADNI (NIH grant U01 AG024904) and DOD ADNI (Department of Defense award W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, by the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc; Biogen; Bristol-Myers Squibb Co; CereSpir, Inc; Cogstate; Eisai Inc; Elan Pharmaceuticals, Inc; Eli Lilly and Co; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc; Fujirebio; GE Healthcare; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co, Inc; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corp; Pfizer Inc; Piramal Imaging; Servier; Takeda Pharmaceutical Co; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. M.E.F. is funded by the BrightFocus Foundation Postdoctoral Fellowship (2018A015289). K.A.J. receives funding from NIH grants R01EB014894, R21 AG038994, R01 AG026484, R01 AG034556, P50 AG00513421, U19 AG10483, P01 AG036694, R13 AG042201174210, R01 AG027435, and R01 AG037497 and the Alzheimer's Association grant ZEN-10-174210. R.A.S. receives research support from the following grants: P01 AG036694, U01 AG032438, U01 AG024904, R01 AG037497, R01 AG034556, K24 AG035007, P50 AG005134, U19 AG010483, R01 AG027435, Fidelity Biosciences, Harvard NeuroDiscovery Center, and the Alzheimer's Association. R.F.B. is funded by the NIH Pathway to Independence Award (K99AG061238), the Charlestown Conference for Alzheimer's Disease, and the Brigham and Women's Hospital Women's Brain Initiative. H.I.J. receives funding from the NIH–National Institute on Aging R01 AG062559.
Disclosure
Dr. Farrell and Dr. Jiang have no disclosures. Dr. Schultz has been a paid consultant for Janssen Pharmaceuticals and Biogen. Mr. Properzi has no disclosures. Dr. Price serves on the Scientific Advisory Council to director of the Center for Scientific Review. Dr. Becker, Dr. Jacobs, and Dr. Hanseeuw have no disclosures. Dr. Rentz has served as a consultant for Eli Lilly, Biogen Idec, and Digital Cognition Technologies and serves as a member of the Scientific Advisory Board for Neurotrack. Dr. Papp has served as an advisor to Biogen Idec and Digital Cognition Technologies. Dr. Mormino has served as paid consultant for Roche and Alector. Dr. Betensky has no disclosures. Dr. Johnson has served as paid consultant for Bayer, GE Healthcare, Janssen Alzheimers Immunotherapy, Siemens Medical Solutions, Genzyme, Novartis, Biogen, Roche, ISIS Pharma, AZTherapy, GEHC, Lundberg, and Abbvie. He is a site coinvestigator for Eli Lilly/Avid, Pfizer, Janssen Immunotherapy, and Navidea. He has spoken at symposia sponsored by Janssen Alzheimers Immunotherapy and Pfizer. Dr. Sperling has served as a paid consultant for AC Immune, Biogen, Cytox, Janssen, Neurocentria, Prothena, and Roche. She has received research support as an investigator for Eli Lilly, Janssen, and Eisai AD clinical trials. Dr. Buckley has no disclosures. Go to Neurology.org/N for full disclosures.
References
- 1.Sperling R, Mormino E, Johnson K. The evolution of preclinical Alzheimer's disease: implications for prevention trials. Neuron 2014;84:608–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sperling RA, Rentz DM, Johnson KA, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med 2014;6:228fs213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jack CR Jr, Bennett DA, Blennow K, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh compound-B. Ann Neurol 2004;55:306–319. [DOI] [PubMed] [Google Scholar]
- 6.Wong DF, Rosenberg PB, Zhou Y, et al. In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (florbetapir [corrected] F 18). J Nucl Med 2010;51:913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jureus A, Swahn BM, Sandell J, et al. Characterization of AZD4694, a novel fluorinated Abeta plaque neuroimaging PET radioligand. J Neurochem 2010;114:784–794. [DOI] [PubMed] [Google Scholar]
- 8.Mormino EC, Betensky RA, Hedden T, et al. Amyloid and APOE epsilon4 interact to influence short-term decline in preclinical Alzheimer disease. Neurology 2014a;82:1760–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 2010;31:1275–1283. [DOI] [PubMed] [Google Scholar]
- 10.Villeneuve S, Rabinovici GD, Cohn-Sheehy BI, et al. Existing Pittsburgh compound-B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain 2015;138:2020–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mormino EC, Brandel MG, Madison CM, et al. Not quite PiB-positive, not quite PiB-negative: slight PiB elevations in elderly normal control subjects are biologically relevant. Neuroimage 2012;59:1152–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farrell ME, Kennedy KM, Rodrigue KM, et al. Association of longitudinal cognitive decline with amyloid burden in middle-aged and older adults: evidence for a dose-response relationship. JAMA Neurol 2017;74:830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen AD, Mowrey W, Weissfeld LA, et al. Classification of amyloid-positivity in controls: comparison of visual read and quantitative approaches. Neuroimage 2013;71:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.La Joie R, Ayakta N, Seeley WW, et al. Multisite study of the relationships between antemortem [(11)C]PiB-PET Centiloid values and postmortem measures of Alzheimer's disease neuropathology. Alzheimers Dement 2019;15:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jack CR, Jr., Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement 2017;13:205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dagley A, LaPoint M, Huijbers W, et al. Harvard Aging Brain Study: dataset and accessibility. Neuroimage 2017;144:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellis KA, Bush AI, Darby D, et al. The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer's disease. Int Psychogeriatr 2009;21:672–687. [DOI] [PubMed] [Google Scholar]
- 18.Aisen PS, Petersen RC, Donohue MC, et al. Clinical core of the Alzheimer's Disease Neuroimaging Initiative: progress and plans. Alzheimers Dement 2010;6:239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papp KV, Rentz DM, Orlovsky I, Sperling RA, Mormino EC. Optimizing the preclinical Alzheimer's cognitive composite with semantic processing: the PACC5. Alzheimers Dement 2017;3:668–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buckley RF, Mormino EC, Amariglio RE, et al. Sex, amyloid, and APOE epsilon4 and risk of cognitive decline in preclinical Alzheimer's disease: findings from three well-characterized cohorts. Alzheimers Dement 2018;14:1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donohue MC, Sperling RA, Salmon DP, et al. The preclinical Alzheimer cognitive composite: measuring amyloid-related decline. JAMA Neurol 2014;71:961–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papp KV, Buckley R, Mormino E, et al. Clinical meaningfulness of subtle cognitive decline on longitudinal testing in preclinical AD. Alzheimers Dement 2019;16:552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson KA, Schultz A, Betensky RA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol 2016;79:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Desikan RS, Segonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 2006;31:968–980. [DOI] [PubMed] [Google Scholar]
- 25.Properzi MJ, Buckley RF, Chhatwal JP, et al. Nonlinear distributional mapping (NoDiM) for harmonization across amyloid-PET radiotracers. Neuroimage 2019;186:446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Landau SM, Mintun MA, Joshi AD, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol 2012;72:578–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klunk WE, Koeppe RA, Price JC, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement 2015;11:1–15.e1–e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Su Y, Flores S, Hornbeck RC, et al. Utilizing the Centiloid scale in cross-sectional and longitudinal PiB PET studies. Neuroimage Clin 2018;19:406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Navitsky M, Joshi AD, Kennedy I, et al. Standardization of amyloid quantitation with florbetapir standardized uptake value ratios to the Centiloid scale. Alzheimers Dement 2018;14:1565–1571. [DOI] [PubMed] [Google Scholar]
- 30.Mormino EC, Betensky RA, Hedden T, et al. Synergistic effect of beta-amyloid and neurodegeneration on cognitive decline in clinically normal individuals. JAMA Neurol 2014b;71:1379–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akaike H. Information theory and an extension of the maximum likelihood principle. Proceedings of the 2nd International Symposium on Information Theory ;1973; Budapest: 267–281.
- 32.Schwarz G. Estimating the dimension of a model. Ann Stat 1978;6:461–464. [Google Scholar]
- 33.Jack CR, Jr., Wiste HJ, Lesnick TG, et al. Brain beta-amyloid load approaches a plateau. Neurology 2013;80:890–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol 2013;12:357–367. [DOI] [PubMed] [Google Scholar]
- 35.Farrell ME, Chen X, Rundle MM, Chan MY, Wig GS, Park DC. Regional amyloid accumulation and cognitive decline in initially amyloid-negative adults. Neurology 2018;91:e1809–e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller R, Siegmund D. Maximally selected chi square statistics. Biometrics 1982;38:1011–1016. [Google Scholar]
- 37.Sperling RA, Jack CR, Jr., Aisen PS. Testing the right target and right drug at the right stage. Sci Transl Med 2011;3:111cm133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297:353–356. [DOI] [PubMed] [Google Scholar]
- 39.Jack CR, Jr., Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010;9:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer's disease. Brain 2008;131:1630–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leal SL, Lockhart SN, Maass A, Bell RK, Jagust WJ. Subthreshold amyloid predicts tau deposition in aging. J Neurosci 2018;38:4482–4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Landau SM, Horng A, Jagust WJ, Alzheimer's Disease Neuroimaging Initiative: memory decline accompanies subthreshold amyloid accumulation. Neurology 2018;90:e1452–e1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bischof GN, Jacobs HIL. Subthreshold amyloid and its biological and clinical meaning: long way ahead. Neurology 2019;93:72–79. [DOI] [PubMed] [Google Scholar]
- 44.Leuzy A, Chiotis K, Hasselbalch SG, et al. Pittsburgh compound B imaging and cerebrospinal fluid amyloid-beta in a multicentre European memory clinic study. Brain 2016;139:2540–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rowe CC, Dore V, Jones G, et al. (18)F-florbetaben PET beta-amyloid binding expressed in Centiloids. Eur J Nucl Med Mol Imaging 2017;44:2053–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Battle MR, Pillay LC, Lowe VJ, et al. Centiloid scaling for quantification of brain amyloid with [(18)F]flutemetamol using multiple processing methods. EJNMMI Res 2018;8:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salvado G, Molinuevo JL, Brugulat-Serrat A, et al. Centiloid cut-off values for optimal agreement between PET and CSF core AD biomarkers. Alzheimers Res Ther 2019;11:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hanseeuw BJ, Betensky RA, Jacobs HIL, et al. Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol 2019;76:915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jack CR Jr, Bennett DA, Blennow K, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016;87:539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Insel PS, Mormino EC, Aisen PS, Thompson WK, Donohue MC. Neuroanatomical spread of amyloid beta and tau in Alzheimer's disease: implications for primary prevention. Brain Commun 2020;2:fcaa007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palmqvist S, Scholl M, Strandberg O, et al. Earliest accumulation of beta-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat Commun 2017;8:1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grothe MJ, Barthel H, Sepulcre J, et al. In vivo staging of regional amyloid deposition. Neurology 2017;89:2031–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data for ADNI and PET data for AIBL are publicly available online at ida.loni.usc.edu. AIBL cognitive data and HABS data are available on request.